Introduction
Acute lung injury (ALI) is a life-threatening
disease which presents with activated inflammatory response,
increased penetrability of the alveoli and pulmonary edema, which
can deteriorate into acute respiratory distress syndrome (ARDS) and
even mortality unless it is promptly controlled (1,2).
Infection and sepsis serve as critical causes contributing to ALI,
as well as ischemia reperfusion, trauma and drug toxicity (3). The annual mortality for ARDS and ALI
remains high worldwide, leading to a significant healthcare burden
although significant achievements have been made in the therapeutic
strategy for ARDS and ALI (4).
Alveolar epithelial cells act as the primary host defense of the
alveolus, forming a robust barrier against various insult including
infectious and noninfectious factors in the pathogenesis of ALI
(5,6). Hence, maintaining the normal function
of alveolar epithelial cells is of great importance to retard the
progression of ALI.
Ferroptosis is a recently discovered form of cell
death, first defined in 2012 and characterized by excessive iron
accumulation as well as lipid peroxidation (7). Ferroptosis is an iron-dependent death,
which is differs from other regulated cell death (RCD) including
apoptosis, pyroptosis, necroptosis and autophagic death (8). Recent studies have revealed that
ferrostatin-1, a typical ferroptosis inhibitor, can effectively
relieve lipopolysaccharide (LPS)-induced ALI in mice and human
bronchial epithelial cell, indicating that ferroptosis is involved
in sepsis-induced ALI (9–11). Additionally, in ALI caused by other
pathogenesis including ischemia reperfusion (12), seawater drowning (13) and acute radiation (14), alveolar epithelial ferroptosis also
participates in the development of ferroptosis. Nuclear factor E2
related factor 2 (Nrf2) is the core transcription factor essential
for cells to keep redox homeostasis in the context of high
oxidative stress (15). Nrf2
transcriptionally activates numerous anti-ferroptotic genes by
binding to certain antioxidant response element in the nucleus,
helping to maintain cellular survival (16). During ALI, Nrf2 blocks alveolar
epithelial ferroptosis and prevents lung injury by upregulating
solute carrier family 7, membrane 11 (SLC7A11) and heme oxygenase 1
(10). Based on these studies,
genes or exogenous drugs that could activate Nrf2 in alveolar
epithelial cells may possess the potential to inhibit ferroptosis
and combat ALI.
Jumonji domain-containing 3 (JMJD3) is a histone
demethylase which specifically demethylates trimethylated H3 lysine
27 (17). In the context of
pathological stimulus, cytoplasm JMJD3 can be recruited around the
chromatin and interact with certain transcription factors,
activating the transcription of inflammatory genes, oncogenes,
developmental genes and oxidative genes (17,18). A
previous study showed that JMJD3 promoted the activation of
Nod-like receptor family pyrin domain-containing 3 inflammasome and
exacerbated colitis in mice by regulating Nrf2 (19). Upregulation of JMJD3 can give rise
to the removal of the repressive histone methylation mark on
NFκB-mediated inflammatory gene promoters, thus triggering
macrophage-mediated inflammation (20). These studies suggest that JMJD3 also
serves a critical role in inflammatory response. However, the
potential roles of JMJD3 in ALI have not been reported. The present
study first investigated the expression levels of JMJD3 in
sepsis-induced mice and LPS-treated A549 cells. Next, the potential
roles of JMJD3 in modulating ALI were observed by using alveolar
epithelial cell-specific knockout of JMJD3 mice. Finally, the
possible mechanisms contributing to the protection of JMJD3
silencing were explored. The present study revealed that the
expression of JMJD3 significantly increased during ALI. Alveolar
epithelial cell-specific knockout of JMJD3 clearly ameliorated
sepsis-induced lung injury and ferroptosis. In addition, Nrf2
overexpression further strengthen the anti-ferroptotic effect from
JMJD3 silence. The findings suggested that JMJD3 functions as a
possible target against ALI.
Materials and methods
Reagents
Anti-GAPDH (cat. no. ab8245), anti-NRF2 (cat. no.
ab62352), anti-JMJD3 (cat. no. ab169197), anti-STA1 (cat. no.
ab105220), anti-glutathione peroxidase 4 (GPX4; cat. no. ab125066),
anti-prostaglandin-endoperoxide synthase 2 (PTGS2; cat. no.
ab179800) and anti-SLC7A11 (cat. no. ab175186) antibodies were
obtained from Abcam. Fetal bovine serum (FBS), trypsin-EDTA (0.25%)
phenol red and Dulbecco's modified Eagle's medium, Nutrient Mixture
F-12 (DMEM/F-12) were purchased from Gibco (Thermo Fisher
Scientific, Inc.). The peroxidase-conjugated secondary antibody was
obtained from LI-COR Biosciences. The QuantiPro Bicinchoninic Acid
Protein Assay kit from Sigma-Aldrich (Merck KGaA) was employed to
detect protein concentrations. All chemical reagents used in the
present study were of analytical grade.
Animals and animal models
All procedures in this study were in accordance with
the Guide for the Care and Use of Laboratory Animals of National
Institutes of Health for live animals (21). All animal experiments were approved
by the Ethics Committee of the Wuhan University (approval no.
YXLL-2020-102). A total of 30 male C57/B6J mice (8–10 weeks, 21–23
g) were acquired from Chinese Academy of Medical Sciences (Beijing,
China). A total of 16 Jmjd3-flox mice and five
Sftpc-cre+ mice with C57/B6J background were provided by
Model Animal Research Center of Nanjing University. The surfactant
protein C (SPC) is exclusively expressed in the type II alveolar
epithelial cells. Sftpc-cre; 25 male Jmjd3+/flox mice
(genotype Sftpc-cre+/−; Jmjd3+/flox; age,
8–10 weeks; weight, 20–24 g) were generated through breeding
Jmjd3flox/flox mice and Sftpc-cre+/− mice.
All animals housed under specific pathogen-free conditions in a
humidity-(40–60%)-and temperature (18–25°C)-controlled environment
with a 12-h light/dark cycle and free access to water and food.
JMJD3 expression was detected via western blotting. In subsequent
experiments, male mice as well as their wild-type littermates (8–10
weeks, 21–23 g) were used. The sepsis-induced ALI model was
constructed by administering LPS intratracheally (5 mg/kg) for 12 h
as previously reported (22). The
control groups were given an isovolumetric sterile saline. Then, 12
h after LPS installation, the animals were sacrificed by cervical
dislocation under deep anesthesia with an intraperitoneal injection
of 2% sodium pentobarbital (60 mg/kg). Next, the left lungs from 5
mice from each group were excised and placed in formalin (10%) for
3 days to expand the alveoli at room temperature. The left lungs
from the other 5 mice in each group were dried in an 80°C oven for
3 days. The lung wet/dry (W/D) ratio was calculated to assess
edema. The right lung tissues were stored at −80°C for biochemical
analysis.
Cell culture and treatment
A549 alveolar epithelial cells were purchased from
Kunming Cell Bank of Typical Cultures Preservation Committee,
Chinese Academy of Sciences (Kunming, China). The A549 alveolar
epithelial cells were incubated at 37°C with DMEM supplemented with
10% FBS in a 5% CO2 incubator. JMJD3 small interfering
RNA (siRNA) and the negative control were obtained from Shanghai
GenePharma Co. Ltd. The primers were as follows: JMJD3 forward,
5′-GACACUATCTACTAACTATACTTA−3′ and reverse,
5′-CCCUAGCTAGTCUUUAAACC-3′ and negative control forward,
5′-CACATGCTGATGCTAUCTU-3′ and reverse,
5′-ACUTTCTATCUTACTACAA-3′.
The epithelial cells were transfected using
Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.) and siRNA (50 nmol/m) based on the manufacturer's
instructions at room temperature for 20 h. Subsequently, to mimic
sepsis-induced lung injury, the A549 alveolar epithelial cells were
treated with LPS at the concentration of 100 ng/ml with Ad-Nrf2
(MOI=25) or adenovirus harboring no overexpression sequence
(Ad-vector) for 6 h. Based on a previous study (23), mouse AT2 cells in
Jmjd3flox/flox mice and Sftpc-cre;
Jmjd3+/flox mice were isolated. In brief, mice were
perfused with phosphate buffer solution and then injected with 0.5
ml 1% low-melting agarose and 2 ml dispase into lung lobe. Then the
lung tissue was incubated with AT2 isolation medium supplemented
with 0.01% DNase I after removing trachea and airways for 0.5 h at
37°C. The mixture was subsequently filtered and re-suspended with
AT2 isolation medium containing a 1:1 mixture of DMEM/F-12
supplemented with 10% FBS, 100 U/ml sodium penicillin G, and 100
µg/ml streptomycin for 1 h. Cells were centrifuged for 10 min at
4°C at 300 × g and incubated on pre-coated dishes with mouse IgG at
4°C. After 2 h, non-adherent cells were collected, centrifuged for
5 min at 4°C at 200 × g and re-suspended with AT2 isolation
medium.
Hematoxylin and eosin (H&E) and
immunofluorescence staining
To evaluate the LPS-induced lung injury model and
therapeutic potential of JMJD3, the left lungs were removed from
mice in each group and subsequently fixed in 4% formalin overnight
at room temperature. Lung tissue samples were dehydrated in
ascending ethanol series and embedded in paraffin. After the
paraffin blocks were cut into 3 µm-thick sections, they were fixed
in ethanol as follows: Immersed in 70, 80 and 90% ethanol for 4–5
sec, then immersed in anhydrous ethanol for 5 min at room
temperature. Subsequently, hematoxylin staining was performed for 5
min and eosin staining was performed for 1 min at room temperature
to assess morphological abnormalities and inflammation score in a
blinded fashion as described previously (22). In brief, the severity of the lung
injury was graded from 0 to 4, on the basis of five independent
variables: Neutrophils in the alveolar space, hemorrhage,
pertinacious debris filling the airspaces, hyaline membranes, and
septal thickening. Scoring was as follows: 4, >75; 3, 51–75; 2,
25–50; l, <25 and 0, 0% damage.
To observe the protein expression of JMJD3,
immunofluorescence staining was performed based on previous study
(24). In brief, the medium in the
24-well plate containing A549 alveolar epithelial cells was
discarded, following by fixation with 4% paraformaldehyde at room
temperature for 5 min. Then 0.1% Triton X-100 was used to
permeabilize cells. Cells were incubated with primary JMJD3
antibody (1:200) overnight at 4°C; fluorescein
isothiocyante-conjugated goat anti-rabbit IgG (1:300) was added to
incubate for 1 h at 37°C away from light. Sections were incubated
with DAPI to stain the nuclei at room temperature. The protein
expression of JMJD3 was observed under an immunofluorescence
microscope (magnification, ×400).
Detection of bronchoalveolar lavage
fluid (BALF)
The BALF was acquired via intratracheal injections
of 1.0 ml of cooled phosphate buffer saline (PSB) three times after
the mice were sacrificed. Then the BALF was centrifuged at 1,200 ×
g for 10 min at 4°C. Total cells, macrophages and neutrophil were
calculated using a hemocytometer as well as Wright-Giemsa staining
as described previously (25). In
brief, a drop of BALF was placed onto a slide to make a smear.
Smears were air dried and then fixed in methanol (100%) for 3 min
at room temperature. Slides were stained with Wright-Giemsa working
solution for 1 min at room temperature and subsequently visualized
using a light microscope (magnification, ×400).
Reverse transcription-quantitative
(RT-q) PCR
Total RNA was extracted from the A549 alveolar
epithelial cells (3×106) using the Transcriptor First
Strand cDNA Synthesis kit (Norgen) according to the protocol. RNA
concentration was detected using a Biodrop µliTe Pc
spectrophotometer. Then the total RNA was synthesized into cDNA
using Prime Script RT Master Mix; the RT reaction conditions were
37°C for 15 min, followed by 85°C for 5 sec. PCR was performed
using the LightCycler 480 SYBR Green 1 Master Mix (Roche
Diagnostics GmbH). The thermocycling conditions were as follows:
Denaturation for 10 sec at 95°C, 20 sec annealing at 60°C and final
extension for 20 sec at 72°C. All primers used in this study are
given in Table I. The mRNA levels
were calculated with the 2−∆∆Cq method and normalized to
GAPDH (26).
 | Table I.Primers. |
Table I.
Primers.
| Species | Gene | Forward primer | Reverse primer |
|---|
| Human | SLC7A11 |
CGTAGCTGTGATAGTAGCTG |
CAGTCGTATAGTCGTGATCCCCC |
| Human | GPX4 |
AGCTAGTGAATATGCTGTACCC |
CGATAGATGCTGTAGCTGATGCC |
| Human | JMJD3 |
CGATCGTAGTAGATCGTAGTC |
CGATGCTGATGCTGATGCTGAA |
| Human | GAPDH |
CGATGTCGATAGTGCTAGTCA |
CAGGCTGATCGTAGTCGTGTAGCTA |
| Mice | JMJD3 |
CGATGCTGATGTCGTAGTCGTA |
CAGCTAGTGCTCGTAGTCGTGATC |
| Mice | GAPDH |
AACAGTCGTAGTCGTAGATGTT |
CAGTCGTAGTCGTAGTCGTATAAC |
Western blot analysis
Total proteins in iced cell lysates and right lung
tissues were extracted after cells and tissues were lysed by a RIPA
buffer. The protein concentrations were then measured using a
commercial BCA Protein Assay kit. Subsequently, the total proteins
(80–100 g) were loaded into 10% SDS-PAGE gel, which were next
transferred on a PVDF membrane. After blocking with milk (5%) for 1
h at room temperature, the membranes were incubated with JMJD3
(cat. no. ab169197, 1:500, Abcam.), GAPDH (cat. no. ab8245,
1:1,000, Abcam.), PTGS2 (cat. no. ab179800, 1:1,000, Abcam.), SAT1
(cat. no. ab105220, 1:1,000, Abcam), GPX4 (cat. no. ab125066,
1:1,000, Abcam), SLC7A11 (cat. no. ab175186, 1:200, Abcam) and NRF2
(cat. no. ab62352, 1:500, Abcam) overnight at 4°C and
peroxidase-conjugated goat anti-rabbit IgG (cat. no. SA00001-2;
1:5,000; ProteinTech Group, Inc.) for 2 h at room temperature.
Finally, the membranes were screened and visualized via an Odyssey
Imaging System (LI-COR Biosciences). Blots were quantitated using
Image Lab software 4.0 (Bio-Rad Laboratories, Inc.). The protein
levels in this study were normalized to GAPDH.
Statistical analysis
The data are presented as mean ± standard error of
mean and were analyzed by SPSS 23.0 (IBM Corp.). Comparisons
between two groups were performed by Student's unpaired t-test.
Multiple comparisons among ≥3 groups were performed by one-way
ANOVA, followed by post hoc Tukey's test. P<0.05 was considered
to indicate a statistically significant difference.
Results
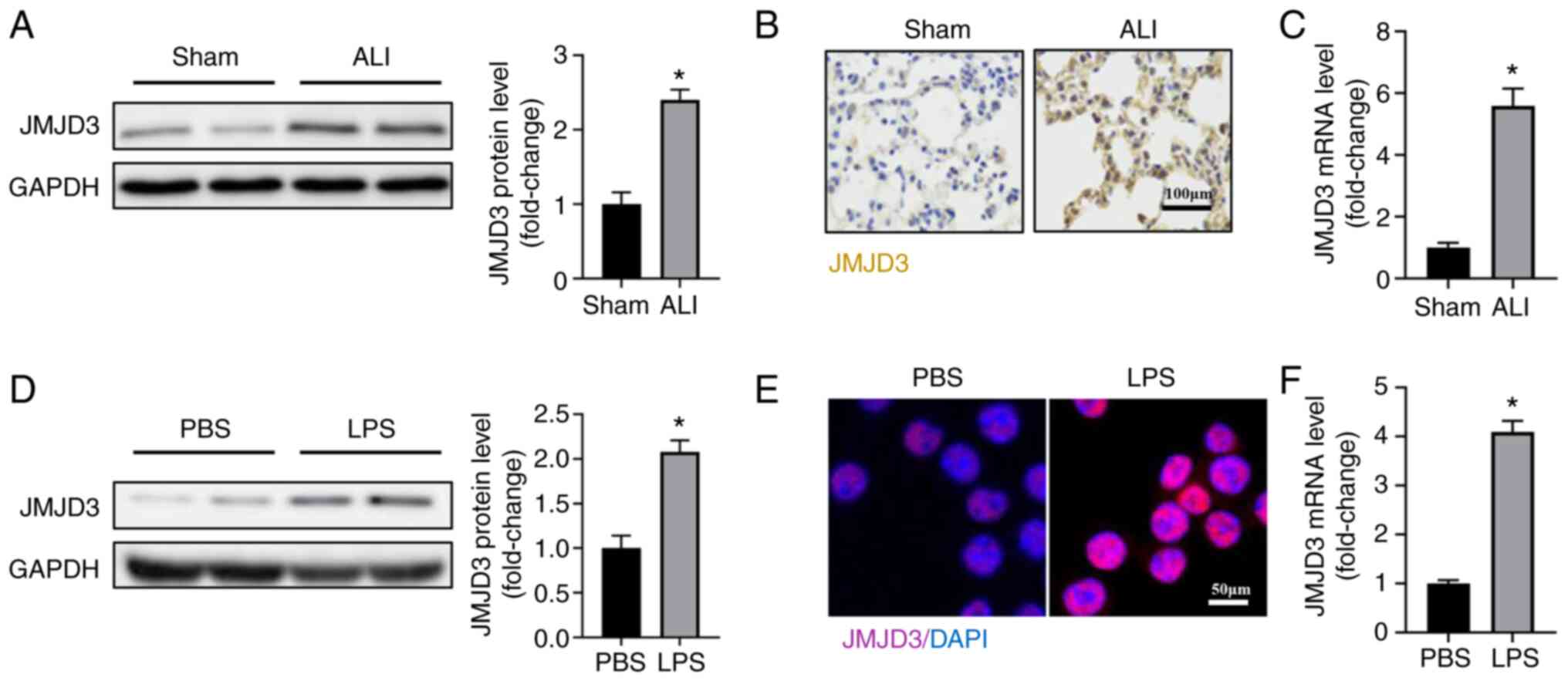
The expression of JMJD3 in LPS-treated
lung tissues and alveolar epithelial cells
To explore the association between JMJD3 and
sepsis-induced ALL, the protein and mRNA levels of JMJD3 were
detected in a murine model of ALI. As shown in Fig. 1A-C, the protein and mRNA levels of
JMJD3 were significantly upregulated in mice with ALI compared with
control group. LPS stimulation also increased the protein and mRNA
levels of JMJD3 in A549 alveolar epithelial cells (Fig. 1D-F). It was also found that JMJD3
mainly expressed in nucleus of alveolar epithelial cells. Taken
together, these data showed that the expression of JMJD3 was
upregulated in response to LPS stimulation.
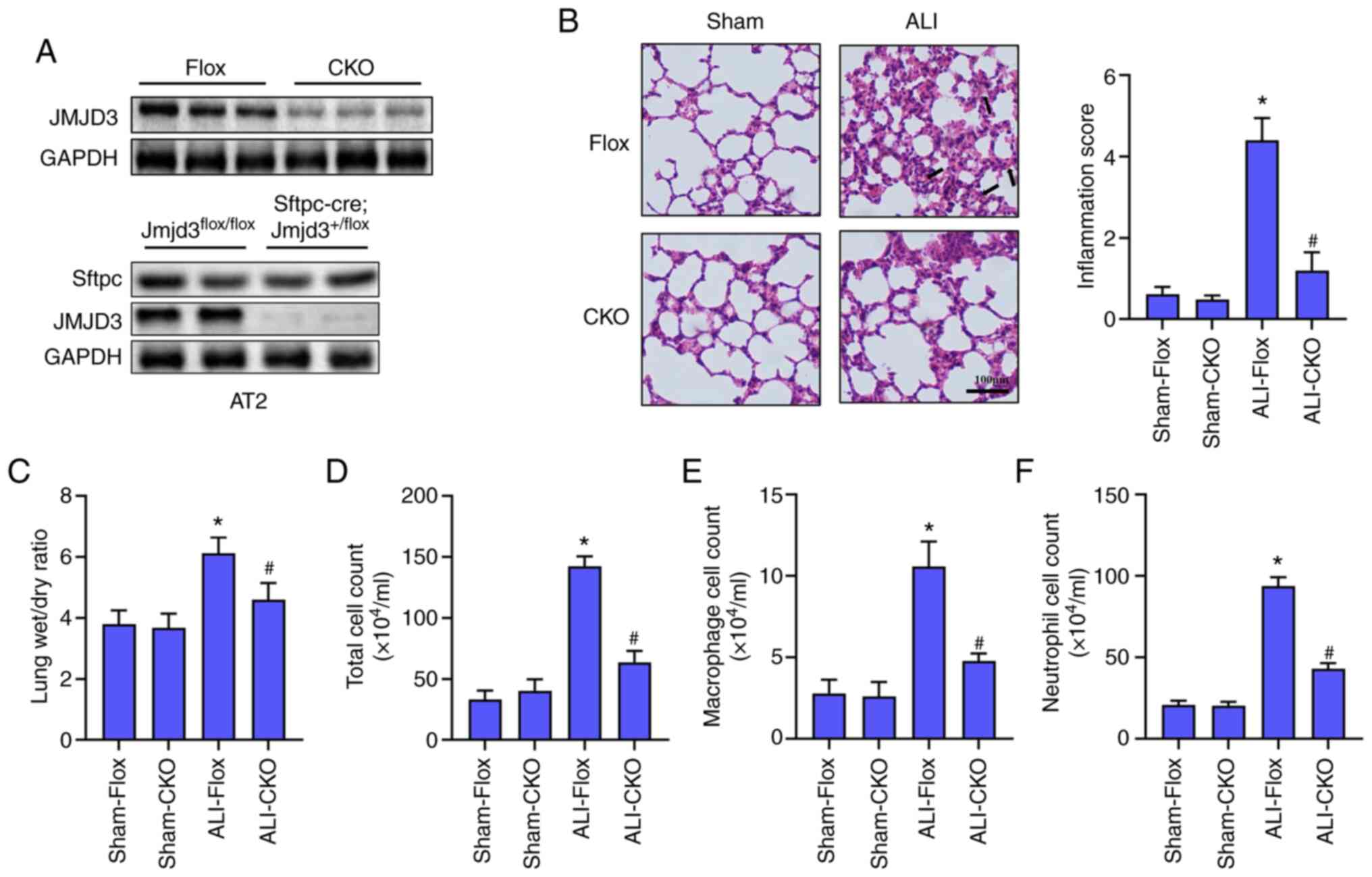
JMJD3 deficiency relieved LPS-induced
ALI
To understand the effect of JMJD3 on LPS-induced
ALI, alveolar epithelial cell-specific knockout of Jmjd3 mice
(JMJD3-CKO) was constructed. Alveolar epithelial cell-specific
deletion of JMJD3 was verified by western blotting (Fig. 2A). H&E staining (Fig. 2B) showed that mice challenged with
LPS developed marked lung injury, as evidenced by thickening of
alveolar walls, lung edema, alveolar hemorrhage and a higher
inflammation score. The lung wet/dry ratio and cells count
including total cell, macrophage and neutrophil in BALF were
significantly higher in ALI-Flox group than those in Sham-Flox
group (Fig. 2C-F). However, JMJD3
knockout significantly alleviated LPS-induced lung edema and
infiltration of inflammatory cells in BALF. Collectively, these
data demonstrated that JMJD3 deficiency contributed to the
improvement of LPS-induced ALI.
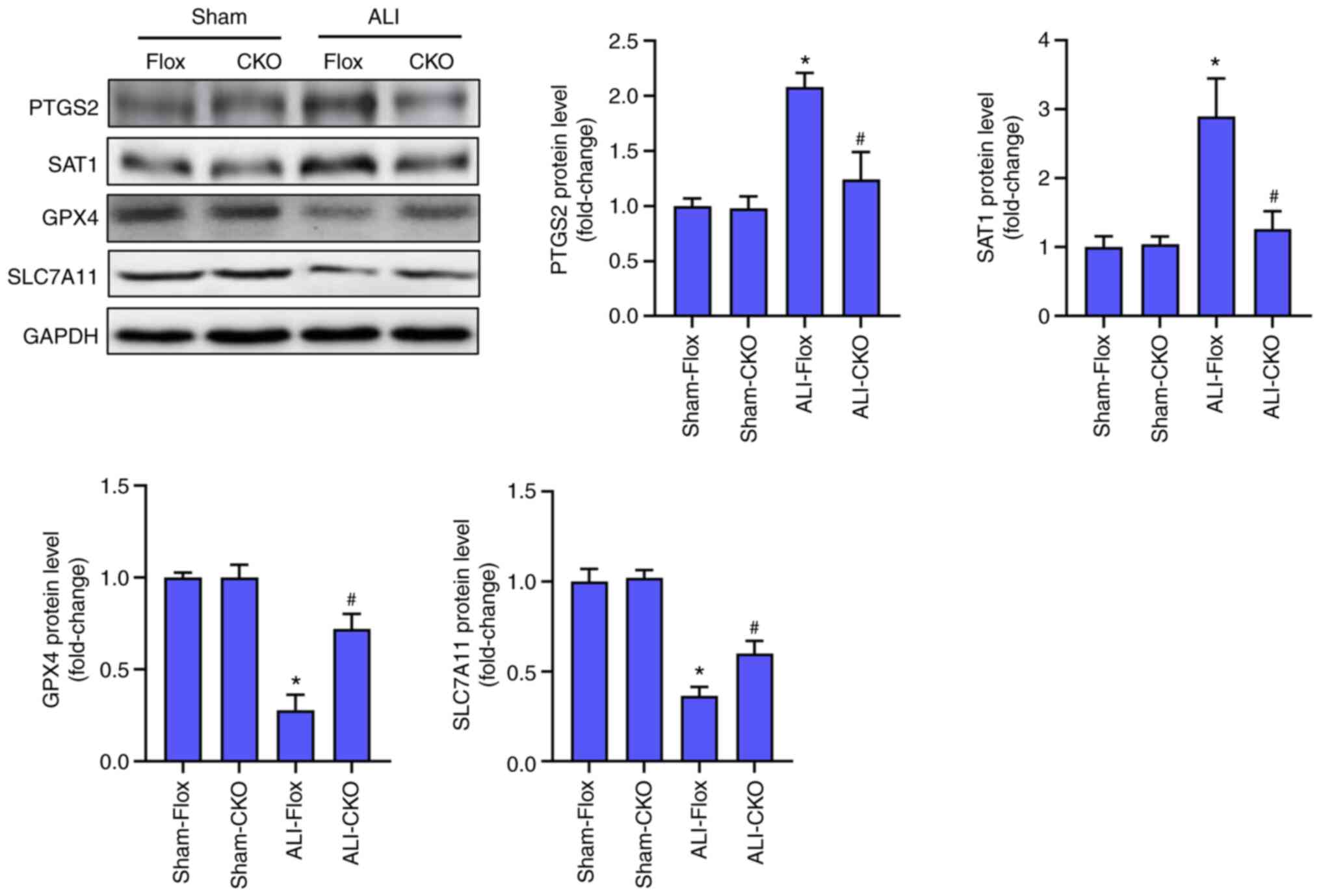
JMJD3 deficiency inhibited ferroptosis
in LPS-treated lung
Alveolar epithelial ferroptosis could aggravate ALI
induced by LPS. The protein markers of ferroptosis in were next
detected the indicated four groups. As shown in Fig. 3, LPS stimulation significantly
increased the protein levels of PTGS2 and SAT1 and decreased the
protein levels of GPX4 and SLC7A11, indicating that LPS triggered
ferroptosis in lung. Compared with the ALI-Flox group, the mice in
ALI-CKO group displayed lower level of ferroptosis, which was
evidenced by the decreased levels of PTGS2 and SAT1 and higher
levels of GPX4 as well as SLC7A11. These results hinted that
alveolar epithelial cell-specific knockout of JMJD3 could repress
the level of ferroptosis induced by LPS.
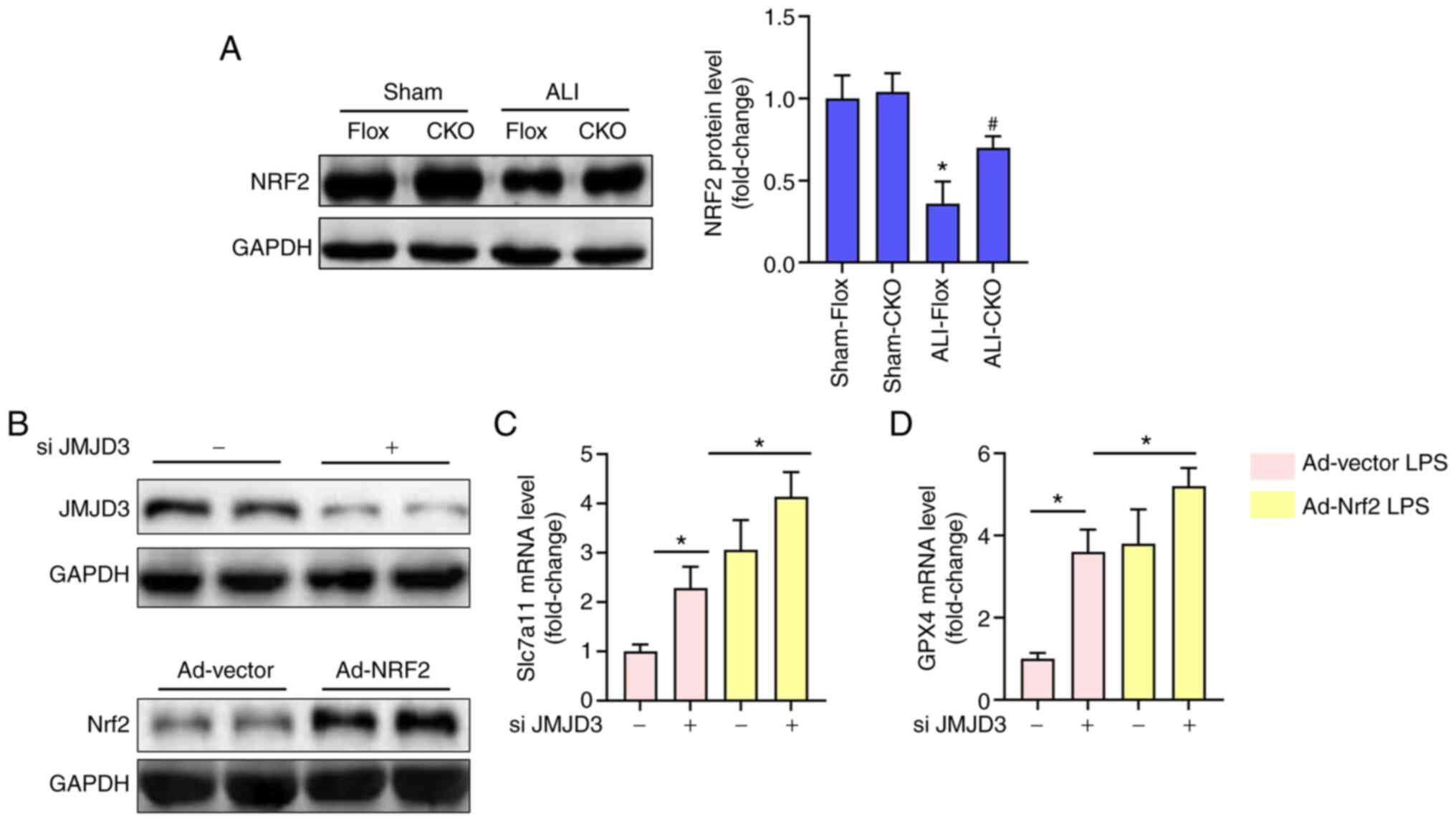
Nrf2 is involved in the protection of
JMJD3 deficiency in vivo and in vitro
Previous studies have illustrated that Nrf2 could
transcriptionally activate certain anti-ferroptotic genes including
Gpx4 and Slc7a11 (16,27). Meanwhile, the expression of Nrf2
could be also modulated by JMJD3 (19). Therefore, the protein level of Nrf2
was detected in the 4 groups. The results (Fig. 4A) showed that alveolar epithelial
cell-specific knockout of JMJD3 inhibited the decrease of Nrf2 in
lung tissues challenged with LPS. To further verify the
relationship between Nrf2 and JMJD3, Nrf2 was overexpressed using
adenovirus and the level of JMJD3 inhibited using siRNA in A549
alveolar epithelial cells. Fig. 4B
showed that the expression of JMJD3 protein was significantly
inhibited following JMJD3 siRNA treatment while the expression of
Nrf2 protein was significantly upregulated after transfection with
Ad-Nrf2. As shown in Fig. 4C and D,
JMJD3 inhibition significantly increased the mRNA levels of Slc7a11
and Gpx4 in alveolar epithelial cells, the effect of which could be
enhanced after Nrf2 was overexpressed. These data suggested that
JMJD3 inhibition could decrease alveolar epithelial ferroptosis in
a Nrf2-dependent manner.
Discussion
The present study found that LPS stimulation led to
lung injury and inflammatory response by triggering alveolar
epithelial ferroptosis and alveolar epithelial cell-specific
knockout of JMJD3 significantly alleviated lung injury caused by
sepsis through blocking ferroptosis. Mechanistically, JMJD3
deficiency could reverse the decrease of Nrf2 during ALI, hence
increasing the transcription levels of anti-ferroptotic genes
including GPX4 and SLC7A11. Based on these findings, it was
hypothesized that JMJD3 may serve as a potential target against
sepsis-induced ALI.
Cells can die from accidental cell death (such as
necrosis) and RCD (such as pyroptosis, ferroptosis, apoptosis,
necroptosis and autophagy), of which ferroptosis is closely
involved in infection and solid organ injury (28,29).
Initiation of ferroptosis demands three critical foundations
including abundant redox-active iron in cytoplasm, vital substrates
undergoing peroxidation and the failure of the lipid peroxide
repair network. SLC7A11 is one of the subunits of system
Xc−, which is in essence a cystine/glutamate reverse
transporter. SLC7A11 is mainly expressed on cell membrane and
responsible for cystine import, which is important for maintaining
cellular redox homeostasis. The intracellular cystine could be
degraded to cysteine, which is then synthesized into antioxidant
GSH (16,30).
As with system Xc-, GPX4 can also negatively
regulate ferroptosis via exerting antioxidant effects (31). In detail, GPX4 can convert reduced
glutathione to oxidized glutathione through reducing lipid
hydroperoxides to lipid hydroxy derivative or converting free
hydrogen peroxide to water, maintaining the redox homeostasis in
cell (16). A number of studies
have reported that ferroptosis is involved in the development of
lung injury. For instance, Liu et al (9) reported that LPS treatment
significantly decreases the viability of a human bronchial
epithelial cells and the levels of SLC7A11 and GPX4, which could be
reversed by Ferrostatin-1. Additionally, intestinal
ischemia/reperfusion-induced ALI also promotes alveolar epithelial
ferroptosis, which can in turn be aggravated by erastin (32). As a basic leucine zipper
transcription factor modulating the expression of antioxidant genes
associated with redox homeostasis, Nrf2 can inhibit ferroptosis by
regulating ferroptotic core genes indirectly, including GPX4 and
SLC7A11, thus exerting protection in ALI (10,32).
In parallel with these studies, the present study also revealed
that Nrf2 overexpression by adenovirus could promote the protective
effects of JMJD3 deficiency in LPS-treated alveolar epithelial
cells.
JMJD3 is a histone demethylase which can demethylate
trimethylated H3 lysine 27 (33).
LPS can upregulate the expression of JMJD3 by activating
NF-κB in macrophages and vascular endothelial cells,
eventually increasing the expression of inflammatory genes
(34,35). Consistent with previous studies, the
present study also found that LPS could increase the protein and
mRNA levels of JMJD3 in murine lung tissues and alveolar epithelial
cells. In addition, JMJD3 deficiency in alveolar epithelial cells
not only alleviated LPS-induced lung injury and inflammatory
response, but also inhibited ferroptosis in lung tissues.
Furthermore, JMJD3 deficiency enhanced the protein expression of
Nrf2 in murine lung tissues challenged with LPS. In vitro
experiments also showed that Nrf2 overexpression increased the
anti-ferroptotic ability of JMJD3 siRNA in alveolar epithelial
cells by upregulating the expression levels of GPX4 and SLC7A11.
However, there are still some limitations to the present study. For
instance, the A549 cell used is in essence a tumor cell line.
Although it also possessed the characteristics of type II alveolar
epithelial cells, A549 cells could not represent alveolar
epithelial cells completely. Hence, future study should further
explore the roles of JMJD3 in primary alveolar epithelial
cells.
In summary, the present study proposed that LPS
stimulation not only gave rise to lung injury by inducing alveolar
epithelial ferroptosis, but also increased the level of JMJD3.
JMJD3 deficiency in alveolar epithelial cells could protect against
LPS-induced ALI, the potential mechanism of which involves the
activation of the Nrf2 in epithelial cells. The present study for
the first time, to the best of the authors' knowledge, revealed
that JMJD3 deficiency may prevent the harmful effects of LPS on
lung tissues, which is expected to serve as a possible candidate
against lung injury caused by sepsis.
Acknowledgements
Not applicable.
Funding
The present study was supported by Scientific
Research Program Guiding Project of Education Department, Hubei
Province (grant no. B2017482).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JP and BF conceived the study and revised the
manuscript. CB and CJ performed the experiments and data analysis.
All authors confirm the authenticity of all the raw data. All
authors have read and approved the final manuscript.
Ethics approval and consent to
participate
All animal experiments were approved by the Ethics
Committee of the Wuhan University (approval no. YXLL-2020-102).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Santa Cruz R, Villarejo F, Irrazabal C and
Ciapponi A: High versus low positive end-expiratory pressure (PEEP)
levels for mechanically ventilated adult patients with acute lung
injury and acute respiratory distress syndrome. Cochrane Database
Syst Rev. 3:CD0090982021.PubMed/NCBI
|
|
2
|
Bian S, Cai H, Cui Y, Liu W and Xiao C:
Nanomedicine-based therapeutics to combat acute lung injury. Int J
Nanomedicine. 16:2247–2269. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shi J, Yu T, Song K, Du S, He S, Hu X, Li
X, Li H, Dong S, Zhang Y, et al: Dexmedetomidine ameliorates
endotoxin-induced acute lung injury in vivo and in vitro by
preserving mitochondrial dynamic equilibrium through the
HIF-1a/HO-1 signaling pathway. Redox Biol. 41:1019542021.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fan E, Brodie D and Slutsky A: Acute
respiratory distress syndrome: Advances in diagnosis and treatment.
JAMA. 319:698–710. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kamio N, Hayata M, Tamura M, Tanaka H and
Imai K: Porphyromonas gingivalis enhances pneumococcal adhesion to
human alveolar epithelial cells by increasing expression of host
platelet-activating factor receptor. FEBS Lett. 595:1604–1612.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mu M, Gao P, Yang Q, He J, Wu F, Han X,
Guo S, Qian Z and Song C: Alveolar epithelial cells promote igf-1
production by alveolar macrophages through TGF-β to suppress
endogenous inflammatory signals. Front Immunol. 11:15852020.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Geng Z, Guo Z, Guo R, Ye R, Zhu W and Yan
B: Ferroptosis and traumatic brain injury. Brain Res Bull.
172:212–219. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dixon S, Lemberg K, Lamprecht M, Skouta R,
Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et
al: Ferroptosis: An iron-dependent form of nonapoptotic cell death.
Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu P, Feng Y, Li H, Chen X, Wang G, Xu S,
Li Y and Zhao L: Ferrostatin-1 alleviates
lipopolysaccharide-induced acute lung injury via inhibiting
ferroptosis. Cell Mol Biol Lett. 25:102020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dong H, Qiang Z, Chai D, Peng J, Xia Y, Hu
R and Jiang H: Nrf2 inhibits ferroptosis and protects against acute
lung injury due to intestinal ischemia reperfusion via regulating
SLC7A11 and HO-1. Aging. 12:12943–12959. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu W, Deng H, Hu S, Zhang Y, Zheng L, Liu
M, Chen Y, Wei J, Yang H and Lv X: Role of ferroptosis in lung
diseases. J Inflam Res. 14:2079–2090. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu Y, Li X, Cheng Y, Yang M and Wang R:
Inhibition of ACSL4 attenuates ferroptotic damage after pulmonary
ischemia-reperfusion. FASEB J. 34:16262–16275. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Qiu YB, Wan BB, Liu G, Wu YX, Chen D, Lu
MD, Chen JL, Yu RQ, Chen DZ and Pang QF: Nrf2 protects against
seawater drowning-induced acute lung injury via inhibiting
ferroptosis. Respir Res. 21:2322020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li X, Zhuang X and Qiao T: Role of
ferroptosis in the process of acute radiation-induced lung injury
in mice. Biochem Biophys Res Commun. 519:240–245. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Song M, Lee D, Chun K and Kim E: The Role
of NRF2/KEAP1 signaling pathway in cancer metabolism. Int J Mol
Sci. 22:43762021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li N, Jiang W, Wang W, Xiong R, Wu X and
Geng Q: Ferroptosis and its emerging roles in cardiovascular
diseases. Pharmacol Res. 166:1054662021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang X, Liu L, Yuan X, Wei Y and Wei X:
JMJD3 in the regulation of human diseases. Protein Cell.
10:864–882. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shentu Y, Tian Q, Yang J, Liu X, Han Y,
Yang D, Zhang N, Fan X, Wang P, Ma J, et al: Upregulation of KDM6B
contributes to lipopolysaccharide-induced anxiety-like behavior via
modulation of VGLL4 in mice. Behav Brain Res. 408:1133052021.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Huang M, Wang Q, Long F, Di Y, Wang J,
Zhun Zhu Y and Liu X: Jmjd3 regulates inflammasome activation and
aggravates DSS-induced colitis in mice. FASEB J. 34:4107–4119.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Davis F, denDekker A, Joshi AD, Wolf SJ,
Audu C, Melvin WJ, Mangum K, Riordan MO, Kunkel SL and Gallagher
KA: Palmitate-TLR4 signaling regulates the histone demethylase,
JMJD3, in macrophages and impairs diabetic wound healing. Eur J
Immunol. 50:1929–1940. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Deng W, He J, Tang XM, Li CY, Tong J, Qi D
and Wang DX: Alcohol inhibits alveolar fluid clearance through the
epithelial sodium channel via the A2 adenosine receptor in acute
lung injury. Mol Med Rep. 24:7252021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ning L, Wei W, Wenyang J, Rui X and Qing
G: Cytosolic DNA-STING-NLRP3 axis is involved in murine acute lung
injury induced by lipopolysaccharide. Clin Transl Med. 10:e2282020.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Do DC, Zhang Y, Tu W, Hu X, Xiao X, Chen
J, Hao H, Liu Z, Li J, Huang SK, et al: Type II alveolar epithelial
cell-specific loss of RhoA exacerbates allergic airway inflammation
through SLC26A4. JCI Insight. 6:e1481472021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yu Y, Jiang P, Sun P, Su N and Lin F:
Pulmonary coagulation and fibrinolysis abnormalities that favor
fibrin deposition in the lungs of mouse antibody-mediated
transfusion-related acute lung injury. Mol Med Rep. 24:6012021.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Huang X, Liu W, Zhou Y, Sun M, Yang HH,
Zhang CY and Tang SY: Galectin-1 ameliorates
lipopolysaccharide-induced acute lung injury via AMPK-Nrf2 pathway
in mice. Free Radic Biol Med. 146:222–233. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Song X and Long D: Nrf2 and ferroptosis: A
new research direction for neurodegenerative diseases. Front
Neurosci. 14:2672020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li N, Wang W, Zhou H, Wu Q, Duan M, Liu C,
Wu H, Deng W, Shen D and Tang Q: Ferritinophagy-mediated
ferroptosis is involved in sepsis-induced cardiac injury. Free
Radic Biol Med. 160:303–318. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xu S, He Y, Lin L, Chen P, Chen M and
Zhang S: The emerging role of ferroptosis in intestinal disease.
Cell Death Dis. 12:2892021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wu X, Li Y, Zhang S and Zhou X:
Ferroptosis as a novel therapeutic target for cardiovascular
disease. Theranostics. 11:3052–3059. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yao Y, Chen Z, Zhang H, Chen C, Zeng M,
Yunis J, Wei Y, Wan Y, Wang N, Zhou M, et al: Selenium-GPX4 axis
protects follicular helper T cells from ferroptosis. Nat Immunol.
22:1127–1139. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li Y, Cao Y, Xiao J, Shang J, Tan Q, Ping
F, Huang W, Wu F, Zhang H and Zhang X: Inhibitor of
apoptosis-stimulating protein of p53 inhibits ferroptosis and
alleviates intestinal ischemia/reperfusion-induced acute lung
injury. Cell Death Differ. 27:2635–2650. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu Z, Cao W, Xu L, Chen X, Zhan Y, Yang
Q, Liu S, Chen P, Jiang Y, Sun X, et al: The histone H3 lysine-27
demethylase Jmjd3 plays a critical role in specific regulation of
Th17 cell differentiation. J Mol Cell Biol. 7:505–516. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yu S, Chen X, Xiu M, He F, Xing J, Min D
and Guo F: The regulation of Jmjd3 upon the expression of NF-κB
downstream inflammatory genes in LPS activated vascular endothelial
cells. Biochem Biophys Res Commun. 485:62–68. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
De Santa F, Narang V, Yap ZH, Tusi BK,
Burgold T, Austenaa L, Bucci G, Caganova M, Notarbartolo S, Casola
S, et al: Jmjd3 contributes to the control of gene expression in
LPS-activated macrophages. EMBO J. 28:3341–3352. 2009. View Article : Google Scholar : PubMed/NCBI
|