Introduction
Developmental glaucoma, also known as congenital
glaucoma, is an ocular defect and is often associated with the
abnormal development of the anterior chamber angle (1). Glaucomatous phenotypes including
elevated intraocular pressure (IOP) are generally displayed at
birth, but the onset may be delayed to adolescence (2). For the latter, the patients only
present with elevated IOP, trabeculodysgenesis and glaucomatous
optic neuropathy, but without the tearing, photophobia,
blepharospasm, enlarged cornea and Haab's striae that is generally
observed in congenital glaucoma (3).
Developmental glaucoma is an inherited disorder
predominantly transmitted in an autosomal recessive manner
(3). Although the pathogenic
mechanism has not been completely elucidated in all cases, genetic
defects appear to be the most crucial risk factor in developmental
glaucoma (3). Several
disease-associated loci have been previously identified, including
glaucoma 3, primary congenital, A (GLC3A) (4), glaucoma 3, primary infantile, B
(GLC3B) (5), glaucoma 3,
primary congenital, C (GLC3C) and glaucoma 3, primary
congenital, D GLC3D (6); in
addition, two genes located in these genetic loci, cytochrome P450
family 1 subfamily B member 1 (CYP1B1) and
latent-transforming growth factor β-binding protein 2
(LTBP2), have been implicated in the mechanisms underlying
developmental glaucoma. CYP1B1 mutations are the most
commonly identified genetic defect causing developmental glaucoma
(6,7). To date, >150 CYP1B1
mutations have been identified in patients with developmental
glaucoma worldwide (8). Of these,
43 variants have been reported in Chinese patients, including
p.R390H, which is a common mutation identified in patients with
primary congenital glaucoma (PCG) from all ethnic groups, and
p.L107V, which is unique to Chinese patients with PCG (9).
The current study aimed to investigate the genetic
background of developmental glaucoma in a Chinese family. Compound
heterozygous mutations in the CYP1B1 gene were identified in
a patient, of which one of the mutations (c.3G>A) has not been
previously reported, to the best of our knowledge.
Patients and methods
Patients
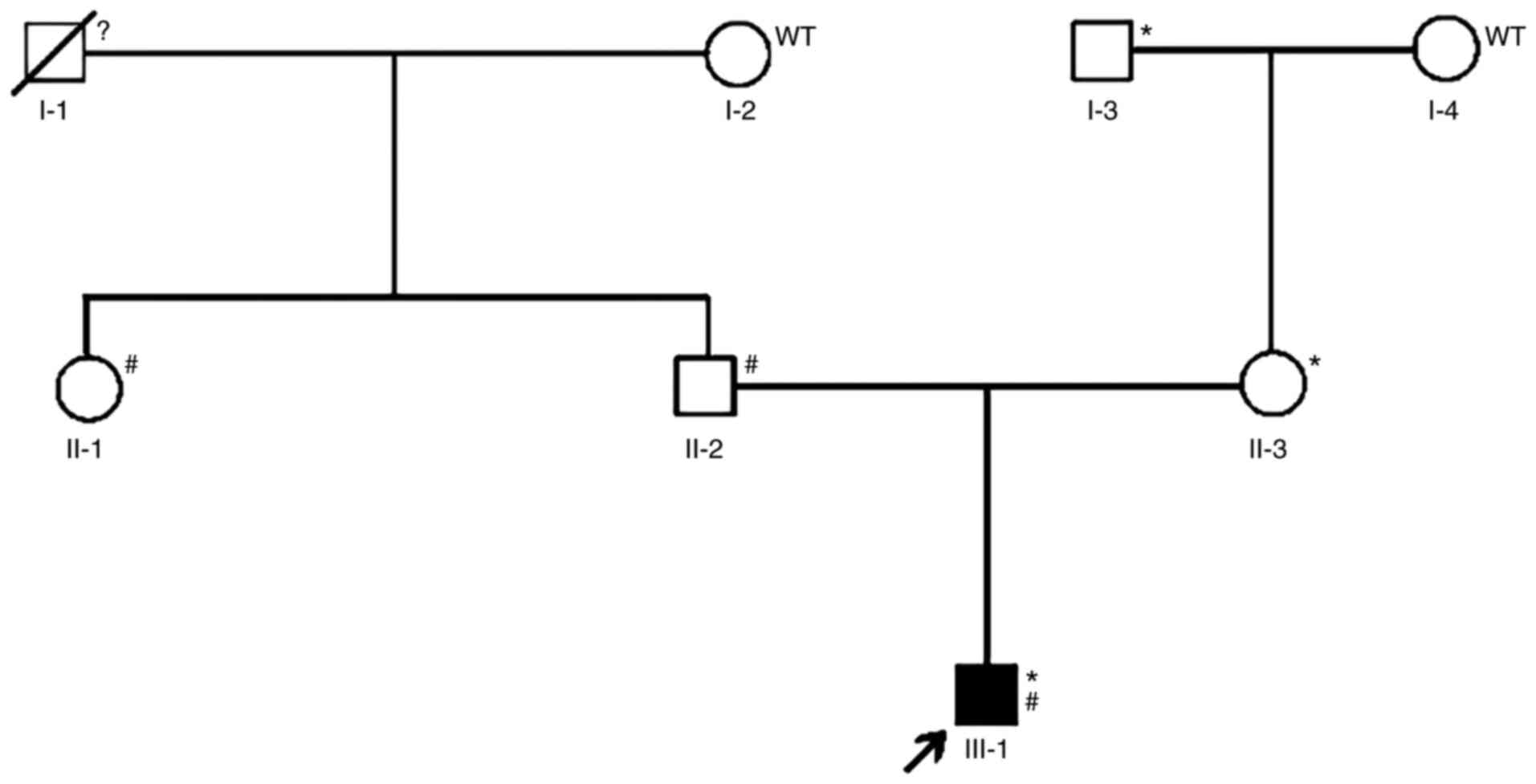
A three-generation pedigree with developmental
glaucoma was recruited to this study Fig. 1, including the 14-year-old male
proband, his parents and four other family members (the proband's
three grandparents and his aunt). The study was conducted in
accordance with the Declaration of Helsinki and the experimental
protocol was approved by the Ethics Committee of The Shenzhen Eye
Hospital (Shenzhen, China). The parents provided consent for
participation and also provided consent for the minor patient.
Detailed ophthalmic examinations, including visual
acuity tests, measurement of IOP, slit lamp biomicroscopy, corneal
size and measurement of cup/disc ratio were performed on each of
the individuals; additional examinations, including a Humphrey
visual fields test and anterior-segment optical coherence
tomography, were exclusively performed on the proband.
Genetic analysis
A total of 2 ml peripheral blood samples were
collected from of each subject. Genomic DNA was extracted using a
Massive Whole Blood Genomic DNA Extraction kit (Tiangen Biotech
Co., Ltd.; cat. no. DP348-03). Second-generation sequencing was
used to screen the candidate glaucoma causal genes in the proband's
genome as previously described (10) (data not shown). The gene panel used
in the present study was obtained from Shanghai Wickhams
Bio-Pharmaceutical Technology Co., Ltd. The panel comprises 2,067
genes known to be involved in 2,486 types of ocular-related
hereditary diseases, including corneal abnormalities, cataracts,
glaucoma, nystagmus, optic nerve abnormalities, retinitis
pigmentosa, strabismus and refractive errors. The 2,067 genes were
investigated using targeted capture and high-throughput sequencing
technologies (Illumina, Inc.) as previously described (10). The average sequencing depth was
>150X, and 30X coverage was achieved for >98.5% of the genes
screened. The test contents and classification criteria were based
on authoritative disease phenotype databases Online Mendelian
Inheritance in Man (omim.org/, updated 2020.05), DECIPHER
(deciphergenomics.org/, updated 2019.04),
Orphanet (orpha.net/, updated 2020.01) and Human
Phenotype Ontology (hpo.jax.org/,
updated 2020.01) and literature reports (3–9). The
panel contained 95 congenital glaucoma-related genes, including TEK
receptor tyrosine kinase (TEK), CYP1B1, and a number
of other genes related to glaucoma and other ocular diseases, such
as neurotrophin 4, optineurin, ankyrin repeat- and SOCS
box-containing 10, WD repeat domain 36, myocilin, OPA1
mitochondrial dynamin-like GTPase, paired box 6, forkhead box C1
and paired-like homeodomain 2.
PCR products were electrophoresed on 2% agarose
gels, visualized following staining with GoldView (Beijing Solarbio
Science & Technology Co., Ltd.) and photographing using a
Tanon-1600 gel camera (Tanon Science and Technology Co., Ltd.). The
PCR products were used to verify the results of the panel screening
in the whole pedigree. Intronic primers flanking the exons
(Table I) were designed based on
gene sequences of CYP1B1 (GenBank: U56438) and synthesized
by BGI Genomics. DNA fragments were amplified by PCR using a
MyCycler thermocycler (Bio-Rad Laboratories, Inc.). The PCR
reaction was performed in a 25 µl reaction mixture containing 0.1
µg genomic DNA, 40 µmol/l forward and reverse primers, 3 mmol/l
magnesium chloride and 2X Taq Master Mix (Sino Biological, Inc.).
The following thermocycling conditions were used for the PCR:
Initial denaturation step at 95°C for 5 min; followed by 35 cycles
of denaturation at 95°C for 30 sec, annealing at 55°C for 30 sec
and extension at 72°C for 30 sec; then a final extension step of 10
min at 72°C.
 | Table I.Primers used for the PCR
amplification of the CYP1B1 gene. |
Table I.
Primers used for the PCR
amplification of the CYP1B1 gene.
| Exon | Primer sequence
(5′→3′) | Product size
(bp) |
|---|
| CYP1B1 | F:
CATTTCTCCAGAGAGTCAGC | 1,260 |
| Exon 2 | R:
GCTTGCAAACTCAGCATATTC |
|
| CYP1B1 | F:
ACCCAATGGAAAAGTCAGCC | 927 |
| Exon 3 | R:
GCTTGCCTCTTGCTTCTTATT |
|
The PCR products were subjected to 1% agarose gel
electrophoresis, and the target PCR fragments were extracted using
the QIAquick Gel Extraction kit (Qiagen China Co., Ltd.). The
products were sequenced using an ABI 377XL automated DNA sequencer
(Applied Biosystems; Thermo Fisher Scientific, Inc.). Sequencing
data were compared with the published CYP1B1 sequences.
Bioinformatics analysis
The wild-type CYP1B1 protein sequence was downloaded
from the RCSB Protein Data Bank (https://www.rcsb.org) and saved as a PDB file. The
amino acids in the CYP1B1 protein were changed manually using
ICM-Pro (molsoft.com/icm_pro.html, v3.5) software, and the PDB
files were uploaded into PyMOL (pymol.org/2/,
v2.5.1) and Swiss Model online (swissmodel.expasy.org, updated 2021.07) to generate
the 3D structure of the protein (the unaltered wild-type protein
was used as a template and the PDB ID is Q16678). The tertiary
structure of protein was predicted by the Swiss Model (swissmodel.expasy.org, updated 2021.07). The altered
amino acids and key ligands were labeled in different colors. The
functional effects of these two mutations were predict by
PolyPhen-2 online software (genetics.bwh.harvard.edu/pph2/, updated
2016.01.05).
Results
Clinical findings
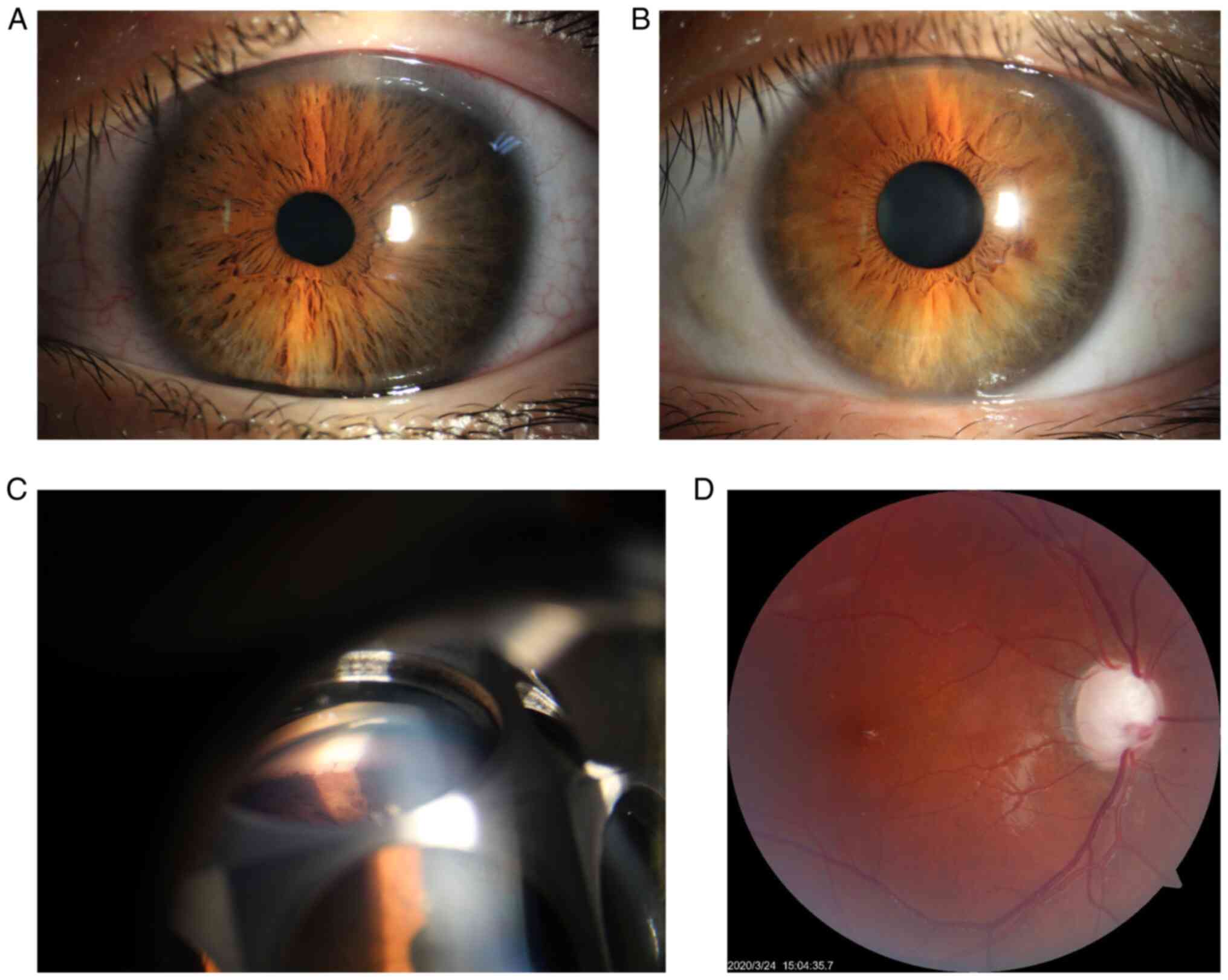
The best-corrected Snellen visual acuity of the
proband was 20/25 in the right eye and 20/20 in the left eye, and
the intraocular pressure without antiglaucoma medications was 42
mmHg for the right eye and 43 mmHg for the left eye. Both
horizontal corneal diameters were 13 mm. The slit lamp examination
showed a clear cornea, a deep anterior chamber, and a loose and
atrophic iris in both eyes (Fig.
2A). Gonioscopic examination showed the presence of
trabeculodysgenesis in both eyes (Fig.
2C). Fundoscopic examination of both eyes showed advanced optic
disc cupping with a C/D ratio of 0.95 in the right and 0.85 in the
left eye (Fig. 2D). B-scan
ultrasonography showed that the axial length was 25.9 mm in the
right eye and 26.7 mm in the left eye. A 360-degree circumferential
trabeculotomy procedure was performed on the proband's right eye
and controlled IOP was achieved. His left eye responded well to two
antiglaucoma drugs, such as prostaglandin analogues and a β
blocker. No significant ocular abnormalities were noticed in other
family members and the relatives, except that loose irises were
found in the proband's mother (Fig.
2B).
Genetics and bioinformatics
analyses
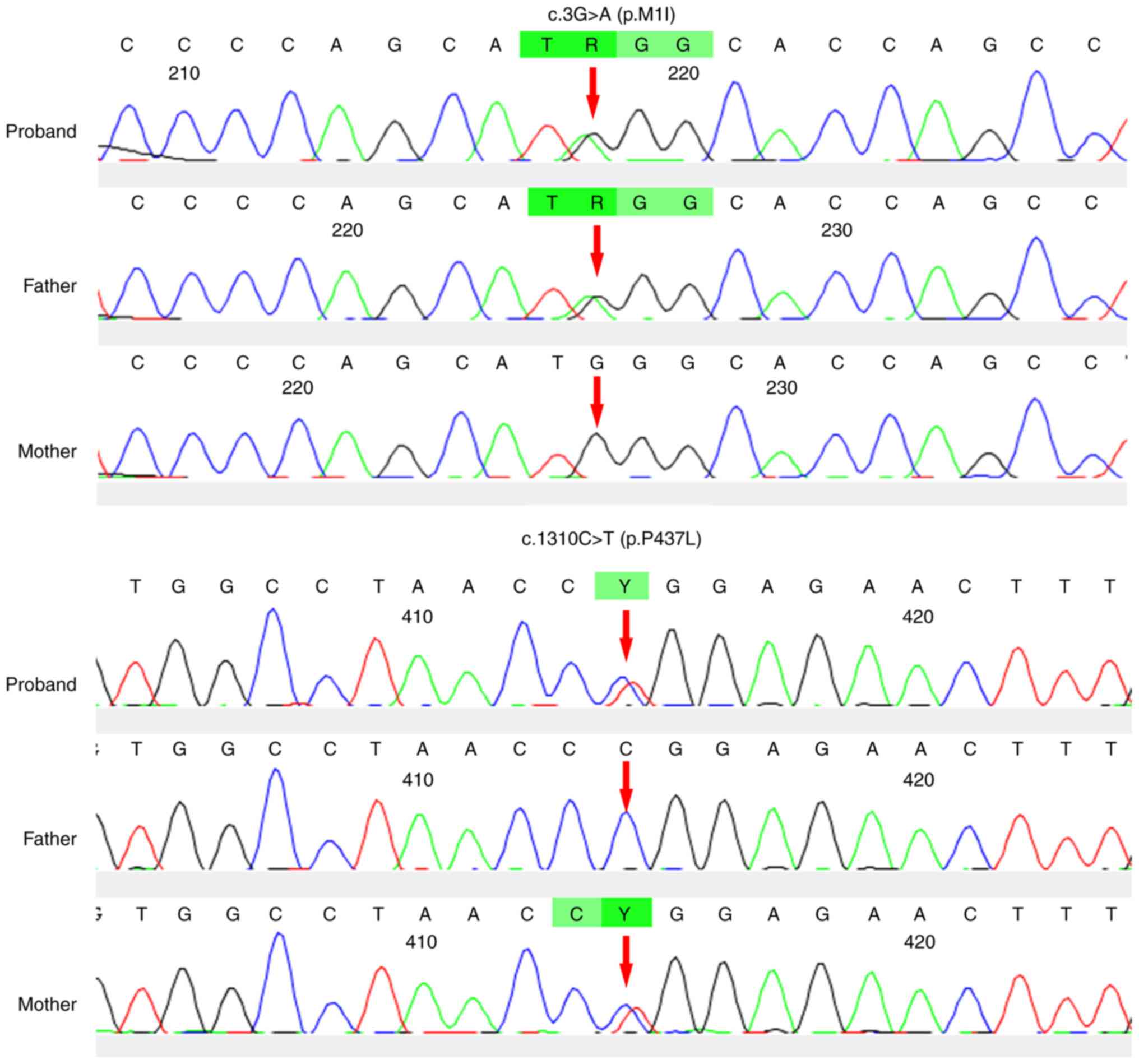
Compound heterozygous mutations for CYP1B1
c.3G>A (p.M1I) and c.1310C>T (p.P437L) were identified by the
panel screening. The novel heterozygous mutation c.3G>A (p.M1I)
and a previously reported mutation (8), c.1310C>T (p.P437L), in
CYP1B1 were identified in individual III-1 (Fig. 1), with the former inherited from his
father and the latter from his mother. The novel variant, c.3G>A
(p.M1I), was found to alter the ATG start codon of exon 1 of
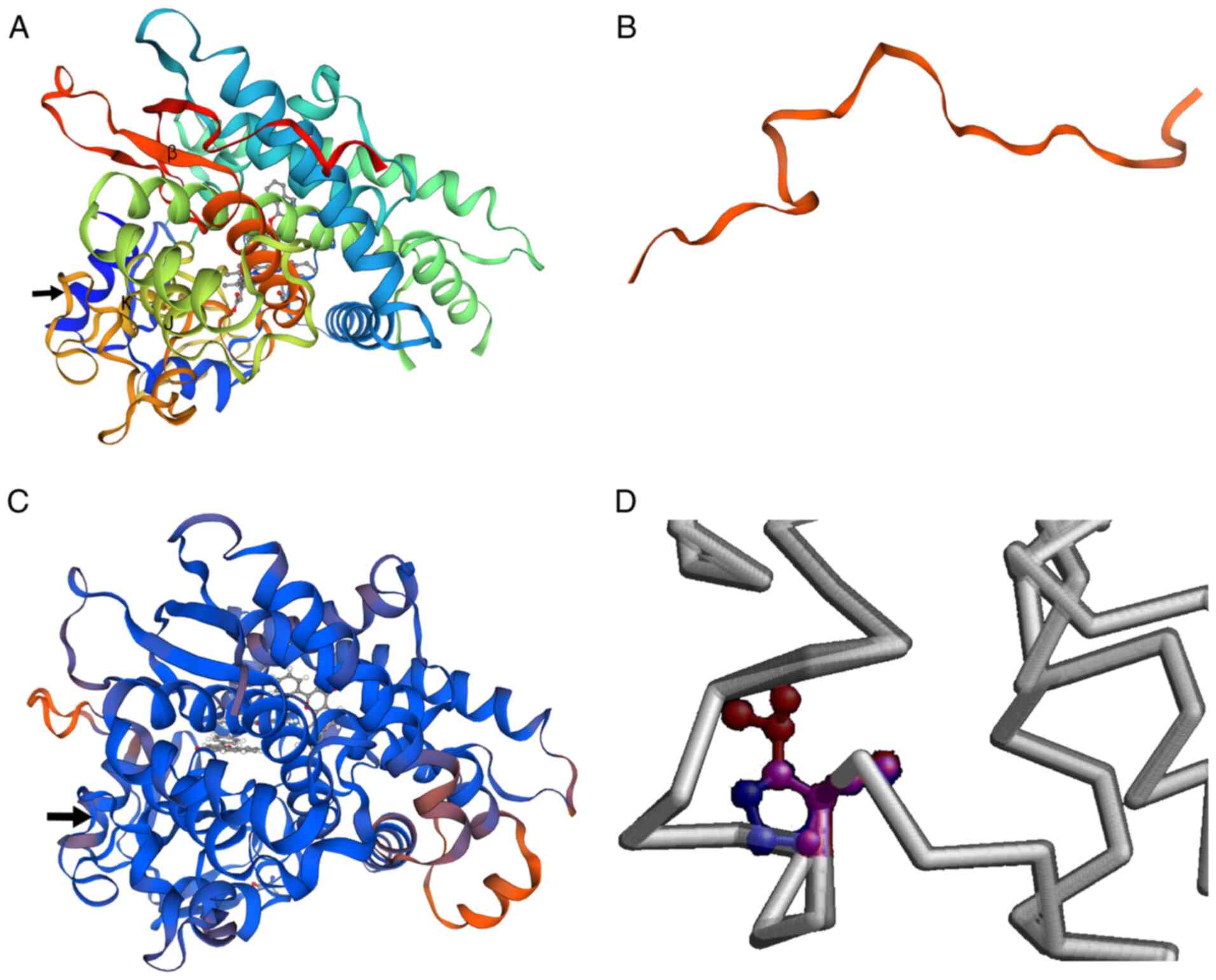
CYP1B1, which disrupted the translation start site (Fig. 3). The two mutations were predicted
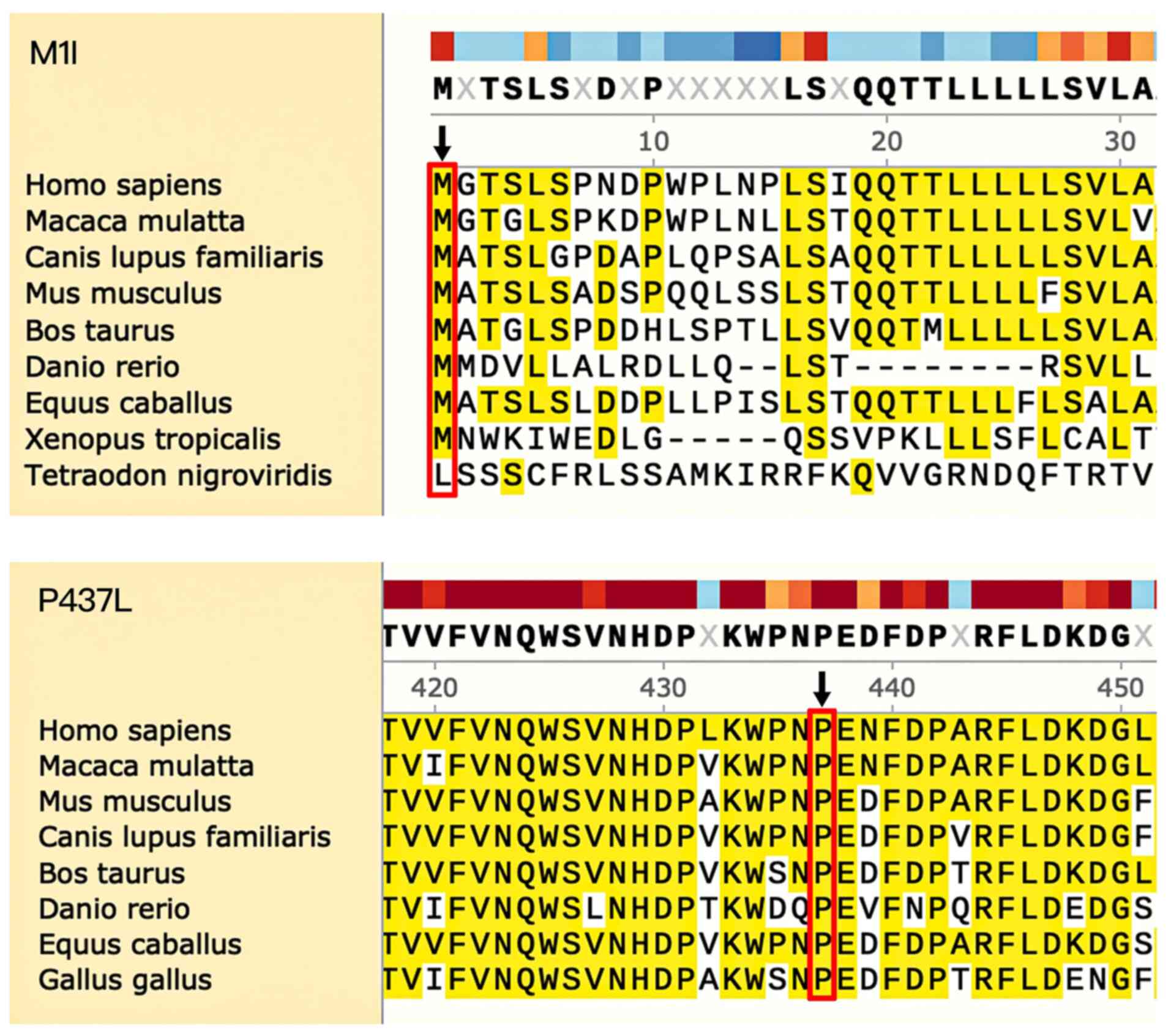
to be ‘possibly damaging’ based on PolyPhen-2 analysis. Both P437
and M1 amino acid positions were found to be highly conserved in
CYP1B1, from Homo sapiens through to Danio
rerio (Fig. 4).
To predict the putative structural and functional
impact of these mutations on the CYP1B1 protein, the protein
structure was predicted using PyMOL (Swiss Model). The novel
c.3G>A mutation was found to be located in the AUG start codon
for methionine, changing it to AUA, which encodes isoleucine
(p.M1I). The major effect of this change was predicted to be the
elimination of the initiation codon of translation, with the next
downstream AUG being at position 293 and encoding a truncated
frameshifted peptide of 53 amino acids. The c.1310C>T (p.P437L)
mutation in the CYP1B1 gene is located in the meander region
of the translated protein (Fig. 5).
As shown in Fig. 5A and C, the
substitution caused minimal distortion of the protein fold and did
not result in regional crowding, but the change from a small
hydrophilic amino acid with obligate torsion of the backbone to a
medium-sized aliphatic residue would be expected to have
significant effects on activity, as implied in the in silico
analysis.
These findings indicated that both p.M1 and p.P437
are important amino acids required to maintain the normal function
of CYP1B1, and both were implicated in the clinical
glaucomatous phenotype of the proband.
Discussion
Even though most cases of developmental glaucoma
seem to be sporadic, up to 40% of cases are considered to be
genetically inherited, demonstrating an autosomal recessive
inheritance pattern with variable penetrance (11,12).
Mutations in the genes, CYP1B1, LTBP2 and TEK have
been reported in patients with developmental glaucoma, and among
these, mutations in CYP1B1 are the most commonly reported
(13–15). CYP1B1 is a drug-metabolizing
enzyme of the cytochrome P450 gene family, which is expressed in a
wide spectrum of tissues, including the iris, trabecular meshwork
(TM), ciliary body and anterior uveal tract of the eye (16). CYP1B1 serves a crucial
catalytic role in the synthesis of cholesterol, steroids and other
lipids (17–19). These metabolic reactions and
products are important for the differentiation and growth of
multiple tissues including liver, cardiovascular and eye (19–21).
In PCG, CYP1B1 mutations interfere with metabolism of
retinol, a key metabolite required for TM development (22,23).
Another major cause of TM pathogenesis is abnormal oxidation status
owing to improper CYP1B1 gene activity. The presence of
oxidative stress during early development can cause TM hypoplasia,
leading to developmental glaucoma (24). Teixeira et al (23) reported that
Cyp1b1−/− mice had progressively decreased levels
of collagen in the TM, increased TM atrophy, an elevated IOP and
increased glaucomatous lesions, with the latter very similar to the
manifestations of human PCG.
The structure of the CYP1B1 protein comprises four
conserved helix bundles, including the J- and K-helices, β-sheets,
a meander region and the heme-binding region (25). In addition, the N-terminal hinge
region and C-terminal conserved core structures (CCSs) are known to
be important regions for maintaining enzymatic activity (2,26).
Notably, clinical reports on a variety of different ethnic groups
have suggested that patients with developmental glaucoma with
CYP1B1 mutations in the N-terminal hinge region or CCSs,
such as the missense mutations c.517G>C (26) and c.1439G>T (27), present with more severe glaucomatous
phenotypes.
To date, >150 mutations in the CYP1B1 gene
have been described in cases of developmental glaucoma, with some
predominantly associated with severe glaucomatous pathology
(7,28). In the current study, a novel
heterozygous mutation, c.3G>A (p.M1I), within the CYP1B1
gene of a Chinese family with developmental glaucoma was
identified. This heterozygous mutation caused the loss of the
primary AUG start codon for methionine, which was replaced by a
triplet AUA encoding isoleucine (p.M1I) and was predicted to affect
the initiation of translation, as the initial methionine codon
surrounded by a Kozak sequence is crucial for ribosomal recognition
(29). The next conserved Kozak
sequence where translation could be initiated is at the c.293 AUG
and ends at the 452 UAA position on the mRNA. The new ORF shifted
protein is predicted to be shortened to only 53 amino acids and
would include different amino acids from the wild-type CYP1B1
protein as it starts in a new open reading frame. Translation can
be initiated at non-AUG codons, including ACG, but this is
extremely rare and requires a favorable mRNA secondary structure
(29). In the current study, this
mutation in the proband was inherited from his father, who was
clinically asymptomatic, indicating the probable functional null
nature of this mutation.
The second variant detected in the present study,
c.1310C>T (p.P437L), was previously reported in different ethnic
groups worldwide (10,11,30–32). A
report from Pakistan reported cases of PCG caused by variants in
CYP1B1, including c.1310C>T. It was predicted that this
mutation may alter the protein backbone conformation at this
position, as the proline residue at position 437 is rigid and
located on the folded protein's surface, and the substitution of
proline with leucine at this position may influence the special
conformation and distort the interactions with other molecules
(33). The modeling results of the
current study suggested that distortion of the protein fold, was
minimal and would favor decreased CYP1B1 enzyme activity as a
mechanism. A previous study using sequence analysis and homology
modeling reported that PCG resulting from CYP1B1 mutations
disrupted either the hinge region or the conserved core structures
of cytochrome P4501B1 (24). By
contrast, the p.P437L variant may affect the meander region. The
segregation of the mutant CYP1B1 alleles was consistent with
autosomal recessive inheritance of the disease in five pedigrees
investigated (26). An analysis of
CYP1B1 in Brazilian patients with PCG showed that four of
the nine mutations were present as compound heterozygotes, two in
homozygotes and only one mutant allele was identified in three of
the cases. In one patient, the c.8147C>T (p.P437L) and
c.8182delG mutations were identified in a compound heterozygote,
and clinical examination revealed a highly compromised phenotype
with low visual acuity and difficultly controlling IOP (13). Screening of CYP1B1 and
LTBP2 in Saudi families with PCG showed that PCG cases with
CYP1B1 variants, including p.P437L, had a more severe
subepithelial haze in cornea and a greater C/D ratio compared with
those cases with no identified mutation (32). Moreover, in a Pakistani family with
PCG, two affected individuals carrying the c.1310C>T mutation in
CYP1B1 manifested PCG symptoms during the first year after
birth and subsequently underwent bilateral trabeculectomy. One had
an elevated IOP with bilateral megalocornea, with opacities and
decreased visual acuity with the perception of light only; the
other also had megalocornea with increased lacrimation and
photophobia (34). In addition to
the aforementioned reports, the c.1310C>T mutation in
CYP1B1 has also been previously reported in families with
PCG in Spain and India (31,35),
but not in China, to the best of our knowledge.
In autosomal recessive phenotypes, heterozygous
carriers are generally asymptomatic. However, parents carrying the
pathogenic variant in a heterozygous state may present a mild
phenotype (36). In the present
study, the father carrying one of the compound heterozygous
mutations (c.3G>A) appeared asymptomatic, whereas the mother
carrying the second mutation (c.1310C>T) presented with loose
and mildly atrophic irises, similar to, but less severe than, the
proband but without other developmental disorders, such as
trabeculodysgenesis, suggesting that a single copy of this mutation
may cause a relatively mild form of the disease. However, there is
no evidence showing that one of these heterozygous mutations
contributes more to the pathogenesis of the child.
Previous studies indicated an association between
specific mutations and the severity of anterior chamber angle
abnormalities (8). The C-terminus
of the CYP1B1 protein includes a substrate binding region and CCS,
whereas the N-terminal of the CYP1B1 protein includes a
membrane-spanning domain and a hinge region (19). Mutations leading to protein variants
p.E229K (37) and p.S239R (38) have also been shown to disrupt the
three-dimensional structures of the I-helix, and subsequently lead
to severe glaucomatous phenotypes. By contrast, the CYP1B1 protein
structure showed that p.P437L is located in the meander region
(26,39). Taken together, these data indicate
that in addition to the hinge region and CCSs, the meander region
can also be important for maintaining the function of the
CYP1B1 gene.
In conclusion, the results of the present study
revealed, for the first time to the best of our knowledge, the
coexistence of the novel c.3G>A mutation alongside the
c.1310C>T mutation in a Chinese pedigree of developmental
glaucoma. In addition, our data indicated the importance of
additional regions, such as the meander region, suggesting that
they can also affect the structure of the CCSs and are crucial in
regulating the function of CYP1B1 and avoiding TM
abnormalities. However, future studies are required to determine
the influence of this mutation on metabolism and selection of key
metabolites, which may lead to the development of potential
therapeutic strategies for PCG.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from The
National Natural Science Foundation of China (grant nos. 81770924
and 82070963), The Sanming Project of Medicine in Shenzhen (grant
no. SZSM201512045) and The Science and Technology Innovation
Committee of Shenzhen (grant no. JCYJ20170306140823343). The
funders had no role in the study design, data collection and
analysis, decision to publish or preparation of the manuscript.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XL and JFH conceptualized and designed the
experiments and supervised the study. DZ, TW and MF collected the
samples and clinical information. SC, YW and XJ performed the
genetic analysis and bioinformatics evaluations. SC conducted the
molecular biology experiments and drafted the manuscript. XL and
JFH reviewed and edited the manuscript. All authors have read and
approved the final manuscript. SC and XL confirmed the authenticity
of all the raw data.
Ethics approval and consent to
participate
The study was conducted in accordance with the
Declaration of Helsinki and the experimental protocol was approved
by the Ethics Committee of the Shenzhen Eye Hospital (Shenzhen,
China). The parents provided consent for participation and also
provided consent for the minor patient.
Patient consent for publication
The mother provided consent for the publication of
images of her iris as well as consent for her child in Fig. 2.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
deLuise VP and Anderson DR: Primary
infantile glaucoma (congenital glaucoma). Surv Ophthalmol. 28:1–19.
1983. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Babu M, Thangaraj V, Vadivelu SD, Arumugam
S and Murali M: A study on developmental glaucoma. J Evid Based
Med. 5:922–926. 2018.
|
|
3
|
Faiq M, Sharma R, Dada R, Mohanty K,
Saluja D and Dada T: Genetic, biochemical and clinical insights
into primary congenital glaucoma. J Curr Glaucoma Pract. 7:66–84.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sarfarazi M, Akarsu AN, Hossain A, Turacli
ME, Aktan SG, Barsoum-Homsy M, Chevrette L and Sayli BS: Assignment
of a locus (GLC3A) for primary congenital glaucoma (Buphthalmos) to
2p21 and evidence for genetic heterogeneity. Genomics. 30:171–177.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Akarsu AN, Turacli ME, Aktan SG,
Barsoum-Homsy M, Chevrette L, Sayli BS and Sarfarazi M: A second
locus (GLC3B) for primary congenital glaucoma (Buphthalmos) maps to
the 1p36 region. Hum Mol Genet. 5:1199–1203. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Narooie-Nejad M, Paylakhi SH, Shojaee S,
Fazlali Z, Rezaei Kanavi M, Nilforushan N, Yazdani S, Babrzadeh F,
Suri F, Ronaghi M, et al: Loss of function mutations in the gene
encoding latent transforming growth factor beta binding protein 2,
LTBP2, cause primary congenital glaucoma. Hum Mol Genet.
18:3969–3977. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li N, Zhou Y, Du L, Wei M and Chen X:
Overview of cytochrome P450 1B1 gene mutations in patients with
primary congenital glaucoma. Exp Eye Res. 93:572–579. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chouiter L and Nadifi S: Analysis of
CYP1B1 gene mutations in patients with primary congenital glaucoma.
J Pediatr Genet. 6:205–214. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yuan R, Zhao J, Zhao YY, Xu MA, Dai QY and
Wang B: Systematic review of CYP1B1 gene mutations in patients with
primary congenital glaucoma in China. J Evid Based Med. 1:42–47.
2015.PubMed/NCBI
|
|
10
|
Bao Y, Yang JM, Chen L, Chen MH, Zhao PQ,
Qiu SP, Zhang L and Zhang GM: A novel mutation in the NDP gene is
associated with familial exudative vitreoretinopathy in a southern
Chinese family. Genet Test Mol Bioma. 23:850–856. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sarfarazi M and Stoilov I: Molecular
genetics of primary congenital glaucoma. Eye (Lond). 14:422–428.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sarfarazi M, Stoilov I and Schenkman JB:
Genetics and biochemistry of primary congenital glaucoma.
Ophthalmol Clin North Am. 16:543–554.vi. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Faiq M, Mohanty K, Dada R and Dada T:
Molecular diagnostics and genetic counseling in primary congenital
glaucoma. J Curr Glaucoma Pract. 7:25–35. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Della Paolera M, de Vasconcellos JP,
Umbelino CC, Kasahara N, Rocha MN, Richeti F, Costa VP, Tavares A
and de Melo MB: CYP1B1 gene analysis in primary congenital glaucoma
Brazilian patients: Novel mutations and association with poor
prognosis. J Glaucoma. 19:176–182. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sitorus R, Ardjo SM, Lorenz B and Preising
M: CYP1B1 gene analysis in primary congenital glaucoma in
Indonesian and European patients. J Med Genet. 40:e92003.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Abdolrahimzadeh S, Fameli V, Mollo R,
Contestabile MT, Perdicchi A and Recupero SM: Rare diseases leading
to childhood glaucoma: Epidemiology, pathophysiogenesis, and
management. Biomed Res Int. 2015:7812942015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Choudhary D, Jansson I, Sarfarazi M and
Schenkman JB: Characterization of the biochemical and structural
phenotypes of four CYP1B1 mutations observed in individuals with
primary congenital glaucoma. Pharmacogenet Genomics. 18:665–676.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kaur K, Mandal AK and Chakrabarti S:
Primary congenital glaucoma and the involvement of CYP1B1. Middle
East Afr J Ophthalmol. 18:7–16. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vasiliou V and Gonzalez FJ: Role of CYP1B1
in glaucoma. Annu Rev Pharmacol Toxicol. 48:333–358. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pedro BJ, Climent E and Benaiges D:
Familial hypercholesterolemia: Do HDL play a role? Biomedicines.
9:810–811. 2021. View Article : Google Scholar
|
|
21
|
Irina A and Nathalie C: Cholesterol
Hydroxylating cytochrome P450 46A1: From mechanisms of action to
clinical applications. Front Aging Neurosci. 13:6967782021.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Prat C, Belville C, Comptour A, Marceau G,
Clairefond G, Chiambaretta F, Sapin V and Blanchon L: Myocilin
expression is regulated by retinoic acid in the trabecular
meshwork-derived cellular environment. Exp Eye Res. 155:91–98.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Teixeira LB, Zhao Y, Dubielzig RR,
Sorenson CM and Sheibani N: Ultrastructural abnormalities of the
trabecular meshwork extracellular matrix in Cyp1b1-deficient mice.
Vet Pathol. 52:397–403. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhao Y, Wang S, Sorenson CM, Teixeira L,
Dubielzig RR, Peters DM, Conway SJ, Jefcoate CR and Sheibani N:
Cyp1b1 mediates periostin regulation of trabecular meshwork
development by suppression of oxidative stress. Mol Cell Biol.
33:4225–4240. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang A, Savas U, Stout CD and Johnson EF:
Structural characterization of the complex between
alpha-naphthoflavone and human cytochrome P450 1B1. J Biol Chem.
286:5736–5743. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Stoilov I, Akarsu AN, Alozie I, Child A,
Barsoum-Homsy M, Turacli ME, Or M, Lewis RA, Ozdemir N, Brice G, et
al: Sequence analysis and homology modeling suggest that primary
congenital glaucoma on 2p21 results from mutations disrupting
either the hinge region or the conserved core structures of
cytochrome P4501B1. Am J Hum Genet. 62:573–584. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hollander DA, Sarfarazi M, Stoilov I, Wood
IS, Fredrick DR and Alvarado JA: Genotype and phenotype
correlations in congenital glaucoma. Trans Am Ophthalmol Soc.
104:183–195. 2006.PubMed/NCBI
|
|
28
|
Chen Y, Jiang D, Yu L, Katz B, Zhang K,
Wan B and Sun X: CYP1B1 and MYOC mutations in 116 Chinese patients
with primary congenital glaucoma. Arch Ophthalmol. 126:1443–1447.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kozak M: Downstream secondary structure
facilitates recognition of initiator codons by eukaryotic
ribosomes. Proc Natl Acad Sci USA. 87:8301–8305. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Stoilov IR, Costa VP, Vasconcellos JP,
Melo MB, Betinjane AJ, Carani JC, Oltrogge EV and Sarfarazi M:
Molecular genetics of primary congenital glaucoma in Brazil. Invest
Ophthalmol Vis Sci. 43:1820–1827. 2002.PubMed/NCBI
|
|
31
|
Reddy AB, Kaur K, Mandal AK, Panicker SG,
Thomas R, Hasnain SE, Balasubramanian D and Chakrabarti S: Mutation
spectrum of the CYP1B1 gene in Indian primary congenital glaucoma
patients. Mol Vis. 10:696–702. 2004.PubMed/NCBI
|
|
32
|
Abu-Amero KK, Osman EA, Mousa A, Wheeler
J, Whigham B, Allingham RR, Hauser MA and Al-Obeidan SA: Screening
of CYP1B1 and LTBP2 genes in Saudi families with primary congenital
glaucoma: Genotype-phenotype correlation. Mol Vis. 17:2911–2919.
2011.PubMed/NCBI
|
|
33
|
Rashid M, Yousaf S, Sheikh SA, Sajid Z,
Shabbir AS, Kausar T, Tariq N, Usman M, Shaikh RS, Ali M, et al:
Identities and frequencies of variants in CYP1B1 causing primary
congenital glaucoma in Pakistan. Mol Vis. 25:144–154.
2019.PubMed/NCBI
|
|
34
|
Waryah YM, Iqbal M, Sheikh SA, Baig MA,
Narsani AK, Atif M, Bhinder MA, Ur Rahman A, Memon AI, Pirzado MS,
et al: Two novel variants in CYP1B1 gene: A major contributor of
autosomal recessive primary congenital glaucoma with allelic
heterogeneity in Pakistani patients. Int J Ophthalmol. 12:8–15.
2019.PubMed/NCBI
|
|
35
|
Campos-Mollo E, López-Garrido MP,
Blanco-Marchite C, Garcia-Feijoo J, Peralta J, Belmonte-Martínez J,
Ayuso C and Escribano J: CYP1B1 mutations in Spanish patients with
primary congenital glaucoma: Phenotypic and functional variability.
Mol Vis. 15:417–431. 2009.PubMed/NCBI
|
|
36
|
Wang D, Pascual JM and De Vivo DD: Glucose
transporter type 1 deficiency syndrome. GeneReviews™ PubMed.
1993.
|
|
37
|
Hollander DA, Sarfarazi M, Stoilov I, Wood
IS, Fredrick DR and Alvarado JA: Genotype and phenotype
correlations in congenital glaucoma: CYP1B1 mutations,
goniodysgenesis, and clinical characteristics. Am J Ophthalmol.
142:993–1004. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Achary MS, Reddy AB, Chakrabarti S,
Panicker SG, Mandal AK, Ahmed N, Balasubramanian D, Hasnain SE and
Nagarajaram HA: Disease-causing mutations in proteins: Structural
analysis of the CYP1B1 mutations causing primary congenital
glaucoma in humans. Biophys J. 91:4329–4339. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhao Y, Sorenson CM and Sheibani N:
Cytochrome P450 1B1 and primary congenital glaucoma. J Ophthalmic
Vis Res. 10:60–67. 2015. View Article : Google Scholar : PubMed/NCBI
|