Introduction
Transfusion-related acute lung injury (TRALI) is an
acute respiratory distress syndrome that occurs within 6 h
following blood transfusion and is the main origin of
transfusion-associated fatalities (1). The pathophysiological mechanism of
TRALI is poorly understood. A ‘two-event’ pathogenesis model has
been suggested (2). Predisposing
factors in transfused recipients, including systemic inflammation,
shock, surgery and mechanical ventilation, comprise the priming
event (1). The second event in
TRALI occurs as a result of blood transfusion. Anti-leukocyte
antibody or non-antibody factors, such as aged erythrocytes and
soluble CD40 ligand that accumulate in stored blood, can activate
primed polymorphonuclear leukocytes and further trigger lung injury
(3). Previous studies on TRALI have
mainly focused on cells, including polymorphonuclear leukocytes,
macrophages, platelets, lymphocytes and endothelial cells (1,4).
Therefore, its detailed pathophysiology remains to be elucidated.
Notably, currently TRALI can only by managed through symptom
alleviation via oxygen and mechanical ventilation and no specific
therapeutic approaches are available. Elucidating the pathogenesis
could aid the development of novel specific therapeutic strategies
for TRALI.
The core pathophysiology of acute lung injury (ALI)
as well as acute respiratory distress syndrome (ARDS) is the
occurrence of fibrin effusion in the lung alveoli (5,6).
Fibrin is deposited in the lungs following a wide range of insults
to the lungs in both clinical and preclinical models, including
severe acute respiratory syndrome coronavirus 2 infection-induced
ALI (7), idiopathic pulmonary
fibrosis (8), alcohol-induced ALI
(9) and trauma-induced ALI
(10). Fibrin deposition can
activate endothelial cells, promote the production and release of
proinflammatory mediators and increase vascular permeability, thus
aggravating lung injury (11).
Fibrin turnover in the lungs originates from enhanced coagulation
and impaired fibrinolysis, which may in turn aggravate inflammation
by influencing immunomodulation, vascular permeability and
chemotaxis (12–15). Therapeutic strategies targeting a
reduction in fibrin (8,16), inhibition of coagulation (17,18)
and improvement of fibrinolysis (19) hold great potential for lung injury.
Vlaar et al (20) reported
that both aged erythrocytes and their supernatant worsen lung
injury via procoagulant activity and soluble lipids, respectively,
both of which are involved in non-antibody-mediated TRALI. However,
whether pulmonary fibrin deposition occurs in non-antibody-mediated
TRALI is unclear. A case-control study showed that restrictive
transfusion of erythrocytes or multiple transfusions can lead to
TRALI and systemic imbalances in coagulation (21). The role of fibrin turnover in the
pathogenesis of antibody-mediated TRALI, which accounts for ~80% of
cases, is poorly understood. Knowledge of the pulmonary
abnormalities that favor fibrin deposition is critical to explore
the pathophysiology of antibody-mediated TRALI.
The present study explored fibrin turnover in a
mouse model of TRALI established using liposaccharide (LPS) priming
followed by an anti-major histocompatibility complex class I
(MHC-I) antibody challenge. It demonstrated that multiple local
abnormalities contributed to fibrin deposition in the lungs.
Therefore, therapies aimed at the prevention of pulmonary fibrin
deposition or turnover of fibrin in TRALI might be a potential
approach to improving survival.
Materials and methods
Reagents
Antibodies for injection into mice, namely,
anti-mouse H-2Kd and H-2Dd IgG2a MHC-I molecules (cat. no. BE0180;
clone 34-1-2S) and mouse isotype control IgG2a (cat. no. BE0085;
clone C1.18.4), were obtained from Bio X Cell. For fibrin
immunofluorescence staining, rabbit polyclonal anti-fibrinogen was
purchased from Abcam (cat. no. ab34269). A CD41a monoclonal
antibody for immunofluorescence detection of CD41 was purchased
from Invitrogen (cat. no. 2072475; Thermo Fisher Scientific, Inc.).
LPS (Escherichia coli 0111: B4) was obtained from
Sigma-Aldrich (Merck KGaA). Bovine serum (cat. no. 22012-8612) was
purchased from Zhejiang Tianhang Biotechnology Co., Ltd. Both
rhodamine-conjugated goat anti-mouse antibody (cat. no. ZF-0316)
and rhodamine-conjugated goat anti-rabbit antibody (cat. no.
ZF-0313) were provided with Beijing Zhongshan Jinqiao Biotechnology
Co., Ltd. Cytometric bead array (CBA) Flex Set kit and BCA assay
kits were provided by BD Biosciences and Thermo Fisher Scientific,
Inc., respectively. Mouse ELISA kits for myeloperoxidase (MPO; cat.
no. ml002070), thrombin-antithrombin complex (TATc; cat. no.
ml001941), tissue factor pathway inhibitor (TFPI; cat. no.
ml001878) and plasminogen activator inhibitor-1 (PAI-1; cat. no.
ml037410) were purchased from Miblo.
Mice
A total of 30 7-week-old male wild-type Balb/c mice
were obtained from Dashuo Animal Laboratory (license no.
SCXK-2015-030). The mice were housed in a standard laboratory
environment at a constant temperature (20±2°C) and humidity
(50±10%) with 12-h/12-h light/dark cycle with free access to food
and water for at least 1 week before use in experiments at 8–10
weeks of age (weight, 23.93±1.880 g). The animal studies were
conducted following approval of the Ethics Committee of the
Institute of Blood Transfusion, Chinese Academy of Medical Science
and Peking Union Medical College (approval no. 201934).
Mouse antibody-mediated TRALI model
establishment
Mice were weighed and assigned to one of three
groups in a blinded manner. Two-hit TRALI models were induced as
previously reported (22). For the
TRALI and isotype control groups, low-dose LPS (0.1 mg/kg) was
injected intraperitoneally as the first hit, and, after 18 h,
either TRALI-inducing antibody clone 34-1-2S (4.5 mg/kg) or the
isotype control antibody (4.5 mg/kg) was injected via the tail vein
as the second hit. The third group comprised untreated control
mice.
At 2 h after challenge with the 34-1-2S and isotype
control antibody, the mice were anesthetized with 2.5% Avertin (15
ml/kg) intraperitoneally. An aliquot of blood (200 µl) from
inferior vena cava was drawn in a tube containing ETA-K
anticoagulant to count platelets with automated device (XT-1800i;
Sysmex). The mice were then sacrificed by cervical dislocation and
the lungs collected. The survival rate was evaluated by dividing
the number of survivors in each group by the total number of ten
animals in each group. Lung wet-to-dry (W/D) weight ratio, lung
total protein, lung MPO activity, lung cytokines and lung histology
were determined to assess the development of TRALI.
Preparation of lung tissue
homogenates
At the experimental endpoint, the lungs were
harvested. The left lung was cut into pieces that were homogenized
in ice-cold phosphate-buffered saline (0.01 M, pH=7.2–7.4) using a
semi-automatic tissue homogenizer. Equivalent volumes of lung
tissue homogenates were stored at −80°C for further detection.
Determination of lung W/D weight
ratio
The upper and middle parts of the right lung were
collected to determine the lung W/D ratio. The fresh lung tissue
was weighed to obtain the wet weight. After drying in an oven at
65°C for 48 h, the lung tissue was weighed again to determine the
dry weight. The W/D weight ratio was calculated as net wet
weight/dry weight.
Determination of lung total
protein
BCA kits, containing standard, Reagent A and Reagent
B, were used to detect lung total protein. The working reagent was
prepared by mixing 50 parts of BCA Reagent A with 1 parts of BCA
Reagent B. A total of 25 µl each standard or lung tissue homogenate
and 200 µl working reagent were pipetted into the wells of a
microplate, which was then incubated at 37°C for 30 min. When the
plate was cooled to room temperature, lung total protein was
determined by measuring the optical density at 562 nm on a
microplate reader.
Determination of lung MPO
activity
Standard or lung homogenate (50 µl) and enzyme
conjugate (100 µl) was added to the wells of a microplate and the
plate was incubated at 37°C for 60 min. The plate was then washed.
Next, substrate A and substrate B were added into the appropriate
wells and the plate was incubated for another 15 min. After stop
solution was added into the wells, the absorbance at 450 nm was
measured within 15 min using a microtiter plate reader.
Cytokine analysis
The concentrations of TNF-α, IL-1β and IL-6 in
aliquots of lung tissue homogenates were determined using a CBA
Flex Set kit in accordance with the manufacturer's protocol. All
measurements were performed using a flow cytometer (FACSCanto II;
BD Biosciences). The data were analyzed using FCAP Array 3.0
software (BD Biosciences).
Coagulation, anticoagulation and
fibrinolysis assays
To assess coagulation, anticoagulation and
fibrinolysis functions, the levels of TATc, TFPI and PAI-1 in lung
tissue homogenates, respectively, were determined using commercial
ELISA kits in accordance with the manufacturer's protocols.
Detection of coagulation factor
activity and fibrin degradation product (FDP)
Lung tissue homogenates were analyzed for
coagulation factor activity and FDP using a CA-1500 automated
system (Sysmex) in accordance with the manufacturer's instructions.
Reagents for coagulation factor activity and FDP detection were
obtained from Siemens AG and were prepared accordance with the
manufacturer's guidelines. To detect the activities of coagulation
factor V (FV), FVII, FVIII and FIX, the samples were incubated with
factor-deficient human plasma and mixed with prothrombin time (PT)
and partial thromboplastin time (aPTT) reagents. Coagulation factor
activity was calculated based on PT and aPTT and a calibration
curve. FDP levels were measured by immunoturbidimetry.
Histopathology
Right lung lobes were immersed in 4% formalin at
room temperature for at least 24 h. Then, the tissues were
dehydrated in alcohol (75% alcohol for 4 h, 85% alcohol for 2 h,
95% alcohol for 1 h, 100% alcohol for 0.5 h, and 100% alcohol for
0.5 h), decolored (xylene for 10 min, xylene for 10 min), and
embedded in soft wax for 1 h and paraffin for 2 h. Sections were
cut at 5-µm and placed on microscope slides, deparaffinized,
hydrated and stained with hematoxylin for 5 min and eosin for 3 min
at room temperature. The following four pathological variables were
graded: Hemorrhage, white blood cell infiltration, thickness of
alveolar wall and lung epithelial cell necrosis. Scores of 0, 1, 2
and 3 represented normal, mild, moderate and severe lung injury,
respectively. Total scores for all variables were calculated to
evaluate the severity of lung injury.
Immunofluorescence
A part of right lung tissues was fixed in 4%
paraformaldehyde, dehydrated, embedded and cut into sections; these
procedures were consistent with those described in the
histopathology section. The sections were incubated with sodium
citrate solution (0.01 M) for antigen retrieval and then with 10%
bovine serum to block antigens for 30 min at room temperature. The
sections were incubated with rabbit polyclonal anti-fibrinogen
(dilution 1:50) and CD41a monoclonal antibody (dilution 1:50) at
4°C overnight and then with rhodamine-conjugated goat anti-rabbit
antibody and rhodamine-conjugated goat anti-mouse antibody for 30
min at 37°C. Nuclei were counterstained with
4′,6-diamidino-2-phenylindole. Images were acquired using a
fluorescence scanning microscope (P250 FLASH microscope; Danjier
Electronic Co., Ltd.). Average fluorescent luminosity was
determined using Image-Pro Plus 6.0 software (Media Cybernetics,
Inc.). The signal intensity in each image was normalized to the
area. The average luminosity in each sample was calculated based on
the average luminosity in three images.
Statistical analysis
Data were expressed as the mean ± standard
deviation. Statistical analysis was performed with GraphPad Prism 8
software for MacOS (GraphPad Software, Inc.). Statistical
comparisons among different treated groups were evaluated by ANOVA
with Tukey's multiple-comparison test and Newman-Keuls
multiple-comparison test. Data not conformed as continuous
variables were assessed by Kruskal-Wallis test followed by Dunn's
multiple-comparison test and expressed as median and interquartile
range. The Kaplan-Meier method was used for survival rate
comparisons. P<0.05 was considered to indicate a statistically
significant difference.
Results
Histological evidence and severity of
antibody-mediated TRALI
Following LPS priming and challenge with an anti-MHC
class I molecule (34-1-2S), the mice developed TRALI. Lung damage
in the mice was assessed by histological examination (Fig. 1A). As shown in Fig. 1A, the lung injury score was
significantly higher in TRALI mice compared with the normal control
group. However, there were no significance in the lung injury score
between TRALI group and isotype control group (Fig. 1A). Additionally, TRALI mice showed
severe lung injury, with a 60% mortality at 2 h, whereas all mice
in the control groups survived (Fig.
1B). These results showed that the TRALI mouse model was
successfully established.
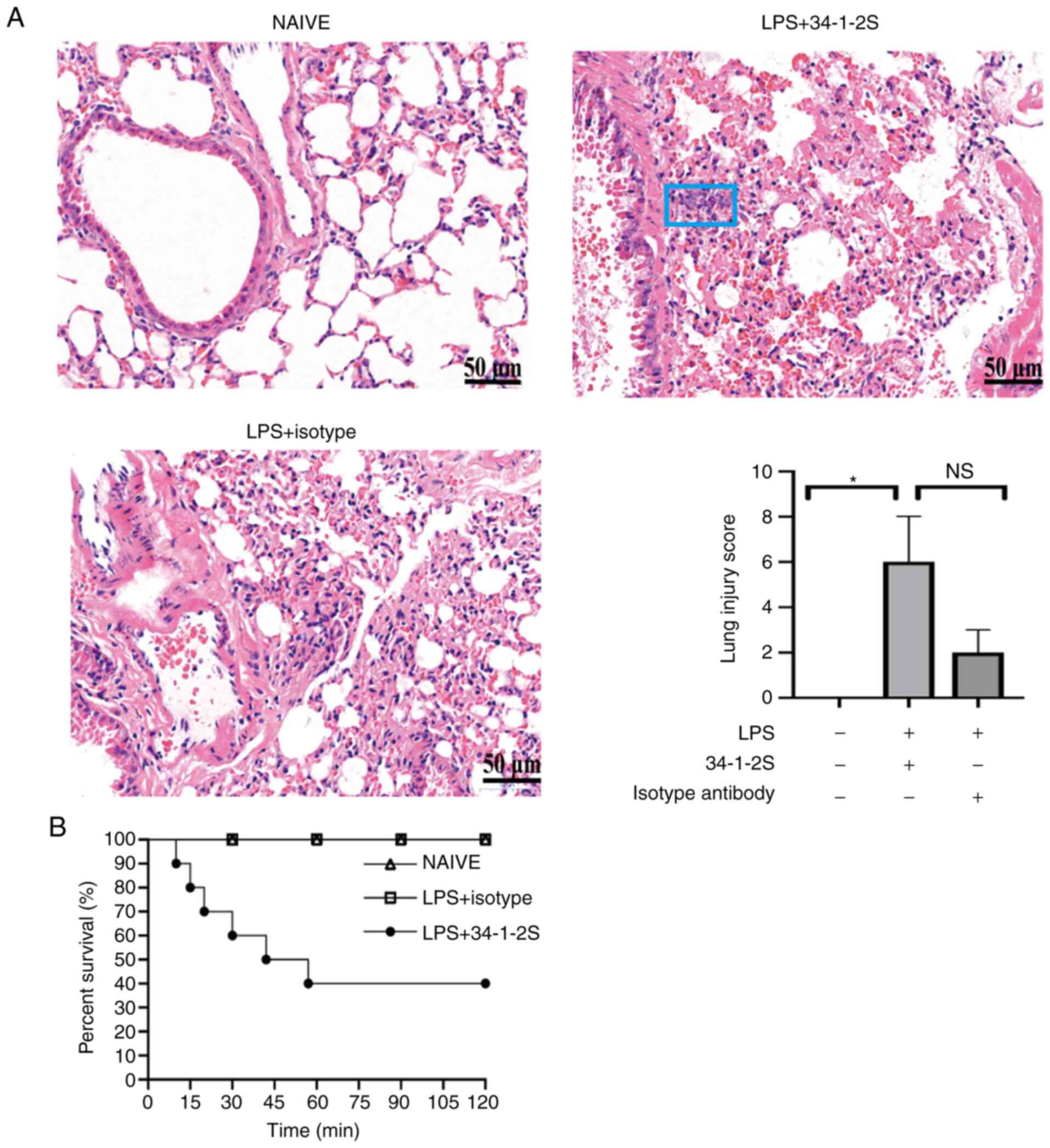 | Figure 1.Histological evidence and severity of
antibody-mediated TRALI. (A) Hematoxylin and eosin stained lung
histology sections photographed (magnification, ×400) and lung
injury scores. Data are presented as the median and interquartile
range (n=3/group). NS, no significance, *P<0.05, by
Kruskal-Wallis test followed by Dunn's multiple comparisons test.
The most representative of two replicate experiments, which were in
good agreement, is shown. The blue rectangle indicates inflammatory
cell accumulation. Scale bar, 50 µm. (B) Kaplan-Meier survival
analysis of the treatment groups (n=10 mice/group). TRALI,
transfusion-related acute lung injury; LPS, lipopolysaccharide;
34-1-2S, anti-MHC-I molecule. |
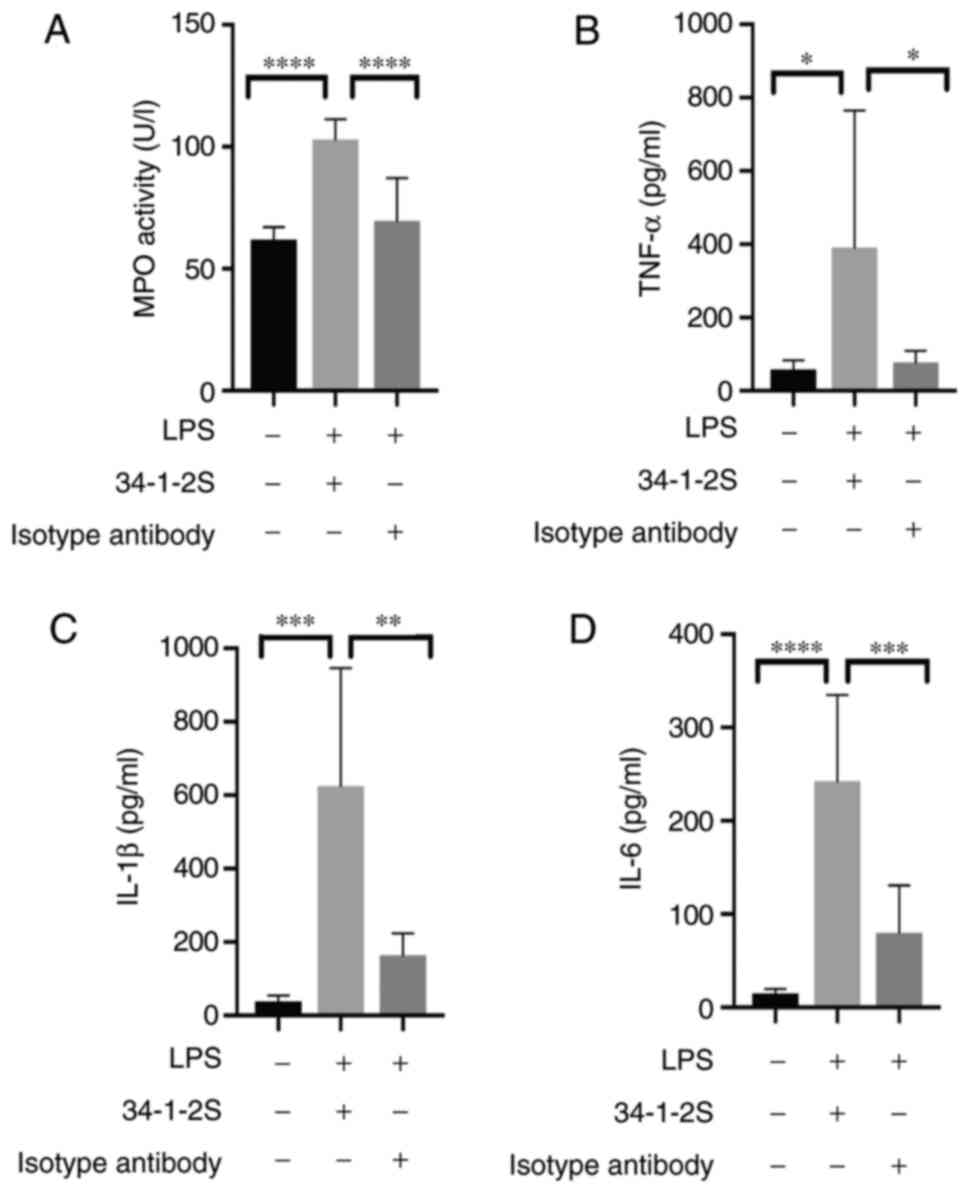
Inflammatory responses induced by
antibody-mediated TRALI
As TRALI occurs, neutrophils accumulated in the
lungs serve an important role in triggering inflammatory responses
(23). MPO activity was measured to
assess inflammation in the lung. Consistent with previous studies
(24,25), pulmonary neutrophil infiltration, as
evidenced by increased MPO activity, was typically observed in
TRALI mice (Fig. 2A).
To investigate inflammatory responses of TRALI, the
current study detected the level of inflammatory cytokines in the
lungs. Compared with control group, the levels of TNF-α (Fig. 2B), IL-1β (Fig. 2C) and IL-6 (Fig. 2D) were elevated in TRALI mice. These
results clearly indicated the presence of pulmonary
inflammation.
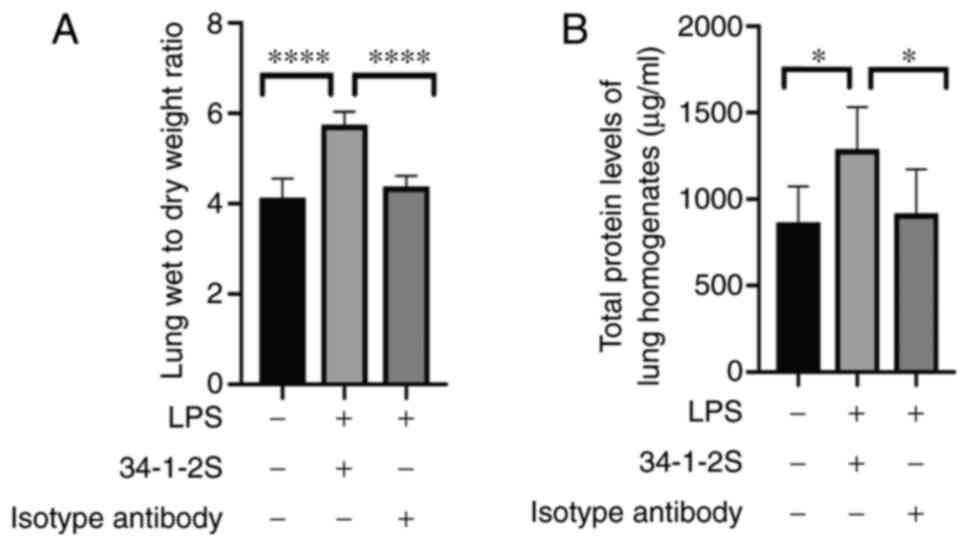
Antibody-mediated TRALI induces
alteration of the alveolar-capillary barrier
Lung injury damages the alveolar-capillary barrier,
leading to protein-rich fluid leakage into the alveolus. Changes in
the pulmonary W/D ratio and total protein level can indicate an
altered alveolar-capillary barrier. The W/D ratio and total protein
level were significantly higher in TRALI mice compared with control
mice (Fig. 3A and B).
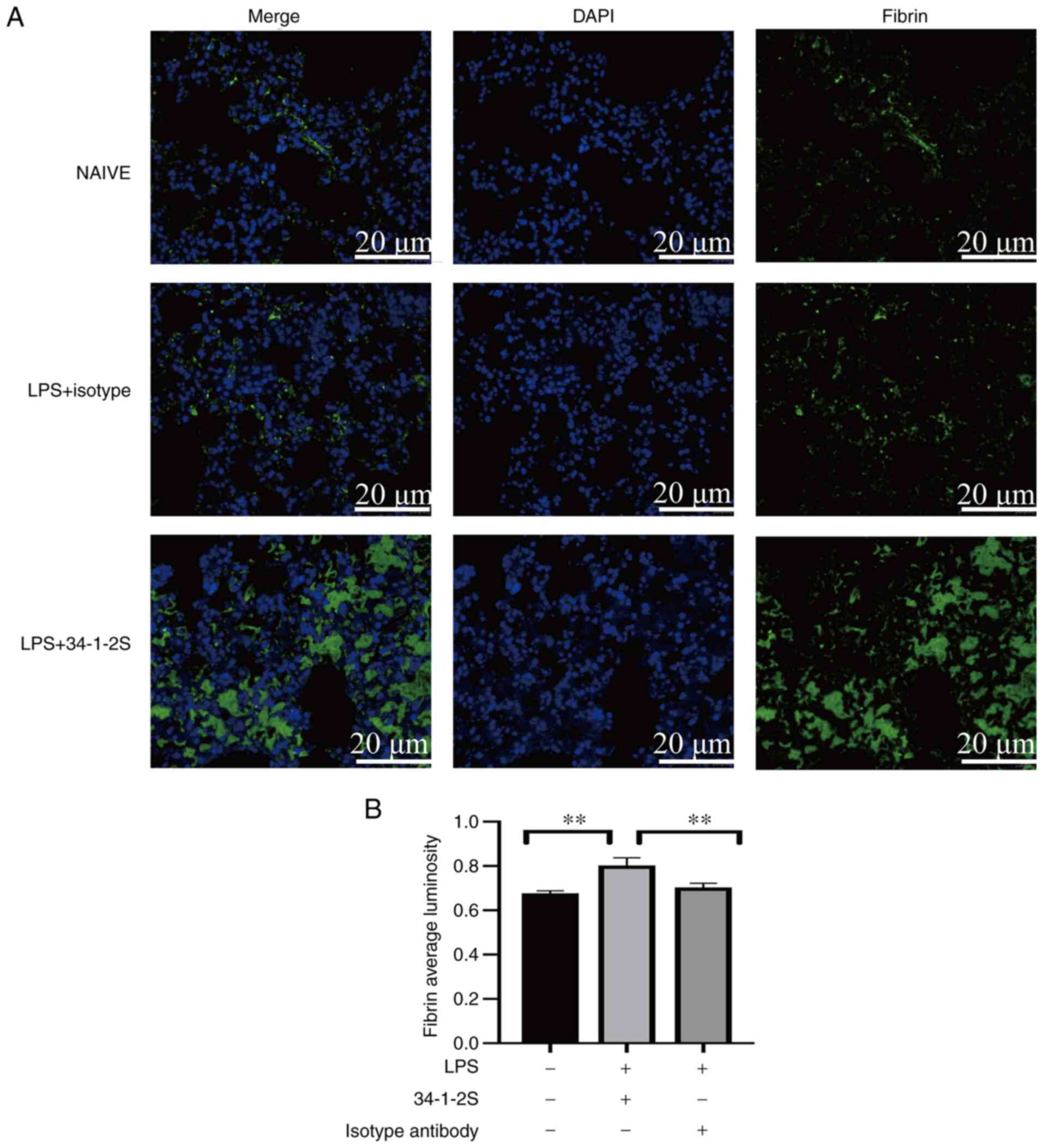
Antibody-mediated TRALI induces
pulmonary fibrin deposition in mice
Preclinical and clinical studies have reported that
coagulopathy has a primary role in non-antibody-mediated TRALI
(20,21). In addition, pulmonary coagulopathy
causes fibrin deposition in the lungs (26). Fibrin can activate endothelial
cells, promote inflammatory responses and increase vascular
permeability, thus aggravating tissue damage (11). However, no study has focused on
pulmonary coagulopathy in antibody-mediated TRALI, to the best of
the authors' knowledge. Therefore, fibrin deposition in the lungs
was detected by immunofluorescence. The model mice showed strongly
enhanced fibrin deposition in the lungs compared with those in the
two control groups (Fig. 4).
Whether the enhanced fibrin deposition in
antibody-mediated TRALI resulted from enhanced coagulation,
inhibited anticoagulation, or impaired fibrinolysis was next
determined. Pulmonary TATc was significantly increased in the model
mice, indicating the activation of coagulation (Fig. 5A). Enhanced coagulation may
exacerbate pulmonary fibrin deposition (27). Additionally, tissue factor (TF)
initiates exogenous coagulation and serves an important role in
inflammation and lung injury (28).
As a natural inhibitor, TFPI was significantly elevated in the
TRALI group compared with that in the normal control group, whereas
there was no significant difference with the isotype control group
(Fig. 5B). PAI-1 is the main
modulator of fibrinolysis. The primary function of PAI-1 is to
inhibit plasminogen activator (PA) activity and thus prevent fibrin
degradation induced by plasmin. In the present study, the PAI-1
levels in the lungs were significantly higher in the TRALI group
than those in the other groups (Fig.
5C).
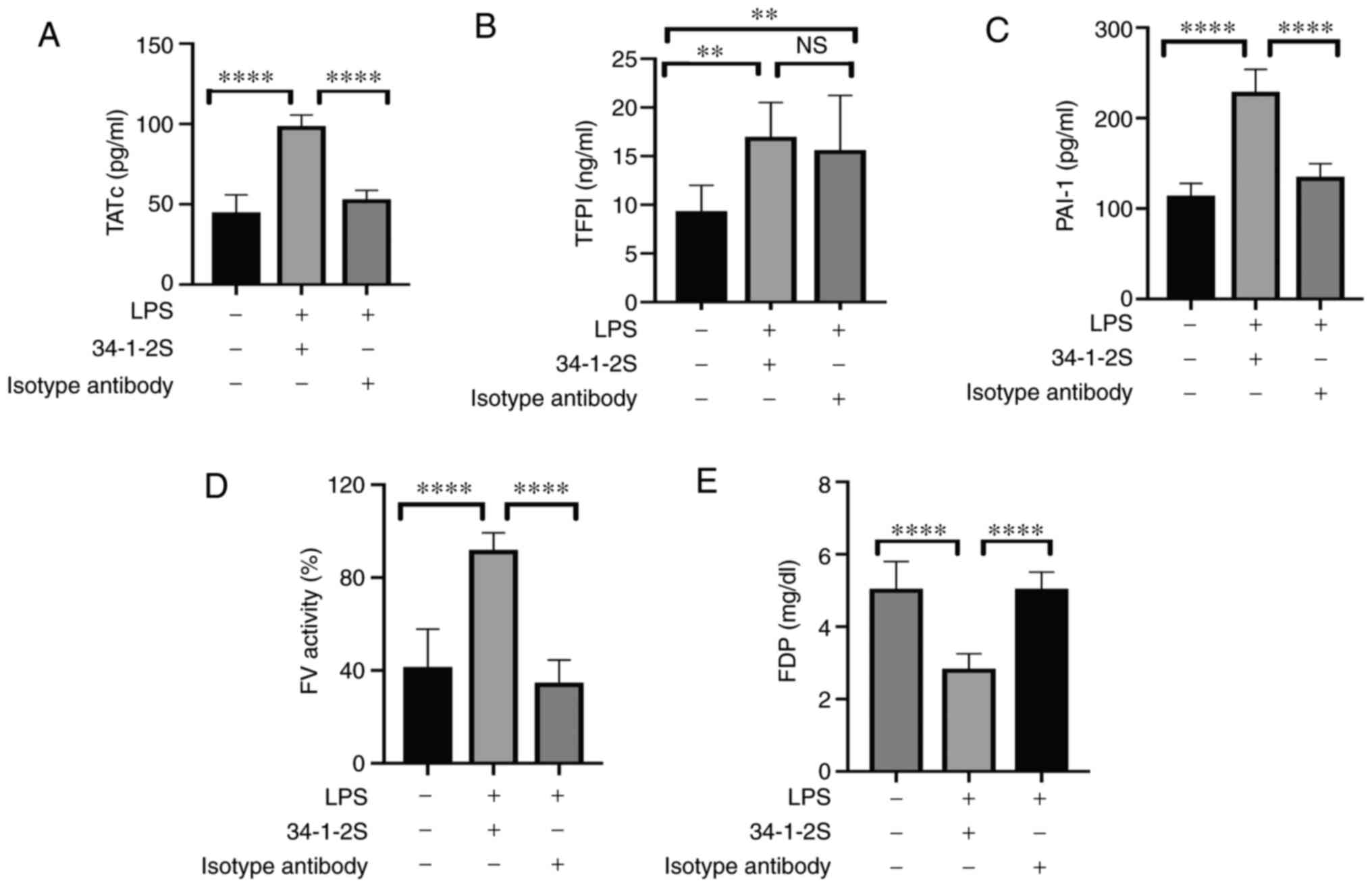 | Figure 5.Levels of TATc, TFPI, PAI-1, FV
activity and FDP in lung homogenates of the three treatment groups.
Levels of (A) TATc, (B) TFPI, (C) PAI-1, and (E) FDP as well as (D)
FV activity in lung homogenates. Data are shown as the mean ±
standard deviation (n=5–9 mice/group). **P<0.01, ****P<0.0001
and NS = no significance, by one-way ANOVA followed by Tukey's
multiple-comparison test. TATc, thrombin-antithrombin complex;
TFPI, tissue factor pathway inhibitor; PAI-1, plasminogen activator
inhibitor-1; FV, factor V; FDP, fibrin degradation product. |
Increased FV and FVII activities and FVIII and FIX
activities, indicate the activation of the extrinsic and intrinsic
coagulation pathways, respectively. Therefore, FV, FVII, FVIII and
FIX activity was measured using an automated device. As shown in
Fig. 5D, the activity of FV was
elevated in TRALI mice compared with that in control mice. However,
no differences in FVII, FVIII and FIX activities were observed
among the three groups (data not shown). In addition, FDP levels
were measured and were markedly decreased in the lungs of TRALI
mice compared with those in control mice (Fig. 5E). These results demonstrated that
pulmonary fibrinolysis was impaired in antibody-mediated TRALI
mice.
Antibody-mediated TRALI induces
thrombocytopenia and pulmonary platelet accumulation
Thrombocytopenia is a clinical manifestation of
TRALI (29,30). As with a previous study (2), mild thrombocytopenia was observed in
the model mice (Fig. 6A).
Additionally, platelets are capable to trigger coagulation
(31). Thus, platelet accumulation
in the lungs was investigated by quantitative immunofluorescence
analysis of the platelet-specific marker CD41a. Although platelet
staining was observed in the untreated and isotype control mouse
lungs, platelet aggregation was enhanced in TRALI mouse lungs
(Fig. 6B and C).
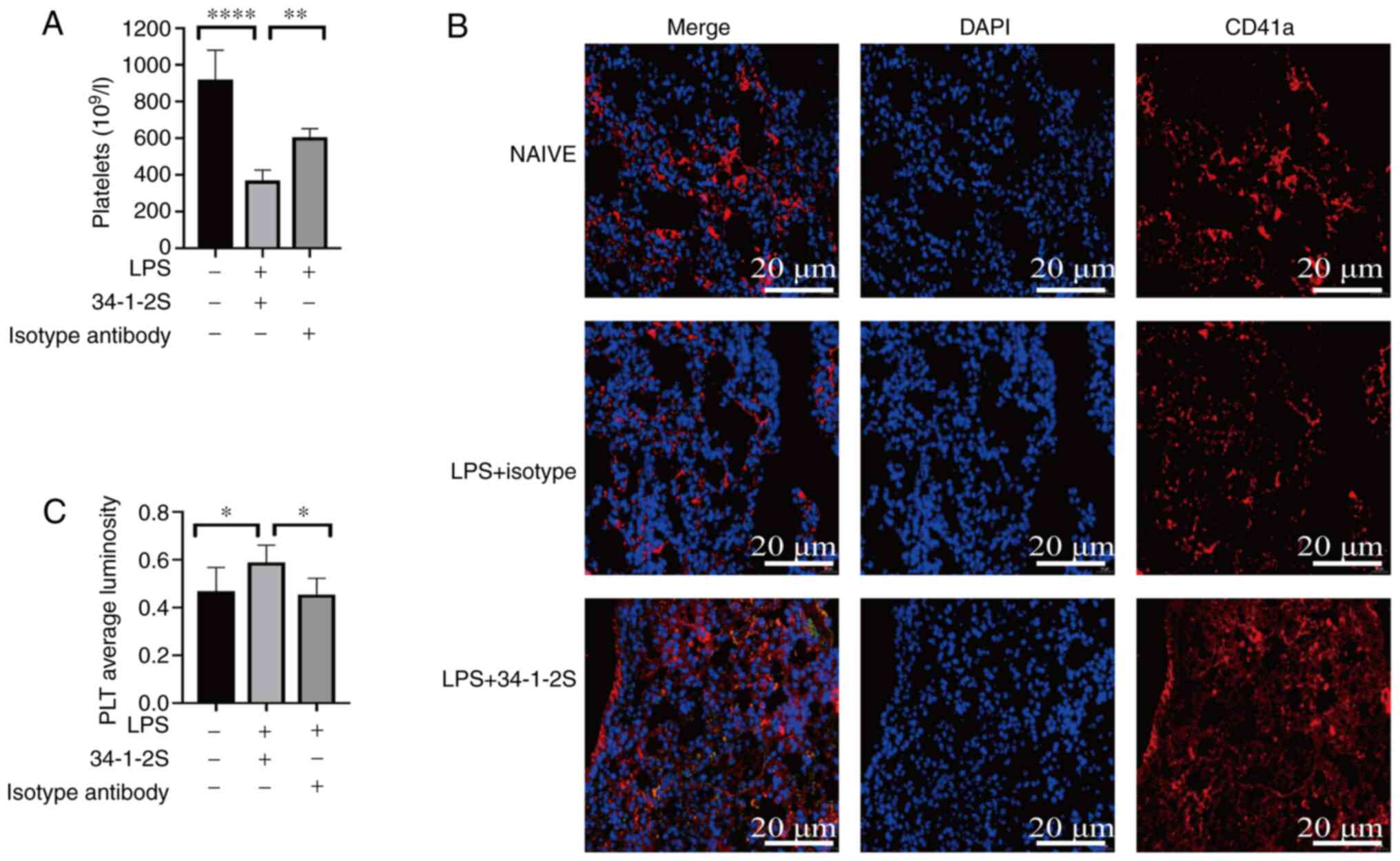 | Figure 6.Antibody-mediated TRALI reduces
platelets in the peripheral blood and platelets accumulation in the
lungs. (A) Numbers of peripheral blood platelets in untreated
control, isotype control and TRALI mice. (B) Representative
immunostaining for CD41a in untreated, isotype control and TRALI
mice. Red, CD41a; blue, DAPI. Scale bar, 20 µm; original
magnification, ×400. (C) The average CD41a immunofluorescence
luminosity was determined using Image-Pro Plus 6.0 software. A
total of six replicates were included in each group and the most
representative images are shown. Data are shown as the mean ±
standard deviation (n=6 mice/group). *P<0.05, **P<0.01,
****P<0.0001. One-way ANOVA variance of peripheral blood
platelets and pulmonary platelets followed by Tukey's
multiple-comparison test and Newman-Keuls multiple-comparison test,
respectively. TRALI, transfusion-related acute lung injury; PLT,
platelet. |
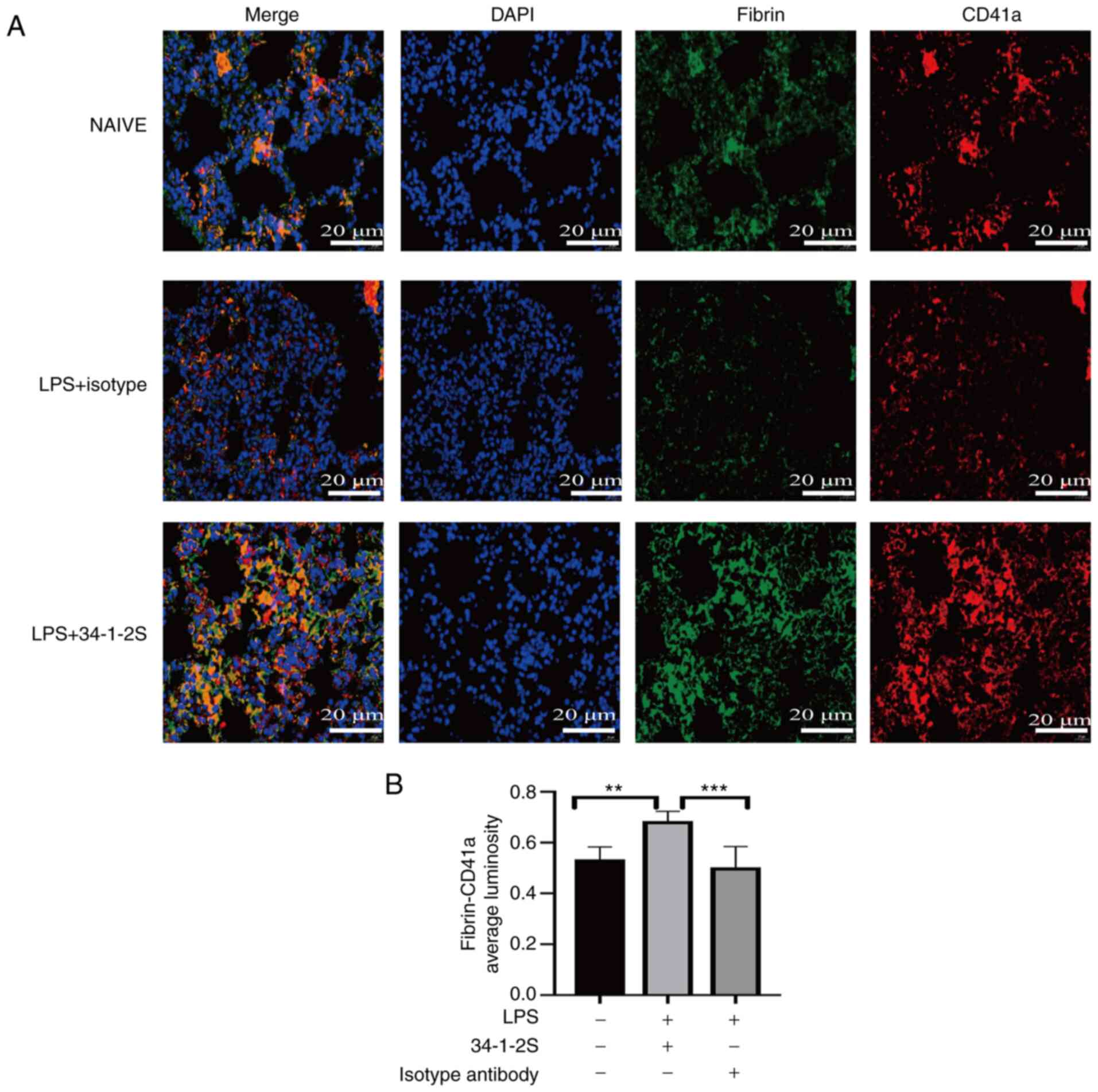
Antibody-mediated TRALI enhances
pulmonary fibrin-platelet interactions
Fibrin, which exerts a proinflammatory effect,
drives platelet accumulation in injured lungs, which in turn can
amplify the injury (32). In
addition, platelets bind fibrin, thus promoting thrombin generation
(33). Therefore, the
fibrin-platelet interactions in the lungs was measured via
immunofluorescence. The results showed that these interactions were
increased in antibody-mediated TRALI mice compared with those in
control mice (Fig. 7).
Discussion
An antibody-mediated TRALI mouse model was
successfully established by challenging the animals with LPS plus
an anti-MHC-I antibody. Preexisting clinical risk factors that
confer an inflammatory state appear to mediate a first hit
(34). In TRALI patients, the
proportions of antibodies against human leukocyte antigen and human
neutrophil antigen are ~50 and 30%, respectively and these
antibodies serve as the second hit (35–37).
Overall, the two-hit mechanism underlying disease pathology in the
antibody-mediated TRALI mouse model is similar to the pathogenesis
of TRALI in patients and thus, the model is representative of human
disease. Antibody-mediated TRALI is the major phenotype of clinical
TRALI; it accounts for ~80% of TRALI cases. In the present study,
the main characteristics of ALI were confirmed by histological
evidence, inflammatory responses and alterations to the
alveolar-capillary barrier. Significant fibrin accumulation was
observed in TRALI lungs, which was associated with enhanced
coagulation and impaired fibrinolysis. Furthermore, platelet
sequestration was observed in the TRALI lungs, which might have
been caused by fibrin capturing platelets. Additionally, the
enhanced coagulation could be related to the binding of platelets
to the polymerized fibrin, pulmonary platelet accumulation and
enhanced FV activity. Indeed, both increased PAI-1 and decreased
FDP in the lungs of the TRALI group indicated that fibrinolysis was
inhibited.
Fibrin accumulation in the lungs can induce
inflammatory injury, create a proinflammatory environment, reduce
gas exchange and aggravate hypoxemia (9,38,39).
Corticosteroids ameliorate smoke inhalation-induced ALI by
attenuating fibrin deposition (40). Furthermore, PAI-1 deficiency
markedly suppresses pulmonary fibrin accumulation and ameliorates
alcohol-enhanced ALI (9). Chen
et al (41) showed that
inhaled NO mitigates endotoxin-induced ALI by suppressing elevated
PAI-1 and fibrin deposition. Khalaj et al (16) reported that extracellular
vesicle-based therapies can alleviate severe pulmonary conditions
by reducing fibrin production. Thus, reduced fibrin deposition in
the lungs seems to hold therapeutic potential for lung injury.
However, the therapeutic effects of fibrin-lowering drugs in TRALI
remain to be elucidated.
In addition to enhanced inflammatory injury, fibrin
provides a chemotactic substrate for blood cells, including
platelets and leukocytes (42).
Platelet integrin αIIbβ3 is considered an important functional
receptor for fibrin (33). In
addition, fibrin-platelet interactions serve a pathogenic role in
lung injury. Activated platelets trigger the secretion of adenosine
59-diphosphate and thromboxane A2, which activate integrin αIIbβ3
on platelets, facilitating their binding to fibrin and expanding
the thrombus (33). β3 deficiency
impairs the capacity of platelets to bind fibrin, thus preventing
alcohol-induced ALI (9). Smyth
et al (43) demonstrated
that in a model of microvascular thrombosis, β3-null mice exhibited
less fibrin and thrombi in the lungs than wild-type mice. These
results indicated that integrin αIIbβ3 on platelets is pivotal for
fibrin binding and serves a pathogenic role. Indeed, the present
study observed enhanced platelet binding to fibrin in the
antibody-mediated TRALI model. However, no evident thrombosis was
observed in the model (data not shown). Of note, fibrin-platelet
binding promotes platelet aggregation (9). Upon endothelial injury, platelets can
be recruited, activated and aggregated via integrin αIIbβ3 binding
to fibrin (44). Then, platelets
that adhere to the lung endothelium exacerbate lung injury by
releasing potent proinflammatory molecules and mediators that
increase the expression of chemokines and vascular adhesion
molecules in endothelial cells (9,44).
Pulmonary platelet activation and aggregation reportedly serve
pathogenic roles in influenza A-induced pneumonia (44), acid-induced ALI (45), endotoxemia (46) and TRALI (2) and inhibition of platelet function
protects against these diseases. Looney et al (2) demonstrated that platelet sequestration
following 34-1-2S challenge was associated with the sequestration
of platelet-capturing neutrophils in the lungs. Adhesive
interactions between neutrophils and platelets are mediated by
platelet glycoprotein Ibα and P-selectin binding of glycoprotein
ligand-1 and multifunctional leukocyte integrin expressed on
leukocytes, respectively (47). Of
note, the present study observed increased fibrin deposition in the
lungs of TRALI model mice, which could capture platelets and result
in platelet sequestration in the lungs. Thrombocytopenia is a
common symptom of ARDS and is associated with poor prognosis
(48,49). Preclinical models and clinical
observations have both shown that thrombocytopenia is a pathogenic
manifestation of TRALI (2). In line
with previous studies, the present study overserved peripheral
blood thrombocytopenia in antibody-mediated TRALI mice. Looney
et al (2) demonstrated that
thrombocytopenia was related to dynamic neutrophil changes in mouse
antibody-mediated TRALI model. It would be worthwhile to
investigate whether the decrease in platelets in the systemic
circulation of antibody-mediated TRALI is associated with pulmonary
fibrin deposition.
Coagulation imbalance commonly occurs in pulmonary
disorders and is often accompanied with hypercoagulable conditions,
even resulting in pulmonary embolism (12). The present study observed enhanced
fibrin deposition in TRALI mice and detected the levels of TATc,
TFPI and PAI-1 in the lungs, which are indicators of coagulation,
anticoagulation and fibrinolysis, respectively. A hypercoagulable
state, enhanced anticoagulation and impaired fibrinolysis was
detected in the TRALI group. TATc is an important marker of
thrombin and its increased levels indicate a hypercoagulable state.
Mammadova-Bach et al (33)
reported that the binding of platelet glycoprotein VI (GPVI) to
fibrin promotes thrombin generation. In the current study,
increased fibrin-platelet binding might have elevated the levels of
TATc to some degree, resulting in the hypercoagulable state in
TRALI mice. In addition to GPVI, integrin αIIbβ3 also mediates
fibrin-platelet interaction (33).
Whether integrin αIIbβ3 mediating fibrin-platelet interaction and
triggering thrombin generation in TRALI mice need further
validation is for future research. The increased extrinsic
coagulation FV activity also indicated an increase in pulmonary
procoagulant activity. Activated platelets offer a procoagulant
surface to coagulation factors, resulting in thrombin production
(50). In the present study,
pulmonary platelet accumulation could have promoted pulmonary
thrombin generation by providing a procoagulant surface in TRALI
mice. Lung tissue is rich in TF and FVII (51). TFPI inhibits thrombin production and
activation by inactivating TF-VII and TF-X conjugate activity
(52). Of note, TFPI was increased
in the antibody-mediated TRALI group compared with the untreated
control group, but not the isotype control group. This result
implied that anticoagulation activity increases in the presence of
a first hit. Elevated TFPI might diminish FVII and FX procoagulant
activities, which could explain why no changes in these activities
were observed (data not shown). Although TFPI was able to inhibit
thrombin production and activation, the elevated TATc levels
indicated that the increase in TFPI was insufficient to control
coagulation. PAI-1 facilitates the stable clot formation following
injury and participates in posttraumatic coagulopathy (53). FDP originates from fibrin
degradation and elevated levels are a manifestation of fibrinolysis
(54). In the present study,
increased levels of PAI-1 combined with decreased FDP in the lungs
following 34-1-2S challenge indicated the inhibition of
fibrinolysis. In addition to PAI-1, fibrinolytic system also
includes urokinase-type PA, tissue-type PA, plasminogen and
plasmin. Continuing experiments are investigating the expression
levels of aforementioned fibrinolytic system components and explain
fibrinolysis disorder in detail. In non-antibody-mediated TRALI,
the PAI-1 level in bronchoalveolar lavage fluid was elevated,
whereas the FDP level was not significantly altered (20). This discrepancy could be explained
by differences in the animal species and disease type. In a model
of aged erythrocyte transfusion-mediated non-antibody TRALI,
increased coagulation and decreased fibrinolysis are observed
(20). However, coagulopathy might
be induced by aged erythrocytes, which exhibit procoagulant
activity by enhancing thrombin generation. The present study
extended previous findings by confirming, for the first time to the
best of the authors' knowledge, that increased coagulation and
anticoagulation, as well as impaired fibrinolysis, would enhance
fibrin deposition in the lungs and result in platelet accumulation
in antibody-mediated TRALI.
Antibody-mediated TRALI is the primary clinical
phenotype of TRALI. The findings of enhanced coagulation, increased
anticoagulation and impaired fibrinolysis expand and deepen our
knowledge of TRALI. However, further research on the complex
mechanism of the coagulation-anticoagulation-fibrinolytic system
with respect to TRALI are needed. The present study determined the
levels of TATc, TFPI, PAI-1, FDP and coagulation factor activity in
the lungs instead of in plasma following antibody-mediated TRALI
establishment. Nevertheless, assessing these factors in the lungs
reveals the pathogenesis in the lungs and is more reliable than
assessing systemic levels. The present study discovered that a
local coagulation-fibrinolysis imbalance was associated with
pulmonary fibrin deposition in antibody-mediated TRALI. The data
provide a therapeutic rationale and indicate the feasibility of
strategies based on modulating abnormalities in either the
coagulation or the fibrinolysis pathway for antibody-mediated
TRALI.
Acknowledgements
Not applicable.
Funding
The present study was funded by the National Natural
Science Foundation of China (grant no. 31801192) and CAMS
Innovation Fund for Medical Sciences (grant no.
2016-12M-3-024).
Availability of data and materials
The datasets used and analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YY designed the study. YY, PJ, PS and NS performed
the experiments. FL collected, analyzed and interpreted the data.
YY drafted the manuscript and prepared the figures. PJ edited the
manuscript and the figures. FL edited the manuscript and provided
financial resources. YY, PJ and FL confirmed the authenticity of
all the raw data. All authors reviewed and approved the final
manuscript.
Ethics approval and consent to
participate
The animal studies were conducted following the
Ethics Committee approval of the Institute of Blood Transfusion,
Chinese Academy of Medical Science and Peking Union Medical College
(approval no. 201934).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Semple JW, Rebetz J and Kapur R:
Transfusion-associated circulatory overload and transfusion-related
acute lung injury. Blood. 133:1840–1853. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Looney MR, Nguyen JX, Hu Y, Van Ziffle JA,
Lowell CA and Matthay MA: Platelet depletion and aspirin treatment
protect mice in a two-event model of transfusion-related acute lung
injury. J Clin Invest. 119:3450–3461. 2009.PubMed/NCBI
|
|
3
|
Hu A, Chen W, Wu S, Pan B, Zhu A, Yu X and
Huang Y: An animal model of transfusion-related acute lung injury
and the role of soluble CD40 ligand. Vox Sang. 115:303–313. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang L, Wu T, Yan S, Wang Y, An J, Wu C,
Zhang Y, Ma Y, Fu Q, Wang D, et al: M1-polarized alveolar
macrophages are crucial in a mouse model of transfusion-related
acute lung injury. Transfusion. 60:303–316. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sebag SC, Bastarache JA and Ware LB:
Therapeutic modulation of coagulation and fibrinolysis in acute
lung injury and the acute respiratory distress syndrome. Curr Pharm
Biotechnol. 12:1481–1496. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ware LB and Matthay MA: The acute
respiratory distress syndrome. N Engl J Med. 342:1334–1349. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
De Michele S, Sun Y, Yilmaz MM, Katsyv I,
Salvatore M, Dzierba AL, Marboe CC, Brodie D, Patel NM, Garcia CK,
et al: Forty Postmortem Examinations in COVID-19 Patients. Am J
Clin Pathol. 154:748–760. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Johnson S, Shaikh SB, Muneesa F, Rashmi B
and Bhandary YP: Radiation induced apoptosis and pulmonary
fibrosis: Curcumin an effective intervention? Int J Radiat Biol.
96:709–717. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Poole LG, Massey VL, Siow DL,
Torres-Gonzáles E, Warner NL, Luyendyk JP, Ritzenthaler JD, Roman J
and Arteel GE: Plasminogen activator inhibitor-1 is critical in
alcohol-enhanced acute lung injury in mice. Am J Respir Cell Mol
Biol. 57:315–323. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yasui H, Donahue DL, Walsh M, Castellino
FJ and Ploplis VA: Early coagulation events induce acute lung
injury in a rat model of blunt traumatic brain injury. Am J Physiol
Lung Cell Mol Physiol. 311:L74–L86. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vadász I, Morty RE, Olschewski A,
Königshoff M, Kohstall MG, Ghofrani HA, Grimminger F and Seeger W:
Thrombin impairs alveolar fluid clearance by promoting endocytosis
of Na+,K+-ATPase. Am J Respir Cell Mol Biol.
33:343–354. 2005. View Article : Google Scholar
|
|
12
|
Mitchell WB: Thromboinflammation in
COVID-19 acute lung injury. Paediatr Respir Rev. 35:20–24.
2020.PubMed/NCBI
|
|
13
|
Tuinman PR, Schultz MJ and Juffermans NP:
Coagulopathy as a therapeutic target for TRALI: Rationale and
possible sites of action. Curr Pharm Des. 18:3267–3272. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Idell S, James KK, Levin EG, Schwartz BS,
Manchanda N, Maunder RJ, Martin TR, McLarty J and Fair DS: Local
abnormalities in coagulation and fibrinolytic pathways predispose
to alveolar fibrin deposition in the adult respiratory distress
syndrome. J Clin Invest. 84:695–705. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tian LQ, Guo ZH, Meng WZ, Li L, Zhang Y,
Yin XH, Lai F, Li YY, Feng LL, Shen FF, et al: The abnormalities of
coagulation and fibrinolysis in acute lung injury caused by gas
explosion. Kaohsiung J Med Sci. 36:929–936. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Khalaj K, Figueira RL, Antounians L,
Lauriti G and Zani A: Systematic review of extracellular
vesicle-based treatments for lung injury: Are EVs a potential
therapy for COVID-19? J Extracell Vesicles. 9:17953652020.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lan CC, Peng CK, Huang SF, Huang KL and Wu
CP: Activated protein C attenuates ischemia-reperfusion-induced
acute lung injury. Exp Lung Res. 41:241–250. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lin L, Lu L, Cao W and Li T: Hypothesis
for potential pathogenesis of SARS-CoV-2 infection - a review of
immune changes in patients with viral pneumonia. Emerg Microbes
Infect. 9:727–732. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu C, Ma Y, Su Z, Zhao R, Zhao X, Nie HG,
Xu P, Zhu L, Zhang M, Li X, et al: Meta-Analysis of Preclinical
Studies of Fibrinolytic Therapy for Acute Lung Injury. Front
Immunol. 9:18982018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vlaar AP, Hofstra JJ, Levi M, Kulik W,
Nieuwland R, Tool AT, Schultz MJ, de Korte D and Juffermans NP:
Supernatant of aged erythrocytes causes lung inflammation and
coagulopathy in a ‘two-hit’ in vivo syngeneic transfusion model.
Anesthesiology. 113:92–103. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tuinman PR, Vlaar AP, Cornet AD, Hofstra
JJ, Levi M, Meijers JC, Beishuizen A, Schultz MJ, Groeneveld AJ and
Juffermans NP: Blood transfusion during cardiac surgery is
associated with inflammation and coagulation in the lung: A case
control study. Crit Care. 15:R592011. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kapur R, Kasetty G, Rebetz J, Egesten A
and Semple JW: Osteopontin mediates murine transfusion-related
acute lung injury via stimulation of pulmonary neutrophil
accumulation. Blood. 134:74–84. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Khoy K, Nguyen MVC, Masson D, Bardy B,
Drouet C and Paclet MH: Transfusion-related acute lung injury:
Critical neutrophil activation by anti-HLA-A2 antibodies for
endothelial permeability. Transfusion. 57:1699–1708. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Qiao J, He R, Yin Y, Tian L, Li L, Lian Z,
Fang P and Liu Z: rIL-35 prevents murine transfusion-related acute
lung injury by inhibiting the activation of endothelial cells.
Transfusion. 60:1434–1442. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
He R, Li L, Kong Y, Tian L, Tian X, Fang
P, Bian M and Liu Z: Preventing murine transfusion-related acute
lung injury by expansion of CD4+ CD25+
FoxP3+ Tregs using IL-2/anti-IL-2 complexes.
Transfusion. 59:534–544. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Glas GJ, Van Der Sluijs KF, Schultz MJ,
Hofstra JJ, Van Der Poll T and Levi M: Bronchoalveolar hemostasis
in lung injury and acute respiratory distress syndrome. J Thromb
Haemost. 11:17–25. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chaudhry R, Usama SM and Babiker HM:
Physiology, Coagulation Pathways. In: StatPearls StatPearls
Publishing Copyright© 2021. StatPearls Publishing LLC;
Treasure Island, FL: 2021
|
|
28
|
van der Poll T: Tissue factor as an
initiator of coagulation and inflammation in the lung. Crit Care.
12 (Suppl 6):S32008. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yomtovian R, Kline W, Press C, Clay M,
Engman H, Hammerschmidt D and McCullough J: Severe pulmonary
hypersensitivity associated with passive transfusion of a
neutrophil-specific antibody. Lancet. 1:244–246. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Leger R, Palm S, Wulf H, Vosberg A and
Neppert J: Transfusion-related lung injury with leukopenic reaction
caused by fresh frozen plasma containing anti-NB1. Anesthesiology.
91:1529–1532. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li J, Tong D, Chen F, Song B, Wang Y, Liu
Y, Zhang X, Liu N, Xu Y, Li Y, et al: Inflammatory cytokines
enhance procoagulant activity of platelets and endothelial cells
through phosphatidylserine exposure in patients with essential
hypertension. J Thromb Thrombolysis. 51:933–940. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zarbock A and Ley K: The role of platelets
in acute lung injury (ALI). Front Biosci. 14:150–158. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mammadova-Bach E, Ollivier V, Loyau S,
Schaff M, Dumont B, Favier R, Freyburger G, Latger-Cannard V,
Nieswandt B, Gachet C, et al: Platelet glycoprotein VI binds to
polymerized fibrin and promotes thrombin generation. Blood.
126:683–691. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kapur R, Kim M, Aslam R, McVey MJ, Tabuchi
A, Luo A, Liu J, Li Y, Shanmugabhavananthan S, Speck ER, et al: T
regulatory cells and dendritic cells protect against
transfusion-related acute lung injury via IL-10. Blood.
129:2557–2569. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Peters AL, Van Stein D and Vlaar AP:
Antibody-mediated transfusion-related acute lung injury; from
discovery to prevention. Br J Haematol. 170:597–614. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kosmaczewska A: Low-dose interleukin-2
therapy: A driver of an imbalance between immune tolerance and
autoimmunity. Int J Mol Sci. 15:18574–18592. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bayat B, Tjahjono Y, Sydykov A, Werth S,
Hippenstiel S, Weissmann N, Sachs UJ and Santoso S: Anti-human
neutrophil antigen-3a induced transfusion-related acute lung injury
in mice by direct disturbance of lung endothelial cells.
Arterioscler Thromb Vasc Biol. 33:2538–2548. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gallelli L, Zhang L, Wang T and Fu F:
Severe acute lung injury related to COVID-19 infection: A review
and the possible role for Escin. J Clin Pharmacol. 60:815–825.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lau CL, Zhao Y, Kim J, Kron IL, Sharma A,
Yang Z, Laubach VE, Linden J, Ailawadi G and Pinsky DJ: Enhanced
fibrinolysis protects against lung ischemia-reperfusion injury. J
Thorac Cardiovasc Surg. 137:1241–1248. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Song LC, Chen XX, Meng JG, Hu M, Huan JB,
Wu J, Xiao K, Han ZH and Xie LX: Effects of different
corticosteroid doses and durations on smoke inhalation-induced
acute lung injury and pulmonary fibrosis in the rat. Int
Immunopharmacol. 71:392–403. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen Y, Lu ZJ, Yang Y, Lu GP, Chen WM and
Zhang LE: Suppression of plasminogen activator inhibitor-1 by
inhaled nitric oxide attenuates the adverse effects of hyperoxia in
a rat model of acute lung injury. Thromb Res. 136:131–138. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Perez RL and Roman J: Fibrin enhances the
expression of IL-1 beta by human peripheral blood mononuclear
cells. Implications in pulmonary inflammation. J Immunol.
154:1879–1887. 1995.PubMed/NCBI
|
|
43
|
Smyth SS, Reis ED, Väänänen H, Zhang W and
Coller BS: Variable protection of beta 3-integrin--deficient mice
from thrombosis initiated by different mechanisms. Blood.
98:1055–1062. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lê VB, Schneider JG, Boergeling Y, Berri
F, Ducatez M, Guerin JL, Adrian I, Errazuriz-Cerda E, Frasquilho S,
Antunes L, et al: Platelet activation and aggregation promote lung
inflammation and influenza virus pathogenesis. Am J Respir Crit
Care Med. 191:804–819. 2015. View Article : Google Scholar
|
|
45
|
Zarbock A, Singbartl K and Ley K: Complete
reversal of acid-induced acute lung injury by blocking of
platelet-neutrophil aggregation. J Clin Invest. 116:3211–3219.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kiefmann R, Heckel K, Schenkat S, Dörger M
and Goetz AE: Role of p-selectin in platelet sequestration in
pulmonary capillaries during endotoxemia. J Vasc Res. 43:473–481.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Simon DI, Chen Z, Xu H, Li CQ, Dong J,
McIntire LV, Ballantyne CM, Zhang L, Furman MI, Berndt MC, et al:
Platelet glycoprotein ibalpha is a counterreceptor for the
leukocyte integrin Mac-1 (CD11b/CD18). J Exp Med. 192:193–204.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Greinacher A and Selleng S: How I evaluate
and treat thrombocytopenia in the intensive care unit patient.
Blood. 128:3032–3042. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wei Y, Tejera P, Wang Z, Zhang R, Chen F,
Su L, Lin X, Bajwa EK, Thompson BT and Christiani DC: A missense
genetic variant in LRRC16A/CARMIL1 improves acute respiratory
distress syndrome survival by attenuating platelet count decline.
Am J Respir Crit Care Med. 195:1353–1361. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Tohidi-Esfahani I, Lee CS, Liang HP and
Chen VM: Procoagulant platelets: Laboratory detection and clinical
significance. Int J Lab Hematol. 42 (Suppl 1):59–67. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Idell S, Peters J, James KK, Fair DS and
Coalson JJ: Local abnormalities of coagulation and fibrinolytic
pathways that promote alveolar fibrin deposition in the lungs of
baboons with diffuse alveolar damage. J Clin Invest. 84:181–193.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wood JP, Ellery PE, Maroney SA and Mast
AE: Biology of tissue factor pathway inhibitor. Blood.
123:2934–2943. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Griemert EV, Schwarzmaier SM, Hummel R,
Gölz C, Yang D, Neuhaus W, Burek M, Förster CY, Petkovic I, Trabold
R, et al: Plasminogen activator inhibitor-1 augments damage by
impairing fibrinolysis after traumatic brain injury. Ann Neurol.
85:667–680. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wang L, He WB, Yu XM, Hu DL and Jiang H:
Prolonged prothrombin time at admission predicts poor clinical
outcome in COVID-19 patients. World J Clin Cases. 8:4370–4379.
2020. View Article : Google Scholar : PubMed/NCBI
|