Introduction
The Brca1 and Brca2 genes play central
roles in homologous recombination repair (HRR) (1,2).
Mutations of Brca1 and Brca2 are normally observed in
many familial cases; for example, mutations in rare
high-penetrating breast cancer account for 16–25% of the inherited
cancer type (3). Given the high
prevalence of polymorphisms of these two genes, some agents are
unable to target the translated proteins and poly(ADP-ribose)
polymerase (PARP) provides an alternative approach by targeting the
HRR pathway (4). In recent years,
several PARP inhibitors have been approved by the US Federal Drug
Administration (FDA) as effective chemotherapy reagents for various
cancers with mutations in Brca1 and Brca2 (5). Acting as substitutes for
Brca1, a series of genes have been identified as DNA repair
regulators, such as Nbn, Fancd2, Rad51, Lig3, Rad51C, Rad21
and Esco1 (6). All of
these genes and related pathways are closely linked to the HRR
pathway and the response to PARP inhibitors; however, only 40% of
patients who receive PARP inhibitor treatment respond effectively.
Although platinum sensitivity has been suggested as a hallmark of
response to PARP inhibitors, patients with platinum-resistant
disease still respond to Olaparib (1). Therefore, a thorough understanding
of the genetic determinants that contribute to the sensitivity of
PARP inhibitors will increase their clinical utility.
A central player in the HRR pathway is the RAD51
recombinase that binds to single-stranded DNA at break sites
(7). RAD51 can bind to the break
sites participating with BRCA2 and five other paralogs, namely,
RAD51B, RAD51C, RAD51D, XRCC2, and XRCC3 (8). Cruz et al regarded Rad51
nuclear foci as an indicator of PARP inhibitor resistance in breast
or ovarian cancer with germline Brca1/2 (gBRCA) mutations
(9) while others have considered
them regulators of sensitivity to PARP inhibitors beyond
Brca1/2-related cancers (10). Olaparib-RAD51 inhibitor conjugates
have also been reported to break resistance mechanisms to olaparib
treatment in triple-negative breast cancer cells regardless of BRCA
status (11). RAD51 nuclear foci
formation is also closely correlated with TOPBP1-dependent
phosphorylation and contributes to PARP inhibitor resistance
(12). Some mutation signatures
of Rad51 and the participating proteins are closely
correlated with the resistance to PARP inhibitors, although other
Rad51 paralogs and participating protein mutants are also
sensitive to PARP inhibitors (13–15). Therefore, more studies are
necessary to identify the genetic polymorphisms of Rad51
that may contribute to sensitivity to the PARP inhibitor.
Clustered regular interval short palindromic repeat
(CRISPR)-directed Cas9-mediated endonuclease activity has been
described as an effective strategy to induce saturated mutations in
a specific genome locus, and mutations have been identified that
contribute to drug resistance (16,17). Compared to a random screening
strategy, a site-specific saturated mutation approach has been more
effective and specific in discovering drug resistance mutations
(16–18). Moreover, mutations near specific
loci are more easily inherited as haplotypes. Compared to
individual single nucleotide polymorphism (SNP) sites, the
haplotype represents a more clinically effective biomarker
(19).
An α/β ATPase core domain similar to those present
in helicases is also found in the RAD51 protein. WALKER A and B
motifs in the α/β ATPase domain are key to ATP binding and
hydrolysis (20,21). HRR-related protein complex
assembly and DNA strand exchange activity are coupled to the
binding and hydrolysis of ATP by RAD51 (22). Therefore the WALKER A and B motifs
signaling between binding sites for DNA and nucleotide ligands are
critical for RAD51 recombination activity. WALKER A and B domains
of Rad51 have been identified as key players in the HRR
pathway (23). Previous studies
have also presented series mutations which are requirements for the
WALKER A and B motifs for Rad51D function in response to DNA
damage (22,24). In terms of technical feasibility,
the relatively short length of WALKER A and B motifs (8 and 5 amino
residues, respectively) is within the limit of saturated mutational
library content and suitable for saturated mutagenesis. All of the
above evidence indicates that investigation of all the potential
mutations in the WALKER domain is valuable and feasible.
Identification of these functional mutations and the highly
correlated variations may lead to a high throughput approach for
the selection of pharmacogenomics targets. This study aimed to
identify olaparib sensitivity-related mutations of Rad51
WALKER domain which were induced by a CRISPR/Cas9-based saturated
mutational library in breast cancer cells, and to assess their
clinical value. We selected Rad51 because previous
resistance to PARP inhibitors is associated with the number of
RAD51 nuclear foci (9) and hazard
ratio (HR) efficiency mutations of Rad51 and its paralog
gene are observed in the WALKER domain (13), which is highly conserved in this
gene family. To identify clinically valuable mutations, we chose to
target an exon in a clinically relevant domain in which known
mutations are associated with poor outcomes. Previous molecular
studies of three nonsense mutations located in the WALKER motif
greatly contributed to HRR efficiency (15). We hypothesized that saturation
genome editing would result in a wide range of outcomes.
Materials and methods
Cell line sensitivity to olaparib
(Pilot study)
The five candidate breast cancer cell lines
(HCC1599, BT-549, Hs578T, MDA-MB-231 and MDA-MB-453) were seeded in
6-well plates (4,000 cells/well) and incubated in Dulbecco's
modified Eagle's medium (DMEM) or Roswell Park Memorial
Institute-1640 (RPMI-1640) medium with 10% fetal bovine serum
(FBS). Drug-containing medium (50 µM olaparib) was refreshed weekly
(25). A total of 106
cells were digested with trypsin after 24 h of culturing, followed
by centrifugation at 110 × g for 5 min, and then cleaned twice and
centrifuged at 110 × g for a further 5 min. The collected cells
were fixed with 1 ml ethanol at 4°C overnight. Then, 100 µl of
RNase A (Solarbio) was added to incubate the cells at 37°C in the
dark for 30 min. Propidium iodide (PI) (50 µl) was then added.
After further incubation in the dark for 1 h at 25°C, the cells
were detected by flow cytometry (Becton, Dickinson and Company).
Finally, the flow cytometric data were analyzed using FlowJo
software version 10 (Tree Star, Inc.). All the cell culture media
and related reagent were purchased from Thermo Fisher Scientific,
Inc.
Construction of the Cas9 and sgRNA
vectors
Five sgRNAs (sgRNA1-1, sgRNA1-2, sgRNA2-1, sgRNA2-2,
and sgRNA2-3) were selected in the WALKER A and B Rad51
domains using an online predictor tool CCTOP (http://crispr.cos.uni-heidelberg.de)
(26). The sgRNAs were then
cloned into a human codon-optimized Streptococcus (S.
pyogenous) Cas9-sgRNA vector (pX330-U6-Chimeric_BB-CBh-hSpCas9;
Addgene plasmid #42230, Addgene). There were three reasons to
choose this cell line. i) The function of Rad51 is closely
linked with Brca1/2 whose gene products act upstream of
Rad51 in HR (27). In
order to eliminate interference influence of the BRCA gene
status, the candidate cell lines do not carry the Brca1/2
mutations, as indicated by the American Type Culture Collection
(ATCC) document (characterization data and other information
relating to this material are available at www.atcc.org). ii) Given the high cost and off-target
ratio of CRISPR/Cas9-based experiments, it is sensible to choose
cells which have been used in similar experiments, as is the case
for BT-549 cells (28). iii)
Cells should have high sensitivity to olaparib, and in the pilot
study (described above; see also Results and Fig. S1) BT-549 cells showed the highest
apoptosis ratio induced by olaparib. Thus, BT-549 cells were
selected as the in vitro cell model for this study. Five
sgRNAs (the top sgRNAs scored by CCTOP) for four targeted sites
were selected for each predicted locus and were cloned into the
CRISPR/Cas9 vector (Table I). The
constructed vector was packaged in lentiviruses, which were then
transfected into BT-549 cells. One microgram of the Cas9-sgRNA
vector was used to transfect BT-549 cells by electroporation.
Briefly, the 2×106 cells with Cas9-sgRNA vector complex
per reaction were used for electroporation following the standard
instruction of the Neon™ Transfection System (cat. no. MPK10025,
Thermo Fisher Scientific, Inc.). The program (1600 V/10 msec/3
pulses) was adopted for electroporation at room temperature. After
the electroporation, the cells were immediately transferred to a
24-well plate containing 0.5 ml of pre-warmed culture medium for
recovery at 37°C. After 48 h of recovery, genomic DNA was extracted
for polymerase chain reaction (PCR). The cleavage efficiency was
evaluated using the T7E1 cleavage assay (NEB) as described
previously (27). Potential
off-target sites (OTS) were predicted using the CCTOP online
prediction tool, amplified from the genomic DNA of the Rad51
KO BT_549, and sequenced. Bulk PCR products can also be sequenced
using a next generation sequencing (NGS) approach followed by
software analysis. The off-target ratios were indicated by the
off-target/total reads count. Potential off-target sites were
predicted using the CCTOP online predicting tool and then amplified
from the genomic DNA extracted from Rad51-knockout (KO)
BT-549 cells. The variants were identified by sequence alignment.
The primers used to amplify the OTS are listed in Table SI.
 | Table I.sgRNAs for the WALKER domain A and
B. |
Table I.
sgRNAs for the WALKER domain A and
B.
| Name | Target domain | DNA strand | Sequence
(5′-3′) |
|---|
| sgRNA-1-1 | WALKER domain
A | (+) |
GGATCTATCACAGAAATGTTTGG |
| sgRNA-1-2 | WALKER domain
A | (−) |
AGCTAGCGTATGACAGATCTGGG |
| sgRNA-2-1 | WALKER domain
B | (+) |
AAGGGACCAGAATCTGACACAGG |
| sgRNA-2-2 | WALKER domain
B | (−) |
GTCTGTTCTGTAAAGGGCGGTGG |
| sgRNA-2-3 | WALKER domain
B | (+) |
AGAACAGACTACTCGGGTCGAGG |
Western blot analysis
Total proteins were extracted by using RIPA Lysis
Buffer (cat. no. P0013E; Beyotime Institute of Biotechnology). The
BCA Protein Assay kit (cat. no. P0012S; Beyotime Institute of
Biotechnology) was used to determine the concentration of protein
extracted from each sample. SDS-PAGE (on 10% gel) was then adopted
to isolate the protein (40 µg) from each sample after which samples
were removed to a polyvinylidene fluoride (PVDF) membrane. The film
was then blocked for 1 h in 5% nonfat milk at 4°C. The membrane was
then incubated in primary monoclonal and secondary antibodies (all
from Abcam) against GAPDH (cat. no. ab8245, dilution 1:1,000),
pro-caspase-3 (cat. no. ab179517, dilution 1:1,000), cleaved
caspase 3 (cat. no. ab2302, dilution 1:500), Bcl-2 (cat. no.
ab32124, dilution 1:500), Bax (cat. no. ab205718, dilution 1:500)
for 2 h at 4°C. The secondary antibodies were anti-rabbit (cat. no.
ab205718, dilution 1:1,000) conjugated with horseradish peroxidase
for 1 h at room temperature. Lastly, the signals were visualized
using an enhanced ECL Western blotting kit (Abcam), and the optical
density of each band was analyzed by Gel-Pro-Analyze software
(version 4.5; Media Cybernetics, Inc.).
Homology-directed repair (HDR) library
construction
Given the high number of variants in the
homology-directed repair (HDR)-edited loci, evaluation of each
variant in gDNA from a population of unedited cells would require a
large amount of sequencing and sufficient cells. To improve the HDR
efficiency, we introduced mutations between the protospacer
adjacent motif (PAM) and the protospacer sequences, which would
prevent Cas9 from recutting HDR-edited genomes. Based on a previous
study, we designed PCR-selective sites in the sequence (17). An HDR library containing all 6,859
possible DNA residues substituted at positions +37 to +40 and +52
to 55 of Rad51 exon 5 and +17 to +19 of exon 8 was
constructed using series of oligonucleotide. The oligonucleotide
was then cloned through the In-Fusion reaction (Clontech; Takara
Bio USA) into a pUC19 vector containing a preinserted 1000 bp
fragment as homologous arms (Table
II). Finally, the amplification products were purified and
ligated in a pRNDM vector in frame as described above (28).
| Table II.List of primer sequences used for the
HDR donor library construction. |
Table II.
List of primer sequences used for the
HDR donor library construction.
| Name | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| WALKERA-1 |
ACAGGTGCCCATCACCAGTTCC |
CTTGGGAGGCCAAGGTGGGT |
| WALKERA-2 |
TTGAAACAGGGTCTCACTGTG |
TTACGCCTATAATTCCAGCAC |
| WALKERB-1 |
TGACACTCTTGCTTTGTAG |
TGCATGGGCGATGATATTTCC |
| WALKERB-2 |
TGCCTCAGATGTTAGTACTGT |
ATGTCTTCACAGTTAATATGA |
| WALKERB-3 |
AAACATCTATATACAGGCCGG |
ACAGTAGCTTTAACAGGGCT |
Lentiviral particle production
Thirteen million 293(T) cells (ATCC) were cultured
in a 15-cm dish with 15 ml Opti-MEM (Thermo Fisher Scientific,
Inc.) in a 5% CO2 incubator at 37°C with 95% humidity.
The third-generation packaging system was used in this experiment.
Transfection of 16 µg genome-editing vectors (donor template
library, lenti-Cas9) with 16 µg packaging plasmid psPAX2 and 6 µg
envelope plasmid pMD2.G (Thermo Fisher Scientific, Inc.) were mixed
in 1 ml Opti-MEM. About 76 µl of 1 mg/ml polyethylenimine
(Polysciences Inc.) was mixed with the plasmids in 1 ml Opti-MEM
for 15 min at room temperature. The DNA/PEI mixture was then added
to the 293(T) cells in Opti-MEM. Forty-eight hours after
transfection, cell culture medium was collected and centrifuged at
200 × g at 4°C for 10 min. The supernatant was then filtered
through Φ 0.45-µm low-protein binding HV/PVDF membrane (Merck
KGaA). The viral dose (multiplicity of infection ≈0.5) was
identified and used to infect the cells. Aliquots of the
supernatant containing viral particles were stored at −80°C until
required.
BT-549 cell culture condition
The human breast cell line BT-549 (ATCC) was used as
the cell model, as explained above. All cell lines were subjected
to confirmation of cell line identity. Cells were cultured in a
medium composed of 10% FBS and 90% RPMI-1640 medium in a 5%
CO2 incubator at 37°C with 95% humidity. Cells were
harvested at 80% confluence and were used for subsequent
experiments. All media were supplemented with 100 U/ml penicillin
and 100 µg/ml streptomycin (Thermo Fisher Scientific, Inc.).
Drug treatment, library construction,
and next-generation sequencing
Construction of the donor libraries was as described
in a previous study (29). Cell
cultures were co-infected with equivalent amounts of amplified
lentiviral library and CRISPR/Cas9-sg Rad51. Cells were
seeded in 100-mm dishes with 3×106 cells per dish.
Puromycin (Merck KGaA) was added into the selection media after
lentiviral infection and cultured at 2 µg/ml for one day. After
selection, the surviving cells were pooled and used for
amplification culture for 7 days. During the amplification culture,
selection medium (containing 2 µg/ml puromycin) was replaced every
2–3 days. The surviving cells were then split into three individual
groups that were treated with DMSO (Merck KGaA), 5 or 10 µM
olaparib for 1 week. For each treatment, 1×107 cells per
replicate were used. Genomic DNA was extracted and real-time PCR
(RT-PCR) was performed using the specific primers listed in
Table III. PCR product was
separated using agarose gel electrophoresis, then an QIAquick Gel
Extraction Kit (cat. no. 28704, Qiagen, Inc.) was used to isolate
DNA. The concentration of the DNA was quantified using a Qubit 2.0
Fluorometer (Thermo Fisher Scientific, Inc.). DNA quality was
determined by measuring 260/280 and 230/260 absorbance ratios on a
spectrophotometer (Nanodrop ND-1000, Peqlab Biotechnologie). The
DNA integrity number was determined using the Lab-on-a-Chip-System
Bio-analyzer 2100 (Agilent Technologies, Inc.). The PCR products
were used to prepare the NGS library. The library for sequencing
was constructed using the NEB Next Ultra II DNA Library Prep Kit
(cat. no. NEB#E7645L, NEB) in line with the manufacturer's
instructions. The average insert fragment in the library
(approximately 292 bp) was measured and controlled using the
Bioanalyzer 2100 (Agilent Technologies, Inc.). The final library
concentration was measured and adjusted to approximately 10 ng/µl
using the Qubit 2.0 Fluorometer (Thermo Fisher Scientific, Inc.).
The library was then sequenced using HiSeq 2500 (Illumina, Inc.)
(PE=125; data are available at DOI 10.4121/19086689). Data analysis
was performed using the MAGeCK analysis (http://www.bioconductor.org/packages/release/bioc/html/MAGeCKFlute.html)
(29). The sequencing was
performed using a paired-end 125 (PE125) sequencing model. HRR
efficiencies were evaluated according to the selective PCR results.
Sequencing reads containing the selective PCR site and at least one
variant in the donor library were used to calculate HRR
efficiency.
| Table III.List of primer sequences used for
selective PCR. |
Table III.
List of primer sequences used for
selective PCR.
| Name | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| WALKERA-1 |
ATCTATCACAGAAGCCGGATG |
TATTAACAGCTATAAGATT |
| WALKERA-2 |
TAAGCTTCCTAAAGTGCT |
GCTAGCGTATGACATCAGCTG |
| WALKERB-1 |
GGACCAGAATCTGAGACTCG |
TCTCTCTTCTTCCTAAGAGCT |
| WALKERB-2 |
TCCGGAGGCTGAAGCAGGAG |
CTGTTCTGTAAATGACACTG |
| WALKERB-3 |
ACTGCACTCCAGCCTGGGCG |
CAGCTCTGAAAGCTCACCTCGA |
Patients and clinical samples
A total of 772 breast cancer patients were admitted
to the Affiliated Hospital of Hebei University (China) from August
2012 to September 2019. Among these, 538 patients (females; age
range, 28 to 69 years; median age ± SD, 51.4±3.5 years) diagnosed
with breast cancer were enrolled in this study. The study was
approved by the Review Board of the Affiliated Hospital of Hebei
University (approval number AHHU20200930). The inclusion criteria
included the following: i) newly diagnosed cases; ii)
BRAC1/2 mutation detected; iii) completion of PAPRP
inhibitor (olaparib) treatment and 5 years of follow-up.
Information on patient treatment, recurrence, and survival status
was well documented. Exclusion criteria included: i) recurrent
breast cancer; ii) therapy performed prior to admission; and iii)
comorbidities due to other clinical disorders. All patients were
informed of the purpose of the experiment and signed informed
consent. The clinical characteristics of the study subjects are
shown in Table SII. Among the
538 patients who received the PARP inhibitor treatment, 68 were
cases with a positive family history. The remaining 234 patients
who failed to meet the inclusion criteria underwent surgical
resection and adjuvant chemotherapy between August 2012 and
September 2019. Of this group, those who did not receive PARP
inhibitor treatment were also invited and accepted to participate
as a population control. In the control group, the volunteers
received standard medical and nursing care, such as vital sign
observation, chemotherapy or surgical resection treatment and wound
and pain management. Five patients withdrew voluntarily from
follow-up in the next five years, but they agreed to our use of the
data gathered during their follow-up.
Based on clinical findings, imaging information,
histopathological tests, and other laboratory indicators, patients
(cases) were graded according to the 8th edition AJCC standards
(30). There were 70, 258, and
210 cases at clinical stages I, II, and III–IV, respectively.
Patients were treated with surgical resection accompanied by
chemotherapy. From the day of admission, all patients were followed
for five years by telephone call or outpatient visits. Patients
(n=2) who died from causes unrelated to breast cancer were excluded
from the survival analysis.
Genotyping methods
DNA was extracted from all blood taken from the
patients using a GenElut™ Blood Genomic DNA Kit (Merck KGaA,
Darmstadt, Germany). Five variants of Rad51 (SNPs and repeat
polymorphisms) (Table IV) were
genotyped using SNAPshot (Thermo Fisher Scientific, Inc.).
| Table IV.Three haplotypes of Rad51
WALKER domain in this study. |
Table IV.
Three haplotypes of Rad51
WALKER domain in this study.
| Haplotype | Alternative nuclei
acids | Amino acids | Location (CDS) | Domain | Coefficient (95%
CI)a |
|---|
| Haplotype-1 | CC>AC;
T>A | 129L, 221E | 387-388; 664 | A and B | 1.22 |
| Haplotype-2 | AA>GU | 133V | 397-398 | A | 0.83 |
| Haplotype-3 | GA>CC;
UC>GU | 128P, 129C | 382–383;
386–387 | A | 0.75 |
Haplotype analysis
Haplotype frequencies of Rad51 variants (five
alternative amino acids listed in Table IV) were estimated using the
Expectation-Maximization algorithm in the haplo.stats R software
package (http://cran.r-project.org/web/packages/haplo.stats).
The three assembled haplotypes were estimated separately in breast
cancer patients at the above clinical stages. The R haplo.stats
package was used to estimate the posterior probabilities of the
haplotypes. The STATA haplologit command was used to model the
association between haplotypes and case status as in a previous
study (31).
Cell malignant behavior-related
experiment
Cell Counting Kit-8 assay
The effects of cell transfections on cell
proliferation were analyzed using a CCK-8 assay (Dojindo
Laboratories, Inc.). A total of 5×103 cells were seeded
per well in 0.1 ml medium (containing 5 µM olaparib) in a 96-well
cell culture plate. Three wells were set for each experiment. Cells
were grown at 37°C and collected at 24, 48, 72, and 96 h after the
initiation of cell culture. The CCK-8 solution was added to each
well to reach a final concentration of 10% at 4 h before harvesting
cells. A microplate reader was used to read the OD values at 450
nm.
Colony formation assays
BT-549 cell suspension (1×103 cells per
well) with/without the selected haplotype were obtained by
trypsinization and filtration through a 40-µm filter, after which
the cells were seeded in a 6-well plate (4,000 cells/well)
containing 0.35% soft agar. The plates were then incubated in DMEM
with 10% FBS for 1 day at 37°C. Drug-containing medium (5 µM
olaparib) was refreshed weekly. After two weeks, the cells were
washed twice with PBS and fixed with methanol at 4°C for 15 min,
following which the colonies were stained with 0.1% crystal violet
for 10 min at 25°C.
Statistical analysis
All experiments were conducted in three technical
replicates and three biological replicates, respectively. All data
and survival curves were analyzed using GraphPad Prism (v.6)
(GraphPad Software, Inc.) unless otherwise indicated. The
quantitative data are presented as mean ± standard deviation (SD).
All single nucleotide polymorphism strategies were employed to
construct the haplotype. Haplotype analysis was performed using
PHASE v2.1.1 software (31) and
the frequencies of haplotypes were compared between all induced
mutation cases vs. the wild-type. A logistic multivariate
regression model was used to look for interactions between SNPs of
the WALKER A domain and the genotype of WALKER B domain.
Correlation analysis was carried out using Pearson's correlation
coefficient. K-M plotter was used to plot survival curves, which
were compared using a Mantel-Cox test. Log-rank test was performed
to evaluate the hazard ratio (HR) and 95% confidence interval (CI).
The non-parametric Mann-Whitney test was utilized for comparisons
between two groups. For multiple groups, the Kruskal-Wallis test
was utilized followed by Newman-Keuls test or Tukey's HSD.
P<0.05 and P<0.01 were considered statistically significant
and highly significant, respectively.
Results
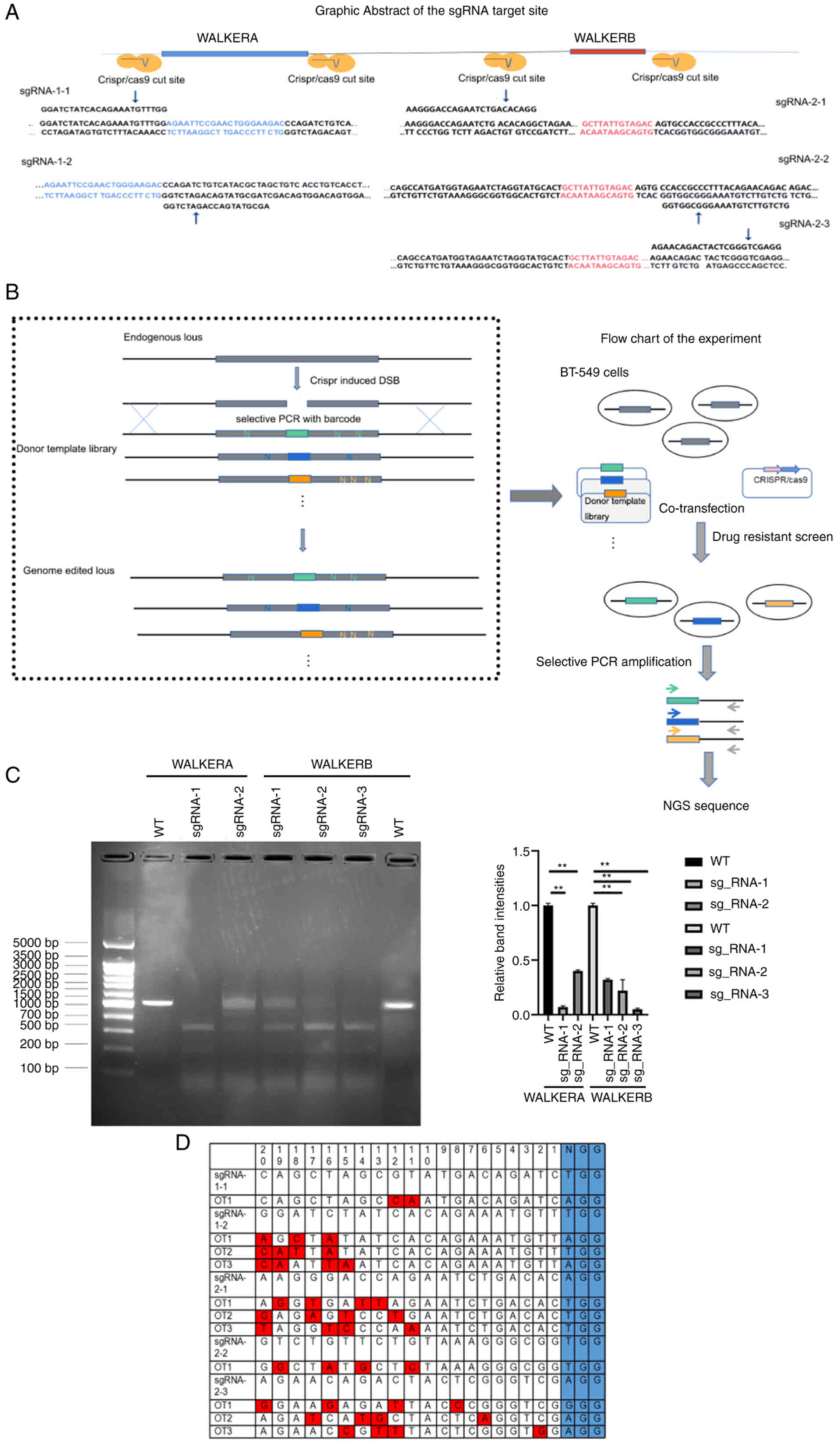
sgRNA design and evaluation
In the pilot study, using flow cytometry we found
that BT-549 cells displayed the highest sensitivity to olaparib
(Fig. S1A and B). The sgRNAs,
which were then designed according to the WALKER A and B
domain-related PAM sequence, were scored using the CCTOP online
prediction tool. The HDR library containing all possible missense
mutations was constructed using a vector library containing a 7-bp
selective PCR site mutation site at the PAM position and the
selective PCR products were then identified by sequencing (Fig. 1A and B). The knockdown
efficiencies for sgRNA1-1, sgRNA1-2, sgRNA2-1, sgRNA2-2, and
sgRNA2-3 were 73.2, 54.9, 33.65, 68.05, and 71.4%, respectively;
the off-target ratios were 1.03, 3.55, 2.71, 0.88, and 2.53%,
respectively. The sgRNA-1-1 and sgRNA-2-2, which displayed
relatively high knock-out efficiency and the lowest off-target
ratios (73.2, 1.03; 68.05, and 0.88%, respectively) were then
chosen for the subsequent experiments (Fig. 1C and D). Eight and five amino
acids were found in the WALKER A and B domains, respectively. The
saturated mutation of this region was obtained by residue
substitutions of all 6,859 possible mutations (Fig. 1). Fig. 1C shows the selective PCR product
region and evaluation of the off-target ratio.
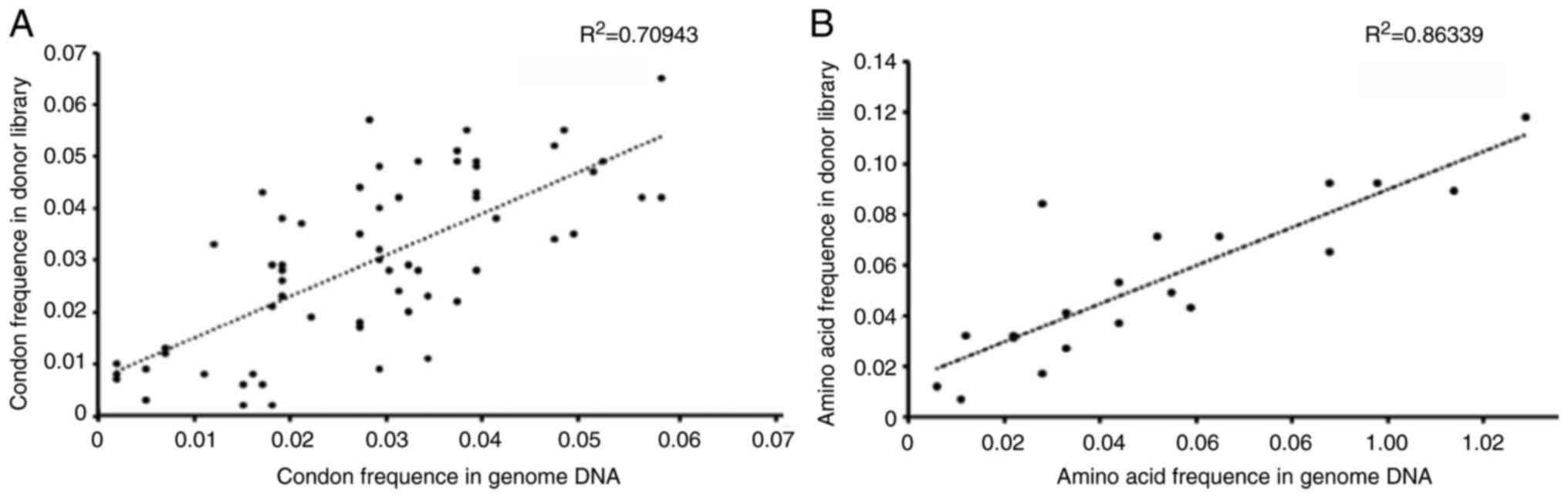
Correlation between the induced
mutagenesis on the target genomic region and the donor
template
Fig. 2A shows that
the frequency of the genomic region mutation was highly correlated
with the donor template library. Five amino acid mutations (128P,
129C, 129L, 133V, and 221E) were associated with high resistance to
olaparib at the amino acid level (Fig. 2B). The alternated wild-type amino
acids for these residues are shown in Table IV. The missense mutation
frequencies of the five alternative amino acids were determined.
According to the targeted NGS results, these SNPs were also
determined at the RNA level using RT-PCR. A good biomarker for
clinical diagnosis should have high signal to noise ratio. Single
missense mutations are rare and have low signal to noise ratio
(32). Several contradictory
studies for the association of Rad51 genetic polymorphisms
with cancer development have supported this viewpoint (33–35), although haplotyping can
significantly overcome this drawback and improve the accuracy of
the genetic polymorphisms (32,36). Therefore we assembled the SNPs
into haplotypes using frequently co-occurring SNPs.
Haplotype assembly and effect
The following three haplotypes were assembled and
identified in the olaparib-resistant cells (number indicates
location of substitutions in cDNA): Haplotype 1
(387–388;664):CC>AC, T>A; Haplotype 2 (397–398):AA>GU;
Haplotype 3 (382–383, 386–387):GA>CC, UC>GU (Fig. 2 and Table IV). Ten single clones were
screened out by olaparib and evaluated by sequencing. All clones
harbored at least one of the three haplotypes. Compared with the
SNP results, it appeared that these haplotypes had greater
discrimination ability in terms of cell survival, and cells with
haplotype 2 and 3 showed the lowest sensitivity to olaparib
(Fig. 3A and B) (P<0.05).
Since RAD51 was a key HRR regulator which directly results in cell
cycle arrest, we then investigated the effect of mutations in this
gene on the cell proliferation and colony formation. The effects of
haplotypes 1, 2 and 3 on the proliferation and cell viability of
BT-549 cells were analyzed using the CCK-8 assay and colony
formation assays. The former showed that harboring of the three
haplotypes led to an increased proliferation of breast cancer cells
(P<0.05) (Fig. 3C). In order
to detect the tumorigenesis of tumor cells with/without the
selected haplotype, colony formation assays was performed.
Similarly, colony formation capacity was also enhanced in cells
with the three haplotypes (Fig.
3D, P<0.05). Since the function of RAD51 is not directly
related to apoptosis and cell migration, we did not investigate its
effect on those cell behaviors. Furthermore, the cells were
screened out in the olaparib-resistant experiment, indicating that
the apoptosis ratio of this type of cell is very low and that the
apoptosis difference would be difficult to determine
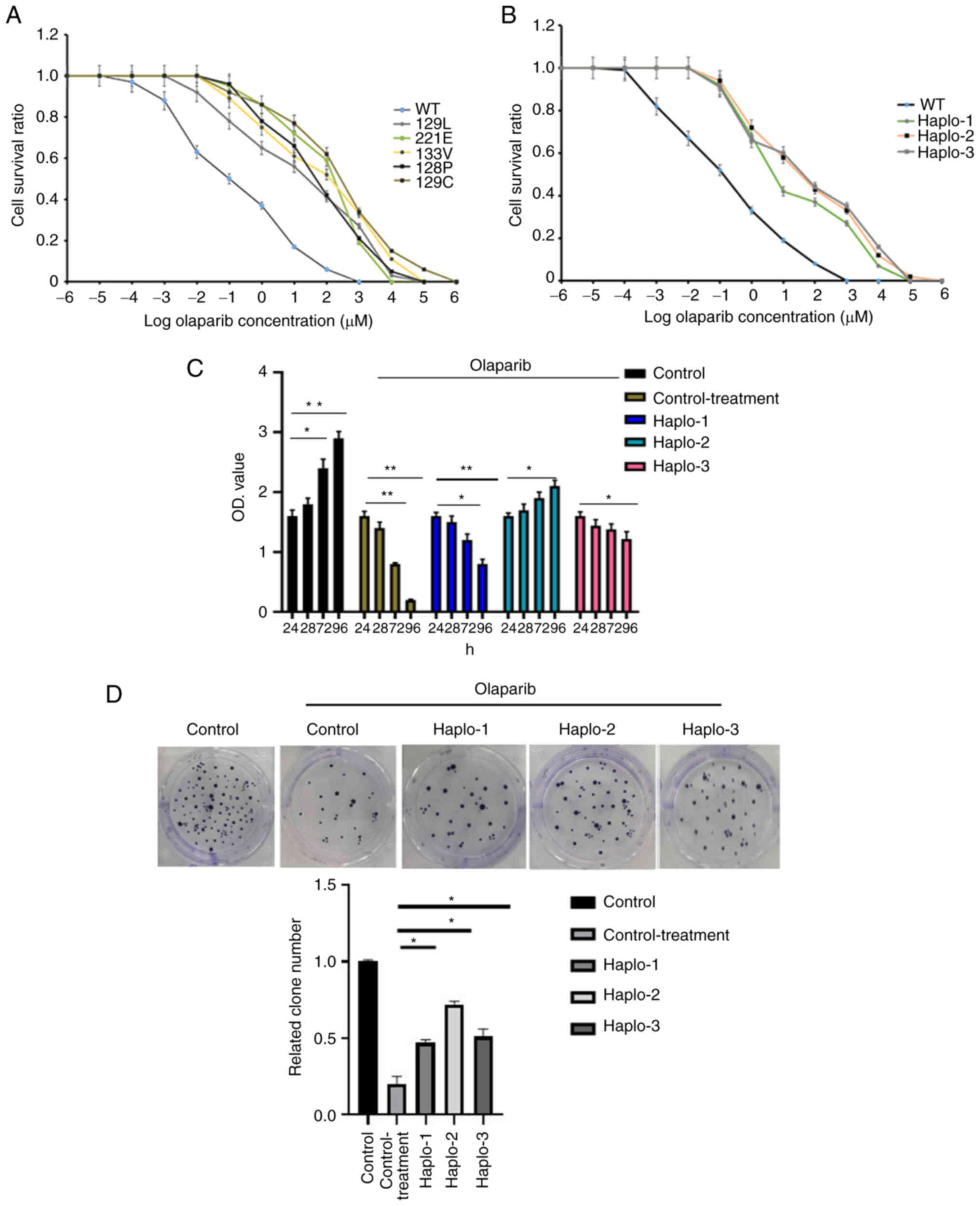 | Figure 3.Olaparib resistance of Rad51
mutants in BT-549 cells. (A) The survival analysis of BT-549 cells
containing five amino acid mutations at increasing concentrations
of olaparib. The mean IC50 values (µM) for wild-type,
129L, 221E, 133V, 128P and 129C were 0.61, 2.46, 2.8, 5.7 and 7.4
µM, respectively. One cell type originating from one single clone
with triplicate repeats was analyzed. (B) The three haplotypes
associated with olaparib resistance in BT-549 cells. The mean
IC50 for wild-type, haplotype 1 (Haplo-1), haplotype 2
(Haplo-2), and haplotype 3 (Haplo-3) were 0.42, 1.74, 2.55, and
2.82 µM, respectively. Each x-axis indicates the Log drug
concentration, and the y-axis indicates the survival rate. One cell
type originating from one single clone with triplicate repeats was
analyzed. The Kruskal-Wallis test was utilized followed by
Newman-Keuls test to determine statistical significance. (C) The
effects of haplotype 1 (Haplo-1), haplotype 2 (Haplo-2), and
haplotype 3 (Haplo-3) carriers on cell proliferation were examined
using the CCK-8 assay. The x-axis indicates the duration of
olaparib treatment. (D) The colony formation capacities of BT-549
cells with haplotype 1 (Haplo-1), haplotype 2 (Haplo-2), and
haplotype 3 (Haplo-3) were also analyzed. The olaparib
concentration was 5 µM for both (C) and (D). All experiments were
performed in triplicate and mean values were compared.
Kruskal-Wallis test was utilized followed by Tukey's HSD to
determine statistical significance. *P<0.05, **P<0.01. |
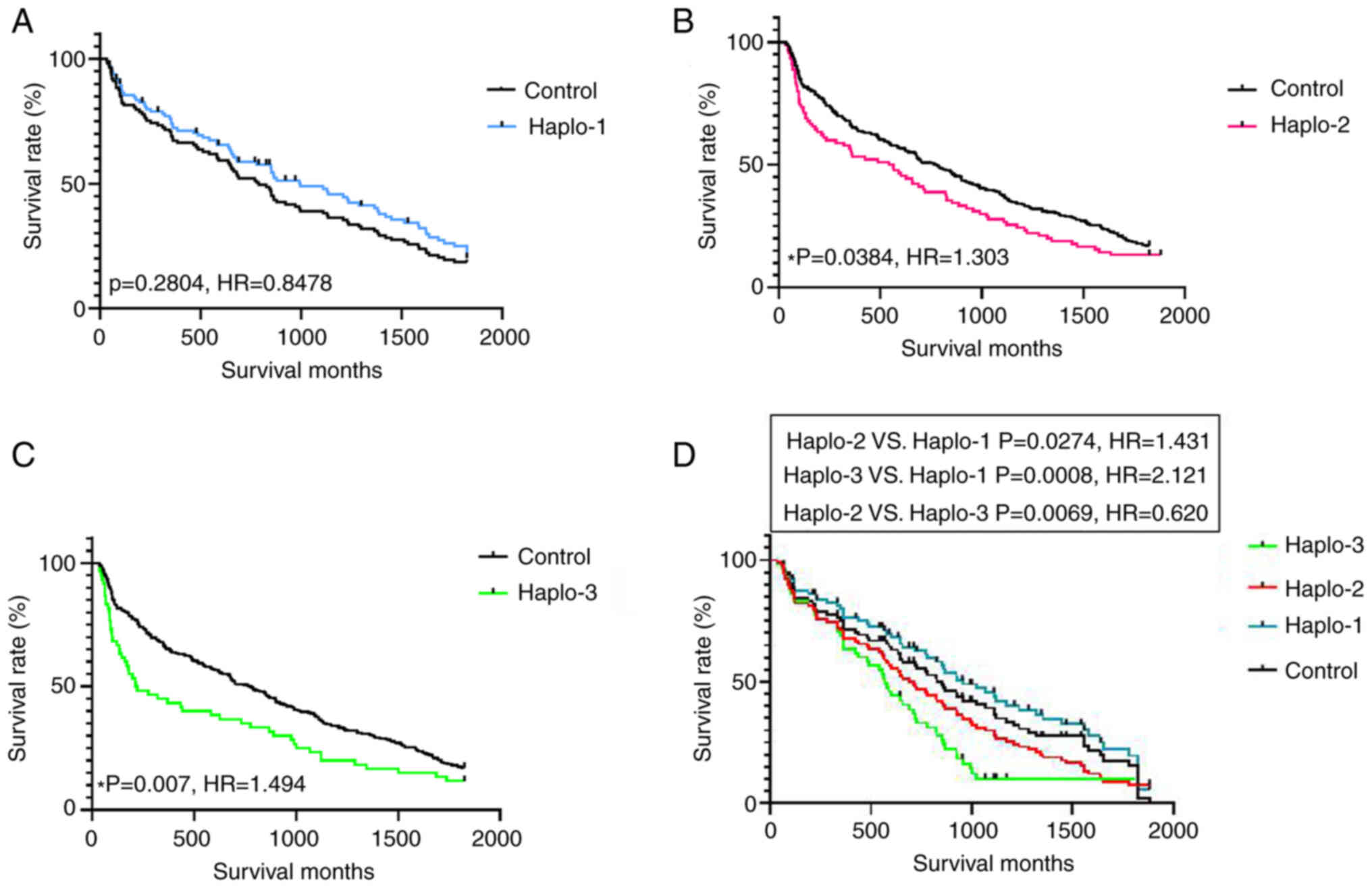
Association between haplotype and
survival
The association between Rad51 haplotypes and
olaparib treatment was evaluated in 538 cases treated with PARP
inhibitors and 234 controls (Table
V). The distribution of the three Rad51 haplotypes was
predicted and identified among the patient samples. The results
indicated that the positive ratio of the three haplotypes was
significantly higher in the olaparib treatment group compared to
the controls (Table V).
Haplotypes 2 and 3 showed a lower positive ratio in both groups
compared to haplotype 1. As shown in Fig. 3A and B, haplotypes 2 and 3 were
likely more sensitive and specific to the resistance of PARP
inhibitors. To further identify the potential drug resistance of
haplotypes 1, 2 and 3 in clinical samples, we performed five-year
survival analyses to compare 245 patient cases carrying any of
these haplotypes (Fig. 4). Two
haplotypes (haplotypes 2 and 3) were closely associated with breast
cancer survival (HR=1.303, 95% CI=0.9922-1.711, for haplotype-2 VS
control; HR=1.494, 95% CI=1.063-2.100 for haplotype-3 VS control;
HR=0.6197, 95% CI=0.4211–0.9120 for haplotype 2 VS haplotype 3).
Haplotype-1 was not associated with survival (HR=0.8478, 95%
CI=0.5848-1.077). The results indicated poorer prognosis with
haplotype 3 than with haplotype 2 or 1.
| Table V.Two-variant DAT haplotype frequency
distributions. |
Table V.
Two-variant DAT haplotype frequency
distributions.
|
| Control
(n=234) | Treated with PARP
inhibitors (n=538) |
|---|
|
|
|
|
|---|
| Haplotype | Positive case | Negative case | Positive ratio
(%) | Positive case | Negative case | Positve ratio
(%) |
|---|
| Haplotype-1 | 113 | 121 | 48.3 | 345 | 193 | 64.1a |
| Haplotype-2 | 86 | 148 | 36.7 | 228 | 310 | 42.3a |
| Haplotype-3 | 23 | 211 | 9.8 | 92 | 446 | 17.1a |
Discussion
Several mechanisms have been proposed for the
resistance to poly(ADP-ribose)polymerase (PARP) inhibitors. Many
gene and protein mutations, as well as abnormal gene expression
reportedly contribute to this cell function, including
Brca1/2 mutation, overexpression of 53bp1, and
increased drug transporter P-glycoprotein levels (37–39). RAD51 is central to three main
double strand break repair pathways: gene conversion,
synthesis-dependent strand annealing, and RAD51-dependent
break-induced replication (40)
and can enroll many other proteins to assemble RAD51 nuclear foci.
According to previous research, the number of RAD51 nuclear foci is
positively correlated with the resistance to PARP inhibitor
(10), and the present findings
confirm this correlation. A 10% cutoff point of the proportion of
RAD51 nuclear foci score has previously been shown to predict the
response to PARP inhibitors with high specificity and sensitivity
(9). In the present study, the
WALKER domain was the key structure in the homologous recombination
repair (HRR) process. These results showed for the first time a
saturation mutation genesis approach using targeted amino acid
changes defined by single nucleotide mutations in this region
adapted to explore the sensitivity to PARP inhibitors.
The CRISPR/Cas9-based system has the advantage of
introducing random potential drug resistance mutations on a genome
scale (41,42). Similarly, its role in saturation
mutation screening is effective in loss-of-function screening using
mammalian cells as well as animal models (16,17,43). We adopted this approach to
identify olaparib resistance single nucleotide polymorphisms (SNPs)
which are limited to a 13-amino acid window. Although the saturated
mutation spectrum was random, the codon bias distribution was
always selected for drug resistance (44,45). Next-generation sequencing (NGS) of
selective PCR products from genomic DNA was used to identify
functional mutation sites.
Our in vitro model experiments allowed us to
observe the effects on cell proliferation to better understand the
molecular evolution of drug resistance in breast cancers without
Brca1/2 mutations. Because RAD51 is a key HRR regulator
which directly results in cell cycle arrest, we investigated the
effect of mutations in this gene on the cell proliferation and
colony formation. After selecting clones exposed to olaparib, cells
with fit mutations remained viable. Our findings showed that a
single nucleotide mutation achieved similar effects on cell
survival as the combination of multiple nucleotide mutations.
Multiple nucleotide substitutions as a group appeared to have a
stronger capacity to distinguish mutations from wild-type codons in
drug resistance during the evaluation of olaparib resistance in
Rad51. Haplotype assembly was used to identify the possible
combined effect of these SNPs. Three haplotypes were assembled and
drug resistance was assessed at the cell level. To evaluate the
clinical value of these haplotypes, a series of clinical samples
were screened to identify the potential predictive ability of the
mutational pathway. The frequency distributions of these haplotypes
were also investigated in all the clinical subjects (Table V), and they were also observed in
control cohorts suggesting that they may be natural variations
which were enriched after PARP treatment. Finally, two haplotypes
were selected as potential prognostic biomarkers for olaparib
resistance in patients with breast cancer chemoresistance.
The mutations found to cause drug resistance in
patients are often not clear until drugs are used clinically.
Multi-drug resistance is frequently observed during cancer
treatment with a PARP inhibitor (46,47) and the related pathways involve
several genes, each of which always contains several mutations
(48). Mutations are highly
variable and are strongly influenced by sex, race, ethnicity, and
even individual differences (49,50). Furthermore, some functional
mutations frequently co-occur with other underlying mutations
making it difficult to screen and accurately identify valuable
drug-resistant mutations for a specific drug (51). In recent years, the
CRISPR/Cas9-based negative screening strategy with high-throughput
random screening has been applied to identify potential drug
resistance mutations. Due to the limits of library construction,
the induced mutation is identified within a limited region of the
gene sequence, while randomly induced mutations are rarely observed
in clinical samples and have limited clinical validity (52–54). The CRISPR/Cas9-based saturation
mutation strategy to identify the drug resistance pathway hub gene
displays high efficiency and clinical utility. It may provide a new
approach to identify clinically useful mutations from in
vitro studies. Finally, to improve the ‘resolving power’ of the
induced mutations, we assembled mutations according to haplotype
and evaluated these haplotypes in clinical samples. Two haplotypes
(haplotypes-2 and −3) were identified as prognostic markers for
breast cancer with olaparib treatment.
Compared to traditional methods, the targets
identified using this method have been determined with high
efficiency and low cost. Given the limits of the editing efficiency
and individual differences, the single mutation site randomly
induced by the CRISPR/Cas9 system is rarely used to evaluate the
response to drug treatment and lacks clinical value. Therefore, a
saturation mutagenesis method for specific oncogene or tumor
suppressor genes seems more efficient to screen for valuable
mutations. The bottleneck of this method is the paradox between the
limited content of the donor library and the number of targets.
Improvement of the payload information stored in the donor library
and the efficiency of the genome editing may be key to resolving
this issue. The present study was also limited by the size and lack
of diversity of the sample, restricting the clinical applicability
of the findings. The explored markers therefore need to be verified
in a larger and more diverse clinical cohort. Overall, the results
of our study provide a new strategy to identify functional drug
resistance mutations.
In conclusion, the present study assessed a series
of saturated mutations of the Rad51 WALKER domain induced
using a CRISPR/Cas9-based approach. Five SNPs were screened and
were associated with the response to olaparib treatment. Finally,
two haplotypes were assembled and evaluated in clinical samples.
Haplotypes 2 and 3 were associated with breast cancer survival and
provide a potential hallmark for breast cancer chemoresistance to
PARP inhibitor. The patients with these haplotypes may have
relatively poor prognosis.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
This work was supported by the Scientific Research Fund of the
Affiliated Hospital of Hebei University (China) (no.
8207102536).
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
Authors' contributions
HY, XL and LR designed the present study. HY, YW and
QZ conducted the experiments. YY and XL conducted the experiments
and the statistical analysis. XB and LY analyzed and interpreted
the patient data. NX interpreted the results. HY wrote the
manuscript. AZ was involed in drafting and revising the manuscript
and also contributed to analysis and interpretation of halpotype
assemble data. XL was involved in designing and drafting the
framework of the study. XL was also responsible for the layout of
the figures. XL and LR revised the final version and confirm the
authenticity of all the raw data. All authors read and approved the
final manuscript for publication.
Ethics approval and consent to
participate
The authors obtained appropriate institutional
review board approval from the Ethics Committee of the Affiliated
Hospital of Hebei University (Hebei, China) (no. AHHU20200930).
Informed consent was obtained from the participants involved.
Patient consent for publication
The authors confirm that they have obtained written
consent from each patient to publish the manuscript.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CRISPR
|
clustered regular interval short
palindromic repeats
|
|
HDR
|
homology-directed repair
|
|
HRR
|
homologous recombination repair
|
|
OTS
|
off-target sites
|
|
PAM
|
protospacer adjacent motif
|
|
PARP
|
poly(ADP-ribose)polymerase
|
References
|
1
|
Mavaddat N, Antoniou AC, Easton DF and
Garcia-Closas M: Genetic susceptibility to breast cancer. Mol
Oncol. 4:174–191. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lord CJ and Ashworth A: BRCAness
revisited. Nat Rev Cancer. 16:110–120. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Apostolou P and Fostira F: Hereditary
breast cancer: The era of new susceptibility genes. Biomed Res Int.
2013:7473182013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Noordermeer SM and van Attikum H: PARP
Inhibitor Resistance: A tug-of-war in BRCA-mutated cells. Trends
Cell Biol. 29:820–834. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Risdon EN, Chau CH, Price DK, Sartor O and
Figg WD: PARP Inhibitors and prostate cancer: To infinity and
beyond BRCA. Oncologist. 26:e115–e129. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bajrami I, Frankum JR, Konde A, Miller RE,
Rehman FL, Brough R, Campbell J, Sims D, Rafiq R, Hooper S, et al:
Genome-wide profiling of genetic synthetic lethality identifies
CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity.
Cancer Res. 74:287–297. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Grundy MK, Buckanovich RJ and Bernstein
KA: Regulation and pharmacological targeting of RAD51 in cancer.
NAR Cancer. 2:zcaa0242020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kolinjivadi AM, Sannino V, de Antoni A,
Técher H, Baldi G and Costanzo V: Moonlighting at replication
forks-a new life for homologous recombination proteins BRCA1, BRCA2
and RAD51. FEBS Lett. 591:1083–1100. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cruz C, Castroviejo-Bermejo M,
Gutiérrez-Enríquez S, Llop-Guevara A, Ibrahim YH, Gris-Oliver A,
Bonache S, Morancho B, Bruna A, Rueda OM, et al: RAD51 foci as a
functional biomarker of homologous recombination repair and PARP
inhibitor resistance in germline BRCA-mutated breast cancer. Ann
Oncol. 29:1203–1210. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Castroviejo-Bermejo M, Cruz C,
Llop-Guevara A, Gutiérrez-Enríquez S, Ducy M, Ibrahim YH,
Gris-Oliver A, Pellegrino B, Bruna A, Guzmán M, et al: A RAD51
assay feasible in routine tumor samples calls PARP inhibitor
response beyond BRCA mutation. EMBO Mol Med. 10:e91722018.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Malka MM, Eberle J, Niedermayer K, Zlotos
DP and Wiesmüller L: Dual PARP and RAD51 inhibitory drug conjugates
show synergistic and selective effects on breast cancer cells.
Biomolecules. 11:9812021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Moudry P, Watanabe K, Wolanin KM, Bartkova
J, Wassing IE, Watanabe S, Strauss R, Troelsgaard Pedersen R,
Oestergaard VH, Lisby M, et al: TOPBP1 regulates RAD51
phosphorylation and chromatin loading and determines PARP inhibitor
sensitivity. J Cell Biol. 212:281–288. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Garcin EB, Gon S, Sullivan MR, Brunette
GJ, Cian A, Concordet JP, Giovannangeli C, Dirks WG, Eberth S,
Bernstein KA, et al: Differential requirements for the Rad51
paralogs in genome repair and maintenance in human cells. PLoS
Genet. 15:e10083552019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Abreu CM, Prakash R, Romanienko PJ, Roig
I, Keeney S and Jasin M: Shu complex SWS1-SWSAP1 promotes early
steps in mouse meiotic recombination. Nat Commun. 9:39612018.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fang P, De Souza C, Minn K and Chien J:
Genome-scale CRISPR knockout screen identifies TIGAR as a modifier
of PARP inhibitor sensitivity. Commun Biol. 2:3352019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ma L, Boucher JI, Paulsen J, Matuszewski
S, Eide CA, Ou J, Eickelberg G, Press RD, Zhu LJ, Druker BJ, et al:
CRISPR-Cas9-mediated saturated mutagenesis screen predicts clinical
drug resistance with improved accuracy. Proc Natl Acad Sci USA.
114:11751–11756. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Findlay GM, Boyle EA, Hause RJ, Klein JC
and Shendure J: Saturation editing of genomic regions by multiplex
homology-directed repair. Nature. 513:120–123. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Burger A, Lindsay H, Felker A, Hess C,
Anders C, Chiavacci E, Zaugg J, Weber LM, Catena R, Jinek M, et al:
Maximizing mutagenesis with solubilized CRISPR-Cas9
ribonucleoprotein complexes. Development. 143:2025–2037.
2016.PubMed/NCBI
|
|
19
|
Pelttari LM, Kiiski JI, Ranta S, Vilske S,
Blomqvist C, Aittomäki K and Nevanlinna H: RAD51, XRCC3, and XRCC2
mutation screening in Finnish breast cancer families. Springerplus.
4:922015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Short JM, Liu Y, Chen S, Soni N,
Madhusudhan MS, Shivji MK and Venkitaraman AR: High-resolution
structure of the presynaptic RAD51 filament on single-stranded DNA
by electron cryo-microscopy. Nucleic Acids Res. 44:9017–9030.
2016.PubMed/NCBI
|
|
21
|
Xu J, Zhao L, Xu Y, Zhao W, Sung P and
Wang HW: Cryo-EM structures of human RAD51 recombinase filaments
during catalysis of DNA-strand exchange. Nat Struct Mol Biol.
24:40–46. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen J, Villanueva N, Rould MA and
Morrical SW: Insights into the mechanism of RAD51 recombinase from
the structure and properties of a filament interface mutant.
Nucleic Acids Res. 38:4889–4906. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bonilla B, Hengel SR, Grundy MK and
Bernstein KA: Rad51 gene family structure and function. Annu Rev
Genet. 54:25–46. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Baldock RA, Pressimone CA, Baird JM,
Khodakov A, Luong TT, Grundy MK, Smith CM, Karpenshif Y,
Bratton-Palmer DS, Prakash R, et al: Rad51D splice variants and
cancer-associated mutations reveal XRCC2 interaction to be critical
for homologous recombination. DNA Repair (Amst). 76:99–107. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Corrales-Sánchez V, Noblejas-López MDM,
Nieto-Jiménez C, Pérez-Peña J, Montero JC, Burgos M, Galán-Moya EM,
Pandiella A and Ocaña A: Pharmacological screening and
transcriptomic functional analyses identify a synergistic
interaction between dasatinib and olaparib in triple-negative
breast cancer. J Cell Mol Med. 24:3117–3127. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Stemmer M, Thumberger T, Del Sol Keyer M,
Wittbrodt J and Mateo JL: CCTop: An intuitive, flexible and
reliable CRISPR/Cas9 target prediction tool. PloS One.
10:e01246332015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kamel D, Gray C, Walia JS and Kumar V:
PARP inhibitor drugs in the treatment of breast, ovarian, prostate
and pancreatic cancers: An update of clinical trials. Curr Drug
Targets. 19:21–37. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chu CY, Lee YC, Hsieh CH, Yeh CT, Chao TY,
Chen PH, Lin IH, Hsieh TH, Shih JW, Cheng CH, et al: Genome-wide
CRISPR/Cas9 knockout screening uncovers a novel inflammatory
pathway critical for resistance to arginine-deprivation therapy.
Theranostics. 11:3624–3641. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang B, Wang M, Zhang W, Xiao T, Chen CH,
Wu A, Wu F, Traugh N, Wang X, Li Z, et al: Integrative analysis of
pooled CRISPR genetic screens using MAGeCKFlute. Nat Prot.
14:756–780. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Giuliano AE, Edge SB and Hortobagyi GN:
Eighth edition of the AJCC cancer staging manual: Breast cancer.
Ann Surg Oncol. 25:1783–1785. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shen B, Zhang J, Wu H, Wang J, Ma K, Li Z,
Zhang X, Zhang P and Huang X: Generation of gene modified mice via
Cas9/RNA-mediated gene targeting. Cell Res. 23:720–723. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Stephens M and Scheet P: Accounting for
decay of linkage disequilibrium in haplotype inference and
missing-data imputation. Am J Hum Genet. 76:449–462. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li P, Guo M, Wang C, Liu X and Zou Q: An
overview of SNP interactions in genome-wide association studies.
Brief Funct Genomics. 2:143–155. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tulbah S, Alabdulkarim H, Alanazi M,
Parine NR, Shaik J, Pathan AA, Al-Amri A, Khan W and Warsy A:
Polymorphisms in Rad51 and their relation with breast cancer in
Saudi females. Onco Targets. 9:269–277. 2016.PubMed/NCBI
|
|
35
|
Vral A, Willems P, Claes K, Poppe B,
Perletti G and Thierens H: Combined effect of polymorphisms in
Rad51 and Xrcc3 on breast cancer risk and chromosomal
radiosensitivity. Mol Med Rep. 4:901–912. 2011.PubMed/NCBI
|
|
36
|
Brooks J, Shore RE, Zeleniuch-Jacquotte A,
Currie D, Afanasyeva Y, Koenig KL, Arslan AA, Toniolo P and Wirgin
I: Polymorphisms in RAD51, XRCC2, and XRCC3 are not related to
breast cancer risk. Cancer Epidemiol Biomark Prev. 17:1016–1019.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kim JH and Hong YC: Modification of PARP4,
XRCC3, and RAD51 gene polymorphisms on the relation between
Bisphenol A exposure and liver abnormality. Int J Environ Res
Public Health. 17:27942020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Marchenko YV, Carroll RJ, Lin DY, Amos CI
and Gutierrez RG: Semiparametric analysis of case-control genetic
data in the presence of environmental factors. Stata J. 8:305–333.
2008. View Article : Google Scholar
|
|
39
|
Hurley RM, Wahner Hendrickson AE, Visscher
DW, Ansell P, Harrell MI, Wagner JM, Negron V, Goergen KM, Maurer
MJ, Oberg AL, et al: 53BP1 as a potential predictor of response in
PARP inhibitor-treated homologous recombination-deficient ovarian
cancer. Gynecol Oncol. 153:127–134. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nacson J, Krais JJ, Bernhardy AJ, Clausen
E, Feng W, Wang Y, Nicolas E, Cai KQ, Tricarico R, Hua X, et al:
Brca1 mutation-specific responses to 53bp1 loss-induced homologous
recombination and PARP inhibitor resistance. Cell Rep.
24:3513–3527. e72018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jaspers JE, Kersbergen A, Boon U, Sol W,
van Deemter L, Zander SA, Drost R, Wientjens E, Ji J, Aly A, et al:
Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated
mouse mammary tumors. Cancer Discov. 3:68–81. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Symington LS and Gautier J: Double-strand
break end resection and repair pathway choice. Annu Rev Genet.
45:247–271. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shalem O, Sanjana NE, Hartenian E, Shi X,
Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG and
Zhang F: Genome-scale CRISPR-Cas9 knockout screening in human
cells. Science. 343:84–87. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Koike-Yusa H, Li Y, Tan EP,
Velasco-Herrera Mdel C and Yusa K: Genome-wide recessive genetic
screening in mammalian cells with a lentiviral CRISPR-guide RNA
library. Nat Biotechnol. 32:267–273. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yamauchi T, Masuda T, Canver MC, Seiler M,
Semba Y, Shboul M, Al-Raqad M, Maeda M, Schoonenberg VAC, Cole MA,
et al: Genome-wide CRISPR-Cas9 screen identifies leukemia-specific
dependence on a Pre-mRNA metabolic pathway regulated by DCPS.
Cancer Cell. 33:386–400. e52018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ali M, Kaltenbrun E, Anderson GR, Stephens
SJ, Arena S, Bardelli A, Counter CM and Wood KC: Codon bias imposes
a targetable limitation on KRAS-driven therapeutic resistance. Nat
Commun. 8:156172017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Morio F, Lombardi L and Butler G: The
CRISPR toolbox in medical mycology: State of the art and
perspectives. PLoS Pathog. 16:e10082012020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gout J, Perkhofer L, Morawe M, Arnold F,
Ihle M, Biber S, Lange S, Roger E, Kraus JM, Stifter K, et al:
Synergistic targeting and resistance to PARP inhibition in DNA
damage repair-deficient pancreatic cancer. Gut. 70:743–760. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Dallavalle S, Dobričić V, Lazzarato L,
Gazzano E, Machuqueiro M, Pajeva I, Tsakovska I, Zidar N and
Fruttero R: Improvement of conventional anti-cancer drugs as new
tools against multidrug resistant tumors. Drug Resist Update.
50:1006822020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Laucht M, Skowronek MH, Becker K, Schmidt
MH, Esser G, Schulze TG and Rietschel M: Interacting effects of the
dopamine transporter gene and psychosocial adversity on
attention-deficit/hyperactivity disorder symptoms among
15-year-olds from a high-risk community sample. Arch Gen
Psychiatry. 64:585–590. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Caspi A, Sugden K, Moffitt TE, Taylor A,
Craig IW, Harrington H, McClay J, Mill J, Martin J, Braithwaite A
and Poulton R: Influence of life stress on depression: Moderation
by a polymorphism in the 5-HTT gene. Science. 301:386–389. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Aguiar D, Wong WS and Istrail S: Tumor
haplotype assembly algorithms for cancer genomics. Pac Symp
Biocomput. 3–14. 2014.PubMed/NCBI
|
|
53
|
Azam M, Latek RR and Daley GQ: Mechanisms
of autoinhibition and STI-571/imatinib resistance revealed by
mutagenesis of BCR-ABL. Cell. 112:831–843. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ma Y, Zhang J, Yin W, Zhang Z, Song Y and
Chang X: Targeted AID-mediated mutagenesis (TAM) enables efficient
genomic diversification in mammalian cells. Nat Methods.
13:1029–1035. 2016. View Article : Google Scholar : PubMed/NCBI
|