Introduction
Sepsis, a systemic inflammatory syndrome, which is
induced in response to infection, is considered to be a significant
clinical issue in intensive and critical care medicine (1). It is estimated that ~14,000
individuals die from sepsis-induced multiple organ dysfunction
every day in the world (2). Acute
lung injury (ALI), one of the most severe complications of sepsis,
is characterized by high complication and mortality rates, while
its treatment is considered to be challenging (3). In response to endotoxins, such as
lipopolysaccharides (LPS), chemokines and inflammatory mediators,
the overactivation of inflammatory responses in the lungs may lead
to lung dysfunction, which in turn promotes oxidative stress
injury, the main pathological mechanism of ALI in sepsis (4–6).
Tranilast was developed and launched to the market
in the 1980s. It is used as an allergic mediator by promoting the
secretion of blockers that stabilize mast cell membranes and basal
cells to prevent the release of allergic reaction-related
substances, such as histamine and 5-hydroxymycin, eventually
protecting against acute asthma (7,8).
In addition, tranilast was originally used to treat type-I allergic
diseases, such as bronchial asthma and allergic rhinitis, while it
was later found to be effective against other inflammatory
diseases, such as acute colitis and atherosclerosis (9–11).
It has been reported that tranilast regulates the inflammatory
response by upregulating the anti-inflammatory factors IL-4, IL-10
and heme oxygenase 1 and inhibiting the levels of the
pro-inflammatory cytokines, IL-1β, IL-6 and TNF-α in serum
(10,12,13). Tranilast can improve the effect of
lung injury induced by cyclophosphamide (14) and decrease TGFβ-induced pulmonary
fibrosis (15), indicating that
tranilast has an important protective effect on lung injury.
However, whether tranilast could reduce lung injury caused by
sepsis, as well as the underlying mechanism, remains unknown. A
recent study demonstrated that pre-treatment of Ldlr−/−
mice with tranilast, fed on a high-fat and high-sugar diet, could
prevent the progression of atherosclerosis and vascular
inflammation (16). The
aforementioned study also reported that tranilast could suppress
the activation of Nod-like receptor family pyrin domain-containing
3 inflammasome in LPS-induced murine macrophages. In addition,
another study showed that treatment of breast cancer cell lines
with tranilast could inhibit C-X-C motif chemokine receptor 4
(CXCR4) signaling, a chemokine receptor involved in lung cancer and
sepsis-induced ALI (17–19). Based on the aforementioned
findings, the present study aimed to investigate whether tranilast
could also ameliorate sepsis-induced ALI by attenuating the
LPS-induced secretion of pro-inflammatory cytokines via the CXCR4
signaling pathway.
Materials and methods
Cell culture and treatment
The human non-cancerous pulmonary epithelial cell
line BEAS-2B (Procell Life Science and Technology Co., Ltd.) was
maintained in 55% bronchial epithelial growth medium (BEGM;
Scienbio; Beijing Sunbio Biotech Co., Ltd.), supplemented with 40%
FBS (Alphacell) and 5% DMSO (Beijing Solarbio Science and
Technology Co. Ltd.) in a humidified incubator with 5%
CO2 at 37°C. The cells were pre-treated with tranilast
at different concentrations (50, 100 or 200 µM) for 8 h prior to
cell stimulation with 10 mg/l LPS (both from Abmole Bioscience
Inc.) for 16 h.
Cell transfection
The CXCR4 overexpression plasmid (ov-CXCR4) and the
corresponding negative control plasmid (ov-NC) with lentiviral
expression vector GV 493 were synthesized by Shanghai GenePharma
Co., Ltd. The BEAS-2B cells were transfected with 20 nM ov-CXCR4 or
ov-NC using Lipofectamine® 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.) at 37°C with 5% CO2 followed by
incubation in serum-free medium for 4 h and then in complete
culture medium for 16 h. The transfection efficacy was determined
using reverse transcription-quantitative PCR (RT-qPCR) after 48
h.
Cell Counting Kit (CCK)-8 assay
The BEAS-2B cells in the logarithmic growth phase
were seeded into 96-well plates, at a density of 1×105
cells/well and cultured in the presence of 100 µl/well complete
culture medium. Each well was then supplemented with 10 µl CCK-8
solution (Beijing Solarbio Science and Technology Co., Ltd.) and
the cells were then incubated for 3 h. The optical density was
measured at 450 nm using a microplate reader.
RT-qPCR analysis
Total RNA was extracted from cells using the
RNAMolPure® Cell RNA kit (Shanghai Yeasen Biotechnology
Co., Ltd.), then reverse transcribed into cDNA using the
TransScript® First-Strand cDNA Synthesis SuperMix
(Beijing Transgen Biotech Co., Ltd.), according to the
manufacturer's instructions. cDNA amplification was performed using
the TransStart® Green qPCR SuperMix (Beijing Transgen
Biotech Co., Ltd.) according to the manufacturer's recommendations.
The following thermocycling conditions were used for qPCR: Initial
denaturation at 95°C for 10 min; followed by 40 cycles at 95°C for
10 sec and 60°C for 60 sec. mRNA expression levels were quantified
using the 2−ΔΔCq method and normalized to the internal
reference gene GAPDH (20). The
following primers were used: TNF-α forward,
5′-CGAGTGACAAGCCTGTAGCC-3′ and reverse,
5′-TTGAAGAGGACCTGGGAGTAG-3′; IL-1β forward,
5′-AAAAGCTTGGTGATGTCTGG-3′ and reverse, 5′-TTTCAACACGCAGGACAGG-3′;
IL-6 forward, 5′-GGAGACTTGCCTGGTGAAA-3′ and reverse,
5′-CTGGCTTGTTCCTCACTACTC-3′; CXCR4 forward,
5′-AGCTGTTGGTGAAAAGGTGGTCTATG-3′ and reverse,
5′-GCGCTTCTGGTGGCCCTTGGAGT-3′; GAPDH forward,
5′-TGACTTCAACAGCGACACCCACT-3′ and reverse,
5′-GACTGAGTGTGGCAGGGACT-3′.
Western blot analysis
The cells from each group were lysed on ice for 30
min using RIPA lysis buffer (Beyotime Institute of Biotechnology).
The supernatant was then collected following centrifugation at 300
× g for 15 min at 4°C. The concentration of the extracted proteins
was measured using a BCA kit. Subsequently, a total of 20 µg
protein/sample were mixed with 12% SDS buffer (Shanghai Uteam
Biotechnology Co., Ltd.) and incubated at 100°C in a water bath for
10 min for protein denaturation. The protein samples were then
separated using SDS-PAGE and electrophoresed until the bromophenol
blue reached the bottom of the separating gel. The proteins were
transferred to PVDF membranes, then washed three times. Following
blocking with 5% skimmed milk for 2 h at room temperature, the
membranes were first incubated with diluted primary antibodies
overnight at 4°C, then with secondary HRP-conjugated anti-rabbit
antibody (1:5,000; cat. no. ab205718; Abcam) at room temperature
for 1 h. Protein bands were visualized using an enhanced
chemiluminescence detection system (EMD Millipore). The gray values
of the proteins were quantified using ImageJ software (v146;
National Institutes of Health). The following primary antibodies
were used: cytochrome c oxidase subunit II (COX2; 1:1,000;
cat. no. ab179800), inducible nitric oxide (iNOS; 1:1,000; cat. no.
ab178945), Bax (1:1,000; cat. no. ab32503), cleaved caspase 3
(1:1,000; cat. no. ab32042), cleaved caspase 3 (1:1,000; cat. no.
ab32351), Bcl2 (1:1,000; cat. no. ab32124), CXCR4 (1:1,000;
ab124824), phosphorylated (p)-Janus kinase 2 (JAK2; 1:1,000; cat.
no. ab32101), p-STAT3 (1:1,000; cat. no. ab76315), JAK2 (1:1,000;
cat. no. ab108596), STAT3 (1:1,000; cat. no. ab68153) and GAPDH
(1:1,000; cat. no. ab8245) (all from Abcam).
TUNEL assay
The cells were seeded onto a glass slide
(dimensions, 1×1 cm), then placed into a 24-well plate. After
transfection of LPS induced cells, the cells were washed three
times with PBS and fixed in 4% paraformaldehyde for 15 min at 37°C.
The TUNEL Detection kit (Shanghai Yeasen Biotechnology Co., Ltd.)
was utilized to stain the apoptotic cells for 1 h at 37°C. The cell
nuclei were counterstained with DAPI for 10 min at room
temperature. Five random fields of views were selected for analysis
and the apoptotic cells were observed under a fluorescence
microscope (magnification, ×200). The percentage of apoptotic cells
was calculated as the ratio of the number of TUNEL-positive nuclei
to that of DAPI-stained cells.
Statistical analysis
SPSS v22.0 statistical software (IBM Corp.) was used
to analyze and process the experimental data. The results are
expressed as the mean ± SD. To compare the differences among
multiple groups, one-way ANOVA followed by Tukey's post hoc test
was used, while comparisons between two groups were assessed using
unpaired Student-t test. P<0.05 was considered to indicate a
statistically significant difference. Each experiment was repeated
three times.
Results
Tranilast attenuates LPS-induced
BEAS-2B cell viability
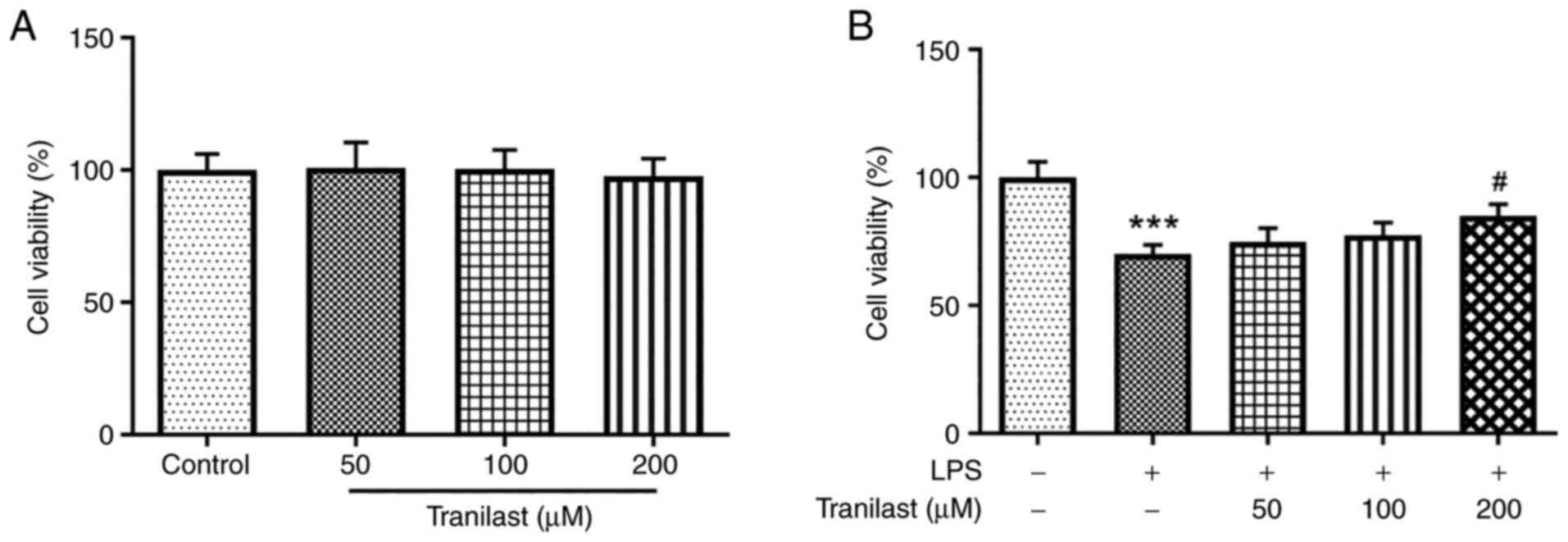
Treatment with different concentrations of tranilast
exerted no significant effect on untreated BEAS-2B cells with
respect to cell viability (Fig.
1A). However, pre-treatment of the LPS-induced BEAS-2B cells
with tranilast significantly increased cell viability in a
dose-dependent manner (Fig. 1B).
Therefore, these findings indicated that tranilast could attenuate
LPS-induced BEAS-2B cell viability.
Tranilast attenuates the release of
pro-inflammatory cytokines in LPS-induced BEAS-2B cells
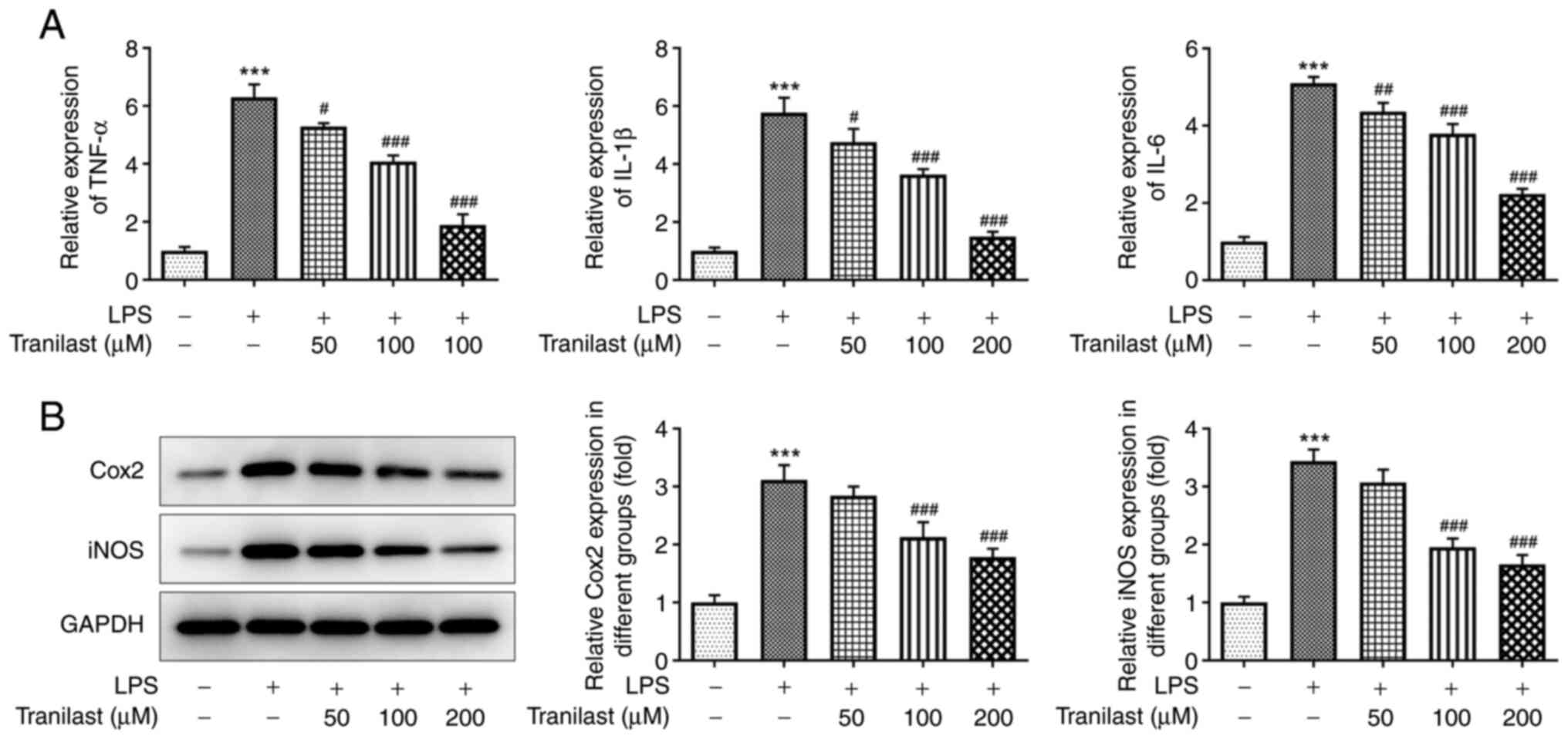
As shown in Fig.
2A, treatment with LPS increased the expression levels of TNF-α
(P<0.001), IL-1β (P<0.001) and IL-6 (P<0.001) in the
BEAS-2B cells, whereas pre-treatment with 50 µM tranilast reduced
them (P=0.0104, P=0.0271 and P=0.00521, for TNF-α, IL-1β and IL-6
respectively). In addition, pre-treatment with 100 and 200 µΜ
tranilast restored TNF-α, IL-1β and IL-6 expression levels
(P<0.001 in all cases). Similarly, western blot analysis
revealed that LPS increased the protein expression levels of COX2
and iNOS (P<0.001). When the cells were pre-treated with 100 µΜ
tranilast, the protein expression levels of COX2 (P=0.0009) and
iNOS (P<0.001) were decreased (Fig. 2B). Similar results were found with
200 µM. The aforementioned results suggested that tranilast could
reduce the LPS-induced secretion of pro-inflammatory cytokines in
the BEAS-2B cells.
Tranilast alleviates LPS-induced
BEAS-2B cell apoptosis
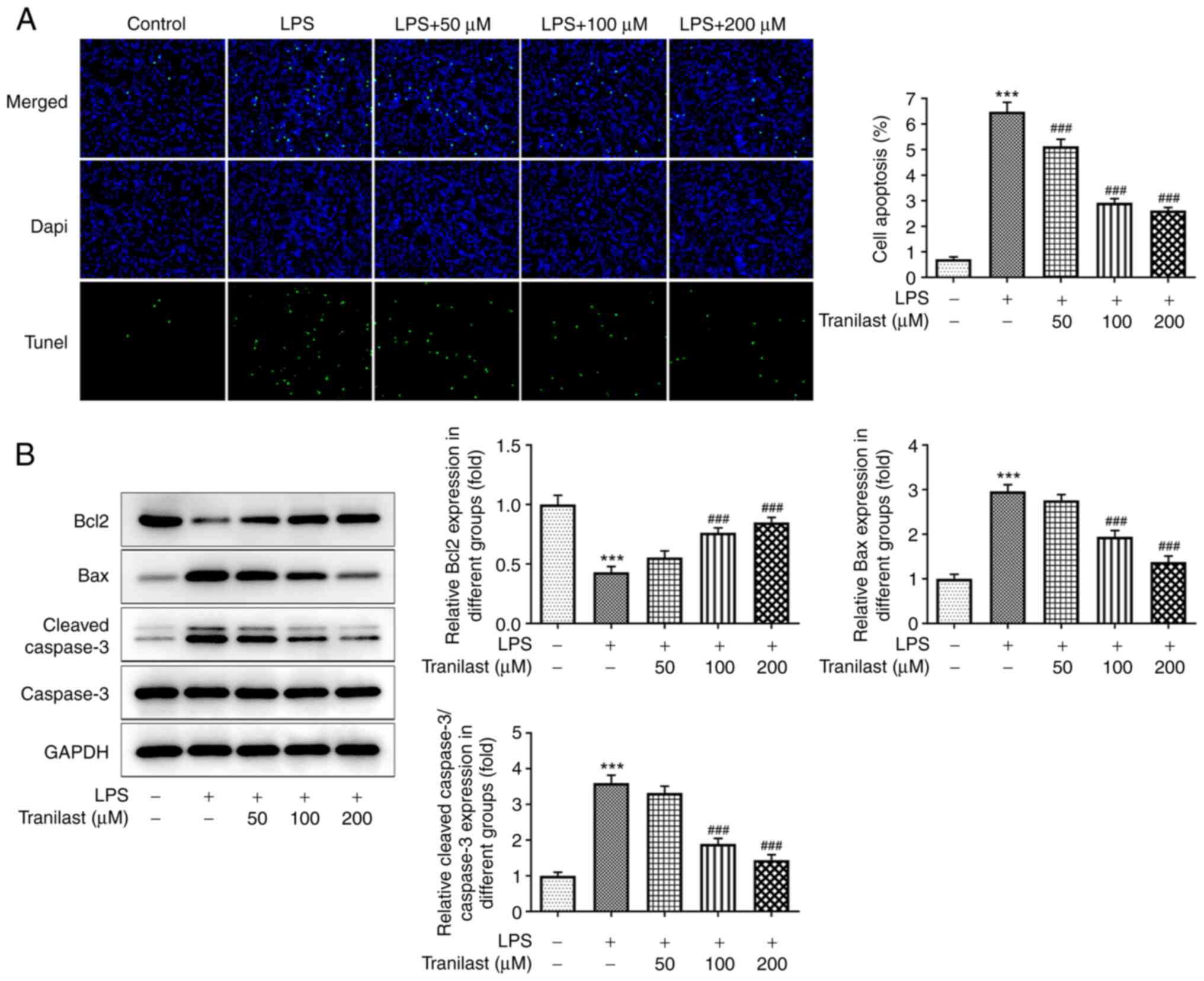
As shown in Fig.
3A, the number of TUNEL-positive apoptotic cells was increased
in the LPS group compared with that in the control group
(P<0.001). However, the number of apoptotic cells was reduced in
the tranilast pre-treatment group with 50 (P=0.0002), 100
(P<0.001) and 200 µM (P<0.001) compared with that in the LPS
group. Consistently, western blot analysis further confirmed the
TUNEL assay results. Pre-treatment of the BEAS-2B cells with
tranilast reversed the LPS-mediated decreased Bcl2 and increased
Bax and cleaved caspase-3 protein expression levels (Fig. 3B), indicating that tranilast could
alleviate LPS-induced BEAS-2B cell apoptosis.
Tranilast suppresses LPS-induced
activation of the CXCR4/JAK2/STAT3 signaling pathway
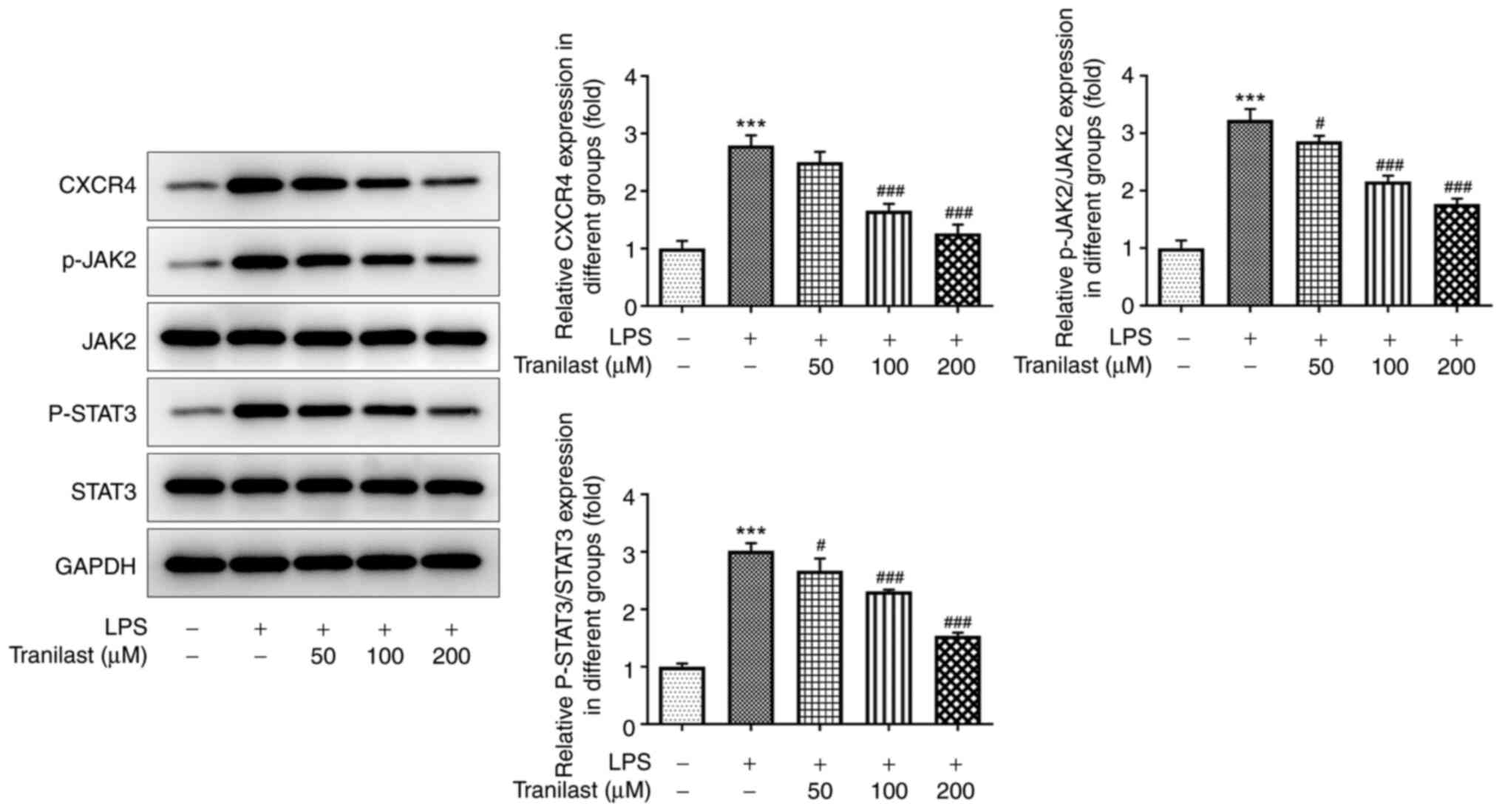
To confirm the association between tranilast and
CXCR4 in ALI, the protein expression levels of CXCR4 and those of
its downstream targets, JAK-2 and STAT3 were evaluated using
western blot analysis. The results showed that CXCR4, p-JAK2 and
p-STAT3 (P<0.001) levels were upregulated in the LPS group
compared with those in the control group. The protein expression
level of CXCR4 was not significantly different following treatment
with 50 µM tranilast (P=0.2226), while the protein expression level
of CXCR4 was decreased following treatment with 100 and 200 µM
tranilast (both P<0.001). Compared with that in the LPS group,
the protein expression level of p-JAK2 decreased following
treatment with 50 (P=0.0318), 100 (P=0.0318) and 200 µM tranilast
(P<0.001). Compared with that in the LPS group, the protein
expression level of p-STAT3 decreased with 50 (P=0.0346), 100
(P=0.0002) and 200 µM tranilast (P<0.001) (Fig. 4). The improvement of LPS-induced
lung injury was significant when 200 µM tranilast was used;
therefore, this concentration was used for subsequent experiments.
These results suggested that tranilast could attenuate LPS-induced
activation of the CXCR4/JAK2/STAT3 signaling pathway in BEAS-2B
cells.
CXCR4 overexpression diminishes the
alleviative effect of tranilast on LPS-induced BEAS-2B cell
viability
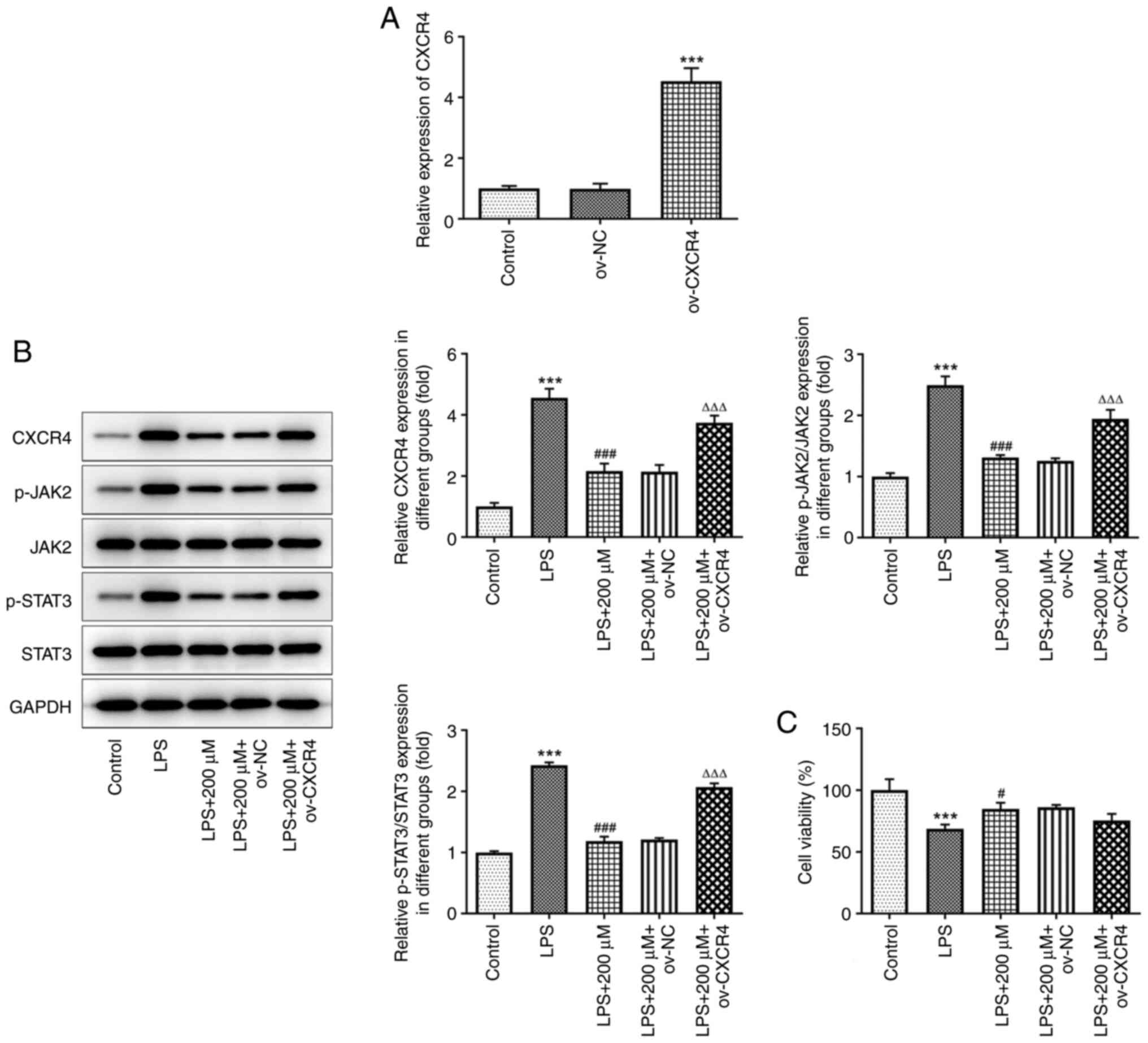
The transfection efficiency of the BEAS-2B cells
with ov-CXCR4 was confirmed using RT-qPCR. The results confirmed
that CXCR4 was successfully overexpressed in the BEAS-2B cells
transfected with ov-CXRC4 (P<0.001) (Fig. 5A). As shown in Fig. 5B, LPS notably elevated the protein
expression levels of CXCR4, p-JAK2 and p-STAT3, while pre-treatment
with tranilast (200 µM) significantly reduced their expression
levels (P<0.001). However, CXCR4 overexpression upregulated the
protein expression level of CXCR4, p-JAK2 and p-STAT3 in
LPS-stimulated BEAS-2B cells pre-treated with tranilast. In
addition, CXCR4 overexpression attenuated cell viability in
LPS-stimulated BEAS-2B cells pre-treated with tranilast (Fig. 5C). These results suggested that
tranilast could attenuate LPS-induced cell viability by inhibiting
the CXCR4/JAK2/STAT3 signaling.
CXCR4 overexpression abrogates the
suppressive effect of tranilast on LPS-induced secretion of
inflammatory cytokines in the BEAS-2B cells
Cell pre-treatment with tranilast could decrease the
mRNA expression level of TNF-α, IL-1β and IL-6 in LPS-stimulated
BEAS-2B cells; however, the results showed that CXCR4
overexpression could reverse this effect (all P<0.001) (Fig. 6A). Compared with that in the LPS +
200 µM + ov-NC group, the protein expression level of COX2
(P=0.0315) and iNOS (P=0.0019) was increased in the LPS + 200 µM +
ov-CXCR4 group (Fig. 6B). This
finding suggested that tranilast could suppress LPS-induced
pro-inflammatory cytokine release by inhibiting the activation of
CXCR4/JAK2/STAT3 signaling.
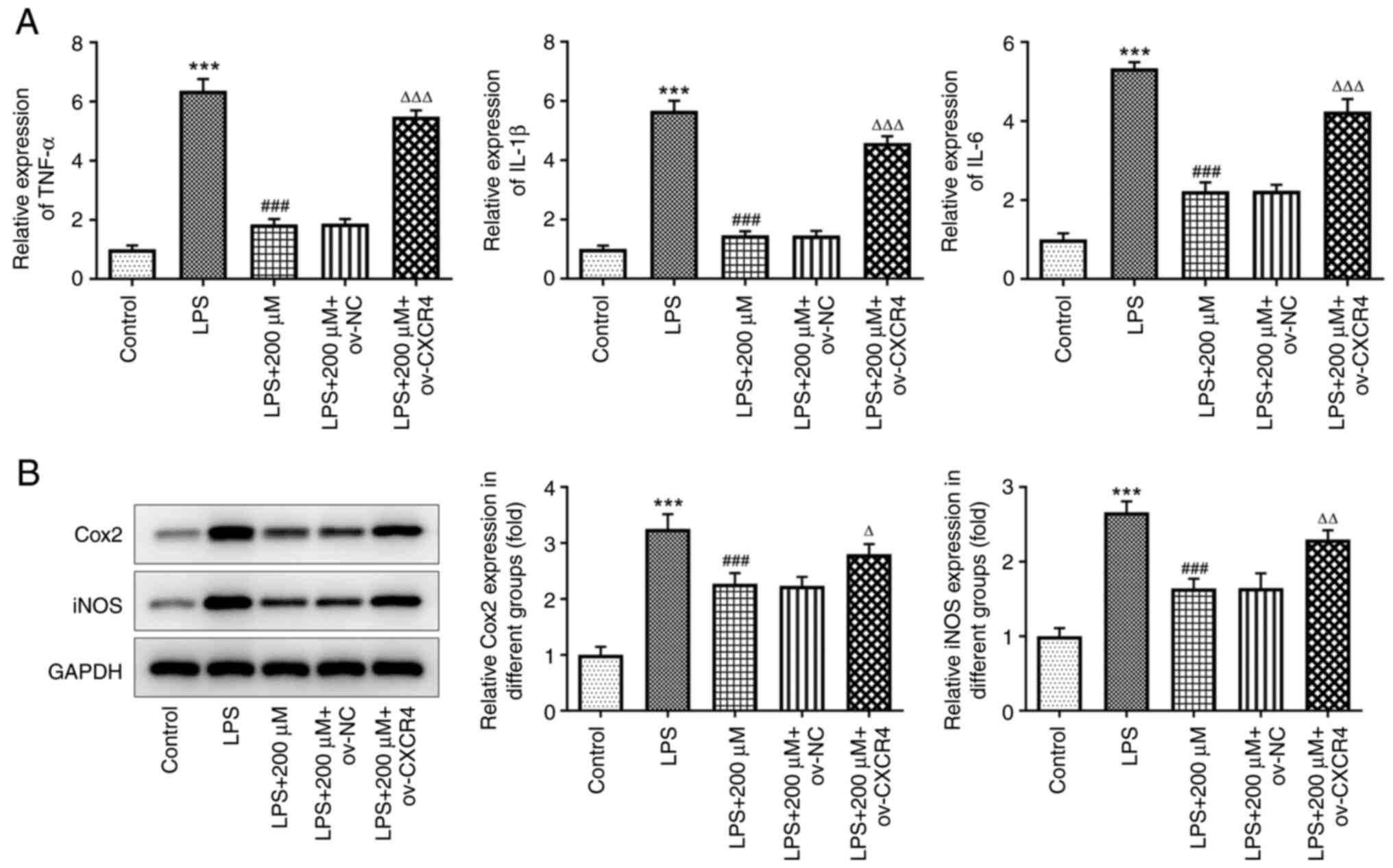 | Figure 6.CXCR4 overexpression reduces the
suppressive effect of tranilast on LPS-induced inflammatory
cytokine release. (A) mRNA expression level of TNF-α, IL-1β, IL-6,
and (B) protein expression level of COX2 and iNOS in LPS-stimulated
BEAS-2B cells, pretreated with tranilast (200 µM) and transfected
with ov-NC or ov-CXCR4. ***P<0.001 vs. control;
###P<0.001 vs. LPS; ∆P<0.05,
∆∆P<0.01, ∆∆∆P<0.001 vs. ov-NC. LPS,
lipopolysaccharide; ov, overexpression; NC, negative control;
CXCR4, C-X-C motif chemokine receptor 4; COX2, cytochrome c
oxidase subunit II; iNOS, inducible nitric oxide. |
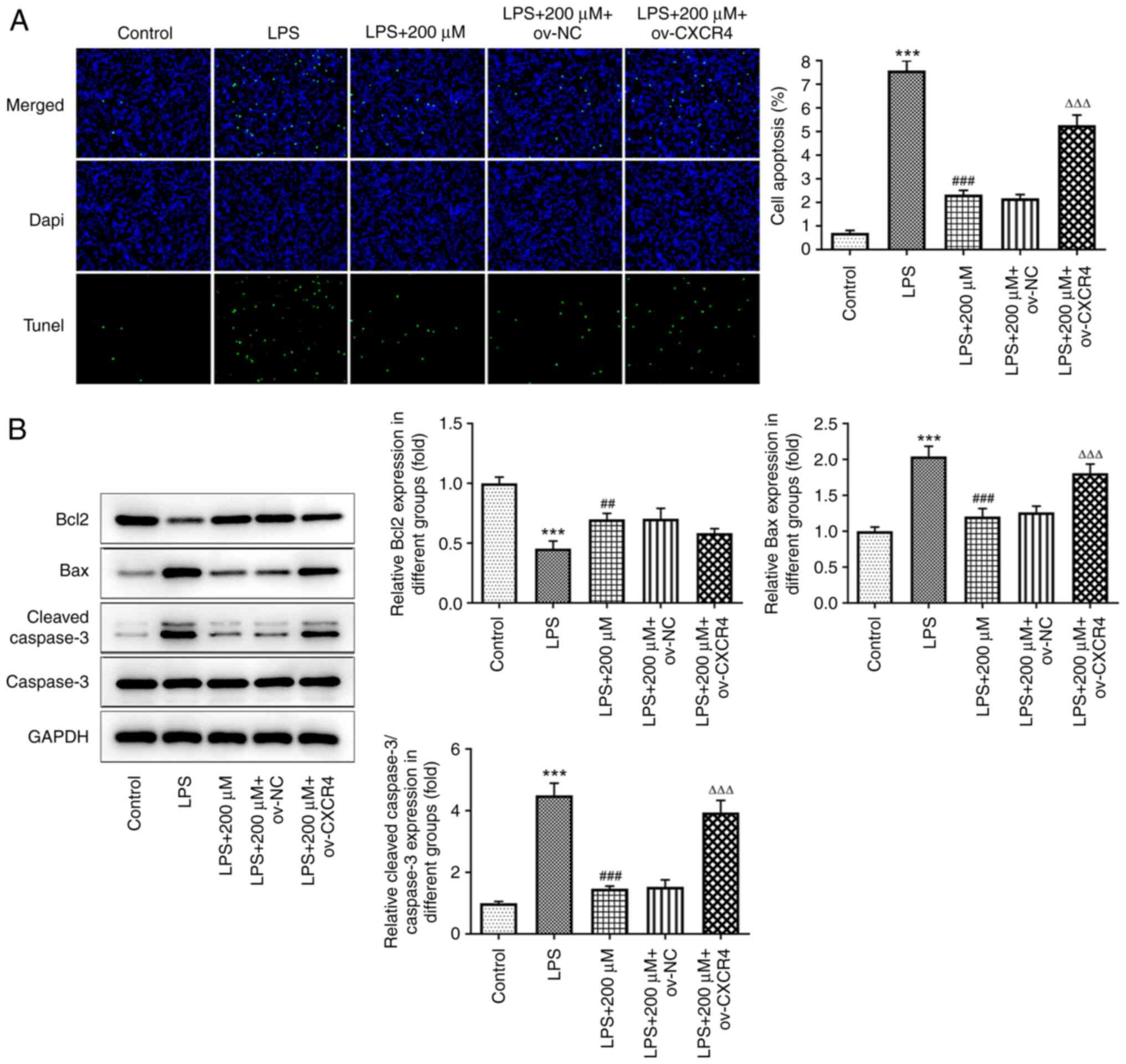
CXCR4 overexpression attenuates the
alleviative effect of tranilast on LPS-induced BEAS-2B cell
apoptosis
TUNEL assay results revealed that CXCR4
overexpression reversed the LPS-mediated increased apoptosis rate
in the BEAS-2B cells pre-treated with tranilast (P<0.001)
(Fig. 7A). Western blot analysis
also demonstrated that the protein expression levels of Bcl2 were
reduced, while those of Bax and cleaved caspase-3 were enhanced
following CXCR4 overexpression in LPS-stimulated BEAS-2B cells
pre-treated with tranilast (Fig.
7B). Therefore, tranilast could alleviate LPS-induced cell
apoptosis via inhibiting CXCR4/JAK2/STAT3 signaling.
Discussion
Sepsis, a common clinical complication of severe
infectious diseases, is characterized by multi-organ failure, as a
result of LPS-induced systemic inflammatory responses (21). Furthermore, ALI is a serious
complication that can develop in the early stages of sepsis, with
an incidence rate of 70% and a mortality rate of 50–70%, without
timely treatment (22,23). Therefore, it is crucial to
identify novel agents for protecting the lungs from exacerbated
sepsis-induced inflammatory injury.
The anti-allergic drug tranilast could inhibit the
release of chemical mediators, such as histamine, thereby exerting
an anti-inflammatory effect (24,25). Emerging evidence suggests that
tranilast exhibits a broad range of anti-inflammatory properties.
Previous studies have shown that tranilast preconditioning can
attenuate the LPS-induced release of pro-inflammatory cytokines
from macrophages (12,26). An in vivo study also
demonstrated that tranilast effectively attenuated inflammation in
rats with spinal cord injury (27). In addition, tranilast was shown to
improve inflammasome-triggered gouty arthritis by directly
targeting NLRP3 (28).
Importantly, tranilast was shown to exert a significant protective
effect against lung injury by ameliorating cyclophosphamide-induced
lung injury and TGF-β-mediated pulmonary fibrosis (14,15). In the present study, preliminary
experiments revealed a dose-dependent improvement in cell viability
in LPS-stimulated BEAS-2B cells following tranilast
preconditioning. To the best of our knowledge, the results
confirmed, for the first time, the potential protective effect of
tranilast on LPS-induced lung injury.
LPS acts as the main ‘mediator’ of sepsis-induced
multi-organ failure by stimulating neutrophils and macrophages to
release large quantities of inflammatory cytokines, such as TNF-α,
IL-1β and IL-6, as well as inflammation-related enzymes, such as
COX2 and iNOS (29,30). TNF-α not only directly damages
vascular endothelial cells in the lungs, but also induces immune
stress by binding to TNF receptors of other inflammatory cells,
such as macrophages and mast cells, thus promoting the secretion of
inflammatory cytokines, leading to the initiation of the
inflammatory cascade (31). IL-1β
can further promote immune stress and inflammation, while IL-6
leads to the release of oxygen-derived free radicals, such as
reactive oxygen species. COX2 and iNOS have been associated with
the inflammatory response in several pulmonary diseases, including
pulmonary fibrosis, ALI and lung cancer (32–34). The results of the current study
showed that pre-treatment of LPS-induced BEAS-2B cells with
tranilast notably downregulated the mRNA expression levels of
TNF-α, IL-1β, IL-6, COX2 and iNOS, suggesting that tranilast could
exert an anti-inflammatory effect on sepsis-induced ALI.
To further verify the aforementioned results, the
mechanism underlying the effects of tranilast on ALI was
investigated. CXCR4, a chemokine receptor, is a highly conserved
7-pass transmembrane G-protein-coupled receptor, that plays a vital
regulatory role in morphogenesis, angiogenesis and immune responses
(35,36). A previous study revealed an
aberrant upregulation of CXCR4 in an in vitro ALI model
(37). In addition, a study
showed that the reduced expression levels of CXCR4 mRNA and the
decreased number of neutrophils could largely improve ALI in
sepsis, thus suggesting that CXCR4 knockdown could be used in ALI
treatment (18). A previous study
also showed that combination treatment of tamoxifen and tranilast
could downregulate CXCR4 mRNA expression in breast cancer cell
lines (17). Kyoto Encyclopedia
of Genes and Genomes pathway enrichment analysis also predicted
that chemokine signaling could activate JAK2/STAT3 signaling,
thereby affecting cell survival and apoptosis (38). In the present study, pre-treatment
with tranilast attenuated LPS-induced activation of the
CXCR4/JAK2/STAT3 signaling pathway, while CXCR4 overexpression
diminished the alleviative effect of tranilast on LPS-induced
BEAS-2B cell viability, the inflammatory response and apoptosis,
which is the novelty of the present study. Those were what make our
paper innovative.
The present study examined the effect of tranilast
on LPS-induced lung injury at the cellular level; however, further
studies using animal models are required to confirm the
results.
In conclusion, the present study showed that
tranilast alleviated LPS-induced BEAS-2B cell viability,
inflammatory response and apoptosis by attenuating CXCR4/JAK2/STAT3
signaling. Therefore, the results confirmed the curative potential
of tranilast in treating sepsis-induced ALI and partly uncovered
its underlying mechanism of action. However, more experiments are
required to verify the results of the current study in
vivo.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YL and YZ wrote the manuscript and analyzed the
data. ZH and HW performed experiments, supervised the present
study, searched the literature and revised the manuscript. All
authors have read and approved the final manuscript. YL and YZ
confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Singer M, Deutschman CS, Seymour CW,
Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche
JD, Coopersmith CM, et al: The third international consensus
definitions for sepsis and septic shock (Sepsis-3). JAMA.
315:801–810. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fleischmann C, Scherag A, Adhikari NK,
Hartog CS, Tsaganos T, Schlattmann P, Angus DC and Reinhart K;
International Forum of Acute Care Trialists, : Assessment of global
incidence and mortality of hospital-treated sepsis. Current
estimates and limitations. Am J Respir Crit Care Med. 193:259–272.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen X, Wang T, Song L and Liu X:
Activation of multiple toll-like receptors serves different roles
in sepsis-induced acute lung injury. Exp Ther Med. 18:443–450.
2019.PubMed/NCBI
|
|
4
|
Liu X, Chen H, Hou Y, Ma X, Ye M, Huang R,
Hu B, Cao H, Xu L, Liu M, et al: Adaptive EGF expression sensitizes
pancreatic cancer cells to ionizing radiation through activation of
the cyclin D1/P53/PARP pathway. Int J Oncol. 54:1466–1480.
2019.
|
|
5
|
Hong G, Zheng D, Zhang L, Ni R, Wang G,
Fan GC, Lu Z and Peng T: Administration of nicotinamide riboside
prevents oxidative stress and organ injury in sepsis. Free Radic
Biol Med. 123:125–137. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yanling Q, Xiaoning C, Fei B, Liyun F,
Huizhong H and Daqing S: Inhibition of NLRP9b attenuates acute lung
injury through suppressing inflammation, apoptosis and oxidative
stress in murine and cell models. Biochem Biophys Res Commun.
503:436–443. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Darakhshan S and Pour AB: Tranilast: A
review of its therapeutic applications. Pharmacol Res. 91:15–28.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Abe S, Makimura S, Kawakami Y, Yamamoto H,
Nojima T and Inoue K: Cytomorphologic features of antiallergic
drug-induced eosinophilic cystitis with bronchial asthma. Hokkaido
Igaku Zasshi. 62:907–912. 1987.
|
|
9
|
Nader MA, Gameil N, Abdelaziz RR, Zalata
KR, Osman A, Zedan MM, Abo-Elkheir N, Elsiddig AA and Zedan M:
Effect of tranilast in comparison with beclomethasone in chronic
murine model of asthma. Exp Lung Res. 42:296–306. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sun X, Suzuki K, Nagata M, Kawauchi Y,
Yano M, Ohkoshi S, Matsuda Y, Kawachi H, Watanabe K, Asakura H and
Aoyagi Y: Rectal administration of tranilast ameliorated acute
colitis in mice through increased expression of heme oxygenase-1.
Pathol Int. 60:93–101. 2010. View Article : Google Scholar
|
|
11
|
Chen S, Pan Y, Zheng S, Liu Y, Xu J, Peng
Y, Zhang Z, Wang Y, Xiong Y, Xu L, et al: Novel role for tranilast
in regulating NLRP3 ubiquitination, vascular inflammation, and
atherosclerosis. J Am Heart Assoc. 9:e0155132020. View Article : Google Scholar
|
|
12
|
Pae HO, Jeong SO, Koo BS, Ha HY, Lee KM
and Chung HT: Tranilast, an orally active anti-allergic drug,
up-regulates the anti-inflammatory heme oxygenase-1 expression but
down-regulates the pro-inflammatory cyclooxygenase-2 and inducible
nitric oxide synthase expression in RAW264.7 macrophages. Biochem
Biophys Res Commun. 371:361–365. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Suzawa H, Kikuchi S, Ichikawa K and Koda
A: Inhibitory action of tranilast, an anti-allergic drug, on the
release of cytokines and PGE2 from human monocytes-macrophages. Jpn
J Pharmacol. 60:85–90. 1992. View Article : Google Scholar
|
|
14
|
Said E, Elkashef WF and Abdelaziz RR:
Tranilast ameliorates cyclophosphamide-induced lung injury and
nephrotoxicity. Can J Physiol Pharmacol. 94:347–358. 2016.
View Article : Google Scholar
|
|
15
|
Kato M, Takahashi F, Sato T, Mitsuishi Y,
Tajima K, Ihara H, Nurwidya F, Baskoro H, Murakami A, Kobayashi I,
et al: Tranilast inhibits pulmonary fibrosis by suppressing
TGFβ/SMAD2 pathway. Drug Des Devel Ther. 14:4593–4603. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen S, Wang Y, Pan Y, Liu Y, Zheng S,
Ding K, Mu K, Yuan Y, Li Z, Song H, et al: Novel role for tranilast
in regulating NLRP3 ubiquitination, vascular inflammation, and
atherosclerosis. J Am Heart Assoc. 9:e0155132020. View Article : Google Scholar
|
|
17
|
Darakhshan S, Bidmeshkipour A, Mansouri K,
Saeid HM and Ghanbari A: The effects of tamoxifen in combination
with tranilast on CXCL12-CXCR4 axis and invasion in breast cancer
cell lines. Iran J Pharm Res. 13:683–693. 2014.PubMed/NCBI
|
|
18
|
Hirano Y, Ode Y, Ochani M, Wang P and Aziz
M: Targeting junctional adhesion molecule-C ameliorates
sepsis-induced acute lung injury by decreasing CXCR4(+) aged
neutrophils. J Leukoc Biol. 104:1159–1171. 2018. View Article : Google Scholar
|
|
19
|
Liang T, Wang B, Li J and Liu Y: LINC00922
Accelerates the proliferation, migration and invasion of lung
cancer via the miRNA-204/CXCR4 axis. Med Sci Monit. 25:5075–5086.
2019. View Article : Google Scholar
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dickson RP, Singer BH, Newstead MW,
Falkowski NR, Erb-Downward JR, Standiford TJ and Huffnagle GB:
Enrichment of the lung microbiome with gut bacteria in sepsis and
the acute respiratory distress syndrome. Nat Microbiol.
1:161132016. View Article : Google Scholar
|
|
22
|
Rubenfeld GD, Caldwell E, Peabody E,
Weaver J, Martin DP, Neff M, Stern EJ and Hudson LD: Incidence and
outcomes of acute lung injury. N Engl J Med. 353:1685–1693. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Villar J, Sulemanji D and Kacmarek RM: The
acute respiratory distress syndrome: Incidence and mortality, has
it changed? Curr Opin Crit Care. 20:3–9. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Huang L, Dong Y, Wu J, Wang P, Zhou H, Li
T and Liu L: Sinomenine-induced histamine release-like
anaphylactoid reactions are blocked by tranilast via inhibiting
NF-κB signaling. Pharmacol Res. 125:150–160. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shimizu T, Kanai K, Kyo Y, Asano K,
Hisamitsu T and Suzaki H: Effect of tranilast on matrix
metalloproteinase production from neutrophils in-vitro. J Pharm
Pharmacol. 58:91–99. 2006. View Article : Google Scholar
|
|
26
|
Hiraide S, Yanagawa Y and Iizuka K:
Tranilast inhibits interleukin-33 production by macrophages. Eur J
Pharmacol. 818:235–240. 2018. View Article : Google Scholar
|
|
27
|
Hanada M, Tsutsumi K, Arima H, Shinjo R,
Sugiura Y, Imagama S, Ishiguro N and Matsuyama Y: Evaluation of the
effect of tranilast on rats with spinal cord injury. J Neurol Sci.
346:209–215. 2014. View Article : Google Scholar
|
|
28
|
Huang Y, Jiang H, Chen Y, Wang X, Yang Y,
Tao J, Deng X, Liang G, Zhang H, Jiang W and Zhou R: Tranilast
directly targets NLRP3 to treat inflammasome-driven diseases. EMBO
Mol Med. 10:e86892018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jiang WY, Ren J, Zhang XH, Lu ZL, Feng HJ,
Yao XL, Li DH, Xiong R, Fan T and Geng Q: CircC3P1 attenuated
pro-inflammatory cytokine production and cell apoptosis in acute
lung injury induced by sepsis through modulating miR-21. J Cell Mol
Med. 24:11221–11229. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xu J, Zhao Y and Aisa HA:
Anti-inflammatory effect of pomegranate flower in
lipopolysaccharide (LPS)-stimulated RAW264.7 macrophages. Pharm
Biol. 55:2095–2101. 2017. View Article : Google Scholar
|
|
31
|
Li P and Schwarz EM: The TNF-alpha
transgenic mouse model of inflammatory arthritis. Springer Semin
Immunopathol. 25:19–33. 2003. View Article : Google Scholar
|
|
32
|
Liu R, Xu KP and Tan GS: Cyclooxygenase-2
inhibitors in lung cancer treatment: Bench to bed. Eur J Pharmacol.
769:127–133. 2015. View Article : Google Scholar
|
|
33
|
Choi S, Lim JW and Kim H: Effect of thiol
antioxidants on lipopolysaccharide-induced cyclooxygenase-2
expression in pulmonary epithelial cells. J Physiol Pharmacol.
69:127–133. 2018.
|
|
34
|
Zheng H, Liang W, He W, Huang C, Chen Q,
Yi H, Long L, Deng Y and Zeng M: Ghrelin attenuates sepsis-induced
acute lung injury by inhibiting the NF-κB, iNOS, and Akt signaling
in alveolar macrophages. Am J Physiol Lung Cell Mol Physiol.
317:L381–L391. 2019. View Article : Google Scholar
|
|
35
|
Strien L, Joensuu K, Heikkila P and
Leidenius MH: Different expression patterns of CXCR4, CCR7, maspin
and FOXP3 in luminal breast cancers and their sentinel node
metastases. Anticancer Res. 37:175–182. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pozzobon T, Goldoni G, Viola A and Molon
B: CXCR4 signaling in health and disease. Immunol Lett. 177:6–15.
2016. View Article : Google Scholar
|
|
37
|
McClendon J, Jansing NL, Redente EF,
Gandjeva A, Ito Y, Colgan SP, Ahmad A, Riches DW, Chapman HA, Mason
RJ, et al: Hypoxia-inducible factor 1α signaling promotes repair of
the alveolar epithelium after acute lung injury. Am J Pathol.
187:1772–1786. 2017. View Article : Google Scholar
|
|
38
|
Kanehisa M and Goto S: KEGG: kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|