Introduction
Vascular wall shear stress (SS) is crucially
important in regulating vascular physiology and pathobiology of the
vessel wall in atherosclerosis (1).
Low (L)SS promotes vascular inflammation and atherosclerosis,
whereas high (H)SS is protective (2–7).
Although there have been a number of studies on endothelial cell
(EC) responses in LSS environments, little attention has been
devoted to understanding the role of HSS in atherosclerosis or
atherosclerotic plaque formation (8).
HSS, usually regarded as >2.5Pa, has been
reported to contribute to adaptive outward remodeling, pathological
remodeling leading to saccular aneurysm initiation and instability
of atherosclerotic plaques (9).
Cheng et al (10) developed
a perivascular SS modifier, referred to as a cast, that induced
changes in SS patterns in apolipoprotein E (ApoE)-deficient mice.
They found that atherosclerotic lesions developed in the LSS
region, whereas no atherosclerotic lesions were found in the HSS
region.
Vascular ECs are exposed to hemodynamic forces under
normal physiological conditions. A previous study demonstrated that
early-stage atherosclerosis is characterized by endothelial
dysfunction (11). ECs are sensors
of blood fluid SS and transduce the frictional force from blood
flow into biochemical signals that regulate gene expression and
cell behavior through specialized pathways, such as caveolae or
caveolin-1 (8). Endothelial
dysfunction in atherosclerosis impairs endothelium-dependent
vasodilation and may lead to other pathophysiological consequences,
such as increased chemotactic and adhesion molecule expression
(12), increased
monocyte/macrophage recruitment and accumulation, decreased EC
regeneration, and increased vascular smooth muscle cells (VSMCs)
proliferation and migration. However, it is unknown whether VSMC
phenotype affects EC properties during vascular remodeling and
endothelium recovery. The aim of the present study was to
investigate the effects of HSS on ECs and VSMCs and to explore the
potential mechanism of HSS on the vessel remodeling.
Materials and methods
Animal models
Male ApoE−/− mice (n=40; age 8 weeks;
weight, 25–30 g) were obtained from the Key Laboratory of
Cardiovascular Remodeling and Function Research of Shandong
University. Among them, 15 mice were used for histological section,
16 mice were used for molecular biological detection and nine mice
were used for Oil Red of vessel. The specific-pathogen free mice
were housed at a constant temperature (24°C) and humidity (50-60%),
fed a high-fat Western-type diet containing 0.25% cholesterol and
provided water ad libitum. All experiments were performed in
compliance with the Guide for the Care and Use of Laboratory
Animals published by the US National Institutes of Health (NIH
Publication No. 85–23, revised 1985) and Shandong University. The
experimental protocol was approved by the institutional animal
ethics committee at the Second Hospital of Shandong University
[Shandong, China; KYLL-2019(KJ)P-0161].
SS modifier placement
A cast was used to induce standardized changes in SS
in vivo as previously described (10,13).
Mice were anesthetized by intraperitoneal injection of 0.08% sodium
pentobarbital (40 mg/kg) prior to SS modifier placement surgery.
The cast was implanted in the right carotid artery of
ApoE−/− mice and imposed a fixed geometry on the vessel
wall that caused gradual stenosis. Consequently, the blood flow in
the vessel segment inside the cast increased and SS was elevated
(HSS region). Conversely, the blood flow upstream from the cast
decreased and SS was lowered (LSS region). The blood flow in a
straight segment of the contralateral left carotid artery without
cast was undisturbed SS (USS region); this served as a control.
Tissue harvesting
To compare the effects of the three types of SS on
the carotid lesion, 40 mice were humanely killed at 10 weeks after
cast implantation. The mice were deeply anesthetized with 5.0%
sevoflurane and then euthanized by exsanguination by aspirating the
blood from the left ventricle. After collecting blood through heart
puncture, a reservoir perfusion apparatus that allowed direct
manipulation of perfusion pressure was used to perfuse the vessel.
Vessels were perfused with PBS at a constant physiological pressure
of 100 mmHg, then constant pressure perfusion in situ with
4% polyformaldehyde. The bilateral common carotid arteries were
carefully removed and fixed them in 4% polyformaldehyde overnight
at 4°C. After carefully removing the cast, the three different SS
regions of each mouse were classified, and the tissues (n=15) were
embedded in paraffin compound for serial cryosections at room
temperature.
Human umbilical vein endothelial cells
(HUVECs) in HSS
An in vitro HSS model was established HUVECs
[purchased from the American Type Culture Collection (ATCC)] in
endothelial cell medium containing 5% FBS (cat. no. 1001; Sciencell
Research Laboratories, Inc.), as previous described (13–15).
Controlled with USS, HSS (2.5 Pa) was imposed to HUVECs for 1–2 h
using a parallel-plate flow system after HUVECs were cultured up to
the fourth passage with 1×106 per plate. The viability
of HUVECs was assessed using an MTT assay (Sigma-Aldrich; Merck
KGaA; M2128-1G) with DMSO for dissolving crystals and a microplate
reader at the wavelength 570 nm, according to the manufacturer's
instructions.
Histological and immunofluorescence
staining
Serial cryosections (6 µm) were stained with
hematoxylin and eosin (Harris 5 g/l; Sigma-Aldrich; Merck KGaA) at
room temperature for 30 min. Consecutive sections were stained with
Sirius red (1.0 g/l; Beyotime Institute of biotechnology) for
collagen and muscle fibers were stained using Masson Trichrome
staining solution (10 g/l; Sigma-Aldrich; Merck KGaA) at room
temperature for 60 min. Oil-red O (5.0 g/l; Sigma-Aldrich; Merck
KGaA) staining was used to identify lipid-rich lesions at room
temperature for 30 min. Aortic arches and carotid arteries were
stained with Oil Red O for atherosclerotic lesions in the LSS
region. The stained cryosections were imaged and analyzed using the
Image-Pro Plus 6.0 automated image analysis system (Media
Cybernetics, Inc.) attached to a color CCD video camera
(fluorescent; ×50-1,000; Olympus BX51; Olympus Corporation). Intima
was defined as the area bounded by the endothelium and the internal
elastic lamina and media were defined as the area bounded by the
internal and external elastic laminae (16).
Tissue sections were also used for
immunofluorescence staining using antibodies against mouse CD31
(1:500; cat. no. sc-376764; Santa Cruz Biotechnology, Inc.),
collagen α1(XVIII) chain (COL18A1; 1:500; cat. no. ab275746;
Abcam), matrix metalloproteinase (MMP-8; 1:500; cat. no. PA5-79687;
Invitrogen; Thermo Fisher Scientific, Inc.) overnight at 4°C. The
secondary antibodies were Alexa 488-conjugated donkey anti-rabbit
IgG (1:2,000; cat. no. A32790; Invitrogen; Thermo Fisher
Scientific, Inc.) and Alexa 568-conjugated donkey anti-goat IgG
(1:2,000; cat. no. A11057; Invitrogen; Thermo Fisher Scientific,
Inc.). Images were acquired using an LSM710 laser scanning confocal
microscope (Zeiss AG).
Following HSS exposure, cultured HUVECs were fixed
with 4% paraformaldehyde and probed with rabbit anti-CD31 (1:1,000
dilution; cat. no. sc-376764; Santa Cruz Biotechnology, Inc.), and
VSMCs (ATCC) with rabbit anti-COL18A1 (1:500 dilution; cat. no.
ab275746; Abcam), MMP-8 (1:800 dilution; cat. no. PA5-79687;
Invitrogen; Thermo Fisher Scientific, Inc.) overnight at 4°C. VSMCs
were cultured with the supernatant of HUVECs. Then, they were
stained with FITC-conjugated goat anti-rabbit IgG (1:200; cat. no.
TA130021; OriGene Technologies, Inc.) polyclonal antibodies for 30
min at 37°C. Counterstaining for the nuclei was performed by
incubating with DAPI (cat. no. D3571; Invitrogen; Thermo Fisher
Scientific, Inc.) for 5 min at room temperature. Images were
acquired with a laser scanning confocal microscope.
Western blot assay
Proteins were extracted from 50 mg carotid artery
specimens of LSS, HSS and USS regions using lysis buffer (cat. no.
P0013; Beyotime Institute of Biotechnology). Equal amounts of
protein (20 µl) were separated on 10% SDS-PAGE and transferred to a
nitrocellulose membrane (Bio-Rad Laboratories, Inc.). After being
blocked with 5% non-fat milk for 2 h at room temperature, the blots
were washed in TBS + 0.05% Tween-20 (TBS-T) three times for 10 min
each and incubated at 4°C overnight with an appropriate primary
antibody: Rabbit anti-β-actin (1:500; cat. no. AF5006; Beyotime
Institute of Biotechnology), rabbit anti-CD31 (1:1,000; cat. no.
sc-376764; Santa Cruz Biotechnology, Inc.), rabbit anti-α-smooth
muscle actin (SMA; 1:1,000; cat. no. 144-60525-100; RayBiotech,
Inc.), rabbit anti-osteopontin (OPN; 1:500; cat. no. 119-10057;
RayBiotech, Inc.), rabbit anti-COL18A1 (1:500; cat. no. ab275746;
Abcam), rabbit anti-MMP8 (1:1,000; cat. no. PA5-79687; Invitrogen;
Thermo Fisher Scientific, Inc.). Following incubation, the blots
were washed with TBS-T and incubated with horseradish
peroxidase-conjugated secondary antibody (1:5,000; cat. no.
sc-2357; Santa Cruz Biotechnology, Inc.) for 2 h at room
temperature. The membranes were washed three times with TBS-T, and
protein expression were visualized by Digital Gel Imaging system
(AlphaEaseF™ software; serial 2200; ProteinSimple) with enhanced
chemiluminescence detection reagents (Merk KGaA).
ELISA
HSS was used on HUVECs for 2 h. The supernatant
obtained from HUVECs with different shear stress (USS, HSS, LSS)
was harvested for culturing the VSMCs. The concentrations of
endostatin (ES; a fragment of COL18A1) and MMP-8 were determined by
using commercially available ELISA kits (cat. no. ab-281282; Abcam)
following the manufacturer's protocol. Either a monoclonal antibody
of ES or MMP-8 rom the kit was added to the assay diluents (100
µl). The plates were incubated for 2 h at 37°C. Each well was
washed three times with wash buffer. The substrate solution (200
µl) was added for 30 min to stop the reaction. The optical density
was determined with a microplate reader at the wavelength 450
nm.
Statistical analysis
The experiments were repeated three times. SPSS
version 16.0 (SPSS Inc.) was used for statistical analysis. Data
are expressed as the mean ± standard deviation. Statistical
evaluation was carried out by two-tailed unpaired Student's t-test
between two groups or by one-way ANOVA with Bonferroni's post hoc
test among three groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
HSS reduces atherosclerotic plaque
formation in ApoE−/− mice
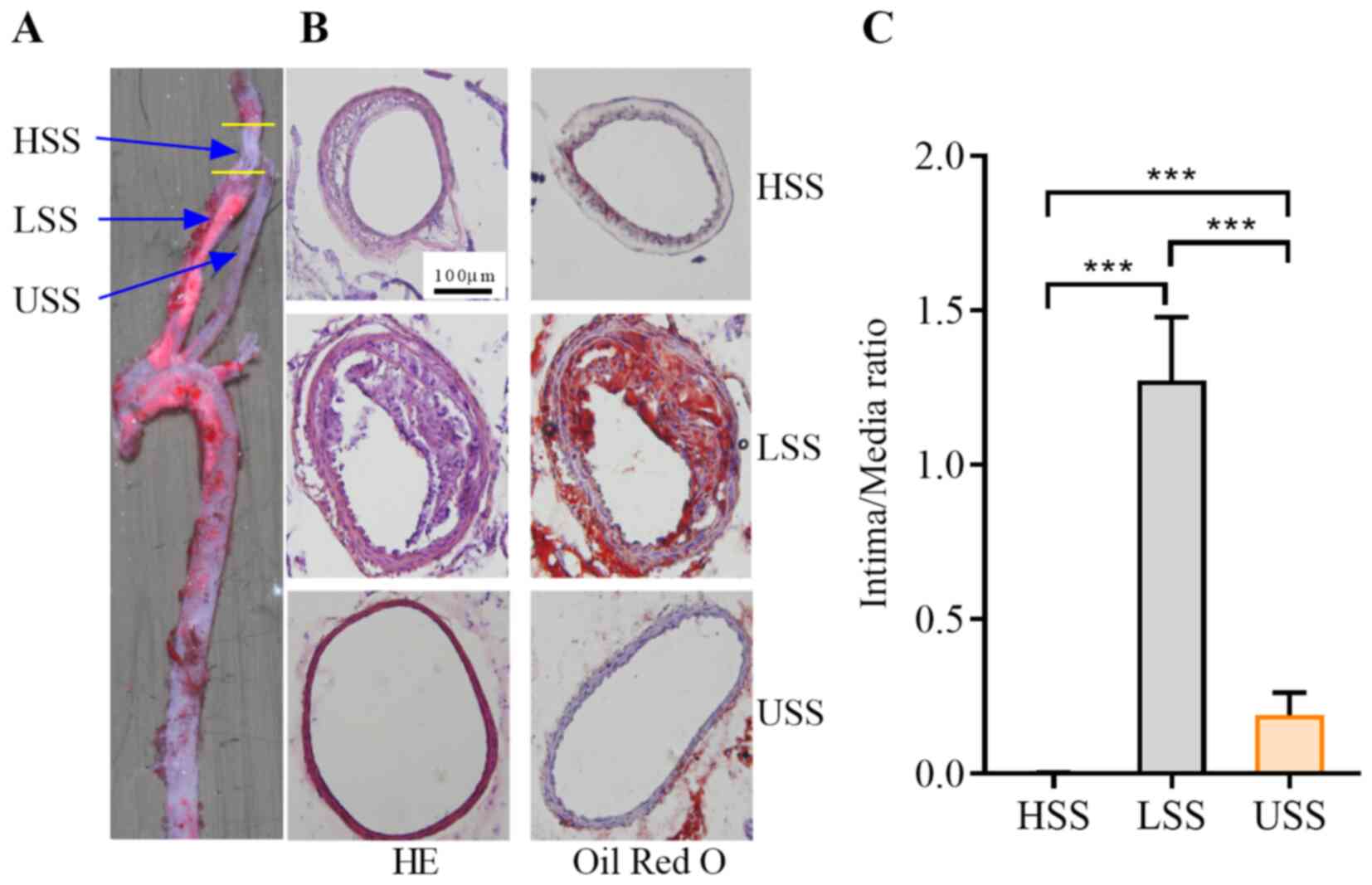
Plaque formation was assessed by HE and Oil Red O
staining (Fig. 1A). No plaque
formations were detected in the HSS and USS regions, whereas plaque
formation and fat content were observed in the LSS region in all
15ApoE−/− mice (Fig. 1A and
B). The intima-media ratio in the HSS region was significantly
lower compared with the USS region, and decreased to near zero
(P<0.001; Fig. 1C). The ratio in
the LSS region was significantly higher compared with that in the
USS or HSS regions (both P<0.05).
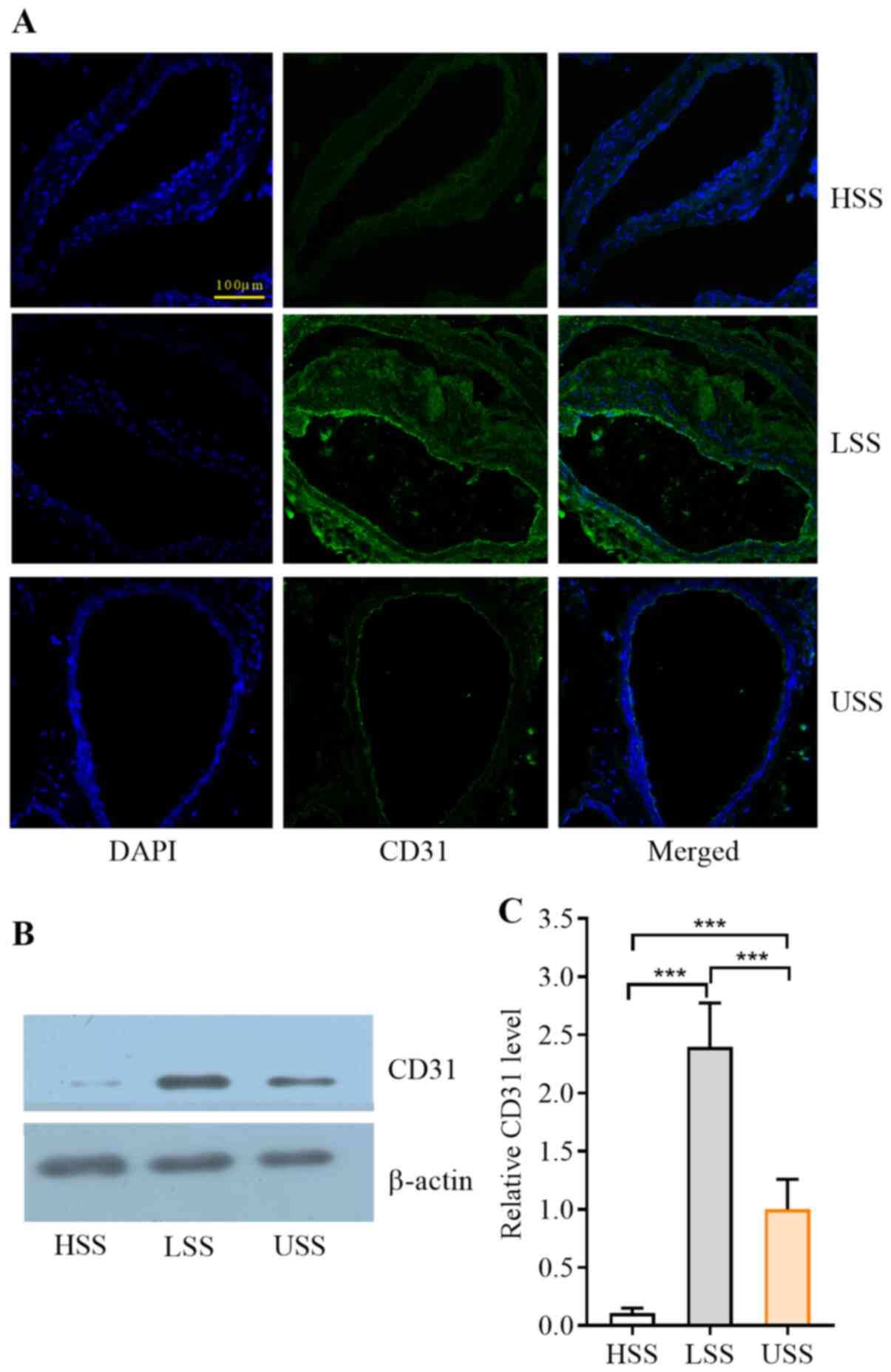
HSS attenuates the vascular ECs and
CD31 expression in mice
CD31, also known as platelet endothelial cell
adhesion molecule, has been used as an EC marker. Compared with the
normal expression of CD31 in USS regions with intact endometrium,
the expression levels of CD31 were increased in LSS (Fig. 2A). Compared with the normal
expression of ECs in the LSS and USS regions, there were fewer ECs,
or almost no ECs detected by immunofluorescence in the HSS regions
(Fig. 2A). CD31 protein expression
levels were assessed by western blotting and were significantly
decreased in the HSS region and significantly increased in the LSS
region compared to the USS region (P<0.001; Fig. 2B and C).
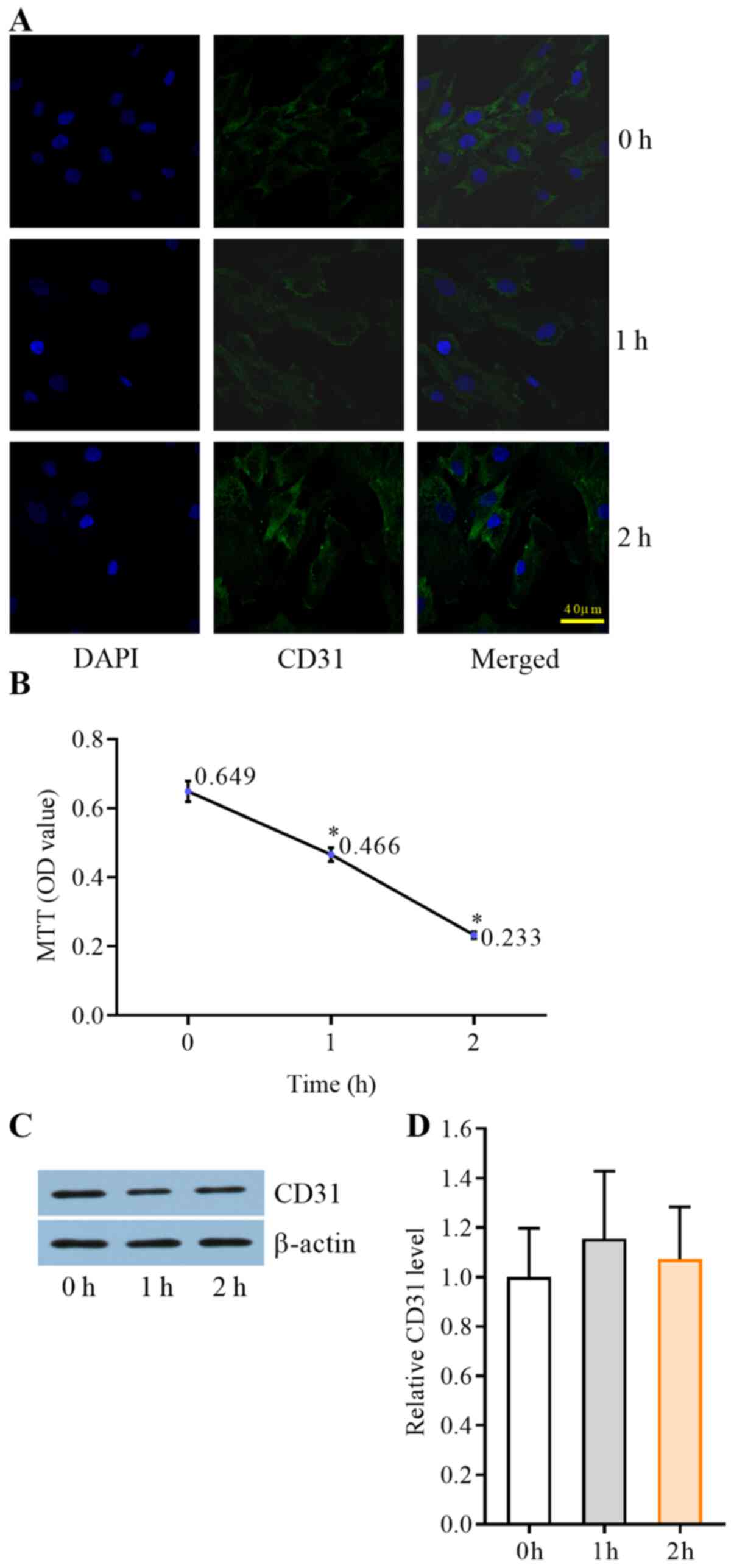
HSS decreases the number of HUVECs in
a time-dependent manner
HUVECs were stimulated by HSS for 0, 1 and 2 h. The
CD31 expression in intensity over time was detected via western
blotting using equal numbers of ECs, while EC numbers were counted.
CD31 immunofluorescence and MTT demonstrated that the counts of
HUVECs were markedly decreased in a time-dependent manner (Fig. 3A and B, respectively). CD31 protein
expression levels in ECs were assessed using western blotting after
the HUVECs were stimulated by HSS for 2 h; no significant
difference was identified (P>0.05; Fig. 3C and D).
Collagen content and synthetic
phenotype of VSMCs are enhanced by HSS
Given the crucial role in atherogenesis, collagen
content and VSMC phenotype were examined in the HSS, LSS and USS
regions. The collagen contents and VSMCs were determined using
Sirius red and Masson's Trichrome staining. Collagen content was
increased and the VSMC content were decreased in the HSS region
compared with the USS region, although the differences were not
significant (P>0.05; Fig. 4A-C).
To explore the effects of HSS on the VSMC phenotype, the expression
of α-SMA and OPN, which are widely used to determine VSMC phenotype
(17,18), were examined by western blotting.
Fig. 4D and F showed the α-SMA
expression in contractile phenotype of VSMCs, and OPN expression in
synthetic phenotype of VSMCs. α-SMA protein expression level was
significantly lower and OPN expression was higher in the HSS region
compared with expression in the USS region (both P<0.001;
Fig. 4D-F). It indicated that HSS
could induce the conversion of VSMCs from the contractile phenotype
(α-SMA) to synthetic phenotype (OPN).
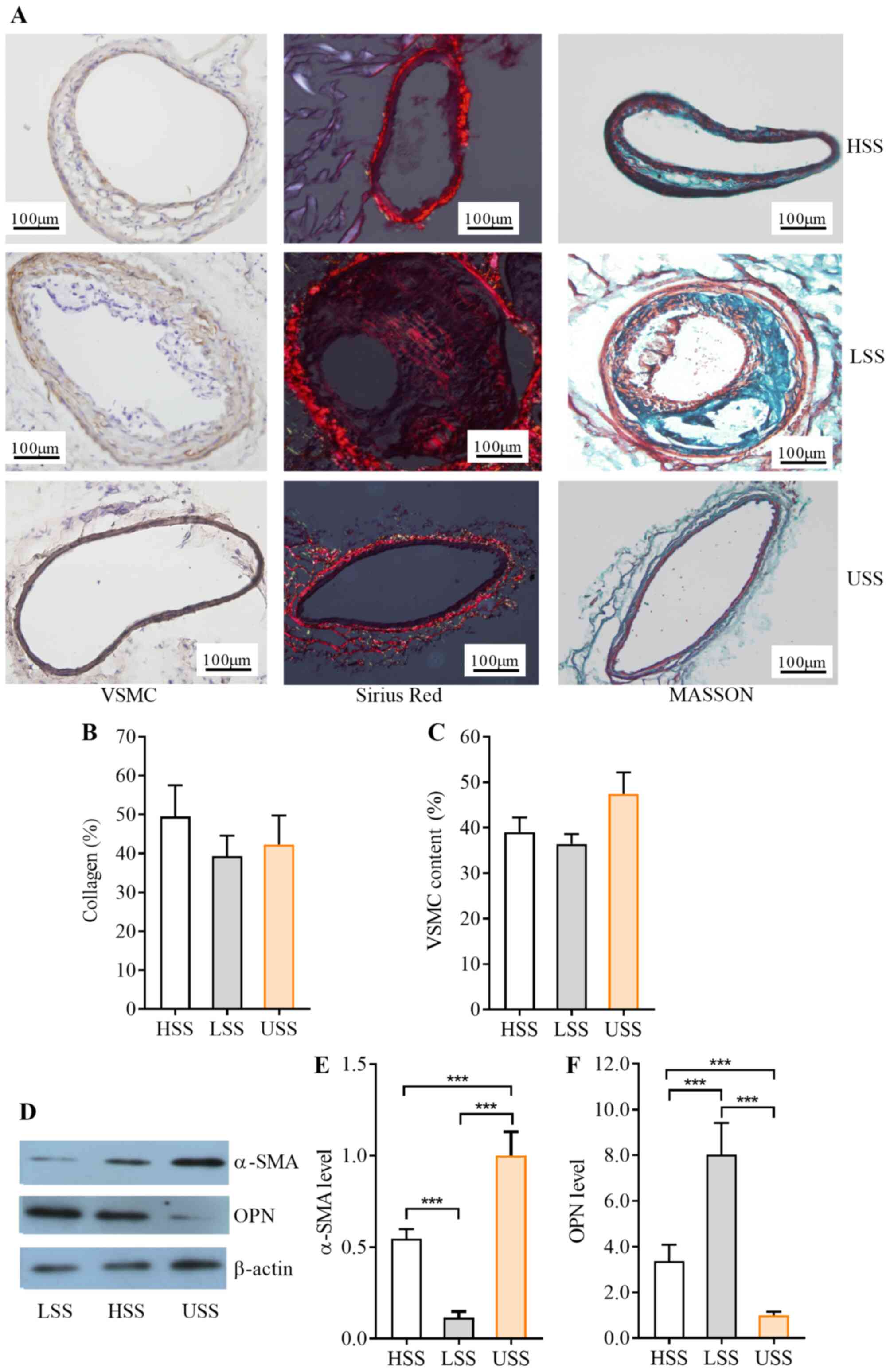 | Figure 4.Contents and phenotype of VSMCs and
collagen in different SS regions. (A) Representative images of
carotid artery sections immunostained with α-SMA, or stained with
sirius red or Masson's trichrome compound solution for VSMC and
collagen in HSS, LSS and USS regions. Scale bar, 100 µm. Histograms
showed the quantification of (B) collagen and (C) VSMCs content
(%). Data are expressed as the mean ± standard deviation; n=15. (D)
Representative western blots and semi-quantification of (E) α-SMA
protein expression levels, as an indicator for SMC contractile
phenotype, and (F) OPN protein expression levels, as an indicator
for SMC synthetic phenotype in the three regions; β-actin was used
as a loading control. Data are expressed as the mean ± standard
deviation, n=16. ***P<0.001. α-SMA, α-smooth muscle actin; HSS,
high shear stress; LSS low shear stress; OPN, osteopontin; SS,
shear stress; USS, undisturbed shear stress; VSMC, vascular smooth
muscle cell. |
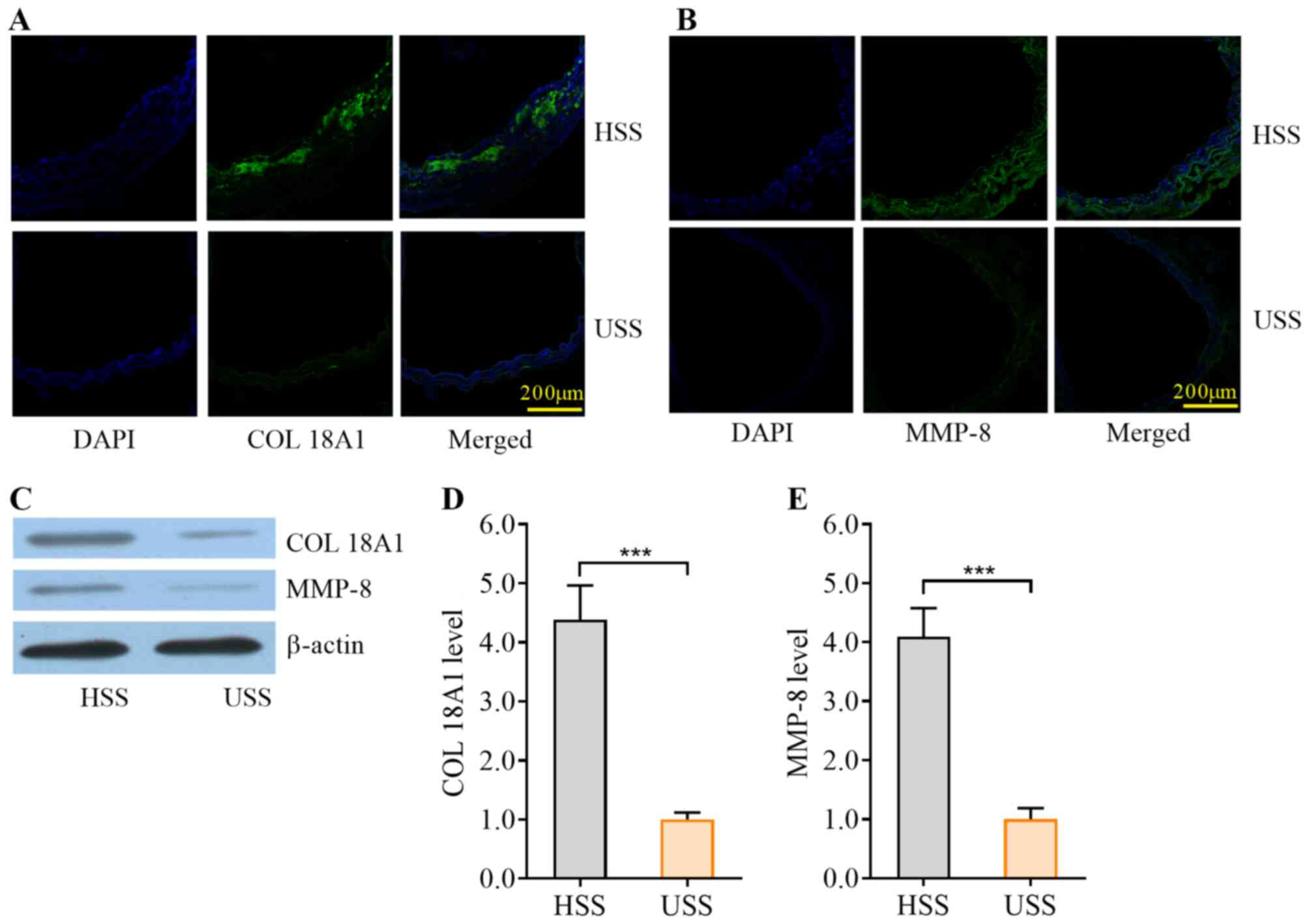
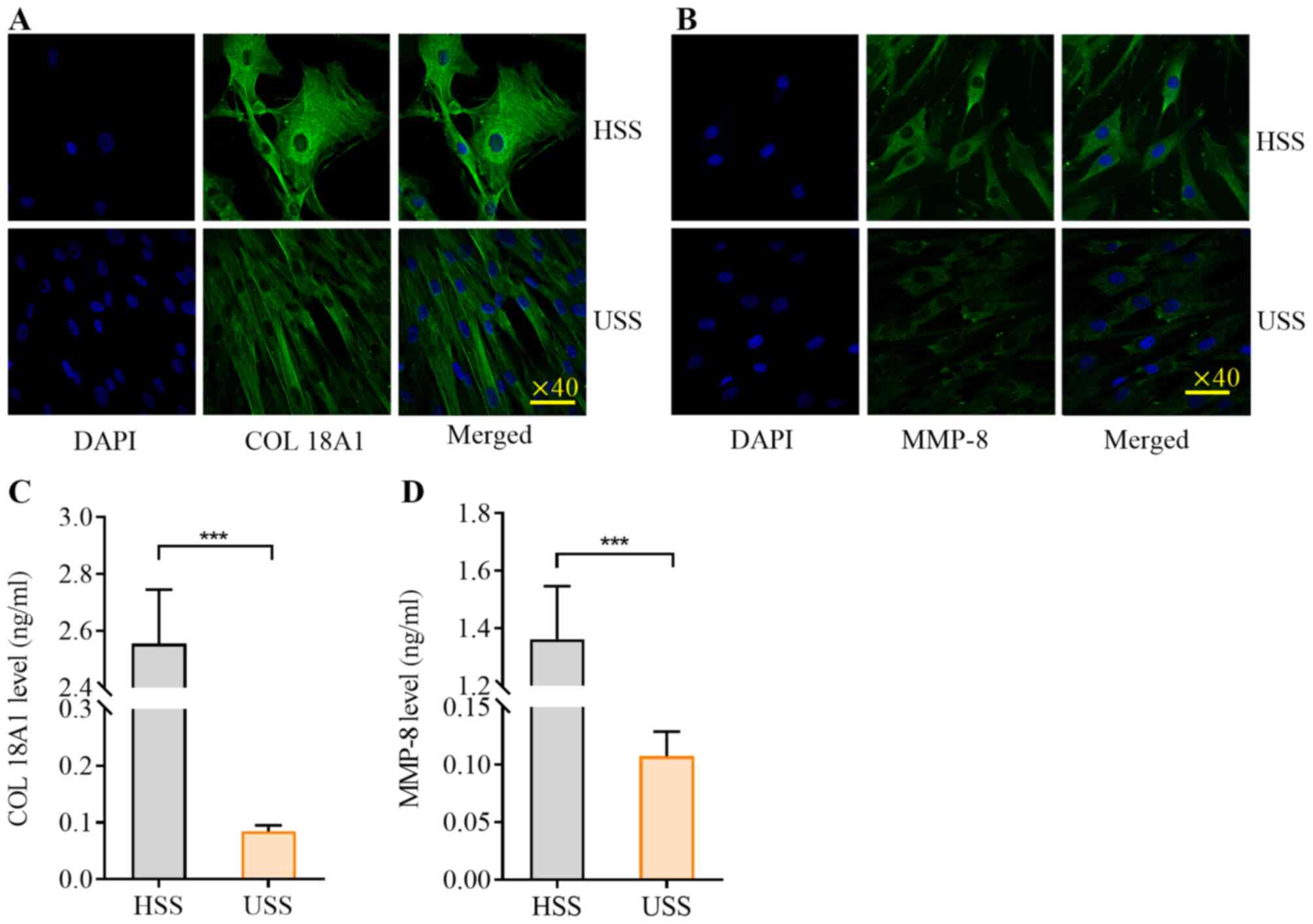
HSS promotes the expression of
vascular COL18A1 and MMP-8 in mouse carotid arteries
To verify the activity of synthetic VSMCs induced by
HSS, the protein expression levels of COL18A1 and MMP-8 in carotid
arteries were determined using immunofluorescence (Fig. 5A and B) and western blotting
(Fig. 5C). The expression levels of
COL18A1 and MMP-8 were significantly higher in the HSS region
compared with the USS region, as detected by western blotting
(P<0.001; Fig. 5D and E).
HSS induces higher levels of
COL18A1/ES and MMP-8 in VSMCs
To further confirm the activity of synthetic VSMCs,
VSMCs were exposed to HSS for 2 h and the expression levels of
COL18A1 and MMP-8 were detected using immunofluorescence and the
supernatant concentrations of ES and MMP-8 were measured using
ELISA. ES is a fragment of COL18A1 and regarded as a possible
surrogate marker for subclinical atherosclerosis with basement
membrane degradation (19,20). The expression levels of COL18A1 and
MMP-8 in the VSMCs were increased by HSS (Fig. 6A and B). The supernatant
concentrations of COL18A1 and MMP-8 were significantly higher after
HSS 2 h compared with USS (P<0.001; Fig. 6C and D).
Discussion
Physiological laminar SS serves a crucial role in
vascular function (1). To
investigate the possible role of HSS on vascular remodeling, EC
function and VSMC phenotype conversion induced by HSS was
determined. The major findings of the present study were that HSS
i) reduced/inhibited carotid plaque formation in ApoE−/−
mice; ii) decreased EC counts and attenuated CD31 expression; and
iii) induced the conversion of VSMCs from the contractile phenotype
to synthetic phenotype.
In this study, no atherosclerotic plaque and low
intima-media ratio were found in the HSS region as well as in the
USS region in the carotid artery of ApoE−/− mice. These
data indicated that HSS may prevent atherosclerotic plaque
formation. In addition, experimental results showed that ECs were
not detected and the expression of CD31 was low in the HSS region
of the carotid artery of ApoE−/− mice. However, in
vitro experiments showed that HSS did not induce changes in
CD31 expression of HUVECs, although the number of HUVECs was
significantly decreased by HSS. These results, at least to some
extent, coincide with previous studies (4,21,22).
Thus, it was suggested that preventing atherosclerotic plaque
formation by HSS resulted from endothelial denudation rather than
the conversion of the ECs phenotype. Lipid infiltration or
deposition, which is a distinguishing feature of atherosclerosis
and atherosclerotic plaque, is closely related to endothelial
dysfunction (2). After endothelial
denudation, the EC monolayer is removed and the subintimal
lipid-binding may be prevent (23).
Finally, the formation of atherosclerosis and atherosclerotic
plaque is limited.
Conversely, results from the present study found
that LSS increased CD31 expression, which was consistent with our
previous study (13). Results from
the previous study demonstrated that CD31 may be a major
mechanoreceptor of LSS for activation of JNK followed by NF-κB and
vascular cell adhesion protein 1 (13). LSS is known to increase EC
inflammatory response, proliferation and apoptosis leading to the
occurrence and development of atherosclerosis. CD31, also called
platelet endothelial cell adhesion molecule1, is one of important
member of the immunoglobulin superfamily (13). Thus, LSS may induce the upregulation
of CD31 expression in ECs.
VSMCs and ECs are in close contacted with each other
in blood vessels (24). VSMCs could
be modulated by the ECs, which are directly exposed to the blood
flow. Normal laminar SS modulate neointima accumulation (25). However, HSS was previously
demonstrated to promote certain cellular responses, such as
metalloproteinase activity to degrade matrix components, VSMC
apoptosis and reduced matrix synthesis (26). In the present study, α-SMA
expression was found to be significantly decreased whereas OPN
expression was remarkably increased by HSS compared with USS. It
indicated that HSS may induce VSMCs to convert from a contractile
to a synthetic phenotype.
In the present study, the secretory activity of
VSMCs was activated by HSS. Moreover, the concentrations of
COL18A1/ES and MMP-8 were significantly elevated by HSS. ES is
universally regarded as a circulating angiostatic factor derived
from the cleavage of COL18A1 (27–29)
and has dramatic effects on EC gene expression as well as the
capacity to inhibit EC migration, proliferation and survival
(29–33). Elevated circulating ES closely
correlates with adverse pulmonary hemodynamics in pulmonary
arterial hypertension (28,34). COL18A1/ES expression was reported to
be significantly increased in the intima and media of remodeled
vessels (34). MMP-8 expression is
correlated with atherosclerotic lesion formation and progression
(35–38). A previous study demonstrated that
the knockdown of MMP-8 in HUVECs results in a decrease in
capillary-like network formation, cell proliferation and migration,
and impaired the capacity of in vivo angiogenesis (38). Elevated ES levels may result from
vasculature basement membrane degradation (39).
In the present study, a cast was used to induce HSS,
which may have limited carotid vasodilative activity. This might be
the reason why there was no carotid artery lumen enlargement,
aneurysm or dissection in ApoE−/− mice. However, the
results are consistent with previous studies showing that HSS and
elevated COl18A1/ES are risk factors for artery aneurysm and
dissection with pathological increscent of synthetic phenotype
VSMCs (40–42), which showed the same results of
pathologic intimal denudation in arterial vessels. A major
limitation to the present study is that the potential pathways of
COL18A1/ES and MMP-8 in EC denudation and VSMC phenotypic
conversion induced by HSS were not investigated. In addition, the
apoptosis of ECs stimulated by HSS has not been discussed in the
current study. These questions will be the focus of further
research.
In summary, the results of the present study
indicated that HSS may prevent atherosclerotic plaque formation.
The probable mechanism is that HSS may induce endothelium
denudation and contractile-to-synthetic phenotypic conversion of
VSMCs. An improved understanding of the underlying mechanisms of SS
in vessel remodeling would potentially lead to improving strategies
for atherosclerotic disease management and novel targets for
pharmacological intervention.
Acknowledgements
Not applicable.
Funding
This work was supported by The National Natural
Science Foundation of China (grant nos. 81500232, 81670432 and
81973139); The Key Technology Research and Development Project of
Shandong Province (grant nos. 2017GSF18169 and 2018GSF118044); The
Shandong Provincial Natural Science Foundation (grant nos.
ZR2020MH047 and ZR2017PH050). Funding bodies did not have any role
in the design or performance of the study, nor in the
interpretation of the results or writing of the manuscript.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JW and ZL confirm the authenticity of all the raw
data, and designed and conducted the study. JW, YW, LS, TH, XN, AX,
HZ and ZL collected and analyzed the data. JW, HZ and ZL
interpreted the data and wrote the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent to publication
Not applicable.
Competing interests
The authors declare that they have not competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ApoE
|
apolipoprotein E
|
|
EC
|
endothelial cell
|
|
ES
|
endostatin
|
|
HSS
|
high shear stress
|
|
HUVEC
|
human umbilical vein endothelial
cell
|
|
MMP
|
matrix metalloproteinase
|
|
SS
|
shear stress
|
|
LSS
|
low shear stress
|
|
USS
|
undisturbed shear stress
|
|
VSMC
|
vascular smooth muscle cell
|
References
|
1
|
Cunningham KS and Gotlieb AI: The role of
shear stress in the pathogenesis of atherosclerosis. Lab Invest.
85:9–23. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gijsen F, van der Giessen A, van der Steen
A and Wentzel J: Shear stress and advanced atherosclerosis in human
coronary arteries. J Biomech. 46:240–247. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Luong L, Duckles H, Schenkel T, Mahmoud M,
Tremoleda JL, Wylezinska-Arridge M, Ali M, Bowden NP, Villa-Uriol
MC, van der Heiden K, et al: Heart rate reduction with ivabradine
promotes shear stress-dependent anti-inflammatory mechanisms in
arteries. Thromb Haemost. 116:181–190. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Malek AM, Alper SL and Izumo S:
Hemodynamic shear stress and its role in atherosclerosis. JAMA.
282:2035–2042. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu Z, Zhao Y, Wang X, Zhang H, Cui Y,
Diao Y, Xiu J, Sun X and Jiang G: Low carotid artery wall shear
stress is independently associated with brain white-matter
hyperintensities and cognitive impairment in older patients.
Atherosclerosis. 247:78–86. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen Y, Yu H, Zhu J, Zhang H, Zhao Y, Dong
Y, Cui Y, Gong G, Chai Q, Guo Y and Liu Z: Low carotid endothelial
shear stress associated with cerebral vessel disease in an older
population: A subgroup analysis of a population-based prospective
cohort study. Atherosclerosis. 288:42–50. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhao Y, Dong Y, Wang J, Sheng L, Chai Q,
Zhang H and Liu Z: Longitudinal association of carotid endothelial
shear stress with renal function decline in aging adults with
normal renal function: A population-based cohort study. Sci Rep.
9:20512019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brown AJ, Teng Z, Evans PC, Gillar JH,
Samady H and Bennett MR: Role of biomechanical forces in the
natural history of coronary atherosclerosis. Nat Rev Cardiol.
13:210–220. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dolan JM, Kolega J and Meng H: High wall
shear stress and spatial gradients in vascular pathology: A review.
Ann Biomed Eng. 41:1411–1427. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cheng C, Tempel D, van Haperen R, van der
Baan A, Grosveld F, Daemen MJ, Krams R and de Croom R:
Atherosclerotic lesion size and vulnerability are determined by
patterns of fluid shear stress. Circulation. 113:2744–2753. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chiu JJ and Chien S: Effects of disturbed
flow on vascular endothelium: Pathophysiological basis and clinical
perspectives. Physiol Rev. 91:327–387. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zakkar M, Chaudhury H, Sandvik G, Enesa K,
Luong LA, Cann S, Mason JC, Krams R, Clark AR, Haskard DO and Evans
PC: Increased endothelial mitogen-activated protein kinase
phosphatase-1 expression suppresses proinflammatory activation at
sites that are resistant to atherosclerosis. Circ Res. 103:726–732.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang J, An FS, Zhang W, Gong L, Wei SJ,
Qin WD, Wang XP, Zhao YX, Zhang Y, Zhang C and Zhang MX: Inhibition
of c-Jun N-terminal kinase attenuates low shear stress-induced
atherogenesis in apolipoprotein E-deficient mice. Mol Med.
17:990–999. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Namdee K, Carrasco-Teja M, Fish MB,
Charoenphol P and Eniola-Adefeso O: Effect of variation in
hemorheology between human and animal blood on the binding efficacy
of vascular-targeted carriers. Sci Rep. 5:116312015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Moriguchi T and Sumpio BE: PECAM-1
phosphorylation and tissue factor expression in HUVECs exposed to
uniform and disturbed pulsatile flow and chemical stimuli. J Vasc
Surg. 61:481–488. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhu Y, Farrehi PM and Fay WP: Plasminogen
activator inhibitor type 1 enhances neointima formation after
oxidative vascular injury in atherosclerosis-prone mice.
Circulation. 103:3105–3110. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang J, Yan G, Guo H, Zhu Y, Shui X, He Y,
Chen C and Lei W: ITE promotes hypoxia-induced transdifferentiation
of human pulmonary aterial endothelial cells possibly by activating
transforming growth factor-β/Smads and MAPK/ERK pathways. J Cell
Biochem. 120:19567–19577. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nakamura H, Kitazawa K, Honda H and
Sugisaki T: Roles of and correlation between alpha-smooth muscle
actin, CD44, hyaluronic acid and osteopontin in crescent formation
in human glomerulonephritis. Clin Nephrol. 64:401–411. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kato Y, Furusyo N, Tanaka Y, Ueyama T,
Yamasaki S, Murata M and Hayashi J: The relation between serum
endostatin level and carotid atherosclerosis in healthy residents
of Japan: Results from the Kyushu and Okinawa Population Study
(KOPS). J Atheroscler Thromb. 24:1023–1030. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ruge T, Carlsson AC, Kjøller E, Hilden J,
Kolmos HJ, Sajadieh A, Kastrup J, Jensen GB, Larsson A, Nowak C, et
al: Circulating endostatin as a risk factor for cardiovascular
events in patients with stable coronary heart disease: A CLARICOR
trial sub-study. Atherosclerosis. 284:202–208. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Paszkowiak JJ and Dardik A: Arterial wall
shear stress: Observations from the bench to the bedside. Vasc
Endovascular Surg. 37:47–57. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dolan JM, Meng H, Singh S, Paluch R and
Kolega J: High fluid shear stress and spatial shear stress
gradients affect endothelial proliferation, survival, and
alignment. Ann Biomed Eng. 39:1620–1631. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Franck G: Role of mechanical stress and
neutrophils in the pathogenesis of plaque erosion. Atherosclerosis.
318:60–69. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tang R, Zhang G and Chen SY: Smooth muscle
cell proangiogenic phenotype induced by cyclopentenyl cytosine
promotes endothelial cell proliferation and migration. J Biol Chem.
291:26913–26921. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Palumbo R, Gaetano C, Antonini A, Pompilio
G, Bracco E, Rönnstrand L, Heldin CH and Capogrossi MC: Different
effects of high and low shear stress on platelet-derived growth
factor isoform release by endothelial cells: Consequences for
smooth muscle cell migration. Arterioscler Thromb Vasc Biol.
22:405–411. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Slager CJ, Wentzel JJ, Gijsen FJ,
Schuurbiers JC, van der Wal AC, van der Steen AF and Serruys PW:
The role of shear stress in the generation of rupture-prone
vulnerable plaques. Nat Clin Pract Cardiovasc Med. 2:401–407. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Miosge N, Simniok T, Sprysch P and Herken
R: The collagen type XVIII endostatin domain is co-localized with
perlecan in basement membranes in vivo. J Histochem Cytochem.
51:285–296. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Goyanes AM, Moldobaeva A, Marimoutou M,
Varela LC, Wang L, Johnston LF, Aladdin MM, Peloquin GL, Kim BS,
Damarla M, et al: Functional impact of human genetic variants of
collagen 18A1/endostatin on pulmonary endothelium. Am J Respir Cell
Mol Biol. 62:524–534. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hutter R, Sauter BV, Reis ED, Roque M,
Vorcheimer D, Carrick FE, Fallon JT, Fuster V and Badimon JJ:
Decreased reendothelialization and increased neointima formation
with endostatinoverexpression in a mouse model of arterial injury.
Circulation. 107:1658–1663. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
O'Reilly MS, Boehm T, Shing Y, Fukai N,
Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR and Folkman J:
Endostatin: An endogenous inhibitor of angiogenesis and tumor
growth. Cell. 88:277–285. 1997. View Article : Google Scholar
|
|
31
|
Dhanabal M, Ramchandran R, Waterman MJ, Lu
H, Knebelmann B, Segal M and Sukhatme VP: Endostatin induces
endothelial cell apoptosis. J Biol Chem. 274:11721–11726. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Abdollahi A, Hahnfeldt P, Maercker C,
Gröne HJ, Debus J, Ansorge W, Folkman J, Hlatky L and Huber PE:
Endostatin's antiangiogenic signaling network. Mol Cell.
13:649–663. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim YM, Jang JW, Lee OH, Yeon J, Choi EY,
Kim KW, Lee ST and Kwon YG: Endostatin inhibits endothelial and
tumor cellular invasion by blocking the activation and catalytic
activity of matrix metalloproteinase. Cancer Res. 60:5410–5413.
2000.PubMed/NCBI
|
|
34
|
Hoffmann J, Marsh LM, Pieper M, Stacher E,
Ghanim B, Kovacs G, König P, Wilkens H, Haitchi HM, Hoefler G, et
al: Compartment-specific expression of collagens and their
processing enzymes in intrapulmonary arteries of IPAH patients. Am
J Physiol Lung Cell Mol Physiol. 308:L1002–L1013. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Herman MP, Sukhova GK, Libby P, Gerdes N,
Tang N, Horton DB, Kilbride M, Breitbart RE, Chun M and Schönbeck
U: Expression of neutrophil collagenase (matrix
metalloproteinase-8) in human atheroma: A novel collagenolytic
pathway suggested by transcriptional profiling. Circulation.
104:1899–1904. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ye S: Putative targeting of matrix
metalloproteinase-8 in atherosclerosis. Pharmacol Ther.
147:111–122. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Laxton RC, Hu Y, Duchene J, Zhang F, Zhang
Z, Leung KY, Xiao Q, Scotland RS, Hodgkinson CP, Smith K, et al: A
role of matrix metalloproteinase-8 in atherosclerosis. Circ Res.
105:921–929. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang F, Li S, Song J, Liu J, Cui Y and
Chen H: Angiotensin-(1–7) regulates angiotensin II-induced matrix
metalloproteinase-8 in vascular smooth muscle cells.
Atherosclerosis. 261:90–98. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sato Y: Endostatin as a biomarker of
basement membrane degradation. J Atheroscler Thromb. 24:1014–1015.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Spring S, van der Loo B, Krieger E,
Amann-Vesti BR, Rousson V and Koppensteiner R: Decreased wall shear
stress in the common carotid artery of patients with peripheral
arterial disease or abdominal aortic aneurysm: Relation to blood
rheology, vascular risk factors, and intima-media thickness. J Vasc
Surg. 43:56–63. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Osswald A, Karmonik C, Anderson JR,
Rengier F, Karck M, Engelke J, Kallenbach K, Kotelis D, Partovi S,
Böckler D and Ruhparwar A: Elevated wall shear stress in aortic
type B dissection may relate to retrograde aortic type A
dissection: A computational fluid dynamics pilot study. Eur J Vasc
Endovasc Surg. 54:324–330. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Holsti M, Wanhainen A, Lundin C, Björck M,
Tegler G, Svensson J and Sund M: Circulating vascular basement
membrane fragments are associated with the diameter of the
abdominal aorta and their expression pattern is altered in AAA
tissue. Eur J Vasc Endovasc Surg. 56:110–118. 2018. View Article : Google Scholar : PubMed/NCBI
|