Introduction
Sepsis is life-threatening organ dysfunction caused
by an uncontrollable immune response induced by infection (1). Among the multi-organ dysfunction
syndromes induced by sepsis, 40% of patients have reduced cardiac
function and 20% have cardiac dysfunction (2). Sepsis-induced myocardial depression
contributes to the prognosis of patients with sepsis and exploring
the pathogenesis of sepsis-induced cardiac depression may alleviate
the prognosis of these patients (3). The mechanism of cardiac depression
may result from structural effects due to cardiomyocyte death, as
well as functional effects caused by cardiac dysfunction. Calcium
(Ca2+) homeostasis is essential for normal cardiac
excitation contraction coupling (4). Ca2+ also serves an
essential role in the signalling pathways associated with cellular
termination, including induction of apoptosis and damage to
structural proteins (5,6). Lipopolysaccharide (LPS) efficiently
increases the intracellular Ca2+ concentration in
cardiomyocytes (7) and disruption
of Ca2+ homeostasis reduces dystrophin, actin and myosin
expression, but increases the expression of calpain. It also leads
to decreases in left ventricular ejection fraction (LVEF) and left
ventricular fractional shortening (LVFS) (8). Moreover, inhibitors of the
sarcoplasmic reticulum (SR) Ca-ATPase and Ca2+ chelators
decrease cellular Ca2+ levels, which may reduce the
cardiac apoptosis induced by LPS (9). However, the transient
Ca2+ concentrations responsible for contraction
fluctuate several times a minute from 0.1 µM in the diastole period
to 1–10 µM in the systole period, which does not appear to activate
cellular signalling events (10).
Therefore, the activation of signalling pathways by Ca2+
is distinct from the Ca2+ fluctuations responsible for
normal cycles of myocardial excitation, contraction and
relaxation.
Voltage-gated Ca2+ entry is the main
source of elevated intracellular cardiac Ca2+ levels and
originates from Ca2+ influx via the plasma membrane
(11). Store-operated
Ca2+ entry (SOCE) is an additional pathway of
Ca2+ entry originally associated with Ca2+
changes in unexcited cells (12–14). Neonatal and embryonic
cardiomyocytes depleted of Ca2+ in the SR were
previously demonstrated to efficiently induce Ca2+
influx, which was blocked by the SOCE inhibitor SKF-96365, but not
the L-type Ca2+ channel (LTCC) blocker verapamil
(15). SOCE has also been
detected in adult cardiomyocytes using whole-cell patch-clamp
experiments (16). However,
understanding the role of SOCE in cardiomyocytes was limited until
the proteins essential to this signalling pathway were discovered.
SOCE is associated with stromal interaction molecule 1 (STIM1) and
Orai calcium release-activated calcium modulator 1 (Orai1)
(17) and the expression of STIM1
and Orai1 has also been reported in myocardium (18–20). Orai1 is a transmembrane protein of
the cytomembrane and is a Ca2+ channel-mediated
Ca2+ entry point from the internal environment (21). As a protein located in the
cellular membrane, Orai1 does not sense changes in Ca2+
levels in the endoplasmic reticulum (ER) (21). The STIM1 protein is distributed
mainly in the ER membrane and can sense changes to the
Ca2+ level in the ER (22). It has been demonstrated that
depletion of Ca2+ in the ER can induce the clustering
and distribution of STIM1 to the ER-plasma membrane junctions,
which subsequently mediates the activation of Orai1 (22).
Numerous studies have demonstrated the role of
STIM1/Orai1-mediated SOCE in the pathogenesis of cardiac disease
(23–26), especially in cardiac hypertrophy
(27,28). However, to the best of our
knowledge the role of STIM1/Orai1-mediated SOCE in the development
of septic myocardial depression has not been reported. During
septic myocardial depression, inhibition of the SR Ca-ATPase and
enhanced ryanodine receptor leakage induce depletion of
Ca2+ in the ER (29).
Therefore, we hypothesize that STIM1/Orai1-mediated SOCE
contributes to the pathogenesis and development of septic cardiac
depression.
Materials and methods
Reagents
LPS and anti-Orai1 antibody (cat. no. SAB3500127)
were purchased from Sigma-Aldrich (Merck KGaA). Antibodies for
STIM1 (cat. no. ab108994) and Bax (cat. no. ab32503) were purchased
from Abcam. Antibody against Bcl-2 (cat. no. 2870T) was purchased
from Cell Signaling Technology, Inc. GAPDH (cat. no. BS60630) was
purchased from Bioworld Technology, Inc. and secondary antibodies
(cat. no. BL003A) for western blotting was purchased from Biosharp
Life. TRIzol® reagent was obtained from Invitrogen
(Thermo Fisher Scientific, Inc.). QuantiTect Reverse Transcription
(RT) Kit and SYBR Green Master Mix were purchased from Takara
Biotechnology Co., Ltd. DyLightFluor® 488-conjugated
donkey anti-rabbit IgG secondary antibodies (cat. no. BYE026 was
obtained from Shanghai Boyun Biotech Co., Ltd.
Fluo-3AMacetoxymethyl ester (AM; cat. no. s1056) and bovine serum
albumin (BSA) were purchased from Beyotime Institute of
Biotechnology. Thapsigargin used for depleting SR Ca2+
stores (TG; cat. no. mb13319) was purchased from Dalian Melone
Biotechnology Co., Ltd. and terminal deoxynucleotidyl transferase
dUTP nick-end labelling (TUNEL) kits were purchased from Roche
Molecular Diagnostics.
Animals
Male C57BL/6 mice (age, 8 weeks; weight, 20–25 g)
were obtained from Shanghai SLAC Laboratory Animal Co., Ltd. and
were raised under pathogen-free conditions at 25°C at 50% humidity
and 12-h light/dark cycles with free access to food and water. The
present study was performed in accordance with the National
Institutes of Health Guidelines for Care and Use of Animals
(30) and was approved by the
Ethics Committee of Wenzhou Medical University (Wenzhou,
China).
CLP model
In total, 90 mice were randomly assigned to the
following six groups: i) Sham 6 h; ii) Sham 12 h; iii) sham 24 h;
iv) CLP 6 h; v) CLP 12 h; and vi) CLP 24 h. The CLP model was
established as described previously (31,32). Briefly, mice were anesthetized
with pentobarbital sodium (50 mg/kg intraperitoneally) and
following sterilization a midline incision (1 cm) was made to
expose the cecum. The cecum was ligated in half and punctured twice
using an 18-gauge needle before removing a small amount of stool
and placing it in the abdomen. The wound was sutured following the
CLP operation. Mice in the sham group underwent a laparotomy
without CLP operation. All mice were resuscitated with a
subcutaneous injection of a 24 ml/kg sterile saline solution
(0.9%). At the corresponding timepoint, mice were sacrificed by
cervical dislocation after anaesthesia using pentobarbital sodium
(50 mg/kg intraperitoneally). Death was verified by cessation of
heartbeat and pupil dilation.
Survival rate
The survival rate of the mice in each group was
evaluated within 72 h of the sham or CLP operation. Mice had free
access to water and food and were kept under pathogen-free
conditions and were monitored every 6 h. All mice were euthanasia
at the end of the study.
Evaluation of cardiac function by
echocardiography
Cardiac function was assessed via transthoracic
echocardiographic examination. Mice were lightly anesthetized with
isoflurane anaesthesia (3% induction and 1~2% maintenance). The
assessment of left ventricular cardiac function was performed
before and after the operation to assess the influence of CLP on
cardiac function. Echocardiographic images were obtained using an
echocardiography instrument equipped with a 15 MHz phased-array
transducer (GE LOGIQ E9; General Electric Company). The motion mode
(M-mode) images of the left ventricular dimensions were taken and
the LVEF and LVFS were assessed.
Hematoxylin and eosin (H&E)
staining
The hearts of the mice were harvested after the sham
or CLP operations and fixed in 4% paraformaldehyde at 4°C for 24 h.
Paraffin-embedded myocardial tissues were cut into sections (5 µm).
H&E staining was performed at room temperature with 0.25%
hematoxylin for 10 min and 0.5% eosin for 2 min. Morphological
changes were evaluated using the Olympus CKX41SF light microscope
(Olympus Corporation).
TUNEL staining
Myocardial apoptosis was detected using the TUNEL
method. The heart tissues were fixed in a 4% paraformaldehyde at
4°C for 24 h. Paraffin-embedded myocardial tissues were cut into
sections (5 µm) and the slides were stained with 50 µl TUNEL
reagent (50 µl terminal deoxynucleotidyl transferase and 450 µl
biotin-dUTP) for 60 min at 37°C. Subsequently, the samples were
incubated with 50 µl horseradish peroxidase-conjugated streptavidin
solution at room temperature for 30 min and were then developed
with DAB chromogenic reagent (5 µl 20X DAB + 1 µl 30% H2O2+94 µl
PBS) for 5 min at room temperature. The slices were stained with
haematoxylin, dehydrated by ethanol and mounted with neutral balata
fixation. Representative images were taken using a light
microscope. Five random fields were evaluated in each slide.
RT-quantitative (q)PCR
Total RNA was isolated from the myocardium of CLP
and sham mice using TRIzol according to the manufacturer's
protocol. Complementary DNA was synthesized from 1 µg total RNA
using PrimeScript™ RT Master Mix (Takara Biotechnology Co., Ltd)
according to the manufacturer's protocol described. qPCR was
performed using the SYBR Green Master Mix (Takara Biotechnology
Co., Ltd) with the CFX96 Real-Time PCR System (Bio-Rad
Laboratories, Inc.). The following thermocycling conditions were
used: 30 sec at 95°C; followed by 40 cycles of 15 sec at 95°C and 1
min at 60°C. The primers used for qPCR were as follows: Bax forward
(F), 5′-CAGTTGAAGTTGCCATCAGC-3′ and reverse (R)
5′-ATGCGTCCACCAAGAAGC-3′; Bcl-2 F, 5′-GCGACGAGAGAAGTCATCC-3′ and R,
5′-AGCCTGAGAGCAACCCAAT-3′, STIM1 F, 5′-CCCTTCCAGATCCTTCATCA-3′ and
R, 5′-AAGGACTTCATGCTGGTGGT-3′; Orai1 F, 5′-GTGCCCGGTGTTAGAGAATG-3′
and R, 5′-TCCCTGGTCAGCCATAAGAC-3′; and GAPDH F,
5′-AAGAGGGATGCTGCCCTTAC-3′ and R, 5′-TACGGCCAAATCCGTTCACA-3′. The
mRNA expression levels of the target genes were normalized to those
of GAPDH and fold changes were quantified using the
2−ΔΔCq method (33).
Western blotting
Myocardial tissues were harvested and lysed using
RIPA lysis buffer (Beyotime Institute of Biotechnology). The
protein concentration of the supernatant was determined using a BCA
protein assay kit. Equivalent amounts of total protein (30–60 µg
protein/lane) were separated via SDS-PAGE on a 8–12% gel.
Subsequently, separated protein was transferred to a PVDF membrane
(MilliporeSigma). After blocking with 5% non-fat milk at room
temperature for 2 h, the membranes were incubated with primary
antibodies against STIM1 (1:1,000), Bax (1:1,000), Bcl-2 (1:1,000),
or Orai1 (1:500) at 4°C overnight. Following the primary incubation
membranes were washed with Tris-buffered saline containing 0.1%
Tween-20 for three times and then incubated followed with secondary
antibodies (1:5,000; cat. no. BL003A; Biosharp Life Science, Inc.)
at room temperature for 2 h. Membranes were detected with an
enhanced chemiluminescence kit (Thermo Fisher Scientific, Inc.) and
were semi-quantified using Quantity One v4.6.6 software (Bio-Rad
Laboratories, Inc.). GAPDH (cat. no. BS60630; 1:8,000; Bioworld
Technology, Inc.) was used as a loading control.
Cell culture and treatment
H9C2 cells were purchased from The Cell Bank of Type
Culture Collection of The Chinese Academy of Sciences. Cells were
cultured in Dulbecco's Modified Eagle's Medium (Gibco; Thermo
Fisher Scientific, Inc.) supplemented with 10% FBS (Gibco; Thermo
Fisher Scientific, Inc.), 100 IU/ml penicillin and 100 mg/ml
streptomycin at 37°C with 5% CO2. H9C2 cells were
randomly assigned to two groups: i) Control (PBS stimulation); ii)
LPS (10 µg/ml). Cells were seeded into plates at an appropriate
density (1×105/ml) and incubated for 24 h before
incubation with LPS or PBS for 0.5–6.0 h at 37°C and were
subsequently harvested for analysis (34).
Immunofluorescence staining
Cells from each treatment group were fixed with 4%
paraformaldehyde at 4°C for 1 h and then permeabilized with 0.5%
Triton X-100 at room temperature for 10 min. After blocking with 1%
BSA at 4°C for 30 min the cells were incubated with anti-STIM1
(1:100) and anti-Orai1 (1:100) primary antibodies at 4°C overnight.
Samples were then incubated with dyLightFluor®
488-conjugated donkey anti-rabbit (1:400) or anti-mouse (1:400) IgG
secondary antibody for 1 h at room temperature and cells were
stained with 10 µg/ml DAPI for 7 min in the dark at room
temperature. Representative images were captured using the Olympus
BX51 fluorescent microscope (Olympus Corporation).
Store-operated Ca2+ entry
in cardiomyocytes
SOCE was quantified as described previously
(18). Briefly, H9C2 cells were
loaded with 5 µmol/l Fluo-3/AM (excitation at 488 nm) for 40 min at
37°C in normal Tyrode's solution (140 mmol/l NaCl, 5.4 mmol/l KCl,
1 mmol/l MgCl2, 2 mmol/l CaCl2, 5.5 mmol/l
glucose and 5 mmol/l HEPES at pH 7.4) in the dark and LPS (10
µg/ml) was administered for 40 min at 37°C. After dye loading,
cells were washed with PBS three times within 20 min at room
temperature. To prevent store refilling and deletion of
intracellular Ca2+ stores, cells were treated with 4
µmol/l TG in Ca2+-free Tyrode's solution. SOCE was
triggered by the addition of 1.8 mmol/l Ca2+ to the
solution. The cells were captured first before TG or
Ca2+ was added. Real-time fluorescent images (all cells)
were recored every 2 sec using confocal microscopy (Nikon A1; Nikon
Corporation) within 1 h and were analysed using the supporting
software (Nikon NIS-Elements AR 4.XX.00). The cells were still
alive when the images were recorded, thus H9C2 cells were
stimulated by LPS for 1–2 h (40 min dying at 37°C, 20 min washing
and 0–1 h recording at room temprature).
Statistical analysis
All experiments were performed in triplicate. Data
are presented as the mean ± SD. All analyses were performed using
SPSS version 16.0 software (SPSS, Inc.). Statistical significance
was assessed using two-tailed unpaired Student's t-test (two
groups) and one-way ANOVA followed by Tukey's post hoc test (three
or more groups). P<0.05 was considered to indicate a
statistically significant difference.
Results
Establishment of septic myocardial
depression
Sepsis-induced alterations in cardiac structure and
function were analysed according to the survival rate, cardiac
contractility and myocardial injury. The survival rate of mice in
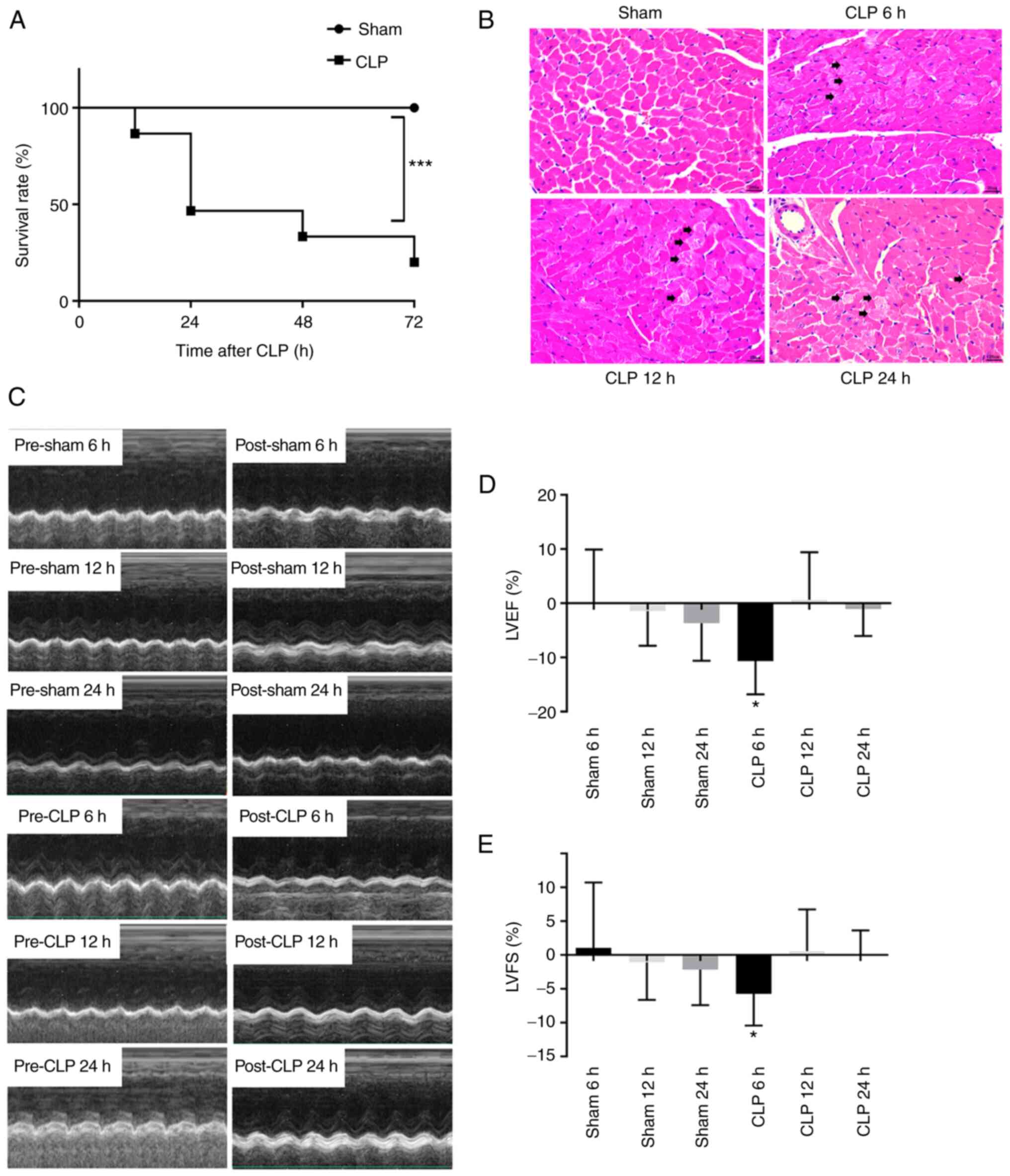
the sham group was 100% following the CLP operation (Fig. 1A). However, compared with the sham
group a marked decrease in survival rate was observed following
CLP, with survival rates of 46.7% at 24 h and 20% at 72 h (vs. sham
group, P<0.001). Changes in survival rate in the CLP group
indicated the success of the septic animal model. Myocardial
sections were stained with H&E to assess the damage to the
myocardium induced by CLP. Cardiomyocytes were demonstrated to be
intact and the cardiac muscle cross striations were clear in the
sham group. In the CLP group, cardiomyocytes were destroyed and the
cardiac muscle cross striations were no longer clear. Damage to the
myocardial tissues by CLP continued following the operation
(Fig. 1B). Changes in cardiac
function resulting from sepsis were analysed using
echocardiography. The results demonstrated significant reductions
in the LVEF (10.7%) and LVES (5.7%) in the CLP group at 6 h
(P<0.05) compared with the sham 6 h group (Fig. 1C-E). No significant change in the
LVEF or LVES was observed at 12 and 24 h in the CLP group compared
with the corresponding time-point sham group.
Myocardial apoptosis is induced in
septic mice
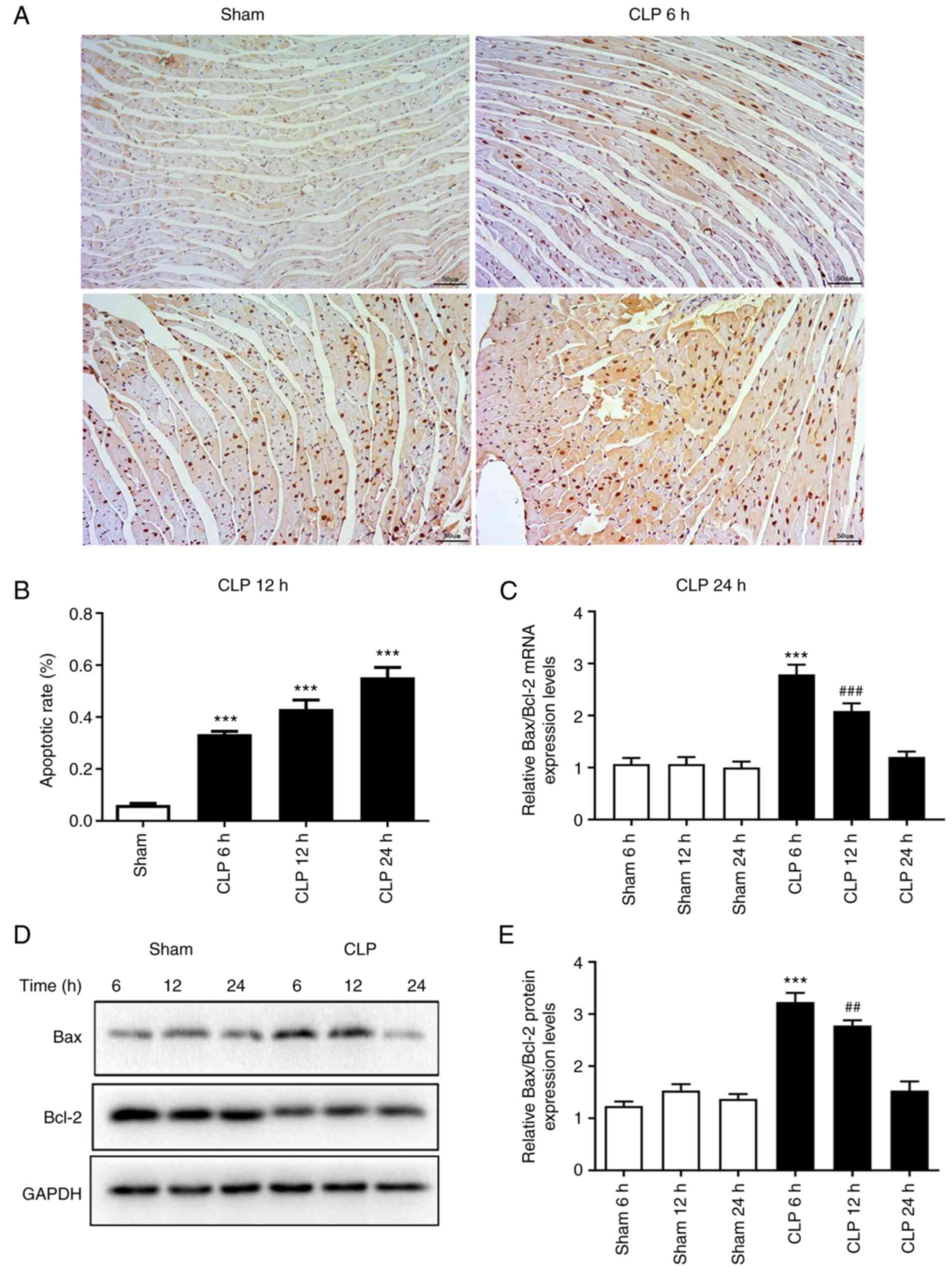
The myocardial sections damaged by the CLP
operation, as confirmed by H&E staining, were used to assess
the rate of cardiomyocyte apoptosis induced by sepsis via TUNEL
staining. The rate of cardiomyocyte apoptosis was 6% in the 24 h
sham group compared with 33.5% at 6 h (P<0.001), 43.1% at 12 h
(P<0.001) and 55.3% at 24 h (P<0.001) following CLP, which
indicated that the myocardial apoptosis rate significantly
increased over time following the operation compared with the sham
group (Fig. 2A and B). The mRNA
and protein expression levels of apoptosis-related Bax and Bcl-2
were assessed using RT-qPCR and western blotting, respectively, to
investigate the activation of sepsis-induced myocardial apoptosis.
The ratio of Bax/Bcl-2 mRNA and protein expression levels were
significantly increased in the CLP group at 6 (P<0.001) and 12 h
(P<0.01) compared with the corresponding sham groups (Fig. 2C-E). The peak Bax/Bcl-2 ratio was
observed at 6 h following the CLP operation and both mRNA and
protein levels returned to normal at 24 h.
Orai1 glycosylation is increased in
septic mice
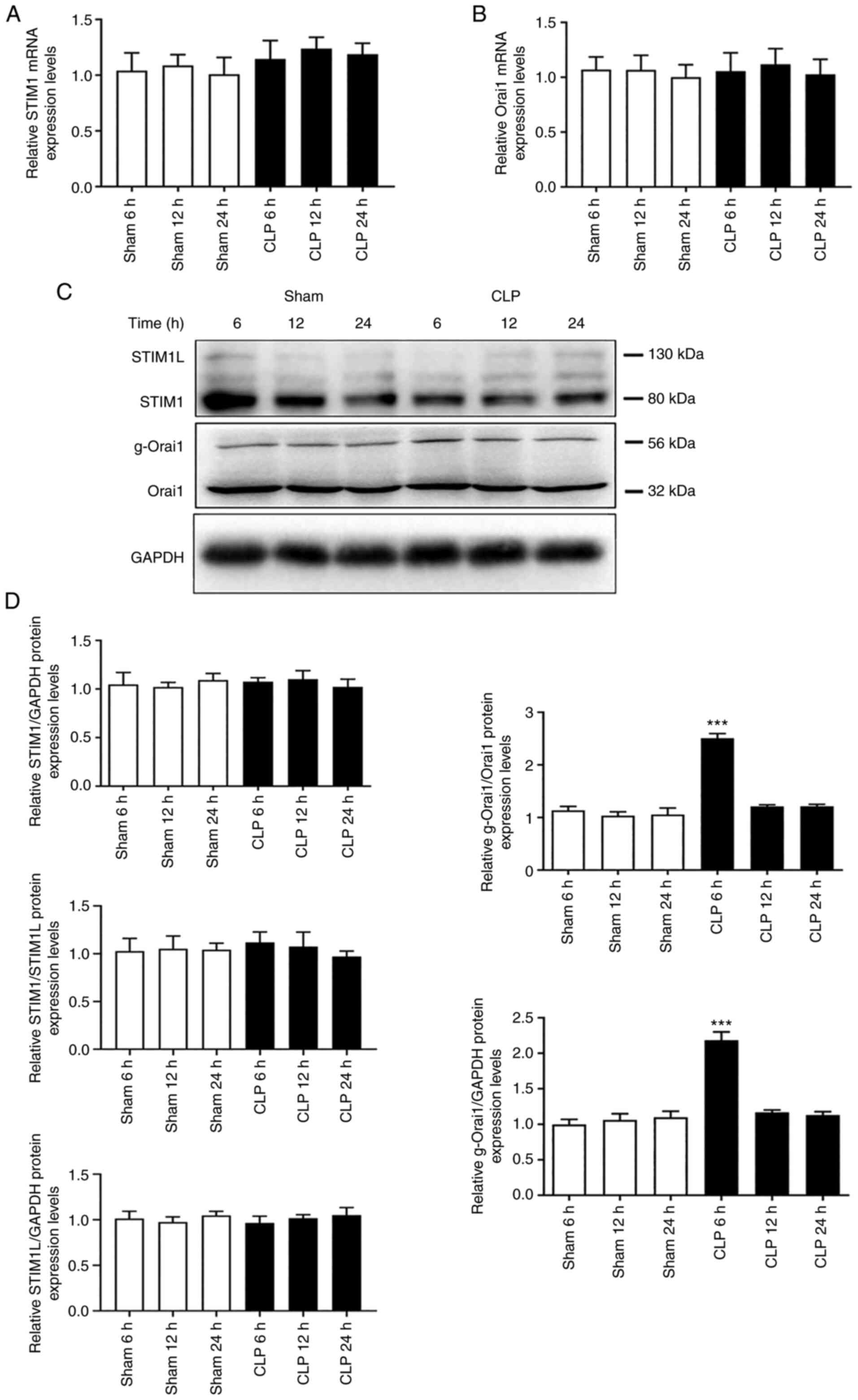
Myocardium exhibiting apoptotic activation were
obtained from septic mice to evaluate changes in STIM1 and Orai1
expression levels via RT-qPCR and western blotting. The results
demonstrated that STIM1 and Orai1 mRNA and protein expression
levels were not significantly different between the CLP and sham
groups at 6, 12, or 24 h (Fig.
3). Interestingly, significant increases in Orai1 glycosylation
were observed at 6 h after the CLP operation compared with the sham
6 h group (P<0.001).
LPS increases cytosolic
Ca2+ levels and induces SOCE in H9C2 cells
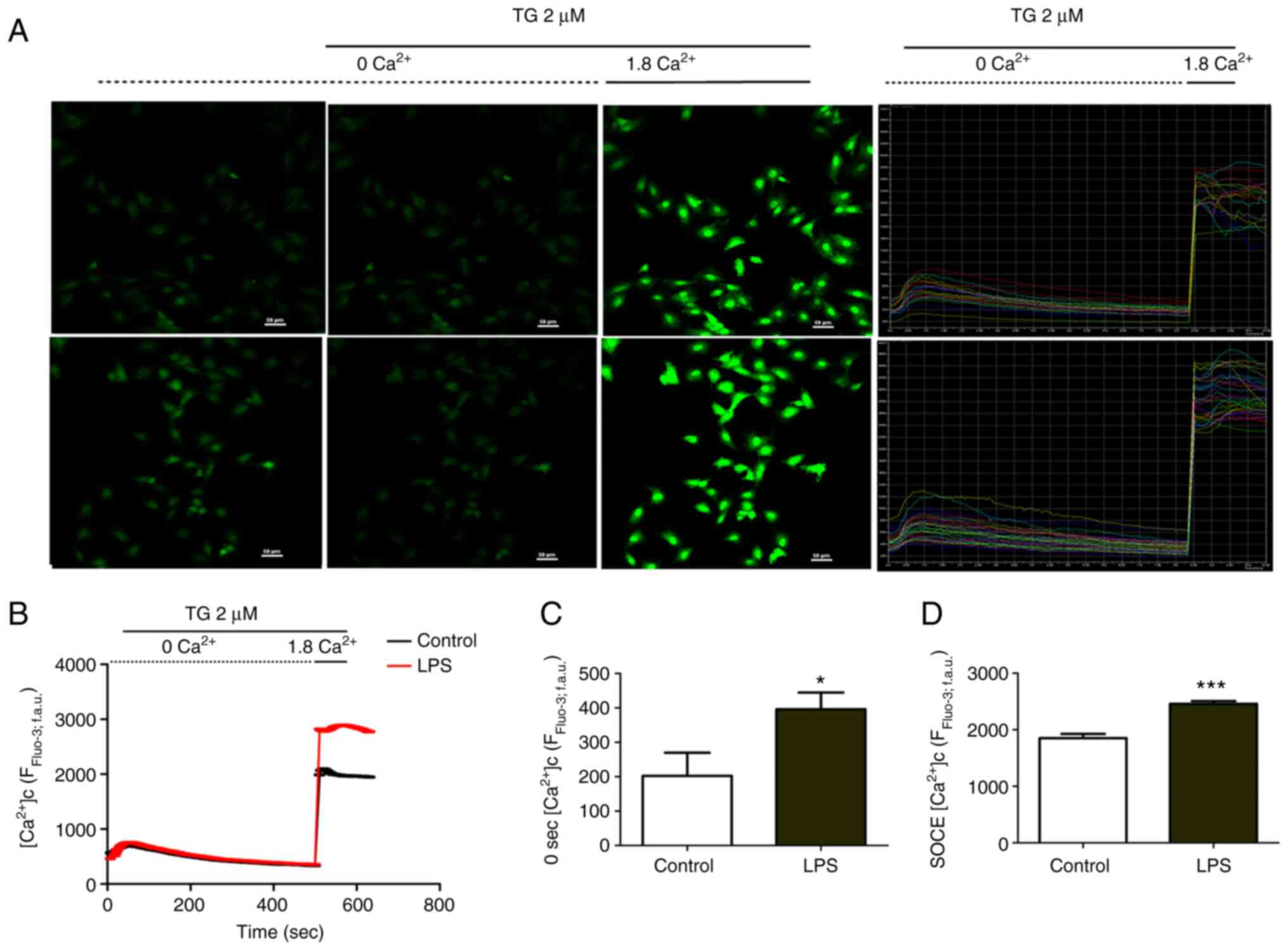
In vitro changes in the cytosolic
Ca2+ concentration and SOCE were observed in septic
cardiomyocytes. The Ca2+ levels in the ER were depleted
by TG in Ca2+-free medium. Ca2+ was re-added
to the medium and fluctuations in the cytosolic Ca2+
concentration were recorded using Fluo-3/AM fluorescence (Fig. 4A). The cytosolic Ca2+
concentration initially decreased before levelling off and then
abruptly and markedly increased after Ca2+ was added to
the medium. SOCE was responsible for the significant gain in
fluorescence intensity after Ca2+ was added compared
with the control (without TG or Ca2+), as observed in
H9C2 cells stimulated with LPS for 1–2 h (P<0.001) (Fig. 4B and C). The cytosolic
Ca2+ concentration in H9C2 cells was also significantly
enhanced following 1–2 h LPS stimulation compared with the control
group (P<0.05) (Fig. 4D).
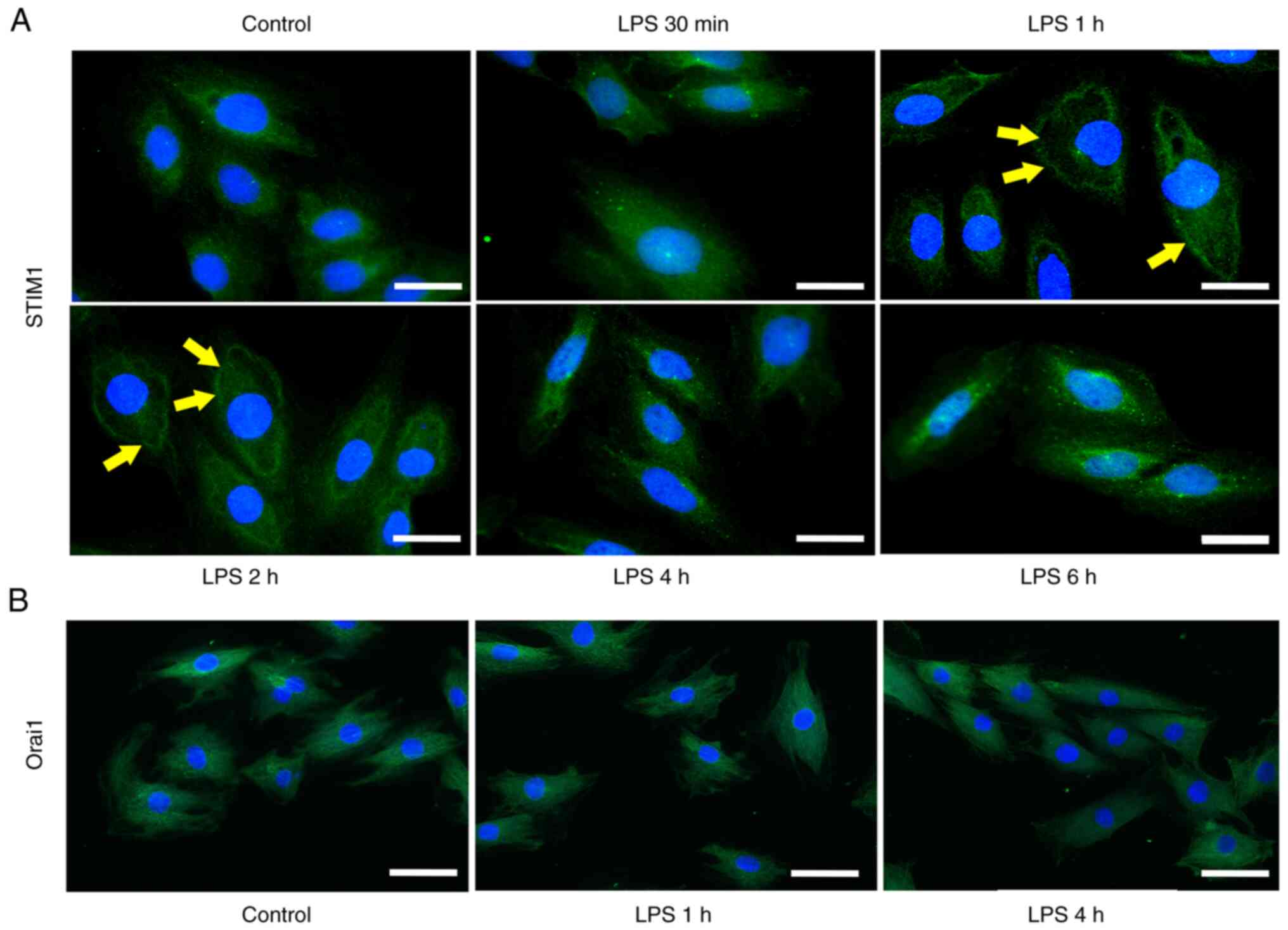
LPS-induced activation of STIM1
clustering and redistribution in H9C2 cells
Immunofluorescent staining of STIM1 was performed in
H9C2 cells following LPS treatment for 30 min, 1, 2, 4 and 6 h. The
distribution of STIM1 at 30 min of LPS treatment was the same as
that in the control group (equivalent PBS used), although changes
in the translocation of STIM1 at the ER-plasma membrane junction
occurred at 1 and 2 h following LPS treatment. The distribution of
STIM1 returned to normal at 4 and 6 h compared with the control
group (Fig. 5A).
Immunofluorescent staining of Orai1 was performed following LPS
stimulation for 1 and 4 h in H9C2 cells. Immunofluorescent analysis
demonstrated that LPS had no effect on the distribution and
expression of Orai1 compared with the control (equivalent PBS used)
(Fig. 5B).
Discussion
The present study demonstrated that myocardial
dysfunction, cardiac injury and myocardial depression may be
associated with increased intracellular Ca2+
concentrations caused by STIM1/Orai1-mediated SOCE. Cardiac
contractile function was significantly reduced at 6 h and
morphological changes in the cardiac tissues and the myocardial
apoptosis rate were increased at 6, 12 and 24 h in the CLP compared
with the corresponding sham groups. The mRNA and protein expression
levels of Bax/Bcl-2 were significantly enhanced at 6 and 12 h and
glycosylation of Orai1 was significantly increased at 6 h in the
myocardium of septic mice. In H9C2 cells treated with LPS, we
detected enhanced intracellular Ca2+ concentration and
SOCE at 1 to 2 h and the change of clustering and distribution of
STIM1 at 1, 2 h.
Septic myocardial depression occurs during the very
early stage of sepsis (35).
Although no consistent diagnostic criteria exist for septic
myocardial dysfunction, it is clearly characterized by reduced
ventricular contractility (36).
Changes in ventricular contractility have been demonstrated in
patients with sepsis, as well as in in vitro and in
vivo experimental studies (37–39). Moreover, myocardium dystfunction
in sepsis is reported to be reversable (40). Echocardiography is an
irreplaceable tool for the evaluation of septic myocardial
dysfunction. LVEF and LVFS are two major parameters used to assess
changes in ventricular contractility. In the present study, LVEF
and LVFS were reduced at 6 h following the CLP operation, before
being restored at 12 h, which indicates ventricular contraction
dysfunction during the early stages of sepsis and reversal of
septic myocardial dysfunction. These results are in accordance with
previous studies (41) and they
have demonstrated the success of this animal model of septic
myocardial dysfunction.
Apoptosis is a type of programmed cell death that
has been demonstrated to contribute to septic myocardial depression
(9). Bax and Bcl-2 belong to the
Bcl-2 family of proteins associated with apoptosis and changes in
these proteins over time have been observed in the heart tissue of
septic animal models (41). Bcl-2
overexpression may prevent myocardial dysfunction and cardiac
apoptosis in rodent models of sepsis (42). Furthermore, certain drugs and
reagents have a protective effect on septic myocardial depression
by inhibiting apoptosis (43–45). In the present study, the
occurrence of myocardial apoptosis in sepsis was detected using
TUNEL staining. The time-dependent expression (Bax/Bcl-2 was first
elevated, reaching to peak, then decreased) of Bax and Bcl-2
occurred at both the mRNA and protein expression levels, which is
consistent with other studies (41,42). These results further demonstrated
the success of this animal model of septic myocardial injury.
The mechanisms underlying septic myocardial
depression include mitochondrial dysfunction, inflammation,
excessive formation of reactive oxygen species and nitric oxide, an
imbalance in Ca2+ homeostasis and regulation of the G
protein receptor kinase (46–48). In a Previous study has shown that
short-term (2 min) stimulation of neonatal cardiomyocytes with
septic serum was demonstrated to be sufficient to increase the
basal Ca2+ concentration and reduce the amplitude of
transient Ca2+ concentrations (8). An increase in the basal
Ca2+ concentration was still detected at 24 h following
stimulation; however, a reduction in the transient Ca2+
amplitude was not observed at this timepoint (8). These previous findings indicated
that cardiac dysfunction is reversible, whereas myocardial cellular
apoptosis is irreversible. Similarly, short-term (3 h) treatment of
adult ventricular cardiomyocytes with LPS increases the mean
cytoplasmic Ca2+ concentration (49). In accordance with other studies
reporting an increase in the basal Ca2+ concentration
after LPS treatment in cardiomyocytes, a significant elevation in
the mean cytosolic Ca2+ concentration was observed in
H9C2 cells stimulated with LPS for 1–2 h.
Ca2+ homeostasis is regulated by
Ca2+ channels, such as LTCCs,
Na+/Ca2+ exchange channels, Orai1 and
transient receptor potential canonical channels, located in the
plasma membrane, as well as the ryanodine receptor and SR
Ca2+ ATPase located in the ER membrane (29,50–52). Ca2+ transporters are
triggered by various stimuli, including voltage changes,
Ca2+ release and depletion of ER (53). Changes in the Ca2+
level in the plasma membrane may contribute to normal physiological
functions, such as the contraction/relaxation cycle, the basal
signalling pathway and various pathological states, such as cell
structural damage, apoptosis and cell hypertrophy. Among the
Ca2+ transporters contributing to Ca2+ entry,
LTCCs function in transient cytoplasm Ca2+ cycling of
the cardiomyocyte contraction/relaxation cycle. SOCE is part of the
signalling pathway responsible for cell survival and death and is
associated with sustained elevation of Ca2+ levels in
the cytoplasm (18,19). The results of the present study
indicated that the Ca2+ levels rise between 1 and 2 h in
the cytoplasm, which suggested that SOCE may contribute to the
occurrence and development of septic myocardial injury.
SOCE is triggered by the depletion of
Ca2+ in the ER, which may be sensed by STIM1 and
Ca2+ entry is activated by Ca2+ channels,
such as Orai1 (17,54). STIM1 and Orai1 are expressed in
the heart; however, their expression levels and SOCE activation are
lower in adult cardiomyocytes compared with embryonic or new-born
cardiomyocytes (18). This may
limit investigations of STIM1/Orai1-mediated SOCE in
cardiomyocytes. STIM1/Orai1-mediated SOCE was initially discovered
in non-excitable cells and was thought to be absent in excitable
cells (55). STIM1/Orai1-mediated
SOCE was later discovered in excitable cells and was reported to
contribute to hyperplasia of smooth muscle cells and hypertrophy of
cardiomyocytes (20,56). However, to the best of our
knowledge the role of STIM1/Orai1-mediated SOCE in myocardial
injury has seldom been reported and the effect of
STIM1/Orai1-mediated SOCE in septic myocardial depression has never
been reported. The present study demonstrated significantly
increased SOCE in cardiomyocytes stimulated by LPS, which indicated
a potential relationship between SOCE and septic myocardial
depression.
Slight changes in the SOCE-related proteins STIM1
and Orai1 were also observed in the present study, which differs
from increased expression of STIM1 and Orai1 previously been found
in the model of cardiac hypertrophy (19,57). The redistribution of STIM1 to the
plasma membrane was observed for a short period following LPS
stimulation and was time-dependent. Translocation of STIM1 was
detected by immunofluorescence and the results indicated the
oligomerized aggregation of STIM1 at the ER-plasma membrane
junction. Moreover, a previous study proposed that short-term TG
stimulation results in changes in the clustering and redistribution
of STIM1, but not in the expression levels (19). Furthermore, two STIM1 subtypes
have been reported, with STIM1L (a long splice variant of STIM1)
exhibiting more prominent changes compared with STIM1 in cardiac
hypertrophy models (18). In the
present study two bands representing STIM1 were detected via
western blotting, which corresponded to STIM1L and STIM1. However,
no significant difference in the protein expression level of either
STIM1L and STIM1were observed in the present study.
No change in Orai1 expression level was observed by
qPCR or western blot analysis in our experiments, while a change
was observed in the myocardial hypertrophy model. The plasma
membrane protein Orai1 was observed at 32 kDa in most protein
expression experiments. However, the SOCE rate was dependent on
glycosylation of Orai1, which produced a 56 kDa marker in
fibroblasts and T cells (58). In
the present study the protein expression of Orai1 at both 32 and 56
kDa was detected and significant changes in Orai1 glycosylation (56
kDa) were demonstrated in the CLP group. Orai1 glycosylation was
significantly increased at 6 h and returned to the baseline, which
was accordance with the change in sepsis-induced cardiac
dysfunction and myocardial apoptosis. Although altered expression
levels of STIM1 and Orai1 were not observed in the present study,
the translocation of STIM1 and significantly increased
glycosylation of Orai1 indicated a potential association between
STIM1/Orai1-mediated SOCE and septic myocardial depression.
In summary, the present study demonstrated that
STIM1/Orai1-mediated SOCE may be associated with septic myocardial
depression. Therefore, the regulation of STIM1/Orai1-mediated SOCE
may have the potential to alleviate septic myocardial depression.
However, further studies are needed to to confirm the exact role of
STIM1/Orai1-mediated SOCE in septic cardiac depression and to
determine its clinical effect and toxicity.
Acknowledgements
Not applicable.
Funding
The present study was funded by the Zhejiang Provincial Natural
Science Foundation (grant no. LQ15H150003), and in part, by grants
from the National Natural Science Foundation of China (grant no.
81772112 and 81871583).
Availability of data and materials
The datasets used and/or analysed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JY, ML GZ and ZL conceived and designed the study.
JY, XH, QL, ZJ, SZ and GH performed the experiments and analysed
the data. JY, GZ and ZL wrote the manuscript. JY, GZ, GH and ZL
confirm the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The experiments involving animals were performed in
accordance with the National Institutes of Health Guidelines for
the Care and Use of Laboratory Animals and were approved by the
Ethics Committee of Wenzhou Medical University (approval no.
wydw2016-0217; Wenzhou, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Singer M, Deutschman CS, Seymour CW,
Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche
JD, Coopersmith CM, et al: The Third International consensus
definitions for sepsis and septic shock (Sepsis-3). JAMA.
315:801–810. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Landesberg G, Gilon D, Meroz Y, Georgieva
M, Levin PD, Goodman S, Avidan A, Beeri R, Weissman C, Jaffe AS and
Sprung CL: Diastolic dysfunction and mortality in severe sepsis and
septic shock. Eur Heart J. 33:895–903. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jeong HS, Lee TH, Bang CH, Kim JH and Hong
SJ: Risk factors and outcomes of sepsis-induced myocardial
dysfunction and stressinduced cardiomyopathy in sepsis or septic
shock: A comparative retrospective study. Medicine (Baltimore).
97:e02632018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Eisner DA, Caldwell JL, Kistamás K and
Trafford AW: Calcium and excitation-contraction coupling in the
heart. Circ Res. 121:181–195. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang HG, Pathan N, Ethell IM, Krajewski S,
Yamaguchi Y, Shibasaki F, McKeon F, Bobo T, Franke TF and Reed JC:
Ca2+-induced apoptosis through calcineurin dephosphorylation of
BAD. Science. 284:339–343. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Winslow RL, Walker MA and Greenstein JL:
Modeling calcium regulation of contraction, energetics, signaling,
and transcription in the cardiac myocyte. Wiley Interdiscip Rev
Syst Biol Med. 8:37–67. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Thompson M, Kliewer A, Maass D, Becker L,
White DJ, Bryant D, Arteaga G, Horton J and Giroir BP: Increased
cardiomyocyte intracellular calcium during endotoxin-induced
cardiac dysfunction in guinea pigs. Pediatr Res. 47:669–676. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Celes MR, Malvestio LM, Suadicani SO,
Prado CM, Figueiredo MJ, Campos EC, Freitas AC, Spray DC, Tanowitz
HB, da Silva JS and Rossi MA: Disruption of calcium homeostasis in
cardiomyocytes underlies cardiac structural and functional changes
in severe sepsis. PLoS One. 8:e688092013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Suzuki J, Bayna E, Li HL, Molle ED and Lew
WY: Lipopolysaccharide activates calcineurin in ventricular
myocytes. J Am Coll Cardiol. 49:491–499. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bers DM: Calcium fluxes involved in
control of cardiac myocyte contraction. Circ Res. 87:275–281. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bers DM: Calcium cycling and signaling in
cardiac myocytes. Annu Rev Physiol. 70:23–49. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Feske S, Gwack Y, Prakriya M, Srikanth S,
Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M and Rao A: A
mutation in Orai1 causes immune deficiency by abrogating CRAC
channel function. Nature. 441:179–185. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zitt C, Strauss B, Schwarz EC, Spaeth N,
Rast G, Hatzelmann A and Hoth M: Potent inhibition of Ca2+
release-activated Ca2+ channels and T-lymphocyte activation by the
pyrazole derivative BTP2. J Biol Chem. 279:12427–12437. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hauser CJ, Fekete Z, Adams JM, Garced M,
Livingston DH and Deitch EA: PAF-mediated Ca2+ influx in human
neutrophils occurs via store-operated mechanisms. J Leukoc Biol.
69:63–68. 2001.PubMed/NCBI
|
|
15
|
Hunton DL, Lucchesi PA, Pang Y, Cheng X,
Dell'Italia LJ and Marchase RB: Capacitative calcium entry
contributes to nuclear factor of activated T-cells nuclear
translocation and hypertrophy in cardiomyocytes. J Biol Chem.
277:14266–14273. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Uehara A, Yasukochi M, Imanaga I, Nishi M
and Takeshima H: Store-operated Ca2+ entry uncoupled with ryanodine
receptor and junctional membrane complex in heart muscle cells.
Cell Calcium. 31:89–96. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shen Y, Thillaiappan NB and Taylor CW: The
store-operated Ca2+ entry complex comprises a small
cluster of STIM1 associated with one Orai1 channel. Proc Natl Acad
Sci USA. 118:e20107891182021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Luo X, Hojayev B, Jiang N, Wang ZV, Tandan
S, Rakalin A, Rothermel BA, Gillette TG and Hill JA:
STIM1-dependent store-operated Ca2+ entry is required
for pathological cardiac hypertrophy. J Mol Cell Cardiol.
52:136–147. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hulot JS, Fauconnier J, Ramanujam D,
Chaanine A, Aubart F, Sassi Y, Merkle S, Cazorla O, Ouillé A,
Dupuis M, et al: Critical role for stromal interaction molecule 1
in cardiac hypertrophy. Circulation. 124:796–805. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Voelkers M, Salz M, Herzog N, Frank D,
Dolatabadi N, Frey N, Gude N, Friedrich O, Koch WJ, Katus HA, et
al: Orai1 and Stim1 regulate normal and hypertrophic growth in
cardiomyocytes. J Mol Cell Cardiol. 48:1329–1334. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Prakriya M, Feske S, Gwack Y, Srikanth S,
Rao A and Hogan PG: Orai1 is an essential pore subunit of the CRAC
channel. Nature. 443:230–233. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck
TJ, Ellisman MH, Stauderman KA and Cahalan MD: STIM1 is a Ca2+
sensor that activates CRAC channels and migrates from the Ca2+
store to the plasma membrane. Nature. 437:902–905. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Luo R, Gomez AM, Benitah JP and Sabourin
J: Targeting Orai1-mediated store-operated Ca2+ entry in
heart failure. Front Cell Dev Biol. 8:5861092020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cacheux M, Strauss B, Raad N, Ilkan Z, Hu
J, Benard L, Feske S, Hulot JS and Akar FG: Cardiomyocyte-Specific
STIM1 (Stromal Interaction Molecule 1) depletion in the adult heart
promotes the development of arrhythmogenic discordant alternans.
Circ Arrhythm Electrophysiol. 12:e0073822019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Correll RN, Goonasekera SA, van Berlo JH,
Burr AR, Accornero F, Zhang H, Makarewich CA, York AJ, Sargent MA,
Chen X, et al: STIM1 elevation in the heart results in aberrant
Ca2+ handling and cardiomyopathy. J Mol Cell Cardiol.
87:38–47. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shiou YL, Lin HT, Ke LY, Wu BN, Shin SJ,
Chen CH, Tsai WC, Chu CS and Lee HC: Very Low-density lipoproteins
of metabolic syndrome modulates STIM1, suppresses store-operated
calcium entry, and deranges myofilament proteins in atrial
myocytes. J Clin Med. 8:8812019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Troupes CD, Wallner M, Borghetti G, Zhang
C, Mohsin S, von Lewinski D, Berretta RM, Kubo H, Chen X, Soboloff
J and Houser S: Role of STIM1 (Stromal Interaction Molecule 1) in
hypertrophy-related contractile dysfunction. Circ Res. 121:125–136.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zheng C, Lo CY, Meng Z, Li Z, Zhong M,
Zhang P, Lu J, Yang Z, Yan F, Zhang Y, et al: Gastrodin inhibits
store-operated Ca2+ entry and alleviates cardiac
hypertrophy. Front Pharmacol. 8:2222017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hobai IA, Edgecomb J, LaBarge K and
Colucci WS: Dysregulation of intracellular calcium transporters in
animal models of sepsis-induced cardiomyopathy. Shock. 43:3–15.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals, . Guide for the care and use of laboratory animals. 8th
edition. National Academies Press (US); Washington, DC: 2011
|
|
31
|
Rumienczyk I, Kulecka M, Ostrowski J, Mar
D, Bomsztyk K, Standage SW and Mikula M: Multi-Organ transcriptome
dynamics in a mouse model of cecal ligation and puncture-induced
polymicrobial sepsis. J Inflamm Res. 14:2377–2388. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gao M, Ha T, Zhang X, Liu L, Wang X,
Kelley J, Singh K, Kao R, Gao X, Williams D and Li C: Toll-like
receptor 3 plays a central role in cardiac dysfunction during
polymicrobial sepsis. Crit Care Med. 40:2390–2399. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li M, Ye J, Zhao G, Hong G, Hu X, Cao K,
Wu Y and Lu Z: Gas6 attenuates lipopolysaccharideinduced TNF-α
expression and apoptosis in H9C2 cells through NF-κB and MAPK
inhibition via the Axl/PI3K/Akt pathway. Int J Mol Med. 44:982–994.
2019.PubMed/NCBI
|
|
35
|
Merx MW and Weber C: Sepsis and the heart.
Circulation. 116:793–802. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Martin L, Derwall M, Al Zoubi S,
Zechendorf E, Reuter DA, Thiemermann C and Schuerholz T: The septic
heart: Current understanding of molecular mechanisms and clinical
implications. Chest. 155:427–437. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Munt B, Jue J, Gin K, Fenwick J and
Tweeddale M: Diastolic filling in human severe sepsis: An
echocardiographic study. Crit Care Med. 26:1829–1833. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ren J, Ren BH and Sharma AC:
Sepsis-induced depressed contractile function of isolated
ventricular myocytes is due to altered calcium transient
properties. Shock. 18:285–288. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Merx MW, Liehn EA, Janssens U, Lütticken
R, Schrader J, Hanrath P and Weber C: HMG-CoA reductase inhibitor
simvastatin profoundly improves survival in a murine model of
sepsis. Circulation. 109:2560–2565. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang H, Bei Y, Shen S, Huang P, Shi J,
Zhang J, Sun Q, Chen Y, Yang Y, Xu T, et al: MiR-21-3p controls
sepsis-associated cardiac dysfunction via regulating SORBS2. J Mol
Cell Cardiol. 94:43–53. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
McDonald TE, Grinman MN, Carthy CM and
Walley KR: Endotoxin infusion in rats induces apoptotic and
survival pathways in hearts. Am J Physiol Heart Circ Physiol.
279:H2053–H2061. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lancel S, Petillot P, Favory R, Stebach N,
Lahorte C, Danze PM, Vallet B, Marchetti P and Neviere R:
Expression of apoptosis regulatory factors during myocardial
dysfunction in endotoxemic rats. Crit Care Med. 33:492–496. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li Z, Yi N, Chen R, Meng Y, Wang Y, Liu H,
Cao W, Hu Y, Gu Y, Tong C, et al: miR-29b-3p protects
cardiomyocytes against endotoxin-induced apoptosis and inflammatory
response through targeting FOXO3A. Cell Signal. 74:1097162020.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu L, Yan M, Yang R, Qin X, Chen L, Li L,
Si J, Li X and Ma K: Adiponectin attenuates
lipopolysaccharide-induced apoptosis by regulating the
Cx43/PI3K/AKT pathway. Front Pharmacol. 12:6442252021. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Su ZD, Wei XB, Fu YB, Xu J, Wang ZH, Wang
Y, Cao JF, Huang JL and Yu DQ: Melatonin alleviates
lipopolysaccharide-induced myocardial injury by inhibiting
inflammation and pyroptosis in cardiomyocytes. Ann Transl Med.
9:4132021. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ravikumar N, Sayed MA, Poonsuph CJ, Sehgal
R, Shirke MM and Harky A: Septic cardiomyopathy: From basics to
management choices. Curr Probl Cardiol. 46:1007672021. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Liu YC, Yu MM, Shou ST and Chai YF:
Sepsis-Induced cardiomyopathy: Mechanisms and treatments. Front
Immunol. 8:10212017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Dal-Secco D, DalBó S, Lautherbach NES,
Gava FN, Celes MRN, Benedet PO, Souza AH, Akinaga J, Lima V, Silva
KP, et al: Cardiac hyporesponsiveness in severe sepsis is
associated with nitric oxide-dependent activation of G protein
receptor kinase. Am J Physiol Heart Circ Physiol. 313:H149–H163.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wang Y, Wang Y, Yang D, Yu X, Li H, Lv X,
Lu D and Wang H: β1-adrenoceptor stimulation promotes
LPS-induced cardiomyocyte apoptosis through activating PKA and
enhancing CaMKII and IκBα phosphorylation. Crit Care. 19:762015.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Gavali JT, Carrillo ED, García MC and
Sánchez JA: The mitochondrial K-ATP channel opener diazoxide
upregulates STIM1 and Orai1 via ROS and the MAPK pathway in adult
rat cardiomyocytes. Cell Biosci. 10:962020. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Numaga-Tomita T and Nishida M: TRPC
channels in cardiac plasticity. Cells. 9:4542020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Johnson MT, Gudlur A, Zhang X, Xin P,
Emrich SM, Yoast RE, Courjaret R, Nwokonko RM, Li W, Hempel N, et
al: L-type Ca2+ channel blockers promote vascular
remodeling through activation of STIM proteins. Proc Natl Acad Sci
USA. 117:17369–17380. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Collins HE, Zhu-Mauldin X, Marchase RB and
Chatham JC: STIM1/Orai1-mediated SOCE: Current perspectives and
potential roles in cardiac function and pathology. Am J Physiol
Heart Circ Physiol. 305:H446–H458. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lewis RS: Store-Operated calcium channels:
From function to structure and back again. Cold Spring Harb
Perspect Biol. 12:a0350552020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Putney JW Jr: A model for
receptor-regulated calcium entry. Cell Calcium. 7:1–12. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Climent B, Santiago E, Sánchez A,
Muñoz-Picos M, Pérez-Vizcaíno F, García-Sacristán A, Rivera L and
Prieto D: Metabolic syndrome inhibits store-operated
Ca2+ entry and calcium-induced calcium-release mechanism
in coronary artery smooth muscle. Biochem Pharmacol.
182:1142222020. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Segin S, Berlin M, Richter C, Flockerzi
RMV, Worley P, Freichel M and Londoño JEC: cardiomyocyte-specific
deletion of orai1 reveals its protective role in
angiotensin-II-induced pathological cardiac remodeling. Cells.
9:10922020. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Kappel S, Borgström A, Stokłosa P, Dörr K
and Peinelt C: Store-operated calcium entry in disease: Beyond
STIM/Orai expression levels. Semin Cell Dev Biol. 94:66–73. 2019.
View Article : Google Scholar : PubMed/NCBI
|