Introduction
As a response to chronic injury, liver fibrosis is
caused by various etiologies, such as viral infection, alcohol,
cholestasis and metabolic diseases (1). The pathological process of liver
fibrosis is produced by the interaction of various cytokines and
growth factors, and it may result from the imbalance between the
synthesis and degradation of extracellular matrix (ECM) in liver
tissue (2). Several studies have
shown that abnormal activation of hepatic stellate cells (HSCs)
promotes the progression of liver fibrosis. Transforming growth
factor-β (TGF-β) is activated from the ECM and released from HSCs
(3–6). In cooperation with other signaling
pathways, such as reactive oxygen species (ROS) and
platelet-derived growth factor (PDGF), TGF-β signaling is
considered the key fibrogenic pathway that drives HSC activation
(7).
Autophagy is a lysosome-dependent cellular
homeostasis mechanism that regulates intracellular homeostasis
(8). Efficient autophagy involves a
series of intracellular signaling cascade events involving mTOR
kinase activation, LC3 processing and the formation of
autolysosomes (9). This selective
removal of autophagic substrates is regulated by P62/SQSMT1 (P62),
which is closely related to the conversion of LC3I to LC3II and the
formation of autolysosomes (10).
Recently, studies have shown that autophagy is implicated in a
variety of liver diseases (11,12).
Induction of autophagy may promote the degradation of ECM
components, such as collagen, and improve the process of hepatic
fibrosis (13). Therefore, it was
hypothesized in the present study that the pathogenic mechanism of
liver fibrosis is the key to finding effective therapeutic targets
to prevent its progression and even reverse liver fibrosis.
However, the regulatory mechanism of autophagy during HSC
activation is still controversial.
MicroRNAs (miRNAs/miRs) are a family of
single-stranded non-coding small molecules (18–25 nucleotides in
length) that are involved in mediating gene expression by cleaving
and destabilizing the targeted mRNAs at the post-transcriptional
level (14). Evidence has shown
that miRNAs are functionally implicated in various cellular
biological mechanisms, such as cell growth, development,
differentiation, proliferation and apoptosis (15–17),
and they have relevance in clinical diagnosis (18). The differential expression of
miRNAs, especially miR221, is upregulated in liver fibrosis tissue.
Roderburg et al (19)
identified 21 downregulated and 10 upregulated miRNAs in fibrotic
livers, especially miR221, which was observed to be upregulated in
CCl4-induced mice. However, the role of miR221 in
hepatic fibrosis remains unclear. In previous years, miR221 has
been widely reported to act as a tumor modulator involved in tumor
growth and invasiveness, especially hepatocellular carcinoma
(20,21). Researchers have increasingly been
concerned about the biological role of miR221 in regulating
hepatocellular carcinoma development (21). According to reports, astrocyte
elevated gene-1 protein/miR221 can regulate the progression of
liver cancer (22), and it also
plays a crucial role in regulating hepatocellular carcinoma
migration by targeting lysine-specific demethylase PHF2 (23). Remarkably, the pathological behavior
of activated HSCs is very similar to that of hepatoma cells
(24). Activated HSCs show enhanced
proliferation and TGF-β1 secretion in large quantities (25), while TGF-β1 is significantly
increased in patients with liver cancer (26).
In the present study, it was determined whether
autophagy regulates hepatic fibrosis by regulating TGF-β1-induced
HSC activation, and then the potential regulatory gene targets
leading to the sequence of events associated with miR221 in HSCs
were investigated.
Materials and methods
Animals and treatment
A total of 10 adult male C57BL/6 mice weighing 20–25
g (age, 6–8 weeks) were obtained from the Animal Research Center of
Hebei Medical University (Shijiazhuang, China). They were housed at
a temperature of 25±2°C and 50% humidity, with free access to food
and water (12 h light and dark cycles). All protocols were approved
by the Hebei Medical University Institutional Animal Experimental
Ethics Committee (approval no. IACUC-Hebmu-2020005) and carried out
according to the provisions of the guidelines for the Care and Use
of Laboratory Animals (27). The
model of liver fibrosis was prepared according to a previous report
by Hyun et al (28). Mice
(n=5) were exposed to CCl4 (Sigma-Aldrich; Merck KGaA)
twice a week for 10 weeks by intraperitoneal injection to establish
the in vivo model. In the control group for comparison, the same
regimen was performed with corn oil. All liver fibrosis model mice
were sacrificed by cervical dislocation, and liver samples were
obtained.
Cell culture and treatment
LX2 cells (a human HSC line; Procell Life Science
& Technology Co., Ltd.) were obtained from cryopreservation.
LX2 cells were cultured in DMEM (Gibco; Thermo Fisher Scientific,
Inc.) containing 10% FBS (Gibco; Thermo Fisher Scientific, Inc.)
and incubated in humidified 5% CO2 at 37°C. The medium
was changed every 24 h. Cultured LX2 cells were used after 3 days
of incubation. Then, LX2 cells (1×105) were treated with
TGF-β1 (Sigma-Aldrich; Merck KGaA) in a 37°C incubator with 5%
CO2 for 24 h to establish the in vitro model.
To verify the role of miR221 in HSC activation and
LAMP2 expression, cultured LX2 cells were transfected with miR221
mimics, miR-negative controls (NCs), miR221 inhibitor and LAMP2
overexpression vector. After transfection, the medium was changed
to fresh medium containing 10% FBS and incubated at 37°C and 5%
CO2 for 24 h. Then, the transfected cells were treated
with TGF-β1 (µM) for 24 h in a 37°C incubator with 5%
CO2. Additionally, to assess the role of autophagy in
regulating HSC activation, the autophagy inducer rapamycin (RAPA; 5
µM; Cell Signaling Technology, Inc.) was added for 1 h prior to
TGF-β1 cotreatment for 24 h in a 37°C incubator with 5%
CO2. All of the aforementioned cells were harvested for
RNA/miRNA isolation, and cell lysates were collected for western
blot analysis.
Cell transfection
LX2 cells were used for cell transfection.
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) was diluted with Optimem I (Gibco; Thermo Fisher
Scientific, Inc.) to a selected optimal concentration and then
transfected into LX2 cells. LX2 cells were well cultured until
60–70% confluence. LX2 cells (1×105) were transfected
with 50 nM miR221 mimics (5′-ACCUGGCAUACAAUGUAGAUUU-3′), 100 nM
miR221 inhibitor (5′-AAAUCUACAUUGUAUGCCAGGU-3′) and their matched
negative controls (25 nM NC-miR mimic, 5′-UUCUCCGAACGUGUCACGUTT-3′
and 25 nM NC-miR inhibitor, 5′-CAGUACUUUUGUGUAGUACAA-3′) using
Lipofectamine® 2000 in a 37°C incubator with 5%
CO2. The cells were collected 48 h after transfection.
miR221 mimics, miR-NCs and miR221 inhibitor were purchased from
Guangzhou RiboBio Co., Ltd. Furthermore, LAMP2 overexpression
plasmids (LAMP2 group; Shanghai GeneChem Co., Ltd.) or empty
pcDNA3.1 plasmids (NC group; Shanghai GeneChem Co., Ltd.) were
transfected into LX2 cells at a multiplicity of infection (MOI) of
50, according to the manufacturer's instructions. The cells were
transfected for 48 h at 37°C with 5% CO2.
Dual-luciferase reporter assay
LAMP2 3′UTR fragments and mutants containing the
miR221 binding site were synthesized and inserted downstream of the
luciferase gene in the vector. Mutant (mut) LAMP2 3′UTR was
constructed using a site-directed mutagenesis kit (Promega
Corporation). The luciferase reporter vector was pGL3 (Shaanxi
Youbio Technology Co., Ltd.). 1×105 LX2 cells were
transfected with wild-type (wt) LAMP2 3′UTR or mut LAMP2 3′UTR
together with miR221 mimics or miR-NC using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.). After transfection for 48 h, a dual-luciferase
reporter assay was performed to measure the luciferase activity
(Promega Corporation). Luciferase activity was normalized against
Renilla luciferase activity (Promega Corporation).
Reverse transcription-quantitative PCR
(RT-qPCR)
Primer sequences were designed and synthesized by
Sangon Biotech Co., Ltd. Total RNA was extracted from liver tissues
or cells by using TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's instructions.
Total RNA was reverse transcribed into cDNA using a PrimeScript™ RT
kit (Takara Bio, Inc.), according to the manufacturer's protocol.
RT-qPCR was performed using SYBR-Green mix (Takara Bio, Inc.) on an
ABI Prism 7500 real-time PCR system. Thermocycling conditions were
as follows: Initial denaturation at 95°C for 10 min, 40 cycles of
95°C for 30 sec and 60°C for 30 sec. Genes of each sample were
amplified in triplicate. The primer sequences were as follows:
LAMP2-forward (F), 5′-CUGGCUUUAAAAAAAGGAGAAAA-3′ and reverse (R),
5′-ACCCATCTCACCCATTCTTG-3′; miR221-F, 5′-CCTGAAACCCAGCAGACAA-3′ and
R, 5′-CAGGTCTGGGGCATGAAC-3′; and U6-F, 5′-GCGCGTCGTGAAGCGTTC-3′ and
R, 5′-GTGCAGGGTCCGAGGT-3′. The 2−ΔΔCq method (29) was used to calculate the expression
of miRNA or LAMP2 normalized to U6 mRNA.
Western blotting assay
LX2 cells were treated according to the experimental
design. LX2 cell lysates were collected in ice-cold RIPA buffer
(Beyotime Institute of Biotechnology). Total protein was quantified
using a BCA protein assay kit (Thermo Fisher Scientific, Inc.).
Proteins (5 µg) were separated via SDS-PAGE using 10% gels, and
electrotransferred to PVDF membranes (MilliporeSigma), which were
incubated with anti-collagen-I (COL-I; cat. no. AF7001; 1:1,000;
Affinity Biosciences), α-smooth muscle actin (α-SMA; cat. no.
19245; 1:1,000; Cell Signaling Technology, Inc.), LC3 (cat. no.
2775; 1:1,000; Cell Signaling Technology, Inc.), P62 (cat. no.
8025; 1:1,000; Cell Signaling Technology, Inc.), LAMP2 (cat. no.
49067; 1:1,000; Cell Signaling Technology, Inc.) and β-actin (cat.
no. 4970; 1:1,000; Cell Signaling Technology, Inc.) primary
antibodies at 4°C overnight. Subsequently, membranes were incubated
with horseradish peroxidase (HRP)-conjugated anti-Rabbit IgG
secondary antibody (cat. no. SA00001-2; 1:2,000; ProteinTech Group,
Inc.) for another 2 h at room temperature. All blots were
visualized using enhanced chemiluminescence (ECL) reagents (Thermo
Fisher Scientific, Inc.), and the scanned band images were
semi-quantified using ImageJ software version 1.8 (National
Institutes of Health).
Immunofluorescence assay
LX2 cells were treated according to the experimental
design. The cells were fixed with 4% ice-cold paraformaldehyde for
20 min at 4°C and permeabilized with 0.05% Triton X-100. Cells were
treated with 3% BSA (Sigma-Aldrich; Merck KGaA) for 1 h at room
temperature and incubated with the primary antibody P62 (cat. no.
7695; 1:100; Cell Signaling Technology, Inc.) overnight at 4°C. The
next day, the cells were stained with FITC-conjugated secondary
antibodies (cat. no. ZF-0311; 1:100; OriGene Technologies, Inc.)
for another 1 h. After washing with PBS, the nuclei were stained
with DAPI for 5 min at room temperature. Fluorescence was
visualized under a microscope (IX-81; Olympus Corporation).
Bioinformatics analysis
Bioinformatics analysis was performed to determine
the target gene of miR221. The possible regulatory genes of miR221
were obtained using TargetScan 3.1 (http://www.targetscan.org/vert_72/), and information
on the predicted autophagy-related genes were chosen by searching
the Human Autophagy-dedicated Database (HADb; autophagy.lu). Then,
the targets from TargetScan and HADb were overlapped, and potential
autophagy-related genes were identified, which were reported to be
closely related to the degradation of autolysosome.
Statistical analysis
Each experiment was performed in triplicate. Data
are expressed as the mean ± SD. The significant differences between
experimental groups were determined by unpaired Student's t-test or
one-way ANOVA followed by the Tukey's test for multiple
comparisons. P<0.05 was considered to indicate a statistically
significant difference.
Results
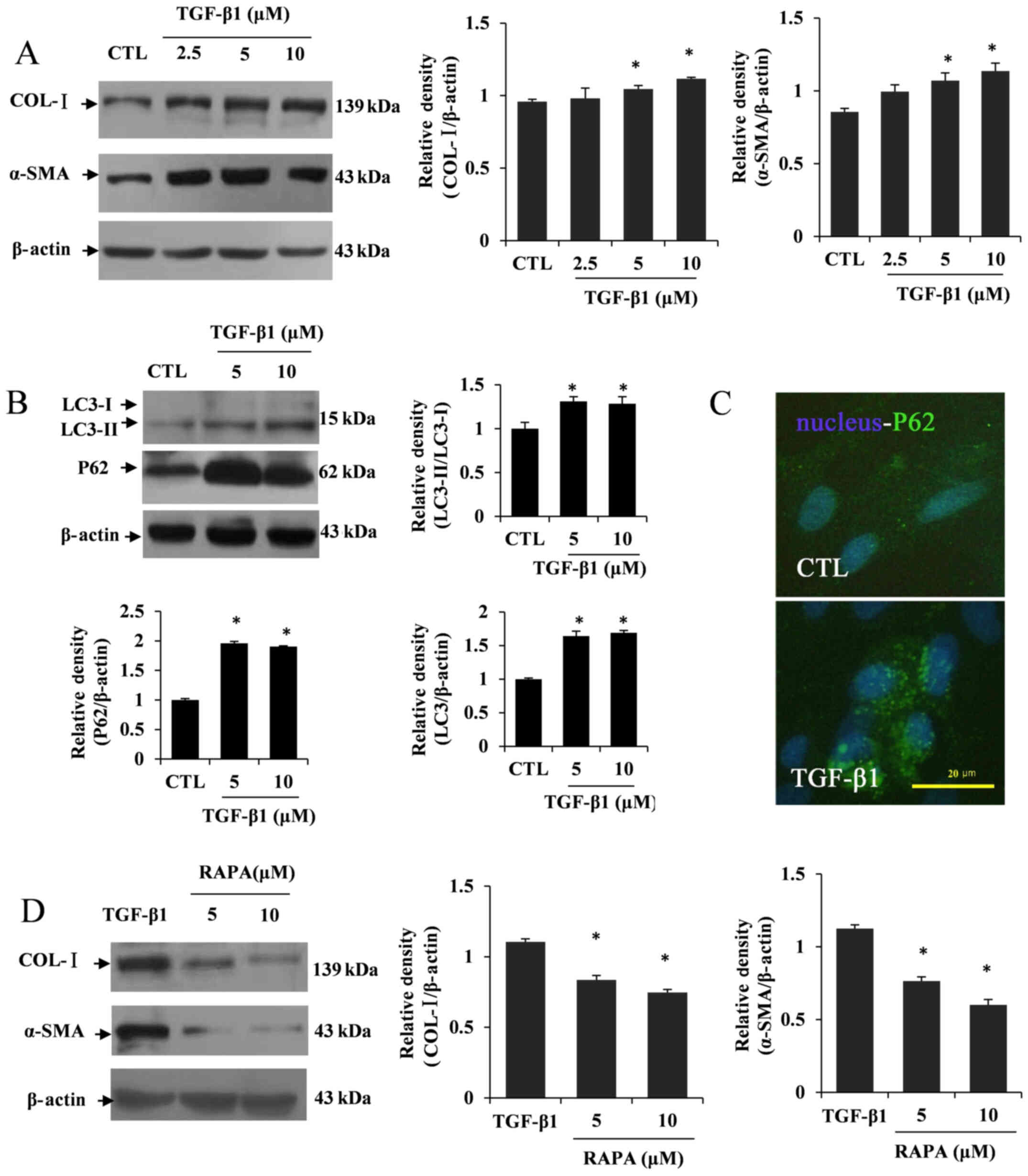
TGF-β1-induced HSC activation is
partially subject to autophagy dysfunction
Considering the potential function of TGF-β1 in
inducing HSC activation, relevant markers for hepatic fibrosis were
detected, including COL-I and α-SMA, which are involved in the
activation of HSCs (30). The
western blotting results revealed that TGF-β1 significantly induced
COL-I and α-SMA expression at concentrations of 5 and 10 µM
compared with the control group (P<0.05; Fig. 1A). Based on these results,
intracellular autophagy changes were also detected by western
blotting and immunofluorescence. The results showed that the
conversion of LC3I to LC3II was increased in TGF-β1-treated LX2
cells. However, the expression of P62 was conversely elevated
(P<0.05; Fig. 1B and C).
Furthermore, LX2 cells were treated with the autophagy inducer RAPA
(5 or 10 µM) following TGF-β1 treatment. The western blotting
results showed that the levels of COL-I and α-SMA expression were
decreased after RAPA treatment in TGF-β1-treated LX2 cells compared
with the TGF-β1-treated group (P<0.05; Fig. 1D). The aforementioned results
implied a potential relationship between HSC activation and
autophagy deficiency in liver fibrosis.
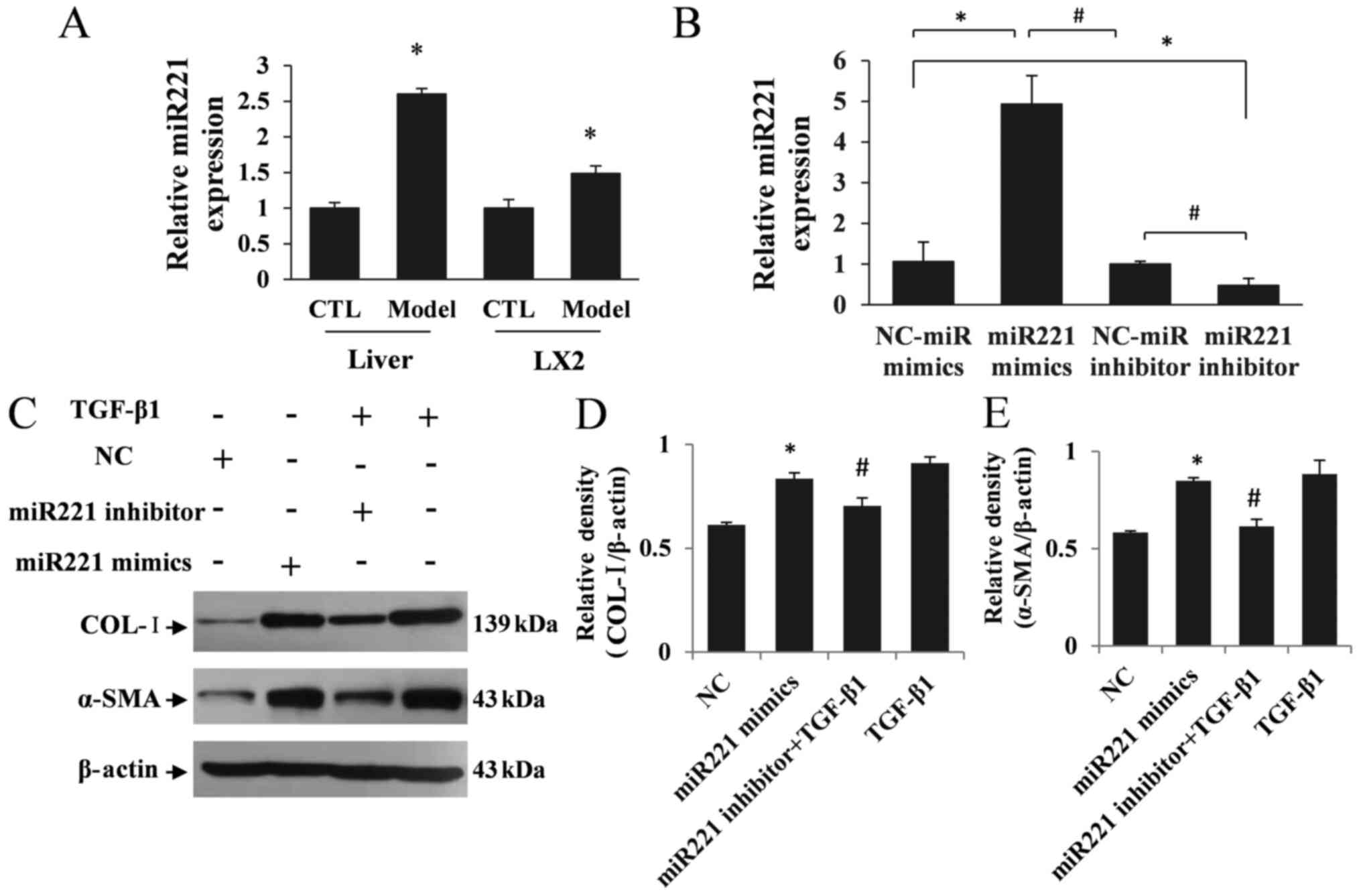
miR221 is upregulated in liver
fibrosis mice and activated HSCs
The differential expression of miRNAs, especially
miR221, has been found to be upregulated in liver fibrosis tissues
(14). To study the function of
miR221 in activated HSCs, a liver fibrosis model was constructed in
vivo and in vitro (21) and the
expression changes of miR221 were verified in liver fibrosis mice
and TGF-β1-treated LX2 cells. Based on the RT-qPCR results, the
expression of miR221 in liver fibrosis mice was significantly
increased (P<0.05; Fig. 2A).
Furthermore, miR221 expression was significantly increased in the
LX2 model group compared with the control group (P<0.05;
Fig. 2A). To evaluate whether
miR221 expression played a role in activated HSCs, miR221 mimics,
miR221 inhibitor and their matched NCs were transfected into LX2
cells. The RT-qPCR results showed that miR221 was increased in LX2
cells transfected with miR221 mimics and decreased in miR221
inhibitor-transfected LX2 cells (P<0.05; Fig. 2B). In addition, COL-I and α-SMA
expression levels were significantly increased in LX2 cells
transfected with miR221 mimics compared with the NC group
(P<0.05; Fig. 2C-E). Whereas,
the expression levels of COL-I and α-SMA were decreased in LX2
cells in the miR221 inhibitor + TGF-β1 group compared with the
TGF-β1-treated group (P<0.05; Fig.
2C-E). These data revealed that miR221 may play a crucial role
in HSC activation.
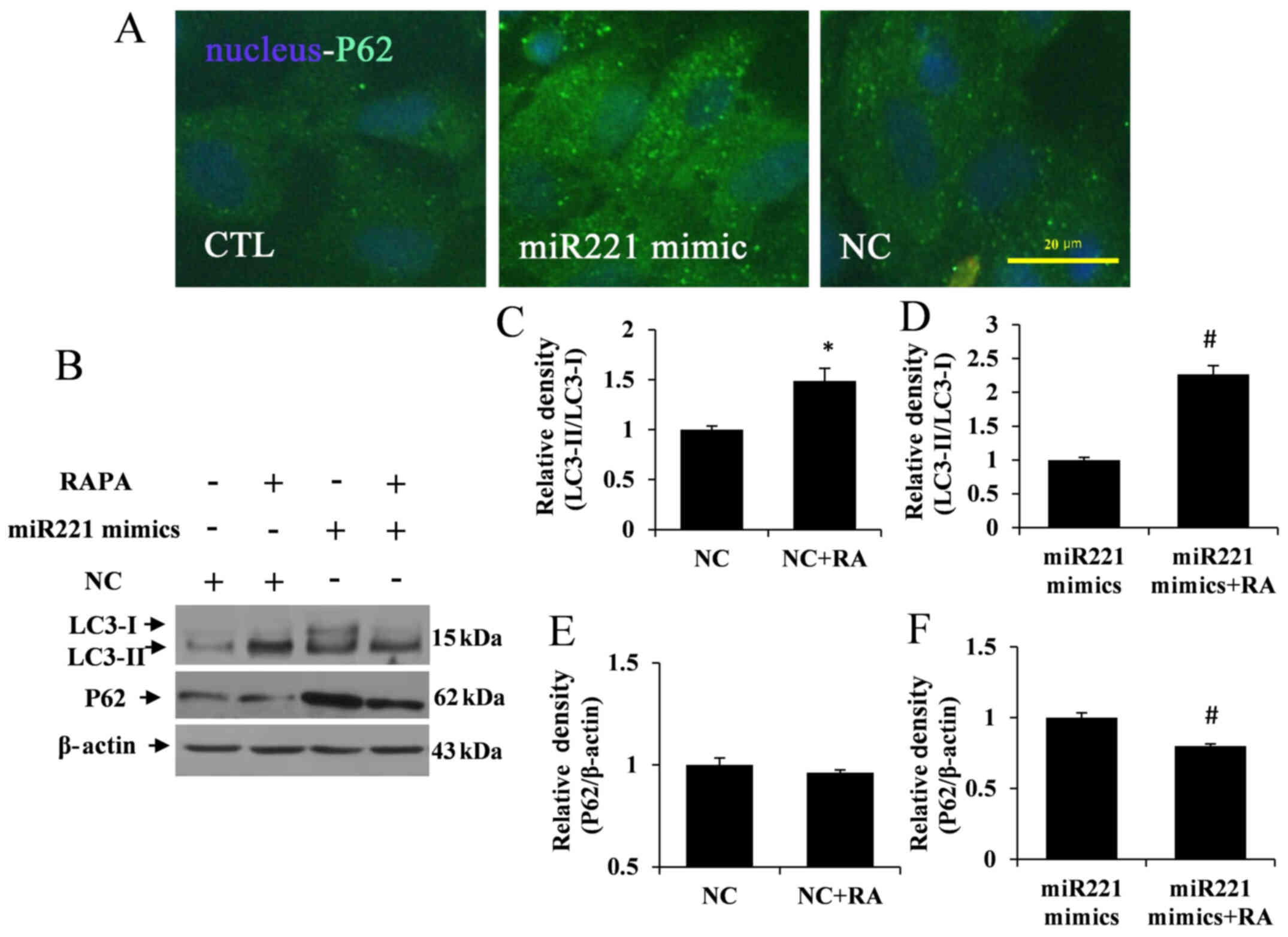
miR221 inhibits autophagy in LX2
cells
To verify the role of miR221 in modulating autophagy
activation or disrupting the process of autophagy initiation,
intracellular autophagy functional changes were detected in
miR221-overexpressing LX2 cells by using immunofluorescence. The
results showed that a mass of P62 was aggregated in miR221
mimic-transfected LX2 cells, but no obvious autolysosome
dysfunction was observed in the control and NC groups (Fig. 3A). Furthermore, to study the
relationship between miR221 expression and autophagy activation,
miR221-overexpressing LX2 cells were treated with RAPA. The western
blotting results showed that the overexpression of miR221 in LX2
cells induced the aggregation of P62 expression and resulted in low
conversion of LC3I to LC3II. Whereas, LC3-I to LC3-II conversion
was significantly increased in the NC + RA and miR221 mimics + RA
groups compared with the NC and miR221 mimics groups, respectively
(P<0.05; Fig. 3B-D).
Furthermore, P62 aggregation was significantly decreased in the
miR221 mimics + RA group compared with the miR221 mimics group
(P<0.05), although there was no significant difference between
the NC + RA and NC groups (Fig. 3B, E
and F). Thus, RAPA appeared to upregulate the autophagic flux
in LX2 cells. These findings implied potential cross-talk between
miR221 expression and autophagic flux.
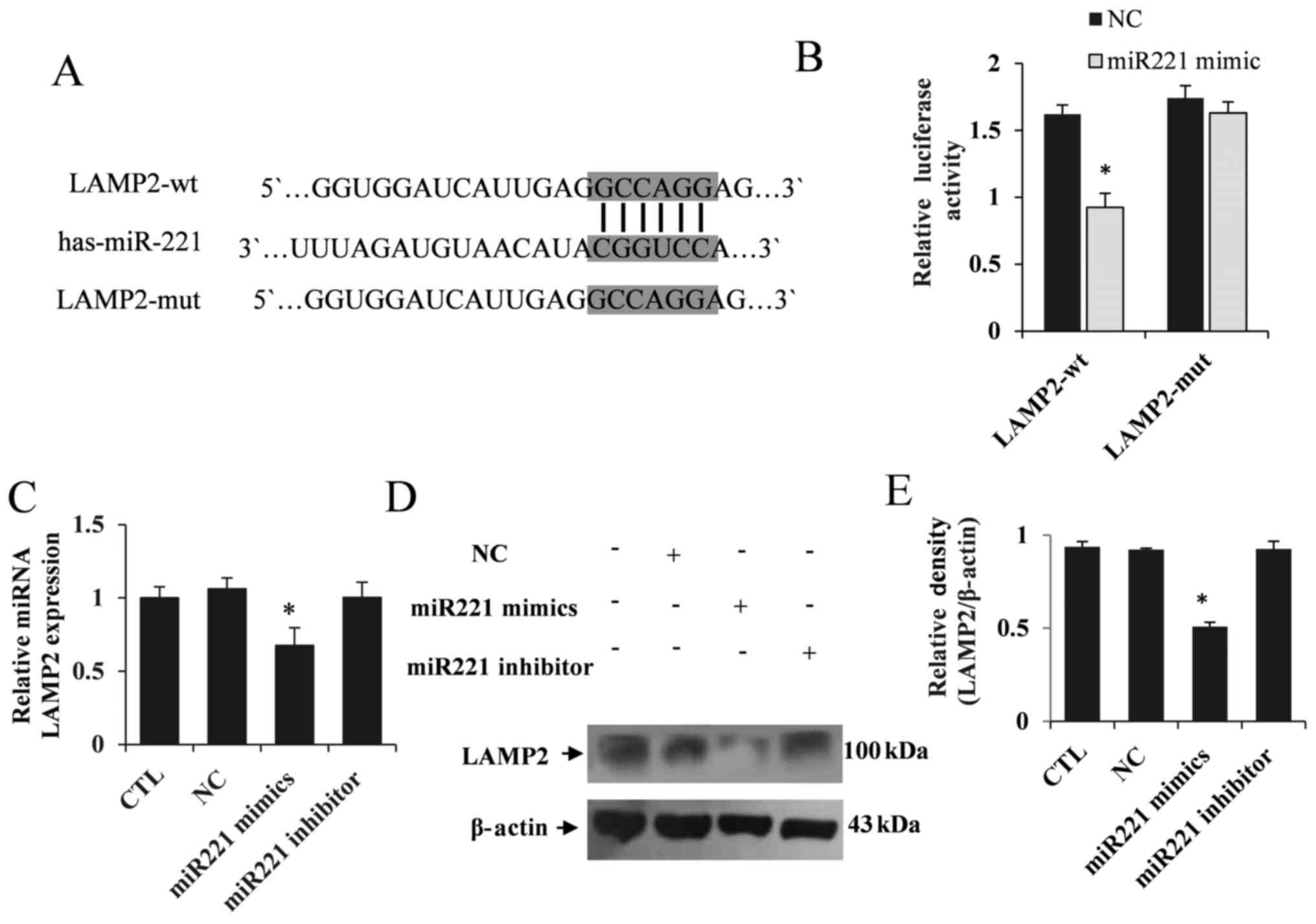
miR221 specifically regulates
autophagy by targeting LAMP2 expression
To explore the molecular mechanism of miR221 in
regulating autophagy, bioinformatics tools were used to predict
potential target genes. Among these potential targets, it was found
that LAMP2, a crucial gene in the regulation of autophagy
activation, was a potential target of miR221 (Fig. 4A). The dual-luciferase reporter
assay showed that miR221 significantly inhibited the fluorescence
activity of LAMP2-wt, but did not regulate LAMP2-mut (P<0.05;
Fig. 4B). These results confirmed
the targeted regulatory effect of miR221 on LAMP2. Furthermore, LX2
cells were transfected with miR221 mimics, NC or miR221 inhibitor
for 48 h, and RT-qPCR and western blotting analyses were performed
to determine LAMP2 expression. It was demonstrated that LAMP2
expression was significantly decreased in the LX2 cells transfected
with miR221 mimics, but the miR221 inhibitor had no effect on LAMP2
expression (Fig. 4C-E).
miR221 regulates TGF-β1-induced HSC
activation by targeting LAMP2 expression
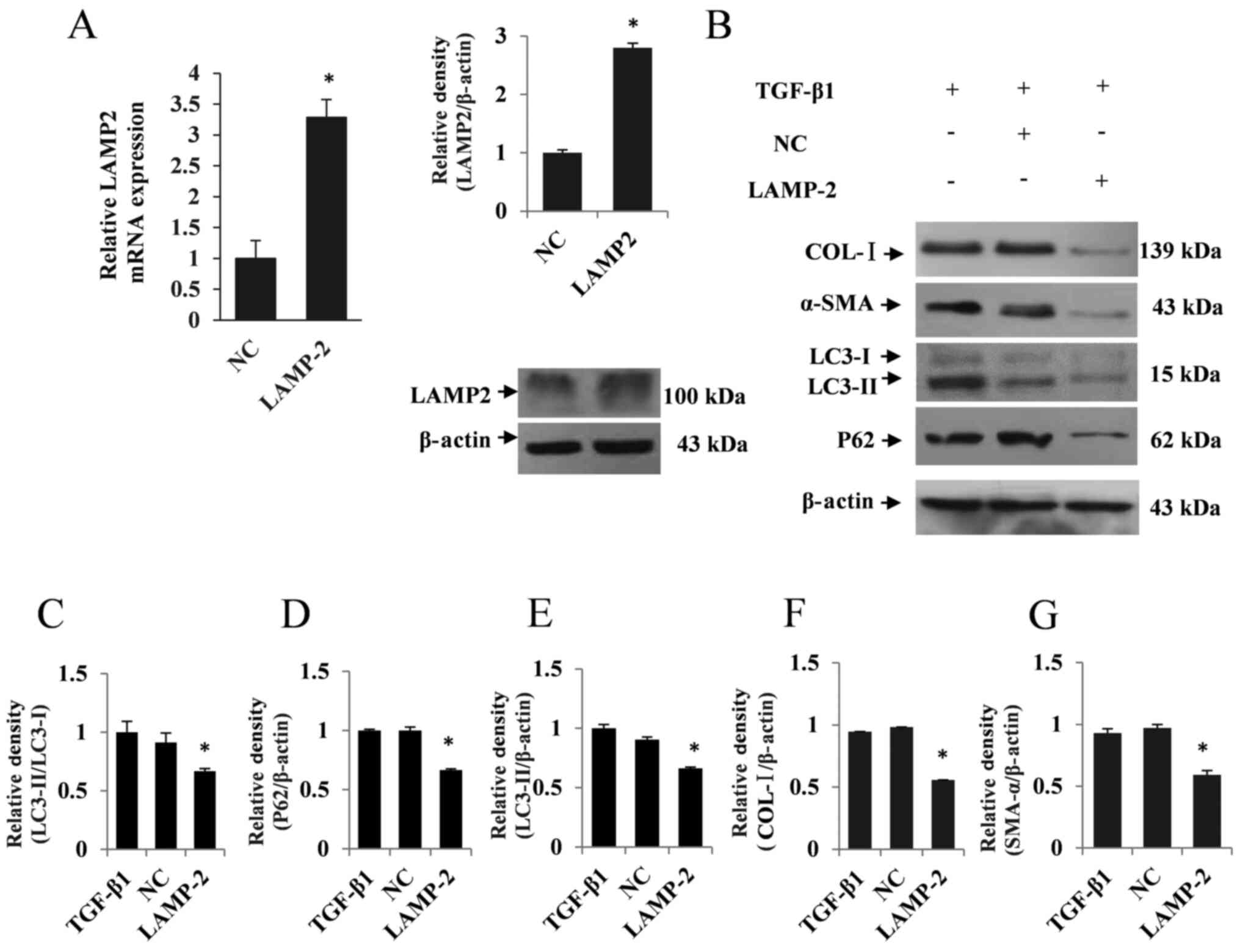
In rescue experiments, LAMP2 expression was
significantly upregulated in LAMP2-overexpressing LX2 cells
compared with the NC group (P<0.05; Fig. 5A). Based on these findings, the
expression of autophagic marker proteins, including LC3II and P62,
were examined by western blotting. As a result, overexpression of
LAMP2 considerably upregulated autophagic flux following TGF-β1
treatment in LX2 cells, as indicated by the decreased expression of
LC3II and P62 compared with the NC group (P<0.05; Fig. 5B-E). Additionally, the expression
levels of COL-I and α-SMA were determined to assess the regulatory
function of LAMP2. The western blotting results showed a
concomitant decrease in the expression levels of COL-I and α-SMA in
LAMP2-overexpressing LX2 cells following TGF-β1 treatment
(P<0.05; Fig. 5B, F and G).
These results indicated that miR221 may regulate TGF-β1-induced HSC
activation by targeting LAMP2 expression.
Discussion
Liver fibrosis is caused by a variety of chronic
pathogenic factors, especially an imbalance in ECM synthesis and
degradation, which lead to abnormal liver structure and
hepatocellular function (2). To
further explore potentially effective targets for liver fibrosis
therapy to solve this problem, elucidating the mechanism of HSC
activation is of importance. Autophagy is a lysosome-dependent
cellular homeostasis mechanism that regulates intracellular
homeostasis. Although the specific mechanism mediating HSC
activation is still unclear, some conclusions have been made,
indicating that the different effects of autophagy on the fate of
HSCs depend on the degree and extent of autophagy (31). In the present study, TGF-β1
activated HSCs and simultaneously caused P62 aggregation, which was
involved in autolysosome degradation, as indicated by the impaired
processing of LC3. These results implied that autolysosome
function, but not autophagy initiation, was blocked. Therefore, it
was hypothesized that its pathogenic mechanism is related to the
abnormal regulation of autophagy. Moreover, induction of autophagic
flux by RAPA reduced TGF-β1-induced COL-I and α-SMA expression.
These results showed that TGF-β1 was able to impair autophagy
function in LX2 cells, thereby further showing that TGF-β1 induced
the activation of HSCs, which was accompanied by dysfunction of
autophagy.
miRNAs play a vital role in the regulation of the
expression of a large number of genes at the posttranscriptional
level. Various microRNAs have been proven to be involved in
epigenetic changes and multiple pathological mechanisms, such as
cancer or fibrosis (32). The
differential expression of miRNAs, especially the upregulation of
miR221, has been observed in liver fibrosis tissue. Roderburg et
al (19) identified 21
downregulated and 10 upregulated miRNAs in fibrotic livers,
especially miR221, which was upregulated in CCl4-induced
mice. Thus, the present study sought to study the function of
miR221 in activated HSCs and elucidate its potential role in
regulating autophagy function. miR221-overexpressing cell models of
liver fibrosis were constructed in the exploration of the possible
molecular physiological and pathogenic mechanisms of miR221 in
hepatic fibrosis. Based on western blotting results, expression
levels of the hepatic fibrosis-relevant markers COL-I and α-SMA
were increased in miR221-overexpressing LX2 cells, while the miR221
inhibitor decreased the induction of COL-I and α-SMA expression
following TGF-β1 treatment. The results revealed that liver
fibrosis progression was associated with increased miR221
expression. Similar studies have reported that miR221 is increased
in liver tissues of patients with hepatocellular carcinoma, and is
involved in the progression of hepatic diseases (20,33).
Previous studies have confirmed the role of miR221
in regulating autophagy (34,35).
TGF-β1-induced HSC activation abnormally decreases autophagy
(36), which seems to be consistent
with the present results showing increased LC3II and P62
expression, indicating disruption of autophagy. To investigate the
possible involvement of miR221 in this process, intracellular
autophagic flux was detected in miR221-overexpressing LX2 cells by
immunofluorescence. Results showed that a mass of P62 was
aggregated in LX2 cells, and the empty vector did not affect P62
expression. Western blot analysis was performed, and the results
indicated that the autophagy inducer RAPA reversed the inhibition
of autophagic flux induced by miR221. Therefore, the aforementioned
findings revealed that miR221 partially regulated hepatic fibrosis
by inhibiting autophagy.
Autophagy maintains cell viability by targeting the
degradation of misfolded proteins in autolysosomes. To explore the
molecular target of miR221 in regulating autophagy, the present
study used bioinformatics tools to predict potential target genes.
Based on these predicted results, the possible regulatory genes of
miR221 were obtained using TargetScan and HADb. Among these
potential autophagy-related gene targets, miR221 was found to
regulate LAMP2 gene expression, which has been reported to be
closely related to the degradation of autolysosome (37). LAMP2 is a crucial gene regulating
cellular autolysosome lysosome fusion during autophagy (38), and was found to be a potential
target of miR221 in the current study. The dual-luciferase reporter
assay verified that miR221 significantly inhibited the fluorescence
activity of LAMP2, but did not regulate the mutant LAMP2. miR221
likely binds to LAMP2 and then recruits and activates autolysosome
function. To further verify the regulation of LAMP2 by miR221 in
activated HSCs, LX2 cells were transfected with miR221 mimics for
48 h. RT-qPCR and western blotting results showed that LAMP2
expression was significantly decreased in miR221-overexpressing LX2
cells. However, there were no significant differences in LAMP2
expression following transfection with the miR221 inhibitor.
The present study demonstrated that autophagic
degradation capacity of lysosomes was impaired due to the elevated
expression of P62 in TGF-β1-treated LX2 cells. Consistent with
these findings, Babuta et al (39) reported the causal role of miR155 in
regulating alcohol-induced LC3-II and p62 accumulation via
targeting LAMP2 in vitro and in vivo. Although LAMP2 has been shown
to mediate autophagy activation (8), the role of LAMP2 in hepatic fibrosis
is still controversial. Considering the potential target gene of
TGF-β1-induced HSC activation in hepatic fibrosis, we intended to
verify the functional role of miR221 in regulating HSC activation
by targeting LAMP2. In rescue experiments, LAMP2-overexpressing LX2
cells were constructed, treated with TGF-β1, and the expression
levels of COL-I and α-SMA were observed to determine the regulatory
function of LAMP2. The results showed that LAMP2 was significantly
increased in LAMP2-overexpressing LX2 cells, while COL-I and α-SMA
were downregulated following TGF-β1 treatment. Based on these
findings, the expression of autophagic marker proteins was examined
by western blotting. Overexpression of LAMP2 considerably
upregulated autolysosome activity in TGF-β1-treated LX2 cells, as
indicated by decreased expression of LC3-II and P62. These results
indicated that upregulated LAMP2-dependent autolysosome activity
blocked TGF-β-induced hepatic fibrosis, which had a similar
fibrosis protective effect in other disease models (40). The current study attempted to
explore miR221 intervention pathways for regulating TGF-β signals.
However, it does not exclude the involvement of miR221 in other
mechanisms of onset of activating HSC (41). In future research, we will verify
the differential expression of miRNA in liver fibrosis by
microarray data and further elucidate the possible mechanism of HSC
activation. Therefore, inhibiting TGF-β1-induced HSC activation by
regulating LAMP2-dependent autolysosome activation could be a
possible therapeutic strategy that exerts antifibrotic effects in
hepatic fibrosis.
Overall, these results demonstrated that miR221
played a regulatory role in hepatic fibrosis and could regulate
TGF-β1-induced HSC activation through inhibiting autophagy by
targeting LAMP2 (Fig. 6). The
molecular mechanism of the involvement of miR221 in regulating the
autophagy-lysosomal pathway may represent a novel therapeutic
approach for liver fibrosis.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
RC and YH contributed to the conception and design
of this study. RC and HX were responsible for the details of the
experimental procedures. RC completed the manuscript and YH revised
the manuscript. RC, HX and YH contributed to data analysis and
interpretation. RC, HX and YH confirm the authenticity of all the
raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
All protocols were approved by the Hebei Medical
University Institutional Animal Experimental Ethics Committee
(approval no. IACUC-Hebmu-2020005; Shijiazhuang, China) and carried
out according to the provisions of the guidelines for the Care and
Use of Laboratory Animals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ellis EL and Mann DA: Clinical evidence
for the regression of liver fibrosis. J Hepatol. 56:1171–1180.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lee YA, Wallace MC and Friedman SL:
Pathobiology of liver fibrosis: A translational success story. Gut.
64:830–841. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Carson JP, Ramm GA, Robinson MW, McManus
DP and Gobert GN: Schistosome-induced fibrotic disease: The role of
hepatic stellate cells. Trends Parasitol. 34:524–540. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Higashi T, Friedman SL and Hoshida Y:
Hepatic stellate cells as key target in liver fibrosis. Adv Drug
Deliv Rev. 121:27–42. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Khomich O, Ivanov AV and Bartosch B:
Metabolic hallmarks of hepatic stellate cells in liver fibrosis.
Cells. 9:92019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wallace MC, Friedman SL and Mann DA:
Emerging and disease-specific mechanisms of hepatic stellate cell
activation. Semin Liver Dis. 35:107–118. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dewidar B, Meyer C, Dooley S and
Meindl-Beinker AN: TGF-β in hepatic stellate cell activation and
liver fibrogenesis-updated 2019. Cells. 8:82019. View Article : Google Scholar
|
|
8
|
Eskelinen EL, Illert AL, Tanaka Y,
Schwarzmann G, Blanz J, Von Figura K and Saftig P: Role of LAMP-2
in lysosome biogenesis and autophagy. Mol Biol Cell. 13:3355–3368.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ravikumar B, Sarkar S, Davies JE, Futter
M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M,
Korolchuk VI, Lichtenberg M, Luo S, et al: Regulation of mammalian
autophagy in physiology and pathophysiology. Physiol Rev.
90:1383–1435. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ke PY: Diverse functions of autophagy in
liver physiology and liver diseases. Int J Mol Sci. 20:202019.
View Article : Google Scholar
|
|
12
|
Qian H, Chao X, Williams J, Fulte S, Li T,
Yang L and Ding WX: Autophagy in liver diseases: A review. Mol
Aspects Med. Jun 10–2021.(Epub ahead of print). doi:
10.1016/j.mam.2021.100973. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu N, Chen L, Yan D, Zhou M, Shao C, Lu Y,
Yao Q, Sun H and Fu Y: Trehalose attenuates TGF-β1-induced fibrosis
of hSCFs by activating autophagy. Mol Cell Biochem. 470:175–188.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang X, He Y, Mackowiak B and Gao B:
MicroRNAs as regulators, biomarkers and therapeutic targets in
liver diseases. Gut. 70:784–795. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mohr AM and Mott JL: Overview of microRNA
biology. Semin Liver Dis. 35:3–11. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Khan MI, Hamid A, Adhami VM, Lall RK and
Mukhtar H: Role of epithelial mesenchymal transition in prostate
tumorigenesis. Curr Pharm Des. 21:1240–1248. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huang PS, Chang CC, Wang CS and Lin KH:
Functional roles of non-coding RNAs regulated by thyroid hormones
in liver cancer. Biomed sJ. 44:272–284. 2021.PubMed/NCBI
|
|
18
|
Jiang Y, He J, Li Y, Guo Y and Tao H: The
diagnostic value of MicroRNAs as a biomarker for hepatocellular
carcinoma: A meta-analysis. BioMed Res Int. 2019:51790482019.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Roderburg C, Urban GW, Bettermann K, Vucur
M, Zimmermann H, Schmidt S, Janssen J, Koppe C, Knolle P, Castoldi
M, et al: Micro-RNA profiling reveals a role for miR-29 in human
and murine liver fibrosis. Hepatology. 53:209–218. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pineau P, Volinia S, McJunkin K, Marchio
A, Battiston C, Terris B, Mazzaferro V, Lowe SW, Croce CM and
Dejean A: miR-221 overexpression contributes to liver
tumorigenesis. Proc Natl Acad Sci USA. 107:264–269. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
He XX, Guo AY, Xu CR, Chang Y, Xiang GY,
Gong J, Dan ZL, Tian DA, Liao JZ and Lin JS: Bioinformatics
analysis identifies miR-221 as a core regulator in hepatocellular
carcinoma and its silencing suppresses tumor properties. Oncol Rep.
32:1200–1210. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kannan M, Jayamohan S, Moorthy RK,
Chabattula SC, Ganeshan M and Arockiam AJ: AEG-1/miR-221 axis
cooperatively regulates the progression of hepatocellular carcinoma
by targeting PTEN/PI3K/AKT signaling pathway. Int J Mol Sci.
20:202019. View Article : Google Scholar
|
|
23
|
Fu Y, Liu M, Li F, Qian L, Zhang P, Lv F,
Cheng W and Hou R: MiR-221 promotes hepatocellular carcinoma cells
migration via targeting PHF2. BioMed Res Int. 2019:43714052019.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Baglieri J, Brenner DA and Kisseleva T:
The role of fibrosis and liver-associated fibroblasts in the
pathogenesis of hepatocellular carcinoma. Int J Mol Sci. 20:202019.
View Article : Google Scholar
|
|
25
|
Ebrahimkhani MR, Oakley F, Murphy LB, Mann
J, Moles A, Perugorria MJ, Ellis E, Lakey AF, Burt AD, Douglass A,
et al: Stimulating healthy tissue regeneration by targeting the
5-HT-B receptor in chronic liver disease. Nat Med. 17:1668–1673.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang R, Gao N, Chang Q, Meng X and Wang W:
The role of IDO, IL-10, and TGF-β in the HCV-associated chronic
hepatitis, liver cirrhosis, and hepatocellular carcinoma. J Med
Virol. 91:265–271. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jones-Bolin S: Guidelines for the care and
use of laboratory animals in biomedical research. Curr Protoc
Pharmacol. Dec 1–2012.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hyun J, Wang S, Kim J, Rao KM, Park SY,
Chung I, Ha CS, Kim SW, Yun YH and Jung Y: MicroRNA-378 limits
activation of hepatic stellate cells and liver fibrosis by
suppressing Gli3 expression. Nat Commun. 7:109932016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ding ZY, Jin GN, Liang HF, Wang W, Chen
WX, Datta PK, Zhang MZ, Zhang B and Chen XP: Transforming growth
factor β induces expression of connective tissue growth factor in
hepatic progenitor cells through Smad independent signaling. Cell
Signal. 25:1981–1992. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shu Y, Liu X, Huang H, Wen Q and Shu J:
Research progress of natural compounds in anti-liver fibrosis by
affecting autophagy of hepatic stellate cells. Mol Biol Rep.
48:1915–1924. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Song B, Wang Y, Xi Y, Kudo K, Bruheim S,
Botchkina GI, Gavin E, Wan Y, Formentini A, Kornmann M, et al:
Mechanism of chemoresistance mediated by miR-140 in human
osteosarcoma and colon cancer cells. Oncogene. 28:4065–4074. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jiang X, Jiang L, Shan A, Su Y, Cheng Y,
Song D, Ji H, Ning G, Wang W and Cao Y: Targeting hepatic
miR-221/222 for therapeutic intervention of nonalcoholic
steatohepatitis in mice. EBioMedicine. 37:307–321. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xu J, Su Y, Xu A, Fan F, Mu S, Chen L, Chu
Z, Zhang B, Huang H, Zhang J, et al: miR-221/222-mediated
inhibition of autophagy promotes dexamethasone resistance in
multiple myeloma. Mol Ther. 27:559–570. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li F, Long TY, Bi SS, Sheikh SA and Zhang
CL: circPAN3 exerts a profibrotic role via sponging miR-221 through
FoxO3/ATG7-activated autophagy in a rat model of myocardial
infarction. Life Sci. 257:1180152020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu N, Feng J, Lu X, Yao Z, Liu Q, Lv Y,
Han Y, Deng J and Zhou Y: Isorhamnetin inhibits liver fibrosis by
reducing autophagy and inhibiting extracellular matrix formation
via the TGF-β1/Smad3 and TGF-β1/p38 MAPK Pathways. Mediators
Inflamm. 2019:61750912019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Eskelinen EL: Roles of LAMP-1 and LAMP-2
in lysosome biogenesis and autophagy. Mol Aspects Med. 27:495–502.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gozuacik D, Akkoc Y, Ozturk DG and Kocak
M: Autophagy-regulating microRNAs and cancer. Front Oncol.
7:652017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Babuta M, Furi I, Bala S, Bukong TN, Lowe
P, Catalano D, Calenda C, Kodys K and Szabo G: Dysregulated
autophagy and lysosome function are linked to exosome production by
Micro-RNA 155 in alcoholic liver disease. Hepatology. 70:2123–2141.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lodder J, Denaës T, Chobert MN, Wan J,
El-Benna J, Pawlotsky JM, Lotersztajn S and Teixeira-Clerc F:
Macrophage autophagy protects against liver fibrosis in mice.
Autophagy. 11:1280–1292. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Fabregat I and Caballero-Díaz D:
Transforming growth factor-β-induced cell plasticity in liver
fibrosis and hepatocarcinogenesis. Front Oncol. 8:3572018.
View Article : Google Scholar : PubMed/NCBI
|