Introduction
The myocardial extracellular matrix (ECM) is an
intricate and dynamic network consisting of extracellular proteins
that provides structural and functional support for the myocardium
(1). The disruption of ECM
homeostasis is a primary driver for the development of cardiac
dysfunction and heart failure (2).
Myocardial fibrosis is a pathological process characterized by
excessive accumulation of ECM in myocardial interstitial spaces and
has been recognized as a common feature in various cardiac diseases
(3). The myocardial architecture is
disorganized during myocardial fibrosis, which facilitates the
progression of cardiac dysfunction, myocardial infarction,
ventricular arrhythmias and eventually heart failure (4). Thus, an effective and safe therapy
that can inhibit myocardial fibrosis from progressing is critical
to prevent heart failure in patients with cardiac diseases
(5).
Transforming growth factor-β1 (TGF-β1) is a central
fibrogenic factor that has been reported to increase ECM
expression, induce the transformation of fibroblasts to
myofibroblasts, and mediate the production of pro-fibrotic
cytokines during myocardial fibrosis (6). Upregulated TGF-β1 can bind to the type
II receptor, which phosphorylates the type I receptor, leading to
translocation of the Smad heterotrimeric complex into the nucleus
and the activation of the Smad-dependent pathway (7). The protein kinase B
(AKT)/extracellular signal-regulated kinase (ERK) signaling pathway
is also downstream of TGF-β1 and can be activated during cardiac
fibrosis (8). TGF-β1 has been
reported to trigger phosphorylation of the AKT pathway in a
RhoA-dependent manner, whereas it induces the activation of ERK via
small GTPase or direct phosphorylation of the ShcA protein
(9). As no efficient anti-fibrotic
treatment is currently available, the exploration of novel
therapeutic approaches targeting these molecular pathways may
provide insights into the management of myocardial fibrosis.
Liquiritigenin (LQ) is a flavanone compound with a
polyphenolic structure (Fig. 1A),
which is primarily found in the roots of licorice (Glycyrrhiza
glabra, Glycyrrhizae radix). Previous evidence has demonstrated
that LQ possesses multiple pharmacological and biochemical
properties, including anti-oxidative, anti-carcinogenic,
anti-inflammatory, hepatoprotective and estrogenic activities
(10). Recently, the
anti-fibrogenic role of LQ has been identified in a murine model of
carbon tetrachloride-induced hepatic fibrosis via regulating the
TGF-β1/Smad pathway (11). It has
also been shown that LQ may attenuate high-glucose-induced ECM
accumulation, inflammatory response and oxidative stress in rat
glomerular mesangial cells (12).
Moreover, LQ has been shown to exhibit cardioprotective effects
against high fructose-induced cardiac injury in mice and cardiac
muscle cells by suppressing the markers of fibrosis and
inflammation, suggesting the therapeutic potential of LQ in
diabetic heart injury (13).
However, whether LQ could attenuate myocardial fibrosis and
preserve cardiac function following heart injury remains
unclear.
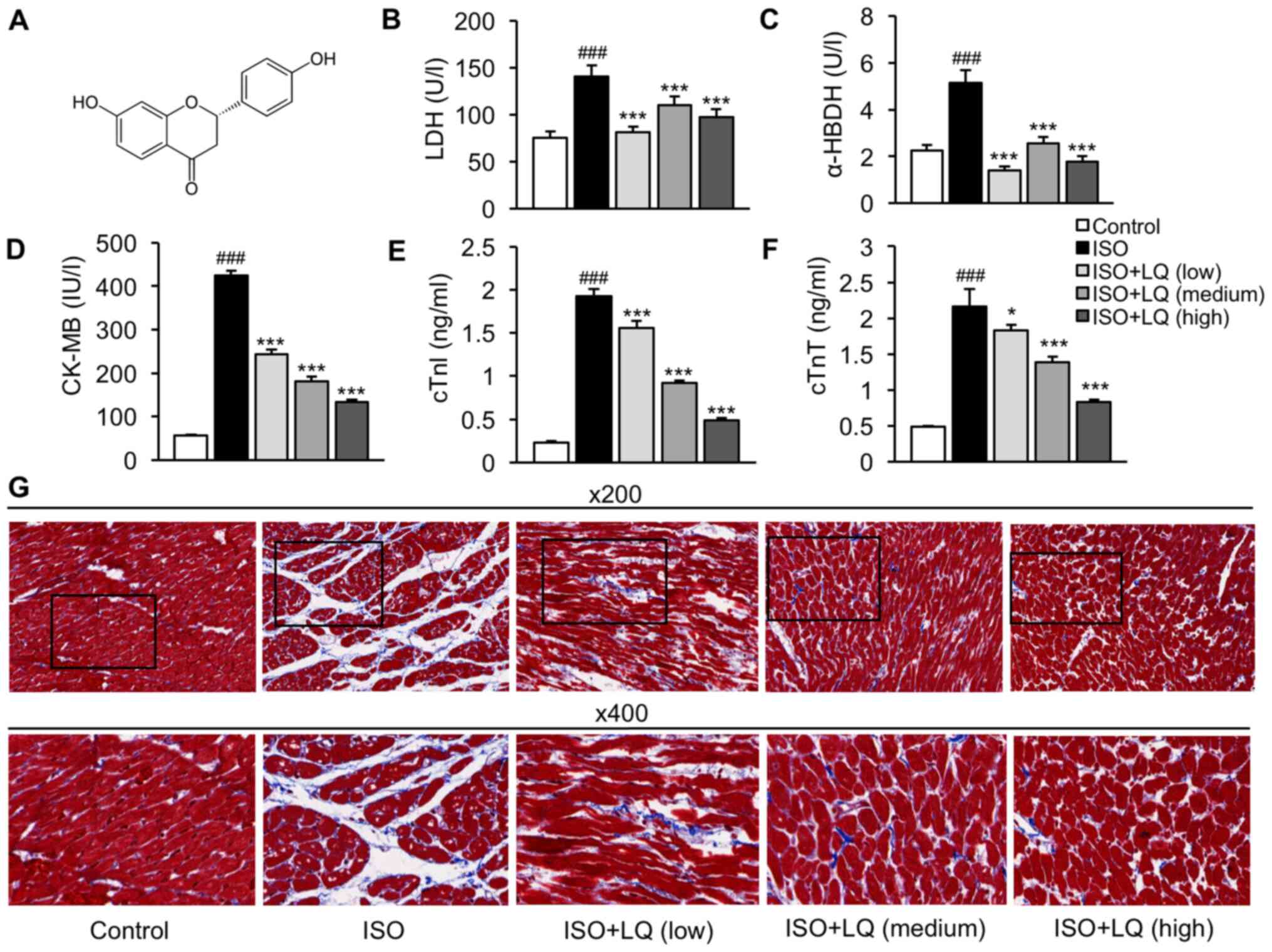 | Figure 1.LQ ameliorates ISO-induced myocardial
fibrosis in mice. (A) Chemical structure of LQ. (B-E) A mouse model
of myocardial fibrosis was established. The serum levels of (B) LDH
(U/l), (C) α-HBDH (U/l), (D) CK-MB (IU/l), (E) cTnl (ng/ml) and (F)
cTnT (ng/ml) were measured by enzyme-linked immunosorbent assay.
###P<0.001 vs. the control group; *P<0.05,
***P<0.001 vs. the ISO group. (G) Mouse heart tissues were
harvested for histopathological examination using Masson's
trichrome staining. Representative images at ×200 and 400
magnification are shown. α-HBDH, α-hydroxybutyrate dehydrogenase;
CK-MB, creatine kinase isoenzyme MB; cTnI, cardiac troponin I;
cTnT, cardiac troponin T; ISO, isoprenaline; LDH, lactate
dehydrogenase; LQ, liquiritigenin. |
In the present study, the potential role of LQ in
myocardial fibrosis was investigated in a mouse model induced by
isoprenaline (ISO) and in an in vitro model stimulated with
angiotensin II (Ang II). The effects of LQ on collagen deposition,
cardiomyocyte damage and cardiac function were examined. In
addition, the regulation of LQ in the TGF-β1/Smad2 and the AKT/ERK
signaling pathways was investigated.
Materials and methods
Animals and study design
Male C57BL/6 mice (age, 6–8 weeks; weight, 23.7±1.2
g; strain code, 027) were purchased from Charles River
Laboratories, Inc. The mice were housed in a pathogen-free facility
under a 12-h light/dark cycle, at a temperature of 23±2°C and at
50% humidity. Animals had ad libitum access to food and
water and were allowed to acclimate for at least 1 week prior to
the study. The present study was approved by the Laboratory Animal
Management and Welfare Ethical Review Committee of Zhejiang
Traditional Chinese Medicine University (Hangzhou, China; approval
no. ZSLL-2019-023) and all experiments were performed following the
Guide for the Care and Use of Laboratory Animals (14).
Animals were randomly divided into the following
five groups (n=6/group): Control, ISO, ISO + LQ (low), ISO + LQ
(medium) and ISO + LQ (high). The model of ISO-induced myocardial
fibrosis was established in all mice, with the exception of mice in
the control group, as previously described (15). Mice subjected to ISO stimulation
were subcutaneously injected with ISO (Sigma-Aldrich; Merck KGaA)
at a dosage of 5 mg/kg on the first day, followed by 2.5 mg/kg/day
for 2 weeks. Mice in the control group were treated with an equal
volume of normal saline (Sigma-Aldrich; Merck KGaA) via
subcutaneous injection following the same procedure. LQ (purity
>98.9%) was purchased from Selleck Chemicals. Mice in the ISO +
LQ (low), ISO + LQ (medium) and ISO + LQ (high) groups were
intragastrically administered with LQ at a dosage of 10, 20 and 30
mg/kg/day, respectively, for 2 weeks prior to ISO stimulation and
were continuously treated with LQ (10, 20 and 30 mg/kg/day,
respectively) via intragastric administration during the period of
ISO treatment.
Echocardiographic measurement
A total of 24 h post-treatment, echocardiography was
performed in all groups of mice using a Vevo 770®
high-resolution imaging system (VisualSonics, Inc.) as previously
described (16). Briefly, the mouse
was positioned on a heating pad in a supine position. Anesthesia
was induced with 5% isoflurane (Sigma-Aldrich; Merck KGaA) and
maintained with 1.5% isoflurane. The chest of the mouse was shaved
and acoustic coupling gel (Olympus Corporation) was applied to the
surface of the thorax. When the heart rate was within the normal
range, an M-mode cursor was positioned perpendicularly to the
posterior walls of the left ventricle (LV). The echocardiograms of
the LV in M mode, and the parameters including LV end-diastolic
dimension (LV EDD, mm), LV end-systolic dimension (LV ESD, mm),
fractional shortening (FS, %) and ejection fraction (EF, %), were
obtained using M-mode ultrasound imaging. FS was calculated as
follows: FS (%)=(LV EDD-LV ESD)/LV EDD ×100.
Assessment of myocardial injury
markers
Following the measurement of echocardiographic
parameters, all mice were anesthetized via intraperitoneal
injection of pentobarbital sodium (35 mg/kg body weight;
Sigma-Aldrich; Merck KGaA). Blood samples (1 ml) were collected
from the abdominal vena cava, transferred to 10-ml centrifuge tubes
and maintained at 4°C overnight. Subsequently, the mice were
sacrificed via exsanguination under anesthesia. The serum samples
were prepared by 30-min centrifugation of blood samples at 2,000 ×
g at 4°C. The serum levels of cardiac injury biomarkers were
measured using commercial ELISA kits, including lactate
dehydrogenase (LDH; cat. no. MBS2018912; MyBioSource, Inc.),
α-hydroxybutyrate dehydrogenase (α-HBDH; cat. no. MBS9303787;
MyBioSource, Inc.), creatine kinase isoenzyme MB (CK-MB; cat. no.
A77886; Antibodies.com), cardiac troponin I
(cTnI; cat. no. B52699; Beckman Coulter, Inc.) and cardiac troponin
T (cTnT; cat. no. MBS 726068; MyBioSource, Inc.).
Histopathological analysis
Mouse heart tissues were harvested immediately after
blood collection and transferred to Petri-dishes filled with cold
normal saline. The adherent connective tissues and atrial appendage
were carefully removed, and the apex was resected. All procedures
were performed on ice. A portion of the apex was used for
histopathological examination and the remaining portion was stored
at −80°C for western blot analysis. For histopathology, heart
tissues were fixed in 4% paraformaldehyde at 4°C for 24 h, embedded
in paraffin and cut into 4-µm sections. Subsequently, Masson's
trichrome staining (cat. no. ab150686; Abcam) was performed to
examine morphological changes and to evaluate collagen deposition
in cardiac tissues, following the manufacturer's instruction.
Slides were observed under a polarized light microscope.
Cell culture
The rat myocardial cell line H9C2 (Merck KgaA;
http://www.sigmaaldrich.com/FR/fr/product/sigma/cb_88092904)
was cultured in DMEM containing 10% fetal bovine serum (Gibco;
Thermo Fisher Scientific, Inc.) and F12 factor (Gibco; Thermo
Fisher Scientific, Inc.) in an incubator containing 5%
CO2 at 37°C. Ang II (Sigma-Aldrich; Merck KGaA) was used
to establish an in vitro model of cardiac dysfunction in
H9C2 cells. When cells had reached 70–80% confluence, they were
digested with 0.25% trypsin (Gibco; Thermo Fisher Scientific, Inc.)
and plated in 12-well plates at a density of 1×105 per
well. Following adhesion, cells in the Ang II + LQ (low), Ang II +
LQ (medium) and Ang II + LQ (high) groups were pretreated with LQ
at 0.1, 1 and 10 µM, respectively, for 6 h at 37°C followed by
stimulation with Ang II (1 µM) for 24 h at 37°C. The Ang II group
was pretreated with an equal volume of phosphate-buffered saline
and then exposed to Ang II (1 µM) for 24 h. The control group
remained untreated.
Flow cytometry
Apoptotic cell death was examined by flow cytometry
as previously described (17).
Briefly, H9C2 cells were digested, washed with cold PBS,
centrifuged at 140 × g for 5 min at room temperature, and
resuspended in 500 µl 1X binding buffer mixed with 5 µl Annexin
V-FITC and 5 µl propidium iodide (apoptosis kit from Abcam) for 10
min at room temperature in the dark. The apoptosis of H9C2 cells
was detected using a Navios flow cytometer and Kaluza software
version 1.3 (both from Beckman Coulter, Inc.). The apoptosis rate
was calculated as the number of early and late apoptotic cells over
the number of total cells observed.
Western blot analysis
Heart tissue homogenates and cell lysates were
prepared in RIPA buffer containing protease and phosphatase
inhibitors (Bio-Rad Laboratories, Inc.). The protein content was
measured using bicinchoninic acid assay (Pierce; Thermo Fisher
Scientific, Inc.). Equal amounts of total protein (50 µg) from each
sample were separated by 10% SDS-PAGE and then transferred to PVDF
membranes (EMD Millipore). After blocking with 5% non-fat milk for
1 h at room temperature, membranes were incubated with the
following primary antibodies at 4°C overnight: α-SMA (1:1,000; cat.
no. ab5694; Abcam), Smad2 (1:800 dilution; cat. no. ab63576;
Abcam), TGF-β1 (1:1,000; cat. no. ab92486; Abcam), collagen III
(1:2,000 dilution; cat. no. ab7778; Abcam), collagen I (1:1,000
dilution; cat. no. ab34710; Abcam), AKT (1:800 dilution; cat. no.
ab8805; Abcam), phosphorylated (p)-AKT (1:1,000 dilution; cat. no.
9271; Cell Signaling Technology, Inc.), ERK (1:1,000 dilution; cat.
no. 4695; Cell Signaling Technology, Inc.), p-ERK (1:1,000
dilution; cat. no. 9101; Cell Signaling Technology, Inc.) and GAPDH
(1:3,000 dilution; cat. no. ab181602; Abcam). After washing with
TBS-0.1% Tween-20 buffer, the membranes were incubated with a
secondary antibody (1:2,000 dilution; cat. no. ab6721; Abcam) at
room temperature for 1 h. Blots were visualized using the BeyoECL
Plus kit (Beyotime Institute of Biotechnology). The protein bands
were semi-quantified using the Alphalmager 2000 Imaging System
(ProteinSimple).
Statistical analysis
All data were analyzed using SPSS software (version
24.0; IBM Corp.) and are presented as the mean ± standard deviation
from three independent experiments. One-way analysis of variance
followed by Tukey's post-hoc analysis was used to determine
statistical significance among the groups. P<0.05 was considered
to indicate a statistically significant difference.
Results
LQ attenuates ISO-induced
cardiomyocyte damage and myocardial fibrosis in mice
To investigate the potential regulatory effects of
LQ on myocardial fibrosis, a murine model of ISO-induced myocardial
fibrosis was established in C57BL/6 mice. Mice subjected to LQ
treatment were intragastrically administered LQ at low (10
mg/kg/day), medium (20 mg/kg/day) and high (30 mg/kg/day) dosages,
before and during the period of ISO stimulation. By measuring the
levels of cardiac injury biomarkers in the serum samples, it was
revealed that the ISO model group exhibited significantly increased
levels of LDH, α-HBDH, CK-MB, cTnI and cTnT compared with those in
the control group, whereas LQ treatment significantly suppressed
the levels of these proteins at all doses tested (Fig. 1B-F). Furthermore, Masson's trichrome
staining was performed to assess the morphological and pathological
features of heart tissue samples from each group. The
cardiomyocytes in the control group were compactly arranged without
intercellular space, whereas in ISO-treated mice, cardiomyocyte
arrangement was disordered and myocardial fibers were ruptured,
which was accompanied by myocardial atrophy and inflammatory cell
infiltration (Fig. 1G). Treatment
with LQ ameliorated fibrosis and inflammation in ISO-induced
hearts; the most robust effect was observed at a dosage of 30
mg/kg/day (Fig. 1G). These results
suggested that LQ treatment attenuated ISO-induced cardiomyocyte
damage and myocardial fibrosis.
LQ improves cardiac function in
ISO-treated mice
To determine whether LQ could improve the cardiac
function of mice treated with ISO, the LV internal dimensions (LV
ESD and LV EDD) and ejection phase indices (FS and EF) of all
groups of mice were measured by echocardiography. Compared with
that in the control group, mice treated with ISO exhibited a
significant increase in LV ESD, but not LV EDD, whereas LQ at
medium and high concentrations significantly decreased LV ESD in
ISO-treated mice (Fig. 2A and B).
In addition, mice treated with a high dosage of LQ showed
significantly smaller LV EDD compared with that in the ISO model
group (Fig. 2B). The FS and EF
values in ISO-treated mice were significantly lower compared with
those in the control group, whereas treatment with LQ effectively
increased FS and EF in a dose-dependent manner (Fig. 2C and D). The M-mode echocardiograms
of the LV also showed that LQ improved myocardial contractile
function in ISO-induced mice (Fig.
2E). These findings indicated that LQ treatment ameliorated
ISO-induced cardiac dysfunction in mice.
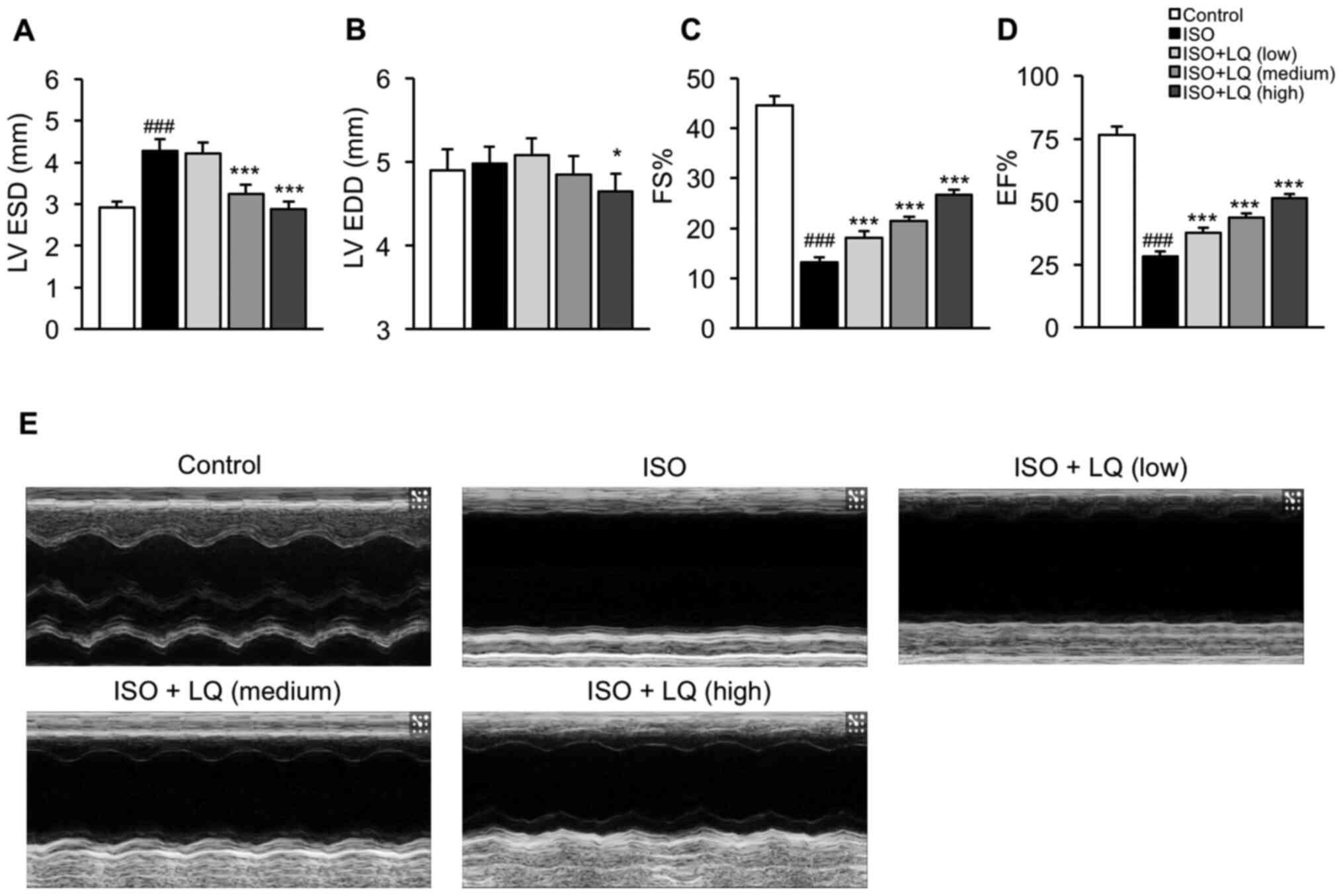 | Figure 2.Effect of LQ on cardiac function in
ISO-treated mice. Echocardiography was performed in all groups of
mice using an M-mode ultrasound imaging system. The parameters
indicating cardiac function were measured, including (A) LV ESD
(mm), (B) LV EDD (mm), (C) FS (%) and (D) EF (%).
###P<0.001 vs. the control group; *P<0.05,
***P<0.001 vs. the ISO group. (E) M-mode echocardiograms of the
LV from each group of mice are shown. EF, ejection fraction; FS,
fractional shortening; ISO, isoprenaline; LQ, liquiritigenin; LV,
left ventricle; LV EDD, LV end-diastolic dimension; LV ESD, LV
end-systolic dimension. |
LQ suppresses activation of the
TGF-β1/Smad2 and AKT/ERK pathways in mice treated with ISO
The present study further investigated the effects
of LQ on regulation of the TGF-β1/Smad2 and AKT/ERK pathways. Mice
in the ISO model group exhibited significantly upregulated α-SMA,
Smad2, TGF-β1 collagen III and collagen I expression levels
compared with those in the control group, indicating that ISO
induced activation of the TGF-β1/Smad2 pathway in the heart
(Fig. 3A and C-G). The intragastric
administration of LQ at 30 mg/kg/day significantly decreased the
expression levels of all these proteins (Fig. 3A and C-G). No significant difference
was observed in the basal levels of AKT and ERK among all groups,
whereas ISO significantly enhanced the expression levels of p-AKT
and p-ERK as compared with those in the control mice, suggesting
that activation of the AKT/ERK signaling pathway was induced in
mice subjected to ISO stimulation (Fig.
3B, H and I). Treatment with LQ at 10, 20 and 30 mg/kg/day
significantly inhibited the phosphorylation of both AKT (Fig. 3B and H) and ERK (Fig. 3B and I) in ISO-treated mice. These
data supported the conclusion that LQ suppressed ISO-induced
activation of the TGF-β1/Smad2 and AKT/ERK pathways.
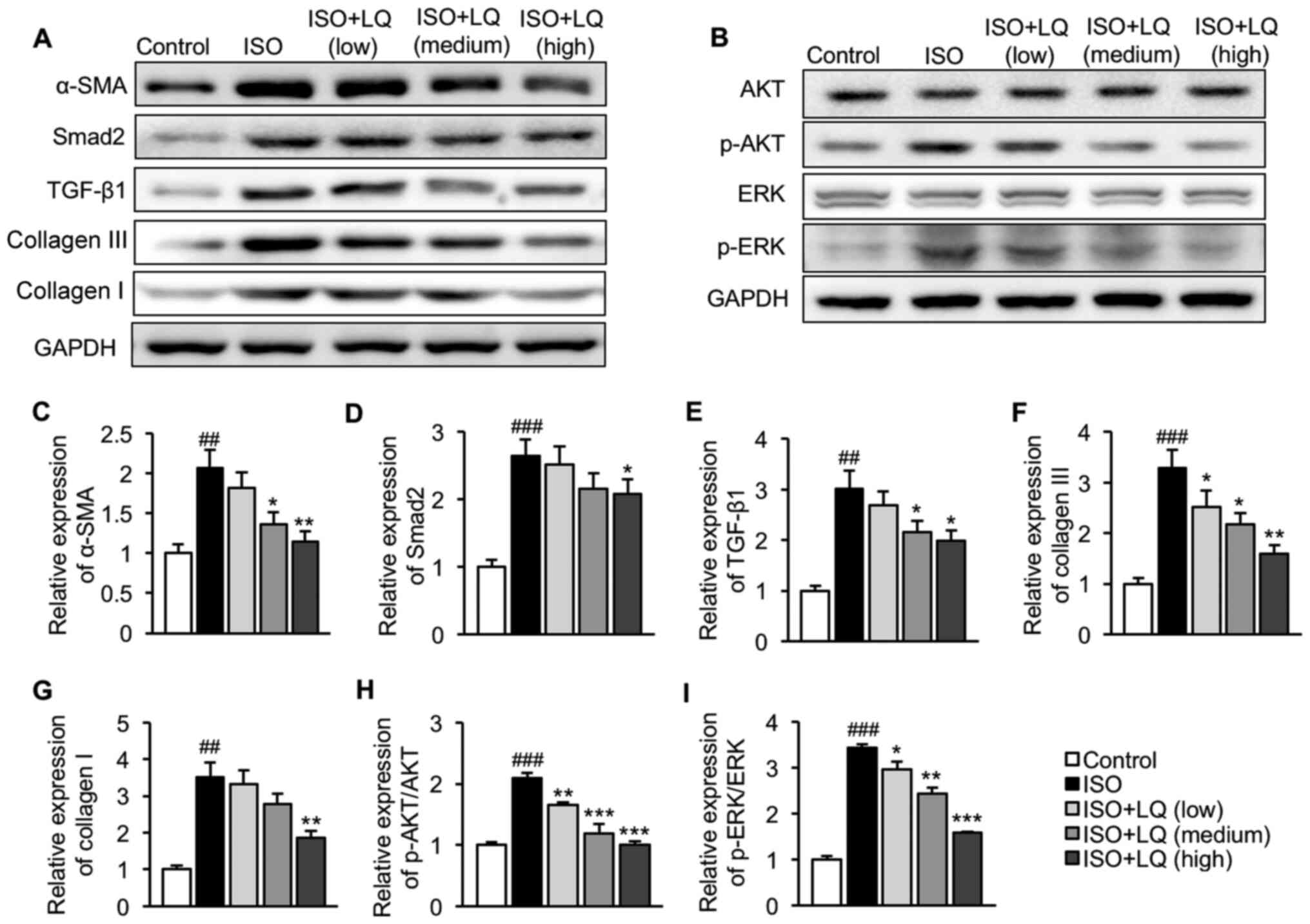 | Figure 3.Effect of LQ on activation of the
TGF-β1/Smad2 and AKT/ERK pathways in ISO-treated mice. Expression
levels of proteins related to the (A) TGF-β1/Smad2 and (B) AKT/ERK
pathways were measured by western blotting. Semi-quantification of
the expression levels of (C) α-SMA, (D) Smad2, (E) TGF-β1, (F)
collagen III, (G) collagen I, (H) p-AKT/AKT and (I) p-ERK/ERK
normalized to the expression of GAPDH. ##P<0.01,
###P<0.001 vs. the control group; *P<0.05,
**P<0.01, ***P<0.001 vs. the ISO group. AKT, protein kinase
B; α-SMA, α-smooth muscle actin; ERK, extracellular
signal-regulated kinase; ISO, isoprenaline; LQ, liquiritigenin; p-,
phosphorylated; TGF- β1, transforming growth factor β1. |
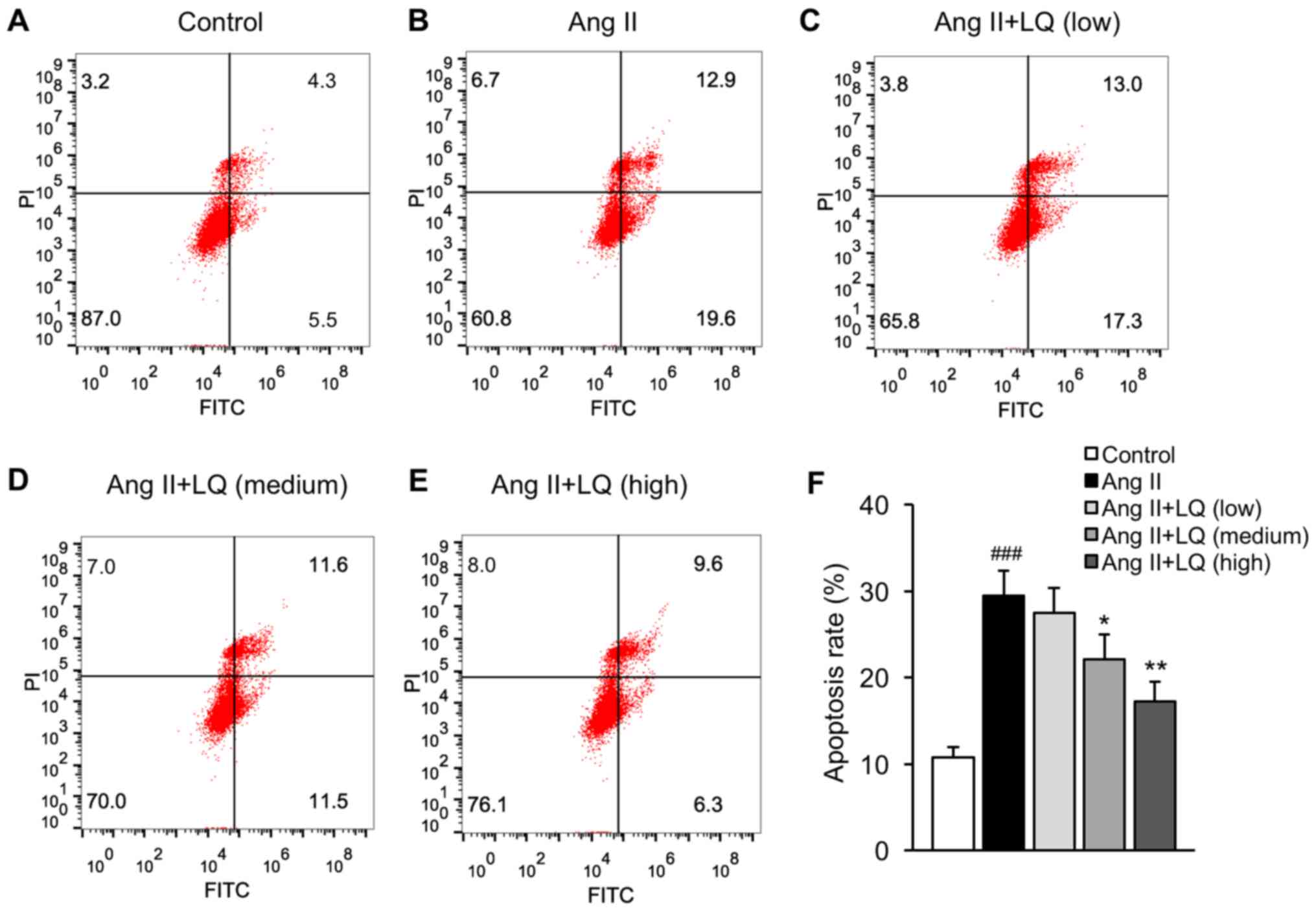
LQ decreases the Ang II-induced
apoptosis of cardiomyocytes
The present study established a model of Ang
II-induced cardiac dysfunction in rat myocardial H9C2 cells to
examine the effects of LQ on cardiomyocyte apoptosis. Cells exposed
to Ang II exhibited significantly increased apoptosis compared with
that in the untreated group (Fig. 4A, B
and F). Pretreatment with LQ at medium and high doses
successfully decreased the apoptosis rate of H9C2 cells following
Ang II treatment (Fig. 4C-F). These
results indicated the anti-apoptotic effects of LQ on cardiomyocyte
apoptosis.
LQ inhibits activation of the
TGF-β1/Smad2 and AKT/ERK pathways in Ang II-induced
cardiomyocytes
Finally, the effect of LQ treatment on regulation of
the TGF-β1/Smad2 and AKT/ERK pathways were evaluated in
vitro. Consistent with the results obtained from the mouse ISO
model, cells treated with Ang II exhibited significantly elevated
expression levels of α-SMA, Smad2, TGF-β1, collagen III and
collagen I compared with those in untreated cells, suggesting
activation of the TGF-β1/Smad2 pathway in Ang II-induced
cardiomyocytes (Fig. 5A and C-G).
Pretreatment with LQ at 1 and 10 µM significantly decreased the
protein expression levels of α-SMA, TGF-β1, collagen III and
collagen I in H9C2 cells following Ang II exposure, whereas the
upregulation of Smad2 in Ang II-treated cells was only
significantly suppressed by LQ at the highest dosage (Fig. 5A and C-G). In addition, Ang II
induced the phosphorylation of AKT and ERK in H9C2 cells without
affecting the basal levels of AKT and ERK (Fig. 5B, H and I). A 6-h pre-incubation
with 0.1, 1 and 10 µM LQ effectively reduced the phosphorylation of
AKT (Fig. 5B and H) and ERK
(Fig. 5B and I) in Ang
II-stimulated H9C2 cells. Taken together, these results suggested
that LQ served an inhibitory role in Ang II-induced activation of
the TGF-β1/Smad2 and AKT/ERK pathways in cardiomyocytes.
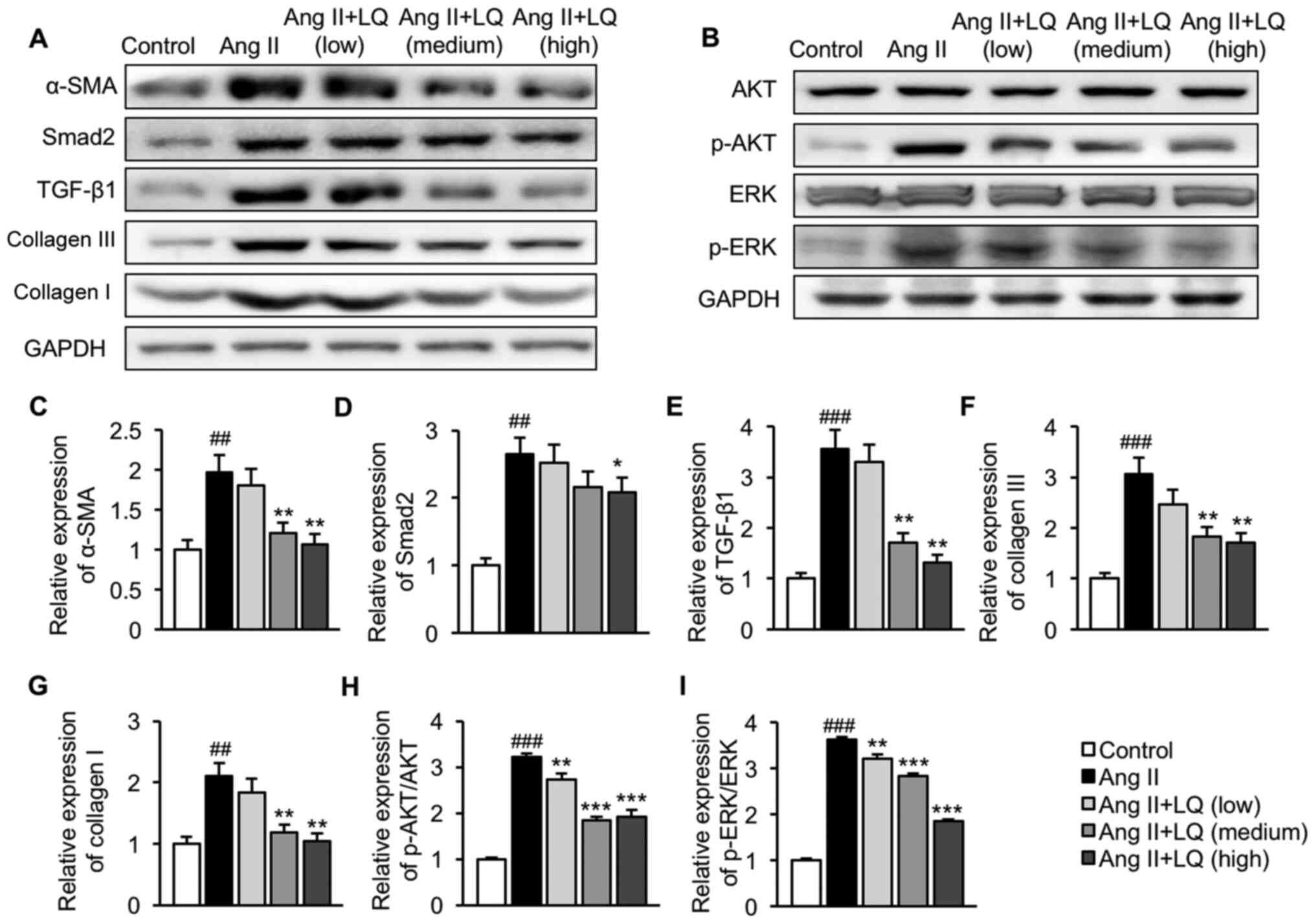 | Figure 5.Effect of LQ on Ang II-induced
activation of the TGF-β1/Smad2 and AKT/ERK pathways. H9C2 cells
were collected following LQ pretreatment and Ang II stimulation.
Cell lysates were prepared for western blot analysis. The
expression levels of proteins related to the (A) TGF-β1/Smad2 and
(B) AKT/ERK pathways were assessed. Semi-quantification of the
protein expression levels of (C) α-SMA, (D) Smad2, (E) TGF-β1, (F)
collagen III, (G) collagen I, (H) p-AKT/AKT and (I) p-ERK/ERK
normalized to the expression of GAPDH. ##P<0.01,
###P<0.001 vs. the control group; *P<0.05,
**P<0.01, ***P<0.001 vs. the Ang II group. AKT, protein
kinase B; Ang II, angiotensin II; α-SMA, α-smooth muscle actin;
ERK, extracellular signal-regulated kinase; LQ, liquiritigenin; p-,
phosphorylated; TGF- β1, transforming growth factor β1. |
Discussion
Myocardial fibrosis is a fundamental process in
ventricular remodeling and is considered a primary contributor to
the progression of heart failure (18). Novel treatment strategies that
collectively target the key signaling pathways and molecular
factors involved in fibrosis need to be considered in the
development of anti-fibrotic therapies (19). The present study demonstrated that
LQ attenuated myocardial fibrosis in mice and cultured myocardial
cells via regulating the TGF-β1/Smad2 and AKT/ERK signaling
pathways, suggesting the protective effects of LQ on cardiac
fibrosis.
The subcutaneous injection of ISO, a β-adrenergic
agonist, has been reported to induce cardiac dysfunction and
fibrosis in mice, which enables the evaluation of anti-fibrotic
agents in vivo (20). CK-MB
and cardiac troponins, as well as LDH and α-HBDH, are classic
diagnostic markers that are highly sensitive and specific for the
detection of myocardial damage in various conditions, including
myocardial fibrosis (21). In the
present study, the serum levels of LDH, α-HBDH, CK-MB, cTnI and
cTnT in the ISO model group were significantly higher compared with
those in the control group, whereas LQ treatment before and during
ISO stimulation downregulated the levels of cardiac injury-related
proteins. Further histological examination of mouse heart tissues
revealed that LQ ameliorated ISO-induced ECM accumulation,
myocardial disarray and inflammatory cell infiltration, further
confirming the cardioprotective role of LQ in cardiac fibrosis.
Previous evidence has suggested an association exists between the
disruption of ECM homeostasis and impaired LV function (22). Using echocardiography, the present
study compared the indicators of LV function in all groups of mice.
The results revealed that the LV ESD and ejection phase indices in
ISO-treated mice were significantly different from those in the
control group, whereas LQ treatment effectively diminished the
effects of ISO on these cardiac function markers.
The fibrotic response in the heart is a dynamic
process in which pro-fibrotic factors, such as TGF-β1, are
upregulated and trigger the activation of downstream signaling,
including Smad, AKT and ERK (23).
These pathways further induce the expression of fibrogenic genes
(i.e. α-SMA and collagen), leading to excessive production of
matrix metalloproteinases and subsequent ECM deposition (24). The present study revealed that ISO
treatment increased cardiac expression of α-SMA, Smad2, TGF-β1,
collagen III and collagen I compared with that in the control
group. The treatment of ISO-treated mice with LQ at 30 mg/kg/day
significantly decreased collagen formation and suppressed the
TGF-β1/Smad2 pathway. Moreover, LQ at all concentrations inhibited
ISO-induced phosphorylation of AKT and ERK with no effects on the
basal levels of these proteins.
The apoptosis of cardiomyocytes is a major
pathological event in myocardial fibrosis and has been regarded as
a key mechanism contributing to impaired LV performance (25). Ang II is a bioactive compound in the
renin-angiotensin system, which has been reported to accelerate
myocardial remodeling and cardiac fibrosis by activating reactive
oxygen species (ROS)-dependent pathways in rat cardiomyocytes
(26). A recent study reported that
Ang II exposure decreased the viability of H9C2 cells and
stimulated pro-apoptotic signaling pathways by enhancing ROS
production (27). The present study
investigated the effect of LQ on the Ang II-induced apoptosis of
cardiomyocytes. Pretreatment with LQ at medium and high dosages
significantly reduced apoptotic cell death in H9C2 cells following
Ang II treatment. Furthermore, the treatment of H9C2 cells with LQ
inhibited Ang II-induced activation of the TGF-β1/Smad2 and AKT/ERK
pathways in cardiomyocytes, which was consistent with the findings
in the animal model. However, the use of the rat myocardial cell
line H9C2 instead of primary cardiac fibroblasts may be considered
as a limitation to the present study. Further investigations using
primary cardiac fibroblasts are required to validate the current
findings.
Taken together, the present study demonstrated that
LQ ameliorated ISO-induced myocardial fibrosis in a mouse model and
inhibited the apoptosis of cardiomyocytes in vitro by
inhibiting both the TGF-β1/Smad2 and AKT/ERK signaling pathways.
These findings elucidated the mechanisms underlying the
anti-fibrotic and cardioprotective potential of LQ in fibrosis, and
provided evidence to support further investigations into LQ for the
management of cardiomyocyte injury and myocardial fibrosis in
patients suffering from heart diseases.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LL, HF, YHY and ZQY conceived and designed the
study. LL, HF, SXL and ZQY performed the experiments and analyzed
the data. LL, HF, YHY and ZQY contributed materials and assisted in
data analysis. LL, YHY, SXL and ZQY wrote and reviewed the paper.
XX and XX confirm the authenticity of all the raw data. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Laboratory
Animal Management and Welfare Ethical Review Committee of Zhejiang
Traditional Chinese Medicine University (Hangzhou, China; approval
no. ZSLL-2019-023).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Valiente-Alandi I, Schafer AE and Blaxall
BC: Extracellular matrix-mediated cellular communication in the
heart. J Mol Cell Cardiol. 91:228–237. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bonnans C, Chou J and Werb Z: Remodelling
the extracellular matrix in development and disease. Nat Rev Mol
Cell Biol. 15:786–801. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gyöngyösi M, Winkler J, Ramos I, Do QT,
Firat H, McDonald K, González A, Thum T, Díez J, Jaisser F, et al:
Myocardial fibrosis: Biomedical research from bench to bedside. Eur
J Heart Fail. 19:177–191. 2017. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Talman V and Ruskoaho H: Cardiac fibrosis
in myocardial infarction-from repair and remodeling to
regeneration. Cell Tissue Res. 365:563–581. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fan Z and Guan J: Antifibrotic therapies
to control cardiac fibrosis. Biomater Res. 20:132016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yue Y, Meng K, Pu Y and Zhang X:
Transforming growth factor beta (TGF-β) mediates cardiac fibrosis
and induces diabetic cardiomyopathy. Diabetes Res Clin Pract.
133:124–130. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gao L, Wang LY, Liu ZQ, Jiang D, Wu SY,
Guo YQ, Tao HM, Sun M, You LN, Qin S, et al: TNAP inhibition
attenuates cardiac fibrosis induced by myocardial infarction
through deactivating TGF-β1/Smads and activating P53 signaling
pathways. Cell Death Dis. 11:442020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kong P, Christia P and Frangogiannis NG:
The pathogenesis of cardiac fibrosis. Cell Mol Life Sci.
71:549–574. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ma ZG, Yuan YP, Wu HM, Zhang X and Tang
QZ: Cardiac fibrosis: New insights into the pathogenesis. Int J
Biol Sci. 14:1645–1657. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Asl MN and Hosseinzadeh H: Review of
pharmacological effects of Glycyrrhiza sp. and its bioactive
compounds. Phytother Res. 22:709–724. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee EH, Park KI, Kim KY, Lee JH, Jang EJ,
Ku SK, Kim SC, Suk HY, Park JY, Baek SY and Kim YW: Liquiritigenin
inhibits hepatic fibrogenesis and TGF-β1/Smad with Hippo/YAP
signal. Phytomedicine. 62:1527802019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhu X, Shi J and Li H: Liquiritigenin
attenuates high glucose-induced mesangial matrix accumulation,
oxidative stress, and inflammation by suppression of the NF-κB and
NLRP3 inflammasome pathways. Biomed Pharmacother. 106:976–982.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xie XW: Liquiritigenin attenuates cardiac
injury induced by high fructose-feeding through fibrosis and
inflammation suppression. Biomed Pharmacother. 86:694–704. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
National Research Council (US), .
Committee for the Update of the Guide for the Care and Use of
Laboratory Animals. 8th edition. National Academies Press; 2011
|
|
15
|
Wang L, Yuan D, Zheng J, Wu X, Wang J, Liu
X, He Y, Zhang C, Liu C, Wang T and Zhou Z: Chikusetsu saponin IVa
attenuates isoprenaline-induced myocardial fibrosis in mice through
activation autophagy mediated by AMPK/mTOR/ULK1 signaling.
Phytomedicine. 58:1527642019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu J, Bu L, Gong H, Jiang G, Li L, Ma H,
Zhou N, Lin L, Chen Z, Ye Y, et al: Effects of heart rate and
anesthetic timing on high-resolution echocardiographic assessment
under isoflurane anesthesia in mice. J Ultrasound Med.
29:1771–1778. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang X, Yang C, Liu X and Yang P: Ghrelin
alleviates angiotensin II-induced H9c2 apoptosis: Impact of the
miR-208 family. Med Sci Monit. 24:6707–6716. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
de Boer RA, De Keulenaer G, Bauersachs J,
Brutsaert D, Cleland JG, Diez J, Du XJ, Ford P, Heinzel FR, Lipson
KE, et al: Towards better definition quantification and treatment
of fibrosis in heart failure. A scientific roadmap by the committee
of translational research of the heart failure association (HFA) of
the european society of cardiology. Eur J Heart Fail. 21:272–285.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Suthahar N, Meijers WC, Silljé H and de
Boer RA: From inflammation to fibrosis-molecular and cellular
mechanisms of myocardial tissue remodelling and perspectives on
differential treatment opportunities. Curr Heart Fail Rep.
14:235–250. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang JJ, Rau C, Avetisyan R, Ren S, Romay
MC, Stolin G, Gong KW, Wang Y and Lusis AJ: Genetic dissection of
cardiac remodeling in an isoproterenol-induced heart failure mouse
model. PLoS Genet. 12:e10060382016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bodor GS: Biochemical markers of
myocardial damage. EJIFCC. 27:95–111. 2016.PubMed/NCBI
|
|
22
|
Scherrer-Crosbie M and Kurtz B:
Ventricular remodeling and function: Insights using murine
echocardiography. J Mol Cell Cardiol. 48:512–517. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dobaczewski M, Chen W and Frangogiannis
NG: Transforming growth factor (TGF)-β signaling in cardiac
remodeling. J Mol Cell Cardiol. 51:600–606. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Giannandrea M and Parks WC: Diverse
functions of matrix metalloproteinases during fibrosis. Dis Mod
Mech. 7:193–203. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Schirone L, Forte M, Palmerio S, Yee D,
Nocella C, Angelini F, Pagano F, Schiavon S, Bordin A, Carrizzo A,
et al: A review of the molecular mechanisms underlying the
development and progression of cardiac remodeling. Oxid Med Cell
Longev. 2017:39201952017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tian H, Yu D, Hu Y, Zhang P, Yang Y, Hu Q
and Li M: Angiotensin II upregulates cyclophilin A by enhancing ROS
production in rat cardiomyocytes. Mol Med Rep. 18:4349–4355.
2018.PubMed/NCBI
|
|
27
|
A P, P SR, M PR and K GR: Apoptosis in
angiotensin II-stimulated hypertrophic cardiac cells -modulation by
phenolics rich extract of Boerhavia diffusa L. Biomed Pharmacother.
108:1097–1104. 2018. View Article : Google Scholar : PubMed/NCBI
|