Introduction
Schnyder's crystalline corneal dystrophy (SCCD; MIM
121800) is a rare autosomal dominant genetic disorder that is
characterized by progressive bilateral corneal opacity, owing to
abnormal accumulation of cholesterol and phospholipids in the
cornea (1). The occurrence is equal
in both sexes and progressive opacity in the cornea leads to visual
loss and eventually blindness (2).
In 1924, French ophthalmologists Van Went and Wibaut first reported
eight patients from a family within this pedigree who recorded
symptoms of corneal opacity in both eyes (3). The Swiss ophthalmologist Schnyder
described the clinical manifestations and genetic characteristics
of this eye disease in 1927, 1929 and 1939 (4,5). On
the basis of his detailed elucidation, this special corneal disease
was named Schnyder's crystalline corneal dystrophy (SCCD) (6). In 2007, Orr et al (7), Weiss et al (8) and Yellore et al (9), for the first time, demonstrated that a
potential prenyltransferase, UbiA prenyltransferase
domain-containing 1 (UBIAD1), was the causal gene responsible for
SCCD, independently. Weiss et al and Yellore et al
also confirmed that the loci of UBIAD1 associated with SCCD were
located on chromosome short arm 1, region 36 (8,9). After
decades, the pathogenesis of SCCD was finally elucidated and linked
to the UBIAD1 gene.
UBIAD1, also known as transitional epithelial
response gene 1 (TERE1), was obtained through reverse transcription
from the human bladder mucosal extracted RNA. This cDNA fragment
was novel and different from the existing full-length gene.
Therefore, it was named TERE1. TERE1 is located at the band 6 short
arm of region 3 of human chromosome 1 (1p36) and expresses two
transcripts (1.5 and 3.5 kb), which are widely present in various
human tissues but absent or down-regulated in bladder
muscle-invasive cell carcinoma (10). These two transcripts were deposited
in NCBI with accession numbers AF117064 and NM_013319. The two
transcripts also harbor the same open reading frame and encode the
UBIAD1 protein. Additionally, formation of these two transcripts is
due to alternative splicing (10).
This newly identified TERE1 gene was mapped to chromosome 1
p36.11-p36.33 between the microsatellite markers D1S2667 and
D1S434, through loss of heterozygosity studies, by McGarvey et
al (11), who also demonstrated
a 61% reduced expression of the TERE1 transcriptome in prostate
cancer cells. After expressing TERE1 in prostate cancer cell lines
LNCAP and PC-3, the cell proliferation rate was reduced by 80%,
indicating that TERE1 was a potential tumor suppression factor
(11).
To further understand the function of TERE1,
McGarvey et al (12) then
isolated TERE1-interacted protein apolipoprotein E through
bacterial two-hybrid assays. Results demonstrated that this gene
reduced the p42/44 MAPK phosphorylation level in human 293 cells.
In 2007, different teams confirmed the molecular basis of SCCD
(7–9). However, the present data are still
elusive in the pathogenesis of this rare hereditary eye
disease.
Therefore, in this review studies the history and
clinical symptoms of SCCD, UBIAD1 mutations causing SCCD and the
pathogenesis of SCCD through diligent literature research. The
present study also elucidates the critical correlations between
SCCD and UBIAD1, to guide treatment and drug development for
SCCD.
History and clinical symptoms of SCCD
History of SCCD
Swiss ophthalmologist Schnyder described the
clinical symptoms and inheritance of the rare autosomal dominant
corneal opacity disease and named it Schnyder's crystalline corneal
dystrophy (SCCD) (4,5). Besides, some scientists also reported
similar cases, for instance, Glees et al (1957) (6) and Garner et al (1972) (13). In 1972, Bron et al (14) reported a family with SCCD whose
proband was a 47-year-old Caucasian male with two siblings having
this eye disease. Bron et al spent 3 years tracking the
causes of SCCD in this family. The patients with SCCD in this
pedigree had hyperlipidemia disease and the level of their blood
cholesterol, triglycerides and phospholipids from tests were higher
compared with that in ordinary individuals. Their blood test
results were also consistent within 3 years to this period. Bron
et al recorded SCCD clinical features in this pedigree, such
as the position of crystalline deposits in the cornea, size and
shape of crystalline deposits and visual acuity level. Meanwhile,
their study summarized reports of SCCD cases from 1924 to 1972 and
was supplemented with patients' personal information and blood
lipid-testing results. Finally, they concluded that SCCD occurrence
correlated with dyslipidemia (14).
Similarly, Michaels (15) reported a disparate SCCD patient in
1974 with normal blood lipid whose crystalline deposits developed
from speckles to circular needle-like crystal precipitates within
20 years. Thiel et al (16)
also reported a 19-year-old male SCCD patient with inherited type
IIA autosomal dominant hyperlipoproteinaemia that developed
progressive deteriorated corneal opacity from 1964 to 1976. However
Burns et al (17) used
14C-cholesterol labeling of blood cholesterol before
giving a penetrating keratoplasty surgery to the SCCD patient. At
the time of surgery, 11 days later, cholesterol levels in the
cornea were much higher compared with those in serum, suggesting
that the cornea was an active site for cholesterol absorption and
storage in this eye disorder. Ingraham et al (18) corrected the misunderstanding that
once SCCD developed, it would never worsen in the cornea.
Meanwhile, Ingraham et al reviewed previous studies of SCCD
and concluded that only few patients with SCCD accompanied with
hyperlipidemia and hypercholesterolemia were at risk of this
disease and that these two indicators alone cannot could not be
used as a biomarker for SCCD diagnosis.
Similarly, McCarthy et al (19) performed quantitative biochemical
analysis on a corneal button obtained from an SCCD patient and
found that accumulated corneal lipids are predominantly composed of
phospholipids, free cholesterols and cholesterol esters. Those
constituents were markedly elevated in the SCCD cornea than in the
cadaveric control cornea. Results were: Phospholipid, 23.6 vs. 4.05
mg/g; free cholesterol, 6.99 vs. 0.52 mg/g and cholesterol ester,
3.16 vs. 0.26 mg/g. The authors concluded that the pathogenesis of
SCCD was due to a primary disorder of lipid metabolism in the
cornea. Weiss (20) collected 33
SCCD cases in 1987. Only 17 (51.5%) out of these demonstrated
crystalline deposits in the cornea. As the name SCCD can mislead
ophthalmologists in diagnosis and treatment, Weiss suggested
renaming the disease Schnyder corneal dystrophy (SCD).
Clinical symptoms of SCCD
Schnyder crystalline corneal dystrophy (SCCD, MIM
121800) is a rare autosomal dominant inherited eye disease
characterized by progressive opacification in the bilateral
corneas, caused by the abnormal accumulation of cholesterols and
phospholipids (1,21–23).
To understand better the visual morbidity and surgical intervention
of SCCD, Weiss et al (8)
examined 115 cases from 34 affected families in 2007. Patients were
divided into three categories on the basis of age for statistical
analysis; i) <26 years, ii) 26–39 years and iii) ≥40 years
(24). To date, the youngest SCCD
patient found has been a 17-month-old and the occurrence age ranged
from 2–81 years, with a mean age of 38.8±20.4 years.
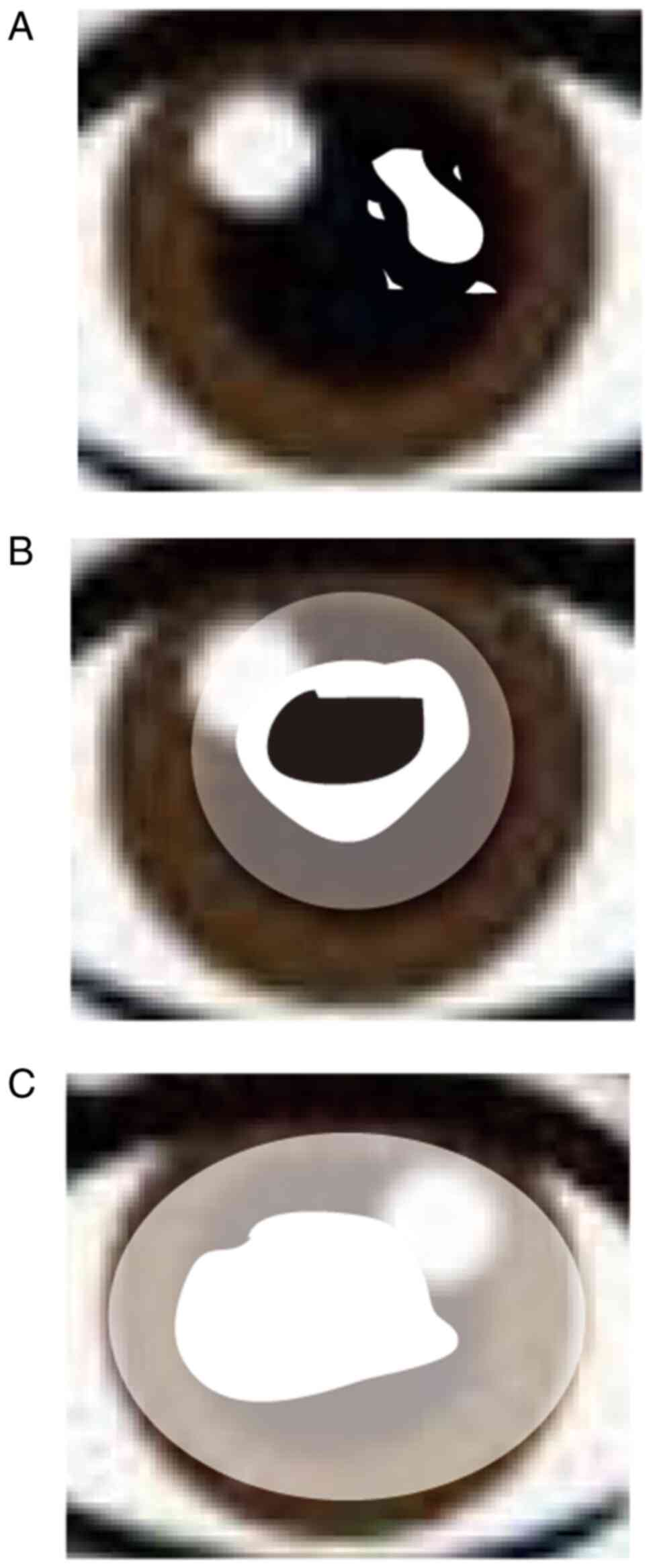
The clinical characteristics include i) early stage
(≤26 years), crystalline deposits in the center stromal epithelium
(Fig. 1A); ii) middle stage (26–39
years), crystalline deposits continue to accumulate and appear to
join together, forming a haze (Fig.
1B); and iii) late stage (≥40 years), which is accompanied by
aging. Here, the degree of crystallization is increasing, which
leads to whole corneal opacity and blindness (Fig. 1C). In addition, ~4% patients with
SCCD developed Genu valgum (24,25).
Patients with SCCD gradually lose their vision as they age, but
their visual acuity was restored to normal through surgery before
they turned 40 years old. When patients with SCCD passed the age of
40, the corneal opacity aggravated to final blindness. To date,
patients with SCCD have had their vision restored mainly through
penetrating keratoplasty and phototherapeutic keratectomy operation
(1,24).
UBIAD1 mutations causing SCCD
UBIAD1 as the molecular basis of
SCCD
The pathogenesis of SCCD remains to be elucidated
and requires urgent attention. Innovative methods in molecular
genetics have been used in uncovering the pathogenesis of corneal
dystrophies (25). The traditional
classification in clinics through histopathological and
electron-microscopical examinations, which reflects an early
disease and immunohistochemical analysis of the deposits, has
already aroused a suspicion that is no longer fulfilled in
dystrophy types (26,27). The BIGH 3 gene point mutation causes
granular dystrophy types I and II, as well as lattice dystrophy
types I and IIIA (28). Point
mutations on the gelsoline gene on chromosome 9 (9q34) result in
the lattice dystrophy type II, which is a manifestation of the
Meretoja syndrome (28). As well as
this mutation, other genes, gene-products and mutations are known
as 16q22 for macular dystrophy, 1p36 for SCCD and 20p11.2-q11.2 for
congenital hereditary endothelial and Schlichting's posterior
polymorphous dystrophies (28). In
another study, Riebeling et al (29) reported the case of a 66-year-old
woman and her son with SCCD. The woman had type IV
hyperlipoproteinemia and hypercholesterolemia, whereas her son had
hypercholesterolemia with elevated LDL-cholesterol levels.
Microsatellite marker analysis demonstrated that D1S228 within the
candidate interval of 1p34.1-p36 led to the observed SCCD. In 1996,
Shearman et al (30)
narrowed the SCCD locus to the 16cM interval between D1S2663 and
D1S228 microsatellite markers localized on 1p34.1-p36 through
haplotype analysis in two large Swede-Finn kindreds in central
Massachusetts.
Similarly, Theendakara et al (31) collected 13 families from Finland,
Germany, Turkey and the USA to refine candidate intervals
correlated with SCCD and they narrowed the putative region to 1.58
Mbp through identity-by-state analysis. To further identify the
genetic basis of SCCD, mutation screening was used to analyze the
15 candidate genes (CORT, CLSTN1, CTNNBIP1, DFFA, ENO1, GPR157,
H6PD, KIF1B, LOC440559, LZIC, MGC4399, PEX14, PGD, PIK3CD and
SSB1), which were localized at putative regions in members of
the two pedigrees affected with SCCD (32). Though none of these 15 genes were
linked to SCCD, Aldave et al (32) reduced the remaining positional
candidate genes by half and led to the identification of the
genetic basis of SCCD.
Orr et al (7)
also confirmed the genetic basis of SCCD, through intensive fine
mapping in a large multigenerational family in Nova Scotia, to
UBIAD1, which encodes a potential prenyltransferase and is involved
in intracellular cholesterol metabolism. Furthermore, Weiss et
al (8) analyzed DNA samples in
six SCCD-affected families and uncovered that, of these six
families, five possessed N102S mutations in UBIAD1 and one family
had a G177R mutation in the UBIAD1 gene. Thus, the molecular basis
of SCCD was clarified and linked to UBIAD1, which participates in
cholesterol synthesis and storage intracellular. After years of
efforts by generations of scientists and ophthalmologists, the
genetic basis of SCCD was elucidated. Their work, therefore,
provided molecular targets and references for SCCD treatment and
drug development for this disease.
UBIAD1 mutations causing SCCD
It is 80 years since the initial description of SCCD
by French ophthalmologists Van Went and Wibaut in 1924 (3) and, finally, the molecular basis of
SCCD has been correlated with UBIAD1, which is located on
chromosome 1 p36 (7–9). Following this, scientists and
ophthalmologists across the world continued to report new cases of
SCCD.
Increasingly, UBIAD1 point mutations leading to SCCD
were also identified (Table I). The
present review has investigated 28 point mutations of UBIAD1 that
cause SCCD through a literature search and the details are
demonstrated in Table I. The
mutation hot spot in UBIAD1 was on 102 (N102S), which had been
demonstrated in a number of SCCD-affected families (7–9,22,33–38).
Similarly, the occurrence of SCCD is higher in European and North
American countries compared with other countries and regions
associated with this gene defect, including Asia and Africa.
Prevalence of SCCD is also reported in the Chinese Han and Japanese
populations (22,36,38,39).
To further understand this gene, Dong et al (40) constructed an SCCD mouse model with
N100S mutation by CRISPR-Cas9 gene editing to further clarify the
pathogenesis and treatment of SCCD. The UBIAD1 mutant mouse line
that was created carried a mis-sense mutation N100S, corresponding
to the human UBIAD1 N102S mutation.
 | Table I.Point mutations of UBIAD1 causing
SCCD (*marks hot spot). |
Table I.
Point mutations of UBIAD1 causing
SCCD (*marks hot spot).
| Author(s)
(year) | Number | Site | Type | (Refs.) |
|---|
| Orr et al
(2007) | 1 | 75 | Ser-Phe | (7) |
| Nickerson et
al (2010), Evans et al (2018) | 2 | 97 | Ala-Thr | (36,87) |
| Jing et al
(2009) | 3 | 98 | Gly-Ser | (39) |
| Orr et al
(2007), Weiss et al (2007), Yellore et al (2007),
Riebeling et al (2003), Nickerson et al (2013),
Al-Ghadeer et al (2011), Du et al (2011), Nickerson
et al (2010), Weiss et al (2010), Meha et al
(2009), Evans et al (2018) | 4 | 102* | Asn-Ser | (7,8,9,29,33,34,35–38,87) |
| Handa et al
(2020), Evans et al (2018) | 5 | 103 | Thr-Ile | (83,87) |
| Orr et al
(2007) | 6 | 112 | Asp-Gly | (7) |
| Nickerson et
al (2013), Nickerson et al (2010), Sarosiak et al
(2018) | 7 | 112 | Asp-Asn | (33,36,84) |
| Riebeling et
al (2003) | 8 | 118 | Asp-Gly | (29) |
| Orr et al
(2007), Yellore et al (2007) | 9 | 119 | Arg-Gly | (7,9) |
| Kitazawa et
al (2018) | 10 | 120 | Thr-Arg | (85) |
| Yellore et
al (2007), Riebeling et al (2003), Al-Ghadeer et
al (2011), Evans et al (2018) | 11 | 121 | Leu-Phe | (9,29,34,87) |
| Nickerson et
al (2010) | 12 | 121 | Leu-Val | (36) |
| Nickerson et
al (2010) | 13 | 122 | Val-Glu | (36) |
| Nickerson et
al (2010) | 14 | 122 | Val-Gly | (36) |
| Riebeling et
al (2003), Nickerson et al (2010), Meha et al
(2009) | 15 | 171 | Ser-Pro | (29,36,38) |
| Riebeling et
al (2003), Nickerson et al (2010) | 16 | 174 | Tyr-Cys | (29,36) |
| Orr et al
(2007), Weiss et al (2007), Riebeling et al (2003),
Nickerson et al (2010) | 17 | 175 | Thr-Ile | (7,8,29,36) |
| Evans et al
(2018) | 18 | 176 | Gly-Glu | (87) |
| Weiss et al
(2007), Nickerson et al (2010) | 19 | 177 | Gly-Arg | (8,36) |
| Nickerson et
al (2013), Kitazawa et al (2018) | 20 | 177 | Gly-Glu | (33,85) |
| Riebeling et
al (2003) | 21 | 181 | Lys-Arg | (29) |
| Riebeling et
al (2003) | 22 | 186 | Gly-Arg | (29) |
| Nickerson et
al (2010) | 23 | 188 | Leu-His | (36) |
| Dudakova et
al (2019) | 24 | 190 | Ile-Thr | (2) |
| Orr et al
(2007), Nickerson et al (2010) | 25 | 232 | Asn-ser | (7,36) |
| Riebeling et
al (2003), Nickerson et al (2010) | 26 | 233 | Asn-His | (29,36) |
| Weiss et al
(2007), Riebeling et al (2003), Nickerson et al
(2010) | 27 | 236 | Asp-Glu | (8,29,36) |
| Weiss et al
(2010) | 28 | 240 | Asp-Asn | (37) |
The genetic basis of SCCD has been demonstrated
since 2007, but the mechanism of SCCD formation is still elusive.
The present study next illustrates the functions of UBIAD1 to build
a connection between abnormal lipid metabolism and the pathogenesis
of SCCD.
Subcellular localization and functions of
UBIAD1
Subcellular localization of
UBIAD1
The GFP-UBIAD1 fusion protein vector was transfected
into a human osteosarcoma cell line MG-63 to investigate the
sub-cellular localization of UBIAD1. The results revealed
endoplasmic reticulum (ER) localization, but not Golgi (41). Nickerson et al (36) also confirmed mitochondrial
sub-localization of UBAID1, but not ER, in cultured corneal cells.
To support their results, immunofluorescence was performed to
ensure wild-type and mutated UBIAD1 (N102S) localization in
excisional human corneal stromal cells. Similarly, Vos et al
(42) reported an orthologous
protein of UBIAD1 in Drosophila that was localized in the
mitochondria, which supported the result of Nickerson et al
(36). Mugoni et al
(43) also indicated that UBIAD1was
a non-mitochondrial localization prenyltransferases that
synthesized CoQ10 in the Golgi. Subsequently, Wang et al
(44) demonstrated that RPWS
residues of UBAID1 proteins were Golgi retention signal peptides.
In mice N2A cells, the UBIAD1 protein was also localized in the
mitochondria, ER and Golgi (45).
Jiang et al (46) examined the subcellular localization
of wild-type UBIAD1 and the SCCD-associated mutants in CHO-K1
cells. They revealed that wild-type UBIAD1 is preferentially
localized at Golgi, whereas the SCCD-associated mutants exhibited a
diffused distribution and were dominantly sequestered in the ER.
Fredericks et al (47) in
his study concluded that the UBIAD1 protein was localized in the
mitochondria, ER and Golgi apparatus with different functions and
exhibited species specificity.
Tumor suppressor
The UBIAD1 is encoded by the UBIAD1 gene and
is involved in significant physiological processes in vivo.
A previous study demonstrated that UBIAD1 inhibited cell
proliferation of transitional cell carcinoma (10). McGarvey et al (11) transduce exogenous UBIAD1 constructs
into two prostate carcinoma cell lines (LNCaP and PC-3), which
markedly decrease cell proliferation by 80%. Fredericks et
al (48) also demonstrated that
UBIAD1 expression decreases in one third of bladder cancer samples
through gene microarray analysis. In constructing bladder carcinoma
nude-mouse model, UBIAD1 expression was induced, which inhibits
tumor occurrence and development. In another study, mutated UBIAD1
did not bind to APOE which causes abnormal cholesterol levels in
vivo (12). Elevated
cholesterol levels also regulated cancer cell apoptosis and
proliferation, thus distorting the balance maintained by
intracellular UBIAD1 (48).
Therefore, although UBIAD1 played important roles in bladder tumor
suppression, the mechanism remains to be elucidated.
UBIAD1 as a novel CoQ10 and vitamin K2
biosynthetic enzyme
The UBIAD1 gene encodes a prenyltransferase
containing 338 amino acids, with a molecular weight of 36.83 kDa
(7,11). Nakagawa et al (41) decreases UBIAD1 expression in human
cells using siRNA, which largely suppresses the conversion of
deuterium-labeled vitamin K derivatives to deuterium-labeled
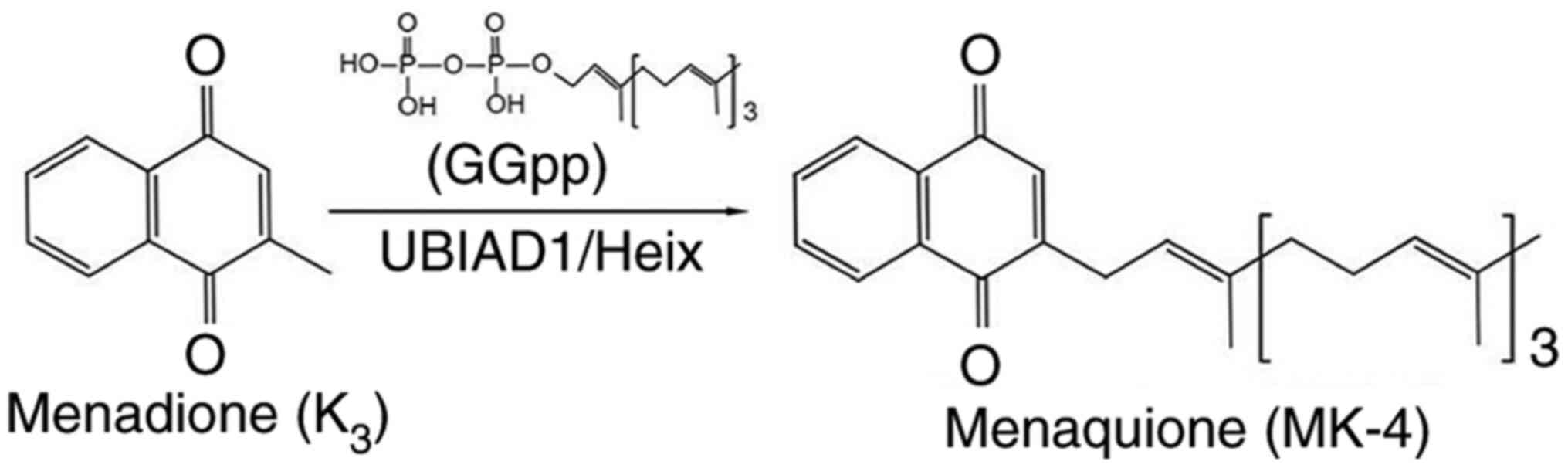
menaquinone-4 (MK-4). To establish UBIAD1 function, Nakagawa et
al (41) also infected insect
cells sf9 with UBIAD1 baculovirus and detected deuterium-labeled
MK-4 formed from vitamin K derivatives. Furthermore, Nakagawa et
al (41) first identified
UBIAD1 as an MK-4 biosynthesis enzyme in humans (Fig. 2), which suggests that vitamin K did
not only existing in plant form, phylloquinone (PK), but also in
bacterial form (MK-4).
Heix (Heixuedian), is an orthologous protein of
UBIAD1 in Drosophila melanogaster, composed of 359 amino
acids with a molecular weight of 39.22 kDa (42). Following Heix mutation, a black dot
phenotype lymphoma appeared in the 3rd to 4th instar larvae in
Drosophila (49,50). UBIAD1 localizes at the mitochondria
and converts vitamin K1 to K2, which is a
significant cofactor in eukaryotic blood coagulation and an
electronic carrier binding to the cell membrane in bacteria
(43). By contrast, vitamin
K2 is necessary and sufficient for electron transport in
Drosophila mitochondria (42). Therefore, when Heix is mutated,
mitochondria dysfunction arise. However, supplementation with
vitamin K2 rescues mitochondrial dysfunction (42). Heix is also a dosage-sensitive
modifier of pink1, which is mutated in Parkinson's disease, as it
affects mitochondrial function. Additionally, Heix converts
menadione to vitamin K2 in Drosophila (Fig. 2) (42).
Mugoni et al (43) found that UBIAD1 is a
non-mitochondrial localized prenyltransferase and it catalyzed
CoQ10 synthesis on the Golgi membrane. Reduced expression of UBIAD1
in vascular cells could decrease antioxidant CoQ10 cytoplasmic
content, resulting in reactive oxygen species (ROS)-mediated lipid
peroxidation. Similarly, the inhibition of endothelial nitric oxide
synthase (eNOS) can prevent UBIAD1-dependent oxidative damage,
revealing the crucial role of UBIAD1 and CoQ10 in nitric oxide (NO)
signaling and cardiovascular system.
To further broaden UBIAD1 function, Mugoni et
al (43) used zebrafish to
investigate intracellular roles of the UBIAD1 ortholog gene,
Barolo (Bar). The zebrafish cardiovascular system fails
after Bar is mutated, resulting from oxidative stress and
ROS-mediated cell damage. Further study also demonstrated that
UBIAD1 is a CoQ10 biosynthesis enzyme localized at the Golgi
apparatus, which regulates ROS levels and the redox status in
vertebrate cardiovascular system. Therefore, increasing oxidative
stress causes heart and vascular cell apoptosis, as well as
cardiovascular system failure when UBIAD1 is deficient (43).
Involvement of UBIAD1 in cellular
lipid metabolism
When UBAID1 mutates, cholesterols and lipids
accumulate abnormally in the cornea, leading to SCCD disease,
indicating that UBIAD1 is involved in lipid metabolism in cells
(7–9,16,17,19,46).
The over-expression of UBIAD1 decreases cholesterol levels in 293
cells and cannot interact with the APOE protein after point
mutation, which causes the abnormal metabolism of intracellular
cholesterol (48). Fredericks et
al (47,51) also constructed a cell line model
based on a castration-resistant prostate cancer cell LNCap-C81 to
reveal the association between UBIAD1 and steroid synthesis.
Intracellular cholesterol synthesis rate, content and correlated
enzymes increased when UBIAD1 was deficient. By contrast, the
down-regulation of UBIAD1 resulted in mitochondrial damage and
considerable cholesterol storage in hepatocellular carcinoma cells,
which indicated the crucial role of UBIAD1 in mitochondrial
function (52). Preliminary
functional studies of UBIAD1 also focused on cell morphology and
biochemical parameter change after abnormal expression of UBAID1
(10–12,41,43,48,52).
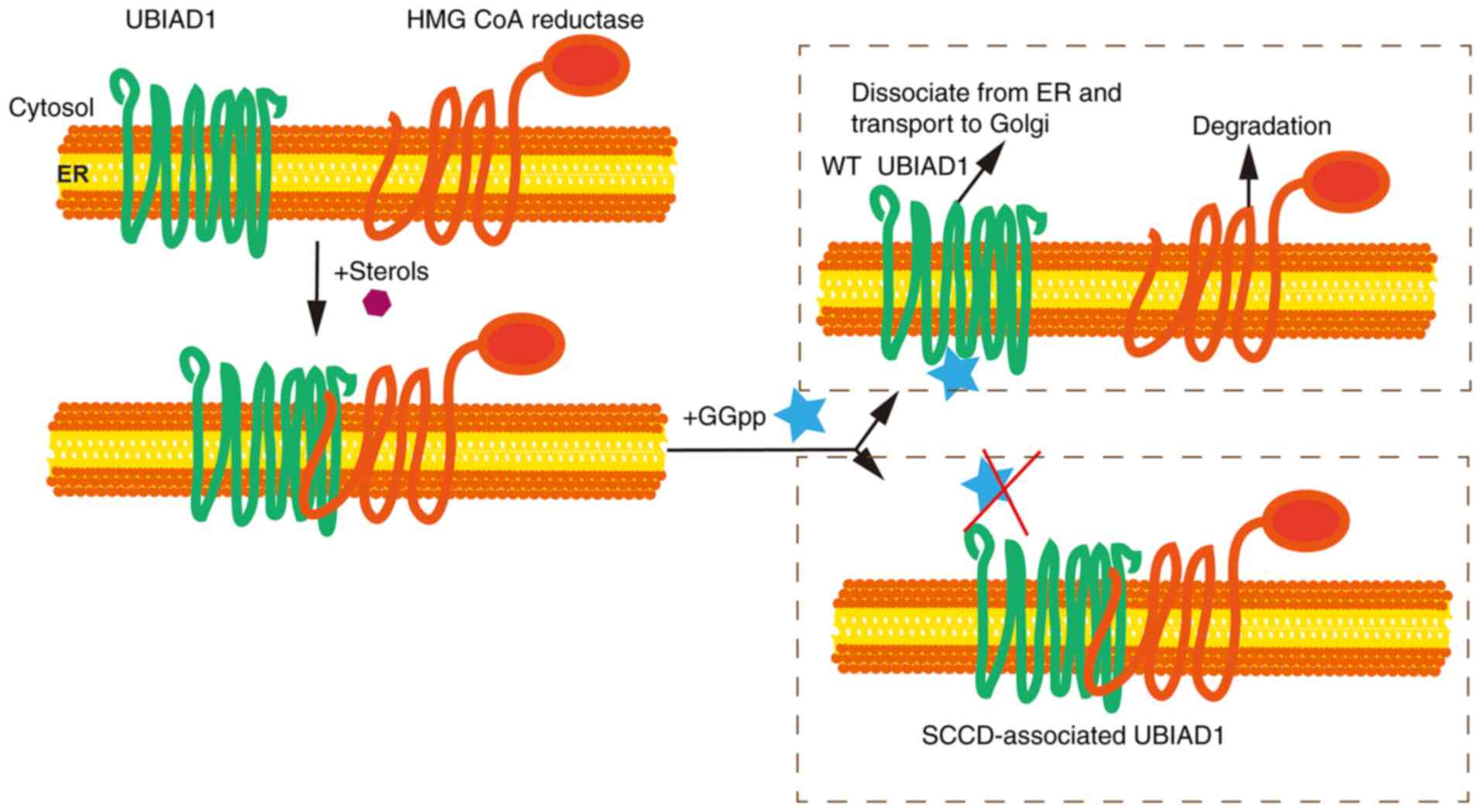
Schumacher et al (53–55)
demonstrated that sterols stimulates UBIAD1 binding to HMG CoA
reductase, which is a cholesterol biosynthetic enzyme and subject
to sterol-accelerated, endoplasmic reticulum-associated degradation
(ERAD) augmented by the non-sterol isoprenoid geranylgeraniol.
Geranylgeraniol then inhibits UBIAD1 binding of
3-hydroxy-3-methyl-glutaryl coenzyme A reductase (HMGCR) that
promotes reductase degradation and transport of UBIAD1 from the ER
to Golgi. This mutation results in a conformation change of mutated
UBAID1, thereby inhibiting the binding of GGpp to UBIAD1 mutants,
which prevents HMGCR degradation and dissociation of UBIAD1, thus
contributing to consistent synthesis and accumulation of
cholesterol in vivo (Fig.
3). HMGCR is an important rate-limiting enzyme in the
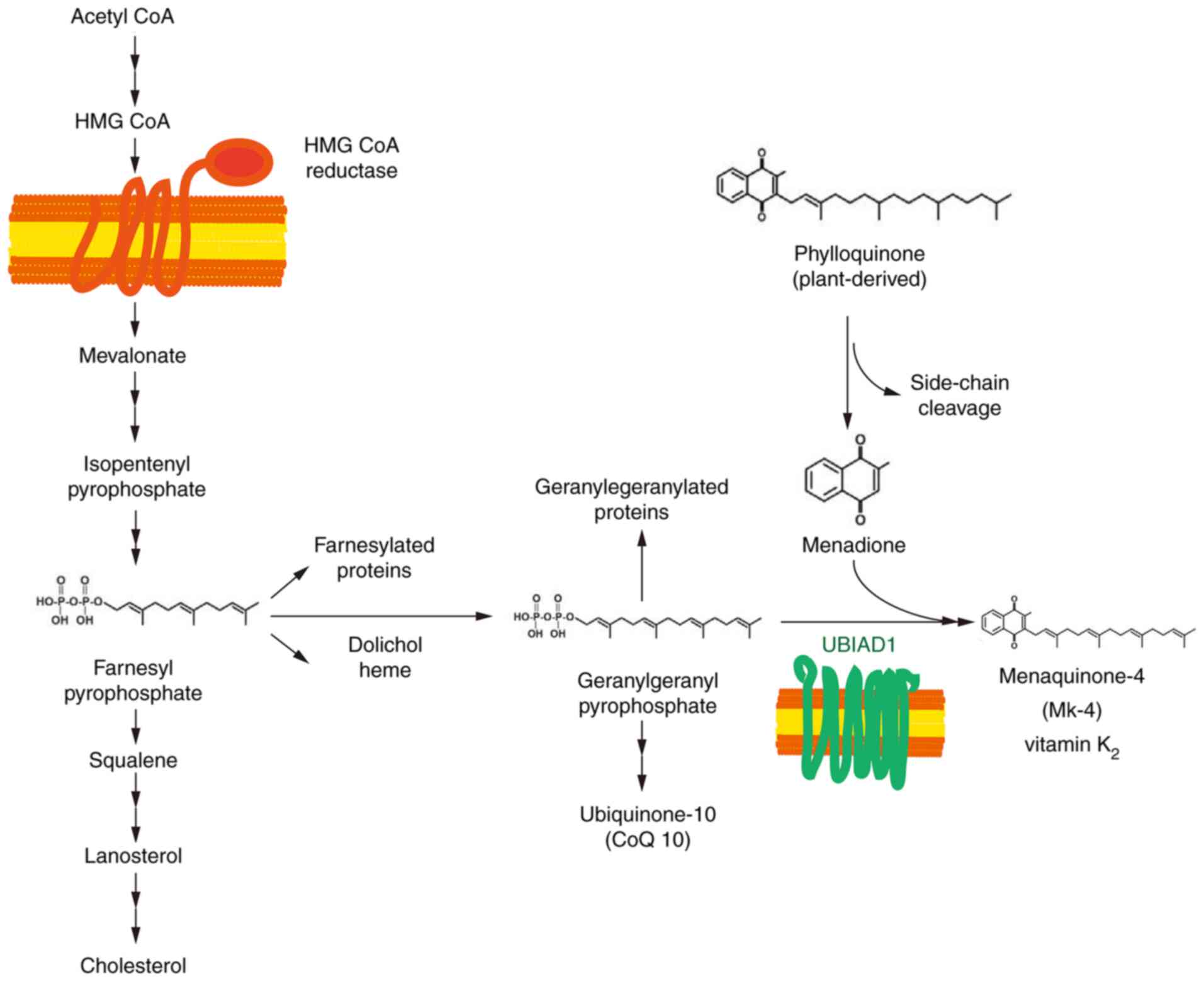
cholesterol and non-sterol isoprenoid biosynthetic pathway, which
is regulated by cholesterol feedback. Mevalonic acid is produced
through the reduction of HMG CoA, which forms farnesyl
pyrophosphate through a series of reactions and is a substrate in
the biosynthesis of GGpp and cholesterol (Fig. 4) (56).
The UbiA superfamily of proteins is a collection of
transmembrane prenyltransferases that catalyze the biosynthesis of
a number of crucial molecules, such as, ubiquinones, menaquinones,
plastoquinones, hemes, chlorophylls, vitamin E and structural
lipids (57). These vital compounds
serve as electron and proton carriers for cellular respiration and
photosynthesis. They also serve as antioxidants to decrease cell
damage and as structural components of microbial cell membranes,
which indicates that the UbiA superfamily of proteins is involved
in significant physiological processes and human diseases (57). Compared with the Golgi localization
of wild-type UBIAD1, SCCD-associated mutants are dominantly
detained in the ER and compete with insulin-induced gene 1 (INSIG1)
for HMGCR binding, hence preventing HMGCR from degradation and
increasing the biosynthesis of cholesterol (46). INSIG1 encodes for a probable six
trans-membrane domain protein of 277 amino acids. Sterols then
induce the ER-anchored INSIG1, which competes with UBIAD1 to bind
HMGCR. Also, INSIG1 is associated with E3 ubiquitin ligase, which
ubiquitinates HMGCR, eventually leading to the degradation of HMGCR
(46).
Jiang et al (46) constructed heterozygous Ubiad1 G184R
(Ubiad1G184R/+) knock-in mice that exhibited an
elevated expression of HMGCR in various tissues. The aged
Ubiad1G184R/+ mice, which exhibited identical
clinical characteristics with patients with SCCD, demonstrated
corneal cholesterol accumulation and opacification. These results
indicated that SCCD-associated mutants impede its ER-to-Golgi
transport and stabilize its interaction with HMGCR. This disturbed
transport then increases cholesterol biosynthesis, causing excess
accumulation of cholesterol in the cornea and, eventually, SCCD.
Previous studies have demonstrated that GGpp triggers the release
of UBIAD1 from HMGCR, allowing ERAD and ER-to-Golgi transport of
UBIAD1 (53–55). A SCCD-related mice model affirms the
physiological significance of UBIAD1 in cholesterol homeostasis and
demonstrates the inhibition of HMGCR ERAD, contributing to SCCD
pathogenesis (46,58). Similarly, Jun et al (59) establishes a biochemical assay for
UBIAD1-mediated synthesis of MK-4 in isolated membranes and intact
cells. The results reveal that mutated UBIAD1 exhibited reduced
Mk-4 biosynthetic activity compared with wild-type UBIAD1.
Sequestration in the ER, therefore, protects
SCCD-associated UBIAD1 from autophagy and allows intracellular
accumulation of the mutated protein, which magnifies the inhibition
of HMGCR ERAD. The results of Jun et al (59) further broaden the understanding of
SCCD pathogenesis and limit the efficacy of cholesterol-lowering
statin therapies.
Conclusion
Mutations in UBIAD1, which synthesizes vitamin
K2 (subtype menaquinone-4, MK-4) and CoQ10, account for
the rare autosomal dominant genetic disorder SCCD, also known as
SCD. It also regulates eNOS activity based on its antioxidant
abilities (36–38). Progressive opaqueness of the cornea,
due to the aberrant accumulation of cholesterol, is one of a number
of symptoms of SCCD (1,13–25).
French ophthalmologists Van Went and Wibaut in 1924 (3) first described SCCD. In the following
years, a number of SCCD-affected families were reported across the
world (13–18,20,23,28–31,60–86).
The search for the pathogenesis and molecular basis
of SCCD lasted for decades until 2007, when three teams uncovered
the conundrum due to UBIAD1 mutations (7–9). The
pathogenesis of SCCD was then investigated and clarified by
consistently exploring the functions of UBIAD1. From research, it
was identified that UBIAD1 regulates HMGCR through GGpp in
cholesterol biosynthesis and metabolism of significant componential
molecules (46,53,54,56,58,59,87,88).
UBIAD1 is also a critical tumor suppressor in the urinary system
(11,44,47,48,51,88)
and regulates cell proliferation through the ras signaling pathway
(88). UBIAD1 homologous proteins
are widely expressed in bacteria and eukaryotes and play vital
roles in different intracellular physiological processes.
The present review clarified the pathogenesis and
functions of the SCCD-associated gene UBIAD1, therefore guiding
effective diagnosis and treatment of the inherent eye disease.
Acknowledgements
This review was written to memorialize Professor
Ling Hong (Huazhong University of Science and Technology) who died
of COVID-19 on 7 February, 2020.
Funding
This study was supported by the Fund of Hubei
Provincial Department of Education (grant no. Q20204508), the
Talent Introduction Project of Hubei Polytechnic University (grant
no. 19XJK02R) and the Medical and Health Science and Technology
Development Program of Shandong Province (grant no. 2019WS208).
Availability of data and materials
Not applicable.
Authors' contributions
JX and LL wrote and revised the manuscript. JX
critically revised and corrected the manuscript. JX and LL
conceived the idea for the review, collected and interpreted the
studies included, reviewed the manuscript and contributed
significantly to the writing the manuscript. Both authors read and
approved the final manuscript. Data authentication is not
applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
SCCD
|
Schnyder's crystalline corneal
dystrophy
|
|
UBIAD1
|
UbiA prenyltransferase
domain-containing 1
|
|
TERE1
|
transitional epithelial response gene
1
|
|
HMGCR
|
3-hydroxy-3-methyl-glutaryl coenzyme
A reductase
|
|
GGpp
|
geranylgeranyl diphosphate
|
|
ROS
|
reactive oxygen species
|
|
ERAD
|
endoplasmic reticulum-associated
degradation
|
References
|
1
|
Weiss JS: Schnyder corneal dystrophy. Curr
Opin Ophthalmol. 20:292–298. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dudakova L, Skalicka P, Davidson AE and
Liskova P: Coincidental occurrence of Schnyder corneal dystrophy
and posterior polymorphous corneal dystrophy type 3. Cornea.
38:758–760. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Van Went JM and Wibaut F: A strange
inherited corneal alteration. Niederl Tijdschr Geneesk.
68:2996–2997. 1924.(In Dutch).
|
|
4
|
Schnyder WF: Report about a new type of
familial corneal disorder. Schweiz Med Wschr. 10:559–571. 1929.(In
German).
|
|
5
|
Schnyder WF: Disk-like inherited
crytstalline inclusions in the corneal center. KIin Monatsbl
Augenheilkd. 103:494–502. 1939.(In German).
|
|
6
|
Glees M: Corneal crystalline dystrophy.
Klin Monbl Augenheilkd Augenarztl Fortbild. 131:721–724. 1957.(In
German). PubMed/NCBI
|
|
7
|
Orr A, Dubé MP, Marcadier J, Jiang H,
Federico A, George S, Seamone C, Andrews D, Dubord P, Holland S, et
al: Mutations in the UBIAD1 gene, encoding a potential
prenyltransferase, are causal for Schnyder crystalline corneal
dystrophy. PLoS One. 2:e6852007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Weiss JS, Kruth HS, Kuivaniemi H, Tromp G,
White PS, Winters RS, Lisch W, Henn W, Denninger E, Krause M, et
al: Mutations in the UBIAD1 gene on chromosome short arm 1, region
36, cause Schnyder crystalline corneal dystrophy. Invest Ophthalmol
Vis Sci. 48:5007–5012. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yellore VS, Khan MA, Bourla N, Rayner SA,
Chen MC, Sonmez B, Momi RS, Sampat KM, Gorin MB and Aldave AJ:
Identification of mutations in UBIAD1 following exclusion of coding
mutations in the chromosome 1p36 locus for Schnyder crystalline
corneal dystrophy. Mol Vis. 13:1777–1782. 2007.PubMed/NCBI
|
|
10
|
McGarvey TW, Nguyen T, Tomaszewski JE,
Monson FC and Malkowicz SB: Isolation and characterization of the
TERE1 gene, a gene down-regulated in transitional cell carcinoma of
the bladder. Oncogene. 20:1042–1051. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
McGarvey TW, Nguyen T, Puthiyaveettil R,
Tomaszewski JE and Malkowicz SB: TERE1, a novel gene affecting
growth regulation in prostate carcinoma. Prostate. 54:144–155.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
McGarvey TW, Nguyen TB and Malkowicz SB:
An interaction between apolipoprotein E and TERE1 with a possible
association with bladder tumor formation. J Cell Biochem.
95:419–428. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Garner A and Tripathi RC: Hereditary
crystalline stromal dystrophy of Schnyder. II. Histopathology and
ultrastructure. Br J Ophthalmol. 56:400–408. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bron AJ, Williams HP and Carruthers ME:
Hereditary crystalline stromal dystrophy of Schnyder. I. Clinical
features of a family with hyperlipoproteinaemia. Br J Ophthalmol.
56:383–399. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Michaels RG: Corneal crystalline dystrophy
of Schnyder. Arch Ophthalmol. 92:64–65. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Thiel HJ, Voigt GJ and Parwaresch MR:
Crystalline corneal dystrophy (Schnyder) in the presence of
familial type IIa hyperlipoproteinaemia (author's transl). Klin
Monbl Augenheilkd. 171:678–684. 1977.(In German). PubMed/NCBI
|
|
17
|
Burns RP, Connor W and Gipson I:
Cholesterol turnover in hereditary crystalline corneal dystrophy of
Schnyder. Trans Am Ophthalmol Soc. 76:184–196. 1978.PubMed/NCBI
|
|
18
|
Ingraham HJ, Perry HD, Donnenfeld ED and
Donaldson DD: Progressive Schnyder's corneal dystrophy.
Ophthalmology. 100:1824–1827. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
McCarthy M, Innis S, Dubord P and White V:
Panstromal Schnyder corneal dystrophy. A clinical pathologic report
with quantitative analysis of corneal lipid composition.
Ophthalmology. 101:895–901. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Weiss JS: Schnyder crystalline dystrophy
sine crystals. Recommendation for a revision of nomenclature.
Ophthalmology. 103:465–473. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Weiss JS: More on Schnyder corneal
dystrophy. Ophthalmology. 116:22602009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Weiss JS, Kruth HS, Kuivaniemi H, Tromp G,
Karkera J, Mahurkar S, Lisch W, Dupps WJ Jr, White PS, Winters RS,
et al: Genetic analysis of 14 families with Schnyder crystalline
corneal dystrophy reveals clues to UBIAD1 protein function. Am J
Med Genet A. 146A:271–283. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lisch W, Weidle EG, Lisch C, Rice T, Beck
E and Utermann G: Schnyder's dystrophy. Progression and metabolism.
Ophthalmic Paediatr Genet. 7:45–56. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Weiss JS: Visual morbidity in thirty-four
families with Schnyder crystalline corneal dystrophy (an American
Ophthalmological Society thesis). Trans Am Ophthalmol Soc.
105:616–648. 2007.PubMed/NCBI
|
|
25
|
Jing Y and Wang L: Morphological
evaluation of Schnyder's crystalline corneal dystrophy by laser
scanning confocal microscopy and fourier-domain optical coherence
tomography. Clin Exp Ophthalmol. 37:308–312. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Weiss JS, Møller HU, Lisch W, Kinoshita S,
Aldave AJ, Belin MW, Kivelä T, Busin M, Munier FL, Seitz B, et al:
The IC3D classification of the corneal dystrophies. Cornea. 27
(Suppl 2):S1–S83. 2008.(In English, Spanish). View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Weiss JS: Corneal dystrophy
classification. Ophthalmology. 116:1013–1014. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Auw-Hädrich C and Witschel H: Corneal
dystrophies in the light of modern molecular genetic research.
Ophthalmologe. 99:418–426. 2002.(In German). View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Riebeling P, Polz S, Tost F, Weiss JS,
Kuivaniemi H and Hoeltzenbein M: Schnyder's crystalline corneal
dystrophy. Further narrowing of the linkage interval at chromosome
1p34.1-p36? Ophthalmologe. 100:979–983. 2003.(In German).
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shearman AM, Hudson TJ, Andresen JM, Wu X,
Sohn RL, Haluska F, Housman DE and Weiss JS: The gene for
Schnyder's crystalline corneal dystrophy maps to human chromosome
1p34.1-p36. Hum Mol Genet. 5:1667–1672. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Theendakara V, Tromp G, Kuivaniemi H,
White PS, Panchal S, Cox J, Winters RS, Riebeling P, Tost F,
Hoeltzenbein M, et al: Fine mapping of the Schnyder's crystalline
corneal dystrophy locus. Hum Genet. 114:594–600. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Aldave AJ, Rayner SA, Principe AH, Affeldt
JA, Katsev D and Yellore VS: Analysis of fifteen positional
candidate genes for Schnyder crystalline corneal dystrophy. Mol
Vis. 11:713–716. 2005.PubMed/NCBI
|
|
33
|
Nickerson ML, Bosley AD, Weiss JS, Kostiha
BN, Hirota Y, Brandt W, Esposito D, Kinoshita S, Wessjohann L,
Morham SG, et al: The UBIAD1 prenyltransferase links menaquinone-4
[corrected] synthesis to cholesterol metabolic enzymes. Hum Mutat.
34:317–329. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Al-Ghadeer H, Mohamed JY and Khan AO:
Schnyder corneal dystrophy in a Saudi Arabian family with
heterozygous UBIAD1 mutation (p.L121F). Middle East Afr J
Ophthalmol. 18:61–64. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Du C, Li Y, Dai L, Gong L and Han C: A
mutation in the UBIAD1 gene in a Han Chinese family with Schnyder
corneal dystrophy. Mol Vis. 17:2685–2692. 2011.PubMed/NCBI
|
|
36
|
Nickerson ML, Kostiha BN, Brandt W,
Fredericks W, Xu KP, Yu FS, Gold B, Chodosh J, Goldberg M, Lu DW,
et al: UBIAD1 mutation alters a mitochondrial prenyltransferase to
cause Schnyder corneal dystrophy. PLoS One. 5:e107602010.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Weiss JS, Wiaux C, Yellore V, Raber I,
Eagle R, Mequio M and Aldave A: Newly reported p.Asp240Asn mutation
in UBIAD1 suggests central discoid corneal dystrophy is a variant
of Schnyder corneal dystrophy. Cornea. 29:777–780. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mehta JS, Vithana EN, Venkataraman D,
Venkatraman A, Yong VH, Aung T and Tan DT: Surgical management and
genetic analysis of a Chinese family with the S171P mutation in the
UBIAD1 gene, the gene for Schnyder corneal dystrophy. Br J
Ophthalmol. 93:926–931. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jing Y, Liu C, Xu J and Wang L: A novel
UBIAD1 mutation identified in a Chinese family with Schnyder
crystalline corneal dystrophy. Mol Vis. 15:1463–1469.
2009.PubMed/NCBI
|
|
40
|
Dong F, Jin X, Boettler MA, Sciulli H,
Abu-Asab M, Del Greco C, Wang S, Hu YC, Campos MM, Jackson SN, et
al: A mouse model of Schnyder corneal dystrophy with the N100S
point mutation. Sci Rep. 8:102192018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Nakagawa K, Hirota Y, Sawada N, Yuge N,
Watanabe M, Uchino Y, Okuda N, Shimomura Y, Suhara Y and Okano T:
Identification of UBIAD1 as a novel human menaquinone-4
biosynthetic enzyme. Nature. 468:117–121. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Vos M, Esposito G, Edirisinghe JN, Vilain
S, Haddad DM, Slabbaert JR, Van Meensel S, Schaap O, De Strooper B,
Meganathan R, et al: Vitamin K2 is a mitochondrial electron carrier
that rescues pink1 deficiency. Science. 336:1306–1310. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mugoni V, Postel R, Catanzaro V, De Luca
E, Turco E, Digilio G, Silengo L, Murphy MP, Medana C, Stainier DY,
et al: Ubiad1 is an antioxidant enzyme that regulates eNOS activity
by CoQ10 synthesis. Cell. 152:504–518. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang X, Wang D, Jing P, Wu Y, Xia Y, Chen
M and Hong L: A novel Golgi retention signal RPWS for tumor
suppressor UBIAD1. PLoS One. 8:e720152013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Huang Y and Hu Z: UBIAD1 protects against
oxygen-glucose deprivation/reperfusion-induced multiple subcellular
organelles injury through PI3K/AKT pathway in N2A cells. J Cell
Physiol. 233:7480–7496. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Jiang SY, Tang JJ, Xiao X, Qi W, Wu S,
Jiang C, Hong J, Xu J, Song BL and Luo J: Schnyder corneal
dystrophy-associated UBIAD1 mutations cause corneal cholesterol
accumulation by stabilizing HMG-CoA reductase. PLoS Genet.
15:e10082892019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Fredericks WJ, Yin H, Lal P,
Puthiyaveettil R and Malkowicz SB, Fredericks NJ, Tomaszewski J,
Rauscher FJ III and Malkowicz SB: Ectopic expression of the TERE1
(UBIAD1) protein inhibits growth of renal clear cell carcinoma
cells: Altered metabolic phenotype associated with reactive oxygen
species, nitric oxide and SXR target genes involved in cholesterol
and lipid metabolism. Int J Oncol. 43:638–652. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Fredericks WJ, McGarvey T, Wang H, Lal P,
Puthiyaveettil R, Tomaszewski J, Sepulveda J, Labelle E, Weiss JS,
Nickerson ML, et al: The bladder tumor suppressor protein TERE1
(UBIAD1) modulates cell cholesterol: Implications for tumor
progression. DNA Cell Biol. 30:851–864. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Xia Y, Midoun SZ, Xu Z and Hong L:
Heixuedian (heix), a potential melanotic tumor suppressor gene,
exhibits specific spatial and temporal expression pattern during
Drosophila hematopoiesis. Dev Biol. 398:218–230. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Dragh MA, Xu Z, Al-Allak ZS and Hong L:
Vitamin K2 prevents lymphoma in Drosophila. Sci Rep. 7:170472017.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Fredericks WJ, Sepulveda J, Lai P,
Tomaszewski JE, Lin MF, McGarvey T, Rauscher FJ III and Malkowicz
SB: The tumor suppressor TERE1 (UBIAD1) prenyltransferase regulates
the elevated cholesterol phenotype in castration resistant prostate
cancer by controlling a program of ligand dependent SXR target
genes. Oncotarget. 4:1075–1092. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Morales CR, Grigoryeva LS, Pan X, Bruno L,
Hickson G, Ngo MH, McMaster CR, Samuels ME and Pshezhetsky AV:
Mitochondrial damage and cholesterol storage in human
hepatocellular carcinoma cells with silencing of UBIAD1 gene
expression. Mol Genet Metab Rep. 1:407–411. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Schumacher MM, Jun DJ, Johnson BM and
DeBose-Boyd RA: UbiA prenyltransferase domain-containing protein-1
modulates HMG-CoA reductase degradation to coordinate synthesis of
sterol and nonsterol isoprenoids. J Biol Chem. 293:312–323. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Schumacher MM, Jun DJ, Jo Y, Seemann J and
DeBose-Boyd RA: Geranylgeranyl-regulated transport of the
prenyltransferase UBIAD1 between membranes of the ER and Golgi. J
Lipid Res. 57:1286–1299. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Schumacher MM, Elsabrouty R, Seemann J, Jo
Y and DeBose-Boyd RA: The prenyltransferase UBIAD1 is the target of
geranylgeraniol in degradation of HMG CoA reductase. Elife.
4:e055602015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Johnson BM and DeBose-Boyd RA: Underlying
mechanisms for sterol-induced ubiquitination and ER-associated
degradation of HMG CoA reductase. Semin Cell Dev Biol. 81:121–128.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Li W: Bringing bioactive compounds into
membranes: The UbiA superfamily of intramembrane aromatic
prenyltransferases. Trends Biochem Sci. 41:356–370. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Jo Y, Hamilton JS, Hwang S, Garland K,
Smith GA, Su S, Fuentes I, Neelam S, Thompson BM, McDonald JG and
DeBose-Boyd RA: Schnyder corneal dystrophy-associated UBIAD1
inhibits ER-associated degradation of HMG CoA reductase in mice.
Elife. 8:e443962019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Jun DJ, Schumacher MM, Hwang S, Kinch LN,
Grishin NV and DeBose-Boyd RA: Schnyder corneal
dystrophy-associated UBIAD1 is defective in MK-4 synthesis and
resists autophagy-mediated degradation. J Lipid Res. 61:746–757.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Wu CW, Lin PY, Liu YF, Liu TC, Lin MW,
Chen WM, Lee FL, Lee SM and Hsu WM: Central corneal mosaic
opacities in Schnyder's crystalline dystrophy. Ophthalmology.
112:650–653. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Köksal M, Kargi S, Gürelik G and Akata F:
Phototherapeutic keratectomy in Schnyder crystalline corneal
dystrophy. Cornea. 23:311–313. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Paparo LG, Rapuano CJ, Raber IM, Grewal S,
Cohen EJ and Laibson PR: Phototherapeutic keratectomy for
Schnyder's crystalline corneal dystrophy. Cornea. 19:343–347. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Vesaluoma MH, Linna TU, Sankila EM, Weiss
JS and Tervo TM: In vivo confocal microscopy of a family with
Schnyder crystalline corneal dystrophy. Ophthalmology. 106:944–951.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Dinh R, Rapuano CJ, Cohen EJ and Laibson
PR: Recurrence of corneal dystrophy after excimer laser
phototherapeutic keratectomy. Ophthalmology. 106:1490–1497. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Meier U, Anastasi C, Failla F and Simona
F: Possibilities of therapeutic photokeratotomy with the excimer
laser in treatment of Schnyder crystalline corneal dystrophy. Klin
Monbl Augenheilkd. 212:405–406. 1998.(In German). View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Battisti C, Dotti MT, Malandrini A,
Pezzella F, Bardelli AM and Federico A: Schnyder corneal
crystalline dystrophy: Description of a new family with evidence of
abnormal lipid storage in skin fibroblasts. Am J Med Genet.
75:35–39. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Takeuchi T, Furihata M, Heng HH, Sonobe H
and Ohtsuki Y: Chromosomal mapping and expression of the human B120
gene. Gene. 213:189–193. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Kohnen T, Pelton RW and Jones DB: Schnyder
corneal dystrophy and juvenile, systemic hypercholesteremia. Klin
Monbl Augenheilkd. 211:135–137. 1997.(In German). View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Santo RM, Yamaguchi T, Kanai A, Okisaka S
and Nakajima A: Clinical and histopathologic features of corneal
dystrophies in Japan. Ophthalmology. 102:557–567. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Chern KC and Meisler DM: Disappearance of
crystals in Schnyder's crystalline corneal dystrophy after
epithelial erosion. Am J Ophthalmol. 120:802–803. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Weiss JS: Schnyder's dystrophy of the
cornea. A swede-finn connection. Cornea. 11:93–101. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Brownstein S, Jackson WB and Onerheim RM:
Schnyder's crystalline corneal dystrophy in association with
hyperlipoproteinemia: Histopathological and ultrastructural
findings. Can J Ophthalmol. 26:273–279. 1991.PubMed/NCBI
|
|
73
|
Rodrigues MM, Kruth HS, Krachmer JH,
Vrabec MP and Blanchette-Mackie J: Cholesterol localization in
ultrathin frozen sections in Schnyder's corneal crystalline
dystrophy. Am J Ophthalmol. 110:513–517. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Freddo TF, Polack FM and Leibowitz HM:
Ultrastructural changes in the posterior layers of the cornea in
Schnyder's crystalline dystrophy. Cornea. 8:170–177. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Wakita M, Kanai A and Nakajima A: Schnyder
crystalline dystrophy. Nippon Ganka Gakkai Zasshi. 93:676–681.
1989.(In Japanese). PubMed/NCBI
|
|
76
|
Kompf J, Ritter H, Lisch W, Weidle EG and
Baur MP: Linkage analysis in granular corneal dystrophy (Groenouw
I), Schnyder's crystalline corneal dystrophy, and reis-bucklers'
corneal dystrophy. Graefes Arch Clin Exp Ophthalmol. 227:538–540.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Rodrigues MM, Kruth HS, Krachmer JH and
Willis R: Unesterified cholesterol in Schnyder's corneal
crystalline dystrophy. Am J Ophthalmol. 104:157–163. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Roth AM, Ekins MB, Waring GO III, Gupta LM
and Rosenblatt LS: Oval corneal opacities in beagles. III.
Histochemical demonstration of stromal lipids without
hyperlipidemia. Invest Ophthalmol Vis Sci. 21:95–106.
1981.PubMed/NCBI
|
|
79
|
Bec P, Arne JL, Secheyron P, Poitevin B
and Hemous JD: Schnyder's crystalline dystrophy of the cornea. Bull
Soc Ophtalmol Fr. 79:1005–1007. 1979.(In French). PubMed/NCBI
|
|
80
|
Ehlers N and Matthiessen ME: Hereditary
crystalline corneal dystrophy of Schnyder. Acta Ophthalmol
(Copenh). 51:316–324. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Delogu A: Contribution to the knowledge of
Schnyder's crystalline corneal dystrophy. Ann Ottalmol Clin Ocul.
93:1219–1225. 1967.(In Italian). PubMed/NCBI
|
|
82
|
Modabber M, Darvish-Zargar M, Breton L,
Chung DD, Duong H, Aldave AJ and Choremis J: Crystalline
keratopathy in post-LASIK ectasia: A case report. Cornea.
38:635–638. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Handa S, Thakur A, Rajneesh D, Kulshrestha
A and Gupta A: Schnyder's crystalline corneal dystrophy. QJM.
113:662020. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Sarosiak A, Udziela M, Ścieżyńska A,
Oziębło D, Wawrzynowska A, Szaflik JP and Ołdak M: Clinical
diversity in patients with Schnyder corneal dystrophy-a novel and
known UBIAD1 pathogenic variants. Graefes Arch Clin Exp Ophthalmol.
256:2127–2134. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Kitazawa K, Wakimasu K, Kayukawa K,
Sugimoto M, Nakai J, Weiss JS, Ueno M, Sotozono C and Kinoshita S:
Long-term outcome after penetrating keratoplasty in a pedigree with
the G177E mutation in the UBIAD1 gene for Schnyder corneal
dystrophy. Cornea. 37:554–559. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Jo Y, Kim SS, Garland K, Fuentes I,
DiCarlo LM, Ellis JL, Fu X, Booth SL, Evers BM and DeBose-Boyd RA:
Enhanced ER-associated degradation of HMG CoA reductase causes
embryonic lethality associated with Ubiad1 deficiency. Elife.
9:e548412020. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Evans CJ, Dudakova L, Skalicka P,
Mahelkova G, Horinek A, Hardcastle AJ, Tuft SJ and Liskova P:
Schnyder corneal dystrophy and associated phenotypes caused by
novel and recurrent mutations in the UBIAD1 gene. BMC Ophthalmol.
18:2502018. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Xu Z, Duan F, Lu H, Dragh MA, Xia Y, Liang
H and Hong L: UBIAD1 suppresses the proliferation of bladder
carcinoma cells by regulating H-Ras intracellular trafficking via
interaction with the C-terminal domain of H-Ras. Cell Death Dis.
9:11702018. View Article : Google Scholar : PubMed/NCBI
|