Introduction
Hepatitis B virus (HBV) is one of the commonest
causes of chronic hepatitis worldwide (1). The chronicity of HBV infection is
related to the fact that HBV produces very stable viral genome in
the host cells (1). The HBV genome
is partially double-stranded relaxed circular DNA (rcDNA), which is
converted to complete double-stranded covalently closed circular
DNA (cccDNA) in the nucleus of infected hepatocytes (2). HBV cccDNA remains the main hurdle in
the eradication of infected HBV as current antiviral agents cannot
eradicate this episomal viral minichromosome (3). HBV cccDNA persists in the liver
throughout the natural course of chronic HBV infection.
Serum HBV DNA, a standard surrogate marker of HBV
replication in hepatocytes, decreases over time during the natural
course of infection in numerous patients with chronic hepatitis B
(CHB) (4). In addition, some of
them lose serum HBsAg which is a sensitive marker of HBV infection
and controlled by a separate promoter (2). These findings suggest that the
transcriptional activity of HBV cccDNA may decrease in the late
stage of infection. Indeed, recent studies confirm transcriptional
suppression of cccDNA in HBeAg-negative chronic hepatitis B
(5,6). The transcriptional activity of HBV
cccDNA is controlled by four promoters (precore/pregenomic, S1, S2
and X) and two enhancer sequences (enhancer I and enhancer II)
(7). These cis-regulatory elements
recruit several ubiquitous and liver-specific transcription factors
and contribute to the regulation of cccDNA transcription (8,9).
In addition to the trans-acting DNA-binding
proteins, structural changes in cccDNA, i.e., epigenetic
modifications, have been implicated as regulatory mechanisms of
cccDNA transcription (10,11). Electron microscopy examination of
cccDNA reveals typical ‘beads-on-a-string’ nucleosomal organization
(12). HBV nucleoprotein is
composed of HBV core protein and histones (13). Similar to the case of mammalian
cellular DNA, posttranslational modifications (PTMs) of histone
proteins modulate the transcription of HBV cccDNA (8). Acetylation of cccDNA-bound H3/H4
histones was first reported to promote HBV replication (14). Thereafter, a genome-wise map of PTMs
revealed that stimulatory PTMs, such as H3K4me3, H3K27ac and
H3K122ac are enriched, whereas suppressive PTMs, including H3K9me3
and H3K27me3, are underrepresented in cccDNA chromatin (15). Recently, succinylation of H3K79 was
reported to upregulate cccDNA transcription (16). Thus, PTMs of histone proteins are
now considered to be major regulators of cccDNA transcription.
Methylation of HBV cccDNA has also been pursued as a
regulatory mechanism for cccDNA transcription (17–20).
There are two CpG island sequences (II and III) which are conserved
across the eight major genotypes (21) and are hotspots for methylation of
cccDNA isolated from human liver tissues (22). In particular, CpG II is located near
the core promoter and methylation densities on CpG II are
correlated with HBeAg status (17),
transcriptional activity of cccDNA (20) and serum HBV DNA levels (20,22).
De novo methylation of mammalian DNA is mediated by DNA
methyltransferases (DNMT) 3A and DNMT3B, but the mechanisms
underlying the methylation process of cccDNA are poorly understood
(23).
Interferon (IFN) α has long been used as an
antiviral therapy for CHB. The antiviral effect of IFN-α is modest
and observed in only a portion of patients compared to that of
nucleos(t)ide analogues (1).
However, patients responding to interferon treatment typically show
durable viral suppression after cessation of therapy (24), suggesting induction of persistent
antiviral milieu in hepatocytes. Diverse interferon-stimulated
genes (ISGs) work at the transcriptional and post-transcriptional
levels to keep HBV replication in check (25–28),
but the downstream intracellular mechanisms responsible for the
durable interferon response remain to be elucidated. Previous
studies have shown that IFN-α induces modification of cccDNA-bound
histones (15,16,29,30).
These epigenetic modifications may explain the transcriptional
suppression of cccDNA and off-treatment sustained HBV suppression
by IFN-α (31). However, while
IFN-α treatment reduces the stimulatory PTMs (H3K4me3, H3K9ac
H3K27ac and H3K122ac), H3K9me3 and H3K27me3, the canonical
repressive PTMs, are not enriched by IFN-α, suggesting that
additional active epigenetic mechanisms may contribute to selective
silencing of the precore/pregenomic HBV promoter by IFN-α (15,28).
Considering the suppressive role of CpG II methylation during the
natural course of HBV infection, it is hypothesized that IFN-α
treatment may induce changes in the methylation status of cccDNA, a
hypothesis not yet tested. Thus, the aim of the present study was
to elucidate the effect of IFN-α on the methylation status of HBV
cccDNA in a cell-based HBV replication system.
Materials and methods
In vitro HBV replication model
Infectious HBV particles were collected from HepAD38
cells (a gift from Professor C. Seeger, Fox Chase Cancer Center,
PA), which allow HBV replication under the control of an inducible
tetracycline promoter (32). The
cells were cultured in Dulbecco's modified Eagle's medium
(DMEM)/F12 medium (Welgene, Inc.) containing 10% fetal bovine
serum, 100 U/ml penicillin, 100 µg/ml streptomycin, 0.3 µg/ml
tetracycline and 400 µg/ml G418 at 37°C in a 5% carbon dioxide
chamber. HBV replication was induced by omitting tetracycline from
the culture medium for 5–7 days. The culture supernatant was
centrifuged at 12,000 × g for 5 min at room temperature to remove
cellular debris and then ultracentrifuged at 25,000 × g for 4 h at
4°C. The pellets were resuspended in PBS and the viral titer was
measured using reverse transcription-quantitative (RT-q) PCR. HepG2
cells, a human liver cancer cell line (cat. no. 88065, passage
number 5–10, Korean Cell Line Bank, Seoul, South Korea), were
infected with HBV particles at a titer of 50 multiplicity of
infection (MOI) in the presence of DMSO (cat. no. H 9268; final
concentration of 0.2%; Sigma-Aldrich; Merck KGaA) because DMSO
facilitates intracellular entry of HBV and enhances intracellular
replication (33,34). The cells were then maintained in
DMEM containing 10% fetal bovine serum, penicillin and
streptomycin. On the following day, IFN-α (Intron A; MSD
International GmbH; 15 MU/ml) was replenished at a final
concentration of 1,500 U/ml for four days before harvest.
Isolation of HBV rcDNA and cccDNA
HBV-infected HepG2 cells that were grown in a 60-mm
tissue culture dish at a density of 5×105 cells/ml were
lysed with 0.4 ml of cell lysis buffer [50 mM Tris-HCl, pH 8.0; 1
mM EDTA, 0.2% (v/v) Nonidet P-40 and 0.15 M NaCl]. The lysate was
centrifuged at 16,000 × g at 4°C for 2 min and the nuclear pellet
was used for cccDNA extraction (as follows) and the supernatant was
used to isolate core particle-associated HBV rcDNA (35). Briefly, the cytoplasmic fraction was
treated with 1/4 volume of 35% PEG8000 in 1.75 M NaCl, incubated on
ice for 30 min and centrifuged at 16,000 × g for 10 min. The
precipitated viral particles were dissolved in DNA extraction
buffer (10 mM Tris-HCl, pH 8.0; 100 mM NaCl; 1 mM EDTA; 0.5% SDS;
200 µg/ml proteinase K) at 45°C for 1 h and viral DNA was recovered
by phenol-extraction and ethanol-precipitation (36). HBV cccDNA was extracted by
dissolving the nuclear pellet in the DNA extraction buffer,
followed by phenol-extraction and ethanol-precipitation.
Contaminating genomic DNA was removed using treatment with
Plasmid-Safe DNase (Epicentre; Illumina, Inc.).
Quantification of HBV using RT-qPCR,
Southern blotting and northern blotting
SYBR Green RT-qPCR was performed to quantify HBV
rcDNA and cccDNA. Primers for HBV rcDNA were GAGTGTGGATTCGCACTCC
(forward) and GAGGCGAGGGAGTTCTTCT (reverse) (37) and for cccDNA were
GCGGCTCCCCGTCTGTGCC, GTCTGTGCCTTCTCATCTGC (forward) and
GTCCATGCCCCAAAGCAACC (reverse) (38) at a final concentration of 200 nM.
The Thermal Cycler Dice Real Time System (Takara Bio, Inc.) was
used according to the manufacturer's instructions. The PCR cycling
program for HBV rcDNA consisted of an initial denaturing step at
95°C for 15 min, followed by 45 amplification cycles at 95°C for 10
sec, 60°C for 20 sec and 72°C for 30 sec. The cycles for HBV cccDNA
were initial denaturation at 95°C for 15 min, followed by 45
amplification cycles at 95°C for 15 sec, 63°C for 10 sec and 72°C
for 25 sec. The mitochondrial DNA gene was used for normalization:
GCCTGCCTGATCCTCCAAAT (forward) and AAGGTAGCGGATGATTCAGCC (reverse).
The Thermal Cycler Dice Real Time System (ver. 6.0.1A, Takara Bio,
Inc.) was used for analysis according to the manufacturer's
instructions.
Southern and northern blots for HBV rcDNA and RNA
were performed using a digoxigenin-labeled RNA probe as previously
reported (39,40). Briefly, 15 µg of HBV rcDNA was
electrophoresed on a 1% agarose/Tris-Borate EDTA gel, which was
depurinated with HCl (0.25 M) for 15 min and denatured in NaOH (0.5
M) for 15 min. DNA was transferred to a Hybond-N+ membrane (Roche
Diagnostics GmbH) by capillary transfer. The membrane was UV
cross-linked and hybridized with digoxygenin-tagged HBV RNA probes
in Dig Easy Hybridization Buffer (Roche Diagnostics GmbH; 20 ng/ml
stock diluted to 1:5,000) overnight at 60°C. The membrane was
washed twice for 5 min at room temperature in 2X SSC, 0.1% SDS and
for 15 min at 60°C in 0.1X SSC, 0.1% SDS. The membrane was blocked
with blocking reagent (Roche Diagnostics GmbH) for 30 min at room
temperature and incubated for 30 min in blocking buffer containing
187.5 mU/ml (1:4,000 v:v) anti-digoxygenin-alkaline phosphate
antibody (Roche Diagnostics GmbH) followed by Immun-Star AP
Substrate (Bio-Rad Laboratories, Inc.) treatment, and exposed to
X-ray film.
For northern blotting, the cytoplasmic fraction was
isolated as described above and treated with TRIzol®
(Thermo Fisher Scientific, Inc.) and 5 µg of RNA was separated on
1% agarose gel. Equal loading was examined by staining the gels
with ethidium bromide and the RNA was transferred onto nylon
membranes. Transferred RNA was detected in the same way as Southern
blotting analysis.
Analysis of methylation profiles of
HBV
The 5-mC dot blot assay was performed to assess the
global methylation of HBV cccDNA using mouse anti-5-methylcytosine
antibody (cat. no. 33D3; Active Motif, Inc.) as previously reported
(41). Briefly, 50 ng of HBV cccDNA
was blotted onto a Hybond-N+ membrane, UV-cross linked and
subjected to immunodetection with 1:5,000 diluted anti-5mC
antibody.
Global methylation of cccDNA was quantified by
MethylFlash Global DNA Methylation (5-mC) ELISA Easy kit (cat. no.
P-1030; EpiGentek Group Inc.) according to the manufacturer's
protocols. A total of 100 ng of cccDNA was used for the assay.
Bisulfite modification of HBV cccDNA was performed
as previously reported (42).
Methyl Primer Express (v1.0; Applied Biosystems; Thermo Fisher
Scientific, Inc.) was used to design primers for
methylation-specific PCR for CpG island II, which is relevant for
transcriptional control of HBV replication as described previously
(21,43): For unmethylated DNA,
GTGGGATGTTTTTTGTTTAT (forward) and AACAAAAAATCCACATAAAA (reverse;
for methylated DNA, GCGGGACGTTTTTTGTTTAC (forward) and
AACGAAAAATCCGCGTAAAA (reverse) were used. The PCR mixture contained
TOPreal qPCR 2X PreMIX (Enzynomics Co., Ltd.), primers (200 nM) and
50 ng of bisulfite-modified DNA in a final volume of 50 µl. PCR
cycles were as follows: Initial activation at 95°C for 5 min,
followed by 35 cycles of 94°C for 30 sec, 55°C for 30 sec, 72°C for
30 sec and a final 5 min extension at 72°C. The PCR amplicons were
quantified via densitometric analysis of 1% ethidium
bromide-stained agarose gels. PCR was performed to measure the
relative amount of methylation using the same primer pairs for
methylation and normalized using the unmethylated primer pair.
Bisulfite sequencing was performed to detect the methylation of CpG
island II (39). The cloned
sequences were analyzed using a BiQ Analyzer (Max Planck Institut
Informatik; http://biq-analyzer.bioinf.mpi-inf.mpg.de/) (44).
Chromatin immunoprecipitation (ChIP)
assay, western blotting and luciferase assay for DNA
methyltransferases
ChIP assay was performed as previously reported
(45), with minor modifications.
Briefly, formaldehyde (cat. no. F8775; Sigma-Aldrich; Merck KGaA)
was added directly to trypsinized HepG2 cells in PBS at a final
concentration of 1% at room temperature for 10 min for fixation.
The process was stopped with the addition of 0.125 M final
concentration of glycine. HepG2 cells were collected using
centrifugation at 300 × g for 3 min at 4°C and rinsed with cold
phosphate-buffered saline. The cell pellets were resuspended in MC
lysis buffer [10 mM Tris-Cl, pH 7.5; 10 mM NaCl; 3 mM MgCl, 0.5%
(v/v) Nonidet P-40], incubated on ice for 15 min and centrifuged at
300 × g for 3 min at 4°C. The nuclei were resuspended in FA lysis
buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 1% Triton X-100, 0.1%
sodium deoxycholate, 0.1% SDS, 1X protease inhibitor) and incubated
at room temperature for 10 min. Prior to sonication, 50 mg of glass
beads (G1277, Sigma-Aldrich; Merck KGaA) was added to each sample.
The samples were sonicated on ice with the Sonic Dismembrator Model
100 (Thermo Fisher Scientific, Inc.) with a microtip probe set to a
power output of 4–6 W for 10 cycles of 10 sec pulse/50 sec rest and
then microcentrifuged at 300 × g for 3 min at 4°C. Dynabeads
Protein G (Thermo Fisher Scientific, Inc.) were blocked with bovine
serum albumin/salmon sperm DNA and incubated for 10 min at room
temperature with 5 µg of appropriate antibodies or negative control
normal mouse IgG (cat. no. sc-2025; Santa Cruz Biotechnology,
Inc.), DNMT1 (cat. no. ab13537; Abcam), DNMT3a (sc-20703; Santa
Cruz Biotechnology, Inc.) and DNMT3b (cat. no. sc-20704, Santa Cruz
Biotechnology). Sonicated chromatin was added to the
Dynabead-antibody complexes and rotated at 4°C for ~16 h. Whole
cell lysate were prepared as positive control. The beads were
washed and cross-linked by addition of NaCl to a final
concentration of 250 mM, followed by boiling for 15 min and
treatment with proteinase K. HBV was detected using PCR with
specific primers as described previously (14). Western blotting was performed as
follows: Total protein was extracted from HepG2cells using RIPA
buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton
X-100, 0.5% Sodium deoxycholate, 0.1% SDS). The protein was
quantified by bicinchoninic acid assay. Protein (100 µg) was
separated per lane via 8% SDS-PAGE and transferred onto a
polyvinylidene difluoride membrane (Roche Diagnostics GmbH). The
membranes were blocked with 2.5% skimmed milk in PBS with 0.1%
Tween20 for 1 h at room temperature. The membranes were incubated
with the following primary antibodies overnight at 4°C: Mouse
monoclonal anti-β-actin (cat. no. sc69879; dilution 1:2,000; Santa
Cruz Biotechnology, Inc.), mouse monoclonal anti-DNMT 3a (cat. no.
ab13888; dilution 1:1,000; Abcam), mouse monoclonal anti-DNMT 3b
(cat. no. ab13604; dilution 1:1,000; Abcam). The membranes were
subsequently incubated with horseradish peroxidase-conjugated goat
anti-mouse IgG secondary antibody (cat. no. sc2005; dilution
1:2,000; Santa Cruz Biotechnology, Inc.) for 1 h at room
temperature. Signals were detected by exposure of the blot to x-ray
film using SuperSignal West Pico Chemiluminescent Substrate (Thermo
Fisher Scientific, Inc.). ImageJ software (version 1.46; the
National Institutes of Health) was used for measuring the intensity
of bands in film image. The promoter of DNMT3b was cloned into the
pGL3-basic vector and used for the luciferase assay with
normalization to Renilla luciferase in the pRL-TK plasmid
according to the instructions of the manufacturer. Briefly, 24 h
after transfection of pGL3-DNMT3b promoter (1 µg) and pRL-TK (100
ng) into HepG2 cells (5×105 cells) using Lipofectamine
2000® (Thermo Fisher Scientific), luciferase activity
was quantified by using a Dual-Luciferase Reporter Assay kit
(Promega Corporation).
Statistical analysis
Continuous data were analyzed by using unpaired
Student's t test except for comparison of real-time PCR data which
were made by pairwise fixed reallocation randomization test using
REST software (REST 2009 version 2.0.13; Qiagen GmbH; http://www.gene-quantification.de/rest-2009.html)
(46). At least three experiments
were repeated and the results were presented as the mean ± standard
deviation. Comparison of bisulfite sequencing data was made by
Fisher's exact test. P<0.05 was considered to indicate a
statistically significant difference.
Results
IFN-α suppresses HBV replication in
HepG2 cells
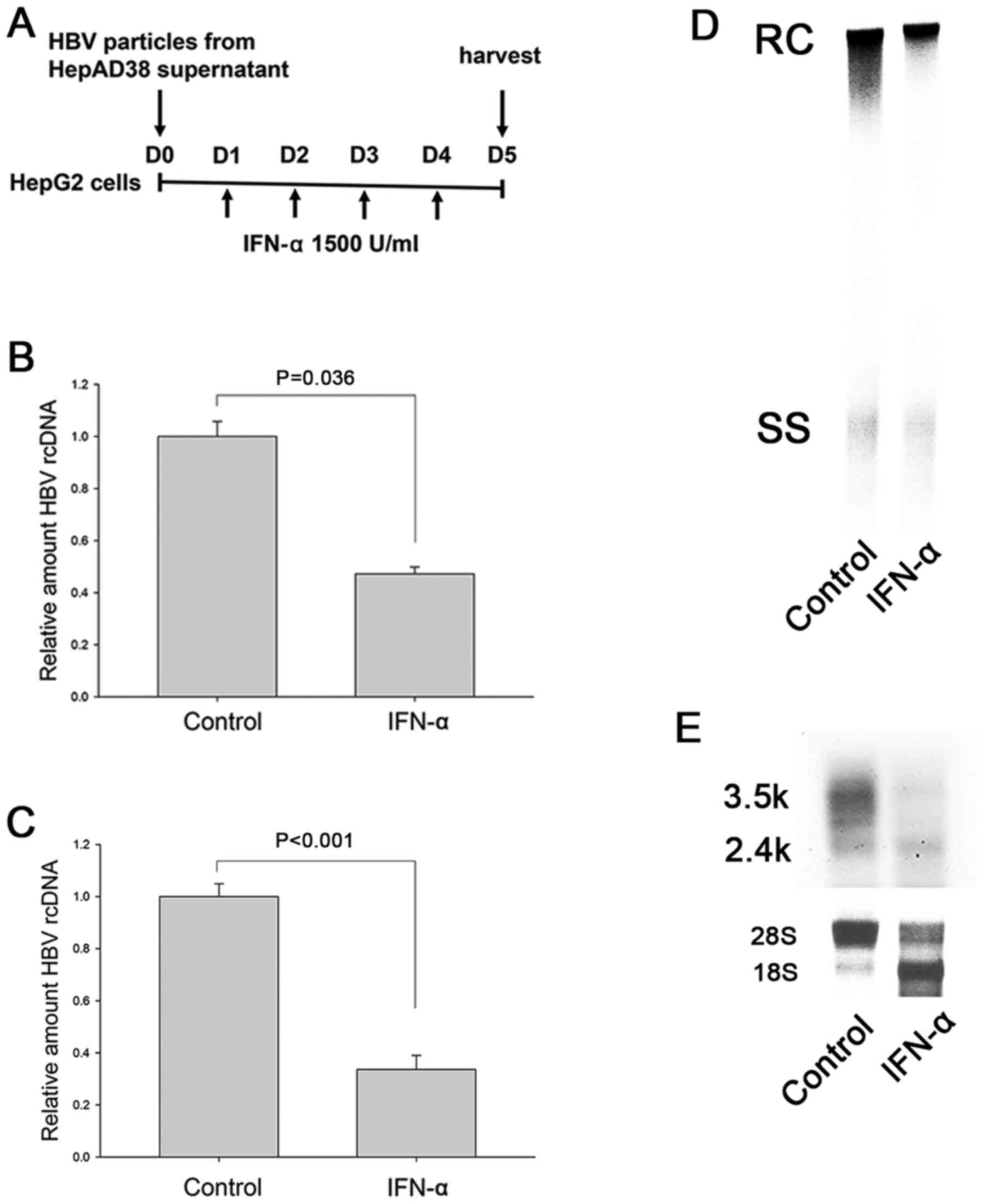
In the cell-based HBV replication model of the
present study HepG2 cells were infected with concentrated HBV
virion particles, rather than HBV-expressing plasmids, in order to
exclude the possibility of carrier plasmid DNA serving as a source
of methylation and of engineered promoters being responsive to IFN
(47). IFN-α was replenished daily
for four days to ensure sufficient time for methylation as that in
our previous study (Fig. 1A)
(42). The model showed that IFN-α
treatment suppressed the intracellular HBV DNA (Fig. 1B and D) and HBV DNA secretion to
culture supernatant (Fig. 1C).
IFN-α also reduced HBV RNA, suggesting the possibility of
transcriptional suppression (Fig.
1E).
IFN-α does not affect methylation
profiles of HBV cccDNA in HepG2 cells
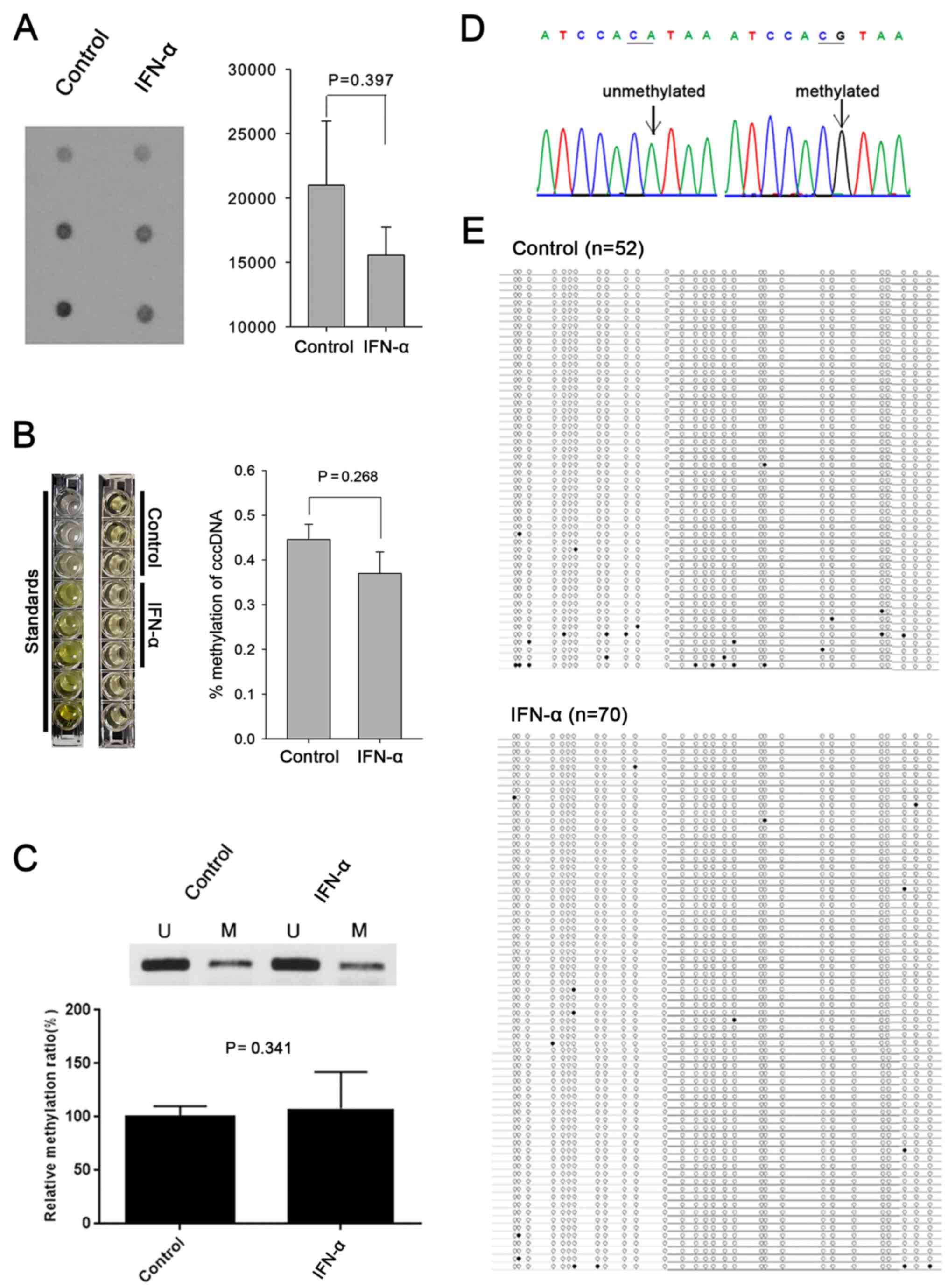
Since methylation of HBV cccDNA has been identified
as a mechanism of regulating HBV replication both in vitro
and in vivo (17–20,48),
the present study sought to determine whether the anti-HBV effect
of IFN-α was also mediated through cccDNA methylation. First,
global methylation level was assessed using 5-mC dot blot assay,
which showed that IFN-α did not affect the overall level of HBV
cccDNA methylation (Fig. 2A).
Global DNA methylation ELISA assay also confirmed no difference of
the degree of methylation between control and IFN-α treated group
(Fig. 2B). Next,
methylation-specific PCR showed that IFN-α did not alter the level
of methylation in CpG island II, which regulates the X promoter of
HBV cccDNA (Fig. 2C) (49). Bisulfite sequencing also confirmed a
similar degree of methylation in CpG island II of HBV cccDNA
regardless of interferon treatment (11/52 for control and 13/70 for
IFN-α; P=0.723; Fig. 2D and E).
IFN-α suppressess DNMT3b binding to
HBV cccDNA
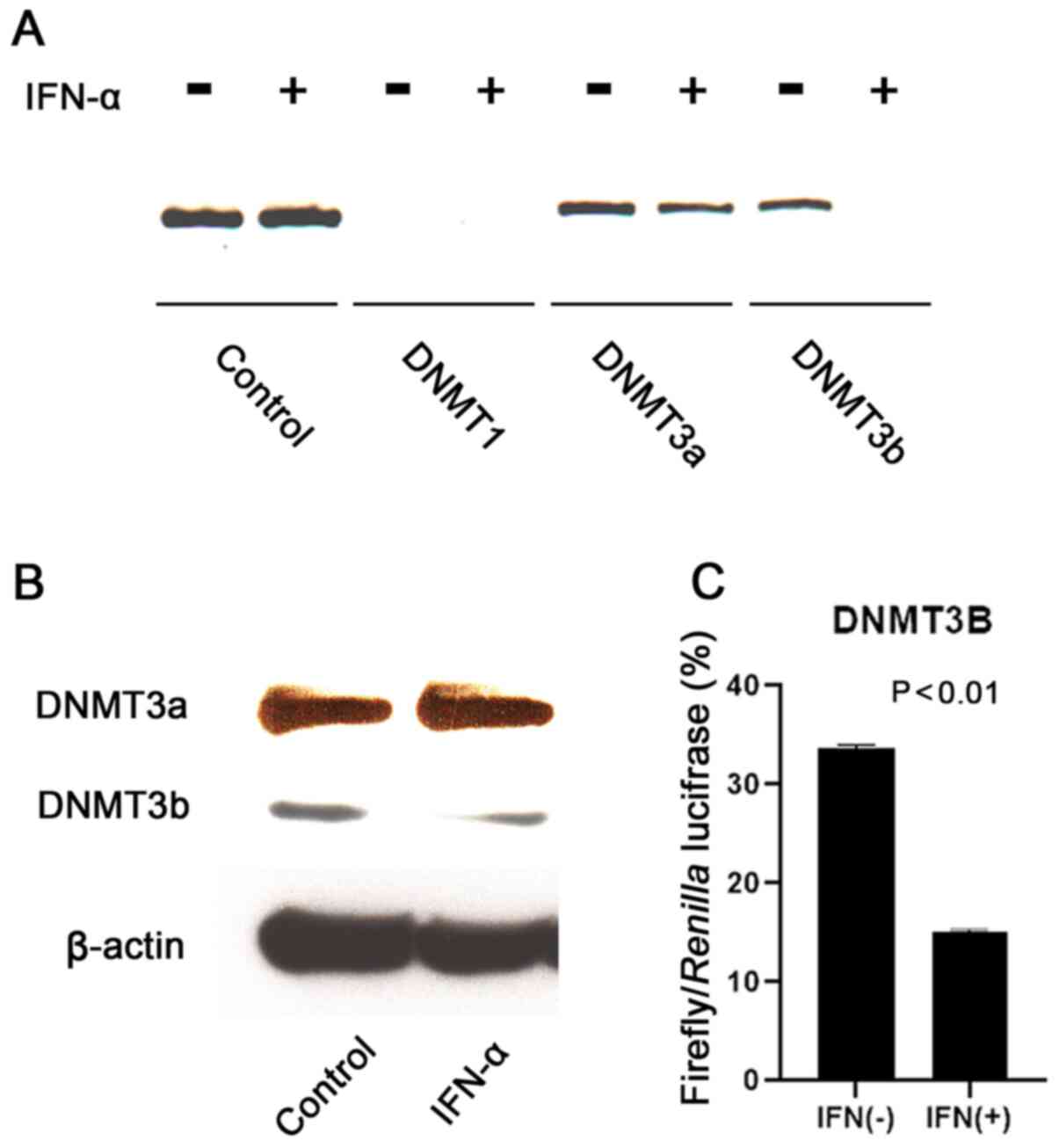
Finally, the present study determined the effect of
IFN-α on the association of DNMTs with HBV cccDNA. ChIP assay
confirmed that DNMT1 did not bind to HBV cccDNA, but DNMT3a and
DNMT3b were associated with cccDNA (Fig. 3A). Notably, IFN-α suppressed the
binding of DNMT3b to cccDNA. Western blotting demonstrated
decreased DNMT3b protein expression induced by IFN-α (Fig. 3B). The luciferase assay indicated
that IFN-α suppressed DNMT3b promoter activity in HepG2 cells
(Fig. 3C).
Discussion
Methylation of cccDNA has recently received
considerable attention as a mechanism of transcriptional regulation
in human HBV infection (10). The
present authors and other researchers have shown that methylation
of cccDNA suppresses the transcriptional activity of cccDNA in
cell-based models (17,18,20).
In addition, cccDNA methylation is frequently observed in the
immunologically controlled stages of chronic HBV infection with low
viremia (20,22). However, the underlying mechanisms of
de novo methylation of cccDNA are largely unknown.
The anti-HBV effect of IFN-α simulates the host
immune response against chronic HBV infection; i.e., sustained
inhibition of viral gene transcription (5), loss of HBeAg and decrease in HBsAg
titers (50). Epigenetic regulation
may serve as a shared mechanism between sustained anti-HBV response
to IFN-α and the low viremia phase of chronic HBV infection
(20,30). While IFN-induced PTMs of histone
proteins have been well established (14,15,29),
only one previous report that assessed the effect of IFN-α on
cccDNA methylation was found, which found a total lack of
methylation in both control and IFN-treated cccDNA (30). In that study, however, CpG island I
was sequenced, which is barely methylated in the advanced stage of
chronic HBV infection and is not related to transcriptional control
of cccDNA in previous studies (22,51).
The present study hypothesized that cccDNA
methylation may also contribute to the IFN-mediated transcriptional
suppression of HBV. To test this hypothesis, our previous
cell-based model was used to generate a condition for cccDNA
methylation, with some modifications (39). First, direct infection with
wild-type HBV virions (52,53) was used instead of plasmid-mediated
in vitro HBV models (30),
because CpG-containing plasmids may trigger inflammatory cytokine
responses and become inadvertently methylated (54). The model of the present study,
although it has low efficacy for HBV infection due to the lack of
sodium taurocholate cotransporting polypeptide (37), has an advantage over transfection of
greater-than-genome HBV plasmid or HBV-producing cell lines in
which newly generated HBV cccDNA cannot be differentiated from the
transfected plasmids or pre-formed cccDNA. Second, conditionally
HBV-producing cell lines are avoided because the
tetracycline-responsive promoter contains functional
interferon-inducible response elements (47). The baseline methylation of HBV
cccDNA was confirmed without interferon treatment (Fig. 2), indicating the presence of
methylation machinery against natural HBV infection, as previously
suggested (20,42).
IFN-α suppressed HBV replication in the cell model
of the present study. Contrary to the authors' prediction, however,
extensive bisulfite sequencing experiments did not detect
significant differences in the level of cccDNA methylation between
control and IFN-α-treated groups. Therefore, the anti-HBV effect of
IFN-α is unlikely to be mediated by DNA methylation. Recently, DNA
methylation has been proposed as a target of epigenetic therapy for
CHB (10). Despite the negative
result of the present study it is hypothesized that modulation of
cccDNA methylation may still be pursued to enhance the therapeutic
efficacy of IFN.
Mammalian de novo DNA methylation is mediated
by DNMT3A and DNMT3B (55). DNMT3a
is suggested to induce de novo HBV cccDNA methylation
(19). The ChIP data confirmed the
association of both DNMT3a and DNMT3b with HBV cccDNA (Fig. 3A). The data also suggested that
innate interferon response is not the main mechanism of HBV
methylation and that ISGs are not sufficient for methylation of
cccDNA. which is in contrast to the fact that downstream signaling
of interferon modifies cccDNA-bound histones (56). Thus, the underlying mechanisms of
de novo cccDNA methylation by DNMTs still remain largely
unknown. The data of the present study does not necessarily exclude
the potential participation of ISGs as a mechanism underlying DNA
methylation in mammalian cells. Whether additional inflammatory
cytokines such as TNFα and IFNγ or small RNAs may cooperate with
IFN signaling to induce cccDNA methylation needs to be clarified by
further studies.
There have been reports demonstrating that HBV
induces de novo methylation of ISGs, leading to suppression
of anti-HBV innate signaling of IFN (57,58).
It is not known whether innate IFN response counter-attacks HBV
epigenetically, but it is plausible considering the fact that IFN-α
may induce reversible DNA demethylation of ISGs (59). The finding of the present study that
IFN-α suppressed DNMT3b expression and DNA binding may also be in
line with the hypothesis, but further studies are warranted.
Since HepG2 cells were maintained in the presence of
fetal bovine serum (FBS) throughout the experiments, the
possibility that various cytokines in FBS may affect the results
cannot be excluded. Serum-free culture condition is difficult to
maintain in our cell model because our previous data showed that ≥5
days were needed before methylation of HBV cccDNA was induced
(39). Other related studies also
used similar study design with serum in culture media (16,30)
and it is hypothesized that possible effect of cytokines may have
been adjusted by comparison with the control with same culture
condition (except for IFN). However, potential interactions between
IFN and other cytokines need to be explored in future studies.
The present study has some limitations. First, it
did not sort HepG2 cells according to intracellular HBV replication
after treatment with HBV particles. However, it was presumed that
the efficacy of HBV infection should be similar between control and
IFN-treated cells. In addition, the degree of methylation was
assessed by normalization to total cccDNA, so that the results are
independent of the efficacy of HBV infection. Second, methylation
of cccDNA was evaluated in liver cancer cells and the possibility
that normal hepatocytes may show different pattern of methylation
cannot be excluded.
In conclusion, methylation of HBV cccDNA was not
induced by IFN-α, which suggested that it is not responsible for
the antiviral effect of IFN-α against HBV in HepG2 cells and that
alternative mechanisms need to be sought to enhance cccDNA
methylation as a novel therapy against HBV.
Acknowledgements
Not applicable.
Funding
This study was supported by the Seoul National
University Bundang Hospital (grant no. 03-2010-001) to Dr Jin-Wook
Kim.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
IYM and JWK designed the study; IYM performed the
experiments; IYM and JWK analyzed the data and contributed to the
final manuscript. Both authors read and approved the final
manuscript and confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
HBV
|
hepatitis B virus
|
|
rcDNA
|
relaxed circular DNA
|
|
cccDNA
|
covalently closed circular DNA
|
|
DNMT
|
DNA methyltransferase
|
|
IFN
|
interferon
|
References
|
1
|
Yuen MF, Chen DS, Dusheiko GM, Janssen
HLA, Lau DTY, Locarnini SA, Peters MG and Lai CL: Hepatitis B virus
infection. Nat Rev Dis Primers. 4:180352018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Seeger C and Mason WS: Hepatitis B virus
biology. Microbiol Mol Biol Rev. 64:51–68. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nassal M: HBV cccDNA: Viral persistence
reservoir and key obstacle for a cure of chronic hepatitis B. Gut.
64:1972–1984. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Trepo C, Chan HL and Lok A: Hepatitis B
virus infection. Lancet. 384:2053–2063. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Volz T, Lutgehetmann M, Wachtler P, Jacob
A, Quaas A, Murray JM, Dandri M and Petersen J: Impaired
intrahepatic hepatitis B virus productivity contributes to low
viremia in most HBeAg-negative patients. Gastroenterology.
133:843–852. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Suslov A, Meier MA, Ketterer S, Wang X,
Wieland S and Heim MH: Transition to HBeAg-negative chronic
hepatitis B virus infection is associated with reduced cccDNA
transcriptional activity. J Hepatol. 74:794–800. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Moolla N, Kew M and Arbuthnot P:
Regulatory elements of hepatitis B virus transcription. J Viral
Hepat. 9:323–331. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xia Y and Guo H: Hepatitis B virus cccDNA:
Formation, regulation and therapeutic potential. Antiviral Res.
180:1048242020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Oropeza CE, Tarnow G, Sridhar A, Taha TY,
Shalaby RE and McLachlan A: The regulation of HBV transcription and
replication. Adv Exp Med Biol. 1179:39–69. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hong X, Kim ES and Guo H: Epigenetic
regulation of hepatitis B virus covalently closed circular DNA:
Implications for epigenetic therapy against chronic hepatitis B.
Hepatology. 66:2066–2077. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dandri M: Epigenetic modulation in chronic
hepatitis B virus infection. Semin Immunopathol. 42:173–185. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bock CT, Schranz P, Schroder CH and
Zentgraf H: Hepatitis B virus genome is organized into nucleosomes
in the nucleus of the infected cell. Virus Genes. 8:215–229. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bock CT, Schwinn S, Locarnini S, Fyfe J,
Manns MP, Trautwein C and Zentgraf H: Structural organization of
the hepatitis B virus minichromosome. J Mol Biol. 307:183–196.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pollicino T, Belloni L, Raffa G, Pediconi
N, Squadrito G, Raimondo G and Levrero M: Hepatitis B virus
replication is regulated by the acetylation status of hepatitis B
virus cccDNA-bound H3 and H4 histones. Gastroenterology.
130:823–837. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tropberger P, Mercier A, Robinson M, Zhong
W, Ganem DE and Holdorf M: Mapping of histone modifications in
episomal HBV cccDNA uncovers an unusual chromatin organization
amenable to epigenetic manipulation. Proc Natl Acad Sci USA.
112:E5715–E5724. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yuan Y, Yuan H, Yang G, Yun H, Zhao M, Liu
Z, Zhao L, Geng Y, Liu L, Wang J, et al: IFN-α confers epigenetic
regulation of HBV cccDNA minichromosome by modulating GCN5-mediated
succinylation of histone H3K79 to clear HBV cccDNA. Clin
Epigenetics. 12:1352020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Guo Y, Li Y, Mu S, Zhang J and Yan Z:
Evidence that methylation of hepatitis B virus covalently closed
circular DNA in liver tissues of patients with chronic hepatitis B
modulates HBV replication. J Med Virol. 81:1177–1183. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Vivekanandan P, Thomas D and Torbenson M:
Methylation regulates hepatitis B viral protein expression. J
Infect Dis. 199:1286–1291. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vivekanandan P, Daniel HD, Kannangai R,
Martinez-Murillo F and Torbenson M: Hepatitis B virus replication
induces methylation of both host and viral DNA. J Virol.
84:4321–4329. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim JW, Lee SH, Park YS, Hwang JH, Jeong
SH, Kim N and Lee DH: Replicative activity of hepatitis B virus is
negatively associated with methylation of covalently closed
circular DNA in advanced hepatitis B virus infection.
Intervirology. 54:316–325. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang Y, Li C, Zhang Y, Zhu H, Kang Y, Liu
H, Wang J, Qin Y, Mao R, Xie Y, et al: Comparative analysis of CpG
islands among HBV genotypes. PLoS One. 8:e567112013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang Y, Mao R, Yan R, Cai D, Zhang Y, Zhu
H, Kang Y, Liu H, Wang J, Qin Y, et al: Transcription of hepatitis
B virus covalently closed circular DNA is regulated by CpG
methylation during chronic infection. PLoS One. 9:e1104422014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang ZM, Lu R, Wang P, Yu Y, Chen D, Gao
L, Liu S, Ji D, Rothbart SB, Wang Y, et al: Structural basis for
DNMT3A-mediated de novo DNA methylation. Nature. 554:387–391. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Konerman MA and Lok AS: Interferon
treatment for hepatitis B. Clin Liver Dis. 20:645–665. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wieland SF, Guidotti LG and Chisari FV:
Intrahepatic induction of alpha/beta interferon eliminates viral
RNA-containing capsids in hepatitis B virus transgenic mice. J
Virol. 74:4165–4173. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xiong W, Wang X, Liu X, Xiang L, Zheng L
and Yuan Z: Interferon-inducible MyD88 protein inhibits hepatitis B
virus replication. Virology. 319:306–314. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lucifora J, Xia Y, Reisinger F, Zhang K,
Stadler D, Cheng X, Sprinzl MF, Koppensteiner H, Makowska Z, Volz
T, et al: Specific and nonhepatotoxic degradation of nuclear
hepatitis B virus cccDNA. Science. 343:1221–1228. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cheng J, Zhao Q, Zhou Y, Tang L, Sheraz M,
Chang J and Guo JT: Interferon alpha induces multiple cellular
proteins that coordinately suppress hepadnaviral covalently closed
circular DNA transcription. J Virol. 94:e00442–20. 2020. View Article : Google Scholar
|
|
29
|
Belloni L, Allweiss L, Guerrieri F,
Pediconi N, Volz T, Pollicino T, Petersen J, Raimondo G, Dandri M
and Levrero M: IFN-α inhibits HBV transcription and replication in
cell culture and in humanized mice by targeting the epigenetic
regulation of the nuclear cccDNA minichromosome. J Clin Invest.
122:529–537. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu F, Campagna M, Qi Y, Zhao X, Guo F, Xu
C, Li S, Li W, Block TM, Chang J and Guo JT: Alpha-interferon
suppresses hepadnavirus transcription by altering epigenetic
modification of cccDNA minichromosomes. PLoS Pathog.
9:e10036132013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Allweiss L, Volz T, Lutgehetmann M,
Giersch K, Bornscheuer T, Lohse AW, Petersen J, Ma H, Klumpp K,
Fletcher SP and Dandri M: Immune cell responses are not required to
induce substantial hepatitis B virus antigen decline during
pegylated interferon-alpha administration. J Hepatol. 60:500–507.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ladner SK, Otto MJ, Barker CS, Zaifert K,
Wang GH, Guo JT, Seeger C and King RW: Inducible expression of
human hepatitis B virus (HBV) in stably transfected hepatoblastoma
cells: A novel system for screening potential inhibitors of HBV
replication. Antimicrob Agents Chemother. 41:1715–1720. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Qiao L, Sui J and Luo G: Robust human and
murine hepatocyte culture models of hepatitis B virus infection and
replication. J Virol. 92:e01255–18. 2018. View Article : Google Scholar
|
|
34
|
Paran N, Geiger B and Shaul Y: HBV
infection of cell culture: Evidence for multivalent and cooperative
attachment. EMBO J. 20:4443–4453. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sohn JA, Litwin S and Seeger C: Mechanism
for CCC DNA synthesis in hepadnaviruses. PLoS One. 4:e80932009.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Garner I: Isolation of
high-molecular-weight DNA from animal cells. The nucleic acid
protocols handbook. Rapley R: Humana Press; Totowa, NJ: pp. 3–7.
2000, View Article : Google Scholar
|
|
37
|
Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z,
Huang Y, Qi Y, Peng B, Wang H, et al: Sodium taurocholate
cotransporting polypeptide is a functional receptor for human
hepatitis B and D virus. Elife. 1:e000492012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bowden S, Jackson K, Littlejohn M and
Locarnini S: Quantification of HBV covalently closed circular DNA
from liver tissue by real-time PCR. Methods Mol Med. 95:41–50.
2004.PubMed/NCBI
|
|
39
|
Moon IY, Choi JH, Chung JW, Jang ES, Jeong
SH and Kim JW: MicroRNA20 induces methylation of hepatitis B virus
covalently closed circular DNA in human hepatoma cells. Mol Med
Rep. 20:2285–2293. 2019.PubMed/NCBI
|
|
40
|
Min BY, Kim NY, Jang ES, Shin CM, Lee SH,
Park YS, Hwang JH, Jeong SH, Kim N, Lee DH and Kim JW: Ethanol
potentiates hepatitis B virus replication through oxidative
stress-dependent and -independent transcriptional activation.
Biochem Biophys Res Commun. 431:92–97. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jia Z, Liang Y, Ma B, Xu X, Xiong J, Duan
L and Wang D: A 5-mC dot blot assay quantifying the DNA methylation
level of chondrocyte dedifferentiation in vitro. J Vis Exp.
555652017.PubMed/NCBI
|
|
42
|
Park HK, Min BY, Kim NY, Jang ES, Shin CM,
Park YS, Hwang JH, Jeong SH, Kim N, Lee DH and Kim JW: Short
hairpin RNA induces methylation of hepatitis B virus covalently
closed circular DNA in human hepatoma cells. Biochem Biophys Res
Commun. 436:152–155. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Vivekanandan P, Thomas D and Torbenson M:
Hepatitis B viral DNA is methylated in liver tissues. J Viral
Hepat. 15:103–107. 2008.PubMed/NCBI
|
|
44
|
Bock C, Reither S, Mikeska T, Paulsen M,
Walter J and Lengauer T: BiQ Analyzer: Visualization and quality
control for DNA methylation data from bisulfite sequencing.
Bioinformatics. 21:4067–4068. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Aparicio O, Geisberg JV, Sekinger E, Yang
A, Moqtaderi Z and Struhl K: Chromatin immunoprecipitation for
determining the association of proteins with specific genomic
sequences in vivo. Curr Protoc Mol Biol. 21 (Suppl
69):21.3.1–21.3.33. 2005.PubMed/NCBI
|
|
46
|
Pfaffl MW, Horgan GW and Dempfle L:
Relative expression software tool (REST) for group-wise comparison
and statistical analysis of relative expression results in
real-time PCR. Nucleic Acids Res. 30:e362002. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Rang A and Will H: The
tetracycline-responsive promoter contains functional
interferon-inducible response elements. Nucleic Acids Res.
28:1120–1125. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhang X, Hou J and Lu M: Regulation of
hepatitis B virus replication by epigenetic mechanisms and
microRNAs. Front Genet. 4:2022013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhong C, Lu H, Han T, Tan X, Li P, Huang
J, Xie Q, Hou Z, Qu T, Jiang Y, et al: CpG methylation participates
in regulation of hepatitis B virus gene expression in host sperm
and sperm-derived embryos. Epigenomics. 9:123–125. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Tan G, Song H, Xu F and Cheng G: When
Hepatitis B virus meets interferons. Front Microbiol. 9:16112018.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Jain S, Chang TT, Chen S, Boldbaatar B,
Clemens A, Lin SY, Yan R, Hu CT, Guo H, Block TM, et al:
Comprehensive DNA methylation analysis of hepatitis B virus genome
in infected liver tissues. Sci Rep. 5:104782015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Bchini R, Capel F, Dauguet C, Dubanchet S
and Petit MA: In vitro infection of human hepatoma (HepG2) cells
with hepatitis B virus. J Virol. 64:3025–3032. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Mabit H, Dubanchet S, Capel F, Dauguet C
and Petit MA: In vitro infection of human hepatoma cells (HepG2)
with hepatitis B virus (HBV): Spontaneous selection of a stable HBV
surface antigen-producing HepG2 cell line containing integrated HBV
DNA sequences. J Gen Virol. 75((Pt 10)): 2681–2689. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Mitsui M, Nishikawa M, Zang L, Ando M,
Hattori K, Takahashi Y, Watanabe Y and Takakura Y: Effect of the
content of unmethylated CpG dinucleotides in plasmid DNA on the
sustainability of transgene expression. J Gene Med. 11:435–443.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Okano M, Bell DW, Haber DA and Li E: DNA
methyltransferases Dnmt3a and Dnmt3b are essential for de novo
methylation and mammalian development. Cell. 99:247–257. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Yang Y, Zhao X, Wang Z, Shu W, Li L, Li Y,
Guo Z, Gao B and Xiong S: Nuclear sensor interferon-inducible
protein 16 inhibits the function of hepatitis B virus covalently
closed circular DNA by integrating innate immune activation and
epigenetic suppression. Hepatology. 71:1154–1169. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lim KH, Park ES, Kim DH, Cho KC, Kim KP,
Park YK, Ahn SH, Park SH, Kim KH, Kim CW, et al: Suppression of
interferon-mediated anti-HBV response by single CpG methylation in
the 5′-UTR of TRIM22. Gut. 67:166–178. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zheng DL, Zhang L, Cheng N, Xu X, Deng Q,
Teng XM, Wang KS, Zhang X, Huang J and Han ZG: Epigenetic
modification induced by hepatitis B virus X protein via interaction
with de novo DNA methyltransferase DNMT3A. J Hepatol. 50:377–387.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Scott R, Siegrist F, Foser S and Certa U:
Interferon-alpha induces reversible DNA demethylation of the
interferon-induced transmembrane protein-3 core promoter in human
melanoma cells. J Interferon Cytokine Res. 31:601–608. 2011.
View Article : Google Scholar : PubMed/NCBI
|