Introduction
Chronic obstructive pulmonary disease (COPD) is
responsible for >1,400,000 annual mortalities worldwide, and is
characterized by persistent airflow limitation and limited
therapeutic options (1). There
are currently 64 million patients with COPD and 3 million cases of
COPD-associated mortality every year estimated by the WHO. COPD is
predicted to be the third leading cause of mortality in the world
by 2030 (2). Annually, COPD
treatment costs ≤€141.4 billion in Europe and ~$50 billion in
America (3,4). Emphysema is the major pathological
diagnostic factor associated with COPD, which affects the distal
space of the terminal bronchioles, and is associated with an
irregular inflammatory response and oxidative imbalance of the
lungs to toxic particles or gases (1,5).
COPD mainly manifests by incomplete reversible airflow limitation,
which significantly decreases the quality of life and exercise
endurance of patients (6).
Therefore, it is important to determine the underlying mechanism
and identify effective preventative measures of COPD to further
improve the survival rate of patients with COPD.
Overwhelming evidence has suggested that cigarette
smoking is the leading cause of COPD worldwide (5). Between 80 and 90% of patients with
COPD are smokers, and 10–15% of smokers will develop COPD (5). It has been shown that there are
>4,500 chemical compounds in the gas mixture generated by
cigarette smoking, most of which are toxic, and 72 compounds are
carcinogenic substances (7,8).
Nicotine, the principal addictive component in cigarette smoking,
is considered to be the main factor driving the pathogenesis and
progression of pulmonary disease (9,10).
Substantial evidence has supported the promoting effect of nicotine
on COPD on a long-term basis (11) and smoking cessation is the top
priority in the treatment of COPD (12); however, the underlying mechanisms
remain largely unknown.
Inflammation and cell death are the two critical
pathological mechanisms of COPD (13,14). Enhanced cell death can be observed
during the destruction of lung tissue in both humans and mice
(14,15). Previously, cell death was
typically ascribed to apoptosis and necrosis; however, several
other forms of cell death have been identified recently, such as
pyroptosis (16). Pyroptosis is a
unique form of inflammatory cell death, which is mediated by the
activation of caspase-1 and the NOD-like receptor protein-3 (NLRP3)
inflammasome (17). The NLRP3
inflammasome is a multiprotein intracellular innate immune sensor
mainly composed of NLRP3, apoptosis-associated speck-like protein
(ASC) and pro-caspase-1 (18).
Caspase-1 activation is responsible for the maturation of pro-IL-18
and pro-IL-1β (19), whereas
NLRP3 activation induces the cleavage of gasdermin D (GSDMD), which
rapidly creates cell membrane rupture and leads to intracellular
protein release, resulting in an inflammatory reaction (20). Non-infectious and infectious
stimuli can both trigger pyroptosis (21). It has been reported that cigarette
smoke can induce the expression of caspase-1 and its downstream
target molecules, IL-1β and IL-18, in the bronchoalveolar lavage,
serum and sputum of patients with COPD (22,23). Furthermore, inflammasome
activators have been shown to be increased in the airway of
patients with COPD, including extracellular ATP, ROS and
damage-associated molecular patterns (24). These previous findings indicate a
critical role for pyroptosis in COPD progression.
Bronchial epithelial cells are the first anatomical
barrier exposed to noxious gases and particles of cigarette smoke,
which can initiate airway remodeling in COPD (25). Apoptosis of epithelial cells
serves a crucial role in the pathogenesis of COPD via airway
remodeling (26,27). Numerous factors associated with
COPD, including cigarette smoke, have the potential to cause
apoptosis of epithelial cells (28,29). Nevertheless, to the best of our
knowledge, it remains to be fully elucidated as to whether
pyroptosis is involved in epithelial cell death upon nicotine
exposure.
The present study aimed to investigate whether
epithelial cells undergo pyroptosis in COPD progression. These
findings may improve understanding of the underlying mechanisms of
COPD and provide valuable information for the diagnosis and
treatment of COPD.
Materials and methods
Cell culture
The normal human bronchial epithelial cell line
16HBE is a common cell line used to study COPD pathogenesis in
vitro (30). 16HBE cells
(American Type Culture Collection) were cultured in RPMI-1640
medium (Gibco; Thermo Fisher Scientific, Inc.) supplemented with
10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.). The
cells were grown in 75-cm2 flasks at 37°C in a
humidified atmosphere containing 5% CO2 and were
passaged 1:3 using 0.25% trypsin when they reached 80–90%
confluence. Nicotine was purchased from Nacalai Tesque, Inc. 16HBE
cells were treated with 0.1 or 1 µM nicotine for 48 h at 37°C, as
previously described (31).
Cell proliferation assay
The proliferation of 16HBE cells was evaluated using
the Cell Counting Kit-8 (CCK-8) assay (Dojindo Laboratories, Inc.).
16HBE cells were seeded in 96-well plates (1.0×104
cells/well), and were exposed to 0, 0.1 or 1 µM nicotine for 0, 48,
72 and 96 h at 37°C (31).
Finally, 10 µl CCK-8 solution was added to 100 µl RPMI-1640 medium
in each well for 2 h. Absorbance values were measured at a
wavelength of 450 nm using an automated microplate reader
(Molecular Devices, LLC).
Flow cytometry
The death of 16HBE cells (1×106/100 µl)
was analyzed using an Annexin V-FITC and PI staining kit (cat. no.
A211-01/02; Vazyme Biotech Co., Ltd.) according to the
manufacturer's instructions. Flow cytometry was performed on a
FACSCalibur flow cytometer (BD Biosciences) and the rate of cell
death (% of PI+ cells) was analyzed with FlowJo software
(FlowJo, LLC, version 10.6.0).
Reverse transcription-quantitative PCR
(qPCR)
Total RNA was isolated from each group of 16HBE
cells using TRIzol® (Thermo Fisher Scientific, Inc.;
cat. no. 15596026). The concentration of RNA was determined using
an ND-2000 Spectrophotometer (Thermo Fisher Scientific, Inc.). RNA
was reverse transcribed into cDNA by M-MLV Reverse Transcriptase
kit (Elk Wuhan Biotechnology Co., Ltd. cat. no. EQ002) with the
following temperature protocol: 42°C for 50 min for the reverse
transcription reaction; 99°C for 5 min to inactivate the reverse
transcriptase; and 4°C to store the reverse transcription product.
qPCR was performed with the KAPA SYBR FAST qPCR Kit (Kapa
Biosystems; Roche Diagnostics) using a 7300 Real-Time PCR System
(Applied Biosystems; Thermo Fisher Scientific, Inc.). Data were
normalized to the housekeeping gene GAPDH. The mRNA levels were
measured using the 2−ΔΔCq method of quantification
(32). All qPCR experiments were
replicated at least three independent times. The thermocycling
conditions used for qPCR were: Initial denaturation at 95°C for 10
min; followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min.
The following primer sequences were used: GAPDH, forward
5′-TGACTTCAACAGCGACACCCA-3′, reverse 5′-CACCCTGTTGCTGTAGCCAAA-3′;
pro-caspase-1, forward 5′-ACAAGACCTCTGACAGCACG-3′, reverse
5′-TTCACTTCCTGCCCACAGAC-3′; IL-18, forward
5′-GCTGAAGATGATGAAAACCTGGA-3′, reverse 5′-GAGGCCGATTTCCTTGGTCA-3′;
and IL-1β, forward 5′-CCAGCTACGAATCTCCGACC-3′, reverse
5′-TATCCTGTCCCTGGAGGTGG-3′.
Western blotting
Proteins were extracted from treated 16HBE cells by
RIPA lysis buffer (Beyotime Institute of Biotechnology; cat. no.
P0013B) and were quantified using the BCA method. Proteins (30 µg
per lane) were separated by SDS-polyacrylamide gel electrophoresis
on 10% gels and were electrophoretically transferred to
polyvinylidene fluoride membranes, which were blocked with 5%
skimmed milk for 1 h at room temperature. The following primary
antibodies were used overnight at 4°C: Rabbit anti-pro-caspase-1
(1:1,000 dilution, cat. no. ab179515; Abcam), rabbit anti-caspase-1
(1:500 dilution, cat. no. 22915-1-AP; Proteintech Group Inc.),
rabbit anti-IL-1β (1:500 dilution, cat. no. A1112; ABclonal Biotech
Co., Ltd.), rabbit anti-IL-18 (1:500 dilution, cat. no. A1115;
ABclonal Biotech Co., Ltd.), rabbit anti-NLRP3 (1:1,000 dilution,
cat. no. ab263899; Abcam), rabbit anti-ASC (1:1,000 dilution, cat.
no. ab283684; Abcam), rabbit anti-cleaved N-terminal GSDMD (1:1,000
dilution, cat. no. ab215203; Abcam) and mouse anti-GAPDH (1:500
dilution, cat. no. 60004-1-lg; Proteintech Group, Inc.).
Horseradish peroxidase-conjugated goat anti-rabbit/mouse IgG
(1:5,000 dilution, cat. no. BA1054/BA1051; Boster Biological
Technology) was used as a secondary antibody for 1 h at room
temperature. Immunoreactive protein bands were detected by ECL
hypersensitive chemiluminescence kit (cat. no. P0018M; Beyotime
Institute of Biotechnology) with an Odyssey Scanning System
(version 3.0, LI-COR Biosciences).
Immunofluorescence analysis
Immunofluorescence staining of 16HBE cells was
performed as previously described (33). 16HBE cells (5×105)
plated onto cover glasses were fixed with 4% (w/v) paraformaldehyde
for 15 min at room temperature. Rabbit anti-caspase-1 (1:50
dilution; cat. no. 22915-1-AP, Proteintech Group Inc.) was added to
the fixed 16HBE cells and incubated at 4°C overnight, followed by
incubation with Goat anti-Rabbit IgG F(ab′)2 secondary antibodies
(dilution, 1:1,000; cat. no. 31234; Invitrogen) for 2 h at room
temperature. 4′,6-diamidino-2-phenylindole (DAPI, dilution,
1:1,000; cat. no. 62248; Thermo Fisher Scientific, Inc.) was used
to stain nuclei at room temperature for 10 min. The images were
acquired using a FV3000 confocal fluorescence microscope (Olympus
Corporation).
TUNEL assay
16HBE cells (4×104 cells/cm2)
were seeded onto cover glasses. After 24 h, the cells were treated
with 0.1 or 1 µM nicotine for another 48 h followed by fixing with
4% (w/v) paraformaldehyde for 15 min at room temperature. Then the
cells were subjected to a TUNEL assay (DeadEnd Fluorometric TUNEL
System; Promega Corporation) according to the manufacturer's
instructions. Briefly, cells were incubated with 250 µl of TUNEL
labeling solution in one well of a 24-well plate for 1 h at 37°C
covered with aluminum foil. Then cells were incubated with PBS
containing DAPI nucleic acid stain (dilution, 1:1,000; 1 mg/ml
solution, cat. no. 62248; Thermo Fisher Scientific, Inc.) for 10
min at room temperature to stain nuclei. TUNEL positive (TUNEL+)
cells were calculated by counting the number of stained cells in
five separate field per slides using a Leica confocal microscope
TCS SP2 (Leica Microsystems GmbH).
Statistical analysis
All experiments were repeated at least three times
in vitro and all data were analyzed using GraphPad Prism 7
(GraphPad Software, Inc.). Data are presented as the mean ± SD.
Differences were analyzed for significance by one-way ANOVA
followed by Duncan's post hoc test. P<0.05 was used to indicate
a statistically significant difference.
Results
Nicotine exposure triggers 16HBE cell
death
The present study used nicotine-treated 16HBE cells
as an in vitro model of COPD. To explore the potential role
of the primary component of cigarette smoke, nicotine, in the
progression of COPD, nicotine was used to treat 16HBE cells. In
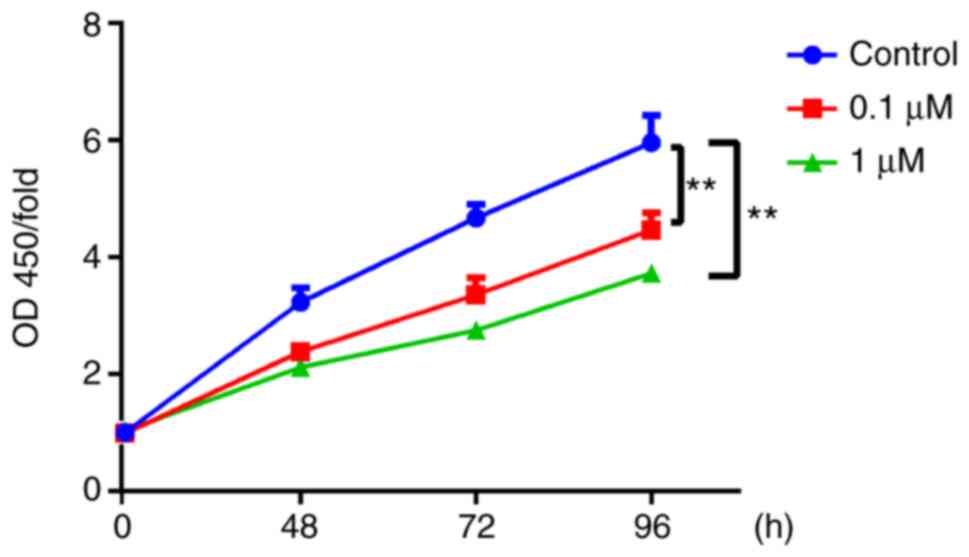
16HBE cells incubated with 0.1 or 1 µM nicotine, the proliferation
rate significantly decreased compared with the control group
(Fig. 1). Conversely, the cell
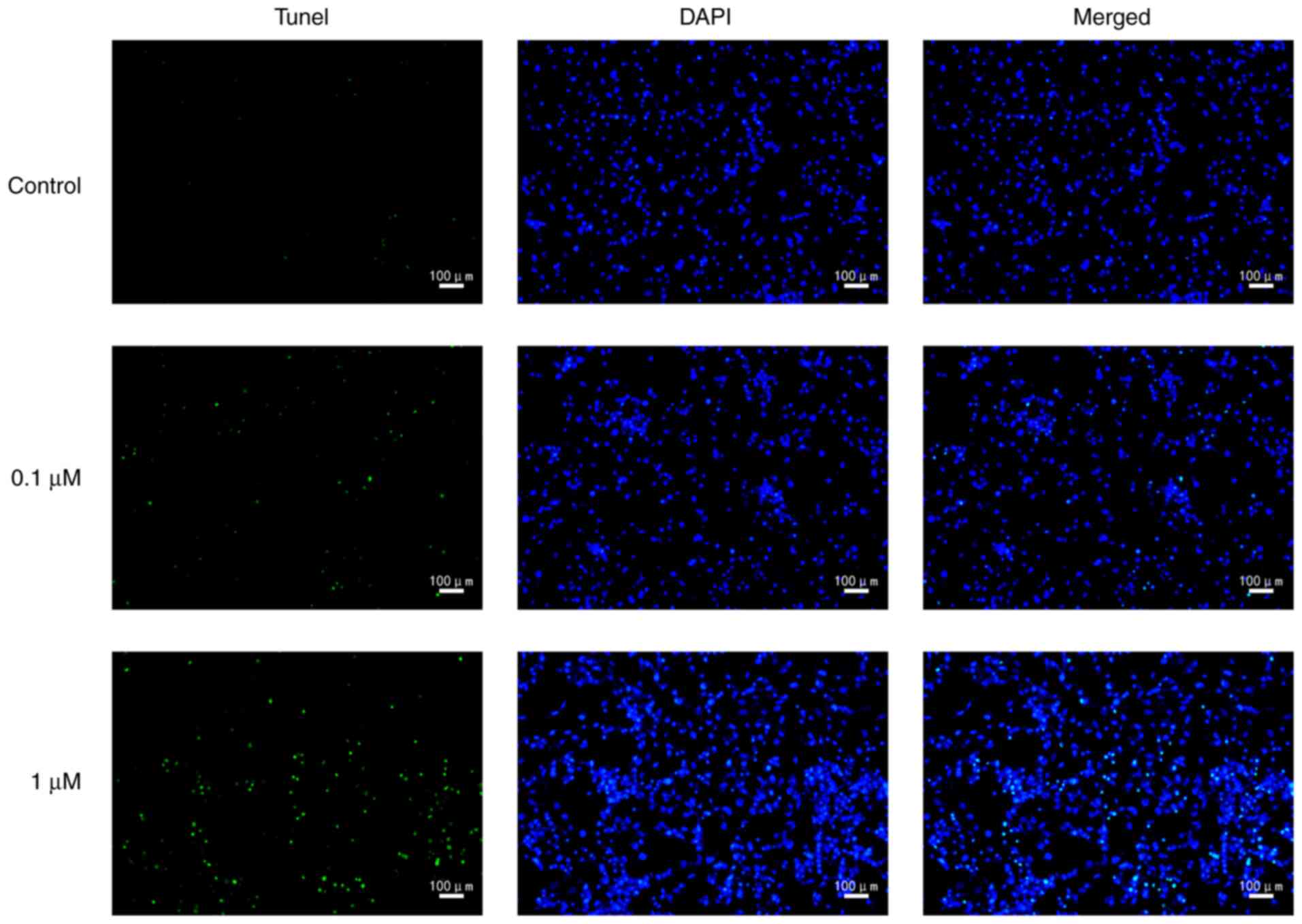
death rate was significantly increased by nicotine (Fig. 2). In order to further characterize
nicotine-induced 16HBE cell death, the cells were stained with
TUNEL. The results revealed that the number of TUNEL-positive cells
was markedly increased in nicotine-treated groups compared with the
control group (Fig. 3).
Epithelial cells display
characteristic features of pyroptosis after nicotine exposure
To elucidate the relationship between nicotine and
pyroptosis, 16HBE cells were used for in vitro experiments.
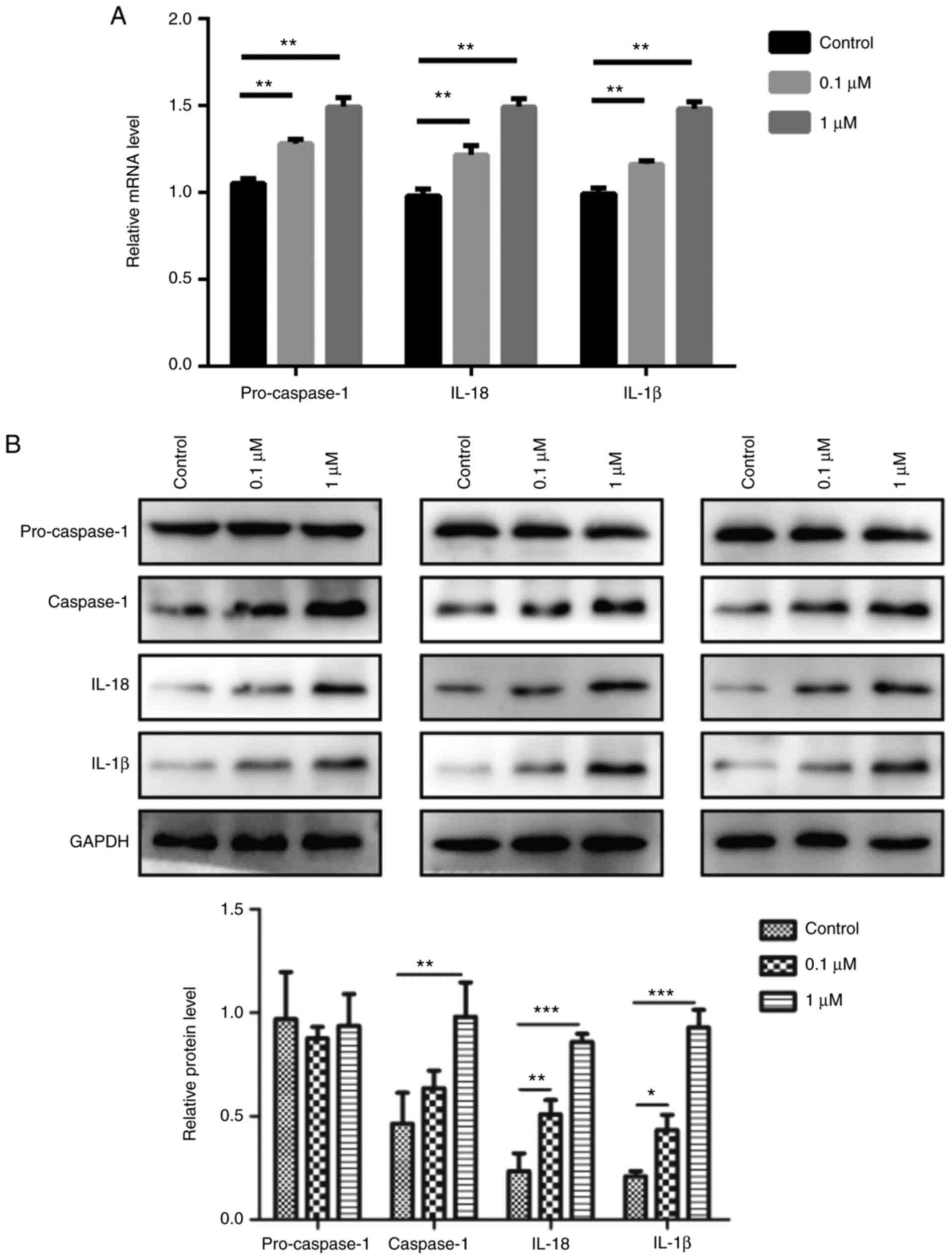
The results revealed that 0.1 and 1 µM nicotine treatment
significantly induced the mRNA expression levels of pro-caspase-1,
IL-1β and IL-18 compared with the control group (Fig. 4A). Furthermore, nicotine promoted
the protein expression level of caspase-1, IL-1β and IL-18 in 16HBE
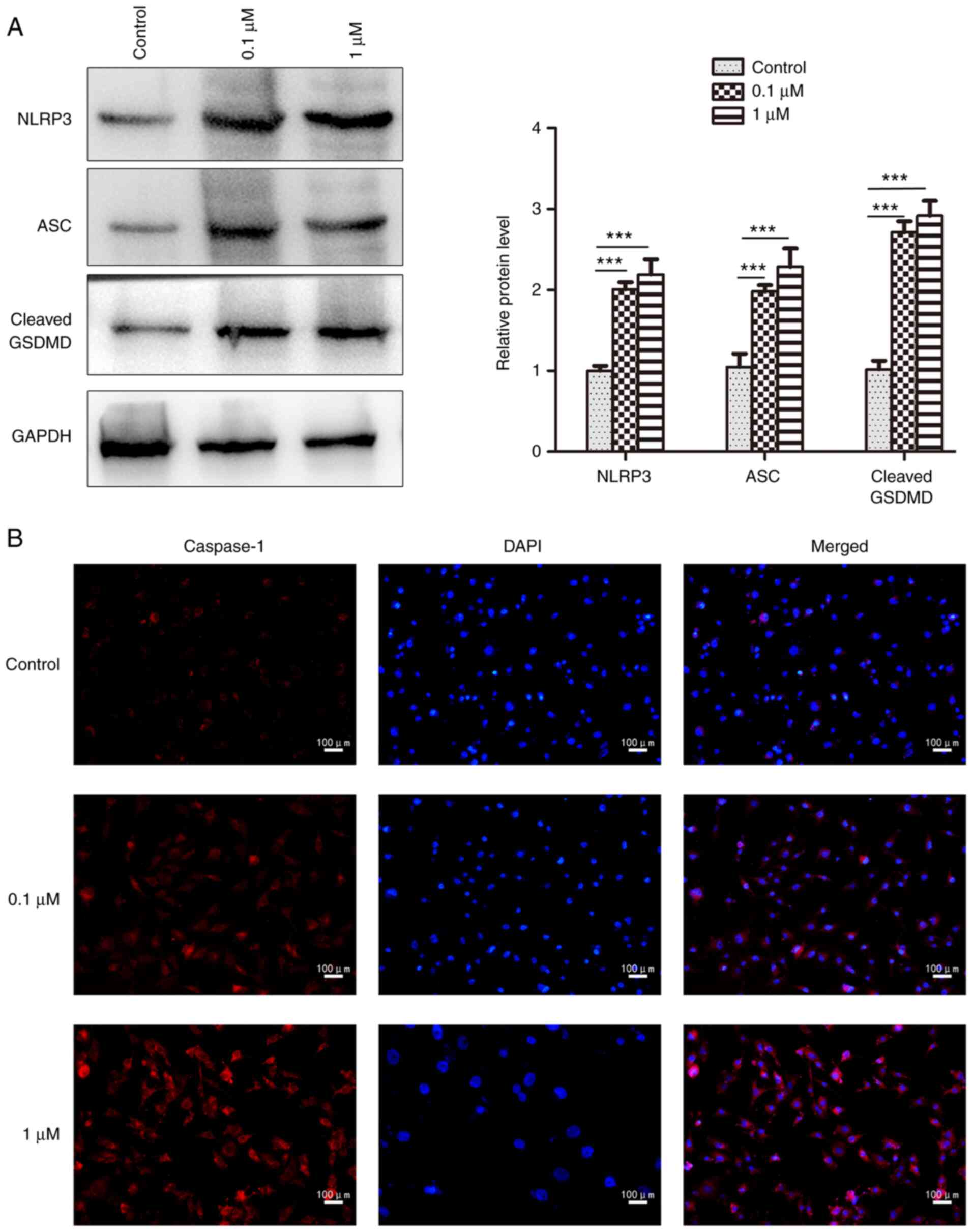
cells; however, it had no obvious effect on pro-caspase-1 (Fig. 4B). In addition, 16HBE cells
exhibited significant increases in the protein expression levels of
NLRP3 and ASC after nicotine administration compared with the
control group, as determined by western blotting (Fig. 5A). The NLRP3, ASC and cleaved form
of GSDMD was also significantly increased by nicotine in 16HBE
cells (Fig. 5A), which is
indicative of pyroptosis (34).
Furthermore, the increased expression of caspase-1 was also
observed in nicotine-treated 16HBE cells by immunofluorescence
analysis (Fig. 5B). Taken
together, these results suggested that pyroptosis was induced in
16HBE cells upon nicotine administration.
Discussion
COPD is a multifactorial disease associated with
numerous mechanisms (35,36). The pathogenesis of COPD involves
several pathophysiological processes, such as inflammatory
response, oxidative stress, apoptosis, protease/anti-proteinase
imbalance, production of autoantibodies, changes in cell
proliferation and aging (37).
The inflammatory response and cell death are two of the most
important pathogenic factors associated with COPD (38). As a novel form of programmed cell
death, pyroptosis is different from apoptosis, which is classically
considered a non-inflammatory form of apoptosis (17,39). Molecularly, apoptosis is executed
by activation of the executioner caspases, caspase-3 and caspase-7,
downstream of the initiator caspases, caspase-8, caspase-9 and
caspase-10 (40). However,
whether pyroptosis is associated with COPD pathogenesis remains to
be determined.
Cigarette smoke can cause inflammation, which is an
important injury factor in lung diseases such as COPD (41). The damage to the lungs caused by
cigarette smoke is most directly reflected in the injury of
bronchial epithelial cells and alveolar epithelial cells (42). Bronchial epithelial cells are one
of the key cell types that mediate inflammation and the 16HBE cell
line is a common cell line used to study COPD pathogenesis in
vitro. When exposed to harmful substances, such as cigarette
smoke, airway epithelial cells are induced to produce various
inflammatory cytokines (e.g., IL-18) leading to the recruitment of
inflammatory cells (e.g., neutrophils) (43). In vitro and in vivo
studies have demonstrated that smoking causes inflammation and cell
death in lung tissue and airway cells (38,41). The present study revealed that
treatment with 0.1 and 1 µM nicotine suppressed the proliferation
and promoted the death of 16HBE cells, as determined by CCK-8
assay, flow cytometry and immunofluorescence analysis. Furthermore,
0.1 and 1 µM nicotine treatment significantly enhanced the
expression levels of caspase-1, IL-1β, IL-18, NLRP3, ASC and GSDMD
in 16HBE cells. During pyroptosis, the activated NLRP3 inflammasome
can induce the cleavage of pro-caspase-1 into mature caspase-1
(44). In the present study,
nicotine treatment significantly increased the mRNA expression
levels of pro-caspase-1 but showed no effect on its protein
expression levels. By contrast, nicotine exposure significantly
increased the protein expression levels of caspase-1. Thus, it was
hypothesized that nicotine promoted the expression of pro-caspase
1, and the increased levels of pro-caspase 1 were then cleaved into
caspase-1. These results indicated that nicotine exposure may
induce pyroptosis in 16HBE cells, suggesting that pyroptosis could
be involved in the progression of COPD. This discovery sheds new
light on the mechanism underlying the pathogenesis of COPD.
Bronchial epithelial cells serve an integral role in
the airway defense mechanism via the mucociliary system and
mechanical barriers, which serve significant roles in the
progression of various lung diseases in addition to COPD, such as
asthma, lung cancer, pneumonia and pulmonary fibrosis (45). Previous studies have revealed that
smoking is significantly prevalent in patients with multiple lung
diseases (46–48). However, to the best of our
knowledge, whether nicotine exposure induces pyroptosis in these
diseases remains to be elucidated. A key mediator of pyroptosis,
GSDMB, has already been highly linked to asthma and non-small cell
lung carcinoma (49–51). Thus, it was hypothesized that
nicotine exposure may induce pyroptosis in bronchial epithelial
cells in these diseases; this hypothesis requires further
study.
Finally, the present study had some limitations. The
in vitro experiments were only performed in 16HBE cells;
therefore, further experiments should be performed in more cell
lines to confirm the findings presented in the current study. Due
to limited experimental conditions, nicotine exposure-induced
pyroptosis has not been confirmed in in vivo experiments.
Thus, further experimental research is needed to confirm the
results in an animal model of COPD. Furthermore, it is still not
clear whether nicotine exposure will induce pyroptosis in patients
with COPD; this will be one of our future research directions.
In conclusion, the results of the present study
suggested that nicotine exposure suppressed the proliferation and
promoted the death of 16HBE cells, which may be associated with the
increased expression levels of caspase-1, IL-1β, IL-18, NLRP3, ASC
and GSDMD. These findings indicated that nicotine treatment induced
pyroptosis in 16HBE cells, which may be associated with the
progression of COPD.
Acknowledgments
Not applicable.
Funding
This work was supported by the National Natural Science
Foundation of China (grant nos. 81660013 and No. 81860015).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YC and YD were responsible for the conception of the
present study. RM, YC and YD confirm the authenticity of all the
raw data. RM and JZ conducted the experiments, analyzed and
interpreted the data. JZ was responsible for statistical analysis.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mouronte-Roibas C, Leiro-Fernandez V,
Fernandez-Villar A, Botana-Rial M, Ramos-Hernandez C and
Ruano-Ravina A: COPD, emphysema and the onset of lung cancer. A
systematic review. Cancer Lett. 382:240–244. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lopez-Campos JL, Tan W and Soriano JB:
Global burden of COPD. Respirology. 21:14–23. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gibson GJ, Loddenkemper R, Lundback B and
Sibille Y: Respiratory health and disease in Europe: The new
European Lung White Book. Eur Respir J. 42:559–563. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mirza S, Clay RD, Koslow MA and Scanlon
PD: COPD Guidelines: A review of the 2018 GOLD Report. Mayo Clin
Proc. 93:1488–1502. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Singh D, Agusti A, Anzueto A, Barnes PJ,
Bourbeau J, Celli BR, Criner GJ, Frith P, Halpin DMG, Han M, et al:
Global strategy for the diagnosis, management, and prevention of
chronic obstructive lung disease: The GOLD science committee report
2019. Eur Respir J. 53:19001642019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sampaio MS, Vieira WA, Bernardino IM,
Herval AM, Flores-Mir C and Paranhos LR: Chronic obstructive
pulmonary disease as a risk factor for suicide: A systematic review
and meta-analysis. Respir Med. 151:11–18. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim EJ, Yoon SJ, Kim YE, Go DS and Jung Y:
Effects of aging and smoking duration on cigarette Smoke-Induced
COPD severity. J Korean Med Sci. 34 (Suppl 1):e902018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hecht SS: Research opportunities related
to establishing standards for tobacco products under the Family
Smoking Prevention and Tobacco Control Act. Nicotine Tob Res.
14:18–28. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gellner CA, Belluzzi JD and Leslie FM:
Self-administration of nicotine and cigarette smoke extract in
adolescent and adult rats. Neuropharmacology. 109:247–253. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hou W, Hu S, Li C, Ma H, Wang Q, Meng G,
Guo T and Zhang J: Cigarette smoke induced lung barrier
dysfunction, EMT, and tissue remodeling: A possible link between
COPD and lung cancer. Biomed Res Int. 2019:20256362019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Garcia-Arcos I, Geraghty P, Baumlin N,
Campos M, Dabo AJ, Jundi B, Cummins N, Eden E, Grosche A, Salathe M
and Foronjy R: Chronic electronic cigarette exposure in mice
induces features of COPD in a nicotine-dependent manner. Thorax.
71:1119–1129. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tonnesen P: Smoking cessation and COPD.
Eur Respir Rev. 22:37–43. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu CH, Chen Z, Chen K, Liao FT, Chung CE,
Liu X, Lin YC, Keohavong P, Leikauf GD and Di YP:
Lipopolysaccharide-mediated chronic inflammation promotes tobacco
carcinogen-induced lung cancer and determines the efficacy of
immunotherapy. Cancer Res. 81:144–157. 2021.PubMed/NCBI
|
|
14
|
Conlon TM, John-Schuster G, Heide D,
Pfister D, Lehmann M, Hu Y, Ertuz Z, Lopez MA, Ansari M, Strunz M,
et al: Inhibition of LTbetaR signalling activates WNT-induced
regeneration in lung. Nature. 588:151–156. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li T, Fanning KV, Nyunoya T, Chen Y and
Zou C: Cigarette smoke extract induces airway epithelial cell death
via repressing PRMT6/AKT signaling. Aging (Albany NY).
12:24301–24317. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fang Y, Tian S, Pan Y, Li W, Wang Q, Tang
Y, Yu T, Wu X, Shi Y, Ma P and Shu Y: Pyroptosis: A new frontier in
cancer. Biomed Pharmacother. 121:1095952020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Riegman M, Sagie L, Galed C, Levin T,
Steinberg N, Dixon SJ, Wiesner U, Bradbury MS, Niethammer P,
Zaritsky A and Overholtzer M: Ferroptosis occurs through an osmotic
mechanism and propagates independently of cell rupture. Nat Cell
Biol. 22:1042–1048. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li N, Zhou H, Wu H, Wu Q, Duan M, Deng W
and Tang Q: STING-IRF3 contributes to lipopolysaccharide-induced
cardiac dysfunction, inflammation, apoptosis and pyroptosis by
activating NLRP3. Redox Biol. 24:1012152019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jiang S, Zhang H, Li X, Yi B, Huang L, Hu
Z, Li A, Du J, Li Y and Zhang W: Vitamin D/VDR attenuate
cisplatin-induced AKI by down-regulating NLRP3/Caspase-1/GSDMD
pyroptosis pathway. J Steroid Biochem Mol Biol. 206:1057892021.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Anderson FL, von Herrmann KM, Andrew AS,
Kuras YI, Young AL, Scherzer CR, Hickey WF, Lee SL and Havrda MC:
Plasma-borne indicators of inflammasome activity in Parkinson's
disease patients. NPJ Parkinsons Dis. 7:22021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
de Zoete MR, Palm NW, Zhu S and Flavell
RA: Inflammasomes. Cold Spring Harb Perspect Biol. 6:a0162872014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Murray LA, Dunmore R, Camelo A, Da Silva
CA, Gustavsson MJ, Habiel DM, Hackett TL, Hogaboam CM, Sleeman MA
and Knight DA: Acute cigarette smoke exposure activates apoptotic
and inflammatory programs but a second stimulus is required to
induce epithelial to mesenchymal transition in COPD epithelium.
Respir Res. 18:822017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kang MJ, Homer RJ, Gallo A, Lee CG,
Crothers KA, Cho SJ, Rochester C, Cain H, Chupp G, Yoon HJ and
Elias JA: IL-18 is induced and IL-18 receptor alpha plays a
critical role in the pathogenesis of cigarette smoke-induced
pulmonary emphysema and inflammation. J Immunol. 178:1948–1959.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Franklin BS, Bossaller L, De Nardo D,
Ratter JM, Stutz A, Engels G, Brenker C, Nordhoff M, Mirandola SR,
Al-Amoudi A, et al: The adaptor ASC has extracellular and
‘prionoid’ activities that propagate inflammation. Nat Immunol.
15:727–737. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Guan R, Wang J, Cai Z, Li Z, Wang L, Li Y,
Xu J, Li D, Yao H, Liu W, et al: Hydrogen sulfide attenuates
cigarette smoke-induced airway remodeling by upregulating SIRT1
signaling pathway. Redox Biol. 28:1013562020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Plataki M, Tzortzaki E, Rytila P,
Demosthenes M, Koutsopoulos A and Siafakas NM: Apoptotic mechanisms
in the pathogenesis of COPD. Int J Chron Obstruct Pulmon Dis.
1:161–171. 2006.PubMed/NCBI
|
|
27
|
Park JW, Ryter SW and Choi AM: Functional
significance of apoptosis in chronic obstructive pulmonary disease.
COPD. 4:347–353. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kim SY, Kim HJ, Park MK, Huh JW, Park HY,
Ha SY, Shin JH and Lee YS: Mitochondrial E3 Ubiquitin Protein
Ligase 1 Mediates Cigarette Smoke-Induced endothelial cell death
and dysfunction. Am J Respir Cell Mol Biol. 54:284–296. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sun Y, An N, Li J, Xia J, Tian Y, Zhao P,
Liu X, Huang H, Gao J and Zhang X: MiRNA-206 regulates human
pulmonary microvascular endothelial cell apoptosis via targeting in
chronic obstructive pulmonary disease. J Cell Biochem.
120:6223–6236. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Waltl EE, Selb R, Eckl-Dorna J, Mueller
CA, Cabauatan CR, Eiwegger T, Resch-Marat Y, Niespodziana K, Vrtala
S, Valenta R and Niederberger V: Betamethasone prevents human
rhinovirus- and cigarette smoke-induced loss of respiratory
epithelial barrier function. Sci Rep. 8:96882018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Takahashi T, Yoshida T, Harada K, Miyagi
T, Hashimoto K, Hide I, Tanaka S, Irifune M and Sakai N: Component
of nicotine-induced intracellular calcium elevation mediated
through α3- and α5-containing nicotinic acetylcholine receptors are
regulated by cyclic AMP in SH-SY 5Y cells. PLoS One.
15:e02423492020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xu R, Li Q, Zhou J, Zhou XD, Perelman JM
and Kolosov VP: The degradation of airway tight junction protein
under acidic conditions is probably mediated by transient receptor
potential vanilloid 1 receptor. Biosci Rep. 33:e000782013.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hu JJ, Liu X, Xia S, Zhang Z, Zhang Y,
Zhao J, Ruan J, Luo X, Lou X, Bai Y, et al: FDA-approved disulfiram
inhibits pyroptosis by blocking gasdermin D pore formation. Nat
Immunol. 21:736–745. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Barnes PJ: Inflammatory mechanisms in
patients with chronic obstructive pulmonary disease. J Allergy Clin
Immunol. 138:16–27. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Polverino F, Celli BR and Owen CA: COPD as
an endothelial disorder: Endothelial injury linking lesions in the
lungs and other organs? (2017 Grover Conference Series). Pulm Circ.
8:20458940187585282018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yao H and Rahman I: Current concepts on
oxidative/carbonyl stress, inflammation and epigenetics in
pathogenesis of chronic obstructive pulmonary disease. Toxicol Appl
Pharmacol. 254:72–85. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hikichi M, Mizumura K, Maruoka S and Gon
Y: Pathogenesis of chronic obstructive pulmonary disease (COPD)
induced by cigarette smoke. J Thorac Dis. 11 (Suppl
17):S2129–S2140. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hsu SK, Li CY, Lin IL, Syue WJ, Chen YF,
Cheng KC, Teng YN, Lin YH, Yen CH and Chiu CC: Inflammation-related
pyroptosis, a novel programmed cell death pathway, and its
crosstalk with immune therapy in cancer treatment. Theranostics.
11:8813–8835. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang Y and Kanneganti TD: From pyroptosis,
apoptosis and necroptosis to PANoptosis: A mechanistic compendium
of programmed cell death pathways. Comput Struct Biotechnol J.
19:4641–4657. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
John G, Kohse K, Orasche J, Reda A,
Schnelle-Kreis J, Zimmermann R, Schmid O, Eickelberg O and Yildirim
AO: The composition of cigarette smoke determines inflammatory cell
recruitment to the lung in COPD mouse models. Clin Sci (Lond).
126:207–221. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sarker RSJ, Conlon TM, Morrone C,
Srivastava B, Konyalilar N, Verleden SE, Bayram H, Fehrenbach H and
Yildirim AO: CARM1 regulates senescence during airway epithelial
cell injury in COPD pathogenesis. Am J Physiol Lung Cell Mol
Physiol. 317:L602–L614. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zou Y, Chen X, He B, Xiao J, Yu Q, Xie B,
Yang S, Dai L, Dai Z and Chen Q: Neutrophil extracellular traps
induced by cigarette smoke contribute to airway inflammation in
mice. Exp Cell Res. 389:1118882020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li H, Lu R, Pang Y, Li J, Cao Y, Fu H,
Fang G, Chen Q, Liu B, Wu J, et al: Zhen-Wu-Tang Protects IgA
nephropathy in rats by regulating exosomes to inhibit NF-κB/NLRP3
Pathway. Front Pharmacol. 11:10802020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Takizawa H: Bronchial epithelial cells in
allergic reactions. Curr Drug Targets Inflamm Allergy. 4:305–311.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kim SY, Sim S and Choi HG: Active,
passive, and electronic cigarette smoking is associated with asthma
in adolescents. Sci Rep. 7:177892017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kalemkerian GP and Schneider BJ: Advances
in small cell lung cancer. Hematol Oncol Clin North Am. 31:143–156.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Margaritopoulos GA, Harari S, Caminati A
and Antoniou KM: Smoking-related idiopathic interstitial pneumonia:
A review. Respirology. 21:57–64. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Das S, Miller M and Broide DH: Chromosome
17q21 Genes ORMDL3 and GSDMB in asthma and immune diseases. Adv
Immunol. 135:1–52. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Peng Z, Wang P, Song W, Yao Q, Li Y, Liu
L, Li Y and Zhou S: GSDME enhances Cisplatin sensitivity to regress
non-small cell lung carcinoma by mediating pyroptosis to trigger
antitumor immunocyte infiltration. Signal Transduct Target Ther.
5:1592020. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Soy M, Keser G, Atagunduz P, Tabak F,
Atagunduz I and Kayhan S: Cytokine storm in COVID-19: Pathogenesis
and overview of anti-inflammatory agents used in treatment. Clin
Rheumatol. 39:2085–2094. 2020. View Article : Google Scholar : PubMed/NCBI
|