Introduction
Coronary artery disease is severe, with high
morbidity and mortality worldwide (1). Early reperfusion with thrombolysis or
percutaneous coronary intervention is an effective strategy for
acute myocardial infarction, and can effectively reduce infarct
size and improve clinical outcome (2). However, further myocardial damage may
occur during the reconstruction of coronary blood flow, called
myocardial ischemia-reperfusion injury (MIRI). Mechanisms involved
in MIRI include oxidative stress, myocardial apoptosis, calcium
overload and mitochondrial dysfunction (3).
Oxidative stress refers to the accumulation of
reactive oxygen species (ROS) and oxidative damage in cells
(4). A previous study reported that
NADPH oxidase (NOX)2 and NOX4 are major sources of cardiac ROS,
playing an important regulatory role in the proliferation and death
of cardiomyocytes (5). Inhibition
of NOX2 and NOX4 contributes to decreased ROS and alleviation of
MIRI (6). However, small quantities
of ROS produced by NOX2 and NOX4 exert an anti-inflammatory
protective effect during ischemia and reperfusion (5). In this scenario, inhibition of both
NOX2 and NOX4 may aggravate reperfusion injury (7). A study on adenosine
5′-monophosphate-activated protein kinase (AMPK) knockout mice
confirmed that the activation of AMPK in MIRI plays an important
role in cardioprotection (8), but
other studies have failed to get the same result (9,10).
Metformin is commonly used for the treatment of type
II diabetes (11). It can
significantly reduce the risk of myocardial infarction and
all-cause mortality in patients with type II diabetes (12). Therefore, it has been suggested that
metformin, besides glycemic control, has an additional role in
improving the prognosis of cardiovascular disease (13). In a rat model of myocardial
infarction, metformin is reported to significantly reduce infarct
size (14), while in a mouse model,
a lower dose of metformin was administered 18 h prior to ischemia,
which also reduced infarct size by 50% (15); however, metformin did not affect
plasma glucose concentration. In a subsequent isolated rat heart
study, intracoronary infusion of metformin within the first 15 min
of the reperfusion period reduced the infarct size by 40–50%
(16). These results have been
validated in vivo in rats and mice (15,16).
Various studies have reported that the cardioprotective effect of
metformin is at least partly dependent on the activation of AMPK
(14,17). Although the effect of metformin on
MIRI has been established, the mechanism involved has not been
clarified. Concerning the antioxidant effects of metformin, studies
have focused on whether it can reduce ROS synthesis, thereby
reducing tissue fibrosis (11,18).
It is reported that metformin inhibits cardiac fibrosis by
inhibiting NOX activity and reducing the production of ROS
(19). For downstream signals,
several studies have shown that AMPK inhibits NOX4 activity and
reduces ROS production in a high-glucose-induced apoptosis model
(20,21). There is no clear explanation for the
relationship between metformin and NOX4 in MIRI. Therefore, it was
hypothesized that metformin inhibits NOX4 activity by activating
AMPK, thereby reducing ROS production, ultimately playing a
protective role against MIRI.
The present study aimed to elucidate whether
metformin decreases MIRI by inhibiting NOX4 in rat MIRI and
neonatal ventricle myocyte (NRVM) models, and to identify potential
drug targets.
Materials and methods
Animals and materials
Sprague-Dawley (SD) male rats (weight, 200–250 g;
age, 6–8 weeks; n=50) and neonatal SD rats (age, 1–3 days; n=30)
were obtained from the Animal Experimental Center of Shandong
University. Protocols for the care and use of laboratory animals
were approved by the Local Ethics Committee at the Medical College
of Shandong University. Laboratory animals were reared according to
the Guide for the Care and Use of Laboratory Animals from the
National Institutes of Health (NIH) (22). At the end of animal experiments,
rats were euthanized using excessive anesthesia administration as
conditionally acceptable (intraperitoneal injection with sodium
pentobarbital at a dosage of 200 mg/kg). Metformin was purchased
from Sigma-Aldrich (Merck KGaA).
Myocardial reperfusion injury
model
Rats were anesthetized by sodium pentobarbital (50
mg/kg) via intraperitoneal injection and then ventilated with a
rodent respirator (ALCV9A; Shanghai Alcott Biotech Co., Ltd.) after
intubation. Then, the heart was exposed, and the left anterior
descending coronary artery (LAD) was ligated by a 6-0 silk suture
for 45 min. Ischemia was determined by blanching of the myocardium,
dyskinesia of the ischemic region and ST segment elevation on the
ECG. Then, the heart was reperfused for 4 h by loosening the knot,
and a marked hyperemic response indicated reperfusion (16).
Experimental protocol in vivo
The rats were randomly divided into three groups: i)
Sham (sham operated) group, in which the heart was exposed but the
LAD was not occluded; ii) ischemia-reperfusion (IR) group, in which
the LAD was occluded for 45 min and reperfused for 4 h; and iii) IR
+ metformin (IR + met) group, in which the LAD was occluded for 45
min, and metformin (5 mg/kg) was intravenously injected via the
jugular vein, subsequently followed by 4 h of reperfusion. The dose
of metformin used in the present study was selected based on a
previous study (16).
Assessment of infarct size
Staining was performed as previously described
(23). At the end of the
reperfusion, the LAD was ligated again, and the heart was
retrogradely infused with Evans blue (0.25% in phosphate buffer;
Sigma-Aldrich; Merck KGaA) from the aorta. The non-ischemic area
was stained blue, indicating the area at risk (AR, non-blue
region). Then, the heart was frozen at −20°C for 30 min and
sectioned into 6 slices (2 mm/slice). The slices were incubated in
triphenyltetrazolium chloride (TTC; 1% in phosphate buffer;
Sigma-Aldrich; Merck KGaA) for 10 min at 37°C and then immersed in
4% paraformaldehyde for 4 h at room temperature. TTC staining can
differentiate the infarct size (IS; white region) from the
non-infarct area at risk (AR; red region). These heart slices were
used only for Evans-blue-TTC staining. Finally, the slices were
arranged from apex to base and digitally photographed. Digital
images of the slices were analyzed using ImageJ software (version
1.47; National Institutes of Health). The final result was
presented as the IS/AR as previously described (24).
Evaluation of apoptosis
The left ventricles of rat hearts were fixed in 4%
paraformaldehyde at room temperature for 24 h and embedded in
paraffin at 56°C for 1.5 h, then sectioned (thickness, 6 µm).
According to the manufacturer's instructions, terminal
deoxynucleotidyl transferase-mediated dUTP nick end labelling
(TUNEL) assays were performed to detect apoptotic cells in heart
tissue sections using an in situ cell death detection kit
(Roche Applied Science). The sections were incubated in TUNEL
reaction mixture for 60 min at 37°C in a humidified atmosphere in
the dark. Sections were then stained with hematoxylin (Beijing
Solarbio Science & Technology, Co., Ltd.) for 2 min at room
temperature, washed with PBS, and mounted with mounting medium
(Abcam). The nuclei were counted in 10 random fields of each
section using a light microscope (magnification, ×400; Leica
Microsystems GmbH), and the results are shown as a percentage of
TUNEL-positive nuclei compared with the total number of cell
nuclei.
Western blotting
RIPA solution (Abcam) was used to homogenize the
frozen tissue samples and cultured cardiomyocytes and BCA assay
(Pierce; Thermo Fisher Scientific, Inc.) was used to detect the
concentration of protein samples. Then, 50 µg/lane extracted
protein was separated via 10% SDS-PAGE and transferred onto PVDF
membranes (EMD Millipore). Non-specific reactivity was blocked with
5% milk for 2 h at room temperature and the membrane was incubated
with primary antibody overnight at 4°C in buffer (10 mM Tris-HCl;
pH 7.5; 150 mM NaCl, 2% Tween-20, 4% bovine serum albumin). The
membrane was then incubated with secondary antibody for 1.5 h at
room temperature. ECL chemiluminescence (Cell Signaling Technology,
Inc.) was used for detection by an imaging system (ChemiDoc XRS;
Bio-Rad Laboratories, Inc.), and densitometry was performed using
ImageJ software (version 1.47; National Institutes of Health)
semi-quantitatively (4). All
protein levels were normalized to that of GAPDH. The antibodies
used in the study were as follows: Phosphorylated (p)-AMPK (Thr172;
1:1,000; cat. no. 50081; rabbit monoclonal antibodies; Cell
Signaling Technology, Inc.), AMPK (1:1,000; cat. no. 2532; rabbit
monoclonal antibodies; Cell Signaling Technology, Inc.) and NOX4
(1:1,000; cat. no. ab109225; rabbit monoclonal antibody; Abcam);
GAPDH (1:10,000; cat. no. AB2302; mouse monoclonal antibody; EMD
Millipore) and corresponding horseradish peroxidase
(HRP)-conjugated secondary antibodies (OriGene Technologies,
Inc.).
Isolation of neonatal rat
cardiomyocytes
NRVMs were isolated from 1–3-day-old SD rats as
previously described (25). The
rats were euthanized by decapitation and the hearts were rapidly
excised, minced and dissociated with 0.1% trypsin and 0.03%
collagen II solution. The dispersed cells were then plated at a
density of 2×105 cells/cm2 on a 6-well plate
with 2 ml/well DMEM supplemented with 10% fetal bovine serum (FBS;
both HyClone; GE Healthcare Life Sciences). Cytosine arabinoside
(10 M) was used to suppress non-cardiomyocytes. After serum-starved
cultivation (37°C, 95% O2, 5% CO2, 24 h), the
myocytes were put in a hypoxia (95% N2, 5%
CO2) incubator for 4 h at 37°C then, the culture medium
was replaced with fresh oxygenated DMEM (10% FBS) and the plates
were transferred to a normoxic incubator (5% CO2) for 6
h of reoxygenation.
Experimental protocol in vitro
(i)
The NRVMs were randomly divided into four groups: i)
Control (CON) group, cells were incubated with normal oxygen; ii)
hypoxia-reoxygenation (HR) group, in which the cells were incubated
in hypoxic conditions for 4 h and reoxygenated for 6 h; iii) HR +
metformin (HR + met) group, at the onset of reoxygenation,
metformin (0.1 mM) was added to the DMEM (the dose of metformin
used in the present study was selected based on a previous study)
(26); and iv) HR + metformin +
compound-C (HR + met + compound-C), at the onset of reoxygenation,
metformin and compound-C (10 nM; cat. no. ab120843; Abcam) were
added into the medium simultaneously (the dose of compound-C used
in the present study was selected based on a previous study)
(26).
MTT assay
Cell viability was assessed by an MTT assay. The MTT
assay was performed according to the manufacturer's protocols (cat.
no. C0009; Beyotime Institute of Biotechnology). Cardiomyocytes
were plated on 96-well dishes at a density of 1×104
cells/well. Following reoxygenation, cells were incubated with MTT
reaction solution (0.5 mg/ml) for 4 h and the medium was removed.
Then, 100 µl DMSO was added to each well for 10 min and mixed
thoroughly with a mechanical plate mixer. At last, the absorbance
was measured with a microplate reader (Molecular Devices, LLC) at a
wavelength of 530 nm.
Plasma creatine kinase-MB form (CK-MB)
and lactate dehydrogenase (LDH) activity
Blood samples were centrifuged at 1,800 × g for 10
min at 4°C. LDH (cat. no. A020-1) and CK-MB (cat. no. E006-1-1)
levels in plasma were detected according to the manufacturer's
protocols (Nanjing Jiancheng Bioengineering Institute). The optical
density of the tetrazolium product was determined
spectrophotometrically (Molecular Devices, LLC) at a wavelength of
490 nm.
Malondialdehyde (MDA) detection
Lipid peroxidation was estimated by measuring the
concentration of MDA in the isolated myocytes using a Lipid
Peroxidation Assay kit (cat. no. MAK085; Calbiochem; Merck KGaA).
Briefly, the cells were lysed and mixed with reagent R1
(N-methyl-2-phenylindole in acetonitrile and methanol) for a total
of 60 min at 45°C. The samples were then centrifuged at 21,130 × g
for 10 min at room temperature. The optical density of the
supernatant was determined spectrophotometrically at a wavelength
of 586 nm (Molecular Devices, LLC). The MDA concentration was
displayed as µmol/g protein.
Immunohistochemistry
Tissue sections (thickness, 6 μm) were
deparaffinized and then blocked with CAS-Block (Invitrogen; Thermo
Fisher Scientific, Inc.) for 1 h at 37°C. The sections were
incubated with anti-4HNE primary antibody (1:200; cat. no. ab48506;
Abcam) for 2 h at 37°C followed by incubation with HRP-conjugated
secondary antibody (1:500; cat. no. 7074; Cell Signaling
Technology, Inc.) for 1 h at 37°C. Sections were developed in
3,3′-diaminobenzidine solution for 3 min at room temperature, then
stained with hematoxylin for 2 min at room temperature, washed with
PBS, and mounted with mounting medium. Sections were visualized
under a light microscope (magnification, ×400; Leica Microsystems
GmbH).
RNA isolation and reverse
transcription-quantitative (RT-q) PCR
Total RNA of myocardial tissues and NRVMs was
extracted using TRIzol® reagent (Invitrogen; Thermo
Fisher Scientific, Inc.). The TaqMan® Reverse
Transcription Reagents (cat. no. N8080234; Applied Biosystems;
Thermo Fisher Scientific, Inc.) was used to generate the
first-strand cDNA using the following thermocycling conditions:
70°C for 5 min, 42°C for 1 h and 70°C for 15 min. A Mx3000
Multi-plex Quantitative PCR System (Stratagene; Agilent
Technologies, Inc.) with SYBR-Green fluorescence (Molecular Probes,
LLC) was used to amplify the cDNA in 35 cycles. Each cycle
consisted of heating denaturation for 30 sec at 94°C, annealing for
30 sec at 56°C and extension for 30 sec at 72°C. All samples were
quantitated using the comparative Cq method for relative
quantitation of gene expression, normalized to GAPDH (27). The primers used in this study were
as follows: NOX4, forward, 5′-TGGCCAACGAAGGGGTTAAA-3′ and reverse,
5′-CACTGAGAAGTTCAGGGCGT-3′; AMPK, forward,
5′-GATCGGACACTACGTGCTGG-3′ and reverse, 5′-TAGTTGCTCGCTTCAAGGGG-3′;
and GAPDH, forward, 5′-TGATGACATCAAGAAGGTGGTGAAG-3′ and reverse,
5′- TCCTTGGAGGCCATGTAGGCCAT-3′.
Small interfering (si)RNA
transfection
Transfection of siRNA was performed with
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocols. NRVM
(6×105 cells per well of a 6-well plate at) were
transfected with siRNA directed to AMPK (cat. no. sc-74461; Santa
Cruz Biotechnology, Inc.) at 10 µM for 6 h in serum-free medium.
Afterwards, it was changed to standard medium containing 10% FBS
for 24 h. Non-specific (NS)-siRNA (cat. no. sc-37007; Santa Cruz
Biotechnology, Inc.) was used as negative control.
Experimental protocol in vitro
(ii)
The NRVMs were randomly divided into four groups: i)
CON + NS-siRNA group, in which the cells were pretreated with
NS-siRNA and then incubated with normal oxygen; ii) HR + NS-siRNA
group, in which the cells were pretreated with NS-siRNA and then
subjected to 4 h of hypoxia and 6 h of reoxygenation; iii) HR +
metformin + NS-siRNA (HR + met + NS-siRNA) group, in which the
cells were pretreated with NS-siRNA and then subjected to 4 h of
hypoxia and 6 h of reoxygenation, and at the onset of
reoxygenation, metformin (0.1 mM) was added; and iv) HR + metformin
+ AMPK-siRNA (HR + met + AMPK-siRNA) group, in which the cells were
pretreated with AMPK-siRNA and then subjected to 4 h of hypoxia and
6 h of reoxygenation, and at the onset of reoxygenation, metformin
(0.1 mM) was added into DMEM.
Statistical analysis
All data are presented as the mean ± SEM (n≥3).
Statistical comparisons between the groups were performed using
one-way ANOVA followed by Newman-Keuls or Tukey's post hoc test.
Kruskal-Wallis test followed by Dunn's post hoc test was used for
non-parametric data. The statistical analyses were performed by
GraphPad Prism 5.0 (GraphPad Software, Inc.). P≤0.05 was considered
to indicate a statistically significant difference.
Results
Cardioprotective effects of
metformin
After 24 h reperfusion in IR rat, the IS and AR were
determined using Evans blue and TTC. The IS/AR was used to evaluate
damage to the heart as previously described (16). The results showed that treatment
with metformin significantly reduced the infarct size in IR rats by
~20% (P<0.05; Fig. 1A and B).
There was no infarct area in the Sham group, so the IS/AR was 0.
Plasma LDH and CK-MB levels in the IR group were increased compared
with the Sham group; however, the increases were significantly
attenuated by metformin (P<0.001; Fig. 1D and E). The viability of
cardiomyocytes decreased significantly after HR treatment compared
with the cells in CON group (P<0.001); treatment with metformin
increased the viability of HR-treated cardiomyocytes (P<0.05;
Fig. 1C).
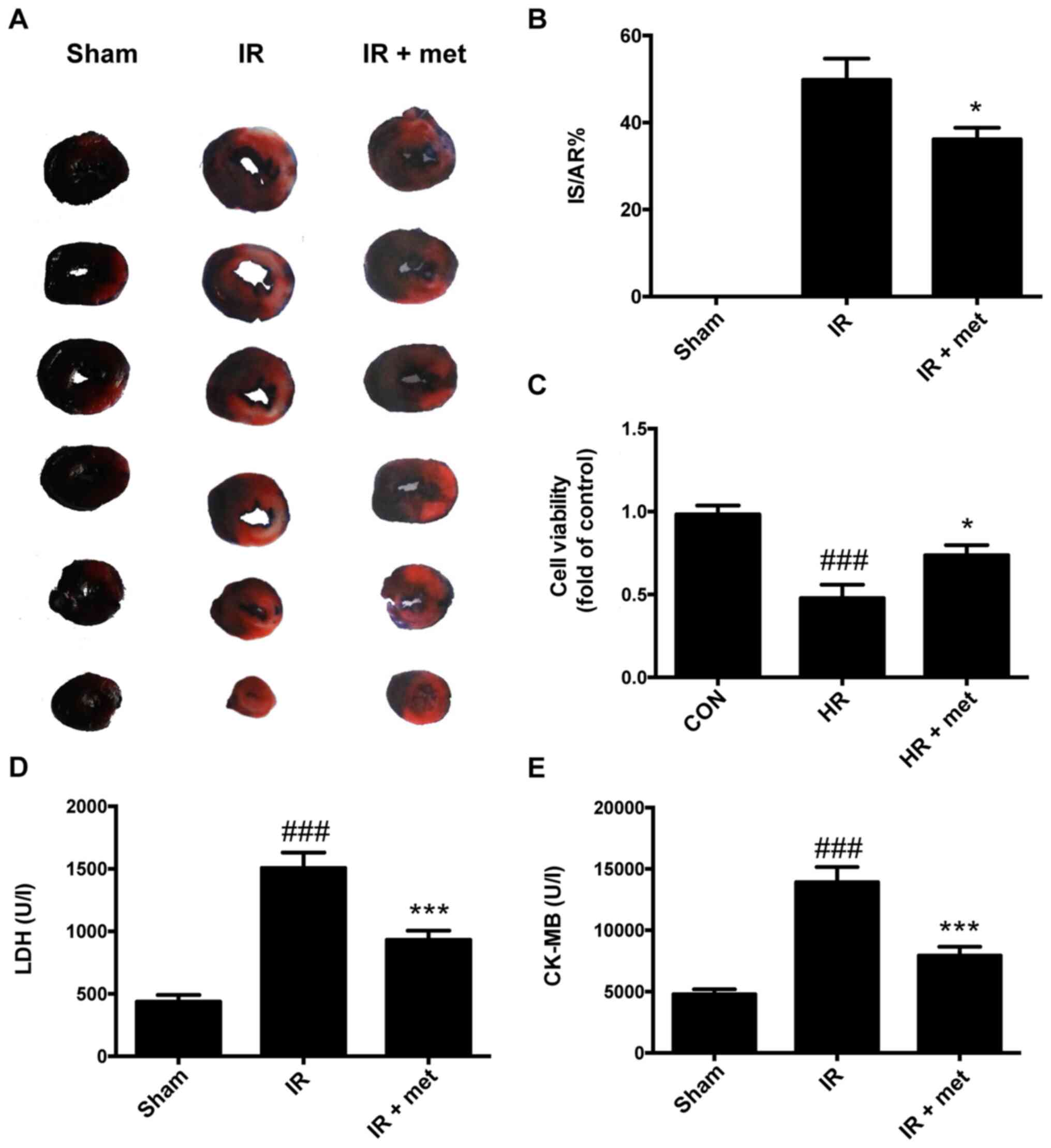 | Figure 1.Cardioprotective effects of metformin
against reperfusion injury. (A) Representative heart slices stained
by Evans blue and triphenyltetrazolium chloride. Blue, non-blue and
white areas represent non-ischemic, AR and IS areas. (B) IS/AR
scores in different groups. (C) Cell viability detected by an MTT
assay. ###P<0.001 vs. CON; *P<0.05 vs. HR. (D)
Plasma LDH concentration. (E) Plasma CK-MB concentration. Data are
shown as the mean ± SEM (n=6). ###P<0.001 vs. Sham;
***P<0.001 vs. IR. Sham, sham-operated control, IR,
ischemia-reperfusion, met, metformin; AR, area at risk; IS, infarct
size; CON, control; HR, hypoxia-reoxygenation; LDH, lactate
dehydrogenase; CK-MB, creatine kinase MB form. |
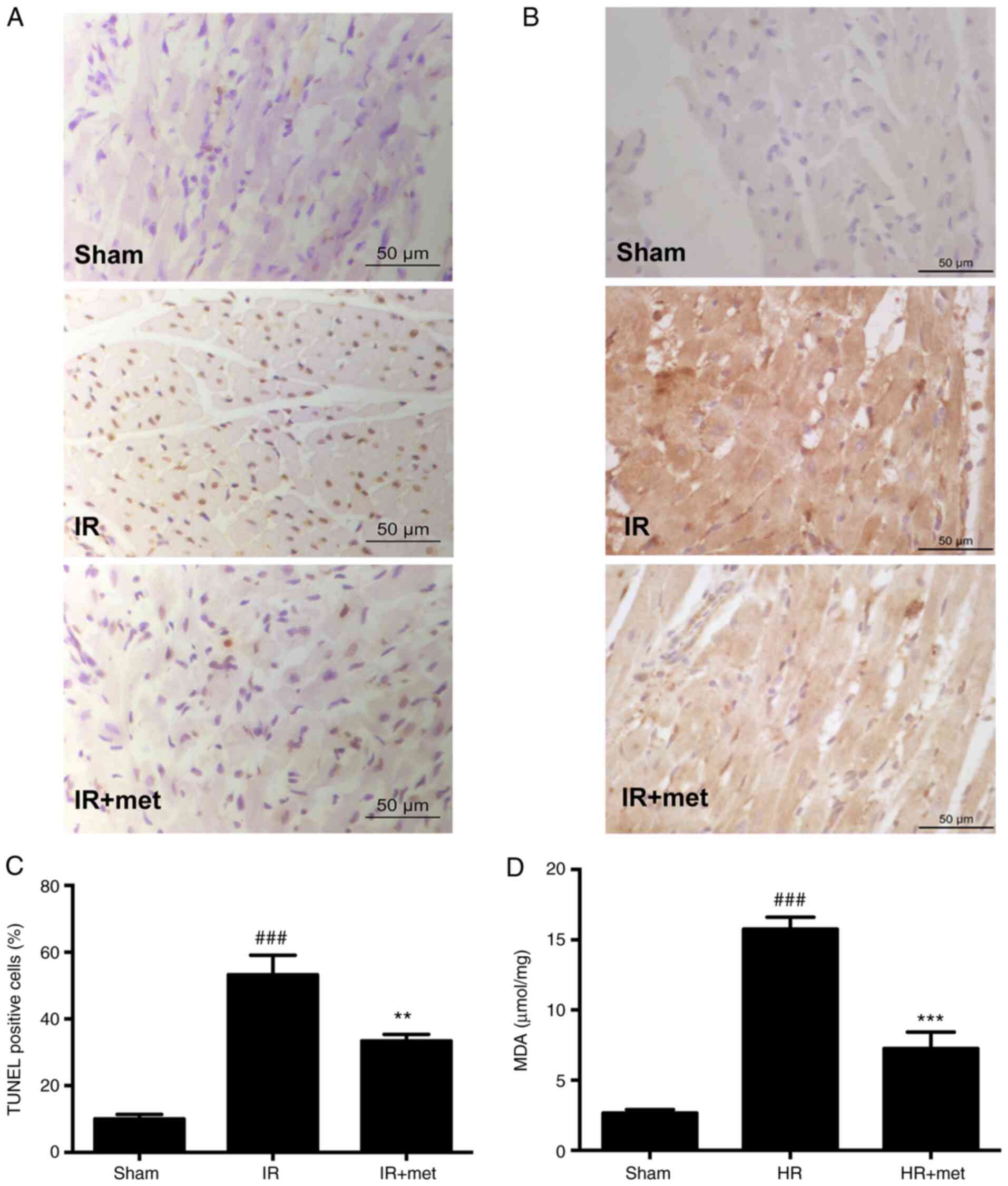
Metformin alleviates oxidative damage
and apoptosis
Oxidative stress plays an important role in the
process of MIRI (14).
4-hydroxynonenal (4HNE) is the final product of lipid oxidation,
the amount of which indicates the severity of oxidative damage
(28). Immunohistochemistry results
showed that the levels of 4HNE in the IR group were notably
increased compared with the Sham group (Fig. 2B). However, the content of 4HNE in
the IR + met group was markedly decreased compared with the IR
group (Fig. 2B). In addition to
4HNE, another lipid peroxidation product, MDA, was measured in
cells following HR. The results showed that the level of MDA in the
HR + met group was significantly decreased compared with the HR
group (Fig. 2D; P<0.001). A
TUNEL assay was performed in tissue sections to evaluate apoptosis.
It was found that apoptosis was increased in the IR group compared
with the Sham group (P<0.05; Fig. 2A
and C). When treated with metformin, the percentage of
TUNEL-positive cells was significantly decreased compared with the
IR group (P<0.01; Fig. 2C).
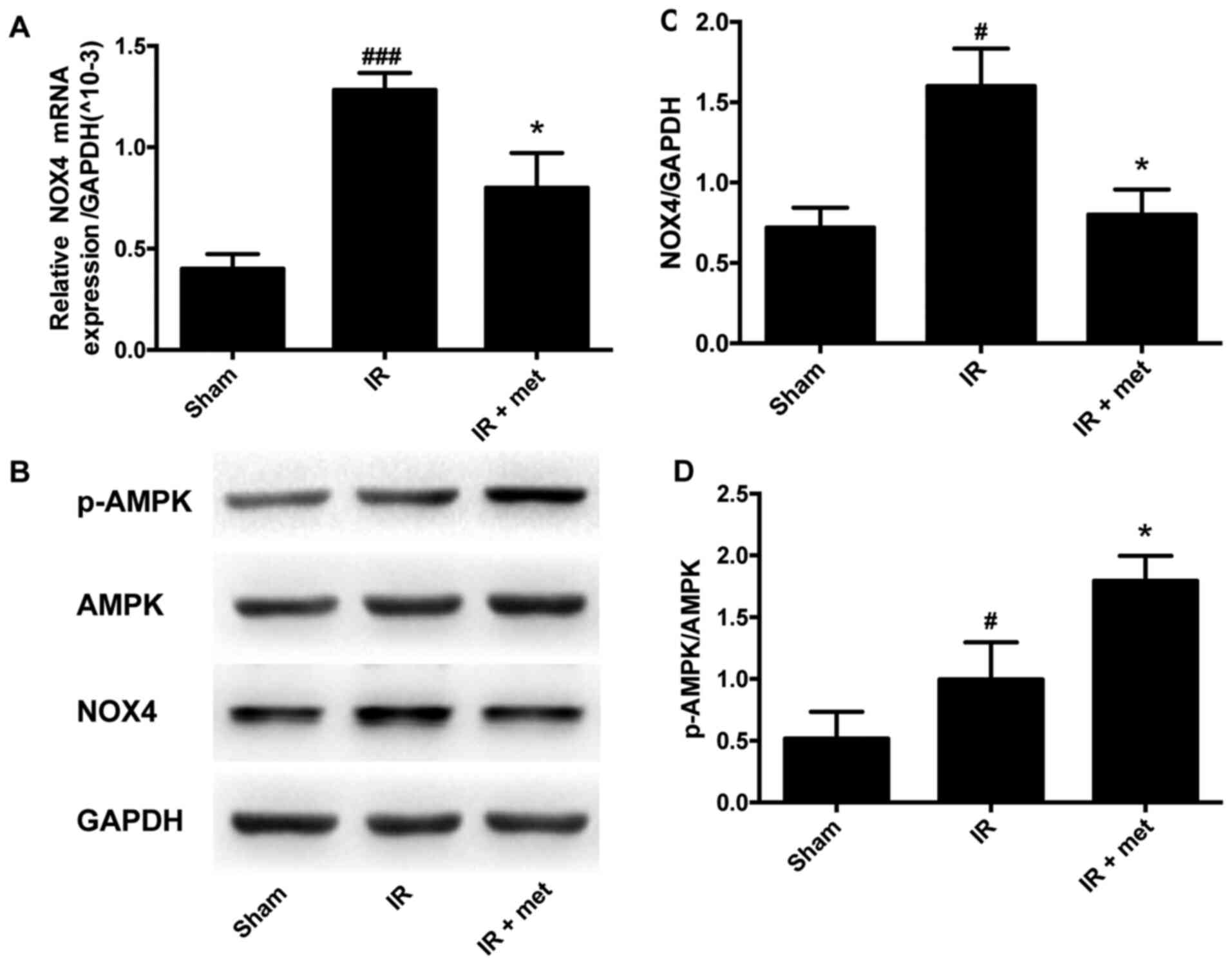
Metformin inhibits the expression of
NOX4 and increases the activation of AMPK
NOX4 plays an important role in regulating the
content of ROS in cardiac myocytes (6). A significant increase in NOX4 mRNA was
observed in the IR group compared with the Sham group; after
metformin was administered, the elevation in NOX4 was significantly
attenuated (P<0.05; Fig. 3A).
The protein level of NOX4 in the IR group was also significantly
increased compared with that in the Sham group; after metformin
treatment, the increase of NOX4 was significantly reversed
(P<0.05; Fig. 3B and C). The
activation of AMPK, which is the downstream factor of metformin
(26), was also evaluated. The
phosphorylation of AMPK in the IR group was significantly increased
compared with that in the Sham group. In the IR + met group,
metformin significantly promoted the phosphorylation of AMPK to a
higher level than that in the IR group (P<0.05; Fig. 3B and D).
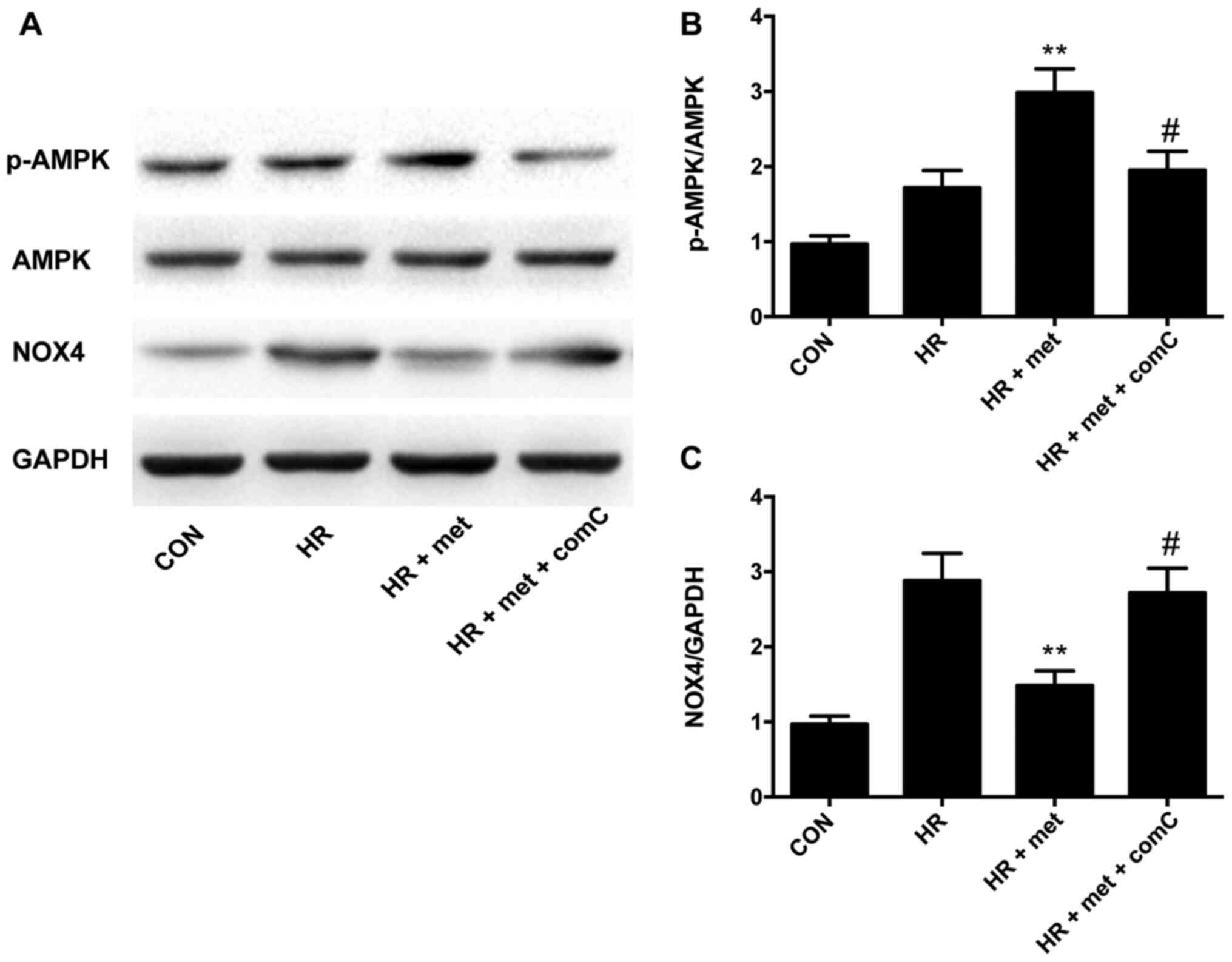
AMPK inhibition upregulates the
expression of NOX4 protein in vitro
In the NRVM HR model, treatment with metformin
increased p-AMPK levels (P<0.01; Fig. 4A and B) and significantly reduced
NOX4 expression (P<0.01; Fig. 4A and
C). The AMPK inhibitor compound-C was used to investigate the
relationship between AMPK and NOX4; following treatment with
compound-C, metformin did not induce a further increase in the
phosphorylation of AMPK (P<0.05; Fig. 4B), and the expression of NOX4 was
significantly increased compared with the HR + met group
(P<0.05; Fig. 4C).
AMPK-siRNA upregulates the expression
of NOX4 protein in vitro
Following transfection with AMPK-siRNA, the
expression of AMPK was significantly downregulated in both the CON
+ AMPK-siRNA group and IR + AMPK-siRNA group compared with the
corresponding NS-siRNA groups (P<0.05; Fig. 5F). In the HR + NS-siRNA NRVM model,
treatment with metformin significantly enhanced AMPK
phosphorylation (P<0.05; Fig. 5A and
B), while significantly reducing NOX4 expression (P<0.01;
Fig. 5C). AMPK-siRNA was used to
explore the relationship between AMPK and NOX4. In the HR + met +
AMPK-siRNA group, the expression of AMPK was significantly
downregulated compared with the HR + met + NS-siRNA group
(P<0.01; Fig. 5A and D). The
p-AMPK/AMPK ratio was used to determine the phosphorylation level
of AMPK; it was found that p-AMPK/AMPK was significantly increased
in the HR + met + NS-siRNA group compared with in the HR + NS-siRNA
group (P<0.05; Fig. 5A and E. No
significant difference, however, was observed between the HR + met
+ NS-siRNA and HR + met + AMPK-siRNA groups (P>0.05; Fig. 5A and E). Following transfection with
AMPK-siRNA, metformin did not induce a further increase in p-AMPK
(P<0.05; Fig. 5B), and the
expression of NOX4 was significantly increased compared with the HR
+ met + NS-siRNA group (P<0.01; Fig.
5A and C).
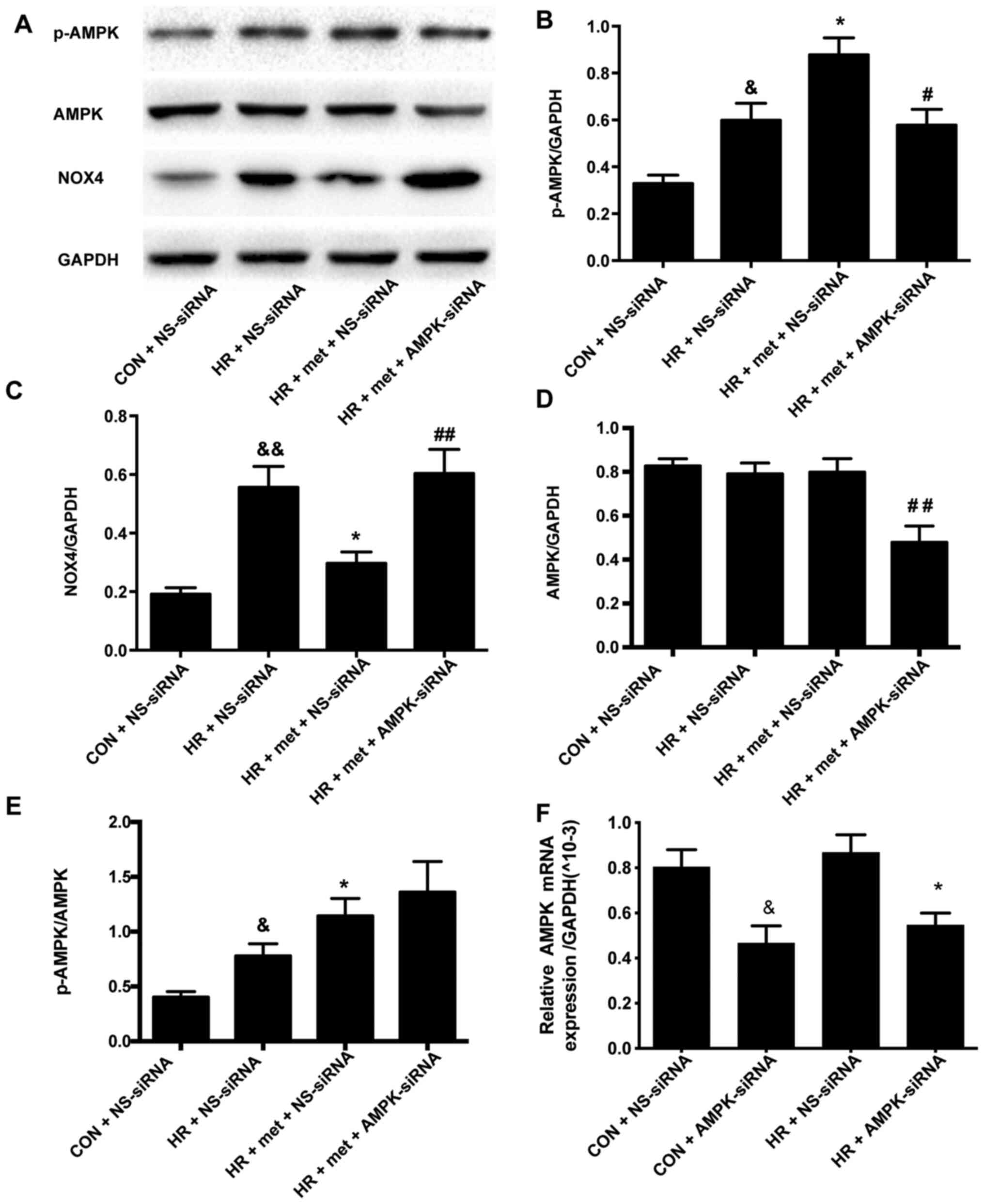 | Figure 5.AMPK siRNA increases the expression
of NOX4 protein in vitro. (A) Representative protein levels
of p-AMPK, AMPK and NOX4 as determined via western blotting. (B)
Semi-quantification of p-AMPK levels. (C) Semi-quantification of
NOX4 protein levels. (D) Semi-quantification of AMPK protein
levels. (E) Semi-quantification of the p-AMPK/AMPK ratio. (F)
Expression of AMPK mRNA as determined via reverse
transcription-quantitative PCR. Protein and mRNA levels were
normalized to GAPDH. Data are shown as the mean ± SEM (n=6).
&P<0.05, &&P<0.01 vs. CON +
NS-siRNA; *P<0.05 vs. HR + NS-siRNA; #P<0.05,
##P<0.01 vs. HR + met + NS-siRNA. CON, control; HR,
hypoxia-reoxygenation; met, metformin; siRNA, small interfering
RNA; NS-siRNA, non-specific siRNA; AMPK-siRNA, AMPK-specific siRNA;
NOX, NADPH oxidase; AMPK, adenosine 5′-monophosphate-activated
protein kinase; p, phosphorylated. |
Discussion
In the present study, the cardioprotective effects
of metformin were examined in animal and cell models. Although
previous studies had indicated that metformin reduces MIRI
(14,17), the specific mechanism remains
unclear. In the present study, it was observed that metformin
reduced myocardial infarct size, alleviated myocardial oxidative
damage and apoptosis, promoted the activation of AMPK and inhibited
the expression of NOX4. In primary NRVMs following HR injury,
metformin inhibited the expression of NOX4, and this inhibition was
reversed using an AMPK inhibitor. These findings indicated that
metformin inhibited the expression of NOX4 via the activation of
AMPK, thereby reducing oxidative damage and ultimately playing a
role in myocardial protection.
It has previously been reported that ischemic
postconditioning could effectively reduce myocardial infarct size
(29); however, its application is
limited. Therefore, there is a motivation to search for medications
with potential cardioprotective effects to simulate the effects of
postconditioning in a manner that may be more accessible to
clinical patients. In the process, it has been found that various
endogenous substances, including adenosine, erythropoietin and cell
growth factors, play crucial roles in reducing MIRI (30). Another option is to look for
existing drugs with cardioprotective effects, expanding their
existing uses and greatly reduces the cost of developing new drugs.
Statins have been found to exert cardioprotective effects outside
of blood lipid regulation, resulting from antioxidant,
anti-apoptotic and other effects (31). Previous studies showed that
metformin may also have a similar cardioprotective effect. For
example, in a prospective diabetes study in the UK, metformin was
found to reduce the risk of acute myocardial infarction (AMI) by
39% in patients with type II diabetes, reducing all-cause mortality
by 36% (32). Regarding patients
with AMI, metformin treatment has also significantly reduced their
mortality compared with sulfonylureas (33,34).
In the present study, it was found that infarct size was
significantly reduced in metformin-treated animals in an IR rat
model. Consistent with a previous study (16), the results showed that 5 mg/kg
metformin was sufficient for myocardial protection and would not
result in changes in blood glucose concentrations.
During MIRI, a large number of intracellular ROS can
be generated, which may cause damage to myocardial tissue through
various pathways, including destruction of various structural
proteins and enzymes, DNA damage, calcium overload and
cardiomyocyte apoptosis (35).
Previous studies have mainly focused on the effect of metformin on
decreasing ROS formation in myocardial fibrosis (19,36).
MDA and 4HNE are the products of lipid peroxidation, which reflect
the levels of ROS production. In the present study. it was found
that metformin could reduce the levels of 4HNE in a rat model of
MIRI, as well as those of MDA in a cell model of HR, thus
indicating a protective role.
Intracellular ROS are produced mainly by NOX and
mitochondrial pathways (35). It
was previously reported that NOX2 and NOX4 are the main sources of
cardiac oxygen free radical production and play important roles in
the growth and death of cardiomyocytes (37,38).
Studies have shown that, following HR treatment, the expression
levels of NOX2 are increased (39,40).
In addition, cardiac-specific knockout of NOX4 aggravates MIRI
(41). In a separate study, there
was no significant reduction of IS in rats with NOX4 knockout,
suggesting that NOX4 did not aggravate MIRI (7). There remains a degree of controversy
concerning the role of NOX4 in MIRI. Therefore, the role of NOX4 in
MIRI was investigated in the present study. It was observed that
the expression level of NOX4 was increased significantly in the IR
group, which is consistent with the finding that 4HNE levels were
significantly increased in the IR group compared with the Sham
group. These findings suggested that NOX4 aggravated MIRI by
promoting oxidative stress.
A previous study had shown that metformin can
inhibit NOX4 activity and alleviate pulmonary fibrosis (36). Similarly, metformin can prevent
cardiac fibrosis by inhibiting the activation of NOX, inhibiting
the production of ROS (19). These
results indicated the potential regulatory association between
metformin and NOX4. Metformin also inhibits intracellular oxidation
by activating AMPK to stimulate myocardial protection (26). A previous study showed that AMPK
alleviated the apoptosis of epithelial cells by inhibiting the
activity of NOX4 (20), indicating
an association between AMPK and NOX4. As a downstream effector of
metformin, increased AMPK phosphorylation has been shown to
decrease cardiomyocyte apoptosis and improve cardiac function
(15). In the present study, it was
found that the phosphorylation of AMPK in the IR group was
increased compared with that in the Sham group. After metformin
treatment, p-AMPK was significantly increased compared with the IR
group, consistent with a previous study (15). In the present study, it was also
found that in the metformin group, the mRNA and protein levels of
NOX4 were significantly reduced compared with the IR group. These
results indicated the association between AMPK and NOX4 in a rat
model of MIRI. In the cell model, pretreatment with the AMPK
inhibitor compound-C significantly reduced NOX4 protein expression
when compared with the group only treated with metformin. To
further investigate the role of AMPK in metformin-induced
inhibition of NOX4, AMPK expression was knocked down by siRNA
transfection. As expected, AMPK-siRNA significantly downregulated
the expression of AMPK. The p-AMPK/AMPK ratio was used to determine
phosphorylation level of AMPK, which was increased by metformin,
but was not affected by transfection with NS-siRNA or AMPK-siRNA in
metformin-treated HR cells. Evaluating p-AMPK in isolation, it was
observed that AMPK-siRNA reduced p-AMPK levels compared with
NS-siRNA, whilst also increasing the expression of NOX4 (Fig. 5C). These findings suggested that the
regulation of NOX4 activity depends on the levels of activated
AMPK. In conclusion, the results of the present study indicated
that metformin inhibited the expression of NOX4 via the activation
of AMPK, leading to a reduction of myocardial oxidative damage,
apoptosis and infarct size, ultimately alleviating MIRI.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
YS and SAH conceived and designed the study. YS
conducted both the in vivo and in vitro studies and
drafted the manuscript. SAH assisted in drafting the manuscript. YS
and SAH confirm the authenticity of all the raw data. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
The Guide for the Care and Use of Laboratory Animals
was followed, and the experimental protocol was approved by the
Local Ethics Committee at the Medical College of Shandong
University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Malakar AK, Choudhury D, Halder B, Paul P,
Uddin A and Chakraborty S: A review on coronary artery disease, its
risk factors, and therapeutics. J Cell Physiol. 234:16812–16823.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Monassier JP: Reperfusion injury in acute
myocardial infarction. From bench to cath lab. Part I: Basic
considerations. Arch Cardiovasc Dis. 101:491–500. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yellon DM and Hausenloy DJ: Myocardial
reperfusion injury. N Engl J Med. 357:1121–1135. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sies H: Oxidative stress: A concept in
redox biology and medicine. Redox Biol. 4:180–183. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Matsushima S, Tsutsui H and Sadoshima J:
Physiological and pathological functions of NADPH oxidases during
myocardial ischemia-reperfusion. Trends Cardiovasc Med. 24:202–205.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Braunersreuther V, Montecucco F, Asrih M,
Pelli G, Galan K, Frias M, Burger F, Quinderé AL, Montessuit C,
Krause KH, et al: Role of NADPH oxidase isoforms NOX1, NOX2 and
NOX4 in myocardial ischemia/reperfusion injury. J Mol Cell Cardiol.
64:99–107. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Matsushima S, Kuroda J, Ago T, Zhai P,
Ikeda Y, Oka S, Fong GH, Tian R and Sadoshima J: Broad suppression
of NADPH oxidase activity exacerbates ischemia/reperfusion injury
through inadvertent downregulation of HIF-1α and upregulation of
PPARα. Circ Res. 112:315–325. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Russell RR III, Li J, Coven DL, Pypaert M,
Zechner C, Palmeri M, Giordano FJ, Mu J, Birnbaum MJ and Young LH:
AMP-activated protein kinase mediates ischemic glucose uptake and
prevents postischemic cardiac dysfunction, apoptosis, and injury. J
Clin Invest. 114:495–503. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Folmes CD, Wagg CS, Shen M, Clanachan AS,
Tian R and Lopaschuk GD: Suppression of 5-AMP-activated protein
kinase activity does not impair recovery of contractile function
during reperfusion of ischemic hearts. Am J Physiol Heart Circ
Physiol. 297:H313–H321. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu B, Clanachan AS, Schulz R and
Lopaschuk GD: Cardiac efficiency is improved after ischemia by
altering both the source and fate of protons. Circ Res. 79:940–948.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang YW, He SJ, Feng X, Cheng J, Luo YT,
Tian L and Huang Q: Metformin: A review of its potential
indications. Drug Des Devel Ther. 11:2421–2429. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nicholls SJ, Tuzcu EM, Kalidindi S, Wolski
K, Moon KW, Sipahi I, Schoenhagen P and Nissen SE: Effect of
diabetes on progression of coronary atherosclerosis and arterial
remodeling: A pooled analysis of 5 intravascular ultrasound trials.
J Am Coll Cardiol. 52:255–262. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Norhammar A, Lindbäck J, Rydén L,
Wallentin L and Stenestrand U; Register of Information and
Knowledge about Swedish Heart Intensive Care Admission (RIKS-HIA),
: Improved but still high short- and long-term mortality rates
after myocardial infarction in patients with diabetes mellitus: A
time-trend report from the Swedish Register of Information and
Knowledge about Swedish Heart Intensive Care Admission. Heart.
93:1577–1583. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang X, Yang L, Kang L, Li J, Yang L,
Zhang J, Liu J, Zhu M, Zhang Q, Shen Y, et al: Metformin attenuates
myocardial ischemia-reperfusion injury via up-regulation of
antioxidant enzymes. PLoS One. 12:e01827772017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Calvert JW, Gundewar S, Jha S, Greer JJ,
Bestermann WH, Tian R and Lefer DJ: Acute metformin therapy confers
cardioprotection against myocardial infarction via
AMPK-eNOS-mediated signaling. Diabetes. 57:696–705. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Paiva M, Riksen NP, Davidson SM, Hausenloy
DJ, Monteiro P, Gonçalves L, Providência L, Rongen GA, Smits P,
Mocanu MM, et al: Metformin prevents myocardial reperfusion injury
by activating the adenosine receptor. J Cardiovasc Pharmacol.
53:373–378. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Solskov L, Løfgren B, Kristiansen SB,
Jessen N, Pold R, Nielsen TT, Bøtker HE, Schmitz O and Lund S:
Metformin induces cardioprotection against ischaemia/reperfusion
injury in the rat heart 24 hours after administration. Basic Clin
Pharmacol Toxicol. 103:82–87. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Melendez GC, Chen Q and Lesnefsky EJ:
Metformin as a modulator of myocardial fibrosis post-myocardial
infarction via regulation of cardiomyocyte-fibroblast crosstalk.
Transl Res. 199:1–3. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bai J, Zhang N, Hua Y, Wang B, Ling L,
Ferro A and Xu B: Metformin inhibits angiotensin II-induced
differentiation of cardiac fibroblasts into myofibroblasts. PLoS
One. 8:e721202013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Eid AA, Ford BM, Block K, Kasinath BS,
Gorin Y, Ghosh-Choudhury G, Barnes JL and Abboud HE: AMP-activated
protein kinase (AMPK) negatively regulates Nox4-dependent
activation of p53 and epithelial cell apoptosis in diabetes. J Biol
Chem. 285:37503–37512. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang WX, Tai GJ, Li XX and Xu M:
Inhibition of neointima hyperplasia by the combined therapy of
linagliptin and metformin via AMPK/Nox4 signaling in diabetic rats.
Free Radic Biol Med. 143:153–163. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
National Research Council, . Guide for the
Care and Use of Laboratory Animals. National Academy Press;
Washington, DC: 1985
|
|
23
|
Lee SY, Ku HC, Kuo YH, Chiu HL and Su MJ:
Pyrrolidinyl caffeamide against ischemia/reperfusion injury in
cardiomyocytes through AMPK/AKT pathways. J Biomed Sci. 22:182015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cheng Y, Zhu P, Yang J, Liu X, Dong S,
Wang X, Chun B, Zhuang J and Zhang C: Ischaemic
preconditioning-regulated miR-21 protects heart against
ischaemia/reperfusion injury via anti-apoptosis through its target
PDCD4. Cardiovasc Res. 87:431–439. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sun HY, Wang NP, Kerendi F, Halkos M, Kin
H, Guyton RA, Vinten-Johansen J and Zhao ZQ: Hypoxic
postconditioning reduces cardiomyocyte loss by inhibiting ROS
generation and intracellular Ca2+ overload. Am J Physiol
Heart Circ Physiol. 288:H1900–H1908. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kobashigawa LC, Xu YC, Padbury JF, Tseng
YT and Yano N: Metformin protects cardiomyocyte from doxorubicin
induced cytotoxicity through an AMP-activated protein kinase
dependent signaling pathway: An in vitro study. PLoS One.
9:e1048882014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Poli G and Schaur JR: 4Hydroxynonenal in
the pathomechanisms of oxidative stress. IUBMB Life. 50:315–321.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ibáñez B, Heusch G, Ovize M and Van de
Werf F: Evolving therapies for myocardial ischemia/reperfusion
injury. J Am Coll Cardiol. 65:1454–1471. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sluijter JP, Condorelli G, Davidson SM,
Engel FB, Ferdinandy P, Hausenloy DJ, Lecour S, Madonna R, Ovize M,
Ruiz-Meana M, et al Nucleus of the European Society of Cardiology
Working Group Cellular Biology of the Heart, : Novel therapeutic
strategies for cardioprotection. Pharmacol Ther. 144:60–70. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hadi NR, Al-Amran F, Yousif M and Zamil
ST: Antiapoptotic effect of simvastatin ameliorates myocardial
ischemia/reperfusion injury. ISRN Pharmacol. 2013:8150942013.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Group UPDS; UK Prospective Diabetes Study
(UKPDS) Group, : Effect of intensive blood-glucose control with
metformin on complications in overweight patients with type 2
diabetes (UKPDS 34). Lancet. 352:854–865. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Schramm TK, Gislason GH, Vaag A, Rasmussen
JN, Folke F, Hansen ML, Fosbøl EL, Køber L, Norgaard ML, Madsen M,
et al: Mortality and cardiovascular risk associated with different
insulin secretagogues compared with metformin in type 2 diabetes,
with or without a previous myocardial infarction: A nationwide
study. Eur Heart J. 32:1900–1908. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Masoudi FA, Inzucchi SE, Wang Y, Havranek
EP, Foody JM and Krumholz HM: Thiazolidinediones, metformin, and
outcomes in older patients with diabetes and heart failure: An
observational study. Circulation. 111:583–590. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bedard K and Krause KH: The NOX family of
ROS-generating NADPH oxidases: physiology and pathophysiology.
Physiol Rev. 87:245–313. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sato N, Takasaka N, Yoshida M, Tsubouchi
K, Minagawa S, Araya J, Saito N, Fujita Y, Kurita Y, Kobayashi K,
et al: Metformin attenuates lung fibrosis development via NOX4
suppression. Respir Res. 17:1072016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Murdoch CE, Grieve DJ, Cave AC, Looi YH
and Shah AM: NADPH oxidase and heart failure. Curr Opin Pharmacol.
6:148–153. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sirker A, Zhang M and Shah AM: NADPH
oxidases in cardiovascular disease: Insights from in vivo models
and clinical studies. Basic Res Cardiol. 106:735–747. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cheng F, Yuan W, Cao M, Chen R, Wu X, Yan
J and Cyclophilin A: Cyclophilin a protects cardiomyocytes against
hypoxia/reoxygenation-induced apoptosis via the AKT/Nox2 pathway.
Oxid Med Cell Longev. 2019:27179862019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jin Q, Jiang Y, Fu L, Zheng Y, Ding Y and
Liu Q: Wenxin granule ameliorates hypoxia/reoxygenation-induced
oxidative stress in mitochondria via the PKC-δ/NOX2/ROS pathway in
H9c2 cells. Oxid Med Cell Longev. 2020:32454832020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Martyn KD, Frederick LM, von Loehneysen K,
Dinauer MC and Knaus UG: Functional analysis of Nox4 reveals unique
characteristics compared to other NADPH oxidases. Cell Signal.
18:69–82. 2006. View Article : Google Scholar : PubMed/NCBI
|