Introduction
Paraquat (PQ) is an effective and widely used
herbicide that is associated with high mortality due to accidental
or voluntary ingestion in China, Japan, Singapore and Sri Lanka,
particularly in developing countries (1–3). PQ
causes fatal damage to multiple organs, particularly the lung,
where the concentrations of PQ may be 6- to 10-fold higher compared
with those in the plasma (4). In
the early stages of PQ poisoning, alveolar epithelial cell damage,
inflammatory cell exudate, alveolar hemorrhaging and edema were all
observed (5,6). Meanwhile, alveolar and interstitial
irreversible pulmonary fibrosis are found to occur in the later
stages (7). However, the mechanism
underlying PQ poisoning-induced pulmonary fibrosis remains unclear.
Thus, it is necessary to explore the possible pathogenesis of
PQ-induced pulmonary fibrosis.
Epithelial-mesenchymal transition (EMT) is a process
by which epithelial cells acquire the phenotype of mesenchymal
cells via signaling pathways mediated by TGF-β, bone morphogenetic
protein, Wnt/β-catenin, Notch, Hedgehog and receptor tyrosine
kinases (8). Downregulated
expression levels of E-cadherin and zona occludens 1, and
upregulated expression levels of vimentin, fibronectin and α-smooth
muscle actin (α-SMA), have been associated with EMT (9–11). A
number of previous studies have reported that EMT played a key role
in the occurrence and development of pulmonary fibrosis (12). In our previous study, PQ induced the
occurrence of EMT in the early stage of PQ poisoning (13,14).
These findings suggested that EMT may play an important role in
PQ-induced pulmonary fibrosis. However, to the best of our
knowledge, the pathogenesis of PQ-induced EMT has not been reported
to date.
The endoplasmic reticulum (ER) is a cellular
organelle in which proteins are synthesized, folded and
glycosylated (15,16). Various disturbances, including
oxidative stress, have been found to adversely affect the
homeostasis of ER and cause the accumulation of unfolded proteins,
resulting in ER stress (17,18).
Under ER stress conditions, the unfolded protein response (UPR) is
activated to attenuate protein translation, increase the production
of protein folding chaperones and upregulate the expression of
protein-degrading enzymes (19).
UPR signaling is initiated by three receptors located in the ER:
Nucleus signaling 1/inositol requiring enzyme-1α, protein kinase
RNA-like ER kinase (PERK) and activating transcription factor 6
(20). Multiple in vivo and
in vitro studies have reported that ER stress was associated
with the development and progression of pulmonary fibrosis induced
by viral infections, cigarette smoke, particulates and aging, among
other factors (21,22). Another previous study demonstrated
that ER stress was involved in cell differentiation, and the
induction of ER stress led to EMT (23). Our group previously reported that
the expression levels of the ER stress-related marker, heat shock
protein family A (Hsp70) member 5 (HSPA5) were upregulated
following PQ poisoning (24).
Notably, sodium tauroursodeoxycholate, a chemical chaperone, was
able to rescue A549 cells from death caused by exposure to PQ
(25). Thus, it was hypothesized
that ER stress may regulate EMT in PQ poisoning-induced pulmonary
fibrosis.
Previous studies have shown that GSK-3β had an
important role in maintaining the epithelial architecture (26–28).
In addition, Snail, one of the key factors regulating EMT,
accumulated in the nucleus following the inhibition of GSK-3β
(29–31). In our previous study, the expression
levels of GSK-3β were found to be downregulated during the early
stages of PQ poisoning (32).
Another previous study reported that the inhibition of ER stress
protected cardiomyocytes against toxicity, potentially through the
inactivation of AKT/GSK-3β signaling (33). The present study aimed to verify
whether ER stress was involved in modulating EMT, and whether ER
stress modulated EMT via the PI3K/AKT/GSK-3β signaling pathway
following PQ poisoning.
Materials and methods
Materials
DMEM (high glucose), trypsin and FBS were purchased
from Gibco; Thermo Fisher Scientific, Inc. Standardized PQ powder
and LY294002 were purchased from Sigma-Aldrich; Merck KGaA.
Anti-PERK (cat. no. ab229912), anti-eukaryotic initiation factor 2α
(eIF2α; cat. no. ab169528), anti-phosphorylated (p)-eIF2α (cat. no.
ab32157), anti-HSPA5 (cat. no. ab108615), anti-α-SMA (cat. no.
ab119952), anti-PI3K (cat. no. ab32089), anti-p-PI3K (cat. no.
ab182651) and anti-GAPDH (cat. no. ab8245) primary antibodies were
obtained from Abcam. Anti-GSK-3β (cat. no. 12456), anti-p-GSK-3β
(cat. no. 5558), anti-AKT (cat. no. 9272), anti-p-AKT (cat. no.
4060) and anti-E-cadherin (cat. no. 14472) primary antibodies were
purchased from Cell Signaling Technology, Inc. HRP-labeled
secondary antibody (cat. nos. A0216 and A0208) and the anti-p-PERK
(Thr982; cat. no. AF5902) antibody were purchased from Beyotime
Institute of Biotechnology. Highly sensitive ECL reagent and PVDF
membranes were obtained from Bio-Rad Laboratories, Inc.
Lipofectamine 2000® reagent was purchased from
Invitrogen; Thermo Fisher Scientific, Inc., while small interfering
RNA (siRNA/si) was obtained from Shanghai GenePharma Co., Ltd.
Cell culture
Human lung adenocarcinoma epithelial cells (A549)
and rat alveolar type II cells (RLE-6TN) were purchased from the
American Type Culture Collection. A549 cells are a suitable cell
model that display numerous properties in common with human
alveolar epithelial cells; therefore, they are often used to
research the mechanism of pulmonary fibrosis (34,35).
A549 cells were cultured in DMEM (high glucose) supplemented with
10% heat-inactivated FBS, while RLE-6TN cells were cultured in F-12
medium (Gibco; Thermo Fisher Scientific, Inc.) supplemented with
10% heat-inactivated FBS. A549 and RLE-6TN cells were maintained at
37°C in a humidified incubator containing 5% CO2. Cells
were treated with PQ (800 µmol/l for A549 cells and 160 µmol/l for
RLE-6TN cells; dissolved in PBS) for 24 h. The morphological
alterations of cells were observed using phase contrast microscopy
(Thermo Fisher Scientific, Inc.).
Western blotting
Following treatment with siRNA or PQ, total protein
was extracted from cells using RIPA lysis buffer (Beyotime
Institute of Biotechnology) and total protein was quantified using
a BCA protein assay kit (Beyotime Institute of Biotechnology).
Proteins (50 µg per lane) were separated via 8% SDS-PAGE and then
transferred onto PVDF membranes, which were blocked with 5% non-fat
milk in TBST (0.05% Tween 20) for 2 h at room temperature. The
membranes were then incubated at 4°C overnight with the following
primary antibodies: Anti-PERK (1:1,000), anti-p-PERK (1:1,000),
anti-eIF2α (1:1,000), anti-p-eIF2α (1:1,000), anti-HSPA5 (1:1,000),
anti-E-cadherin (1:500), anti-α-SMA (1:200), anti-PI3K (1:1,000),
anti-p-PI3K (1:1,000), anti-AKT (1:1,000), anti-p-AKT (1:1,000),
anti-GSK-3β (1:200), anti-p-GSK-3β (1:200) and anti-GAPDH (1:500).
Following the primary antibody incubation, the membranes were
incubated with the appropriate HRP-conjugated secondary antibody
(1:3,000) at room temperature for 2 h. Protein bands were
visualized using ECL reagent and densitometric analysis was
performed using ImageJ software (version 1.52g; National Institutes
of Health). All experiments were repeated at least three times.
Cell transfection
A549 (2×104/well) and RLE-6TN
(5×104/well) cells were seeded into 6-well plates and
divided into four groups: i) sicontrol (transfected with scrambled
RNA); ii) siPERK; iii) sicontrol + PQ; and iv) siPERK + PQ. Cells
were transfected with 200 pmol siRNA (sicontrol or siPERK) using
Lipofectamine 2000® (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol at 37°C
in a humidified incubator containing 5% CO2. For A549
cells, the sequence of siPERK was 5′-GAAGCUACAUUGUCUAUUU-3′ and the
sequence of sicontrol was 5′-AACCACUCAACUUUUUCCCAA-3′. For RLE-6TN
cells, the sequence of siPERK was 5′-AAGUAGAAGAGACCAUGCCUGCUCC-3′
and the sequence of sicontrol was 5′-ACGUGACACGUUCGGAGAATT-3′. At
48 h post-transfection, 800 µmol/l (for A549 cells) or 160 µmol/l
(for RLE-6TN cells) PQ (dissolved in PBS) was added to the
sicontrol + PQ and siPERK + PQ groups for 24 h at 37°C in a
humidified incubator containing 5% CO2. The doses of PQ
were selected according to our previous study (14).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from A549 and RLE-6TN cells
using RNAiso Plus reagent (Takara Biotechnology Co., Ltd.)
according to the manufacturer's protocol. The concentration of
total RNA was determined using an ultraviolet spectrophotometer.
Total RNA was reverse transcribed into cDNA using a HiScript II Q
RT SuperMix for qPCR (Vazyme Biotech Co., Ltd.) according to the
manufacturer's instructions. qPCR was subsequently performed using
a ChamQ™ SYBR qPCR Master mix (Vazyme Biotech Co., Ltd.) on an ABI
ViiA™ 7 Real-Time PCR system (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The following thermocycling conditions were used
for qPCR: 95°C for 2 min, 60°C for 30 sec and 72°C for 30 sec. The
following primer pairs for β-actin and PERK were provided by Sangon
Biotech Co., Ltd.: Human PERK forward, 5′-GGACCTCAAGCCATCCAACA-3′
and reverse, 5′-TCCTGGTCCATTGCAGTCAC-3′; human β-actin forward,
5′-CTGGAACGGTGAAGGTGACA-3′ and reverse,
5′-AAGGGACTTCCTGTAACAATGCA-3′; rat PERK forward,
5′-GGAAACGAGAGCCGGATTTATT-3′ and reverse,
5′-ACTATGTCCATTATGGCAGCTTC-3′; and rat β-actin forward,
5′-AGGATGCAGAAGGAGATTACTGC-3′ and reverse,
5′-AAAACGCAGCTCAGTAACAGTGC-3′. mRNA expression levels were
quantified using the 2−ΔΔCq method (36).
Immunofluorescence assay
A549 (5×105/well) cells were incubated
with anti-E-cadherin (1:50) or anti-α-SMA (1:50) primary antibodies
at 4°C overnight, washed with TBST (0.05% Tween-20) for 5 min at
room temperature thrice, fixed with 4% paraformaldehyde (Beyotime
Institute of Biotechnology) for 10 min at room temperature, blocked
with 5% BSA (Beyotime Institute of Biotechnology) for 2 h at room
temperature and incubated with fluorescent secondary antibodies
(Alexa Fluor 488-labeled Goat Anti-Mouse IgG and Alexa Fluor
647-labeled Goat Anti-Mouse IgG; 1:200) in a humidified chamber for
2 h at 37°C. The cell nuclei were stained with
4′,6-diamidino-2-phenylindole at room temperature for 5 min.
Stained cells were visualized using a laser confocal microscope
(Leica TCS SP8; Leica Microsystems GmbH).
Wound healing
A549 (2×104/well) were cultured in 6-well
plates and transfected with siPERK. After 48 h, PQ (800 µmol/l) was
added to treat the cells in the corresponding groups for 24 h at
37°C in a humidified incubator containing 5% CO2. At 70%
confluence, a scratch was then made in the center of the cell
monolayer with a 10-µl Eppendorf pipette tip. The culture medium
was replaced with FBS-free medium and cells were visualized under a
light microscope. The change in the width of the scratch was
photographed at 0 and 24 h using an EVOS cell imaging system
(Thermo Fisher Scientific, Inc.).
Inhibitor intervention
LY294002 (Selleck Chemicals), a PI3K inhibitor, was
used to block the PI3K/AKT signaling pathway (37). Briefly, 20 µM LY294002 was dissolved
with 0.1% DMSO and added into the cell culture medium of both cell
lines (2×104/well for A549 and 5×104/well for
RLE-6TN) seeded into 6-well plates, while cells exposed to the
corresponding concentration of DMSO (0.1%) were used as the control
at 37°C. Following 24 h of LY294002 or DMSO incubation, the cells
were treated with PQ (800 µmol/l for A549 and 160 µmol/l for
RLE-6TN) for 24 h at 37°C. Subsequently, cells were collected by
digestion with trypsin followed by centrifugation at 1,000 × g for
3 min at 4°C.
Statistical analysis
Statistical analysis was performed using SPSS 16.0
software (SPSS, Inc.). All data are presented as the mean ± SD of
three independent experiments. Statistical comparisons between two
groups were performed using an unpaired Student's t-test.
Multigroup comparisons were performed using a one-way ANOVA
followed by a Bonferroni's correction post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
ER stress participates in the process
of PQ poisoning
Our previous study indicated that the expression
levels of HSPA5, an ER stress-marker protein, were significantly
upregulated following 2 h of PQ poisoning (24). To validate whether ER stress-related
signals exerted a role in PQ-induced lung injury, the present study
performed western blotting to analyze the expression levels of
proteins involved in ER stress in PQ-treated A549 and RLE-6TN
cells. The results revealed that the expression levels of HSPA5
were significantly upregulated following PQ treatment compared with
the control group in both cell lines (Fig. 1A, B, D and E). In addition, the
protein expression levels of PERK, p-PERK/PERK and p-eIF-2α/eIF-2α,
which are all involved in ER stress-related signaling (38,39),
were all significantly upregulated in the PQ group compared with
the control group in both cell lines (Fig. 1A-E). These results suggested that
PERK signaling in ER stress was activated during PQ poisoning.
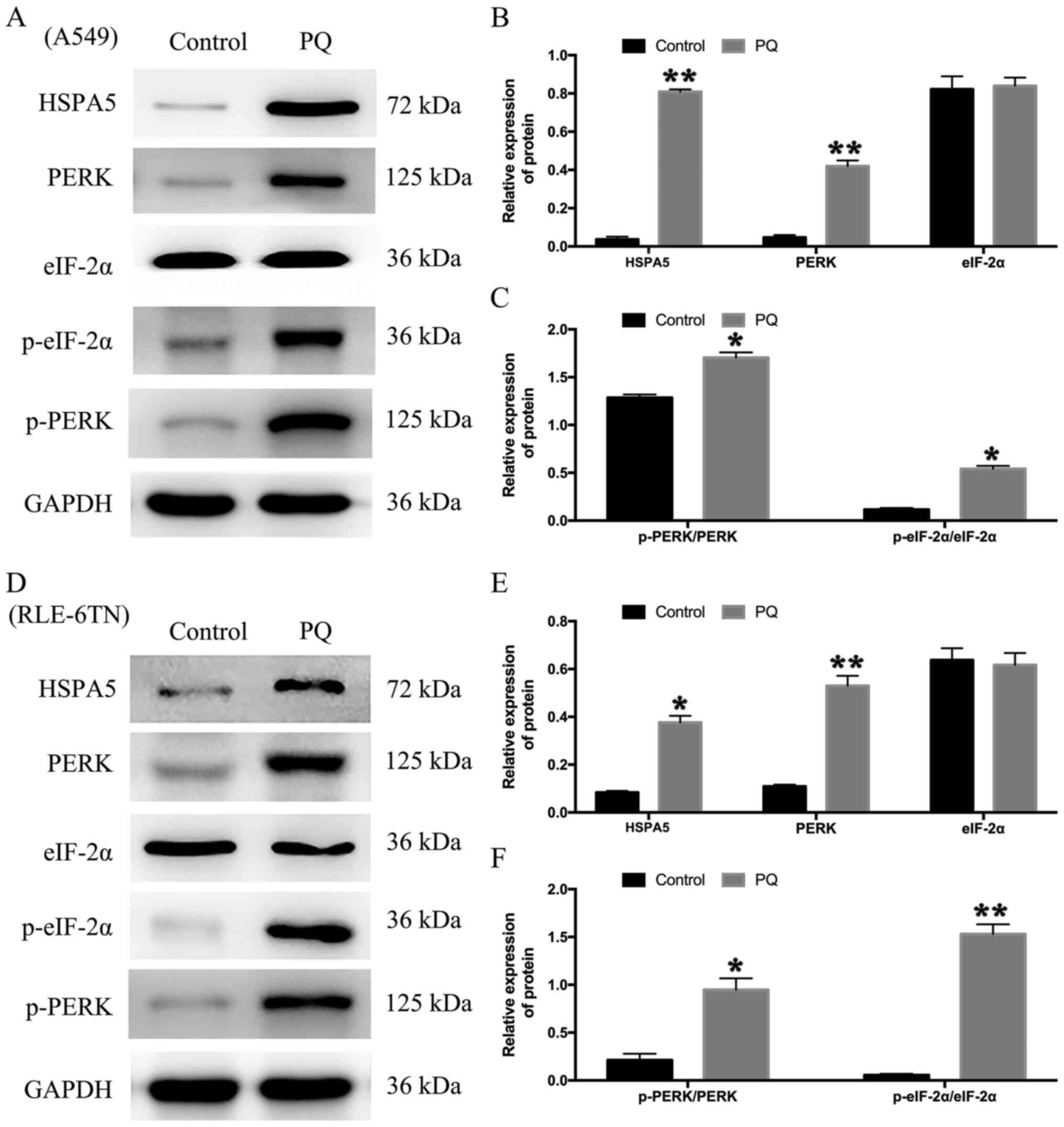 | Figure 1.Endoplasmic reticulum stress
participates in the progression of PQ poisoning. (A) A549 cells
were treated with PQ for 24 h. The expression levels of HSPA5,
PERK, p-PERK, eIF-2α and p-eIF-2α were analyzed using western
blotting. (B) The relative expression levels of HSPA5, PERK and
eIF-2α in A549 cells. (C) The relative expression levels of p-PERK
and p-eIF-2α in A549 cells. (D) RLE-6TN cells were treated with PQ
for 24 h. The expression levels of HSPA5, PERK, p-PERK, eIF-2α and
p-eIF-2α were analyzed using western blotting. (E) The relative
expression levels of HSPA5, PERK and eIF-2α in RLE-6TN cells. (F)
The relative expression levels of p-PERK and p-eIF-2α in RLE-6TN
cells. GAPDH served as the loading control. Data are presented as
the mean ± SD from three independent experiments. *P<0.05,
**P<0.01 vs. Control. PQ, paraquat; HSPA5, heat shock protein
family A (Hsp70) member 5; PERK, protein kinase RNA-like ER kinase;
p-, phosphorylated; eIF-2α, eukaryotic initiation factor 2α. |
Role of PERK signaling in ER stress in
regulating PQ-induced EMT in vitro
A previous study indicated that ER stress may be
involved in cell differentiation and apoptosis (40). To evaluate whether PQ induced
pulmonary fibrosis through the PERK-mediated signaling of ER
stress, the present study blocked PERK signaling by transfection
with siPERK in both cell lines. The expression levels of PERK mRNA
in the siPERK group were significantly decreased compared with
those in the sicontrol group. The mRNA expression levels of PERK
were significantly downregulated in the siPERK + PQ group compared
with the sicontrol + PQ group in both cell lines (Fig. 2A and B). In addition, the protein
expression levels of PERK and p-PERK/PERK were significantly
downregulated in the siPERK + PQ group compared with the sicontrol
+ PQ group (Fig. 2C and D). In A549
cells, the expression of p-eIF-2α/eIF-2α was downregulated in the
siPERK + PQ group compared with the sicontrol + PQ group.
Meanwhile, in the siPERK + PQ group, the protein expression levels
of E-cadherin (an epithelial marker) were upregulated, while the
protein expression levels of α-SMA (a mesenchymal marker) were
downregulated compared with the sicontrol + PQ group (Fig. 2C and D). Similar results were
obtained for RLE-6TN cells (Fig.
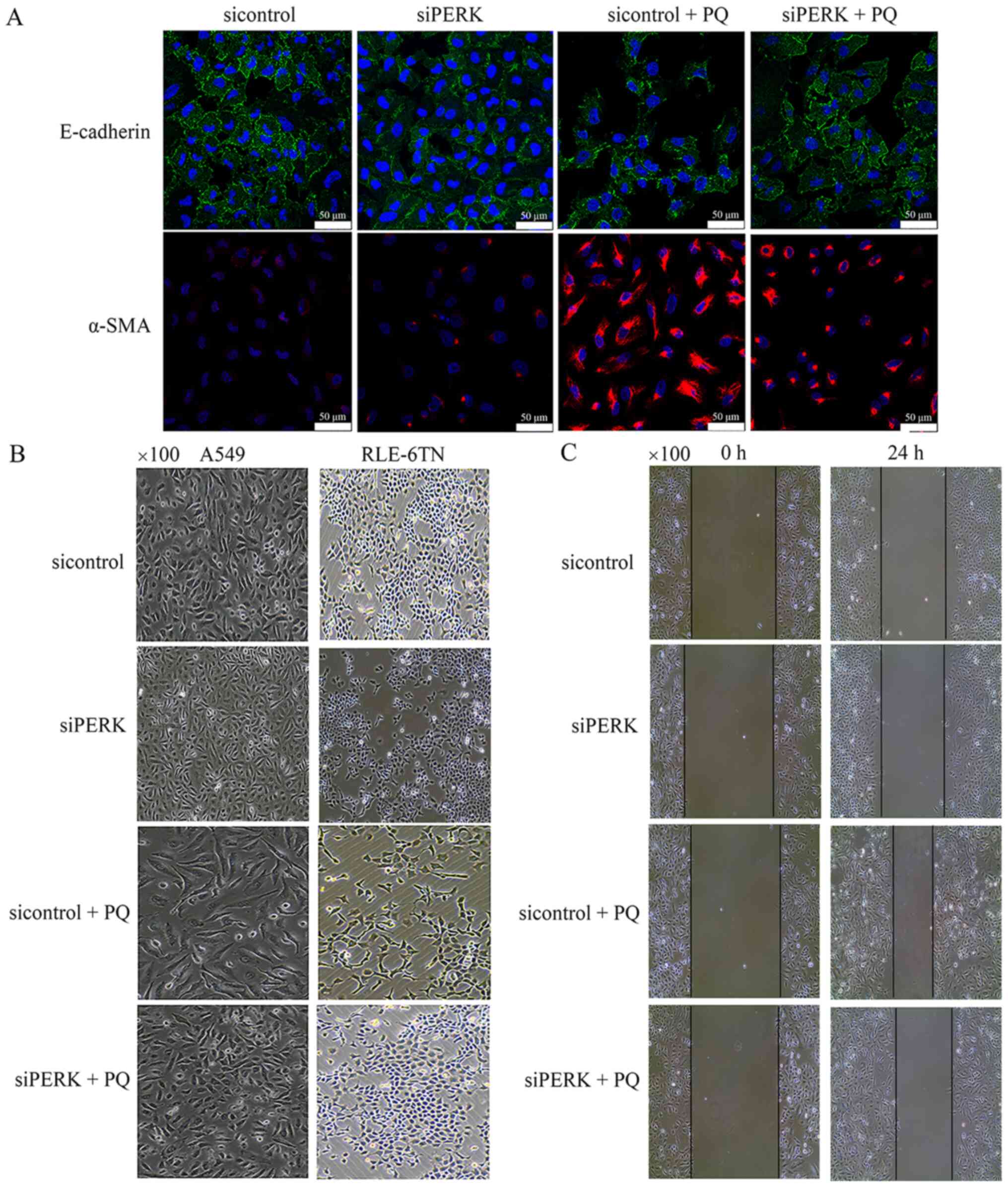
2F-H). Immunofluorescence analysis revealed that the expression
levels of E-cadherin were markedly upregulated in the siPERK + PQ
group compared with the sicontrol + PQ group, wheras the expression
levels of α-SMA were notably downregulated at 24 h in A549 cells in
the siPERK + PQ group compared with the sicontrol + PQ group
(Fig. 3A). A549 and RLE-6TN cells
were observed to undergo transition from a polygonal to a fusiform
morphology in the sicontrol + PQ group compared with the sicontrol
group under phase contrast microscopy; however, this change was
attenuated in the siPERK + PQ group compared with the sicontrol +
PQ groups (Fig. 3B). The results of
the wound healing assay demonstrated that cell migration in the
sicontrol + PQ group was increased compared with the sicontrol
group, while cell migration in the siPERK + PQ group was markedly
decreased compared with the sicontrol + PQ group (Fig. 3C). These data indicated that
PQ-induced EMT may be regulated by PERK signaling, which is an ER
stress-regulated signaling pathway.
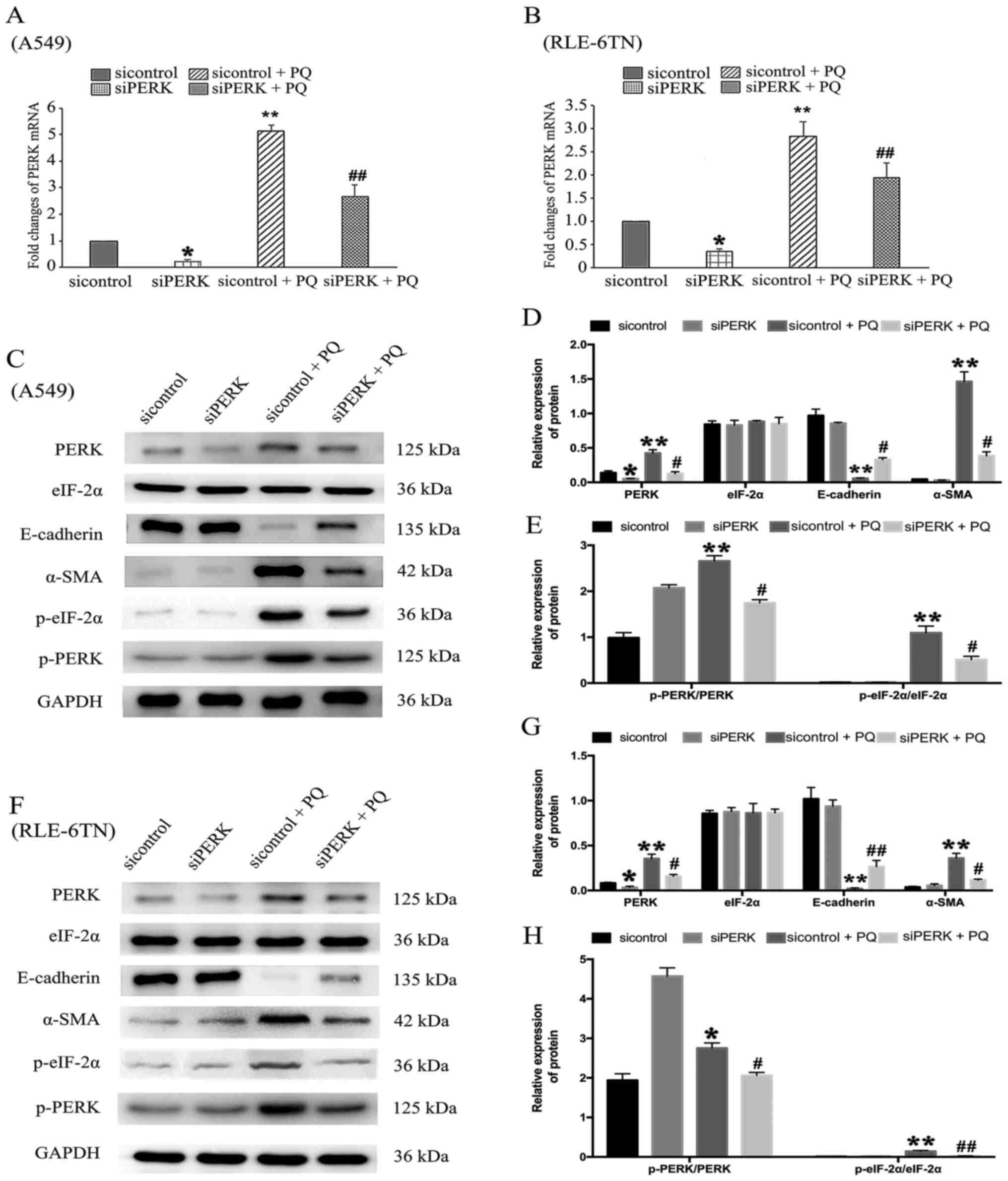 | Figure 2.Role of PERK signaling in endoplasmic
reticulum stress and regulation of PQ-induced
epithelial-mesenchymal transition in vitro. siPERK was
transfected into (A) A549 and (B) RLE-6TN cells for 48 h. Cell
lines were then treated with PQ for 24 h. mRNA expression levels of
PERK were analyzed using reverse transcription-quantitative PCR.
PERK, p-PERK, eIF-2α, p-eIF-2α, E-cadherin and α-SMA protein
expression levels in (C) A549 cells were analyzed using western
blotting. (D) The relative expression levels of PERK, eIF-2α,
E-cadherin and α-SMA in A549 cells. (E) The relative expression
levels of p-PERK and p-eIF-2α in A549 cells. (F) PERK, p-PERK,
eIF-2α, p-eIF-2α, E-cadherin and α-SMA protein expression levels in
RLE-6TN cells were analyzed using western blotting. (G) The
relative expression levels of PERK, eIF-2α, E-cadherin and α-SMA in
RLE-6TN cells. (H) The relative expression levels of p-PERK and
p-eIF-2α in RLE-6TN cells. GAPDH served as a loading control. Data
are presented as the mean ± SD from three independent experiments.
*P<0.05, **P<0.01 vs. sicontrol; #P<0.05,
##P<0.01 vs. sicontrol + PQ. PQ, paraquat; si, small
interfering RNA; PERK, protein kinase RNA-like ER kinase; p-,
phosphorylated; α-SMA, α-smooth muscle actin; eIF-2α, eukaryotic
initiation factor 2α. |
PERK signaling in ER stress may
regulate PQ-induced EMT via the PI3K/AKT/GSK-3β signaling
pathway
The results of our previous study demonstrated that
PERK-regulated ER stress had a significant effect on PQ-induced
EMT; however, the underling mechanism remained unclear (24). The expression levels of proteins
associated with the PI3K/AKT/GSK-3β signaling pathway were
subsequently determined via western blotting following the
knockdown of PERK and PQ poisoning. The total proteins of GSK-3β,
AKT and PI3K in each group displayed no significant differences
(Fig. 4A and B). The results
revealed that PERK silencing significantly downregulated the
protein expression levels of p-GSK-3β/GSK-3β, p-AKT/AKT and
p-PI3K/PI3K in the siPERK + PQ group compared with the sicontrol +
PQ group in both cell lines (Fig. 4A
and C). Thus, it was suggested that PQ may induce EMT through
the PERK-mediated PI3K/AKT/GSK-3β signaling pathway.
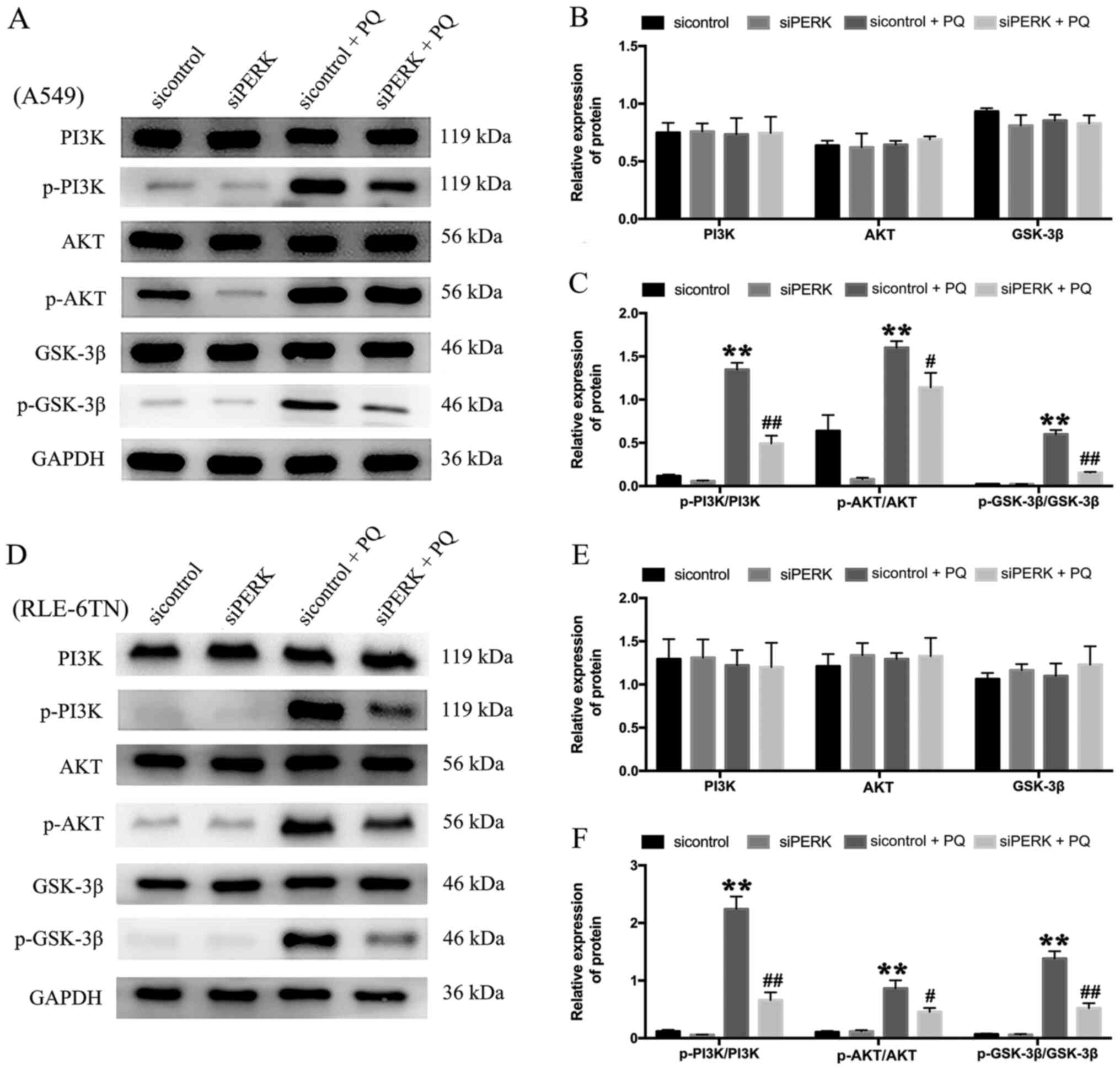 | Figure 4.Expression levels of PI3K/AKT/GSK-3β
signaling pathway-related proteins following the knockdown of PERK.
siPERK was transfected into (A) A549 cells for 48 h and were then
treated with PQ for 24 h. The expression levels of PI3K, p-PI3K,
AKT, p-AKT, GSK-3β and p-GSK-3β were analyzed using western
blotting. (B) The relative expression levels of PI3K, AKT and
GSK-3-β in A549 cells. (C) The relative expression levels of
p-PI3K, p-AKT and p-GSK-3-β in A549 cells. (D) siPERK was
transfected into RLE-6TN cells for 48 h and cells were then treated
with PQ for 24 h. The expression levels of PI3K, p-PI3K, AKT,
p-AKT, GSK-3β and p-GSK-3β were analyzed using western blotting.
(E) The relative expression levels of PI3K, AKT and GSK-3-β in
RLE-6TN cells. (F) The relative expression levels of p-PI3K, p-AKT
and p-GSK-3-β in RLE-6TN cells. GAPDH served as the loading
control. Data are presented as the mean ± SD from three independent
experiments. **P<0.01 vs. Control; #P<0.05,
##P<0.01 vs. sicontrol + PQ. PQ, paraquat; PERK,
protein kinase RNA-like ER kinase; p-, phosphorylated; eIF-2α,
eukaryotic initiation factor 2α; α-SMA, α-smooth muscle actin; si,
small interfering RNA. |
PI3K/AKT/GSK-3β signaling regulates
PQ-induced EMT
A recent study indicated that the PI3K/AKT/GSK-3β
signaling pathway, which can mediate survival, migration and
apoptosis, also played an important role in TGF-β1-induced EMT
(41). Therefore, the present study
investigated the expression levels of proteins related to the
PI3K/AKT/GSK-3β signaling pathway in A549 and RLE-6TN cells
following treatment with a PI3K-specific inhibitor, and
investigated its role in PQ poisoning. The total proteins of
GSK-3β,AKT and PI3K in each group displayed no significant
differences (Fig. 5A, B, D and E).
The present results revealed that the protein expression levels of
p-PI3K/PI3K, p-AKT/AKT and p-GSK-3β/GSK-3β were significantly
upregulated in the DMSO + PQ group compared with the DMSO group in
A549 (Fig. 5A and C)and RLE-6TN
cells (Fig. 5D and F). To verify
the role of the PI3K/AKT/GSK-3β signaling pathway in PQ-induced
EMT, the PI3K-specific inhibitor, LY294002, was used to block
PI3K/AKT signaling. The results revealed that LY294002 partially
reversed the PQ-induced upregulation in p-GSK-3β, p-AKT and p-PI3K
expression levels in both cell lines (Fig. 5A, C, D and F). In addition, LY294002
+ PQ group partially alleviated the downregulated E-cadherin
expression levels and upregulated α-SMA expression levels compared
with the DMSO + PQ group (Fig. 5A, B, D
and E). These results indicated that the PI3K/AKT/GSK-3β
signaling pathway may regulate PQ-induced EMT.
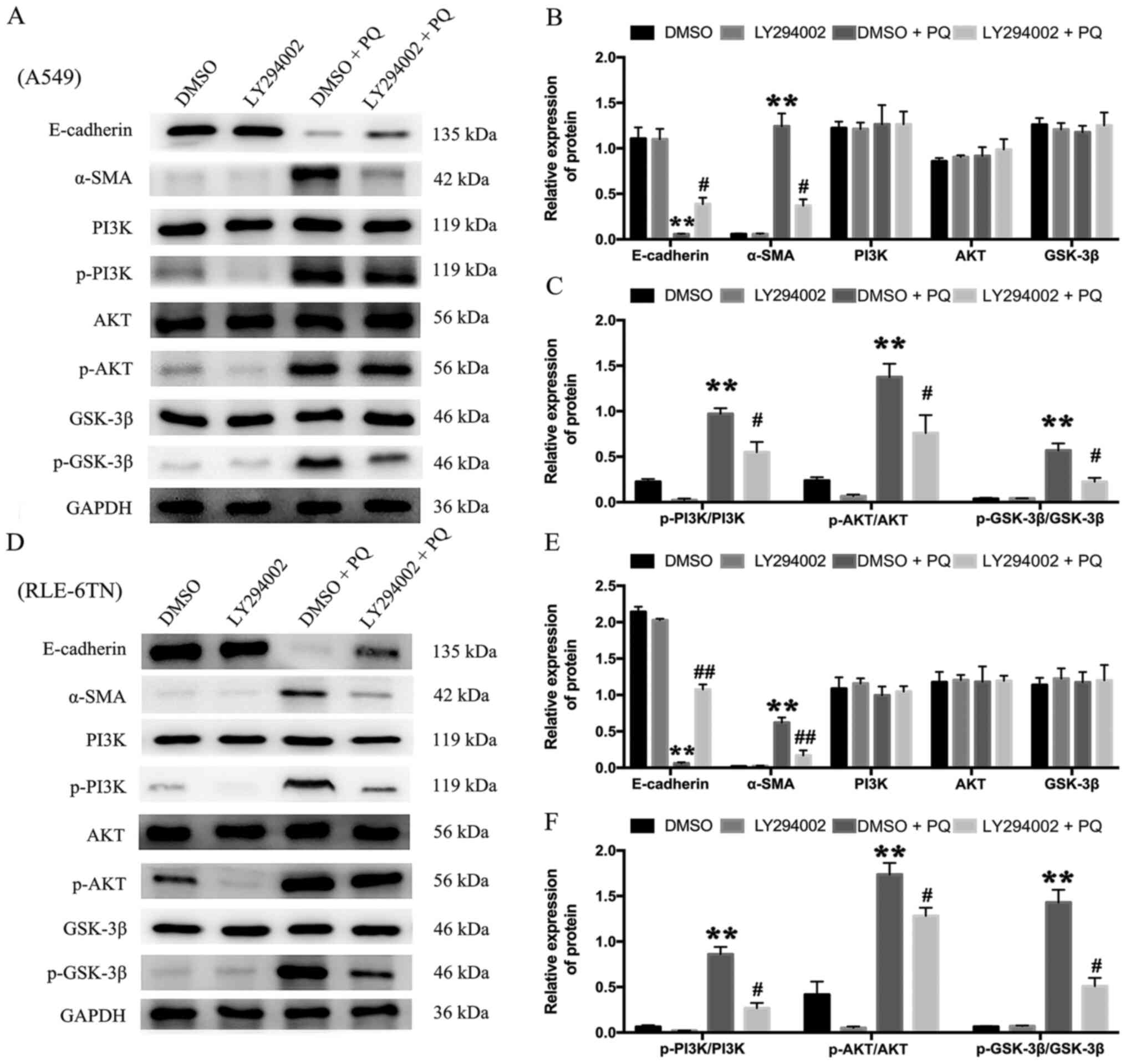 | Figure 5.PI3K/AKT/GSK-3β signaling pathway
regulates PQ-induced epithelial-mesenchymal transition. (A)
Following 24 h of incubation with the PI3K inhibitor, LY294002,
A549 cells were treated with PQ for 24 h. The expression levels of
α-SMA, E-cadherin, PI3K, p-PI3K, AKT, p-AKT, GSK-3β and p-GSK-3β
were analyzed using western blotting. (B) The relative expression
levels of α-SMA, E-cadherin, PI3K, AKT and GSK-3-β in A549 cells.
(C) The relative expression levels of p-PI3K, p-AKT and p-GSK-3-β
in A549 cells. (D) Following 24 h of incubation with the PI3K
inhibitor, LY294002, RLE-6TN cells were treated with PQ for 24 h.
The expression levels of α-SMA, E-cadherin, PI3K, p-PI3K, AKT,
p-AKT, GSK-3β and p-GSK-3β were analyzed using western blotting.
(E) The relative expression levels of α-SMA, E-cadherin, PI3K, AKT
and GSK-3-β in RLE-6TN cells. (F) The relative expression levels of
p-PI3K, p-AKT and p-GSK-3-β in RLE-6TN cells. GAPDH served as the
loading control. Data are presented as the mean ± SD from three
independent experiments. **P<0.01 vs. DMSO;
#P<0.05, ##P<0.01 vs. DMSO + PQ. PQ,
paraquat; PERK, protein kinase RNA-like ER kinase; p-,
phosphorylated; eIF-2α, eukaryotic initiation factor 2α; α-SMA,
α-smooth muscle actin. |
Discussion
PQ poisoning has been reported as a major public
health concern, as it accumulates and induces lung alveolar
epithelial cell damage (4,42). To date, to the best of our
knowledge, there is no effective therapeutic method to prevent PQ
poisoning, and its molecular mechanism in lung toxicity remains
unclear. The final clinical outcome of PQ poisoning is collagen
deposition in the lung; however, the mechanism that leads to
pulmonary fibrosis is still unclear (4). In the present study, the roles of ER
stress on EMT in PQ-induced pulmonary fibrosis were investigated,
as well as the possible underlying signaling pathways involved in
the mechanism.
Fibroblasts accumulate via three mechanisms: i)
Proliferation and differentiation of resident lung fibroblasts; ii)
EMT; and iii) transformation of bone marrow-derived cells or
circulating progenitors to fibroblasts (43,44).
Fibroblasts transformed by EMT account for ~50% of the total number
of fibroblasts (45). EMT is
characterized by the loss of epithelial markers and the acquisition
of mesenchymal markers (46). In
the present study, alveolar epithelial cells exhibited features of
EMT, including downregulated expression levels of the epithelial
marker E-cadherin and upregulated expression of the mesenchymal
marker α-SMA, at the early stage of PQ poisoning in vitro.
The results of the present study suggested that EMT may play a key
role in the progression of PQ-induced pulmonary fibrosis.
The ER is an organelle that provides a unique
environment for protein synthesis, post-translational modifications
and folding (47). ER stress is
induced when cell homeostasis is altered and the unfolding of
proteins increases (48,49). Previous studies have reported that
PQ induces alveolar epithelial cell injury through the ER stress
pathway (46,50,51).
PQ poisoning was also discovered to promote the production of
reactive oxygen species (ROS) (52,53).
In addition to the enhancement of lipid peroxidation and regulation
of apoptosis, ROS can also inactivate key enzymes in metabolic
pathways, which disrupts protein folding, thereby leading to ER
stress (53,54). Under ER stress conditions, the
expression levels of the HSPA5 protein were found to be
upregulated, and PERK was activated, which led to the
phosphorylation of eIF-2α (55). In
our previous study, HSPA5 levels were increased during the early
stages of PQ poisoning (24). In
the present study, PQ upregulated the expression levels of PERK and
p-eIF-2α. In addition, PQ-induced EMT was significantly alleviated
after silencing PERK in alveolar epithelial cells. Thus, these data
indicated that ER stress may be involved in PQ-induced pulmonary
fibrosis, and that the PERK signaling pathway may be activated to
promote EMT.
GSK-3β, which is required for the maintenance of
epithelial architecture, was discovered to serve a fundamental role
in EMT (27,56). Snail, an EMT-promoting factors, was
found to be negatively regulated by GSK-3β (29–31,57).
In our previous study, GSK-3β expression levels were downregulated
following treatment with PQ. GSK-3β is also a downstream target of
AKT, and AKT activity enhances the phosphorylation of GSK-3β,
leading to a reduction in its activity (32). To further investigate the mechanisms
of PI3K/AKT/GSK-3β signaling in PQ-induced EMT, the PI3K inhibitor
LY294002 was used, and its effects on PI3K/AKT/GSK-3β signaling
were determined. The present results demonstrated that LY294002
could partially attenuate the upregulated p-PI3K, p-AKT and
p-GSK-3β expression levels induced by PQ, and the LY294002 + PQ
group alleviated the downregulated E-cadherin expression levels and
upregulated α-SMA expression levels compared with the DMSO + PQ
group. These findings indicated that the PI3K/AKT/GSK-3β signaling
pathway may be associated with EMT in PQ-induced pulmonary
fibrosis.
A previous study indicated that ER stress may
promote cardiomyocyte damage through AKT/GSK-3β/NADPH oxidase 4
signaling (33). To investigate the
potential mechanism of ER stress-induced EMT in PQ poisoning, the
expression levels of PI3K, p-PI3K, AKT, p-AKT, GSK-3β and p-GSK-3β
were analyzed in both A549 and RLE-6TN cells. The present results
revealed that the expression levels of p-GSK-3β, p-AKT and p-PI3K
were downregulated in the siPERK + PQ group compared with the
sicontrol + PQ group. These results suggested that ER stress may
regulate EMT through the PI3K/AKT/GSK-3β signaling pathway in
PQ-induced pulmonary fibrosis.
In conclusion, the results of the present study
further elucidated the molecular mechanism of PQ-induced pulmonary
fibrosis. ER stress was activated in type II alveolar epithelial
cells, and it was discovered to regulate EMT through the
PI3K/AKT/GSK-3β signaling pathway in PQ-induced pulmonary fibrosis.
Therefore, inhibiting ER stress-induced EMT in pulmonary epithelial
cells may represent a promising strategy for treating PQ poisoning
and pulmonary fibrosis in the clinic. However, the present study
only studied the mechanism in vitro, while the results were
not validated in vivo. Therefore, further investigations are
required to validate the current findings.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grants nos. 81502829 and
81171868).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
RW designed the study. HX, YZ, WJ and JL performed
the experiments. XM and KL analyzed the data. XM wrote the
manuscript. All authors read and approved the final manuscript. All
authors confirmed the authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Meredith TJ and Vale JA: Treatment of
paraquat poisoning in man: Methods to prevent absorption. Hum
Toxicol. 6:49–55. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gil HW, Hong JR, Jang SH and Hong SY:
Diagnostic and therapeutic approach for acute paraquat
intoxication. J Korean Med Sci. 29:1441–1449. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Eddleston M: Patterns and problems of
deliberate self-poisoning in the developing world. QJM. 93:715–731.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dinis-Oliveira RJ, Duarte JA,
Sanchez-Navarro A, Remiao F, Bastos ML and Carvalho F: Paraquat
poisonings: Mechanisms of lung toxicity, clinical features, and
treatment. Crit Rev Toxicol. 38:13–71. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li Y, Wang N, Ma Z, Wang Y, Yuan Y, Zhong
Z, Hong Y and Zhao M: Lipoxin A4 protects against paraquat-induced
acute lung injury by inhibiting the TLR4/MyD88-mediated activation
of the NF-κB and PI3K/AKT pathways. Int J Mol Med. 47:862021.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Amin F, Roohbakhsh A, Memarzia A, Kazerani
HR and Boskabady MH: Immediate and late systemic and lung effects
of inhaled paraquat in rats. J Hazard Mater. 415:1256332021.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yoon SC: Clinical outcome of paraquat
poisoning. Korean J Intern Med. 24:93–94. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gonzalez DM and Medici D: Signaling
mechanisms of the epithelial-mesenchymal transition. Sci Signal.
7:re82014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nawshad A, Lagamba D, Polad A and Hay ED:
Transforming growth factor-beta signaling during
epithelial-mesenchymal transformation: Implications for
embryogenesis and tumor metastasis. Cells Tissues Organs.
179:11–23. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ohbayashi M, Kubota S, Kawase A, Kohyama
N, Kobayashi Y and Yamamoto T: Involvement of
epithelial-mesenchymal transition in methotrexate-induced pulmonary
fibrosis. J Toxicol Sci. 39:319–330. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xie H, Tan JT, Wang RL, Meng XX, Tang X
and Gao S: Expression and significance of HIF-1α in pulmonary
fibrosis induced by paraquat. Exp Biol Med (Maywood).
238:1062–1068. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhu Y, Tan J, Xie H, Wang J, Meng X and
Wang R: HIF-1α regulates EMT via the Snail and β-catenin pathways
in paraquat poisoning-induced early pulmonary fibrosis. J Cell Mol
Med. 20:688–697. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhu H and Zhou H: Novel Insight into the
role of endoplasmic reticulum stress in the pathogenesis of
myocardial ischemia-reperfusion injury. Oxid Med Cell Longev.
2021:55298102021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Magallón M, Carrión AE, Bañuls L, Pellicer
D, Castillo S, Bondía S, Navarro-García MM, González C and Dasí F:
Oxidative stress and endoplasmic reticulum stress in rare
respiratory diseases. J Clin Med. 10:12682021. View Article : Google Scholar
|
|
17
|
Hayashi T, Saito A, Okuno S, Ferrand-Drake
M, Dodd RL, Nishi T, Maier CM, Kinouchi H and Chan PH: Oxidative
damage to the endoplasmic reticulum is implicated in ischemic
neuronal cell death. J Cereb Blood Flow Metab. 23:1117–1128. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Laybutt DR, Preston AM, Akerfeldt MC,
Kench JG, Busch AK, Biankin AV and Biden TJ: Endoplasmic reticulum
stress contributes to beta cell apoptosis in type 2 diabetes.
Diabetologia. 50:752–763. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tanjore H, Blackwell TS and Lawson WE:
Emerging evidence for endoplasmic reticulum stress in the
pathogenesis of idiopathic pulmonary fibrosis. Am J Physiol Lung
Cell Mol Physiol. 302:L721–L729. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Feng YX, Sokol ES, Del Vecchio CA, Sanduja
S, Claessen JH, Proia TA, Jin DX, Reinhardt F, Ploegh HL, Wang Q
and Gupta PB: Epithelial-to-mesenchymal transition activates
PERK-eIF2α and sensitizes cells to endoplasmic reticulum stress.
Cancer Discov. 4:702–715. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Burman A, Tanjore H and Blackwell TS:
Endoplasmic reticulum stress in pulmonary fibrosis. Matrix Biol.
68-69:355–365. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Marciniak SJ: Endoplasmic reticulum stress
in lung disease. Eur Respir Rev. 26:1700182017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tanjore H, Cheng DS, Degryse AL, Zoz DF,
Abdolrasulnia R, Lawson WE and Blackwell TS: Alveolar epithelial
cells undergo epithelial-to-mesenchymal transition in response to
endoplasmic reticulum stress. J Biol Chem. 290:32772015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Meng XX, Liu K, Tan JT, Xie H and Wang RL:
The relationship of endoplasmic reticulum stress with paraquat
induced lung fibrosis in rats. Zhonghua Wei Zhong Bing Ji Jiu Yi
Xue. 25:331–334. 2013.(In Chinese). PubMed/NCBI
|
|
25
|
Omura T, Asari M, Yamamoto J, Oka K,
Hoshina C, Maseda C, Awaya T, Tasaki Y, Shiono H, Yonezawa A, et
al: Sodium tauroursodeoxycholate prevents paraquat-induced cell
death by suppressing endoplasmic reticulum stress responses in
human lung epithelial A549 cells. Biochem Biophys Res Commun.
432:689–694. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang D, Liu Z, Yan Z, Liang X, Liu X, Liu
Y, Wang P, Bai C, Gu Y and Zhou PK: MiRNA-155-5p inhibits
epithelium-to-mesenchymal transition (EMT) by targeting GSK-3β
during radiation-induced pulmonary fibrosis. Arch Biochem Biophys.
697:1086992021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zheng H, Yang Z, Xin Z, Yang Y, Yu Y, Cui
J, Liu H and Chen F: Glycogen synthase kinase-3β: A promising
candidate in the fight against fibrosis. Theranostics.
10:11737–11753. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shih JY and Yang PC: The EMT regulator
slug and lung carcinogenesis. Carcinogenesis. 32:1299–304. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bergmann C, Akhmetshina A, Dees C, Palumbo
K, Zerr P, Beyer C, Zwerina J, Distler O, Schett G and Distler JH:
Inhibition of glycogen synthase kinase 3β induces dermal fibrosis
by activation of the canonical Wnt pathway. Ann Rheum Dis.
70:2191–2198. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Peinado H, Portillo F and Cano A:
Switching on-off Snail: LOXL2 versus GSK3beta. Cell Cycle.
4:1749–1752. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhou BP, Deng J, Xia W, Xu J, Li YM,
Gunduz M and Hung MC: Dual regulation of Snail by
GSK-3beta-mediated phosphorylation in control of
epithelial-mesenchymal transition. Nat Cell Biol. 6:931–940. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang J, Zhu Y, Tan J, Meng X, Xie H and
Wang R: Lysyl oxidase promotes epithelial-to-mesenchymal transition
during paraquat-induced pulmonary fibrosis. Mol Biosyst.
12:499–507. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang Q, Wen L, Meng Z and Chen Y: Blockage
of endoplasmic reticulum stress attenuates nilotinib-induced
cardiotoxicity by inhibition of the Akt-GSK3β-Nox4 signaling. Eur J
Pharmacol. 822:85–94. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Foster KA, Oster CG, Mayer MM, Avery ML
and Audus KL: Characterization of the A549 cell line as a type II
pulmonary epithelial cell model for drug metabolism. Exp Cell Res.
243:359–366. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Uhal BD, Dang M, Dang V, Llatos R, Cano E,
Abdul-Hafez A, Markey J, Piasecki CC and Molina-Molina M: Cell
cycle dependence of ACE-2 explains downregulation in idiopathic
pulmonary fibrosis. Eur Respir J. 42:198–210. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jung JK, Jung HI, Neupane S, Kim KR and
Kim JY, Yamamoto H, Cho SW, Lee Y, Shin HI, Sohn WJ and Kim JY:
Involvement of PI3K and PKA pathways in mouse tongue epithelial
differentiation. Acta Histochem. 119:92–98. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kumari N, Reabroi S and North BJ:
Unraveling the molecular Nexus between GPCRs, ERS, and EMT.
Mediators Inflamm. 2021:66554172021. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang Y, Sun Y, Fu Y, Guo X, Long J, Xuan
LY, Wei CX and Zhao M: Calumenin relieves cardiac injury by
inhibiting ERS-initiated apoptosis during viral myocarditis. Int J
Clin Exp Pathol. 10:7277–7284. 2017.PubMed/NCBI
|
|
40
|
Zhong Q, Zhou B, Ann DK, Minoo P, Liu Y,
Banfalvi A, Krishnaveni MS, Dubourd M, Demaio L, Willis BC, et al:
Role of endoplasmic reticulum stress in epithelial-mesenchymal
transition of alveolar epithelial cells: Effects of misfolded
surfactant protein. Am J Respir Cell Mol Biol. 45:498–509. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li CY, Wang Q, Shen S, Wei XL and Li GX:
Oridonin inhibits migration, invasion, adhesion and TGF-β1-induced
epithelial-mesenchymal transition of melanoma cells by inhibiting
the activity of PI3K/Akt/GSK-3β signaling pathway. Oncol Lett.
15:1362–1372. 2018.PubMed/NCBI
|
|
42
|
Sittipunt C: Paraquat poisoning. Respir
Care. 50:383–385. 2005.PubMed/NCBI
|
|
43
|
Chapman HA: Epithelial-mesenchymal
interactions in pulmonary fibrosis. Annu Rev Physiol. 73:413–435.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zavadil J and Bottinger EP: TGF-β and
epithelial-to-mesenchymal transitions. Oncogene. 24:5764–5774.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hashimoto N, Phan SH, Imaizumi K, Matsuo
M, Nakashima H, Kawabe T, Shimokata K and Hasegawa Y:
Endothelial-mesenchymal transition in bleomycin-induced pulmonary
fibrosis. Am J Respir Cell Mol Biol. 43:161–172. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Guarino M, Tosoni A and Nebuloni M: Direct
contribution of epithelium to organ fibrosis:
Epithelial-mesenchymal transition. Hum Pathol. 40:1365–1376. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Phillips BP and Miller EA: Membrane
protein folding and quality control. Curr Opin Struct Biol.
69:50–54. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Selimovic D, Ahmad M, El-Khattouti A,
Hannig M, Haikel Y and Hassan M: Apoptosis-related protein-2
triggers melanoma cell death by a mechanism including both
endoplasmic reticulum stress and mitochondrial dysregulation.
Carcinogenesis. 32:1268–1278. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chen YW, Yang YT, Hung DZ, Su CC and Chen
KL: Paraquat induces lung alveolar epithelial cell apoptosis via
Nrf-2-regulated mitochondrial dysfunction and ER stress. Arch
Toxicol. 86:1547–1558. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Song CQ, Sun DZ, Xu YM, Yang C, Cai Q and
Dong XS: Effect of endoplasmic reticulum calcium on
paraquat-induced apoptosis of human lung type II alveolar
epithelial A549 cells. Mol Med Rep. 20:2419–2425. 2019.PubMed/NCBI
|
|
51
|
Xu Y, Sun D, Song C, Wang R and Dong X:
MnTMPyP inhibits paraquat-induced pulmonary epithelial-like cell
injury by inhibiting oxidative stress. J Toxicol Sci. 43:545–555.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Yamashita M, Yamashita M and Ando Y: A
long-term follow-up of lung function in survivors of paraquat
poisoning. Hum Exp Toxicol. 19:99–103. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Malhotra JD, Miao H, Zhang K, Wolfson A,
Pennathur S, Pipe SW and Kaufman RJ: Antioxidants reduce
endoplasmic reticulum stress and improve protein secretion. Proc
Natl Acad Sci USA. 105:18525–18530. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lewen A, Matz P and Chan PH: Free radical
pathways in CNS injury. J Neurotrauma. 17:871–890. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Davies PF and Civelek M: Endoplasmic
reticulum stress, redox, and a proinflammatory environment in
athero-susceptible endothelium in vivo at sites of complex
hemodynamic shear stress. Antioxid Redox Signal. 15:1427–1432.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Bagnato A and Rosanò L:
Epithelial-mesenchymal transition in ovarian cancer progression: A
crucial role for the endothelin axis. Cells Tissues Organs.
185:85–94. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lamouille S and Derynck R: Emergence of
the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin
axis in transforming growth factor-β-induced epithelial-mesenchymal
transition. Cells Tissues Organs. 193:8–22. 2011. View Article : Google Scholar : PubMed/NCBI
|