Introduction
Cardiovascular complications due to cancer therapy
cause a significant reduction in treatment efficacy for patients
with cancer (1). Doxorubicin (DOX)
is a potent anthracycline that has been confirmed to cause
long-term cardiovascular side effects and decrease the quality of
life of cancer survivors, which eventually reduces the clinical
application of DOX (2). Among the
diverse mechanisms involved in the cardiotoxicity of DOX, the
contribution of ferroptosis has been recently reported by an
increasing number of studies (3–5).
Ferroptosis is a novel type of regulated cell death
that is different from apoptosis and necrosis. It is an iron- and
reactive oxygen species (ROS)-dependent cell death, characterized
by the accumulation of ROS and inactivation of the cellular
antioxidant glutathione (GSH), leading to redox dysregulation
(6). Ferroptosis has been
implicated in the pathological processes associated with
carcinogenesis, degenerative diseases, stroke and kidney
ischemia/reperfusion injury (7).
Recently, ferroptosis was shown to exhibit a crucial role in
DOX-induced cardiotoxicity. For instance, it has been documented
that ferroptosis promoted DOX-induced cardiomyopathy and mortality
in mice, whereas the ferroptosis inhibitor ferrostatin-1 and iron
chelation ameliorate this process and effectively improve the
survival rate of mice (4). In
addition, mitochondrial ferroptosis has been shown to be a major
cause of DOX-induced cardiotoxicity (2). It has also been reported that
overexpression of GSH peroxidase 4 (GPX4) ameliorated cardiac
impairment in mice, whereas GPX4 knockdown exacerbated this process
(3). Therefore, inhibition of
ferroptosis may effectively reduce DOX-induced cardiotoxicity.
Autotaxin (ATX) is a secreted enzyme encoded by the
ectonucleotide pyrophosphatase/phosphodiesterase 2 (ENPP2) gene,
which is important for generating lysophosphatidic acid (LPA). The
ATX/LPA pathway serves a major role in embryonic, vascular and
neuronal development (8).
Disturbances in normal ATX/LPA signaling are associated with the
development of multiple diseases, including cardiovascular disease
(9). A previous study reported that
ENPP2/LPA protected cardiomyocytes from erastin-induced
ferroptosis, indicating the inhibitory effect of ENPP2 on
ferroptosis induction in cardiomyocytes (10). The transcription factor FoxO4, which
is a member of the FoxO transcription factor family, is predicted
to bind to the promoter of ENPP2. The FoxO proteins are involved in
a variety of biological processes, including cell proliferation,
oxidative stress response, metabolism, immunity and apoptosis
(11). It has been suggested that
FoxO4 may be involved in aggravating cardiovascular diseases
(12). FoxO4 was also reported to
regulate DOX-induced toxicity in liver and kidney cells both in
vivo and in vitro (13).
In the present study, it was hypothesized that FoxO4 may be a
transcriptional regulator of ENPP2 by inhibiting ferroptosis, which
in turn can protect cardiomyocyte H9c2 cells against DOX-induced
injury.
Materials and methods
Cell culture and treatment
H9c2 rat cardiomyocytes were purchased from The Cell
Bank of Type Culture Collection of The Chinese Academy of Sciences
and cultured in DMEM (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.)
and 1% penicillin-streptomycin (Gibco; Thermo Fisher Scientific,
Inc.). The cells were incubated at 37°C in a humidified atmosphere
containing 5% CO2. The cells were exposed to 0.625,
1.25, 2.5, 5 or 10 µM DOX (Sigma-Aldrich; Merck KGaA) at 37°C for
12, 24 or 48 h to detect cell viability. Cells were treated with
0.625, 1.25, 2.5, 5 or 10 µM DOX (Sigma-Aldrich; Merck KGaA) for 24
h at 37°C or with 5 µM DOX for 12, 24 and 48 h at 37°C to determine
the mRNA and protein expression of ENPP2. In subsequent
experiments, 5 µM DOX was used for 24 h to treat H9c2 cells.
Cell transfection
ENPP2 and FoxO4 were overexpressed in H9c2 cells to
produce overexpressing (Oe)-ENPP2 and Oe-FoxO4 cell lines. The rat
ENPP2 and FoxO4 cDNA sequences were synthesized by GenScript and
inserted into the pcDNA3.1 vector (Invitrogen; Thermo Fisher
Scientific, Inc.). An empty pcDNA3.1 vector was used as the
negative control (NC). H9c2 cells were seeded into 6-well plates at
the density of 4×105 cells/well 10 h prior to
transfection, and the vectors (2.5 µg) were transfected into cells
using Lipofectamine® 2000 reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions, at 37°C for 48 h. At 48 h post-transfection, the
cells were exposed to 5 µM DOX for 24 h at 37°C and collected for
subsequent analysis. Control cells were devoid of treatment.
Cell viability assessment
Cell viability was determined using the MTT assay
kit (Beyotime Institute of Biotechnology). Briefly, control or
transfected H9c2 cells were seeded into 96-well plates at the
density of 2×103 cells/well. Following treatment with
DOX for 12, 24 or 48 h, the culture medium was replaced with DMEM
without DOX and then added to 10 µl MTT solution at 37°C for 4 h.
The cells were incubated with formazan lysis solution at room
temperature for 10 min until the purple crystals were completely
dissolved. Finally, the absorbance was measured at 570 nm using a
spectrophotometer.
Oxidative stress measurement
To detect cellular total ROS status, the
2×103 cells/well were seeded into 96-well plates and
stained with 2′,7′-dichlorodihydrofluorescein diacetate (Abcam) at
37°C for 45 min. Fluorescence was measured at excitation and
emission wavelengths of 485 and 535 nm using a multimode plate
reader (PerkinElmer, Inc.).
The increase in ROS levels was also examined using a
commercially available thiobarbituric acid reactive substances
(TBARS) assay kit (cat. no. 700870; Cayman Chemical Company)
according to the manufacturer's instructions. GSH peroxidase
(GSH-Px) activity was measured using a GSH-PX assay kit (cat. no.
A005-1-2; Nanjing Jiancheng Bioengineering Institute) in accordance
with the manufacturer's instructions.
Measurement of Fe2+
activity
Cellular Fe2+ activity was measured using
the Iron Assay kit (Abcam) according to the manufacturer's
instructions. Briefly, control or transfected H9c2 cells were
seeded into 96-well plates at the density of 1×103
cells/well. Following treatment with 5 µM DOX for 24 h at 37°C, 5
µl assay buffer was added to each sample and incubated at 37°C for
30 min. Following addition of 100 µl Iron Probe to each well and
incubation at 37°C for 60 min in the dark, the optical density (593
nm) was measured using a colorimetric microplate reader.
Dual-luciferase reporter assay
The JASPAR database (http://jaspar.genereg.net/) was searched and FoxO4 was
identified as a potential target that could bind to the promoter of
ENPP2. To confirm the interaction between FoxO4 and the ENPP2
promoter, a dual-luciferase reporter assay was performed. Wild-type
or mutant ENPP2 E1 and E2 sequences were amplified and cloned
downstream of the luciferase reporter gene in pMIR-REPORT
luciferase vectors (Thermo Fisher Scientific, Inc.). Subsequently,
they were co-transfected with pcDNA3.1-FoxO4 (Oe-FoxO4; 2.5 µg) or
Oe-NC (2.5 µg) using Lipofectamine 2000 reagent. At 48 h
post-transfection, luciferase activity was measured with a
dual-luciferase reporter system (Promega Corporation).
Renilla luciferase activity was used for normalization.
Chromatin immunoprecipitation (ChIP)
assay
H9c2 cells were crosslinked with 1% formaldehyde for
10 min at 37°C and a ChIP assay was performed with a
high-sensitivity kit (Abcam). Cells were washed with cold PBS and
suspended in 1 ml PBS containing 5 µl protease inhibitors. Cells
were centrifuged at 4°C at 716 × g for 5 min. A total of 300 µl SDS
lysis buffer (1% SDS, 10 mM EDTA and 50 mM Tris-HCl pH 8.0) was
then used to lyse the cells, which were subsequently sonicated at
150 Hz and sheared with four sets of 10 sec pulses on wet ice using
a high intensity ultrasonic processor. An equal amount of chromatin
(100 µl) was immunoprecipitated at 4°C overnight. The antibodies
used in this assay included anti-FoxO4 (cat. no. ab128908; 1:50;
Abcam) and IgG (as the NC; cat. no. ab172730; 1:50; Abcam).
Immunoprecipitated products were collected after incubation with
magnetic beads coupled with anti-IgG or anti-FoxO4. The beads were
washed using a magnetic separation rack and the bound chromatin was
eluted in ChIP Elution Buffer with Proteinase K mixer. The primer
sequences used for ENPP2 detection were as follows: Forward
5′-TTCCAATGTACCCCGCCTTC-3′ and reverse 5′-AGCTGCCTTCCACATACTGTT-3′.
The recovered DNA fragments were evaluated via RT-qPCR. The
relative level of ENPP2 was normalized according to the average
level of the IgG group.
Lipid ROS measurement
Lipid ROS generation was measured by adding
C11-BODIPY (Invitrogen; Thermo Fisher Scientific, Inc.) at a final
concentration of 1.5 µM for 20 min at 37°C before cell harvesting,
according to the manufacturer's instructions. Lipid ROS-positive
cells were finally assessed using a FACSCanto II flow cytometer (BD
Biosciences). The lipid ROS can be differentiated by adding
C11-BODIPY, which is a lipid soluble, ratio type fluorescent probe
used to indicate lipid peroxidation and antioxidant properties in
model membrane systems and in living cells (14).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was isolated from H9c2 cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). The sample concentration was quantified via
spectrophotometry. Total RNA was reverse transcribed into cDNA
using the PrimeScript RT reagent kit (Takara Bio, Inc.) according
to the manufacturer's instructions. qPCR was performed using a
SYBR® Premix Ex Taq kit (Takara Bio, Inc.) in accordance
with the manufacturer's instructions. The thermocycling conditions
were as follows: Initial denaturation for 95°C for 5 min, followed
by 40 cycles of denaturation at 95°C for 15 sec and annealing at
60°C for 30 sec; and a final extension of 10 min at 72°C. The fold
difference in gene expression was calculated using the
2−ΔΔCq method (15)
following normalization to the expression levels of GAPDH mRNA. The
primer sequences were as follows: ENPP2 forward,
5′-TTCCAATGTACCCCGCCTTC-3′ and reverse,
5′-AGCTGCCTTCCACATACTGTT-3′; FoxO4 forward,
5′-AGCGACTGACACTTGCCCAGAT-3′ and reverse, 5′-
AGGGTTCAGCATCCACCAAGAG-3′; and GAPDH forward,
5′-CGTGCCGCCTGGAGAA-3′ and reverse, 5′-CCCTCAGATGCCTGCTTCAC-3′.
Western blot analysis
Total proteins were isolated from H9c2 cells using
RIPA lysis buffer (Beyotime Institute of Biotechnology). Protein
concentration was determined using a BCA protein assay kit
(Beyotime Institute of Biotechnology) and proteins (40 µg/lane)
were separated via 12% SDS-PAGE and subsequently transferred to a
PVDF membrane. The membranes were blocked with 10% non-fat dry milk
at room temperature for 2 h and incubated with primary antibodies
at 4°C overnight. Following the primary antibody incubation, the
membranes were incubated with HRP-conjugated anti-rabbit or
anti-mouse IgG secondary antibodies. The immunoreactive bands were
visualized using DAB (Wuhan Boster Biological Technology, Ltd.).
GAPDH was used as a loading control, which was in accordance with
the previous studies showing that GAPDH expression could be used as
a loading control in for DOX investigation (16–18).
Densitometric analysis was performed using ImageJ software (version
1.52r; National Institutes of Health). Anti-LC3I/II (cat. no.
4108S; 1:1,000), anti-Beclin1 (cat. no. 3495T; 1:1,000),
anti-autophagy related 5 (ATG5; cat. no. 12994T; 1:1,000), anti-p62
(cat. no. 23214S; 1:1,000) and anti-GAPDH (cat. no. 5174T; 1:1,000)
antibodies were purchased from Cell Signaling Technology, Inc.
Anti-FoxO4 (cat. no. ab128908; 1:1,000), anti-ENPP2 (cat. no.
ab140915; 1:1,000), anti-solute carrier family 7 member 11
(SLC7A11; cat. no. ab175186; 1:1,000), anti-GPX4 (cat. no.
ab125066; 1:1,000), anti-ferroportin 1 (FPN1; cat. no. ab239511;
1:1,000), anti-NADPH oxidase 4 (NOX4; cat. no. ab133303; 1:1,000),
HRP-conjugated anti-mouse (cat. no. ab6728; 1:5,000) and
HRP-conjugated anti-rabbit IgG (cat. no. ab6721; 1:5,000)
antibodies were provided by Abcam.
Statistical analysis
All experiments were repeated three times and the
results are presented as the mean ± SD. Data were analyzed using
GraphPad Prism 8 software (GraphPad Software, Inc.). Comparisons
between two groups were conducted using an unpaired Student's
t-test, while comparisons among multiple groups were performed
using one-way ANOVA followed by Tukey's post hoc test. P<0.05
was considered to indicate a statistically significant
difference.
Results
ENPP2 expression is downregulated
following DOX treatment
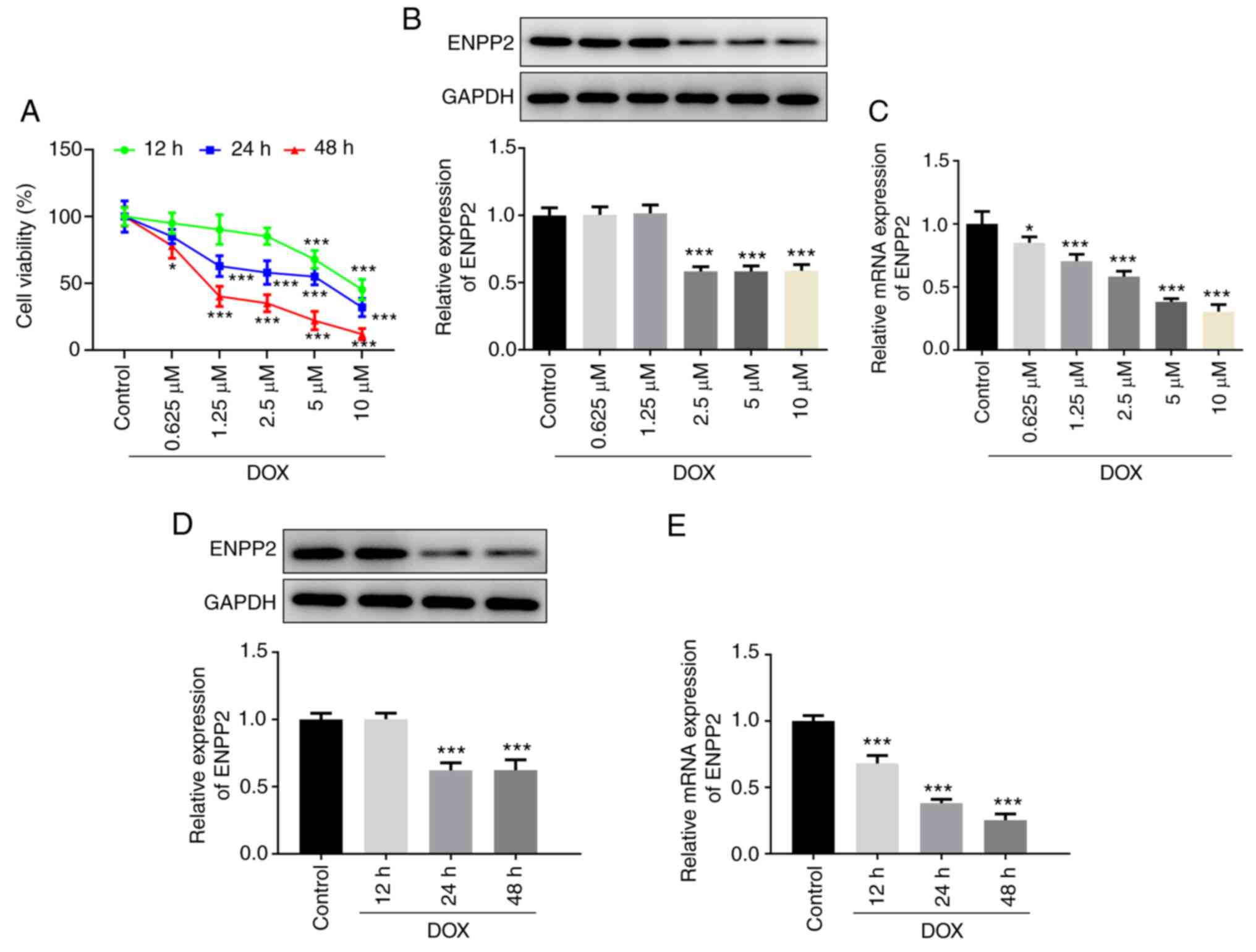
Different concentrations (0.625, 1.25, 2.5, 5 and 10
µM) of DOX were used to treat H9c2 cells for 12, 24 and 48 h,
respectively. Cell viability was significantly reduced following
DOX treatment in a time- and dose-dependent manner (Fig. 1A). In addition, the protein
expression level of ENPP2 were significantly decreased compared
with those of the control cells, following treatment of the cells
with 2.5, 5 and 10 µM DOX for 24 h and treatment of the cells with
5 µM DOX for 24 and 48 h. In addition, the mRNA expression levels
of ENPP2 were downregulated in cells following DOX treatment in a
dose- and time-dependent manner (Fig.
1B-E). Moreover, 5 µM DOX treatment was used for 24 h to treat
H9c2 cells in subsequent experiments.
Overexpression of ENPP2 enhances
autophagy and inhibits ferroptosis induced by DOX in H9c2
cardiomyocytes
Overexpression of ENPP2 was achieved in H9c2 cells
and the results verified the effective increase of ENPP2 protein
and mRNA expression levels (Fig. 2A and
B). Subsequently, control or ENPP2-Oe cells were exposed to DOX
in order to examine the changes noted in cell viability, oxidative
stress and autophagy. The reduction in cell viability caused by DOX
induction was effectively rescued by ENPP2 overexpression (Fig. 2C). As shown in Fig. 2D, DOX induction significantly
elevated in the level of total ROS compared with the control group,
which was notably restored by ENPP2 overexpression. Moreover, DOX
treatment resulted in a significant increase in TBARS content and a
decrease in GSH-Px expression, which were both partially recovered
by ENPP2 overexpression (Fig. 2E and
F).
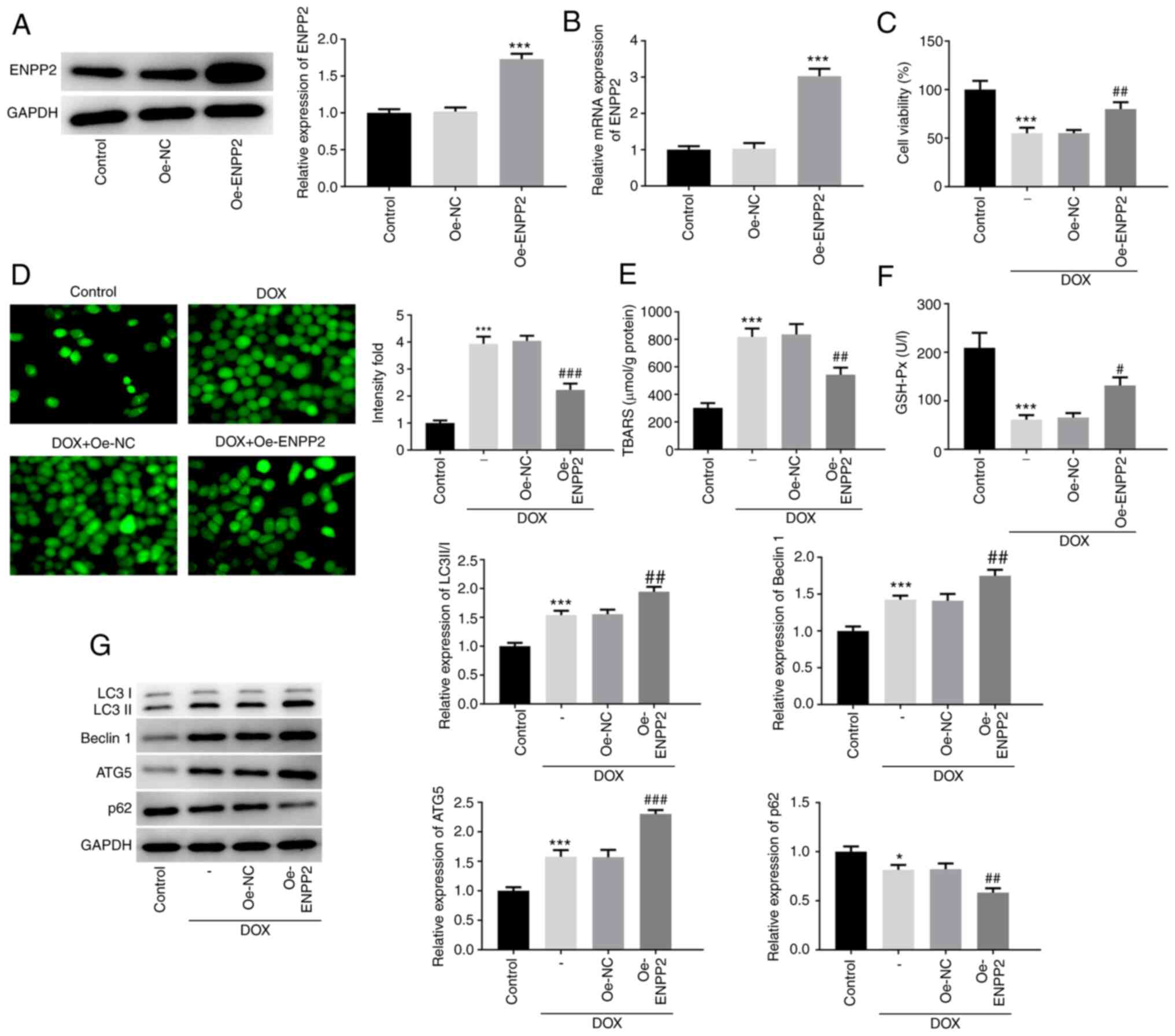 | Figure 2.Effects of ENPP2 overexpression on
DOX-induced autophagy in H9c2 cardiomyocytes. H9c2 cells were
transfected with ENPP2 vector in order to obtain ENPP2-Oe cells.
The transfection efficiency was detected via (A) western blotting
and (B) reverse transcription-quantitative PCR. ***P<0.001 vs.
Oe-NC. (C) ENPP2-Oe H9c2 cells were exposed to 5 µM DOX for 24 h
and cell viability was measured. (D) The total reactive oxygen
species production was assessed using a
2′,7′-dichlorodihydrofluorescein diacetate kit (magnification,
×200). ENPP2-Oe H9c2 cells were exposed to 5 µM DOX for 24 h, and
subsequently (E) TBARS concentration levels and (F) GSH-Px
expression levels were measured. (G) ENPP2-Oe H9c2 cells were
exposed to 5 µM DOX for 24 h and the expression levels of the
proteins involved in autophagy were evaluated. *P<0.05 and
***P<0.001 vs. control; #P<0.05,
##P<0.01 and ###P<0.001 vs. Oe-NC. DOX,
doxorubicin; NC, negative control; TBARS, thiobarbituric acid
reactive substances; GSH-Px, glutathione peroxidase; ENPP2,
ectonucleotide pyrophosphatase/phosphodiesterase 2; Oe,
overexpressing; ATG5, autophagy related 5. |
The expression levels of the proteins involved in
autophagy, including LC3II/I, Beclin 1, ATG5 and p62, were also
assessed. The protein expression levels of LC3II/I, Beclin 1 and
ATG5 were significantly increased, but p62 expression was decreased
following DOX treatment, indicating the induction of autophagy by
DOX in H9c2 cells (Fig. 2G).
Concomitantly, ENPP2 overexpression enhanced the changes noted in
these proteins following induction of autophagy by DOX.
To evaluate the induction of ferroptosis in
DOX-treated H9c2 cells, Fe2+ activity, lipid ROS
production and the expression levels of ferroptosis-associated
proteins, including SLC7A11, GPX4, FPN1 and NOX4, were determined.
DOX treatment resulted in a significant increase in cellular
Fe2+ activity and lipid ROS production (Fig. 3A and B), while the cells that
overexpressed ENPP2 exhibited decreased Fe2+ activity
and lipid ROS production compared with the cells transfected with
NC vectors (Fig. 3A and B).
Furthermore, the decreased expression levels of SLC7A11, GPX4 and
FPN1 and increased expression level of NOX4 caused by DOX were
significantly reversed by the overexpression of ENPP2 (Fig. 3C). These data suggested the
inhibitory effects of ENPP2 on DOX-induced ferroptosis in H9c2
cells.
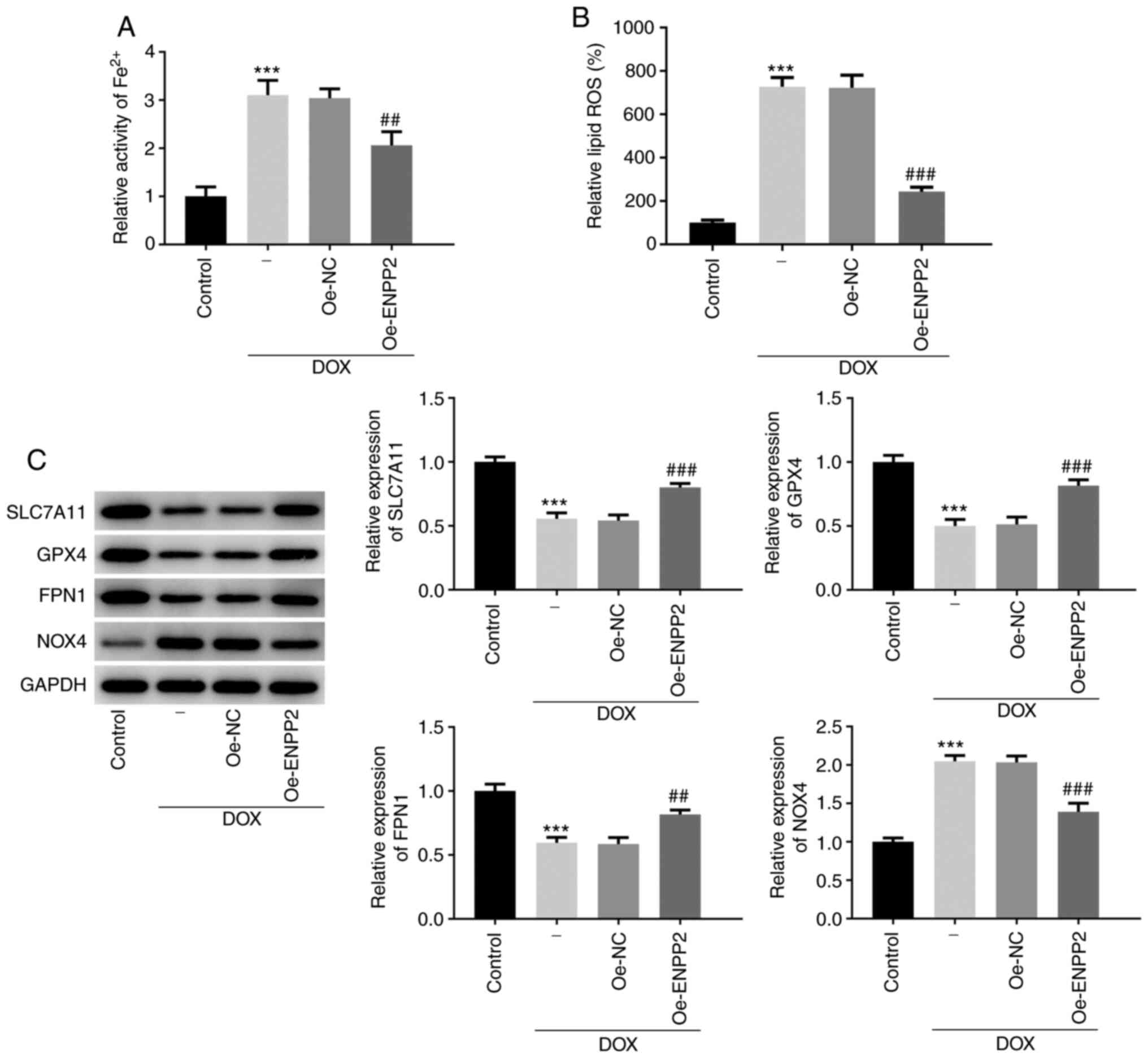 | Figure 3.Effects of ENPP2 overexpression on
DOX-induced ferroptosis in H9c2 cardiomyocytes. (A) H9c2 cells that
overexpressed ENPP2 were exposed to 5 µM DOX for 24 h and cellular
Fe2+ activity was measured using an Iron Assay kit. (B)
The lipid ROS levels were assessed. (C) The expression levels of
the proteins involved in ferroptosis were measured using western
blot analysis. ***P<0.001 vs. control; ##P<0.01
and ###P<0.001 vs. Oe-NC. DOX, doxorubicin; ROS,
reactive oxygen species; DCFDA, 2′,7′-dichlorodihydrofluorescein
diacetate; NC, negative control; ENPP2, ectonucleotide
pyrophosphatase/phosphodiesterase 2; Oe, overexpressing; SLC7A11,
solute carrier family 7 member 11; GPX4, GSH peroxidase 4; FPN1,
ferroportin 1; NOX4, NADPH oxidase 4. |
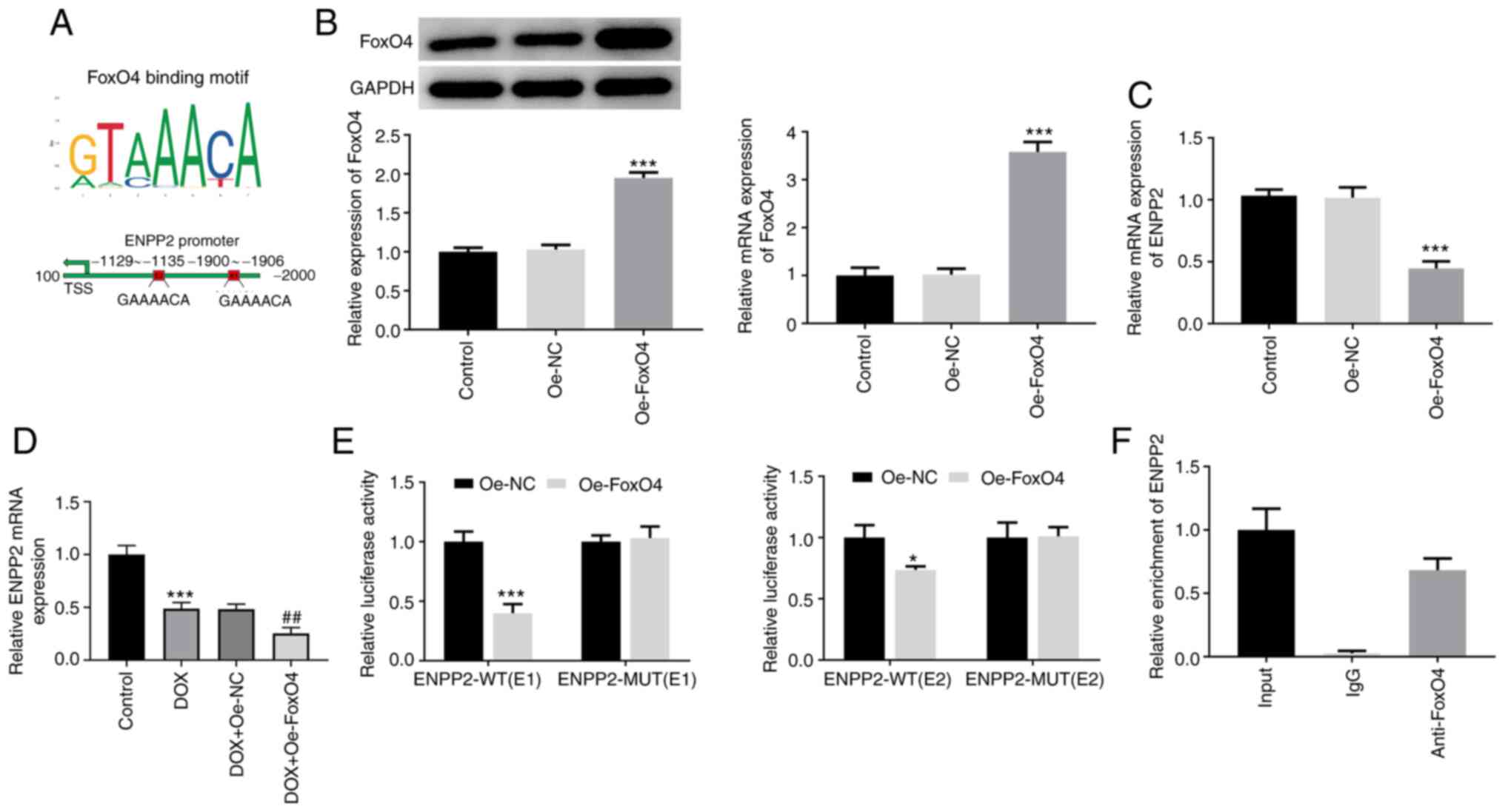
FoxO4 binds to the promoter of ENNP2
and regulates ENPP2 expression in H9c2 cardiomyocytes
FoxO4 was predicted to bind to the E1 and E2
sequences of the ENPP2 promoter (Fig.
4A). As displayed in Fig. 4B,
the protein and mRNA expression levels of FoxO4 were significantly
upregulated after transfection with FoxO4 plasmid. Additionally,
FoxO4 overexpression significantly reduced ENPP2 mRNA expression,
suggesting that FoxO4 may downregulate ENPP2 expression by binding
to the ENPP2 promoter (Fig. 4C).
Then, the expression level of ENPP2 in DOX-induced H9c2 cells with
FoxO4 overexpression was assessed using RT-qPCR. It was found that
gain-function of FoxO4 significantly downregulated the expression
level of ENPP2 compared with the DOX + Oe-NC group (Fig. 4D). Subsequently, the interaction
between FoxO4 and ENPP2 was validated using dual-luciferase
reporter (Fig. 4E) and ChIP
(Fig. 4F) assays. These results
suggested that FoxO4 could bind to the promoter of ENNP2 and
regulate ENPP2 expression in H9c2 cardiomyocytes.
Overexpression of FoxO4 partially
reverses the effects of ENPP2 on DOX-induced autophagy and
ferroptosis in H9c2 cardiomyocytes
Finally, FoxO4 and ENPP2 were co-overexpressed in
H9c2 cells, which had been treated with DOX. Cell viability was
reduced in cells that co-overexpressed FoxO4 and ENPP2 compared
with cells that only overexpressed ENPP2 (Fig. 5A). This indicated that FoxO4
overexpression inhibited the protective effect of ENPP2 on cell
viability, which was impaired by DOX (Fig. 5A). Moreover, the effects of ENPP2
overexpression on the total ROS levels, TBARS concentration and
GSH-Px expression were partially reversed by FoxO4 overexpression
(Fig. 5B-E). Similar results were
observed in Fig. 5F, as the
co-transfection with FoxO4 and ENPP2 plasmids significantly
decreased the levels of LC3II/I, Beclin 1 and ATG5 and increased
the expression of p62 compared with those in the DOX + Oe-ENPP2
group. In addition, the inhibitory effects of ENPP2 on DOX-induced
ferroptosis were significantly reduced by FoxO4 overexpression, as
demonstrated by increased Fe2+ and lipid ROS activity
levels, decreased SLC7A11, GPX4 and FPN1 expression, and increased
NOX4 expression, which were observed following FoxO4 overexpression
(Fig. 6A-C).
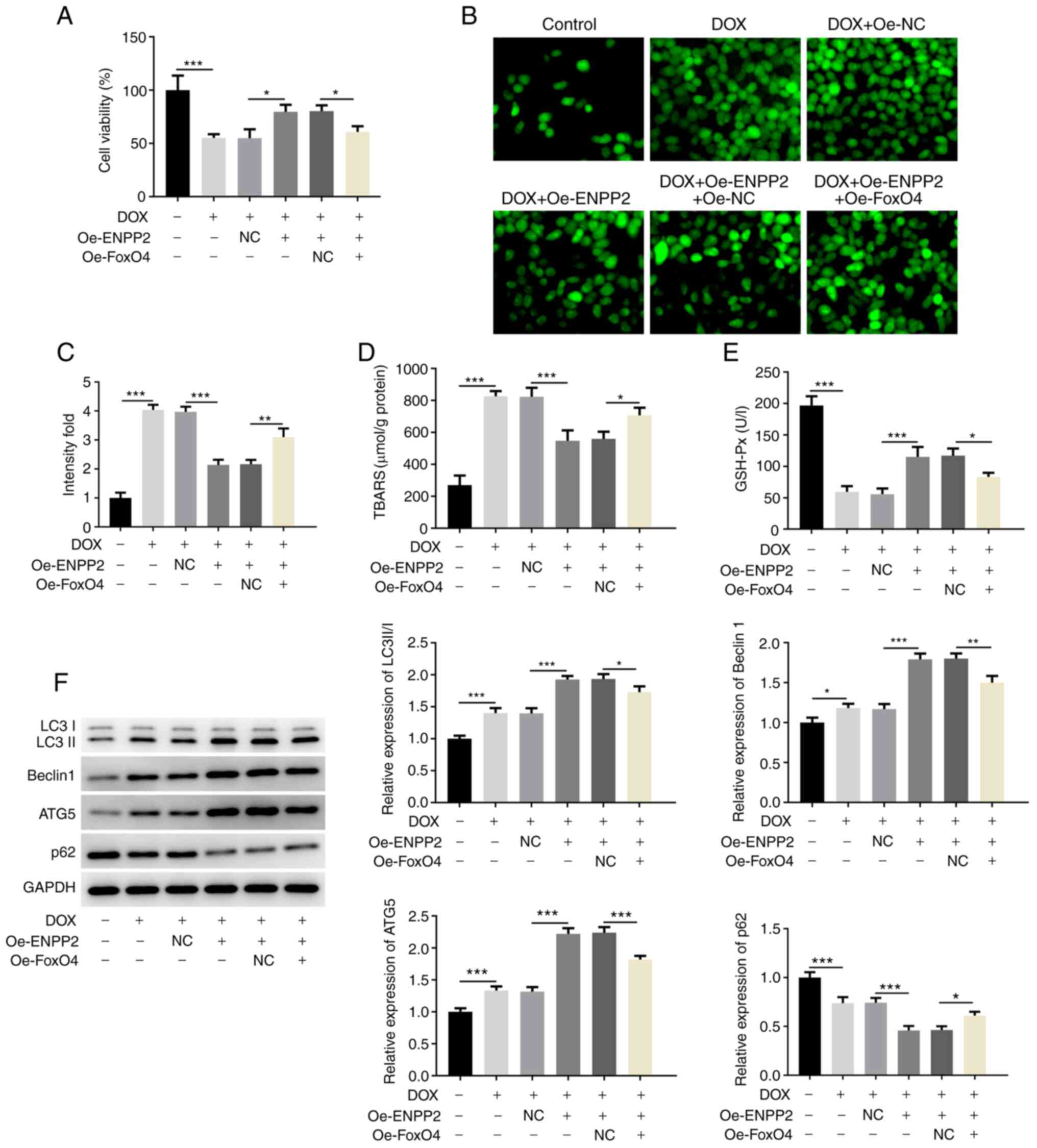 | Figure 5.FoxO4 overexpression inhibits the
effects of ENPP2 overexpression on DOX-induced autophagy in H9c2
cardiomyocytes. H9c2 cells that co-overexpressed FoxO4 and ENPP2
were exposed to 5 µM DOX for 24 h. Subsequently, (A) cell viability
was measured with a MTT assay. (B) Total reactive oxygen species
production was assessed using the DCFDA kit (magnification, ×200),
(C) and the results were quantified. (D) TBARS concentration levels
and (E) GSH-Px expression levels were detected using their
corresponding kits. (F) The expression levels of the proteins
involved in autophagy were evaluated via western blot analysis.
*P<0.05, **P<0.01 and ***P<0.001. DOX, doxorubicin; TBARS,
thiobarbituric acid reactive substances; GSH-Px, glutathione
peroxidase; ENPP2, ectonucleotide pyrophosphatase/phosphodiesterase
2; Oe, overexpressing; NC, negative control; ATG5, autophagy
related 5. |
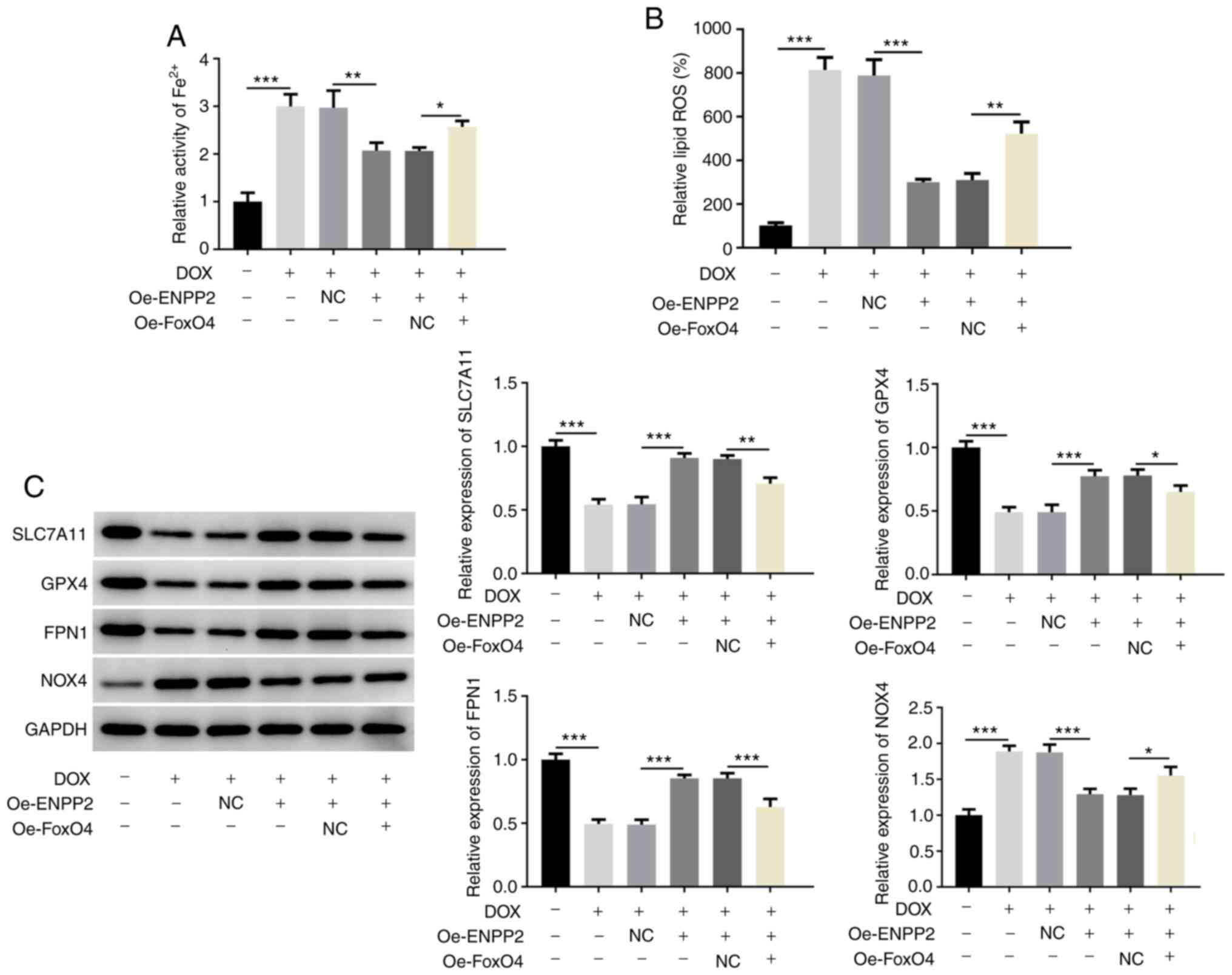 | Figure 6.FoxO4 overexpression inhibits the
effects of ENPP2 overexpression on DOX-induced ferroptosis in H9c2
cardiomyocytes. (A) H9c2 cells co-Oe FoxO4 and ENPP2 were exposed
to DOX (5 µM) for 24 h and cellular Fe2+ activity was
measured using the Iron Assay kit. (B) The lipid ROS level was
assessed. (C) The expression levels of the proteins involved in
ferroptosis were measured using western blot analysis. *P<0.05,
**P<0.01 and ***P<0.001. DOX, doxorubicin; ROS, reactive
oxygen species; ENPP2, ectonucleotide
pyrophosphatase/phosphodiesterase 2; Oe, overexpressing; NC,
negative control; SLC7A11, solute carrier family 7 member 11; GPX4,
GSH peroxidase 4; FPN1, ferroportin 1; NOX4, NADPH oxidase 4. |
Discussion
DOX is a potent anticancer drug clinically applied
for the treatment of various cancer types, such as breast, colon
and bladder cancer (19–21). However, its cardiotoxicity is a
major challenge, thereby limiting its clinical use. Therefore,
further understanding of the molecular mechanisms involved in DOX
function can alleviate cardiotoxicity and improve the clinical
efficacy of this chemotherapeutic drug. Currently, no effective
method exists to relieve the cardiac dysfunction caused by DOX.
Thus, the development of effective treatments is a major focus for
improving the side effects of DOX. The results of the present study
found that ENPP2 mRNA and protein expression levels were
downregulated following DOX treatment. Gene expression can be
divided into two levels: Transcription and translation, that is,
the mRNA level and the protein level. The time and site of
transcription and translation of gene expression are
spatiotemporal. After transcription, there will be
post-transcriptional processing, degradation of transcriptional
products, translation, post-translation processing and
modification. Therefore, transcription levels and translation
levels are not completely consistent, which may explain the
different degrees of decline between the mRNA and protein
expression levels of ENPP2 (22).
In the present study, FoxO4 was found to bind to the ENNP2 promoter
to improve DOX-induced cardiotoxicity via the induction of
ferroptosis.
Oxidative stress and dysregulation of autophagy have
been mainly reported to participate in the cardiotoxicity of DOX
(23,24). In the present study, the results
demonstrated that DOX treatment impaired cell viability, increased
TBARS concentration levels and reduced GSH-Px and p62 expression in
H9c2 cardiomyocytes. Concomitantly, DOX increased LC3II/I, Beclin 1
and ATG5 expression levels. The results of the present study
confirmed that DOX induced oxidative stress and autophagy in
cardiomyocytes. In addition, the relative Fe2+ activity
and lipid ROS production were enhanced, whereas the expression
levels of SLC7A11, GPX4 and FPN1 were decreased following treatment
of the cells with DOX. In contrast to these findings, NOX4
expression was increased following incubation of the cells with
DOX. Thus, ROS accumulation in DOX-treated cardiomyocytes was
accompanied with deregulation of their antioxidant defense system.
GSH serves a major role in the cellular antioxidant defense
(25). Depletion of GSH leads to
accumulation of lipid ROS levels, protein or membrane damage and
subsequent ferroptotic cell death (26). Cellular iron is required in normal
life processes, such as DNA synthesis, metastasis, cell cycle
progression, angiogenesis and/or mitochondrial iron metabolism.
Ferritin is the iron storage protein of the cells. Fe2+
promotes lipid peroxide accumulation via the Fenton reaction and
lipid oxidation (27). GPX4 can
prevent the toxicity of lipid peroxides by its enzyme activity. It
can also maintain the homeostasis of membrane lipid bilayers.
Furthermore, GSH is the co-factor of GPX4 and aids the catalysis of
peroxides into alcohols. Downregulation of GSH expression directly
inactivates GPX4 and leads to subsequent induction of ferroptosis
(28). Therefore, the present
results, when taken together, demonstrated that DOX could induce
ferroptosis.
Ferroptosis was recently reported as a novel
mechanism of iron-dependent regulated cell death (27). It has been proposed that ferroptosis
serves a significant role in DOX-induced cardiotoxicity, and
ferroptosis inhibition protects against this process (4,5).
Moreover, ENPP2 has been shown to inhibit ferroptosis in
cardiomyocytes (10). In the
present study, DOX treatment caused the downregulation of ENPP2
expression. It was hypothesized that ENPP2 may protect
cardiomyocytes from DOX-induced cardiotoxicity by inhibiting
ferroptosis. Therefore, this enzyme was overexpressed in H9c2
cells, which were subsequently treated with DOX. Numerous
regulators, such as GPX4, SLC7A11, FPN1 and NOX4, have been
identified to be involved in regulation of cell ferroptosis
(29–31). It has been reported that ENPP2
overexpression enhances the expression levels of the
ferroptosis-associated gene GPX4 in H9c2 cells (10). The current results indicated that
ENPP2 overexpression partially recovered TBARS, GSH-Px and ROS
levels, which in turn enhanced autophagy and inhibited ferroptosis.
Autophagy exerts a dual function in the regulation and progression
of cardiac dysfunction. The induction of autophagy has been shown
to attenuate DOX-induced cardiotoxicity. In addition, autophagy has
been reported to regulate ferroptosis (32–34).
In the present study, ENPP2 inhibited ferroptosis by blocking the
oxidative stress response, which in turn reduced ROS production and
enhanced the induction of autophagy. These processes then promoted
the elimination of long-lived proteins and damaged organelles.
The JASPAR database was searched and FoxO4 was
identified as a potential target that could bind to the promoter of
ENPP2. Additional analysis validated the direct interaction between
FoxO4 and ENPP2. It was found that FoxO4 overexpression reduced
ENPP2 expression. FoxO4 regulates negatively gene transcription via
post-transcriptional suppression of coding mRNAs (35). The results of the present study
indicated that FoxO4 may negatively control ENPP2 transcription by
binding to the ENPP2 promoter. Notably, FoxO4 has been extensively
investigated with regards to its effects on promoting myocardial
injury, and this transcription factor is involved in myocardial
ischemia/reperfusion, myocardial infarction and DOX-induced
cardiotoxicity (12,13,36).
Subsequently, the overexpression of FoxO4 in ENPP2-Oe H9c2 cells
was assessed with regards to the effects of ENPP2 on DOX-induced
cardiotoxicity. FoxO4 overexpression inhibited all the effects
caused by ENPP2 overexpression on DOX-induced cardiotoxicity, which
supported the initial hypothesis.
However, there are some limitations to the present
study that should be addressed, Firstly, in vivo experiments
were not performed and the effects of FoxO4 overexpression on H9c2
cells in the presence of DOX treatment were not investigated in the
present study. Additionally, the absence of clinical data is
another limitation of this study. Therefore, further experiments
should be performed in future studies to support the present
conclusions.
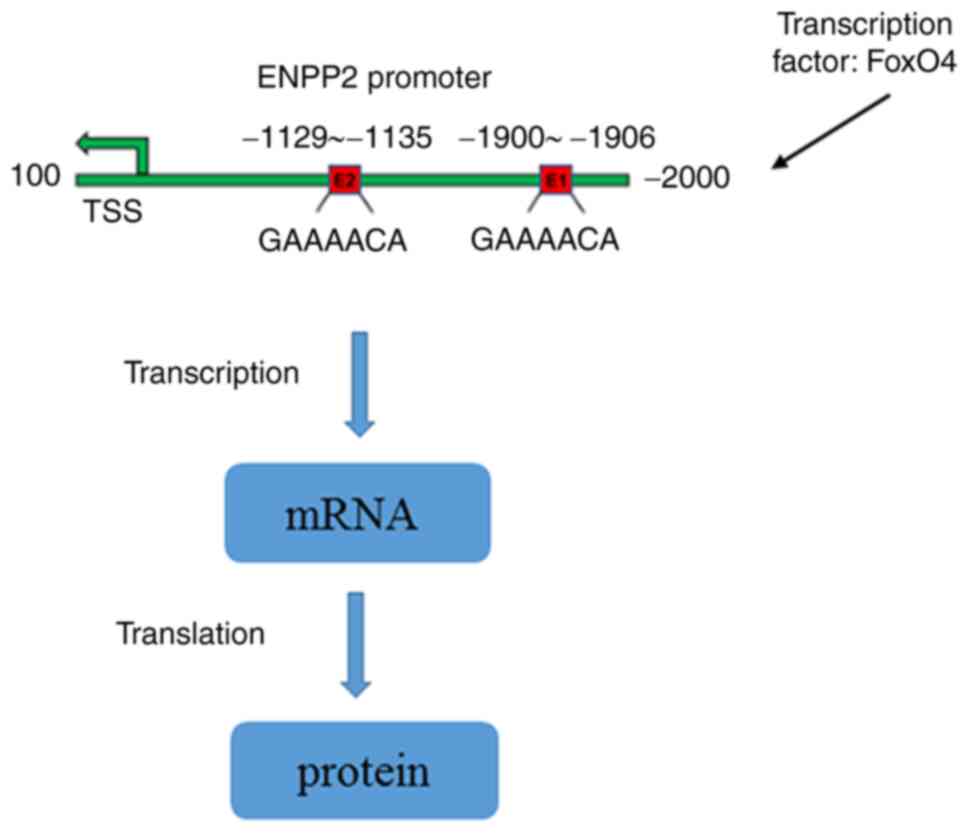
In conclusion, to the best of our knowledge, the
current study was the first to investigate the role of ENNP2 in
DOX-induced cardiomyocyte injury and its potential regulatory
mechanisms. The results of this study demonstrated that FoxO4 could
bind to the ENNP2 promoter to affect the post-transcriptional
suppression of encoding mRNA and therefore negatively control the
transcription of ENNP2 (Fig. 7),
thereby regulating DOX-induced cardiotoxicity via the induction of
ferroptosis. Specific therapies that target FoxO4/ENPP2 to reduce
and increase FoxO4 and ENPP2 expressions, respectively, may provide
potential approaches for ameliorating DOX-induced
cardiotoxicity.
Acknowledgements
Not applicable.
Funding
The present study was funded by the National Natural
Science Foundation of China (grant no. 82060070) and the Science
and Technology Research Project of Jiangxi Provincial Department of
Education (grant no. 161453).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LH, YY and JL contributed to study conception or
design. LH and YY performed the experiments and contributed to the
acquisition of data. JC and PZ contributed to data analysis or
interpretation. JL, LH and YY drafted the work and revised it
critically for important intellectual content. All authors read and
approved the final manuscript. LH and JL confirm the authenticity
of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Babiker HM, McBride A, Newton M, Boehmer
LM, Drucker AG, Gowan M, Cassagnol M, Camenisch TD, Anwer F and
Hollands JM: Cardiotoxic effects of chemotherapy: A review of both
cytotoxic and molecular targeted oncology therapies and their
effect on the cardiovascular system. Crit Rev Oncol Hematol.
126:186–200. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gupta SK, Garg A, Bär C, Chatterjee S,
Foinquinos A, Milting H, Streckfuß-Bömeke K, Fiedler J and Thum T:
Quaking inhibits doxorubicin-mediated cardiotoxicity through
regulation of cardiac circular RNA expression. Circ Res.
122:246–254. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tadokoro T, Ikeda M, Ide T, Deguchi H,
Ikeda S, Okabe K, Ishikita A, Matsushima S, Koumura T, Yamada KI,
et al: Mitochondria-dependent ferroptosis plays a pivotal role in
doxorubicin cardiotoxicity. JCI Insight. 5:e1327472020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fang X, Wang H, Han D, Xie E, Yang X, Wei
J, Gu S, Gao F, Zhu N, Yin X, et al: Ferroptosis as a target for
protection against cardiomyopathy. Proc Natl Acad Sci USA.
116:2672–2680. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu Y, Zeng L, Yang Y, Chen C, Wang D and
Wang H: Acyl-CoA thioesterase 1 prevents cardiomyocytes from
Doxorubicin-induced ferroptosis via shaping the lipid composition.
Cell Death Dis. 11:7562020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mou Y, Wang J, Wu J, He D, Zhang C, Duan C
and Li B: Ferroptosis, a new form of cell death: Opportunities and
challenges in cancer. J Hematol Oncol. 12:342019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hirschhorn T and Stockwell BR: The
development of the concept of ferroptosis. Free Radic Biol Med.
133:130–143. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Magkrioti C, Galaris A, Kanellopoulou P,
Stylianaki EA, Kaffe E and Aidinis V: Autotaxin and chronic
inflammatory diseases. J Autoimmun. 104:1023272019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhao Y, Hasse S, Zhao C and Bourgoin SG:
Targeting the autotaxin-Lysophosphatidic acid receptor axis in
cardiovascular diseases. Biochem Pharmacol. 164:74–81. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bai YT, Chang R, Wang H, Xiao FJ, Ge RL
and Wang LS: ENPP2 protects cardiomyocytes from erastin-induced
ferroptosis. Biochem Biophys Res Commun. 499:44–51. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Maiese K, Hou J, Chong ZZ and Shang YC: A
fork in the path: Developing therapeutic inroads with FoxO
proteins. Oxid Med Cell Longev. 2:119–129. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhu M, Goetsch SC, Wang Z, Luo R, Hill JA,
Schneider J, Morris SM Jr and Liu ZP: FoxO4 promotes early
inflammatory response upon myocardial infarction via endothelial
Arg1. Circ Res. 117:967–977. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
You J, Gao F, Tang H, Peng F, Jia L, Huang
K, Chow K, Zhao J, Liu H, Lin Y and Chen J: A medicinal and edible
formula YH0618 ameliorates the toxicity induced by Doxorubicin via
regulating the expression of Bax/Bcl-2 and FOXO4. J Cancer.
10:3665–3677. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang Z, Zhang X, Tian X, Yang Y, Ma L,
Wang J and Yu Y: CREB stimulates GPX4 transcription to inhibit
ferroptosis in lung adenocarcinoma. Oncol Rep. 45:882021.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jiang Y and Zhang Q: Catalpol ameliorates
doxorubicin-induced inflammation and oxidative stress in H9C2 cells
through PPAR-γ activation. Exp Ther Med. 20:1003–1011. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ye M, Zhang L, Yan Y and Lin H:
Punicalagin protects H9c2 cardiomyocytes from doxorubicin-induced
toxicity through activation of Nrf2/HO-1 signaling. Biosci Rep.
39:BSR201902292019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hu C, Zhang X, Wei W, Zhang N, Wu H, Ma Z,
Li L, Deng W and Tang Q: Matrine attenuates oxidative stress and
cardiomyocyte apoptosis in doxorubicin-induced cardiotoxicity via
maintaining AMPKα/UCP2 pathway. Acta Pharm Sin B. 9:690–701. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shafei A, El-Bakly W, Sobhy A, Wagdy O,
Reda A, Aboelenin O, Marzouk A, El Habak K, Mostafa R, Ali MA and
Ellithy M: A review on the efficacy and toxicity of different
doxorubicin nanoparticles for targeted therapy in metastatic breast
cancer. Biomed Pharmacother. 95:1209–1218. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Abd-Rabou AA, Ahmed HH and Shalby AB:
Selenium overcomes doxorubicin resistance in their nano-platforms
against breast and colon cancers. Biol Trace Elem Res. 193:377–389.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mikhail AS, Negussie AH, Pritchard WF,
Haemmerich D, Woods D, Bakhutashvili I, Esparza-Trujillo J,
Brancato SJ, Karanian J, Agarwal PK and Wood BJ:
Lyso-thermosensitive liposomal doxorubicin for treatment of bladder
cancer. Int J Hyperthermia. 33:733–740. 2017.PubMed/NCBI
|
|
22
|
de Klerk E and ‘t Hoen PA: Alternative
mRNA transcription, processing, and translation: Insights from RNA
sequencing. Trends Genet. 31:128–139. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shabalala S, Muller CJF, Louw J and
Johnson R: Polyphenols, autophagy and doxorubicin-induced
cardiotoxicity. Life Sci. 180:160–170. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xiao B, Hong L, Cai X, Mei S, Zhang P and
Shao L: The true colors of autophagy in doxorubicin-induced
cardiotoxicity. Oncol Lett. 18:2165–2172. 2019.PubMed/NCBI
|
|
25
|
Cappetta D, De Angelis A, Sapio L,
Prezioso L, Illiano M, Quaini F, Rossi F, Berrino L, Naviglio S and
Urbanek K: Oxidative stress and cellular response to doxorubicin: A
common factor in the complex milieu of anthracycline
cardiotoxicity. Oxid Med Cell Longev. 2017:15210202017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wu C, Zhao W, Yu J, Li S, Lin L and Chen
X: Induction of ferroptosis and mitochondrial dysfunction by
oxidative stress in PC12 cells. Sci Rep. 8:5742018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Latunde-Dada GO: Ferroptosis: Role of
lipid peroxidation, iron and ferritinophagy. Biochim Biophys Acta
Gen Subj. 1861:1893–1900. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Seibt TM, Proneth B and Conrad M: Role of
GPX4 in ferroptosis and its pharmacological implication. Free Radic
Biol Med. 133:144–152. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Luo LF, Guan P, Qin LY, Wang JX, Wang N
and Ji ES: Astragaloside IV inhibits adriamycin-induced cardiac
ferroptosis by enhancing Nrf2 signaling. Mol Cell Biochem.
476:2603–2611. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu T, Jiang L, Tavana O and Gu W: The
deubiquitylase OTUB1 mediates ferroptosis via stabilization of
SLC7A11. Cancer Res. 79:1913–1924. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang Z, Ding Y, Wang X, Lu S, Wang C, He
C, Wang L, Piao M, Chi G, Luo Y and Ge P: Pseudolaric acid B
triggers ferroptosis in glioma cells via activation of Nox4 and
inhibition of xCT. Cancer Lett. 428:21–33. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xu H, Yu W, Sun S, Li C, Zhang Y and Ren
J: Luteolin attenuates doxorubicin-induced cardiotoxicity through
promoting mitochondrial autophagy. Front Physiol. 11:1132020.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhou B, Liu J, Kang R, Klionsky DJ,
Kroemer G and Tang D: Ferroptosis is a type of autophagy-dependent
cell death. Semin Cancer Biol. 66:89–100. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu J, Kuang F, Kroemer G, Klionsky DJ,
Kang R and Tang D: Autophagy-dependent ferroptosis: Machinery and
regulation. Cell Chem Biol. 27:420–435. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Oteiza A and Mechti N: FoxO4 negatively
controls Tat-mediated HIV-1 transcription through the
post-transcriptional suppression of Tat encoding mRNA. J Gen Virol.
98:1864–1878. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yu L, Zhang W, Huang C, Liang Q, Bao H,
Gong Z, Xu M, Wang Z, Wen M and Cheng X: FoxO4 promotes myocardial
ischemia-reperfusion injury: The role of oxidative stress-induced
apoptosis. Am J Transl Res. 10:2890–2900. 2018.PubMed/NCBI
|