Introduction
The capacity for the adult brain to repair itself by
axonal regeneration or compensatory fiber growth following
hypoxia/ischemia is extremely limited and incomplete, partly due to
several factors, including glial scars, lack of neurotrophins and
inhibitory proteins that create a non-permissive environment and
limit the structural plasticity of injured neurons (1). It has been reported that three
myelin-associated proteins, Nogo-A, myelin-associated glycoprotein
and oligodendrocyte myelin glycoprotein, are implicated as
inhibitors of axonal growth (2).
Among them, Nogo-A cloned in 2000 has received much attention as a
major obstacle to successful axon regeneration (3). Nogo-A, a reticulon protein family
member, is widely localized in the cell bodies of oligodendrocytes
and in some neuronal populations (4,5).
Researchers have presumed that neuronal Nogo-A may be involved in
normal cell functioning different from the inhibitory role of
oligodendroglial Nogo-A (6). To
date, there is limited information concerning the distribution of
Nogo-A in the intact and lesioned central nervous system (CNS), and
the time-dependent changes of Nogo-A expression after damage to the
CNS remain debatable. For example, Huber et al (7) failed to show a change in Nogo-A
protein after cortical lesions in rats. By contrast, Zhou et
al (8) demonstrated that the
expression of Nogo-A was significantly increased at 6 h, lasting
for 7 days in global ischemic rats. Eslamboli et al
(9) reported the increased level
of Nogo-A up to 2 months following cerebral ischemia in marmoset
monkeys. However, the change of Nogo-A levels has not been well
studied in the middle cerebral artery occlusion (MCAO) model.
Therefore, the purpose of the present study was to systemically
investigate the expression pattern of Nogo-A at different time
points from days 1 to 28 after focal cerebral ischemia.
Nogo-A binds to the Nogo receptor (Ng-R) to activate
intracellular signaling pathways, resulting in growth cone collapse
that contributes to the failure of axon regeneration (10). This inhibition can be reversed by
an anti-Nogo-A antibody (3).
Previously, available data has attempted to elucidate that
inhibition of Nogo-A may promote extensive growth of axons and
behavioral recovery after injury (11). However, the exact mechanism
underlying Nogo-A failure in myelin repair remains unclear. Brain
plasticity has been demonstrated to occur following experimental
injury (12), but no previous
reports have evaluated the effect of myelin inhibitors on
post-ischemic plasticity. Growth-associated binding protein 43
(GAP-43) is a nervous system-specific protein and considered to be
an intrinsic determinant of axonal plasticity (13,14). When the nervous system is damaged,
GAP-43 expression increases until the establishment of complete
synaptic connections (15). A
previous in vivo study reported that the expression of
GAP-43 is upregulated in vulnerable brain lesions after hypoxia
injury (16). The elevated
expression of GAP-43 caused by injury may contribute the
regenerative response of axonal damage and compensate for brain
damage. Microtubule-associated protein 2 (MAP-2) is a cytoskeletal
protein that serves a role in the growth, differentiation and
plasticity of neurons (17).
Several studies have reported a decrease in MAP-2 expression after
ischemia (18), which suggests
that MAP-2 is sensitive to ischemic injury and loss of MAP-2 leads
to neuronal dysfunction and dendritic breakdown (19). Therefore, the expression levels of
GAP-43 and MAP-2 are considered to be the preferred probe for
studying neural and axonal plasticity. In the present study, to
elucidate a potential molecular mechanism underlying the Nogo-A
inhibitor, Nogo extracellular peptide 1-40 (NEP1-40), on myelin
repair, the changes in GAP-43 and MAP-2 expression associated with
brain plasticity in ischemic model rats were investigated.
Therefore, the results of the present study may provide an insight
for the treatment of poststroke deficits by targeting Nogo-A to
promote CNS regeneration through enhancement of neuroanatomical
plasticity.
Materials and methods
MCAO model establishment
Adult male Sprague-Dawley rats (n=146; weight,
260–280 g; age, 6–8 weeks) were purchased from the Department of
Experimental Animal Sciences, Dalian Medical University. The
animals were housed in pairs in standard cages with free access to
food and water under a 12/12-h light/dark cycle and in standard
laboratory conditions (temperature, 20–25°C; relative humidity,
50–70%). Efforts were made to minimize the discomfort of the
animals. All animal-related experiments were approved by the Animal
Welfare Committee of Dalian Medical University (Dalian, China;
approval number, 202106101) and followed the guidelines for Animal
Care and Use adapted from the National Institutes of Health
(20).
MCAO was performed as described by Longa et
al (21). The rats were
anesthetized by the intraperitoneal injection of 10% chloral
hydrate (300 mg/kg). No signs of peritonitis or pain/discomfort
were observed following the injection of 10% chloral hydrate. After
median incision of the neck skin, the right common carotid artery
(CCA), external carotid artery (ECA) and internal carotid artery
(ICA) were carefully isolated from the surrounding nerves. Briefly,
no. 4-0 nylon monofilament suture was inserted into the ECA and
advanced into the ICA to block the origin of the right middle
cerebral artery (MCA). The suture was inserted 18–20 mm beyond the
origin of the ICA. At 2 h after MCAO, the intraluminal filament was
carefully removed. Rats without left forelimb paresis or circling
towards the left side were regarded as unsuccessful models and were
excluded from further experiments. Sham control rats underwent
surgery procedures without filament insertion.
Experimental group and drug
stereotactic delivery
A total of 134 rats were established by MCAO for 2
h, eight rats died primarily due to the trauma of the surgery and
six rats failed to induce successful model establishment.
Successful ischemic rats were randomly allocated into three groups:
i) MCAO model group (n=60, 30 for immunostaining and 30 for western
blotting); ii) MCAO + saline group (received saline; n=30 for
immunostaining); and iii) MCAO + NEP1-40 group (received NEP1-40;
n=30 for immunostaining). Rats of each group were divided into the
following five subgroups by the days of observation: Days 1, 3, 7,
14 and 28 (n=6 each time point). At days 1, 3 and 14 after
transient MCAO, rats were anesthetized by the intraperitoneal
injection of 10% chloral hydrate (300 mg/kg) and placed on a
stereotaxic apparatus. A 2.0-mm hole was drilled through the skull,
and a 10 µl solution containing 10 µg/µl NEP1-40 or an equal volume
of saline was microinjected into the right lateral ventricle
(coordinate, 0.8 mm anterior to the bregma, 1.3 mm lateral to the
midline, 3.5 mm beneath the dura) at a rate of 0.05 µl/min for 10
min using a Hamilton microsyringe (10 µl syringe Hamilton 900
series). After finishing the injection, the needle was left in
place for an additional 10 min to prevent reflux and slowly
withdrawn over 5 min. Penicillin was used to prevent the wound from
infection. After NEP1-40 microinjection, six rats died, which was
most likely due to aseptic inflammation. Animals that did not
receive an injection and only received sham operations were used as
controls (n=12, six for immunostaining and six for western
blotting). The general state of health of the animals was observed
daily. When the rats exhibited body weight loss, reduced activity,
or showed signs of pain, distress or discomfort, it was determined
that humane endpoints had been reached and the animals were
euthanized before the end of experiment. At 12 h after NEP1-40
administration, the rats were anaesthetized by the intraperitoneal
injection of 10% choral hydrate, then sacrificed by decapitation or
cervical dislocation on the observation time points. Animal death
was verified by cessation of carotid artery beats. Subsequently,
the brain tissues were collected for further assessment.
Luxol fast blue (LFB) and Bielschowsky
silver staining
LFB and Bielschowsky silver staining were performed
as previously described (22).
Brain tissues were resected, embedded in paraffin blocks, cut into
continuous coronal sections of 5 µm thickness, and then stained
with LFB or Bielschowsky silver. In brief, for LFB staining,
sections were stained with 0.1% LFB solution for 10 h at 60°C,
washed with distilled water, then placed in 95% alcohol for 10 min.
The slides were differentiated with 0.05% lithium carbonate for
10–30 sec and then rinsed in 70% alcohol solution for 20 sec. The
differentiation step was repeated until the gray matter became
colorless and the white matter remained blue.
For Bielschowsky silver staining, brain sections of
striatum were placed in 20% silver nitrate at 37°C in the dark for
25 min, followed by rinsing with distilled water. Ammoniacal silver
solution was then added dropwise to the slices. After rinsing with
water, slices were immersed in 10% formaldehyde for 10 min at 25°C.
Finally, the sections were fixed in 5% sodium thiosulfate for 5 min
at 25°C. Images were acquired in the lesioned striatum using a
light microscope (Olympus Corporation; magnification, ×200).
Image-Pro Plus software (version 6.0; Media Cybernetics, Inc.) was
used to calculate the staining density to quantify the damage of
white matter nerve fibers.
Immunohistochemical staining
Following the establishment of successful
euthanasia, immunohistochemistry was performed. Rats were perfused
through the aorta with 200 ml normal saline, followed by 300 ml 4%
paraformaldehyde (PFA; pH 7.4). The brains were removed, post-fixed
in 4% PFA for 20–24 h at 25°C and then dehydrated in 30% sucrose.
The brain sections were cut into 5-µm thick serial sections with a
cryostat (Cryocut 1800; Leica Microsystems, Inc.) and mounted on
gelatin/chrome alum-coated glass slides. The slides were processed
for Nogo-A, myelin basic protein (MBP), GAP-43 and MAP-2 staining
using the avidin-biotin technique as described by Tu et al
(23). Sections were blocked with
2% normal goat serum (OriGene Technologies, Inc.) for 30 min at
37°C, and then incubated with a primary polyclonal antibody against
Nogo-A (1:400; catalog no. AB5664P; Chemicon International; Thermo
Fisher Scientific, Inc.), rabbit anti-MBP (1:750; catalog no.
BA0094; Wuhan Boster Biological Technology, Ltd.), rabbit
anti-GAP-43 (1:800; catalog no. sc-17790; Santa Cruz Biotechnology,
Inc.) and rabbit anti-MAP-2 (1:500; catalog no. sc-3279; Santa Cruz
Biotechnology, Inc.) overnight at 4°C. After washing with PBS,
sections were incubated with goat anti-mouse or goat anti-rabbit
secondary antibodies conjugated to HRP (1:100; catalog nos. SP9001
or SP9002; OriGene Technologies, Inc.) for 30 min at 37°C. The
sections were thereafter rinsed and then processed with
diaminobenzidine (1 mg/ml; 0.001% H2O2) for 2
min at 25°C. The stained sections were viewed and images acquired
using a light microscope at ×100 magnification (Olympus
Corporation).
MetaMorph software (version 7.8; Molecular Devices,
LLC) was used for the quantification of cell numbers. A region with
1,400 µm width and 1,200 µm length in the ischemic striatum was
defined for counting Nogo-A+ cells. The slide was viewed
at ×100 magnification in a blinded manner. For each brain, four
slides were obtained from 20-µm thick coronal sections between 1.4
mm anterior and 0.4 mm posterior to the bregma. All counts were
pooled together and results are expressed as the average number per
rat.
For the analysis of MBP, GAP-43 and MAP-2 optical
density in the ischemic striatum of saline- or NEP1-40-treated
rats, four non-continuous brain slides from each brain were
obtained. For each slide, four high-power fields under ×100
magnification were randomly selected. MetaMorph software was used
for the quantification of average intensity in the region of the
ischemic striatum. The average intensity was defined as the
difference between the average gray value of a chosen field within
the ischemic striatum and its background (24).
Western blotting
The ischemic striatum of the brain was removed and
quickly placed into −196°C liquid nitrogen. Frozen tissues were
homogenized in homogenization buffer (50 mmol/l−1 Tris
base, 2 mmol/l−1 EDTA, 40 mmol/l−1 NaF, 1
mmol/l−1 phenylmethylsulfonyl fluoride). The
concentration of protein was quantified using the bicinchoninic
acid assay kit. Proteins (60 µg) were separated via 10% SDS-PAGE
gel and transferred onto a PVDF membrane. The membranes were
blocked with 5% non-fat milk for 1 h at 25°C and then incubated
with a rabbit polyclonal Nogo-A antibody (1:2,000; catalog no.
AB5664P; Chemicon International; Thermo Fisher Scientific, Inc.)
overnight at 4°C. After washing with TBST (0.05% Tween-20), the
membranes were incubated with a goat anti-mouse or goat anti-rabbit
IgG secondary antibody at 4°C (1:1,000; catalog nos. sc-2005 or
sc-2004; Santa Cruz Biotechnology, Inc.) for 1 h and then detected
with ECL reagent (Santa Cruz Biotechnology, Inc.). The blots were
then exposed to X-ray sensitive films for 1–5 min. Mouse anti-rat
β-actin primary antibody (1:8,000; sc-47778; Santa Cruz
Biotechnology, Inc.) served as the loading control.
Blots were scanned with Scanwizard (version 5.0;
ScanWizard, Inc.) and band densities (Nogo-A and β-action) were
measured using TotalLab software (version 1.0; Leica Microsystems
GmbH). Values for Nogo-A were standardized based on the intensity
of β-actin and expressed as the fold of control.
Neurological behavior assessment
Animal health was determined every day throughout
the experiment. The rats were subjected to neurological tests at
days 1, 3, 7, 14 and 28 following MCAO and treatment with saline or
NEP1-40. Neurological deficit scores were analyzed as described
previously (25), using modified
neurological severity score (mNSS). mNSS is a composite of motor,
sensory, reflex and balance tests. Neurological function was graded
on a scale of 0–18 (normal, 0; maximal deficit score, 18). The
higher the score, the more severe the injury. All experiments were
conducted in a blinded manner.
Statistical analysis
The experiments were repeated three times. Data are
expressed as the mean ± SEM. Statistical analyses were performed
using GraphPad Prism 5.0 (GraphPad Software, Inc.). The data
presented in Fig. 1,Fig. 2,Fig.
3 were analyzed using one-way ANOVA followed by Dunnett's post
hoc test. The data presented in Figs.
4 and 5 were analyzed using
one-way ANOVA followed by Tukey's post hoc test or two-way ANOVA
followed by Tukey's post hoc test. The data presented in Fig. 6 were analyzed using one-way ANOVA
followed by Tukey's post hoc test or mixed two-way ANOVA followed
by Tukey's post hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
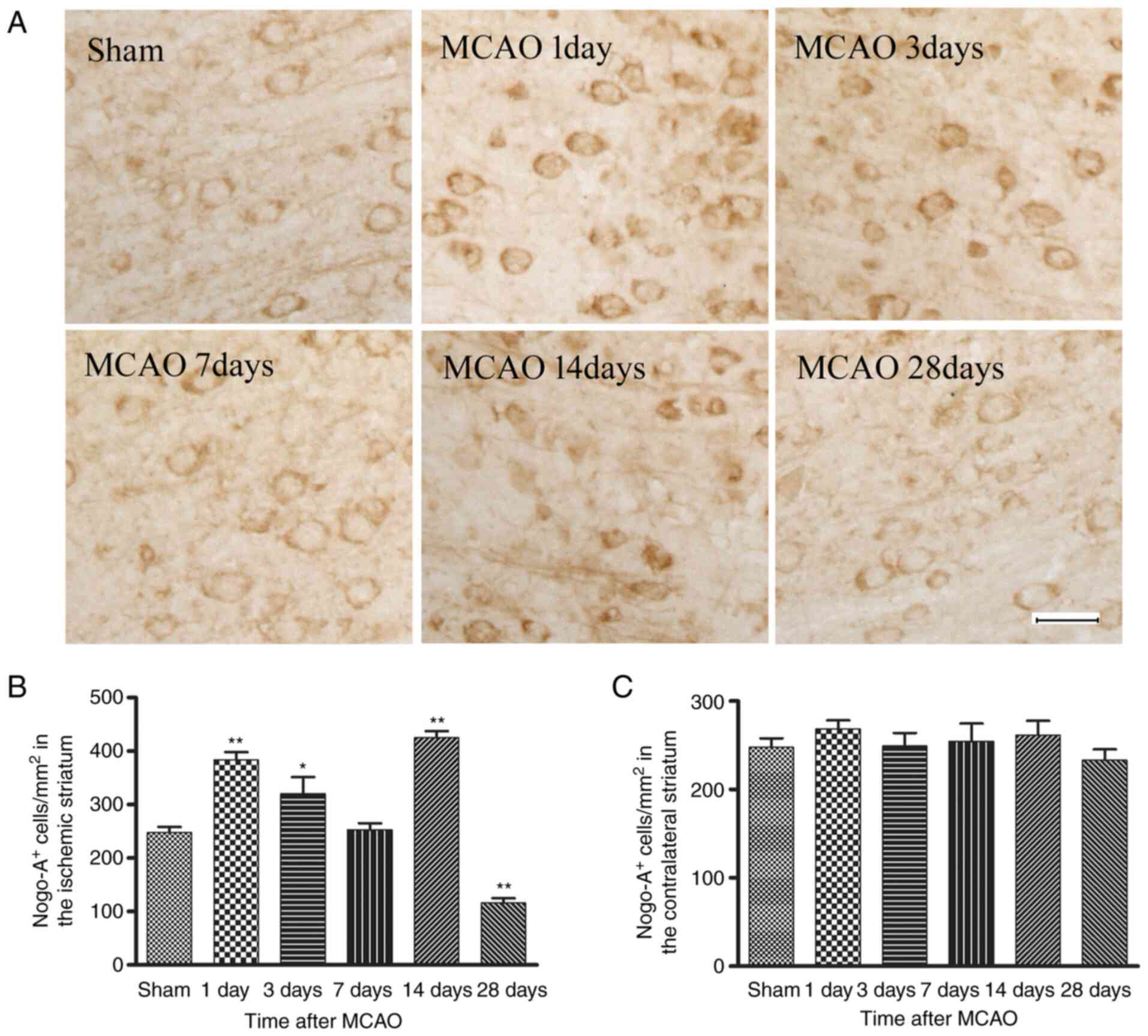
Time course of Nogo-A immunoreactivity
after MCAO
A representative example of immunostaining for
Nogo-A immunoreactivity in the right striatum of the sham-operation
group (control) and on days 1, 3, 7, 14 and 28 after MCAO is
presented in Fig. 1A. The Nogo-A
immunoreactivity was located primarily in the cytoplasm of
oligodendrocytes (small cell bodies and few processes) and observed
predominantly in white matter tracts, typically found in parallel
rows. Compared with the sham group, the number of Nogo-A
immunoreactive cells was significantly elevated from day 1 to 3
post-stroke, then nearly returned to the control level at day 7,
significantly increased again at day 14, but then significantly
decreased at day 28 in MCAO model rats (Fig. 1B). However, there was no
significant change in the number of Nogo-A immunopositive
oligodendrocytes between sham and MCAO model rats in the
contralateral striatum at any time point (Fig. 1C), which suggested that the
greatest inhibitory effect of Nogo-A may be exerted in the white
matter areas immediately adjacent to the lesion.
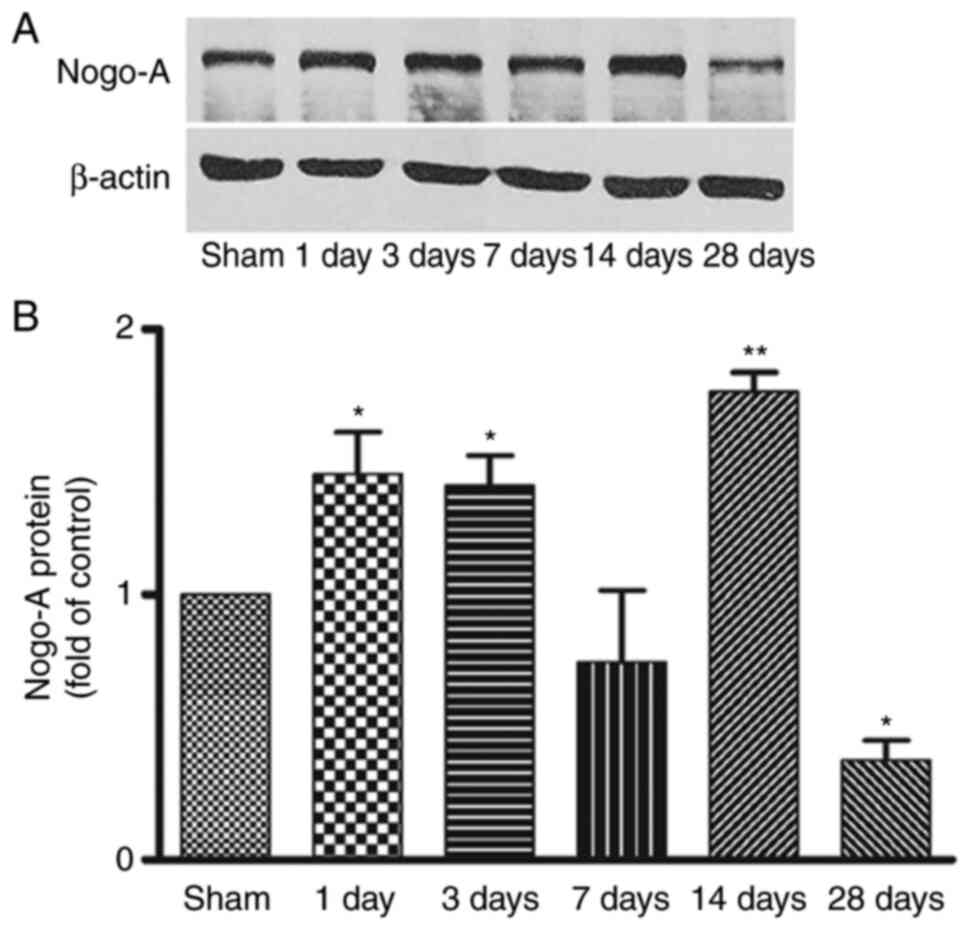
Western blot analysis of Nogo-A
expression in the striatum after MCAO
Representative bands of Nogo-A protein in the
ischemic striatum from day 1 to day 28 after focal cerebral
ischemia and the corresponding β-actin bands are shown in Fig. 2A. Consistent with the
immunohistochemistry findings, compared with the sham group, Nogo-A
expression was significantly increased from day 1 to day 3
following focal ischemia, returned to basal levels at day 7, then
significantly elevated again at day 14 and significantly decreased
at day 28 (Fig. 2B). This
indicated that changes in Nogo-A expression were time-dependent
following ischemia. However, no difference of Nogo-A protein was
detected in the contralateral striatum at any time point (data not
shown).
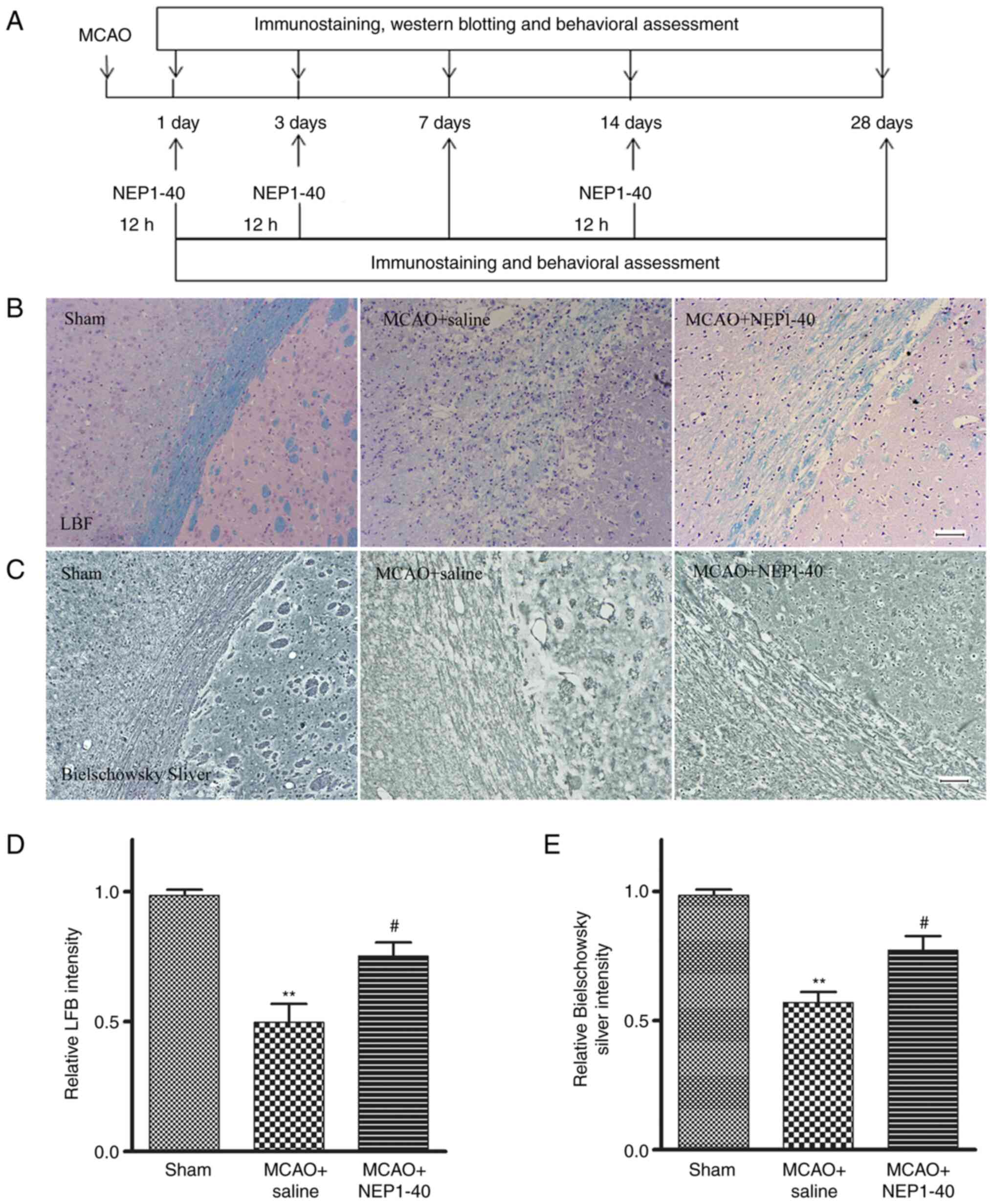
Effects of NEP1-40 on myelin repair in
MCAO model rats
To investigate the effect of Nogo-A inhibitor
NEP1-40 on the extent of demyelination in the white matter of MCAO
model rats, LFB, Bielschowsky silver staining and MBP
immunohistochemistry were performed to assess the post-ischemic
densities of myelinated axons in the striatum (Fig. 3A). LFB immunostaining was
performed to assess the loss of myelin in sham, saline-treated and
NEP1-40-treated rats at day 7 after MCAO (Fig. 3B and D). In the sham group, myelin
exhibited a highly organized and compact appearance. In the MCAO +
saline group, brain tissues exhibited a loose structure with
disrupted morphology, and the staining intensity was significantly
reduced compared with that in the sham group. In the MCAO + NEP1-40
group, the morphological changes were reduced compared with those
in the saline-treated rats, whereas the staining intensity was
significantly increased compared with that in the MCAO + saline
group.
Bielschowsky silver is a marker for axons. Enhanced
Bielschowsky silver staining has been correlated with axonal
regrowth in rodent brains following MCAO (22). Thus, Bielschowsky silver staining
was performed to assess axonal loss (Fig. 3C and E). In the sham group, the
nerve fibers around the striatum were arranged closely and orderly.
In the MCAO + saline group, the nerve fibers were disordered,
suggesting there was white matter damage, and the staining
intensity was significantly reduced compared with that in the sham
group. NEP1-40 treatment inhibited these MCAO-induced effects.
These data indicated that NEP1-40 treatment significantly reduced
the extent of both myelin and axon loss after MCAO.
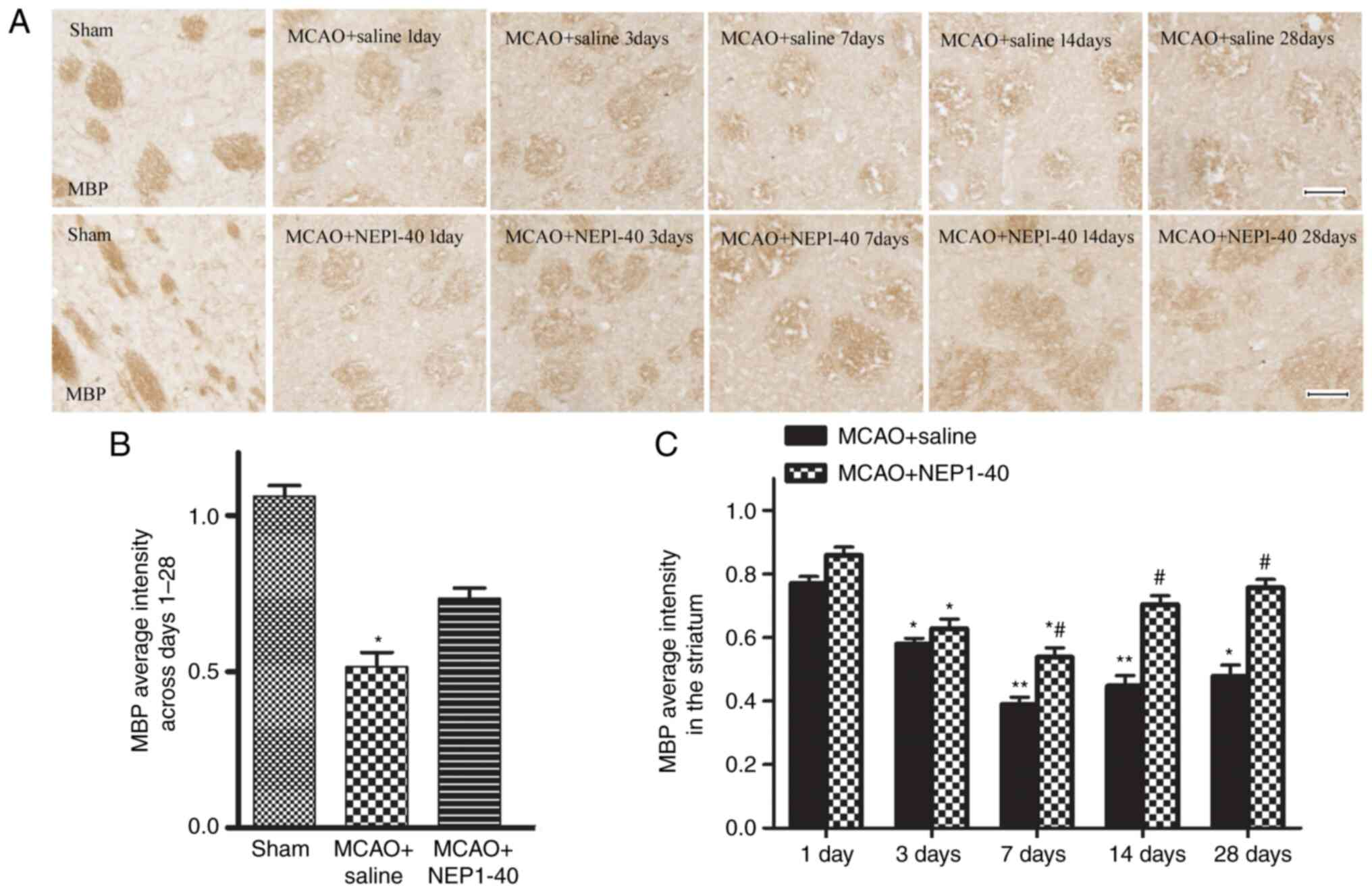
MBP, a major constituent of CNS myelin, has been
used as a marker of post-ischemic white matter injury and repair
(1). Accordingly, demyelination
was also observed in the same region of white matter by
immunostaining evaluation of MBP in the present study.
Representative images of the time course of MBP staining in
affected striatal tissues of rats in the MCAO + saline and MCAO +
NEP1-40 groups are shown in Fig. 4A
and B. A number of myelinated fibers, which were detected with
the anti-MBP antibody, were clearly visible in the cerebral
striatum of the sham group and each fiber tract could be easily
followed. By contrast, these fibers became obscure and spaces
between fiber tracts appeared during reperfusion following MCAO.
Following NEP1-40 administration, the extent of myelin damage was
slightly recovered in the ischemic area. The ratio of MBP optical
density on the ischemic side was also analyzed (Fig. 4C). The average intensity of MBP
immunoreactivity in the MCAO + saline group gradually decreased
from day 1 to day 7, and marginally recovered at days 14 and 28.
However, the difference between days 14 and 28 was not
statistically significant. In addition, MBP expression was
significantly restored by NEP1-40 treatment at days 7, 14 and 28
after MCAO compared with the MCAO + saline group. These results
demonstrated that the myelin sheaths progressively deteriorated
after ischemia, and blocking Nogo-A activity partially enhanced the
extent or efficiency of endogenous remyelination.
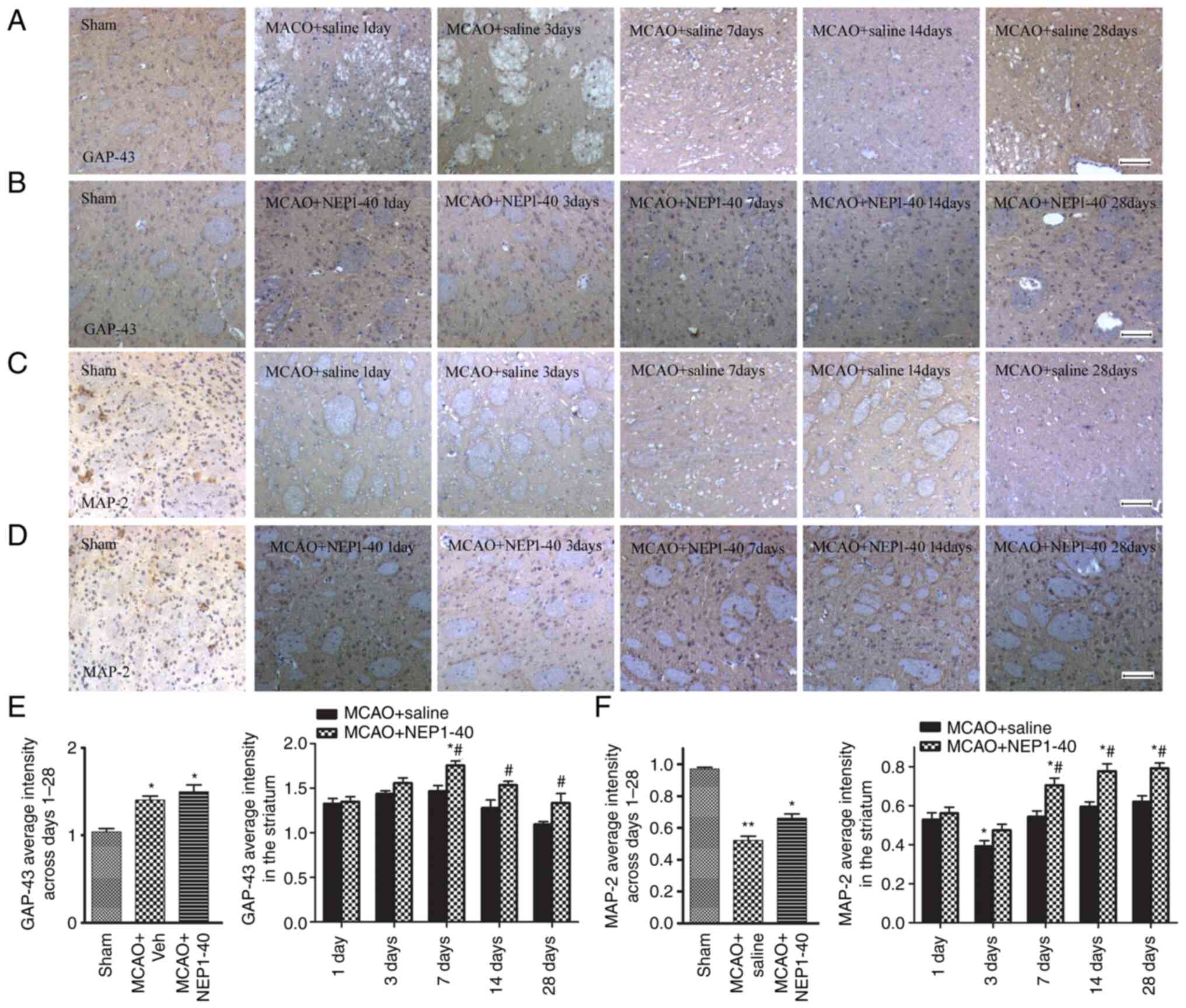
Effects of NEP1-40 on GAP-43 and MAP-2
expression after MCAO
To further confirm a possible mechanism underlying
the remyelinated effect mediated by Nogo-A inhibition, the
expression levels of GAP-43 and MAP-2, which are molecular
indicators of axonal plasticity in the CNS, were determined
(13,17). GAP-43, an important promoter of
axonal outgrowth (14), was
detected via immunohistochemistry. Representative images of GAP-43
staining in the striatum of the MCAO + saline and MCAO + NEP1-40
groups are shown in Fig. 5A and
B. In the sham group, GAP-43+ staining was brown and
present in the axons and cytoplasm of neurons. After MCAO,
GAP-43+ staining was observed in the ipsilateral
striatum. The increase of GAP-43+ staining was markedly
pronounced from day 1 to day 14, and declined to a baseline level
at day 28 after ischemia (Fig.
5E). Treatment with NEP1-40 further increased the expression of
GAP-43 at days 7, 14 and 28 after MCAO. These data indicated that
the increase in GAP-43 expression may be closely related to injury
repair and NEP1-40 may enhance the regenerative response in the
damaged area.
MAP-2 is primarily associated with microtubules in
the dendrite structure and is a sensitive marker of neuronal
dysfunction (19). Thus, the
expression of MAP-2 in the striatum of the ipsilateral hemisphere
in saline- and NEP1-40-treated MCAO model rats was also examined by
immunohistochemical staining (Fig. 5C
and D). MAP-2 immunoreactivity stained brown was located
exclusively in both the soma and dendrites of the neuron. After
MCAO, distorted and disrupted axons alone with extensive loss of
MAP-2 immunoreactivity in the striatum were observed, indicating
that MAP-2 was vulnerable to ischemic injury. Quantification of the
MAP-2 average intensity was also analyzed (Fig. 5F). MAP-2 showed a progressive
decrease from day 1 to day 3 after MCAO. After NEP1-40
administration, the decreased level of MAP-2 was increased at days
7, 14 and 28 compared with the levels observed in the
saline-treated group, but still remained lower compared with those
in the sham group. These results suggested that Nogo-A inhibition
prevented the loss of MAP-2+ dendrites and decreased
neuronal damage.
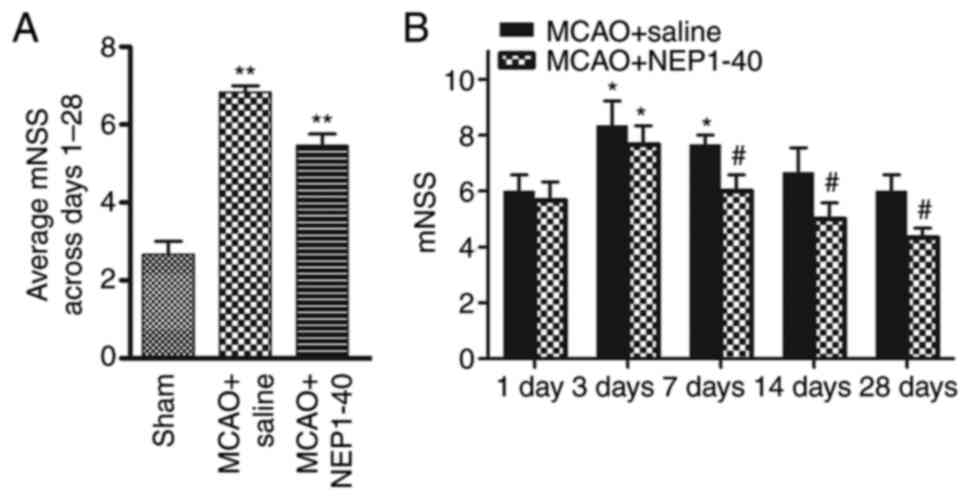
Administration of NEP1-40 attenuates
neurological deficits following ischemic stroke
To examine the effect of NEP1-40 treatment on
behavioral function, the saline- and NEP1-40-treated MCAO model
rats were examined using behavioral tests. After NEP1-40 treatment
in the MCAO model rats, neurological performance was evaluated
using mNSS at days 1, 3, 7, 14 and 28 after MCAO (Fig. 6). The behavior test showed that
the neurological score in the saline-treated group was
significantly increased compared with that in the sham group. In
addition, in the MCAO + NEP1-40 group, neurological deficits were
significantly reduced at days 7, 14 and 28 compared with those in
the MCAO + saline group.
Discussion
Nogo-A is a major factor expressed in adult CNS
myelin that mediates the inhibition of neurite regrowth and axonal
regeneration (3). It has been
reported that Nogo-A binds to the Ng-R to activate intracellular
RhoA signaling pathways, resulting in growth cone collapse
(26,27). In the present study, it was
observed that Nogo-A+ cells were distributed throughout
the white matter tract and primarily located in the cytoplasm of
cells with oligodendroglial morphology, which was in agreement with
a previous study (9). The
localization of Nogo-A in oligodendrocytes was consistent with its
role as a myelin-associated inhibitor of regenerative fiber growth
and structural plasticity.
Next, a systematical observation regarding Nogo-A
protein after MCAO was established via immunostaining and western
blotting. Nogo-A expression began to increase at day 1 after MCAO,
returning to the normal level at day 7. The alterations in Nogo-A
expression during the first 7 days following ischemia were
consistent with a previous study (8), but the present study did not observe
the changes over a long-term period. The finding that Nogo-A
expression increased at the early stages of ischemia suggested that
it may serve an important role in preventing regeneration
immediately following injury. In addition, the expression of Nogo-A
increased again at day 14 after ischemia. It was hypothesized in
the current study that this upregulation may prevent excessive axon
regeneration, in particular oligodendrocyte proliferation, in
response to injury. The expression of Nogo-A significantly
decreased at day 28, leading to speculation that Nogo-A may be
involved in the regulation of structural plasticity at the late
phase of ischemia.
Increased Nogo-A expression after ischemic brain
lesions can inhibit neurite outgrowth and fibroblast spouting
(11), therefore treatment
targeting Nogo-A could provide novel therapeutic strategies for
nerve regeneration and repair. In the present study, it was found
that MBP immunoreactivity staining significantly decreased across
days 1–28 following ischemia, indicating that myelin loss occurred
at an early time point (day 1) and advanced during ischemia. When
the Nogo-A inhibitor was administered following MCAO, it was found
that MBP expression significantly increased at day 7 to day 28
after MCAO, suggesting that NEP1-40 may promote remyelination and
this protective effect occurred early at day 7 following ischemia.
A previous study conducted by Zhu et al (28) also supported this result in
ischemic neonatal rats. Other similar studies also showed that
antibodies against Nogo-A (IN-1) and treatment with NEP1-40
resulted in the formation of new axonal connections and functional
recovery (29). Nogo-A
neutralization has been shown to improve behavioral outcomes and
reduce neuronal damage in stroke model rats (30). Taken together, these results
suggested that Nogo-A is a potent inhibitor of neurite growth
involved in regenerative failure, and neutralization of Nogo-A may
allow for the generation of nerve growth and enhance structural
reorganization in regions surrounding the lesions. However, the
underlying molecular mechanism remains to be elucidated. After
cerebral ischemia, post-acute brain plasticity evolved as an
important mechanism to maintain brain function (31,32). To investigate whether the enhanced
regenerative processes mediated by Nogo-A inhibition were
associated with post-traumatic plasticity, the effect of the myelin
inhibitor NEP1-40 on MAP-2 and GAP-43, which are key markers of
neuronal plasticity proteins (13,17), were determined.
GAP-43 is an intracellular membrane-associated
phosphoprotein that is closely related to synaptic plasticity,
axonal regeneration and neural sprouting (33). Studies have shown that in the
early stage of nervous system development, GAP-43 content is
increased in the neuronal cytoplasm (33,34). As the brain develops, GAP-43
expression gradually decreases; however, when brain injury occurs,
its expression increases again until brain injury is repaired
(14,35–36). Therefore, GAP-43 is regarded as an
important marker for investigating nerve growth, development and
repair. The findings of the current study revealed that
GAP-43+ cells stained brown were present in the
cytoplasm of neurons and were expressed at low levels in the brains
of the sham group. After MCAO, the expression of GAP-43 in the
ischemic striatum continuously increased from day 1 to day 14, then
decreased to a baseline level at day 28. This observation coincided
with a previous finding that the expression of GAP-43 in rat
hippocampal neurons increases at the early phase of hypoxia
(37). Furthermore, changes in
the expression of GAP-43 have been found to be consistent with
synapse formation over time (38), suggesting that GAP-43 may be
involved in events such as axonal outgrowth and the process of
myelin repair. Additional previous studies have found further
evidence for the notable role of GAP-43 in plasticity by
demonstrating that increased GAP-43 synthesis is correlated with
axonal remodeling and behavioral recovery (39–41). Additionally, in transgenic mice,
overexpression of GAP-43 has been observed to lead to the
spontaneous formation of new synapses and enhanced neuronal
sprouting (42). The elevated
expression of GAP-43 caused by ischemia in the early phase of MCAO
may restore a microenvironment permissive for neuronal survival. As
the time following injury increases, GAP-43 expression gradually
decreases in the injured area, leading to the speculation that
restored oligodendrocytes, decreased injured axons and myelin
debris at the late stage of ischemia may in turn reduce GAP-43
expression. These findings support the fact that GAP-43 expression
occurs during periods of accelerated spontaneous recovery and
returns to baseline as recovery plateaus (38). Although ischemic injury can
enhance GAP-43 expression to initiate a self-protection
compensatory mechanism to a certain extent, this effect fails to
fully repair the damaged myelin. Notably, in the present study
GAP-43 expression significantly increased on days 1, 3, 7, 14 and
28 after NEP1-40 administration compared with that of
saline-treated rats, which suggested that Nogo-A inhibition may
have marked effects on white matter plasticity, and the repair of
plasticity damage may be a novel mechanism of regeneration.
MAP-2 is a static structural protein, localized in
neuronal somata and dendrites, that is necessary along with other
cytoskeletal proteins to maintain neuroarchitecture (43,44). Previous investigations revealed
dynamic functions for MAP-2 in the plasticity of neurons and
synaptic activity. MAP-2 has been shown to be vulnerable to
ischemic injury, and it is primarily speculated to be a specific
marker of regeneration of axons and dendrites (45–49). In the present study, MAP-2
immunoreactivity was widely localized in the soma and dendrites of
neurons, which had a smooth, regular appearance in the
sham-operated control rats. After ischemia, the pattern of white
matter in the MAP-2-stained sections showed a rough, globular
appearance, indicating that ischemia induced damage to myelinated
fiber tracts. The expression of MAP-2 was clearly reduced at day 1,
reached a minimum at day 3 and slightly increased from day 7 to day
28, although the differences between days 7, 14 and 28 were not
statistically significant. The loss of MAP-2 immunostaining during
the early initial phase of cerebral ischemia indicated the collapse
of cytoskeletal proteins and axonal disconnection. Despite the late
partial increase in MAP-2, the expression failed to reach the
levels observed in the sham group, suggesting that the intrinsic
recovery mechanism was limited. NEP1-40 administration increased
the expression of MAP-2+ dendrites in the damaged area
and rescued neuronal survival. Increased and sustained MAP-2
expression may be an additional mechanism of overcoming myelin
inhibition of axonal outgrowth. It is highly possible that the
adaptive nature of the CNS environment mediated by Nogo-A
inhibition may heighten structural plasticity and facilitate the
process of axonal regeneration to some extent. However, the
mechanism by which it changes in the MCAO model rat remains
unknown.
In conclusion, the present data demonstrated that
Nogo-A was localized in CNS myelin in a manner that was consistent
with its described function as a neurite growth inhibitor.
Moreover, the alterations in Nogo-A expression at different time
points after MCAO and its potential role in regeneration were
explored. Finally, it was revealed that NEP1-40 treatment at
relevant time points post-injury may contribute to recovery and
repair, most likely through compensatory increased levels of GAP-43
and MAP-2, which are speculated to be associated with brain
plasticity. However, the precise mechanisms underlying the
beneficial effects of Nogo-A inhibition in the presented
experimental stroke model require further exploration.
Acknowledgements
The authors would like to thank Dr Yue Lu at the
Peking University School of Nursing (Beijing, China) for their
technical assistance.
Funding
This work was supported by grants from The National
Natural Science Foundation of China (grant nos. 30570626, 81371355
and 81671191), The Beijing Natural Science Foundation (grant no.
7082028) and The Liaoning Revitalization Talents Program (grant no.
XLYC 1807083).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HZ and YBZ conceived and designed the study, and
finalized the manuscript. ZDL, XYG and XFW performed the
experiments. CW and YL contributed to data collection and analysis.
HZ drafted the manuscript. All authors have read and approved the
final manuscript. HZ and ZDL confirm the authenticity of all the
raw data.
Ethics approval and consent to
participate
The animal-related experiments were approved by the
Animal Welfare Committee of Dalian Medical University (Dalian,
China) and followed the guidelines for Animal Care and Use adapted
from the National Institutes of Health.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Filbin MT: Myelin-associated inhibitors of
axonal regeneration in the adult mammalian CNS. Nat Rev Neurosc.
4:703–713. 2003. View
Article : Google Scholar
|
|
2
|
Baldwin KT and Giger RJ: Insights into the
physiological role of CNS regeneration inhibitors. Front Mol
Neurosci. 8:232015. View Article : Google Scholar
|
|
3
|
Chen MS, Huber AB, van der Haar ME, Frank
M, Schnell L, Spillmann AA, Christ F and Schwab ME: Nogo-A is a
myelin-associated neurite outgrowth inhibitor and an antigen for
monoclonal antibody IN-1. Nature. 403:434–439. 2000. View Article : Google Scholar
|
|
4
|
Liu H, Ng CE and Tang BL: Nogo-A
expression in mouse central nervous system neurons. Neurosci Lett.
328:257–260. 2002. View Article : Google Scholar
|
|
5
|
Jin WL, Liu YY, Liu HL, Yang H, Wang Y,
Jiao XY and Ju G: Intraneuronal localization of Nogo-A in the rat.
J Comp Neurol. 458:1–10. 2003. View Article : Google Scholar
|
|
6
|
Llorens F, Gil V and del Río JA: Emerging
functions of myelin-associated proteins during development,
neuronal plasticity, and neurodegeneration. FASEB J. 25:463–475.
2011. View Article : Google Scholar
|
|
7
|
Huber AB, Weinmann O, Brösamle C, Oertle T
and Schwab ME: Patterns of Nogo mRNA and protein expression in the
developing and adult rat and after CNS lesions. J Neurosci.
22:3553–3567. 2002. View Article : Google Scholar
|
|
8
|
Zhou C, Li Y, Nanda A and Zhang JH: HBO
suppresses Nogo-A, Ng-R, or RhoA expression in the cerebral cortex
after global ischemia. Biochem Biophys Res Commun. 309:368–376.
2003. View Article : Google Scholar
|
|
9
|
Eslamboli A, Grundy RI and Irving EA:
Time-dependent increase in Nogo-A expression after focal cerebral
ischemia in marmoset monkeys. Neurosci Lett. 408:89–93. 2006.
View Article : Google Scholar
|
|
10
|
Fournier AE, GrandPre T and Strittmatter
SM: Identification of a receptor mediating Nogo-66 inhibition of
axonal regeneration. Nature. 409:341–346. 2001. View Article : Google Scholar
|
|
11
|
Otero-Ortega L, Gómez-de Frutos MC,
Laso-García F, Sánchez-Gonzalo A, Martínez-Arroyo A, Díez-Tejedor E
and Gutiérrez-Fernández M: NogoA Neutralization promotes axonal
restoration after white matter injury in subcortical stroke. Sci
Rep. 7:94312017. View Article : Google Scholar
|
|
12
|
Cheatwood JL, Emerick AJ and Kartje GL:
Neuronal plasticity and functional recovery after ischemic stroke.
Top Stroke Rehabil. 15:42–50. 2008. View Article : Google Scholar
|
|
13
|
Benowitz LI and Routtenberg A: GAP-43: An
intrinsic determinant of neuronal development and plasticity.
Trends Neurosci. 20:84–91. 1997. View Article : Google Scholar
|
|
14
|
Frey D, Laux T, Xu L, Schneider C and
Caroni P: Shared and unique roles of CAP23 and GAP43 in actin
regulation, neurite outgrowth, and anatomical plasticity. J Cell
Biol. 149:1443–1454. 2000. View Article : Google Scholar
|
|
15
|
Emery DL, Raghupathi R, Saatman KE,
Fischer I, Grady MS and McIntosh TK: Bilateral growth-related
protein expression suggests a transient increase in regenerative
potential following brain trauma. J Comp Neurol. 424:521–531. 2000.
View Article : Google Scholar
|
|
16
|
Schmidt-Kastner R, Bedard A and Hakim A:
Transient expression of GAP-43 within the hippocampus after global
brain ischemia in rat. Cell Tissue Res. 288:225–238. 1997.
View Article : Google Scholar
|
|
17
|
Matesic DF and Lin RC:
Microtubule-associated protein 2 as an early indicator of
ischemia-induced neurodegeneration in the gerbil forebrain. J
Neurochem. 63:1012–1020. 1994. View Article : Google Scholar
|
|
18
|
Kitagawa K, Matsumoto M, Niinobe M,
Mikoshiba K, Hata R, Ueda H, Handa N, Fukunaga R, Isaka Y, Kimura
K, et al: Microtubule-associated protein 2 as a sensitive marker
for cerebral ischemic damage-immunohistochemical investigation of
dendritic damage. Neuroscience. 31:401–411. 1989. View Article : Google Scholar
|
|
19
|
Johnson GV and Jope RS: The role of
microtubule-associated protein 2 (MAP-2) in neuronal growth,
plasticity, and degeneration. J Neurosci Res. 33:505–512. 1992.
View Article : Google Scholar
|
|
20
|
Zimmermann M: Ethical guidelines for
investigations of experimental pain in conscious animals. Pain.
16:109–110. 1983. View Article : Google Scholar
|
|
21
|
Longa EZ, Weinstein PR, Carlson S and
Cummins R: Reversible middle cerebral artery occlusion without
craniectomy in rats. Stroke. 20:84–91. 1989. View Article : Google Scholar
|
|
22
|
Mao L, Jia J, Zhou X, Xiao Y, Wang Y, Mao
X, Zhen X, Guan Y, Alkayed NJ and Cheng J: Delayed administration
of a PTEN inhibitor BPV improves functional recovery after
experimental stroke. Neuroscience. 231:272–281. 2013. View Article : Google Scholar
|
|
23
|
Tu H, Deng L, Sun Q, Yao L, Han JS and Wan
Y: Hyperpolarization-activated, cyclic nucleotide-gated cation
channels: Roles in the differential electrophysiological properties
of rat primary afferent neurons. J Neurosci Res. 76:713–722. 2004.
View Article : Google Scholar
|
|
24
|
Zhao H, Gao XY, Liu ZH, Lin JW, Wang SP,
Wang DX and Zhang YB: Effects of the transcription factor Olig1 on
the differentiation and remyelination of oligodendrocyte precursor
cells after focal cerebral ischemia in rats. Mol Med Rep.
20:4603–4611. 2019.
|
|
25
|
Mao L, Huang M, Chen SC, Li YN, Xia YP, He
QW, Wang MD, Huang Y, Zheng L and Hu B: Endogenous endothelial
progenitor cells participate in neovascularization via CXCR4/SDF-1
axis and improve outcome after stroke. CNS Neurosci Ther.
20:460–468. 2014. View Article : Google Scholar
|
|
26
|
Huang S, Huang D, Zhao J and Chen L:
Electroacupuncture promotes axonal regeneration in rats with focal
cerebral ischemia through the downregulation of
Nogo-A/NgR/RhoA/ROCK signaling. Exp Ther Med. 14:905–912. 2017.
View Article : Google Scholar
|
|
27
|
Cao Y, Shumsky JS, Sabol MA, Kushner RA,
Strittmatter S, Hamers FP, Lee DH, Rabacchi SA and Murray M:
Nogo-66 receptor antagonist peptide (NEP1-40) administration
promotes functional recovery and axonal growth after lateral
funiculus injury in the adult rat. Neurorehabil Neural Repair.
22:262–278. 2008. View Article : Google Scholar
|
|
28
|
Zhu WW, Ma XL, Guo AL, Zhao HY and Luo HH:
Neuroprotective effects of NEP1-40 and fasudil on Nogo-A expression
in neonatal rats with hypoxic-ischemic brain damage. Genet Mol Res.
10:2987–2995. 2011. View Article : Google Scholar
|
|
29
|
Niederöst B, Oertle T, Fritsche J,
McKinney RA and Bandtlow CE: Nogo-A and myelin-associated
glycoprotein mediate neurite growth inhibition by antagonistic
regulation of RhoA and Rac1. J Neurosci. 22:10368–10376. 2002.
View Article : Google Scholar
|
|
30
|
Papadopoulos CM, Tsai SY, Cheatwood JL,
Bollnow MR, Kolb BE, Schwab ME and Kartje GL: Dendritic plasticity
in the adult rat following middle cerebral artery occlusion and
Nogo-a neutralization. Cereb Cortex. 16:529–536. 2006. View Article : Google Scholar
|
|
31
|
Liu Q, Lv HW, Yang S, He YQ, Ma QR and Liu
J: NEP1-40 alleviates behavioral phenotypes and promote
oligodendrocyte progenitor cell differentiation in the hippocampus
of cuprizone-induced demyelination mouse model. Neurosci Lett.
725:1348722020. View Article : Google Scholar
|
|
32
|
Boghdadi AG, Teo L and Bourne JA: The
involvement of the myelin-associated inhibitors and their receptors
in CNS plasticity and injury. Mol Neurobiol. 55:1831–1846. 2018.
View Article : Google Scholar
|
|
33
|
Jacobson RD, Virag I and Skene JH: A
protein associated with axon growth, GAP43 is widely distributed
and developmentally regulated in rat CNS. J Neurosci. 6:1843–1855.
1986. View Article : Google Scholar
|
|
34
|
Hsu JY, Stein SA and Xu XM: Temporal and
spatial distribution of growth-associated molecules and astroglial
cells in the rat corticospinal tract during development. J Neurosci
Res. 80:330–340. 2005. View Article : Google Scholar
|
|
35
|
Dijk F, Bergen AA and Kamphuis W: GAP-43
expression is upregulated in retinal ganglion cells after
ischemia/reperfusion-induced damage. Exp Eye Res. 84:858–867. 2007.
View Article : Google Scholar
|
|
36
|
Gregersen R, Christensen T, Lehrmann E,
Diemer NH and Finsen B: Focal cerebral ischemia induces increased
myelin basic protein and growth-associated protein-43 gene
transcription in peri-infarct areas in the rat brain. Exp Brain
Res. 138:384–392. 2001. View Article : Google Scholar
|
|
37
|
Gorup D, Bohaček I, Miličević T, Pochet R,
Mitrečić D, Križ J and Gajović S: Increased expression and
colocalization of GAP43 and CASP3 after brain ischemic lesion in
mouse. Neurosci Lett. 597:176–182. 2015. View Article : Google Scholar
|
|
38
|
Zhu X, Wang P, Liu H, Zhan J, Wang J, Li
M, Zeng L and Xu P: Changes and significance of SYP and GAP-43
expression in the hippocampus of CIH rats. Int J Med Sci.
16:394–402. 2019. View Article : Google Scholar
|
|
39
|
Masliah E, Fagan AM, Terry RD, DeTeresa R,
Mallory M and Gage FH: Reactive synaptogenesis assessed by
synaptophysin immunoreactivity is associated with GAP-43 in the
dentate gyrus of the adult rat. Exp Neurol. 113:131–142. 1991.
View Article : Google Scholar
|
|
40
|
Hulsebosch CE, DeWitt DS, Jenkins LW and
Prough DS: Traumatic brain injury in rats results in increased
expression of Gap-43 that correlates with behavioral recovery.
Neurosci Lett. 255:83–86. 1998. View Article : Google Scholar
|
|
41
|
Ceber M, Sener U, Mihmanli A, Kilic U,
Topcu B and Karakas M: The relationship between changes in the
expression of growth associated protein-43 and functional recovery
of the injured inferior alveolar nerve following transection
without repair in adult rats. J Craniomaxillofac Surg.
43:1906–1913. 2015. View Article : Google Scholar
|
|
42
|
Aigner L, Arber S, Kapfhammer JP, Lauz T,
Schneider C, Botteri F, Brenner HR and Caroni P: Overexpression of
the neural growth-associated protein GAP-43 induces nerve sprouting
in the adult nervous system of transgenic mice. Cell. 183:269–278.
1995. View Article : Google Scholar
|
|
43
|
Wiche G: High-Mr microtubule-associated
proteins: Properties and functions. Biochem J. 259:1–12. 1989.
View Article : Google Scholar
|
|
44
|
Dehmelt L and Halpain S: The MAP2/Tau
family of microtubule-associated proteins. Genome Biol. 6:2042005.
View Article : Google Scholar
|
|
45
|
Goodson HV and Jonasson EM: Microtubules
and microtubule-associated proteins. Cold Spring Harb Perspect
Biol. 10:a0226082018. View Article : Google Scholar
|
|
46
|
Rosenstein JM: Diminished expression of
microtubule-associated protein (MAP-2) and beta-tubulin as a
putative marker for ischemic injury in neocortical transplants.
Cell Transplant. 4:83–91. 1995. View Article : Google Scholar
|
|
47
|
Dawson DA and Hallenbeck JM: Acute focal
ischemia-induced alterations in MAP2 immunostaining: Description of
temporal changes and utilization as a marker for volumetric
assessment of acute brain injury. J Cereb Blood Flow Metab.
16:170–174. 1996. View Article : Google Scholar
|
|
48
|
Li Y, Jiang N, Powers C and Chopp M:
Neuronal damage and plasticity identified by microtubule-associated
protein 2, growth-associated protein 43, and cyclin D1
immunoreactivity after focal cerebral ischemia in rats. Stroke.
29:1972–1980; discussion 1980-1. 1998. View Article : Google Scholar
|
|
49
|
Malinak C and Silverstein FS:
Hypoxic-ischemic injury acutely disrupts microtubule-associated
protein 2 immunostaining in neonatal rat brain. Biol Neonate.
69:257–267. 1996. View Article : Google Scholar
|