Introduction
Metastasis-associated protein 1 (MTA1), a
member of the MTA family, was first identified by Toh et al
(1). MTA1 expression is reported
to be associated with tumor malignancy in several cancer types,
including esophageal, gastrointestinal, non-small-cell lung, breast
and ovarian cancer (2–6). In addition, MTA1 promotes tumor cell
proliferation and invasion, and tumor angiogenesis and metastasis
(7–9).
However, the precise functional role of endothelial
MTA1 in angiogenesis remains unclear. Our previous study
examined the function of MTA1 in endothelial cells by
MTA1 knockdown and found that MTA1 small interfering
(si)RNA significantly inhibited tube formation but not the
proliferation and migration of endothelial cells (10). In addition, it was demonstrated
that MTA1 knockdown induced a decrease in S100
calcium-binding protein A4 (S100A4) expression and an increase in
phosphorylated non-muscle myosin heavy chain IIa (NMIIa). This
phosphorylation level of NMIIa may influence the formation of tube
structures via altered cytoskeletal dynamics in endothelial cells
(10). To evaluate the function
of MTA1 further, it is necessary to confirm the inhibition
of tube formation using MTA1-knockout (KO) cells and to
evaluate whether the ability of tube formation is restored by
re-expressing MTA1 in the KO cells.
In the present study, MTA1-KO endothelial
cells (MTA1-KO MSS31 cells) were established and examined to
clarify the role of MTA1 expression in tube formation by
endothelial cells. The relationship between the inhibition of tube
formation in MTA1-KO MSS31 cells and the vascular endothelial
growth factor (VEGF)/VEGF receptor 2 (VEGFR2) pathway was also
investigated.
Materials and methods
Cell culture and reagents
Mouse endothelial cells (MSS31 cells), a gift from
Dr H Endo (The University of Tokyo, Tokyo, Japan), and MTA1-KO
MSS31 cells were maintained in Dulbecco's modified Eagle's medium
(DMEM)/Ham's F-12 medium (Nissui Pharmaceuticals Co., Ltd.)
supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich; Merck
KGaA) at 37°C in a humidified 5% CO2/95% air mixture.
MSS31 cells and MTA1-KO MSS31 cells (3×105) were
transfected with 2 µg MTA1 expression vector or empty vector as a
negative control provided by Dr K Takenaga (Chiba Cancer Center
Research Institute, Chiba, Japan) using Lipofectamine®
2000 (Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol at room temperature for 20 min. The
transfected cells were cultured for 24 h and used in subsequent
experiments. When conducting subsequent experiments, each
experiment had a non-treatment group.
Engineering of guide (g)RNA and
CRISPR-Cas9 vector for MTA1 KO
The gRNA for MTA1 KO targeting the first exon
of MTA1 on the coding strand was engineered using the online
CRISPR design tool (version 1.3) (http://crispr.mit.edu/). The targeted sequence was
5′-CAGGATTGAAGAGCTTAACAAGG-3′. Complementary guide oligonucleotides
(forward, 5′-CACCGCAGGATTGAAGACCTTAACA-3′ and reverse,
5′-AAACTGTTAAGCTCTTCAATCCTGC-3′) were custom synthesized separately
by Sigma-Aldrich (Merck KGaA), annealed and cloned into the
BbsI site of pSpCas9(BB)-2A-GFP (pX458). pX458 was a gift
from Feng Zhang (Addgene; cat. no. 48138; http://n2t.net/addgene:48138; Research Resource
Identifiers (RRID; Addgene_48138) (11).
Electroporation and cell sorting
MSS31 cells were harvested by trypsinization using
0.1% trypsin and counted using a hemacytometer. Thereafter,
1-2×106 cells were resuspended with 3 µg plasmid DNA in
100 ml electroporation buffer, transferred to a 0.2 cm cuvette
(Nepa Gene Co., Ltd.) and electroporated using a square electric
pulse generating electroporator (NEPA21; Nepa Gene Co., Ltd.) with
poring pulse (voltage, 150 V; pulse length, 7.5 msec; pulse
interval, 50 msec; number of pulses, 2; decay rate, 10%; polarity,
+) and transfer pulse (voltage, 20 V; pulse length, 50 msec; pulse
interval, 50 msec; number of pulses, 5; polarity, +/-). The cells
were transferred to DMEM/Ham's F-12 medium (Nissui Pharmaceuticals
Co., Ltd.) supplemented with 10% FBS (Sigma-Aldrich; Merck KGaA)
and seeded in 60 mm dishes. After 24 h of electroporation, the
transfected populations were sorted by GFP expression using MoFlo
XDP cell sorter, equipped with 488 nm blue laser (Beckman Coulter,
Inc.). GFP was excited by 488 nm blue laser and its emission
detected using a 529/28 Band pass (BP) filter. Single cells were
sorted into 384-well microplates and cultured for >1 month to
allow for colony formation.
Genomic DNA isolation, PCR and cloning
for DNA sequencing
Genomic DNA was extracted from cells using the
Gentra Puregene Cell kit (Qiagen GmbH) according to the
manufacturer's instructions and PCR analyses were performed as
follows. The PCR amplification of the targeted regions was
performed with TaKaRa Ex Taq Hot Start (Takara Bio, Inc.) using PCR
gene-specific forward (5′-CTCTCTGGGCTCTGTCCATC-3′) and reverse
(5′-CGGACCCACTCTCAGTCTCT-3′) primers. The following temperature
protocol was used for PCR: 98°C for 1 min; 40 cycles of 98°C for 15
sec, 60°C for 15 sec and 72°C for 40 sec; and 72°C for 7 min. The
PCR reaction mixture contained each 0.25 µM forward and reverse
primer, 1.25 units Ex Taq HS polymerase (Takara Bio, Inc.), 1X Ex
Taq Buffer (Takara Bio, Inc.), 0.2 mM each dNTP mixture (Takara
Bio, Inc.) and 50 ng genomic DNA. PCR products were directly
sequenced using specific primers and cloned into the
pCR™4-TOPO® TA vector (Thermo Fisher Scientific, Inc.)
using the TOPO™ TA Cloning™ Kit for Sequencing, without competent
cells (Thermo Fisher Scientific, Inc.) following the manufacturer's
instructions. Plasmid DNA was isolated using the PureYield™ Plasmid
Miniprep System (Promega Corporation). Plasmids were sequenced
using the M13 forward primer (5′-GTAAAACGACGGCCAG-3′) and M13
reverse primer (5′-CAGGAAACAGCTATGAC-3′).
Tube formation assay
For the tube formation assays, 24-well plates were
coated with Geltrex (Thermo Fisher Scientific, Inc.), which was
allowed to solidify overnight at 4°C. MSS31 cells and MTA1-KO MSS31
cells (8×104 cells/well) were seeded and cultured in
serum-free medium or serum-free medium supplemented with VEGF-A (10
ng/ml; R&D Systems, Inc.). Tube formation was evaluated after
4, 5 or 19 h. Images of two to three random fields for each sample
(magnification, ×4) were captured using an all-in-One Fluorescence
Microscope BZ-X710 microscope (Keyence Corporation). The number of
junctions was quantified using an Angiogenesis Analyzer (developed
by Gilles Carpentier) (https://imagej.nih.gov/ij/macros/toolsets/Angiogenesis%20Analyzer.txt)
in ImageJ [version 1.52v (National Institute of Health)]. Each
experiment was repeated three or four times.
Western blotting
Cells were washed in cold PBS and lysed in lysis
buffer containing 20 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.1% SDS,
1% sodium deoxycholate, 1% Triton X-100, 1 µg/ml aprotinin and 1
µg/ml leupeptin. Lysates were centrifuged at 13,000 × g for 5 min
at 4°C. Protein concentrations were estimated using the Bradford
protein assay (Bio-Rad Laboratories, Inc.) with bovine serum
albumin (Fujifilm Wako Pure Chemical Co.) as the standard. Proteins
were loaded 30 µg per lane and resolved by SDS-PAGE using 8, 10, 12
and 15% gels, and then electrotransferred to PVDF membranes
(MilliporeSigma). After being blocked with 5% fat-free milk for 2 h
at room temperature, the membranes were blotted using primary
antibodies overnight at 4°C, washed using PBS with 1% Tween-20 and
then incubated with secondary antibodies for 20 min at room
temperature. After washing using PBS with 1% Tween-20, the bound
antibodies were detected using ECL Prime Western Blotting Detection
Reagent (RPN2236; Cytiva). The primary antibodies used in the
present study were: Anti-MTA1 polyclonal antibody (1:2,000; cat.
no. ab71153; Abcam), anti-S100A4 polyclonal antibody (1:1,000; cat.
no. 07-2274; MilliporeSigma), anti-NMIIa polyclonal antibody
(1:1,000; cat. no. 3403; Cell Signaling Technology, Inc.),
anti-phosphorylated (p)-NMIIa polyclonal antibody (1:1,000; cat.
no. 5026; Cell Signaling Technology, Inc.), anti-VEGFR2 polyclonal
antibody (1:1,000; cat. no. 9698; Cell Signaling Technology, Inc.),
anti-p-VEGFR2 (Tyr 1175) polyclonal antibody (1:1,000; cat. no.
3770; Cell Signaling Technology, Inc.), anti-β-actin monoclonal
antibody (1:2,000; cat. no. A5441; Sigma-Aldrich; Merck KGaA). The
secondary antibodies used in the present study were: Goat
anti-mouse IgG-HRP (1:2,000; cat. no. PM009-7; Medical and
Biological Laboratories Co., Ltd.) and goat anti-rabbit IgG-HRP
(1:2,000; cat. no. sc-2004; Santa Cruz Biotechnology, Inc.).
Densitometry was performed using ImageJ (version 1.52v; National
Institute of Health). For the western blotting for the
phosphorylation of VEGFR2, wild-type MSS31 cells and MTA1-KO clones
were treated with VEGF (10 ng/ml) for 30 min and were lysed in
lysis buffer. For the western blotting for MTA1 expression by VEGF
treatment, MSS31 cells treated with VEGF (0, 10, 50 or 100 ng/ml)
for 24, 48 or 72 h and were lysed in lysis Buffer. Each experiment
was repeated three or four times.
Reverse transcription-quantitative
(RT-q) PCR
Total RNA was extracted using TRIzol®
reagent (Thermo Fisher Scientific, Inc.) and 1 µg total RNA was
reverse transcribed into cDNA using the TaKaRa PrimeScript RT
master mix (Takara Bio, Inc.) according to the manufacturer's
protocol. Subsequently, 2 µl cDNA was used for quantitative PCR.
qPCR was performed on a Roche LightCycler 480 (Roche Diagnostics)
using SYBR Premix Ex Taq II (Tli RNaseH Plus; Takara Bio, Inc.).
The primer sequences are as follows: MTA1 forward,
5′-GCGGCGAATGAACTGGA-3′ and reverse, 5′-TTGGTTTCTGAGGATGAGAGCA-3′;
and β-actin forward, 5′-AGAGGGAAATCGTGCGTGAC-3′ and reverse,
5′-CAATAGTGATGACCTGGCCGT-3′. The following thermocycling conditions
were used for qPCR: 95°C for 30 sec; 95°C for 10 sec; followed by
45 cycles of 95°C for 10 sec and 60°C for 1 min. β-actin was used
as the internal control. The results are expressed as the fold
change between the expression level of each mRNA and the internal
reference using the 2−ΔΔCq method (12). Each experiment was repeated three
times.
Immunoprecipitation
Cells were washed in PBS and lysed in lysis buffer A
[25 mM Tris-HCl (pH 7.5), 420 mM NaCl, 0.5% Triton X-100, 5 mM
CaCl2, 10 µg/ml aprotinin, 10 µg/ml leupeptin and 1 mM
phenylmethylsulfonyl fluoride (PMSF)] for 30 min at 4°C. Lysates
were diluted 1.8-fold with lysis buffer B [25 mM Tris-HCl (pH 7.5),
0.5% Triton X-100, 5 mM CaCl2, 10 µg/ml aprotinin, 10
µg/ml leupeptin and 1 mM PMSF] and were then centrifuged at 13,000
× g for 10 min at 4°C. Immunoprecipitation assays were performed
using 50 µl of the non-magnetic sepharose beads (nProtein A
Sepharose 4 Fast Flow or nProtein G Sepharose 4 Fast Flow; (cat.
no. 17528001, 17061801; Cytiva) according to the manufacturer's
instructions. Briefly, 500 µl of the supernatants containing
proteins was incubated for 1 h at 4°C with 3 µg anti-MTA1
polyclonal antibody (cat. no. ab71153; Abcam), anti-S100A4
monoclonal antibody (cat. no. CPTC-S100A4-3; Developmental Studies
Hybridoma Bank), normal rabbit IgG (cat. no. 2729; Cell Signaling
Technology, Inc.) or normal mouse IgG (I-8765; Sigma-Aldrich; Merck
KGaA). Immune complexes were recovered on nProtein A Sepharose
beads or nProtein G Sepharose beads for 1 h at 4°C. The
immunoprecipitants were washed five times with TBS [50 mM Tris-HCl
(pH 7.5), 150 mM NaCl] and then 2X additional buffer [1 M Tris-HCl
(pH 6.8), 10% SDS, 100% glycerol, bromophenol blue] was added.
After boiling for 3 min at 95°C, the supernatants were used as
western blotting samples. Western blotting was according to the
aforementioned protocol. The experiment was repeated four
times.
Statistical analysis
The number of samples to be analyzed was
predetermined based on the requirement of the statistical test
used. Statistical analysis was performed using a personal computer
with the program Excel Statistics version 3.0 (Esumi Co. Ltd.)
functioning on the program Excel (Microsoft Corporation). Data are
shown as the mean ± standard deviation. Comparisons between two
groups were analyzed using the unpaired Student's t-test.
Comparisons among multiple groups were analyzed using one-way ANOVA
followed by Dunnett's post hoc test. The researchers involved were
not blinded and the samples were not randomized during sample
collection or data analysis. P<0.05 was considered to indicate a
statistically significant difference.
Results
Engineering of MTA1-KO MSS31
clones
To gain deeper insight into the functions of
MTA1, the present study designed gRNA targeting MTA1
and used the CRISPR-Cas9 system to generate MTA1-KO MSS31 cell
lines. A total of two colonies (#1 and #2) grown from individual
cells were sorted via FACS. These colonies were confirmed as
MTA1 KO by RT-qPCR (data not shown) and western blotting
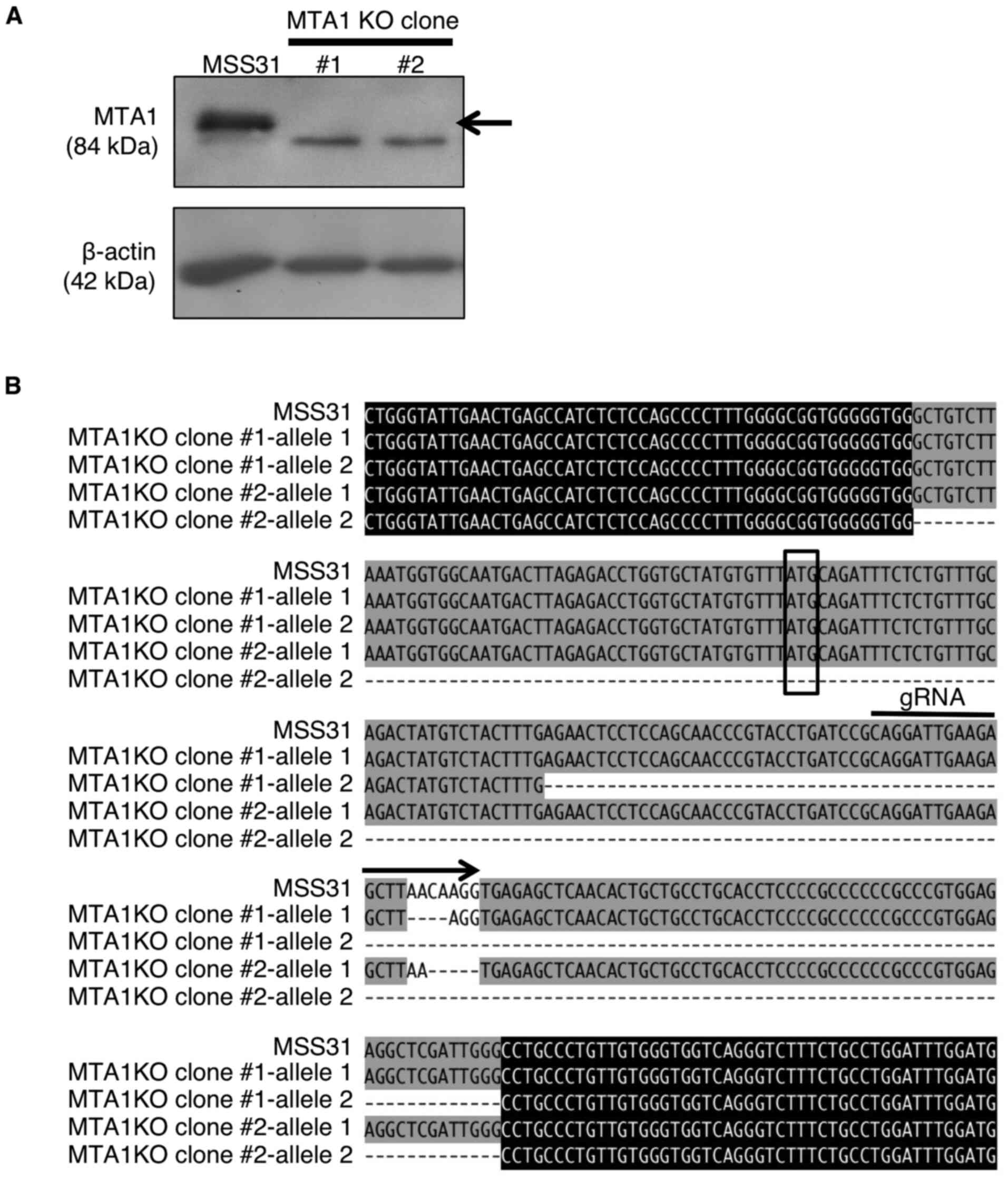
(Fig. 1A). The deletion of
MTA1 in the cellular genome of the two colonies was also
checked by genome sequencing (Fig.
1B). Clone #1 had a four-base deletion in one allele and a
116-base deletion. Clone #2 had a five-base deletion in one allele
and a 201-base deletion. A frameshift mutation due to multiple-base
deletion in each allele resulted in the loss of wild-type MTA1
protein expression. These results indicated the successful
generation of MTA1-KO MSS31 cell clones.
MTA1 expression contributes to tube
formation in vitro
Previously, we reported that suppression of MTA1 in
endothelial cells inhibited tube formation in vitro
(10). The present study
performed a tube formation assay using wild-type MSS31 cells and
two MTA1-KO clones to determine the effect of tube formation in
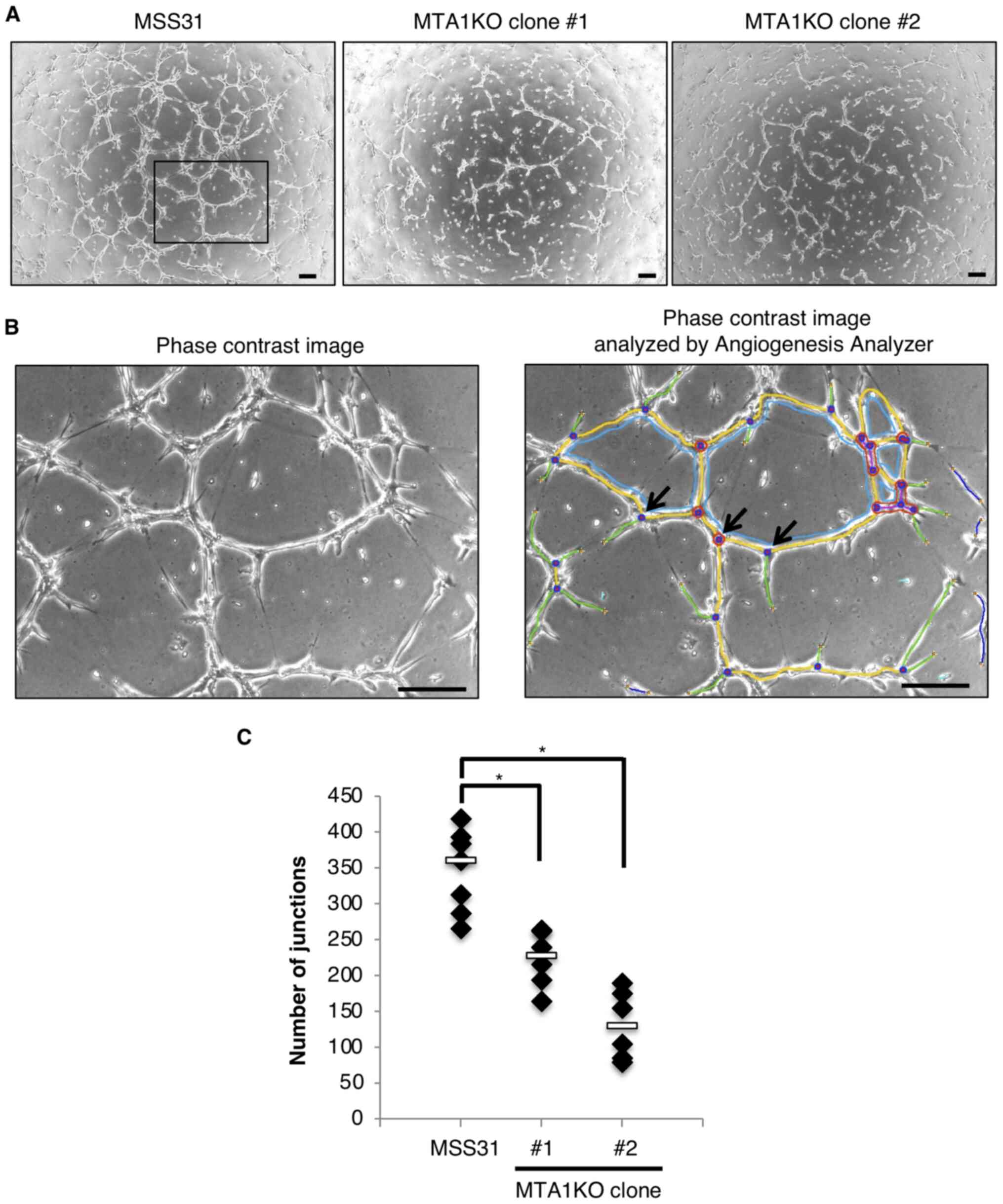
MTA1-KO clones. Images were captured of MSS31 cells and two MTA1-KO
clones at 5 h after the cells were plated onto Geltrex (Fig. 2A) and analyzed using the
Angiogenesis Analyzer. An example of the computer-adjusted image to
evaluate tube formation and junction is shown in Fig. 2B. At 5 h after the cells were
seeded, wild-type MSS31 cells showed tube formation spanning the
entire well. Compared with the wild-type MSS31 cells, the two
MTA1-KO clones showed significantly impaired tube formation
(Fig. 2A and C).
It was also hypothesized that diminished tube
formation in MTA1-KO clones could be recovered by MTA1
overexpression. After MTA1 overexpression in MTA1-KO MSS31 clones
transfected with MTA1 expression vector [MTA1-KO + overexpression
(OE) MSS31 clones] was confirmed by RT-qPCR and western blotting
(Fig. S1A and B), a tube
formation assay was performed using MTA1-KO clones and MTA1-KO + OE
MSS31 clones. At 5 h after the cells were seeded, tube formation
was not observed in the MTA1-KO + OE MSS31 clones (Fig. S2). At 19 h after the cells were
plated onto Geltrex, short and incomplete tube-like structures were
observed in MTA1-KO + OE MSS31 clones; however, no tube formation
was observed in MTA1-KO clones that were transfected with empty
vector or remained untransfected (Fig. 3A and B). These results indicated
that the inhibition of tube formation caused by MTA1 KO was
recovered by MTA1 overexpression.
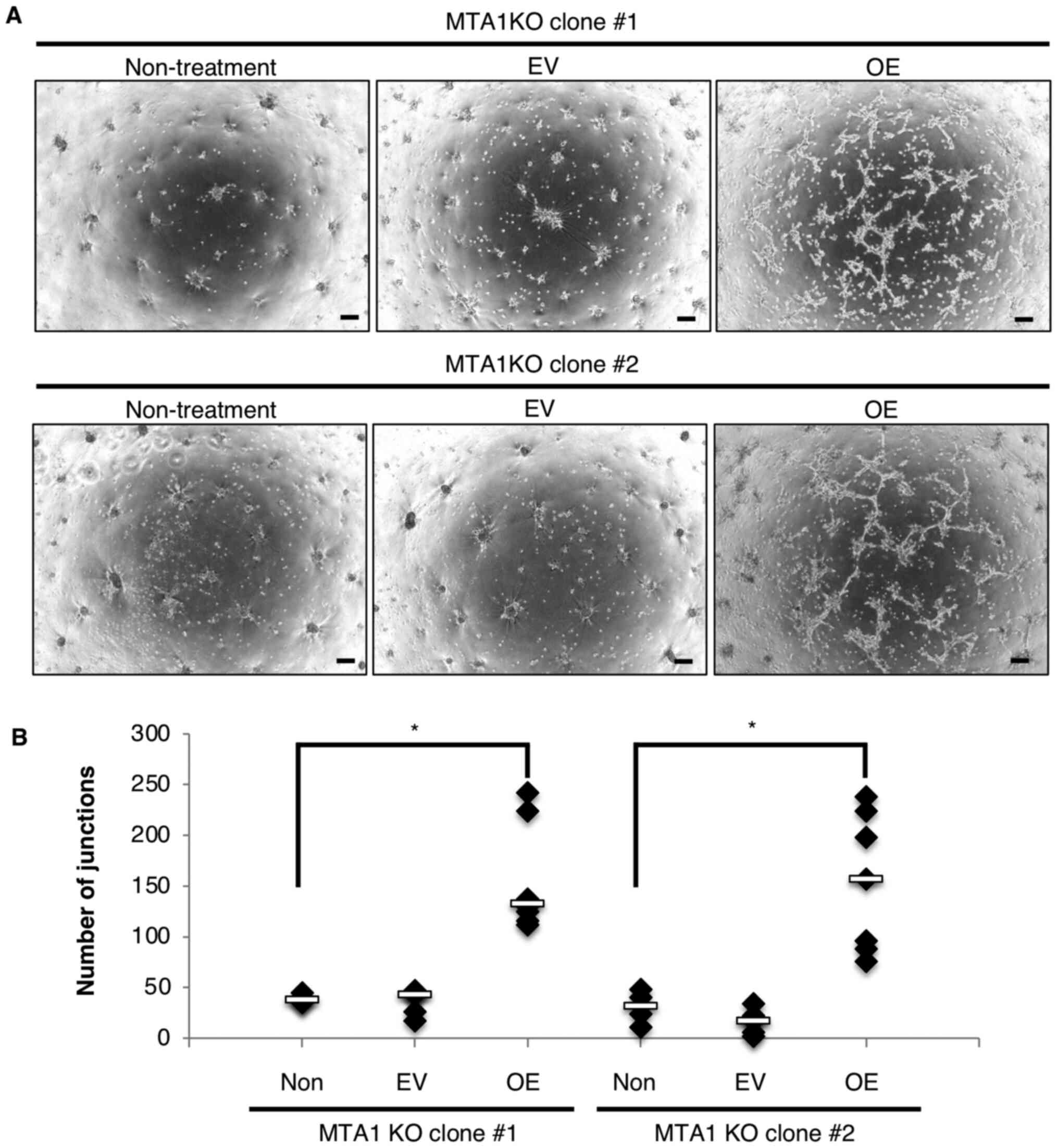 | Figure 3.MTA1-KO MSS31 cells transfected with
MTA1 expression vector (MTA1-KO + OE MSS31 cells) can partly
recover tube formation. (A) Tube formation assays were performed
using MTA1-KO MSS31 cells and MTA1-KO + OE MSS31 clones, and images
were captured after 19 h. Scale bar, 200 µm. (B) Quantification of
the number of junctions in MTA1-KO MSS31 cells (non-treatment),
MTA1-KO cells transfected with EV and MTA1-KO cells transfected
with MTA1 expression vector (MTA1-KO + OE MSS31 cells).
Non-treatment, n=6; EV, n=6; OE, n=8. *P<0.05. MTA1,
metastasis-associated protein 1; KO, knockout; OE, overexpression;
EV, empty vector; non, non-treatment. |
Involvement of MTA1/S100A4 interaction
and p-NMIIa in tube formation in MTA1-KO and MTA1-KO + OE MSS31
cells
We previously reported the mechanism of tube
formation by MTA1, wherein MTA1 interacts with S100A4 and
changes the phosphorylation state of NMIIa (10). Therefore, the present study
examined whether inhibition of tube formation by MTA1 KO and
its restoration by MTA1 overexpression was associated with the
MTA1/S100A4 interaction and the phosphorylation state of NMIIa.
First, the MTA1/S100A4 interaction was found to have disappeared in
MTA1-KO MSS31 cells, but was detected in MTA1-KO + OE MSS31 cells,
as determined via immunoprecipitation using anti-MTA1 or
anti-S100A4 antibodies (Fig. 4A and
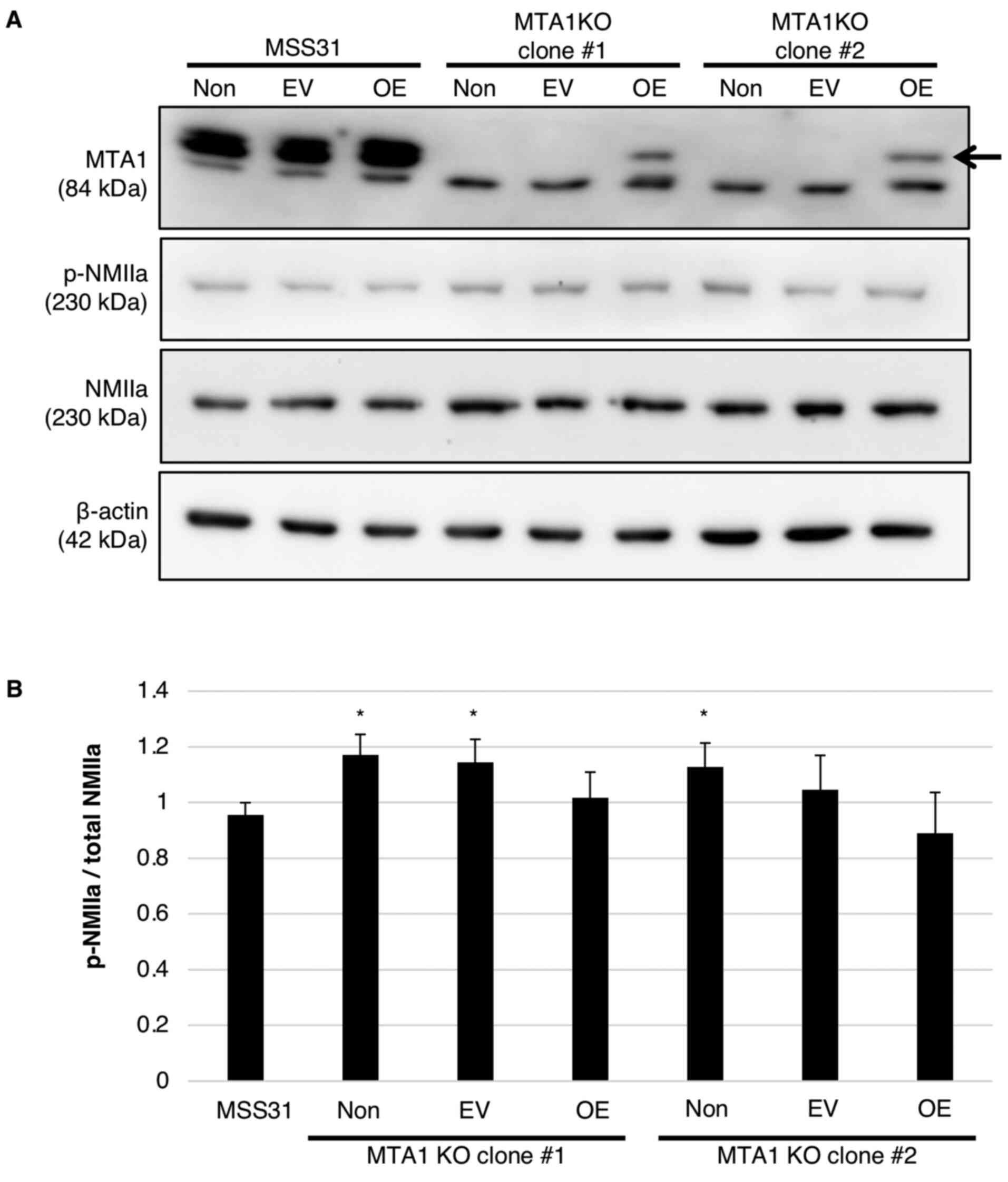
B). Moreover, the phosphorylation of NMIIa was found to be
increased by MTA1 KO compared with wild-type MSS31 cells
(Fig. 5A and B). The increased
NMIIa phosphorylation in MTA1-KO clones was slightly
inhibited by MTA1 overexpression; however, the difference was not
statistically significant (Fig. 5A
and B). Thus, it was proposed that the altered tube formation
abilities of MTA1-KO and -OE cells were caused by
MTA1/S100A4 interaction and NMIIa phosphorylation.
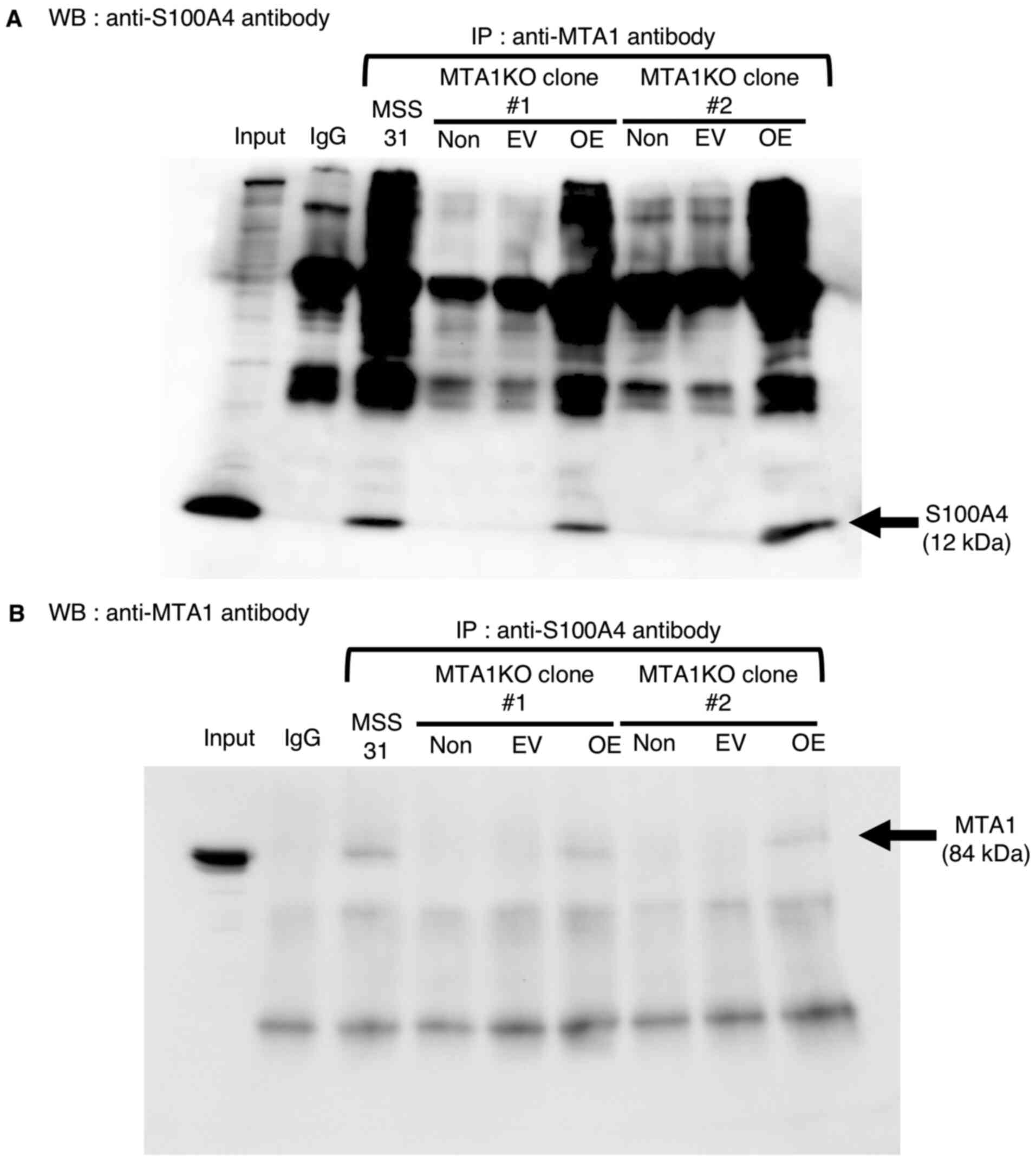 | Figure 4.Formation of the MTA1/S100A4 complex
in MTA1-KO MSS31 cells and MTA1-KO MSS31 cells transfected with the
MTA1 expression vector (MTA1-KO + OE MSS31 cells). (A) WB with
anti-S100A4 antibody after IP with the indicated antibodies using
wild-type MSS31 cells, MTA1-KO MSS31 cells (non-treatment), MTA1-KO
cells transfected with EV and MTA1-KO cells transfected with MTA1
expression vector (MTA1-KO + OE MSS31 cells). Input represents 10%
of the cell lysate. Normal rabbit IgG was used as a negative
control (n=3). (B) WB with anti-MTA1 antibody after IP with the
indicated antibodies using cell lysates. Normal mouse IgG was used
as a negative control (n=4). MTA1, metastasis-associated protein 1;
S100A4, S100 calcium-binding protein A4; KO, knockout; OE,
overexpression; IP, immunoprecipitation; EV, empty vector; non,
non-treatment; WB, western blotting. |
Relationship between MTA1 and VEGF in
MTA1-KO MSS31 cells
Following our previous report that MTA1
knockdown suppressed tube formation (10), the present study revealed that
MTA1 knockout also inhibited tube formation in vitro.
Angiogenesis is influenced by numerous angiogenic pathways,
including the VEGF-VEGFR2 pathway (13–15). Therefore, the present study
examined whether the suppressed tube formation in MTA1-KO MSS31
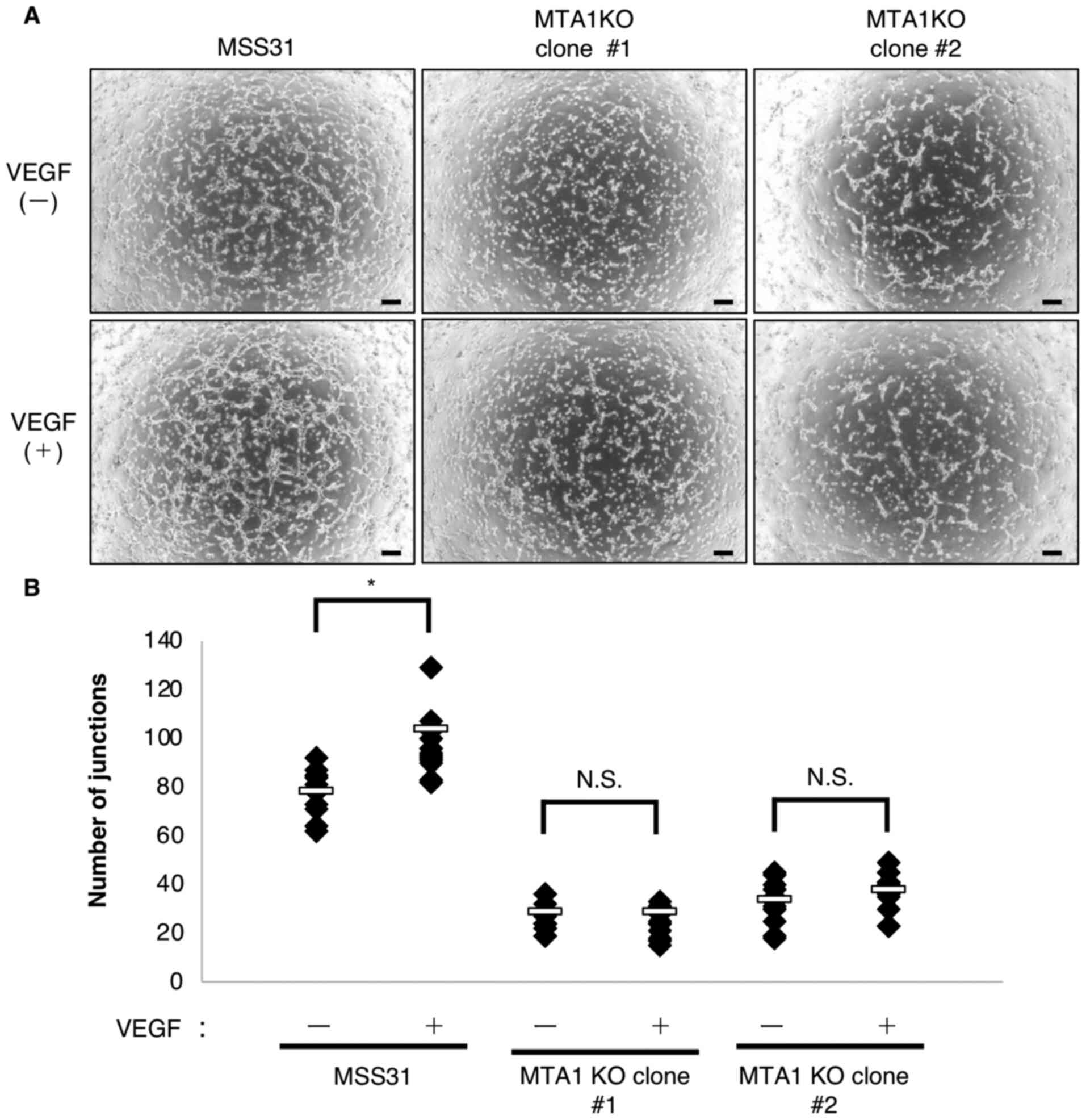
cells could be rescued by stimulation with VEGF. It was first
confirmed that VEGFR2 was phosphorylated following VEGF stimulation
in both wild-type MSS31 cells and MTA1-KO MSS31 cells (Fig. S3A). In addition, VEGFR2
expression levels were not notably altered by MTA1 KO. Tube
formation was promoted in wild-type MSS31 cells treated with VEGF
compared with untreated cells (Figs.
6A, B and S3A). By contrast,
despite the phosphorylation of VEGFR2 by VEGF treatment, tube
formation could not be rescued in MTA1-KO MSS31 cells treated with
VEGF (Figs. 6A, B and S3A). In addition, MTA1 expression was
not affected by VEGF stimulation (Fig. S3B). These results indicated that
VEGF stimulation could not promote tube formation in MTA1-KO MSS31
cells, implying that the role of MTA1 in tube formation may be
independent of VEGF.
Discussion
MTA1 expression in tumor cells is reported to exert
multiple functions associated with cancer progression, including
proliferation, migration, invasion and tumor angiogenesis (7–9).
However, the role of MTA1 in endothelial cells remains to be
elucidated. Our group previously reported that MTA1 is
involved in tumor angiogenesis via the MTA1/S100A4/NMIIa axis in
endothelial cells (10).
Therefore, the present study aimed to confirm the function of
endothelial MTA1 in angiogenesis using MTA1-KO MSS31 cells
generated with the CRISPR-Cas9 system.
The phosphorylation level of NMIIa was decreased in
the MTA1-KO + OE MSS31 cells compared with the corresponding
phosphorylation level in MTA1-KO MSS31 cells and MTA1-KO cells
transfected with an empty vector. Although the protein expression
level of MTA1 in MTA1-KO + OE MSS31 cells was lower than
that in the wild-type MSS31 cells, the interaction between MTA1 and
S100A4 occurred in a similar way to that seen in the wild-type
MSS31 cells. These results indicated that low levels of MTA1 were
sufficient to alter angiogenesis via the interaction with S100A4.
Concerning the association between MTA1, NMIIa and tube formation,
MTA1-KO MSS31 cells showed an increase in NMIIa phosphorylation and
a decrease in tube formation. However, whether the level of the
NMIIa phosphorylation directly affected the ability of tube
formation was not investigated. Therefore, a mutated form of the
phosphorylation site of NMIIa should be generated to investigate
this further.
The present study observed that MTA1-KO MSS31 cells
displayed impaired tube formation at 5 h after seeding cells and
the tube structure disappeared at 19 h. By contrast, MTA1-KO + OE
MSS31 cells partly formed tube-like structures at 19 h, but this
was not observed at 5 h, possibly because the direct use of MTA1
expression vector-transfected cells in the tube formation assay
caused differences in MTA1 expression levels among the MSS31 cells.
These results indicated that endothelial MTA1 may serve an
important role in tube formation and maintenance.
Angiogenesis involves multiple steps, including
protease production, endothelial migration and proliferation,
vascular tube formation and blood vessel maturation, that are
regulated by numerous angiogenic pathways (13,16,17). A number of signaling pathways are
associated with this process, whereby VEGF/VEGFR2 signaling is the
most commonly known and serves as an important key pathway
(14,18,19). Therefore, the present study
examined the association between MTA1 and VEGF in vitro. The
tube formation ability was not recovered by treatment of MTA1-KO
MSS31 cells with VEGF, although the phosphorylation of VEGFR2 was
confirmed in those cells treated with VEGF. These data suggested
that the involvement of the MTA1/S100A4/NMIIa pathway in
angiogenesis was distinct from the regulation by the VEGF/VEGFR2
signaling pathway. However, the proangiogenic factors and pathways,
including the VEGF/VEGFR2 pathway and fibroblast growth factor
(FGF)/FGF receptors pathway, primarily control the survival,
migration and proliferation of vascular endothelial cells in
previous studies (14,15,18).
The present study focused on this difference between
the functions of MTA1 and VEGF in the angiogenic process. The
angiogenic process can be roughly divided into two steps:
Degradation of the basement membrane, proliferation, migration and
sprout formation (step 1); and vascular tube formation and
maturation (step 2; Fig. 7)
(13,16,17). Based on this model, the
VEGF/VEGFR2 signaling pathway controls the first step of the
angiogenic process. Conversely, the MTA1/S100A4/NMIIa axis
primarily regulates tube formation in the second step. In other
words, MTA1 and VEGF are involved in the angiogenesis process, but
the point at which they exert their functions may be different.
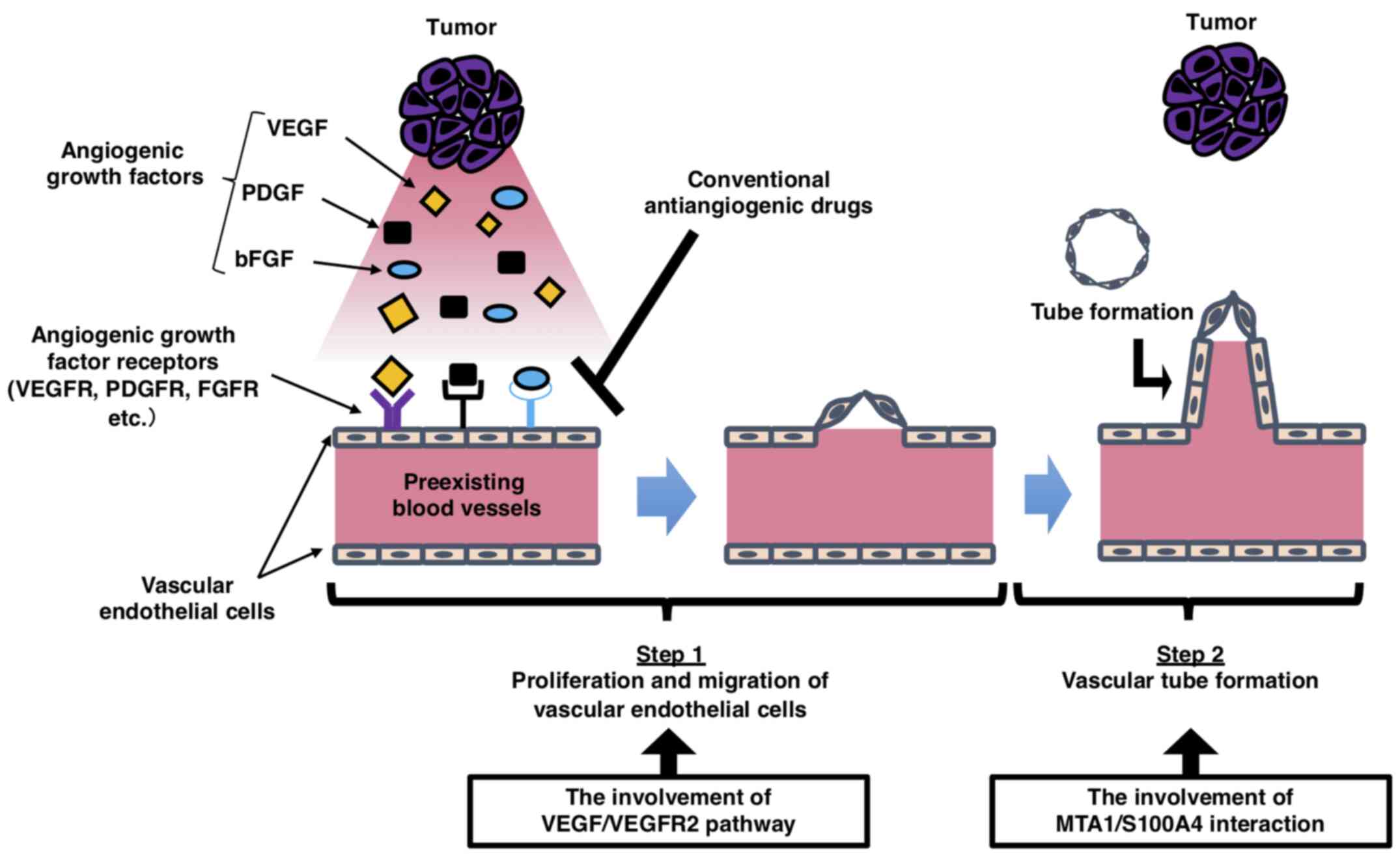 | Figure 7.Schematic diagram of the process of
tumor angiogenesis. New blood vessels are generated from
pre-existing blood vessels through angiogenic growth factors
secreted by tumor cells. The vascular endothelial cells in
pre-existing blood vessels degrade the basement membrane,
proliferate, migrate and form sprouts after the activation of
angiogenic stimuli (step 1). Subsequently, these sprouts lead to
vascular tube formation, followed by blood vessel maturation to
complete the tube structure through which blood can flow (step 2).
VEGF stimulates endothelial proliferation and migration in step 1,
whereas MTA1 is primarily associated with vascular tube formation
in step 2. VEGF, vascular endothelial growth factor; VEGFR, VEGF
receptor 2; PDGF, platelet-derived growth factor; bFGF, basic
fibroblast growth factor; FGFR, FGF receptor; PDGFR,
platelet-derived growth factor receptor; MTA1,
metastasis-associated protein 1; S100A4, S100 calcium-binding
protein A4. |
Tumor angiogenesis is an appealing therapeutic
target (20). Antiangiogenic
therapy has been applied clinically and has emerged as one of the
viable treatment options for cancer (21). Due to the pivotal role of the
VEGF/VEGFR2 pathway in pathological angiogenesis, most approved
antiangiogenic drugs target VEGF-A or its receptors, including
VEGFR2 (22,23). Although these treatment strategies
have provided substantial clinical benefits in cancer therapeutics,
their effects are limited by the development of resistance
(24,25). The present study revealed that the
MTA1/S100A4 axis in endothelial cells may serve as an important
pathway for tube formation and was functionally distinct from the
VEGF/VEGFR2 pathway at the site of action in angiogenesis. Thus,
therapies targeting the endothelial MTA1/S100A4 axis may provide a
novel therapeutic strategy distinct from conventional therapies. In
other words, the axis may serve as a useful target molecule for
treating patients who manifest VEGF-resistance or show poor
responses to VEGF inhibitors because it may be possible to inhibit
angiogenesis by blocking step 2 with or without activation of step
1 (Fig. 7). In addition, the
combination of antiangiogenic drugs targeting the VEGF/VEGFR2
pathway and the MTA1/S100A4 pathway may induce additive or
synergistic effects in the inhibition of tumor angiogenesis and
tumor growth, resulting in improved therapeutic outcomes. These
possibilities, such as the alternative for the treatment of
VEGF-resistance and the enhancement of the antiangiogenic effect
using VEGF inhibitors, could be the benefits of targeting the
MTA1/S100A4 pathway.
However, there are problems with using MTA1 as a
therapeutic target. Since MTA1 forms complexes with other proteins
and regulates a number of genes, such as myeloid differentiation
factor 88 and cryptochrome 1 (26,27), suppressing MTA1 itself using MTA1
inhibitors may have negative effects on other pathways. In
addition, hypertension and impaired wound healing, which are known
side effects of antiangiogenic therapy (14,28), may also occur. Therefore,
suppressing MTA1 function using molecules that specifically act on
the MTA1/S100A4 pathway in antiangiogenic treatment strategies
targeting MTA1 is important.
The present study had several limitations. First,
the investigation of the MTA1/S100A4 interaction was not complete.
The present study reported that this interaction was serial (i.e.,
the two proteins were associated with each other) as it focused on
the formation of the MTA1/S100A4 complex as a mechanism of
angiogenesis. However, the possibilities that this interaction is
serial, parallel (i.e., the two proteins function independently) or
both should be considered. Second, the results are insufficient to
definitively conclude whether MTA1 may be involved in angiogenesis
regulated by the VEGF/VEGFR2 pathway. The present study
demonstrated that the angiogenic process cannot occur normally if
MTA1 is absent in vascular endothelial cells even when the cells
were stimulated by VEGF. With only this result, the question as to
how MTA1 is involved in angiogenesis compared with VEGF cannot be
clearly answered. To answer these points, a screening assay should
be generated to search for the candidate molecules that inhibit the
formation of the MTA1/S100A4 complex. Once the candidate molecules
are determined, it is expected that the MTA1/S100A4 interaction and
the functions of MTA1 and VEGF in angiogenesis could be shown more
clearly by comparing the results of tube formation using MTA1-KO
MSS31 cells, MTA1-S100A4 inhibitors and VEGF inhibitors.
Using MTA1-KO MSS31 cells, the present study not
only reconfirmed that MTA1 expression in endothelial cells served a
crucial role in tube formation and that the mechanism involved the
MTA1/S100A4 axis, but also demonstrated that MTA1 was functionally
distinct from VEGF in its site of action in angiogenesis. These
results were a novel finding and emphasized the importance of MTA1
in angiogenenic process. Based on these findings and the results of
our previous study (10), the
MTA1/S100A4/NMIIa pathway in endothelial cells may serve as a
potential target for suppressing angiogenesis and tumor growth via
a mechanism of action different from that of VEGF inhibitors.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Dr H Endo (The
University of Tokyo, Tokyo, Japan) for kindly providing MSS31 cells
and Dr K. Takenaga (Chiba Cancer Center Research Institute, Chiba,
Japan) for providing the MTA1 expression vector.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MI, MO, NU, TO, HK and FO designed the experiments.
MI, NU, TO and MO performed the experiments and analyzed the data.
MI, MO, NU, TO, HK and FO wrote, reviewed and revised the
manuscript. MI, MO and NU confirm the authenticity of all the raw
data. All authors reviewed and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Toh Y, Pencil SD and Nicolson GL: A novel
candidate metastasis-associated gene, mta1, differentially
expressed in highly metastatic mammary adenocarcinoma cell lines.
cDNA cloning, expression, and protein analyses. J Biol Chem.
269:22958–22963. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Toh Y, Kuwano H, Mori M, Nicolson GL and
Sugimachi K: Overexpression of metastasis-associated MTA1 mRNA in
invasive oesophageal carcinomas. Br J Cancer. 79:1723–1726. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Toh Y, Oki E, Oda S, Tokunaga E, Ohno S,
Maehara Y, Nicolson GL and Sugimachi K: Overexpression of the MTA1
gene in gastrointestinal carcinomas: Correlation with invasion and
metastasis. Int J Cancer. 74:459–463. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhu X, Guo Y, Li X, Ding Y and Chen L:
Metastasis-associated protein 1 nuclear expression is associated
with tumor progression and clinical outcome in patients with
non-small cell lung cancer. J Thorac Oncol. 5:1159–1166. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jang KS, Paik SS, Chung H, Oh YH and Kong
G: MTA1 overexpression correlates significantly with tumor grade
and angiogenesis in human breast cancers. Cancer Sci. 97:374–379.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dannenmann C, Shabani N, Friese K, Jeschke
U, Mylonas I and Brüning A: The metastasis-associated gene MTA1 is
upregulated in advanced ovarian cancer, represses ERbeta, and
enhances expression of oncogenic cytokine GRO. Cancer Biol Ther.
7:1460–1467. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nicolson GL, Nawa A, Toh Y, Taniguchi S,
Nishimori K and Moustafa A: Tumor metastasis-associated human MTA1
gene and its MTA1 protein product: Role in epithelial cancer cell
invasion, proliferation and nuclear regulation. Clin Exp
Metastasis. 20:19–24. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kai L, Wang J, Ivanovic M, Chung YT,
Laskin WB, Schulze-Hoepfner F, Mirochnik Y, Satcher RL Jr and
Levenson AS: Targeting prostate cancer angiogenesis through
metastasis-associated protein 1 (MTA1). Prostate. 71:268–280. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pakala SB, Rayala SK, Wang RA, Ohshiro K,
Mudvari P, Reddy SD, Zheng Y, Pires R, Casimiro S, Pillai MR, et
al: MTA1 promotes STAT3 transcription and pulmonary metastasis in
breast cancer. Cancer Res. 73:3761–3770. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ishikawa M, Osaki M, Yamagishi M, Onuma K,
Ito H, Okada F and Endo H: Correlation of two distinct
metastasis-associated proteins, MTA1 and S100A4, in angiogenesis
for promoting tumor growth. Oncogene. 38:4715–4728. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ran FA, Hsu PD, Wright J, Agarwala V,
Scott DA and Zhang F: Genome engineering using the CRISPR-Cas9
system. Nat Protoc. 8:2281–2308. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Welti J, Loges S, Dimmeler S and Carmeliet
P: Recent molecular discoveries in angiogenesis and antiangiogenic
therapies in cancer. J Clin Invest. 123:3190–3200. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Teleanu RI, Chircov C, Grumezescu AM and
Teleanu DM: Tumor Angiogenesis and Anti-Angiogenic Strategies for
Cancer Treatment. J Clin Med. 9:E842019. View Article : Google Scholar
|
|
15
|
Jászai J and Schmidt MH: Trends and
challenges in tumor anti-angiogenic therapies. Cells. 8:E11022019.
View Article : Google Scholar
|
|
16
|
Rajabi M and Mousa SA: The role of
angiogenesis in cancer treatment. Biomedicines. 5:E342017.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Herbert SP and Stainier DY: Molecular
control of endothelial cell behaviour during blood vessel
morphogenesis. Nat Rev Mol Cell Biol. 12:551–564. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ferrara N, Gerber HP and LeCouter J: The
biology of VEGF and its receptors. Nat Med. 9:669–676. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Apte RS, Chen DS and Ferrara N: VEGF in
signaling and disease: Beyond discovery and development. Cell.
176:1248–1264. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Folkman J: Tumor angiogenesis: Therapeutic
implications. N Engl J Med. 285:1182–1186. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jayson GC, Kerbel R, Ellis LM and Harris
AL: Antiangiogenic therapy in oncology: Current status and future
directions. Lancet. 388:518–529. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ferrara N and Kerbel RS: Angiogenesis as a
therapeutic target. Nature. 438:967–974. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Haibe Y, Kreidieh M, El Hajj H, Khalifeh
I, Mukherji D, Temraz S and Shamseddine A: Resistance mechanisms to
anti-angiogenic therapies in cancer. Front Oncol. 10:2212020.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bergers G and Hanahan D: Modes of
resistance to anti-angiogenic therapy. Nat Rev Cancer. 8:592–603.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
van Beijnum JR, Nowak-Sliwinska P,
Huijbers EJ, Thijssen VL and Griffioen AW: The great escape; the
hallmarks of resistance to antiangiogenic therapy. Pharmacol Rev.
67:441–461. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pakala SB, Reddy SD, Bui-Nguyen TM,
Rangparia SS, Bommana A and Kumar R: MTA1 coregulator regulates LPS
response via MyD88-dependent signaling. J Biol Chem.
285:32787–32792. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li DQ, Pakala SB, Reddy SD, Peng S,
Balasenthil S, Deng CX, Lee CC, Rea MA and Kumar R:
Metastasis-associated protein 1 is an integral component of the
circadian molecular machinery. Nat Commun. 4:25452013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mitchell DC and Bryan BA: Anti-angiogenic
therapy: Adapting strategies to overcome resistant tumors. J Cell
Biochem. 111:543–553. 2010. View Article : Google Scholar : PubMed/NCBI
|