Introduction
Alexander disease (AxD) is a progressive and fatal
neurological disorder characterized by astrocytic cytoplasmic
inclusions (1). These inclusions,
namely Rosenthal fibers, contain glial fibrillary acidic protein
(GFAP) along with several stress proteins, such as small heat shock
proteins 27 and αB-crystallin (2).
In 2001, GFAP was identified as a candidate gene for AxD,
which encodes the major intermediate filament (IF) protein in
astrocytes (3). GFAP plays an
important role in cell migration, motility and mitosis, and has
also been implicated in the mechanical integrity of cells and cell
signaling (4). Unlike most variants
of other IF disorders, which act in a loss-of-function manner, all
known GFAP mutations in AxD are genetically dominant and
appear to produce a toxic gain-of-function effect (5). The downstream consequences include the
sequestration of protein chaperones, the abnormality of IF network
assembly and the predisposition for the GFAP protein to form
aggregates, as well as the hyperactivation of cellular stress
(6). The combination of these
consequences then induces numerous dysfunctions, from intracellular
vesicle regulation to ion homeostasis, to synapses formation and
cellular communication, ultimately causing neurological disorders
(7). However, the specific
mechanism of AxD pathogenesis remains unclear.
In terms of clinical characteristics, AxD is
typically classified into three forms, according to age at onset:
The infantile (<2 years old), juvenile (2-12 years old) and
adult (≥13 years old) forms (8).
However, the heterogeneity of neurological manifestations and wide
variety in onset age render diagnosis challenging. Based on
neurological analysis and magnetic resonance imaging (MRI), Yoshida
et al (9) proposed new
guidelines for diagnosing AxD in 2011. Under these guidelines, AxD
can be classified into three types: The cerebral (type 1),
bulbospinal (type 2) and intermediate (type 3) forms. The cerebral
form AxD is characterized by delayed psychomotor development,
convulsions, macrocephaly and leukoencephalopathy, appearing as
frontal lobe predominance on brain imaging scans. Patients with the
bulbospinal (type 2) form present with muscle weakness,
hyperreflexia and distinct bulbar dysfunction, typically appearing
as medulla oblongata or cervical cord atrophy on MRI scans. The
intermediate (type 3) form is characterized by several of the
symptoms of the cerebral and bulbospinal forms. Although featured
neurological and neuroradiological findings can assist the
diagnosis of AxD, the definitive diagnosis currently relies on a
genetic test or pathological examination (10).
The present study reported two cases of bulbospinal
form AxD, and clinical, functional (f)MRI and functional analyses
were conducted. In addition, bioinformatics analysis of published
data was performed to explore the potential pathogenic mechanisms
of AxD.
Materials and methods
Participants
In total, two identified probands (P3433 and P4288)
with AxD from two unrelated families and 500 healthy subjects as
controls for genetic analysis, as well as 15 normal individuals as
controls for imaging analysis were enrolled from Department of
Neurology, Rui Jin Hospital, Shanghai Jiao Tong University School
of Medicine (Shanghai, China). Diagnostic workups of patients with
AxD included taking history, physical examination and brain imaging
according to diagnosis guidelines (9). Patient P3433 was a 29-year-old female
(recruited March 2018). Patient P4288 was a 33-year-old man
(recruited December 2018). The healthy subjects who did not carry
any disease and had no family history of AxD were enrolled from
March 2018 to May 2019. For genetic analysis, the 500 healthy
controls (234 females and 266 males; age, 15–47 years; mean age,
34.570±9.079 years). For imaging analysis, 15 normal controls were
recruited from January 2019 to May 2019 (10 females and 5 males;
age, 23–45 years; mean age, 30.4±7.5). This study was approved by
the Ethics Committee of Rui Jin Hospital, Shanghai Jiao Tong
University School of Medicine (approval no. 2019-153; Shanghai,
China). All participants provided written informed consent for
participation.
Genetic analysis
Genomic DNA was extracted from peripheral blood
using the phenol-chloroform method (11). EDTA anticoagulated blood and red
blood cell lysis buffer (10 mmol/l NaCl, 10 mmol/l Tris-HCl, 5
mmol/l MgCl2) were mixed and incubated at 4°C for 20
min, then centrifuged at 1,811 × g for 10 min at 4°C. The cell
lysates were digested with nuclei lysis buffer (5 mmol/l NaCl, 10
mmol/l EDTA, 10 mmol/l Tris-HCl), 10% SDS and protease K solution
(20 mg/ml; Thermo Fisher Scientific, Inc.) at 37°C overnight. After
digestion, equal volumes of tris-saturated phenol and
chloroform/isopropanol mixture (24:1) were added to each tube.
Following centrifugation at 1,811 × g for 10 min at 4°C, the upper
aqueous phase was transferred to new tubes and mixed gently with 2
volumes of room temperature absolute ethanol to precipitate the
DNA. After rinsing the DNA pellet with 75% ethanol twice (3,220 ×
g, 1 min), the supernatant was discarded and the DNA pellet was air
dried. Then, the DNA was dissolved in TE buffer for 2 h at 37°C and
stored at −80°C. Whole-exome sequencing was performed in two
probands. DNA quality was verified by the 2200 TapeStation system
(Agilent Technologies, Inc.). A total of 3 µg DNA per sample was
utilized for WES using SureSelectXT Human All Exon V6 kits (cat.
no. 5190-8864; Agilent Technologies, Inc.) according to the
manufacturer's protocol. The concentration and quality of DNA
libraries were detected by Qubit 3.0 Fluorometer (Thermo Fisher
Scientific, Inc.) and 2200 TapeStation system (Agilent
Technologies, Inc.). Using a loading concentration of 2 nM, data
were generated by 150 base paired-end reads on an HiSeq X Ten
platform (Illumina, Inc.) using Hiseq X HD Reagent V2.5 kit (cat.
no. FC-501-2501; Illumina, Inc.). The sequence reads were aligned
to the human genome reference sequence (GRCh37/hg19) with BWA-MEM
software version 0.7.17 (12).
Variant calling and annotation were performed by Genome Analysis
Toolkit (GATK version 4.1.9.0) software and Annotate Variation
(ANNOVAR version 20191024) software, respectively (13,14).
Variants in which the minor allele frequency was >1% were
filtered using public databases, including 1000 Genomes (1000 g;
internationalgenome.org), The Exome
Aggregation Consortium (ExAC; gnomad.broadinstitute.org) and The Genome Aggregation
Database (gnomAD; gnomad.broadinstitute.org). PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2),
Scale-invariant feature transform (SIFT; http://sift.jcvi.org) and MutationTaster (http://www.mutationtaster.org) were used for
pathogenic prediction. The variants were further interpreted and
classified according to the American College of Medical Genetics
and Genomics (ACMG) guidelines (15). Putative pathogenic variants were
subsequently confirmed by Sanger sequencing, and a total of 500
healthy subjects were enrolled as controls. All GFAP
variants were denoted as RefSeq NM_002055.5. In addition, all
previously reported mutations of the GFAP gene were
summarized and labeled in the diagram of GFAP protein domain
structure according to the Human Gene Mutation Database
(hgmd.cf.ac.uk/).
MRI acquisition, preprocessing and
statistical analysis
The two probands were scanned on an MR system
(Ingenia, Philips 3T MR system; Philips Healthcare) with an
8-channel head coil array, using 15 normal individuals as the
controls. The protocol included a three-dimensional high-resolution
turbo field echo T1-weighted sequence for neuroanatomy (sagittal
slice orientation; matrix = 256×256; repetition time = 7.2 msec;
echo time = 3.3 msec; flip angle = 7°; slice thickness = 1 mm;
slice number = 192). Resting-state blood oxygen level-dependent MRI
used T2*-weighted echo-planar imaging sequence (240 functional
images; sagittal slice orientation; 39 slices; slice thickness =
3.5 mm; matrix = 64×64; repetition time =2,000 msec; echo time =30
msec; flip angle = 90°). The two patients also underwent
T2-weighted fluid-attenuated inversion recovery to obtain a more
accurate image of white matter lesions.
The T1-weighted anatomical image was first segmented
into grey matter, white matter and cerebrospinal fluid using
computational anatomy toolbox (CAT)12 (http://www.neuro.uni-jena.de) in Statistical
Parametric Mapping (SPM)12 software (v.6685; http://fil.ion.ucl.ac.uk/spm) in a MATLAB 2014b
environment (https://www.mathworks.com), with reference to tissue
probabilistic maps in Montreal Neurological Institute (MNI) space.
White matter hyperintensity was also estimated using a grey
matter-white matter tissue probability map in CAT12. Voxel-based
morphometry was performed between each patient and normal controls
to analyze for grey and white matter, using unpaired Student's
t-tests with total intracranial volume (TIV) as a covariate. False
discovery rate (FDR) correction was used to correct for multiple
comparisons at an FDR-adj.P<0.05.
With regards to the fMRI, the first 40 fMRI images
of each individual were discarded. The remaining 200 images were
realigned to adjust head motion, co-registered to the anatomical
image and normalized to the MNI space using a modified MATLAB
toolbox [Data Processing & Analysis of Brain Imaging (DPABI);
version 3.0] (16). Mean amplitude
of low-frequency fluctuation (ALFF) was computed to represent
regional neural activity of the individuals, regional homogeneity
(ReHo) and degree centrality (DC) to represent the quantity of
functional connections of a region (17–19).
Individual participants' weighted DC values were obtained from all
voxels in standard space using DPABI. Mean ALFF and voxel-wise
centrality values were also compared between each patient and the
controls using unpaired Student's t-tests with FDR-adj.P<0.05,
with voxel-based morphometry of grey matter volume as a
covariate.
Cell culture and transfection
All plasmids were purchased from GeneCreate. cDNA of
wild-type (WT) or mutant (MUT) GFAP (NM_002055.5) was
inserted into the pcDNA3.1-green fluorescent protein (GFP) plasmids
to express GFP-tagged fusion proteins. The 293T cell line was
obtained from The Cell Bank of Type Culture Collection of The
Chinese Academy of Sciences. 293T cells were grown in DMEM (Gibco;
Thermo Fisher Scientific, Inc.) supplemented with 10% FBS (Gibco;
Thermo Fisher Scientific, Inc.) and 1% penicillin-streptomycin
(Invitrogen; Thermo Fisher Scientific, Inc.) at 37°C in a
humidified incubator with 5% CO2. Next, 2×105
cells/well were seeded into 6-well plates or 5×104
cells/well were seeded into 24-well plates for transfection. Then,
24 h after plating, 293T cells were transiently transfected with WT
or MUT GFAP-GFP (c.214G>A and c.1235C>T) plasmids
using Lipofectamine® 3000 transfection reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) at room temperature. A
hot-spot mutation (c.715C>T, p.R239C) was set as the positive
control (8). All experiments were
independently repeated three times.
Western blotting and
immunofluorescence
A total of 48 h after transfection, 293T cells in
6-well plates were collected to extract proteins for western blot
analysis. For lysosomal inhibitor treatment, bafilomycin AI (BafAI;
5 nM; Merck KGaA) was added to 293T cells 24 h post-transfection,
with DMSO as the vehicle control. 293T cells were then incubated
for 12 h to extract proteins (20).
Protein concentrations were quantified using Pierce BCA Protein
Assay kit (cat. no. 23225; Thermo Fisher Scientific, Inc.). RIPA
buffer (Beyotime Institute of Biotechnology) with protease
inhibitors was used for protein extraction. Following
centrifugation at 13,000 × g for 20 min at 4°C, cell lysates were
separated into two parts, the supernatant as the soluble fraction
and the sedimentation as the insoluble fraction. The insoluble
fraction was dissolved with denaturing protein solubilization
reagents (Invent Biotechnologies, Inc.). A total of 20 µg protein
was loaded per lane. Proteins were separated via 10% SDS-PAGE, and
then subsequently transferred to a PVDF membrane. The membrane was
blocked with 5% BSA (Sangon Biotech Co., Ltd.) for 60 min at room
temperature. Anti-GFP (1:2,500; cat. no. GFP-1010; Aves Labs,
Inc.), anti-autophagy light chain 3 (LC3; 1:1,000; cat. no. 3868;
Cell Signaling Technology, Inc.) and anti-lysosomal-associated
membrane protein 1 (LAMP-1) antibodies (1:1,000; cat. no. 9091;
Cell Signaling Technology, Inc.) were used to detect relative
protein expression levels. GAPDH antibodies (1:1,000; cat. no.
2118; Cell Signaling Technology, Inc.) were used for sample loading
and transfer normalization. PVDF membranes with transferred
proteins were incubated with primary antibodies at 4°C overnight.
Then, blots were incubated with secondary HRP-conjugated antibodies
(1:5,000; cat. no. A0208; Beyotime Institute of Biotechnology; cat.
no. D110203; Sangon Biotech Co., Ltd.) for 60 min at room
temperature and detected by SuperSignal™ Western Blot Enhancer
(cat. no. 46641; Thermo Fisher Scientific, Inc.). Densitometry
analysis of protein bands was performed using ImageJ software
version 1.52p (National Institutes of Health).
For immunofluorescence, 48 h after transfection,
293T cells in 24-well plates were fixed with 4% paraformaldehyde
for 30 min at room temperature, blocked with 10% normal donkey
serum (cat. no. 017-000-121; Jackson ImmunoResearch Laboratories,
Inc.) and 0.3% Triton X-100 in PBS for 60 min at room temperature,
and incubated with primary antibodies mentioned above to detect LC3
and LAMP-1 in blocking solution at 4°C overnight. Next, cells were
stained with a Alexa Fluor® 594-conjugated secondary
antibody (1:1,000; cat. no. A-21442; Thermo Fisher Scientific,
Inc.) for 60 min at room temperature, and nucleic acid was stained
with DAPI (1:10,000; cat. no. 62248; Thermo Fisher Scientific,
Inc.) for 5 min at room temperature. Cells were visualized under a
Zeiss LSM 710 confocal microscope (magnification, ×40; zoom, ×2;
Carl Zeiss AG).
Bioinformatics analysis
The GSE116327 expression profile dataset (21) sequenced on GPL16791 (Illumina HiSeq
2500; Illumina, Inc.) was downloaded from the Gene Expression
Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/). A total of five
AxD and three normal post-mortem human brain samples were selected
(Table SI). All tissues were
frontal cortex tissues. Rosenthal fiber accumulation could be
detected in the tissues of patients with AxD, but not in those of
normal controls (21).
Differential gene expression analysis was performed
using the DESeq2 package (22).
Adjusted (adj.)P<0.05 and the absolute value of log2 fold-change
>1.00 were set as the threshold for differentially expressed
genes (DEGs). The ggplot2 package was used to construct the volcano
plot (23). The clusterProfiler
package was used to perform Gene Set Enrichment Analysis (GSEA),
basing the ‘Biological Process’ terms of the Gene Ontology (GOBP)
database (24–26). The list of the top 100 cell
type-specific genes for microglia, neurons, oligodendrocytes,
oligodendrocyte precursor cells, astrocytes and endothelial cells
was obtained from the study of McKenzie et al (27) (Table
SII). Based on this list, cell type-specific DEGs were
extracted from the selected data. The circlize package was used to
visualize the expression levels of cell type-specific DEGs
(28). Cell type-specific DEGs
lists were uploaded to Metascape (metascape.org/; November 2020) (29) for functional and pathway enrichment
analysis. Based on the Metascape online tool, different genes were
linked in the circos plot if both were associated with the same
function or pathway term. To demonstrate the association between
these terms, a subset of significant representative terms from each
of the 20 top-score clusters were selected (≤15 terms/cluster; ≤250
terms in total). Then the enriched terms were converted into a
network layout by Metascape (29).
Statistical analysis
Results of the cellular experiments are presented as
the mean ± SD and were statistically analyzed using GraphPad Prism
version 8.0.1 software (GraphPad Software, Inc.). One-way ANOVA was
used to compare the expression levels of GFAP protein in different
groups, followed by Tukey's post hoc test. A paired Student's
t-test was used to analyze relative GFAP levels of each group after
two different treatments (DMSO or BafAI. P<0.05 was considered
to indicate a statistically significant difference.
Results
Clinical findings
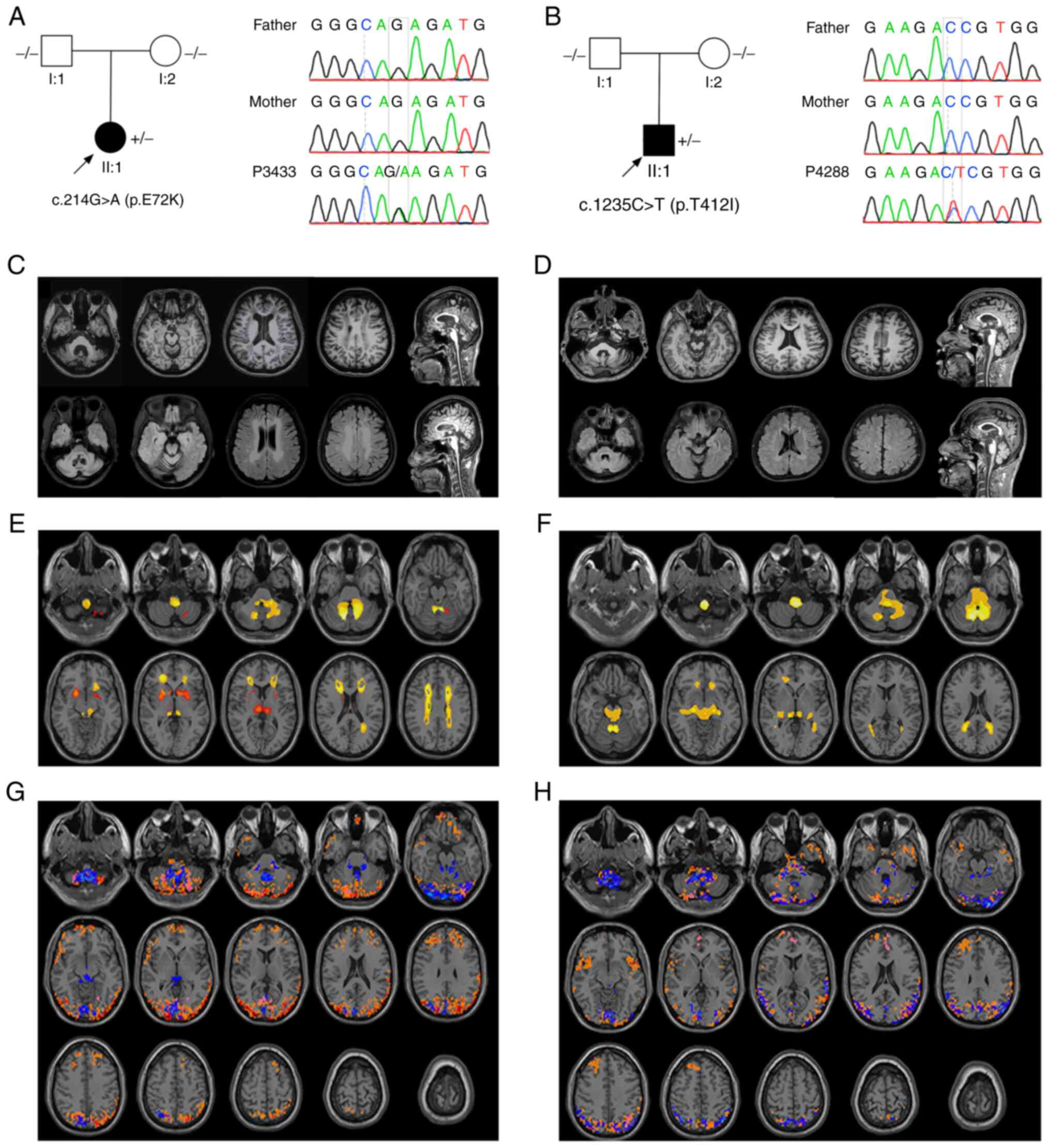
Patient P3433, a 29-year-old woman with no family
history of AxD, had been suffering from gait disturbance since the
age of 10. No issues with developmental retardation or psychomotor
abnormalities had been recorded. She had been diagnosed with
scoliosis at 14 years old (Fig.
S1A), for which she had received corrective surgery (Fig. S1B). At the age of 20 years old, she
was completely wheelchair-bound and began to suffer dysarthria,
urinary dysfunction and aspiration pneumonia. Neurological
examination revealed strabismus, movement disorders of extraocular
muscle and bilaterally horizontal nystagmus. Muscle tension and
strength of lower extremities were notably decreased. The bilateral
pathological reflexes and finger-to-nose tests were positive.
Cognitive function was normal. Laboratory examinations yielded
unremarkable findings. A documented de novo mutation was
identified in the GFAP gene (c.214G>A), which was absent
in the 1000 g, ExAC, gnomAD data and the controls (30). This mutation was also not detected
in her healthy parents; the family pedigree is presented in
Fig. 1A. c.214G>A was predicted
to be damaging by PolyPhen2 (probability score, 0.945), damaging by
SIFT (score, 0.001) and disease-causing by MutationTaster
(probability score, 1.000; data not shown). According to the ACMG
guidelines, this variant was predicted as likely pathogenic.
Patient P4288 was a 33-year-old man without a family
history of AxD. He began having gait disorders and external
rotation of the left foot at the age of 31. Imaging examinations
revealed thoracic spinal cord thinning and tethered cord syndrome.
Surgical decompression of the cauda equina nerve was performed and
symptoms were slightly improved following surgery. However, ~1 year
later, the patient presented with poor coordination, spasticity and
dysphagia, subsequently relying on a wheelchair. Physical
examinations revealed speech disfluency and horizontal nystagmus.
Muscle strength in the lower limbs was decreased, while muscle
tension was significantly increased. Bilateral pathological
reflexes were positive. Coordinated movement tests were unable to
complete. Laboratory investigations were almost normal. The
c.1235C>T variant was detected in the patient, which was absent
in the 1000 g, ExAC and gnomAD data, as well as the controls. In
addition, this mutation was not identified in the patient's parents
(Fig. 1B). SIFT (score, 0.001),
PolyPhen-2 (score, 0.519) and MutationTaster (probability score,
1.000) predicted that the variants were damaging, possibly damaging
and disease-causing, respectively (data not shown). According to
the ACMG guidelines, the c.1235C>T variant was classified as
likely pathogenic.
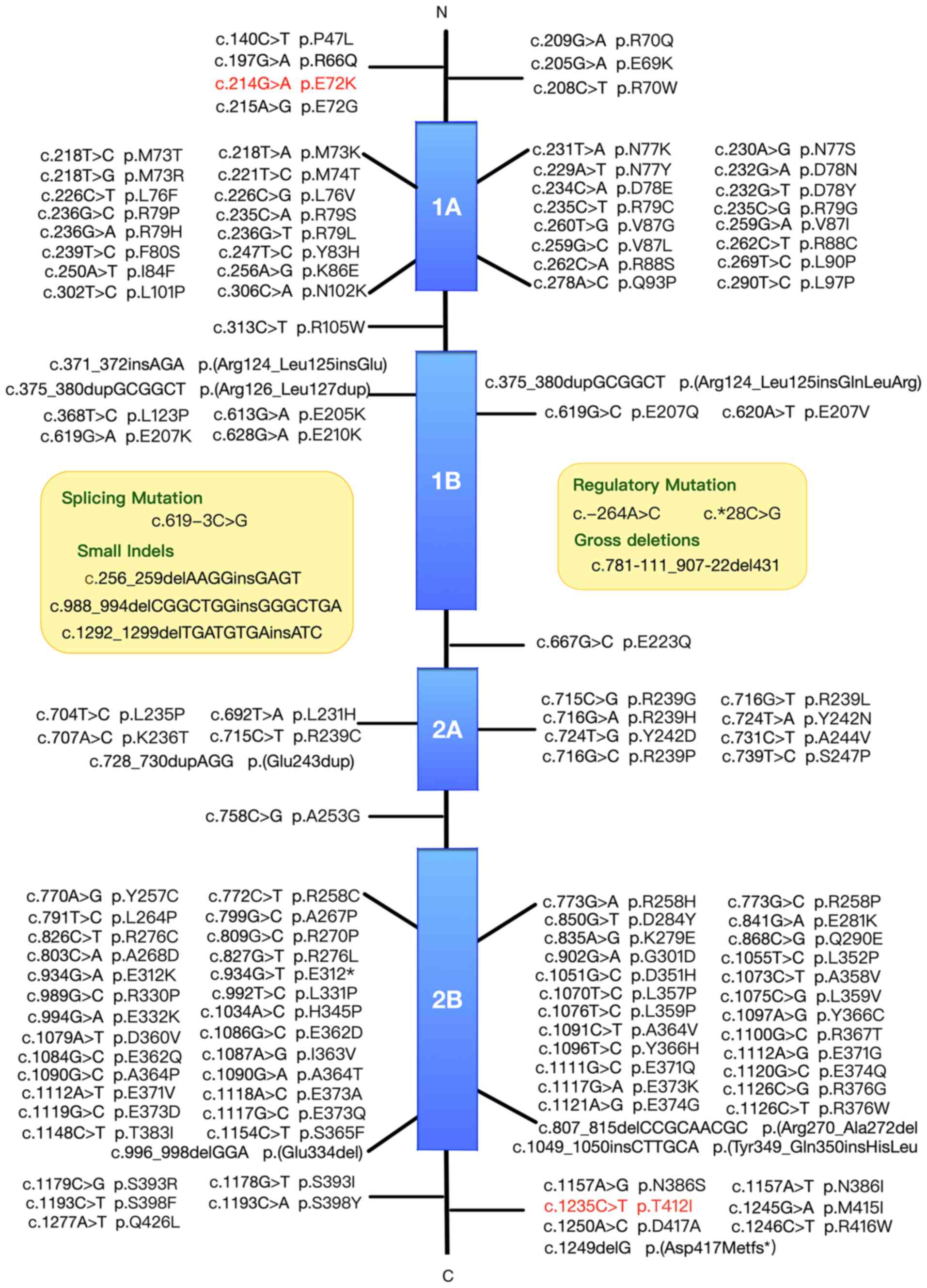
To investigate the association between mutation
sites and protein domains, previously reported GFAP
mutations were summarised according to the Human Gene Mutation
Database. A total of 135 mutations in the GFAP gene
(NM_002055.5) had been previously reported, including 121 missense,
one nonsense, one splicing, two regulatory, three small deletion,
five small insertion and three small indel mutations (Fig. 2).
Neuroimaging findings
The brain MRI scan of the two probands displayed
white matter lesions in bilateral corona radiata, centrum semiovale
and the regions surrounding the 4th ventricle (Fig. 1C and D). Volume estimation based on
a white matter tissue probability map in CAT12 suggested 17.33 ml
white matter hyperintensity in patient P3433, while the TIV was
1,218.15 ml (data not shown). Comparatively minor white matter
lesions were observed in patient P4288.
The average TIV of subjects in the control group was
1,481.34 ml, with a standard deviation of 1,13.83 ml (data not
shown). After adjusting for TIV, patient P3433 exhibited atrophy
following grey matter analysis, mainly in the bilateral putamen,
thalamus and cerebellum. Atrophic white matter was observed in the
corona radiata, centrum semiovale, cerebellopontine angle and
medulla of the patient, which was consistent with leukodystrophy in
AxD. Patient P4288 exhibited a similar pattern of atrophy in the
white matter, while no significant grey matter differences were
observed (Fig. 1E and F).
Regarding neural activity, a higher ALFF in the
cerebellar vermis, cerebellopontine angles, occipital and posterior
parietal cortex was observed in patient P3433. An increased DC
distribution was observed in both the frontal and posterior
parietal cortex, overlapping with ReHo mainly in the cerebellum and
posterior cortex. In P4288, a higher ALFF, DC and ReHo overlapped
in similar regions. An increased DC was also observed in the
bilateral insula (Fig. 1G and
H).
Functional analysis
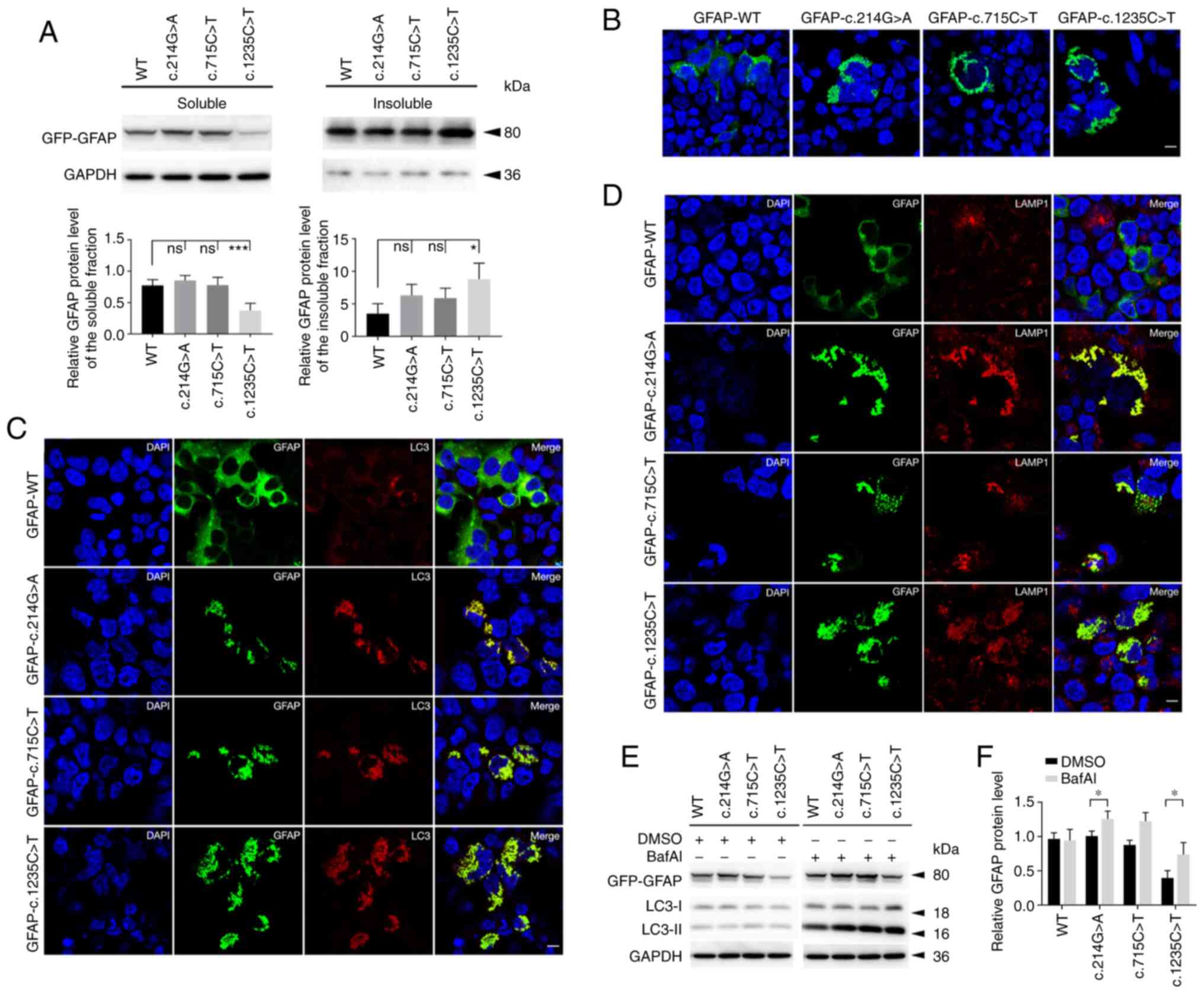
To explore the effects of GFAP variants on
the protein level and localization, WT or MUT GFAP-GFP
plasmids were transiently transfected into 293 cells. In the course
of the experiment, it was noted that the levels of soluble GFAP in
the c.1235G>T group were lower than those in the WT group.
Correspondingly, the c.1235G>T group exhibited a relatively
higher level in the insoluble fraction. No significant difference
in soluble or insoluble fractions was observed among the WT,
c.214G>A and positive control groups (c.715C>T) (Fig. 3A). Immunostaining results showed
that the WT group exhibited diffuse distribution of GFAP proteins
throughout the cytoplasm with a few aggregates, while MUT GFAP
proteins appeared as punctate aggregations in perinuclear areas
(Fig. 3B).
It was also detected whether aberrant GFAP
accumulation was associated with the autophagy-lysosome pathway. As
shown by immunofluorescence, MUT GFAP was clearly co-expressed with
LC3 and lysosome (labeled by LAMP1; Fig. 3C and D). Next, the autophagic flux
was detected using BafAI. Increased levels of LC3-II were observed
in soluble MUT groups when they were treated with BafAI (Fig. 3E). However, the WT group also
exhibited mild autophagy, since GFAP-WT overexpression could partly
contribute to aggregate formation. Of note, under BafAI treatment,
the soluble GFAP levels of MUT groups exhibited an increasing
trend, particularly in the c.1235C>T and c.214G>A groups
(Fig. 3F).
Bioinformatics analysis
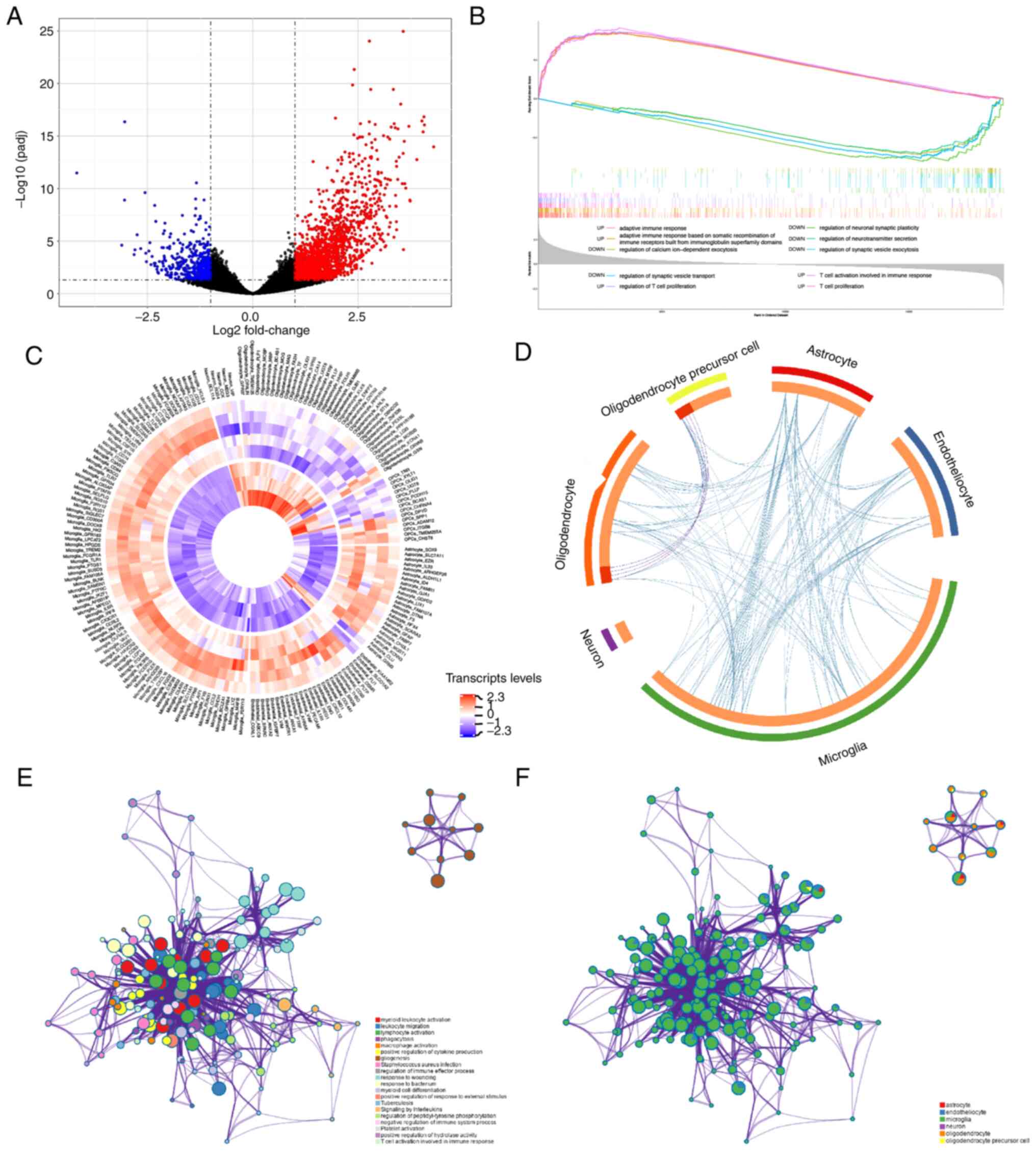
To investigate potential pathway changes in the
pathology of AxD, the RNA-sequencing data of patients with AxD and
healthy controls were downloaded from the GEO database. Compared
with the control brains, a total of 2,100 DEGs were detected in the
AxD brains (Fig. 4A). The overall
expression data were analyzed by GSEA based on the GO-BP gene sets.
A total of 1,315 up- and 125 downregulated ‘Biological Process’
pathways were identified in AxD samples compared with control
samples (adj.P<0.05). The top five up- and downregulated gene
sets are shown in Fig. 4B and
revealed the upregulation of ‘adaptive immune response’,
‘adaptative immune response based on somatic recombination of
immune receptors built from immunoglobin superfamily domains’,
‘regulation of T cell proliferation’, ‘T cell activation involved
in immune response’ and ‘T cell proliferation’, and downregulation
of ‘regulation of calcium ion-dependent exocytosis’, ‘regulation of
neuronal synaptic plasticity’, ‘regulation of neurotransmitter
secretion’, ‘regulation of synaptic vesicle exocytosis’ and
‘regulation of synaptic vesicle transport’ in AxD.
Furthermore, a set of brain cell consensus
signatures were used to screen for cell-specific changes in AxD
brain transcripts (27). Based on
cell-specific gene lists, the expression levels of significant DEGs
are shown in Fig. 4C. The overlap
of functional terms among specific cell types is shown in a circos
plot (Fig. 4D). A subset of the
representative enriched terms from each of the top 20 clusters,
including ‘myeloid leukocyte activation’, ‘lymphocyte activation’,
‘phagocytosis’ and ‘signaling by interleukins’, was converted into
a network layout (Fig. 4E). Most
terms of this network were associated with inflammatory-immune
responses and formed closely functional networks. The details are
shown in Table SIII. In addition,
it was found that microglia may play a crucial role in the
development of inflammatory processes, since the upregulated genes
in microglia were mainly associated with ‘leukocyte activation
involved in immune response’, ‘leukocyte migration’, ‘lymphocyte
activation’, as well as the ‘regulation of cytokine production’
(Figs. 4F and S2A). Considering that astrocytes are
mainly involved in AxD pathogenesis (31), astrocyte gene expression changes
were compared between AxD states and healthy states. GO enrichment
results of astrocytes showed that the GRM3 gene, the only
downregulated cell-specific gene in astrocytes, was involved in
‘synaptic signaling’ (GO:0099536; data not shown). The upregulated
genes in astrocytes were mainly associated with the ‘regulation of
protein catabolic process’, ‘spinal cord injury’ and ‘myeloid
leukocyte activation’ (Fig.
S2B).
Discussion
In the present study, two cases of bulbospinal form
AxD due to de novo GFAP mutations were described. It has
been reported that 98% of patients with a clinical diagnosis of AxD
carry a variant of GFAP, while the cause of AxD in the
remaining 2% of patients remains unknown (32). GFAPα is the predominant isoform,
which is the 432 amino acid protein that accounts for 90–95% of the
total GFAP protein in the human central nervous system (1,33). The
other GFAP isoforms, such as GFAPβ, δ and κ, derive from
alternative RNA start sites (4). To
date, GFAPα is the subject of most published studies (4,33). The
GFAPα protein comprises a central α-helical rod domain flanked with
the non-helical N-terminal head and C-terminal tail domains, which
are important for assembling into the cellular IF (diameter, 10 nm)
(4). The rod domain is divided into
four α-helical segments (1A and B, and 2A and B) and exhibits
higher conservation (8). Pathogenic
mutations are scattered all over the GFAP protein domains, but are
more abundant in the 1A and 2B segments of the rod domain.
However, the clinical severity of AxD varies
markedly and the genotype-phenotype correlation is complicated. The
variants affecting the hot-spot amino acids R79, R88, R239 and R416
account for >50% of mutations identified in patients with AxD,
while R79, R88 and R239 mutations are common in infantile and
juvenile forms AxD (8,30). In contrast to these typical
relations, the phenotype correlations of numerous other mutations
are poorly understood. There exists a variety of clinical
presentations in AxD, even among individuals carrying the same
mutation. For example, the R416W variation can be found in all
three forms of AxD (8). In
addition, it has been found that individuals carrying the same
mutation, such as D78E, S247P, L331P and D417A, show clinical
variability, with mixed infantile-adult or juvenile-adult
manifestations (1,34,35).
Patient P3433 with the c.214G>A mutation in the present study
exhibited similar symptoms to those of adult-onset form, but at a
juvenile-onset age, which may be linked to the fact that the
variant is located near R79 and D78. Patient P4288 carrying the
c.1235C>T mutation in the C-terminal domain exhibited typical
adult-onset symptoms, which rapidly progressed to severe
manifestations. Compared with R416 mutations, these findings were
consistent with previous reports that mutations in the tail domain
can have varied clinical courses and severities (8,36). The
cause of these variations remains unclear. Genetic modifiers or
environmental impactors may affect clinical phenotypes (1).
A novel analysis method based on the fMRI data was
used in the present study to explore the atrophic pattern and
spontaneous brain functional network of AxD. A similar pattern of
white matter atrophy was found in two patients, with involvements
of the medulla and periventricular regions. The subventricular
region has been reported to be the most vulnerable to the
pathogenesis of AxD (37). In
total, ~1/3 of patients with AxD display abnormal signals in the
periventricular rim, which may be associated with the abnormal
aggregation of Rosenthal fibers in subependymal regions (38,39).
Of note, in the current study patient P3433 with major white matter
damages also presented grey matter atrophy, mainly in the bilateral
putamen and thalamus, which was first reported in AxD. Grey matter
volume loss may be linked to long-term disability (40,41),
which could explain the grey matter atrophy in patient P3433.
Furthermore, several mechanisms may underlie grey matter damage,
including iron deposition, mitochondrial failure, white matter
lesion-induced retrograde degeneration and meningeal inflammation
(42,43). To the best of our knowledge, the
present study was the first to evaluate the neural activities and
regional connections in these patients through three different
types of data-driven analysis: ReHo, DC and ALFF. The results
showed increased ReHo, DC and ALFF overlapping in the cerebellum
and posterior parietal cortex, indicating a higher amount of neural
communication among these regions than controls. The cerebellum, as
part of certain largescale networks, participates in communicating
with association areas, such as the frontal lobe and posterior
parietal cortex (44). Severe
atrophies of the white or grey matter could result in the overload
and collapse of brain networks (40). Increased connectivity in these
regions may be a type of compensatory mechanism or reorganization
of the brain network for this disease. Considering the very small
sample size of AxD in the present study, more clinical studies are
required to reach correlational conclusions and explore the
underlying mechanisms.
It is commonly known that the best-known pathology
in AxD is the accumulation of mutant GFAP (31). It is noteworthy that the solubility
of GFAP variant c.1235C>T was significantly decreased in the
present study, similar to the performance of variant c.1178G>T
and c.1246C>T (45,46). These mutations are located in the
tail domain, which is highly conserved and important for
stabilizing filament-filament interactions (47). Filament disorganization may enhance
the stability of the assembled protein, which could result in
increased resistance to salt extraction, and a declined solubility
of GFAP (48). Further studies need
to examine the detailed mechanism of the tail domain that
facilitates the assembly of GFAP. Furthermore, the overlap of LC3
with abnormal GFAP accumulation was clearly observed in the present
study. Aggregate-prone proteins unsuccessfully corrected by
chaperones are generally ubiquitylated and subsequently recognized
by protein degrading pathways, such as the ubiquitin-proteasome
system and the autophagy-lysosomal pathway (49). It has been demonstrated that mutant
GFAP produces a strong inhibition of proteasome activity and leads
to decreased protein turnover rates (50,51).
In the present study, the degradation of GFAP aggregates was
accompanied by LC3-II upregulation, suggesting that the autophagy
pathway may act as a compensatory mechanism for degrading
aggregates in AxD (52).
Nevertheless, it remains to be explored whether any other potential
pathways are associated with GFAP degradation. Collectively, these
findings verified the disease-causing of the variants studied
herein and supported that GFAP mutations can be
distinguished by mutant aggregates and the upregulation of
autophagy.
To further explore the potential pathogenic
mechanisms of AxD, transcriptional alterations in AxD brains were
investigated. Bioinformatics analysis of gene expression profiles
revealed the involvement of inflammatory immune-related reactions
in AxD. It has been demonstrated that AxD astrocytes sustain a
state of cellular stress caused by abnormal aggregates and act as
origins of pathology (31).
Microglia changes may directly result from chemokines released by
activated astrocytes, while damage-associated molecular patterns,
such as small heat shock proteins, which markedly accumulate in AxD
astrocytes, could also function in microglia alterations (2,53). It
is possible that the inflammatory responses are due not only to
astrocyte stress, but also to the reactions of dysfunctional
astrocytes to external stimulations from other cells, particularly
activated microglia (31).
Consistent with the present findings, dysfunctional astrocytes are
less able to maintain the ion transport, synaptic transmission and
neurotransmitter homeostasis required for normal cell-cell
communications (31), thereby
playing an important role in inflammation alongside microglia. In
AxD, astrocyte-derived molecules also inhibit oligodendrocyte
progenitor cell function and myelination formation (21). These disruptions of brain
homeostasis, in turn, influence astrocyte phenotypes and contribute
to inflammation, creating a vicious circle. However, the nature of
these interactions and their consequences are unclear, and future
studies are required to provide novel insights into mechanistic
investigations for AxD.
Although AxD has not been acknowledged as an
inflammatory disease, several studies have revealed a marked
inflammatory environment in both mice and patients with AxD
(21,54,55).
Transgene of WT human GFAP (GFAPTg) mice
and heterozygous R236H knock-in mutation lines crossing with the
GFAPTg lines
(GFAPTg/R236H+/−) mice exhibit
Rosenthal fibers, particularly in the hippocampus, corpus callosum,
olfactory bulbs, subpial tissues and periventricular regions, more
closely resembling adult-onset AxD than infantile AxD (56,57).
Studies have reported clearly upregulated inflammatory processes in
the hippocampus and spinal cord of
GFAPTg/R236H+/− mice, and olfactory
bulb of GFAPTg mice (54,58).
Furthermore, activated inflammatory responses in the brainstem and
spinal cord have also been reported in infantile- and juvenile-form
patients (54). Accordingly,
inflammation may be associated with all three forms of the disease.
We hypothesized that inflammatory responses may also occur in these
central nervous system tissues in patients with bulbospinal form,
particularly in the brainstem and spinal cord, as these regions are
particularly affected in these patients. However, no data from the
brainstem or cerebellar regions of patients with bulbospinal form
were available to be analyzed, and the transcriptional data in the
present study were derived from the frontal lobe cortex of patients
with infantile form. Future research should focus on the specific
brain regions of different subtypes to obtain more detailed
findings.
In conclusion, two de novo variants of
GFAP (c.214G>A and c.1235C>T) were identified in
patients with AxD from unrelated families. The functional analysis
provided essential evidence revealing the pathogenicity of the
identified variants. Increased brain functional connectivity in the
cerebellum and posterior parietal cortex was observed in two
probands, and grey matter atrophy in the patient with the more
severe white matter damage. It was concluded that these changes
might be a type of compensatory mechanism or reorganization of the
collapsed brain network in AxD. Bioinformatics analysis further
indicated that inflammatory immune-related responses play a
critical role in AxD. These findings not only broadened the
clinical and genetic spectrums of AxD, but also provided an
important basis for the study of its pathogenic mechanism.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81870889, 81571086
and 82071258), Ministry of Science and Technology of the People's
Republic of China (grant nos. 2017YFC1310200 and 2016YFC1305804),
Shanghai Municipal Education Commission-Gaofeng Clinical Medicine
Grant Support (grant no. 20161401), Shanghai Municipal Planning
Commission of Science and Research Fund for Youth (grant no.
20184Y0358) and Interdisciplinary Project of Shanghai Jiao Tong
University (grant no. YG2016MS64).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XS, JJ and LC conceived and designed the study. XS
performed the experiments and processed the data. BL and HT were
responsible for MRI data acquisition, preprocessing and statistical
analysis. WT and FZ performed genetic analysis. ZZ performed
bioinformatic analysis. XS, JJ, HT and LC confirmed the
authenticity of all the raw data. XS and JJ wrote the manuscript.
JJ produced figures and tables. BL, HT and LC reviewed and edited
the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
This study was approved by the Ethics Committee of
Rui Jin Hospital, Shanghai Jiao Tong University School of Medicine
(approval no. 2019-153; Shanghai, China). All participants provided
informed consent for participation.
Patient consent for publication
Written informed consent was obtained from all
participants involved in this study.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Messing A: Alexander disease. Handb Clin
Neurol. 148:693–700. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Iwaki T, Kume-Iwaki A, Liem RK and Goldman
JE: α B-crystallin is expressed in non-lenticular tissues and
accumulates in Alexander's disease brain. Cell. 57:71–78. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Brenner M, Johnson AB, Boespflug-Tanguy O,
Rodriguez D, Goldman JE and Messing A: Mutations in GFAP, encoding
glial fibrillary acidic protein, are associated with Alexander
disease. Nat Genet. 27:117–120. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hol EM and Capetanaki Y: Type III
Intermediate Filaments Desmin, Glial Fibrillary Acidic Protein
(GFAP), Vimentin, and Peripherin. Cold Spring Harb Perspect Biol.
9:a0216422017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li R, Messing A, Goldman JE and Brenner M:
GFAP mutations in Alexander disease. Int J Dev Neurosci.
20:259–268. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Quinlan RA, Brenner M, Goldman JE and
Messing A: GFAP and its role in Alexander disease. Exp Cell Res.
313:2077–2087. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jones JR, Kong L, Hanna MG IV, Hoffman B,
Krencik R, Bradley R, Hagemann T, Choi J, Doers M, Dubovis M, et
al: Mutations in GFAP Disrupt the Distribution and Function of
Organelles in Human Astrocytes. Cell Rep. 25:947–958.e4. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li R, Johnson AB, Salomons G, Goldman JE,
Naidu S, Quinlan R, Cree B, Ruyle SZ, Banwell B, D'Hooghe M, et al:
Glial fibrillary acidic protein mutations in infantile, juvenile,
and adult forms of Alexander disease. Ann Neurol. 57:310–326. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yoshida T, Sasaki M, Yoshida M, Namekawa
M, Okamoto Y, Tsujino S, Sasayama H, Mizuta I and Nakagawa M;
Alexander Disease Study Group in Japan, : Nationwide survey of
Alexander disease in Japan and proposed new guidelines for
diagnosis. J Neurol. 258:1998–2008. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Johnson AB and Brenner M: Alexander's
disease: Clinical, pathologic, and genetic features. J Child
Neurol. 18:625–632. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Miller SA, Dykes DD and Polesky HF; MWer
S, : A simple salting out procedure for extracting DNA from human
nucleated cells. Nucleic Acids Res. 16:12151988. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M, et al: The Genome Analysis Toolkit: A MapReduce framework for
analyzing next-generation DNA sequencing data. Genome Res.
20:1297–1303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang K, Li M and Hakonarson H: ANNOVAR:
Functional annotation of genetic variants from high-throughput
sequencing data. Nucleic Acids Res. 38:e164. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al ACMG
Laboratory Quality Assurance Committee, : Standards and guidelines
for the interpretation of sequence variants: A joint consensus
recommendation of the American College of Medical Genetics and
Genomics and the Association for Molecular Pathology. Genet Med.
17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yan CG, Wang XD, Zuo XN and Zang YF:
DPABI: Data Processing & Analysis for (Resting-State) Brain
Imaging. Neuroinformatics. 14:339–351. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang H, Long XY, Yang Y, Yan H, Zhu CZ,
Zhou XP, Zang YF and Gong QY: Amplitude of low frequency
fluctuation within visual areas revealed by resting-state
functional MRI. Neuroimage. 36:144–152. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Eijlers AJ, Meijer KA, Wassenaar TM,
Steenwijk MD, Uitdehaag BM, Barkhof F, Wink AM, Geurts JJ and
Schoonheim MM: Increased default-mode network centrality in
cognitively impaired multiple sclerosis patients. Neurology.
88:952–960. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zang Y, Jiang T, Lu Y, He Y and Tian L:
Regional homogeneity approach to fMRI data analysis. Neuroimage.
22:394–400. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bresciani A, Spiezia MC, Boggio R, Cariulo
C, Nordheim A, Altobelli R, Kuhlbrodt K, Dominguez C, Munoz-Sanjuan
I, Wityak J, et al: Quantifying autophagy using novel LC3B and p62
TR-FRET assays. PLoS One. 13:e01944232018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li L, Tian E, Chen X, Chao J, Klein J, Qu
Q, Sun G, Sun G, Huang Y, Warden CD, et al: GFAP Mutations in
Astrocytes Impair Oligodendrocyte Progenitor Proliferation and
Myelination in an hiPSC Model of Alexander Disease. Cell Stem Cell.
23:239–251.e6. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Love MI, Huber W and Anders S: Moderated
estimation of fold change and dispersion for RNA-seq data with
DESeq2. Genome Biol. 15:5502014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wickham H: ggplot2. Wiley Interdiscip Rev
Comput Stat. 3:180–185. 2011. View Article : Google Scholar
|
|
24
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES, et al: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yu G, Wang L-G, Han Y and He Q-Y:
clusterProfiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al The Gene Ontology Consortium, : Gene ontology: Tool for the
unification of biology. Nat Genet. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
McKenzie AT, Wang M, Hauberg ME, Fullard
JF, Kozlenkov A, Keenan A, Hurd YL, Dracheva S, Casaccia P, Roussos
P, et al: Brain cell type specific gene expression and
co-expression network architectures. Sci Rep. 8:1–19. 2018.
View Article : Google Scholar
|
|
28
|
Gu Z, Gu L, Eils R, Schlesner M and Brors
B: circlize Implements and enhances circular visualization in R.
Bioinformatics. 30:2811–2812. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhou Y, Zhou B, Pache L, Chang M,
Khodabakhshi AH, Tanaseichuk O, Benner C and Chanda SK: Metascape
provides a biologist-oriented resource for the analysis of
systems-level datasets. Nat Commun. 10:1–10. 2019.
|
|
30
|
Prust M, Wang J, Morizono H, Messing A,
Brenner M, Gordon E, Hartka T, Sokohl A, Schiffmann R,
Gordish-Dressman H, et al: GFAP mutations, age at onset, and
clinical subtypes in Alexander disease. Neurology. 77:1287–1294.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Olabarria M and Goldman JE: Disorders of
Astrocytes: Alexander Disease as a Model. Annu Rev Pathol.
12:131–152. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yoshida T: Clinical characteristics of
Alexander disease. Neurodegener Dis Manag. 10:325–333. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Messing A and Brenner M: GFAP at 50. ASN
Neuro. 12:17590914209496802020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Stumpf E, Masson H, Duquette A, Berthelet
F, McNabb J, Lortie A, Lesage J, Montplaisir J, Brais B and
Cossette P: Adult Alexander disease with autosomal dominant
transmission: A distinct entity caused by mutation in the glial
fibrillary acid protein gene. Arch Neurol. 60:1307–1312. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Messing A, Brenner M, Feany MB, Nedergaard
M and Goldman JE: Alexander disease. J Neurosci. 32:5017–5023.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kinoshita T, Imaizumi T, Miura Y, Fujimoto
H, Ayabe M, Shoji H, Okamoto Y, Takashima H, Osame M and Nakagawa
M: A case of adult-onset Alexander disease with Arg416Trp human
glial fibrillary acidic protein gene mutation. Neurosci Lett.
350:169–172. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sawaishi Y: Review of Alexander disease:
Beyond the classical concept of leukodystrophy. Brain Dev.
31:493–498. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Balbi P, Salvini S, Fundarò C, Frazzitta
G, Maestri R, Mosah D, Uggetti C and Sechi G: The clinical spectrum
of late-onset Alexander disease: A systematic literature review. J
Neurol. 257:1955–1962. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
van der Knaap MS, Ramesh V, Schiffmann R,
Blaser S, Kyllerman M, Gholkar A, Ellison DW, van der Voorn JP, van
Dooren SJ, Jakobs C, et al: Alexander disease: Ventricular garlands
and abnormalities of the medulla and spinal cord. Neurology.
66:494–498. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Eshaghi A, Prados F, Brownlee WJ, Altmann
DR, Tur C, Cardoso MJ, De Angelis F, van de Pavert SH, Cawley N, De
Stefano N, et al MAGNIMS study group, : Deep gray matter volume
loss drives disability worsening in multiple sclerosis. Ann Neurol.
83:210–222. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Roosendaal SD, Bendfeldt K, Vrenken H,
Polman CH, Borgwardt S, Radue EW, Kappos L, Pelletier D, Hauser SL,
Matthews PM, et al: Grey matter volume in a large cohort of MS
patients: Relation to MRI parameters and disability. Mult Scler.
17:1098–1106. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Eshaghi A, Marinescu RV, Young AL, Firth
NC, Prados F, Jorge Cardoso M, Tur C, De Angelis F, Cawley N,
Brownlee WJ, et al: Progression of regional grey matter atrophy in
multiple sclerosis. Brain. 141:1665–1677. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Calabrese M, Magliozzi R, Ciccarelli O,
Geurts JJ, Reynolds R and Martin R: Exploring the origins of grey
matter damage in multiple sclerosis. Nat Rev Neurosci. 16:147–158.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ramnani N: Frontal lobe and posterior
parietal contributions to the cortico-cerebellar system.
Cerebellum. 11:366–383. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chen YS, Lim SC, Chen MH, Quinlan RA and
Perng MD: Alexander disease causing mutations in the C-terminal
domain of GFAP are deleterious both to assembly and network
formation with the potential to both activate caspase 3 and
decrease cell viability. Exp Cell Res. 317:2252–2266. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Der Perng M, Su M, Wen SF, Li R, Gibbon T,
Prescott AR, Brenner M and Quinlan RA: The Alexander
disease-causing glial fibrillary acidic protein mutant, R416W,
accumulates into Rosenthal fibers by a pathway that involves
filament aggregation and the association of alpha B-crystallin and
HSP27. Am J Hum Genet. 79:197–213. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yoshida T, Sasayama H and Nakagawa M: The
process of inducing GFAP aggregates in astrocytoma-derived cells is
different between R239C and R416W mutant GFAP. A time-lapse
recording study. Neurosci Lett. 458:11–14. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hsiao VC, Tian R, Long H, Der Perng M,
Brenner M, Quinlan RA and Goldman JE: Alexander-disease mutation of
GFAP causes filament disorganization and decreased solubility of
GFAP. J Cell Sci. 118:2057–2065. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kwon YT and Ciechanover A: The Ubiquitin
Code in the Ubiquitin-Proteasome System and Autophagy. Trends
Biochem Sci. 42:873–886. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Tang G, Xu Z and Goldman JE: Synergistic
effects of the SAPK/JNK and the proteasome pathway on glial
fibrillary acidic protein (GFAP) accumulation in Alexander disease.
J Biol Chem. 281:38634–38643. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Tang G, Perng MD, Wilk S, Quinlan R and
Goldman JE: Oligomers of mutant glial fibrillary acidic protein
(GFAP) Inhibit the proteasome system in alexander disease
astrocytes, and the small heat shock protein alphaB-crystallin
reverses the inhibition. J Biol Chem. 285:10527–10537. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Tang G, Yue Z, Talloczy Z, Hagemann T, Cho
W, Messing A, Sulzer DL and Goldman JE: Autophagy induced by
Alexander disease-mutant GFAP accumulation is regulated by p38/MAPK
and mTOR signaling pathways. Hum Mol Genet. 17:1540–1555. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Singhal G, Jaehne EJ, Corrigan F, Toben C
and Baune BT: Inflammasomes in neuroinflammation and changes in
brain function: A focused review. Front Neurosci. 8:3152014.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Olabarria M, Putilina M, Riemer EC and
Goldman JE: Astrocyte pathology in Alexander disease causes a
marked inflammatory environment. Acta Neuropathol. 130:469–486.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kora K, Kato T, Ide M, Tanaka T and
Yoshida T: Inflammatory neuropathology of infantile Alexander
disease: A case report. Brain Dev. 42:64–68. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Hagemann TL, Connor JX and Messing A:
Alexander disease-associated glial fibrillary acidic protein
mutations in mice induce Rosenthal fiber formation and a white
matter stress response. J Neurosci. 26:11162–11173. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Minkel HR, Anwer TZ, Arps KM, Brenner M
and Olsen ML: Elevated GFAP induces astrocyte dysfunction in caudal
brain regions: A potential mechanism for hindbrain involved
symptoms in type II Alexander disease. Glia. 63:2285–2297. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Hagemann TL, Gaeta SA, Smith MA, Johnson
DA, Johnson JA and Messing A: Gene expression analysis in mice with
elevated glial fibrillary acidic protein and Rosenthal fibers
reveals a stress response followed by glial activation and neuronal
dysfunction. Hum Mol Genet. 14:2443–2458. 2005. View Article : Google Scholar : PubMed/NCBI
|