Introduction
An imbalance in metabolic homeostasis is the primary
cause of various chronic diseases. The number of patients
experiencing various chronic diseases caused by metabolic disorders
has increased significantly in various countries worldwide.
According to the prediction of the World Health Organization,
obesity, hyperlipidaemia, fatty liver disease and other diseases
dominated by lipid metabolism disorders have affected more than one
third of the global population (1,2).
The number of patients with diabetes and glucose metabolism
disorders will reach 642 million by 2040, and more than 90% of
these patients will have type 2 diabetes mellitus (T2DM) (3). Lipid metabolism disorder is a common
metabolic disorder leading to targeted organ damage. For example,
lipid metabolism disorder is one of the risk factors for
cardiovascular disease, which can cause atherosclerosis, unstable
plaque rupture and accelerated thrombosis (4). Disorder of lipid metabolism can also
cause renal injury. High levels of triglycerides (TG) and
low-density lipoprotein cholesterol (LDL-C) are deposited in the
arterial wall and glomerular basement membrane, which may cause
renal endothelial cell injury. In addition, chylomicrons and other
residual lipoproteins in plasma can affect the development of
diabetic nephropathy by damaging the vascular endothelial barrier
and activating the platelets. Abnormal lipid deposition in the
liver can cause hepatocyte steatosis and non-alcoholic
steatohepatitis, accompanied by hepatocyte injury, inflammation,
angiogenesis and varying degrees of fibrosis. Simultaneously, it
can affect the insulin signal pathway and cause insulin resistance
in the liver (5,6). Lipid and glucose metabolisms
interact and regulate each other. High levels of TG can promote the
disorder of insulin signal transduction, resulting in a decrease in
glucose utilization. Furthermore, persistent hyperglycemia in
patients with T2DM can promote fatty acid (FA) synthesis and TG
accumulation, resulting in abnormal lipid metabolism and
deposition. Long-term hyperglycemia will not only lead to diabetes
but also increase the risk of metabolic diseases such as
cardiovascular and cerebrovascular and non-alcoholic fatty liver
diseases (7,8). The research on the influence of the
regulation mechanism of blood glucose homeostasis and lipid
metabolism on the development of chronic diseases is one of the hot
issues concerned for the scholars.
VEGFB (vascular endothelial growth factor B) is a
glycoprotein that induces a series of reactions via its receptors
VEGFR1 and neuropilin-1 (NRP1) through the paracrine pathway
(9). VEGFR1 is highly expressed
in mitochondrial-rich tissues such as the heart, liver, muscle and
brown adipose tissue. Previously, it was suggested that VEGFB is a
survival molecule involved in the regulation of FA metabolism and
glucose homeostasis (10,11). In 2010, Hagberg et al (12) first reported in Nature that VEGFB
can regulate FA absorption in endothelial cells, transfer excess
FAs to tissues with high energy metabolism and improve lipid
transportation in endothelial cells. Targeting VEGFB may be a novel
approach to preventing pathological lipid deposition (12). In 2012, Hagberg et al (10) reported that reducing VEGFB signal
improves insulin sensitivity and glucose tolerance in T2DM animal
models. In 2014, Wagenmakers et al (13) suggested that VEGFB controls the
expression of FA transporters in capillary endothelial cells, which
may prevent the accumulation of lipotoxic FAs. In 2017, Falkevall
et al (14) revealed that VEGFB
gene deletion can not only prevent pathological lipid deposition
but also improve glucose homeostasis and insulin sensitivity by
targeting the lipid transport properties of endothelial cells. In
2020, Moessinger et al (15)
established a VEGFB gene knockout mouse model and observed that the
VEGFB signal pathway can reduce endothelial cell glucose transport
and cardiac glucose utilization. In the same year, Jensen et al
(16) observed that islet β-cell
specific VEGFB deficiency increased insulin secretion by
upregulating the insulin gene. Therefore, numerous scholars have
proposed that VEGFB may be a target for the treatment of metabolic
diseases such as obesity and diabetes.
As one of the members of the VEGFs family, VEGFB
mainly plays a biological role after binding to the membrane
receptor VEGFR1. VEGFR1 consists of seven extracellular
immunoglobulin homologous regions, one transmembrane structural
region and one intracellular tyrosine kinase region (17). VEGFR1 is primarily expressed in
endothelial cells, osteoblasts, monocytes/giant cells, phage cells,
blastoderm trophoblast cells, renal interstitial cells and certain
haematopoietic stem cells (18).
In 2016, Robciuc et al (11)
reported in Cell that in VEGFB transduced adipose tissue and VEGFB
transgenic mouse models, fat vasodilation induced by VEGFB/VEGFR1
can counteract obesity and related metabolic complications
(11). Shen et al (19) observed that VEGFB can reduce
myocardial lipid accumulation and hypertrophy through VEGFR1 and
its related downstream signaling pathways. In 2021, Hu et al
(20) also proved that
VEGFB/VEGFR1 could reduce lipid accumulation by activating the
CaMKK/AMPK/ACC/CPT1 pathway.
At present, studies have reported that VEGFB can
participate in the regulation of lipid metabolism. However, there
are limited reports on whether it can cause changes in glucose
metabolism while regulating lipid metabolism. Whether VEGFB can
participate in the regulation of glucose and lipid metabolism via
the VEGFB/VEGFR1 pathway remains unclear. In the present study, a
VEGFB gene knockout mouse was established to study the effects of
VEGFB gene deletion on glucose and lipid metabolism. In addition,
the role of VEGFB in the signal pathway that regulates glucose and
lipid metabolism was analyzed, and its findings provided evidence
for the pathogenesis of metabolic diseases.
Materials and methods
Animals
All animal experiments were approved by the ethics
committee of Binzhou Medical University (Yantai, China) and
strictly abided by the animal ethics code. All procedures involving
animals were reviewed and approved by the Institutional Animal Care
and Use Committee of the Medical Ethics Committee of Binzhou
Medical University (IACUC protocol no. 2021-300). C57BL/6N male
mice were selected as experimental animals. The VEGFB gene knockout
mouse model was constructed by Saiye (Guangzhou) Biological
Technology Co., Ltd. using Crispr/cas9 system. VEGFB gene ID is
22340, locates on chromosome 19, and NM_ 011697.3 transcript was
selected as reference transcript. This transcript has 7 exons, ATG
is on exon 1 and TAG is on exon 6. In the present experiment, 90%
of the protein coding region of exon 2~6 of VEGFB gene was knocked
out in a large fragment manner, and the knockout fragment was
~2,000 bp. VEGFB gene retains the first 30 amino acids of exon 1
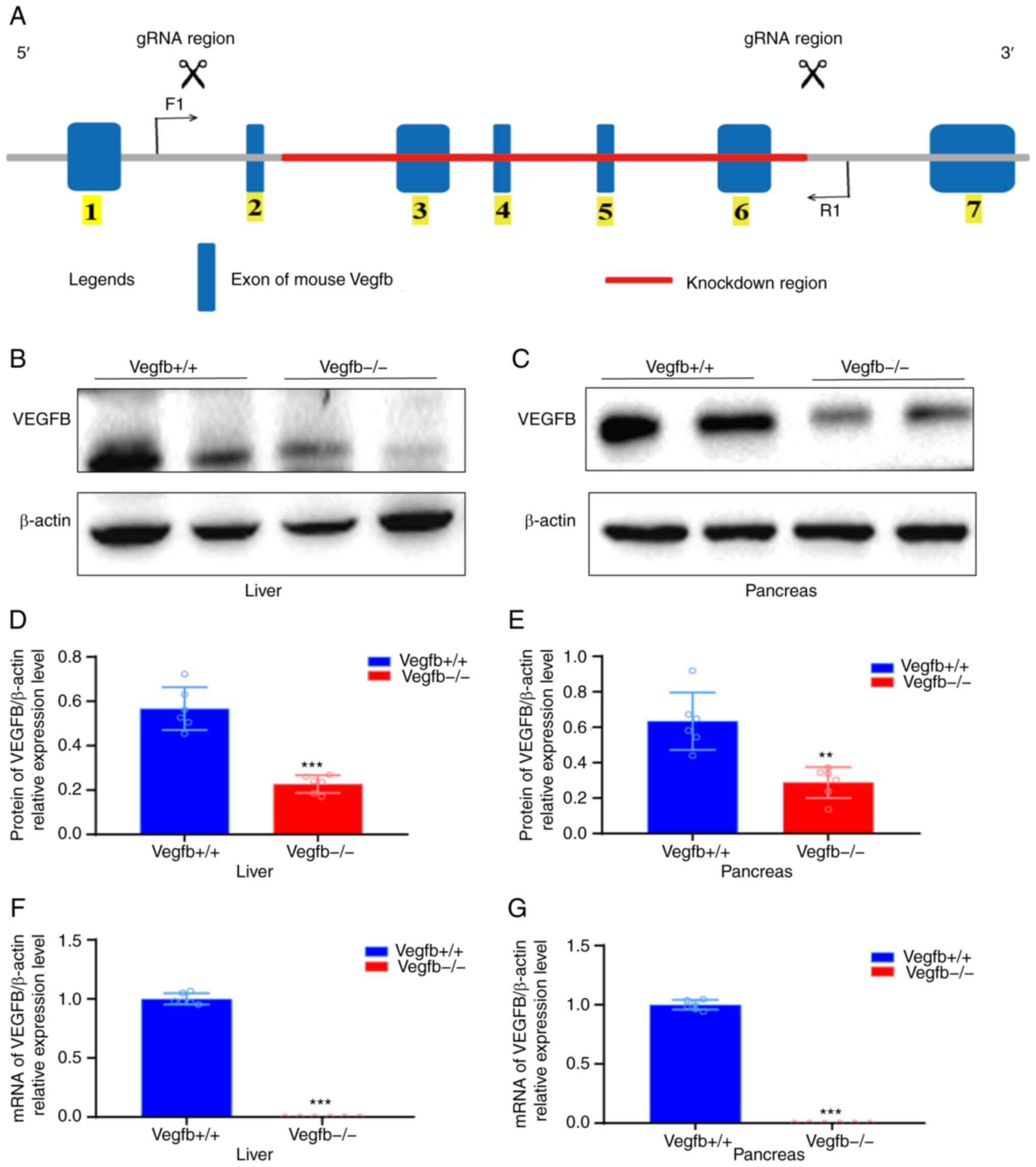
(Fig. 1A). The level of VEGFB
knockdown in fertilized eggs was verified by PCR amplification and
sequencing. Two pairs of primers were used for PCR cycle and the
primer 1 sequence was: forward, 5′-TCTCAAGGTTGGCGGAAGTGG-3′ and
reverse,: 5′-CAAACTCACCATGTCACCAAGGAG-3′. Primer 2 sequence was:
forward, 5′-TCTCAAGGTTGGCGGAAGTGG-3′ and reverse,
5′-TTGGGATCACGCAAGATAAGGG-3′. The gRNA vector and cas9 mRNA were
co-injected into in vitro fertilized eggs and transplanted into the
fallopian tubes of C57BL/6N strain surrogate mice. Positive VEGFB
heterozygous mice (VEGFB+/−) mice of generation 0 (F0
mice) were obtained after inoculation. After sexual maturation, F0
mice were bred and propagated with C57BL/6N mice, and F1 hybrid
knockdown mice were identified. The F1 generation of heterozygous
knockdown mice were selfed to obtain homozygous knockdown mice
(VEGFB−/−), heterozygous knockdown mice and wild mice
(VEGFB+/+). Healthy male mice, aged 4 weeks and weighing
18–22 g were randomly selected for the experiment. In total 9 mice
were selected for each of the VEGFB+/+ and
VEGFB−/− groups. All animals were kept in the SPF animal
room of the Medical Research Center of Binzhou Medical College.
Conditions were maintained at 20°C and a relative humidity of
50±20%, with food and water obtained freely and a 12-h light/dark
cycle. During animal experiment, 6 mice of each group were selected
for anesthesia with 3% isoflurane, and then the mice were
sacrificed by cervical dislocation after blood collection from the
eyeball. The liver and pancreas tissue were received in 4°C
condition for molecular biology experiments. Meanwhile, 3 mice of
each group were sacrificed by cervical dislocation to obtain liver
and pancreas for morphological experiments.
Isolation of mouse islets
Mice in VEGFB+/+ and VEGFB−/−
groups were received after fasting for 12 h, with 6 mice in each
group. After anesthesia, they were disinfected with alcohol, the
abdominal cavity was opened, then the pancreas was extracted and
placed in the culture dish. The adipose tissue was picked out in
Hank's buffer, and then 0.5 mg/ml collagenase P was poured into the
pancreas. After the pancreas was completely expanded, it was
digested in a 37°C water bath for 10 min. The digested pancreas was
shaken quickly until it reached the shape of sediment; then it was
placed into the petri dish and precooled (4°C) Hank's buffer was
added to stop digestion. Under the ×100 visual field of a
stereomicroscope, isolated round and smooth islet cell masses were
selected.
Islet identification
The same amount of dithizone (DTZ) solution was
added to 50 µl islet cell suspension, and staining was performed at
room temperature for 10 min. The islet cell mass was identified
under a ×200 visual field of a fluorescence microscope.
Western blot analysis
Mouse tissue lysates were analyzed by western
blotting to detect changes in related protein expression. The liver
tissue and islets from six mice in each group were selected for
organ extraction. Protease inhibitor (1% PMSF; cat. no. PO100) and
tissue/cell high-efficiency lysate (RIPA; cat. no. R0010; both from
Beijing Solarbio Science & Technology Co., Ltd.) were added
into tissue and then tissue was lysed on ice in 4°C for 30 min
using ultrasonication (Ultrasonic Cell Crusher XC-CD). The
concentration of the extracted proteins was measured using a BCA
kit. The supernatant was collected after centrifugation at 15,300 ×
g and 4°C for 20 min. Sample loading buffer (D1020-5; Beijing
Solarbio Science & Technology Co., Ltd.) was added, boiled in a
pan (Tu-100C) at 95°C for 10 min, and 20 µg sample protein was
dissolved in gel after cooling and then transfered to a polymer
PVDF membrane. VEGFB (22 kDa) and VEGFA (27 kDa) are small
molecular weight proteins and the concentration of gel was 12%.
VEGFR1 (180 kDa), VEGFR2 (151 kda) and NRP1 (103 kDa) are high
molecular weight proteins and the gel concentration was 10%. The
membrane was blocked with 5% skimmed milk at room temperature for 1
h, and then incubated overnight with primary antibody (Table I) at 4°C. The next day, incubation
was performed with anti-rabbit IgG HRP-conjugated antibody (cat.
no. 111-035-003) and anti-mouse IgG HRP-conjugated antibody (cat.
no. 115-035-003) (1:5,000; Jackson ImmunoResearch Laboratories,
Inc.) at room temperature for 2 h. Protein band Biosharp ECL prime
Western blot Reagent (Biosharp Life Sciences; cat. no. BL520b) was
used and detected in the enhanced chemiluminescence system
(Tanon-5200; Tanon Science & Technology). ImageJ software
v1.8.0 (National Institutes of Health) was used to analyze the gray
value of protein.
 | Table I.Antibody information. |
Table I.
Antibody information.
| Antibody name | Dilution | Species/source | Company | Cat. no. |
|---|
| VEGFB | 1:1,000 | Rabbit | Affinity
Biosciences | AF5250 |
| VEGFA | 1:1,000 | Rabbit | Affinity
Biosciences | DF7470 |
| VEGFR1 | 1:1,000 | Rabbit | Affinity
Biosciences | AF6204 |
| VEGFR2 | 1:1,000 | Rabbit | Affinity
Biosciences | AF6281 |
| NRP1 | 1:1,000 | Rabbit | Affinity
Biosciences | DF7877 |
| β-actin | 1:1,000 | Mouse | Affinity
Biosciences | AF7018 |
Reverse transcription-quantitative
(RT-q) PCR
A total of six mice in each group of
VEGFB+/+ group and VEGFB−/− group were
selected to extract liver and islet tissues; total RNA was
extracted with TRIzol® reagent (Invitrogen; Thermo
Fisher Scientific, Inc.). RT was performed according to the
manufacturer's instructions using an RNA Easy Isolation Reagent
(Vazyme Biotech Co., Ltd.). qPCR was performed according to the
manufacturer's instructions using TB Green Premix Ex Taq II (Takara
Bio USA, Inc.) fluorescence quantitative kit. PCR amplification was
performed on PCR machine QuantStudio 3 (Thermo Fisher Scientific,
Inc.). qPCR was performed using the following thermocycling
conditions: Initial denaturation at 95°C for 30 sec; then 40 cycles
were performed at 95°C for 5 sec and 60°C for 34 sec; finally, the
dissolution process was carried out at 95°C, 60°C and 95°C for 15
sec, 1 min and 15 sec, respectively. The 2−ΔΔCq method
was used to quantify the level of mRNA expression using β-actin as
an internal reference gene (21).
Primer sequences are provided in Table II.
| Table II.Primer sequences for quantitative
PCR. |
Table II.
Primer sequences for quantitative
PCR.
| Primer name | Primer sequence
(5′-3′) |
|---|
| VEGFB | F:
AGCCACCAGAAGAAAGTGGT |
|
| R:
GCTGGGCACTAGTTGTTTGA |
| VEGFA | F:
GAGGCTGCTGTAACGATGAA |
|
| R:
TATGTGCTGGCTTTGGTGAT |
| VEGFR1 | F:
TTGGTGGTGGCTGACTCTCA |
|
| R:
TCTCCTTCGGCTGGCATCTT |
| VEGFR2 | F:
TGATTTCACCTGGCACTCTCC |
|
| R:
CCTTGGTCACTCTTGGTCACA |
| NRP1 | F:
CAGGGTTTTCCATCCGCTATG |
|
| R:
ACTCCAGTAGGTGCTGTATAGTT |
| β-actin | F:
CATCCGTAAAGACCTCTATGCCAAC |
|
| R:
ATGGAGCCACCGATCCACA |
Body weight, food intake and blood
glucose measurement
The gene identification of mice started at the 3rd
week, and the weight and food intake of wild and homozygote group
were measured every 2 weeks from the 4th week to draw the growth
and food intake curve. The mice were euthanized until the 32nd week
for tissue section. Experiments in vivo lasted 7 months. Meanwhile,
the fasting blood sugar and postprandial blood sugar of mice were
examined every 2 weeks to draw the blood sugar curve from the 4th
week to the 32nd week. After fasting for 12 h, blood was drawn from
the tail vein by using a Roche blood glucose-meter for fasting
blood glucose (FBG) measurement. Then food was provided for 2 h and
the Roche blood glucose-meter was used to draw blood for
postprandial blood glucose (PBG) measurement and the curve graph of
blood glucose was drawn.
Organ coefficient ratio
Mice (32 weeks old) were sacrificed by cervical
dislocation. The liver and pancreas were immediately dissected, the
fatty tissue around the organ was removed and the organ surface and
residual blood in the cavity were treated. The net weight of the
organ was measured with an electronic balance (BS210S) according to
the following formula: organ coefficient=net organ weight (g)/body
weight (g).
Hematoxylin-eosin (H&E)
staining
The liver and pancreas of the mice were fixed with
4% paraformaldehyde at room temperature for 24–48 h, then
dehydrated with gradient alcohol, transparency was achieved with
xylene, tissues embedded in paraffin and sectioned at a thickness
of 4 µm. The tissue sections were incubated at 60°C in an oven for
2 h for H&E staining at room temperature. The protocol was as
follows: the tissue sections were dewaxed with xylene twice for 10
min each time. Then they were treated with 100, 100, 95, 85 and 75%
alcohol for 5 min each time. After washing with distilled water,
tissue sections were immersed in 0.4% hematoxylin, stained for 5
min and washed with running water for 1 min. Subsequently, the
tissue sections were immersed in 1% hydrochloric acid ethanol
differentiation solution for 10 sec and washed with water until
they turned into blue. Then, the tissue sections were immersed in
0.1% eosin, stained for 1 min, and dehydrated with 75, 85, 95, 100
and 100% inverse concentration alcohol for 5 min each time. After
xylene transparency-treatment twice, the sections were sealed with
neutral gum and observed under the microscope.
Transmission electron microscopy
A total of 3 mice (aged 32 weeks) in each group were
fasted for 12 h and sacrificed under anesthesia. The liver and
pancreas of the mice were isolated and fixed with 2.5%
glutaraldehyde solution at 4°C for 2 h. After rinsing with buffer
solution 3 times at room temperature, the tissues were immediately
fixed with 1% osmic acid at 4°C for 90 min, and rinsed with buffer
solution 3 times again. Sequential gradient ethanol solution
dehydration followed by acetone replacement treatment twice for 15
min each time. The samples were embedded in different proportions
of acetone (V1/V2=1/2; V1/V2=2/1) and mixed and processed for 1 and
2 h, and then embedded with epoxy resin overnight at 70°C. The
samples were sliced with a Reichert ultrathin microtome (70 nm).
The sections were stained with lead citrate solution for 15 min at
25°C and then stained with uranyl acetate 50% ethanol saturated
solution for 15 min at 25°C. Then observed and photographed through
a transmission electron microscope (JEM-1400, Japan), and used
Image-Pro Plus version 6.0 (media cybernetics, Inc.) to
analyze.
ELISA
A total of 6 mice (aged 32 weeks) were selected from
each of the VEGFB+/+ and VEGFB-/- groups. After 12 h of fasting,
they were deeply anesthetized with 3% isoflurane. Eyeball blood was
taken and then the mice were killed by cervical dislocation. Whole
blood was taken in each group. The supernatant was collected using
3,800 × g centrifugation at 4°C for 20 min, and a microplate reader
was used to measure the optical density value. A standard curve was
established based on the measured values of the standard products,
and the level of each group of samples was calculated from the
standard curve obtained. Data results were statistically analyzed.
The mouse ELISA kits used for the detection of TG (cat no:
a110-1-1), total cholesterol (TC; cat. no.: a111-1-1), LDL (cat.
no.: 113-1-1) and high-density lipoprotein (HDL; cat. no.: 112-1-1)
were purchased from Nanjing Jiancheng Bioengineering Institute. The
mouse ELISA kits used for the detection of glycosylated hemoglobin
(HbA1c; cat. no.: ml063816), glucose (cat. no.: ml076701), insulin
(cat. no.: ml001983) and glucagon (cat. no.: ml057708) were all
purchased from Shanghai Enzyme-linked Biotechnology Co., Ltd. The
ELISA tests were performed according to the manufacturers' protocol
and the OD value was measured at 510 or 450 nm on the microplate
reader (BioTek China).
Oral glucose tolerance test (OGTT) and
intraperitoneal insulin tolerance test (IPITT)
In VEGFB+/+ group and VEGFB−/−
group, 9 mice in each group were selected for OGTT and IPITT
experiments. In the OGTT experiment, mice were fed with 40% glucose
at the dose of 2 g/kg after fasting for 12 h, and then the venous
blood glucose was measured with a blood glucose-meter at 0, 15, 30,
60, 90 and 120 min, and the OGTT curve was drawn. In the IPITT
experiment, the mice fasted for 6 h, were injected with insulin
intraperitoneally at the rate of 0.5 µg/kg, the venous blood
glucose of the mice was measured at 0, 15, 30, 60, 90 and 120 min
and the IPITT curve was drawn.
Insulin resistance index (HOMA-IR) and
pancreatic β cell function index (HOMA-β) measurement
Calculating mouse insulin resistance index (HPMA-IR)
and pancreatic β-cell function index (HOMA-β) were determined by
FBG and fasting insulin (FINS). The formula is as follows:
HOMA-IR=FBG × FINS)/22.5; HOMA-β=FINS × 20/(FBG-3.5).
Statistical analysis
All data were analyzed statistically with SPSS 22.0
statistical software (IBM Corp.). The results were expressed as the
mean ± SD. One-way ANOVA followed by Dunnet's post hoc test was
used, while comparisons between two groups were assessed using
paired Student's t-test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Systemic VEGFB-knockdown mouse model
construction and genetic identification
In order to explore the regulatory effect of VEGFB
on glucose and lipid metabolism and its mechanism, systemic
VEGFB-knockdown mice were constructed through CRISPR/cas9-mediated
genomic engineering. The VEGFB gene is located on mouse chromosome
19. A total of 7 exons were identified. The ATG start codon was in
exon 1, the tag stop codon was in exon 6 and exons 2–6 were
selected as targeted sites (Fig.
1A). To determine whether the VEGFB gene was knocked out, liver
and pancreatic tissues with high VEGFB expression were selected for
western blotting and gene transcription level analysis. Compared
with VEGFB+/+ mice, the protein and mRNA levels of VEGFB
in liver and pancreas tissues of VEGFB−/− mice were
significantly downregulated (P<0.05; Fig. 1B-G).
The effect of VEGFB downregulation on
the liver and pancreas
Mice in both groups were in favorable mental health,
lively and active, with bright coat colors, normal diet and water
intake and urine output and dry litter. There was no significant
change in weight gain or average food intake in VEGFB−/−
mice from week 4 to week 32 compared with VEGFB+/+ mice
under the same feeding conditions (P>0.05; Fig. 2A and B). Compared with that of
VEGFB+/+ mice, the liver-to-viscera ratio of
VEGFB−/− mice increased (P<0.001), but the
pancreas-to-viscera ratio was not significantly different
(P>0.05; Fig. 2C and D). The
results of H&E staining showed that the structure of liver
lobules in VEGFB+/+ mice was normal and the liver cells
were arranged neatly. The liver tissue of VEGFB−/− mice
showed obvious steatosis, the volume of hepatocytes was increased,
and circular lipid droplets of varying numbers and sizes were
observed in the cytoplasm. The nuclei of liver cells were squeezed
from the centre to the periphery (Fig. 2E). Compared with that of
VEGFB+/+ mice, the pancreatic tissue of
VEGFB−/− mice showed no obvious changes. Under the
microscope, spherical islet structures scattered among spicules of
different sizes and stains were observed (Fig. 2E). To further observe the effects
of VEGFB gene deletion on mouse liver and pancreas, sections of
32-week-old VEGFB+/+ mice and VEGFB−/− mice
were received to observe ultrastructural changes of liver and islet
cells by transmission electron microscopy. The liver cells of
VEGFB−/− mice had a large number of lipid droplets, up
to 5-µm in diameter, irregular concave nuclei and abnormal
inhomogeneous chromatin in the nuclei. The islet nuclei of
VEGFB+/+ mice and VEGFB−/− mice were round,
the nuclear membrane was clear and complete, the perinuclear space
was normal and the organelles such as mitochondria, ribosomes and
rough endoplasmic reticulum could be clearly observed in the
cytoplasm (Fig. 2F). In addition,
more endocrine granules were observed, most of which were secretory
granules of pancreatic islet β-cells. Insulin-secreting vesicles
were present in two different forms: immature vesicles with a
uniform grey color within and mature vesicles with a dense black
core (Fig. 2F). Compared with
VEGFB+/+ mice, VEGFB−/− mice had
significantly more insulin vesicles. The numbers of mature and
immature vesicles were also significantly higher than those of
VEGFB+/+ mice (P<0.05; Fig. 2G and H).
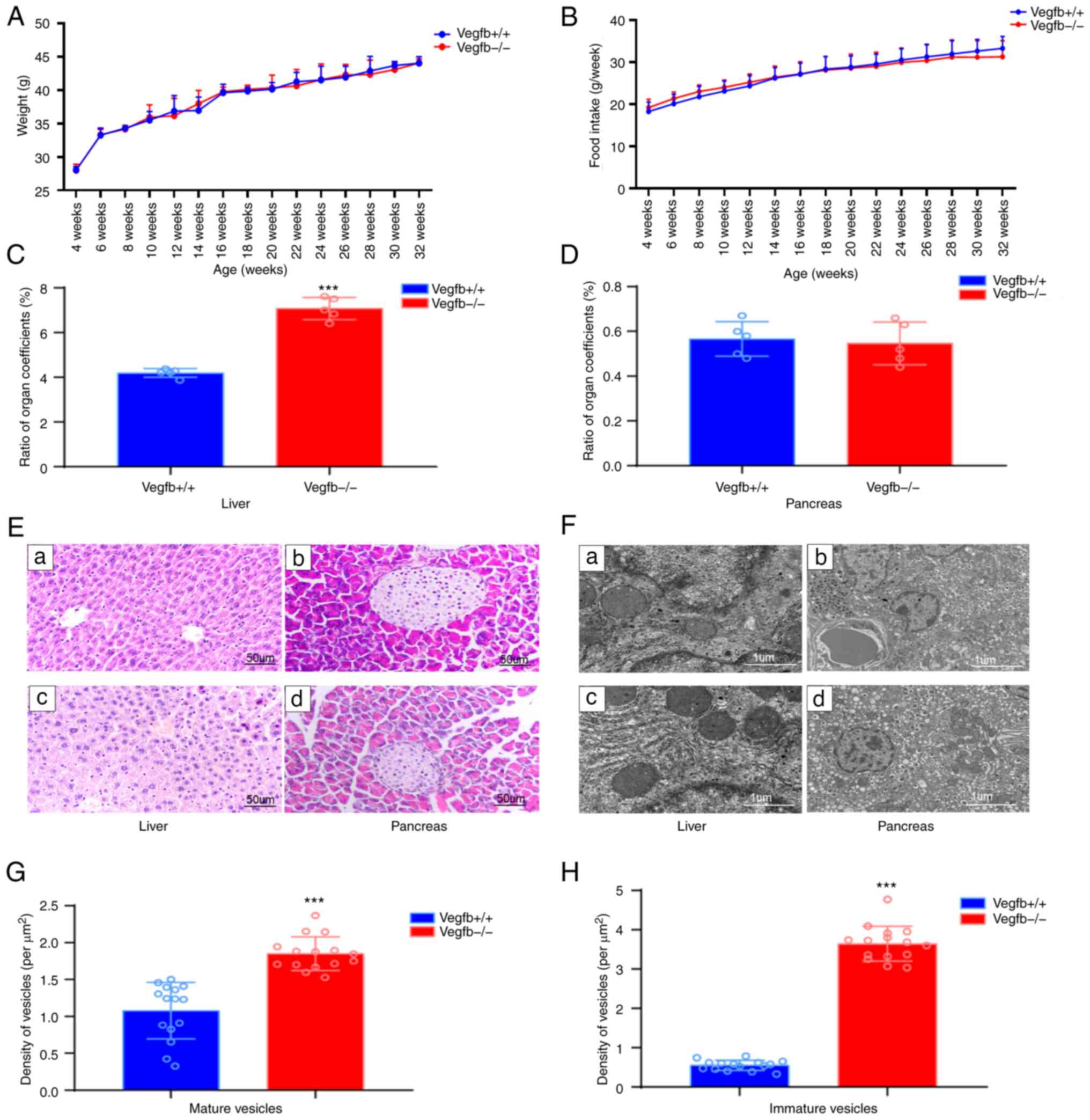 | Figure 2.Effect of VEGFB downregulation on
liver and pancreas. (A) Body weight curve of mice (4–32 weeks). (B)
Food intake curve of mice (4–32 weeks) (C and D) Organ coefficient
ratio between liver and pancreas in mice. (E) Light microscopic
structure of liver and pancreas; a and b: VEGFB+/+ group, c and d:
VEGFB-/- group (scale bar, 50 µm; magnification, ×400). (F)
Electron microscopic structure of liver and pancreas; a and b:
VEGFB+/+ group, c and d: VEGFB-/- group (scale bar, 1 µm; images a
and c, ×15,000 magnification; images b and d, ×8,000
magnification). (G and H) Density of mature and immature vesicles.
***P<0.001 vs. VEGFB+/+ group. VEGFB, vascular endothelial
growth factor B. |
The effect of VEGFB downregulation on
serum glucose and lipid metabolism
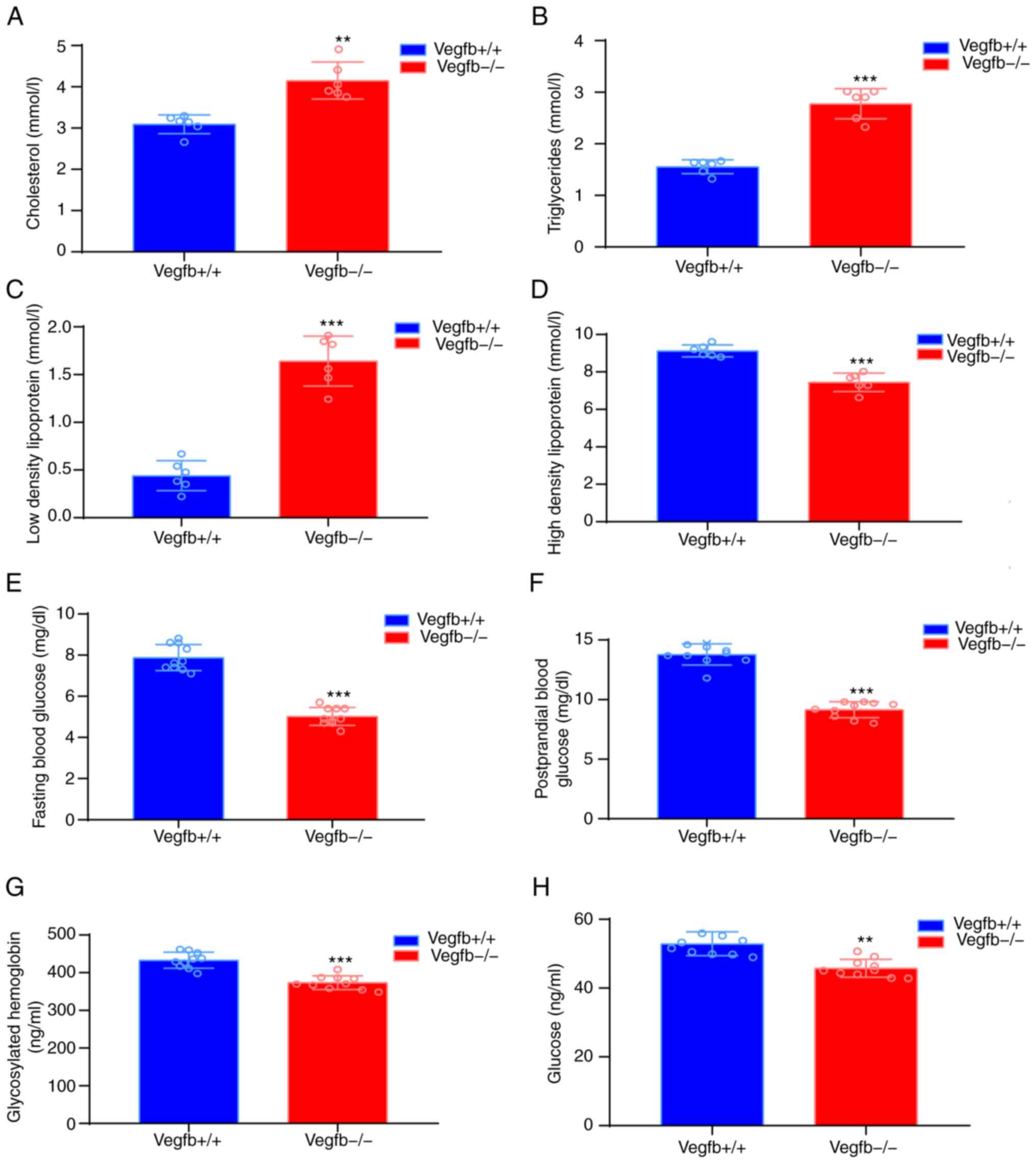
Serological analysis revealed that
VEGFB−/− TC (Fig. 3A),
TG (Fig. 3B) and LDL-C (Fig. 3C) levels were significantly
increased, while HDL-C levels were significantly lower in
VEGFB−/− mice (P<0.001; Fig. 3D). Compared with those of
VEGFB+/+ mice, the FBG and PBF levels of
VEGFB−/− mice were significantly lower (P<0.001;
Fig. 3E and F, respectively).
Serum ELISA results demonstrated that HbA1c and glucose metabolism
in 32-week-old VEGFB−/− mice were significantly lower
than those of VEGFB+/+ mice (P<0.01; Fig. 3G and H).
The effect of VEGFB downregulation on
blood glucose balance and insulin resistance
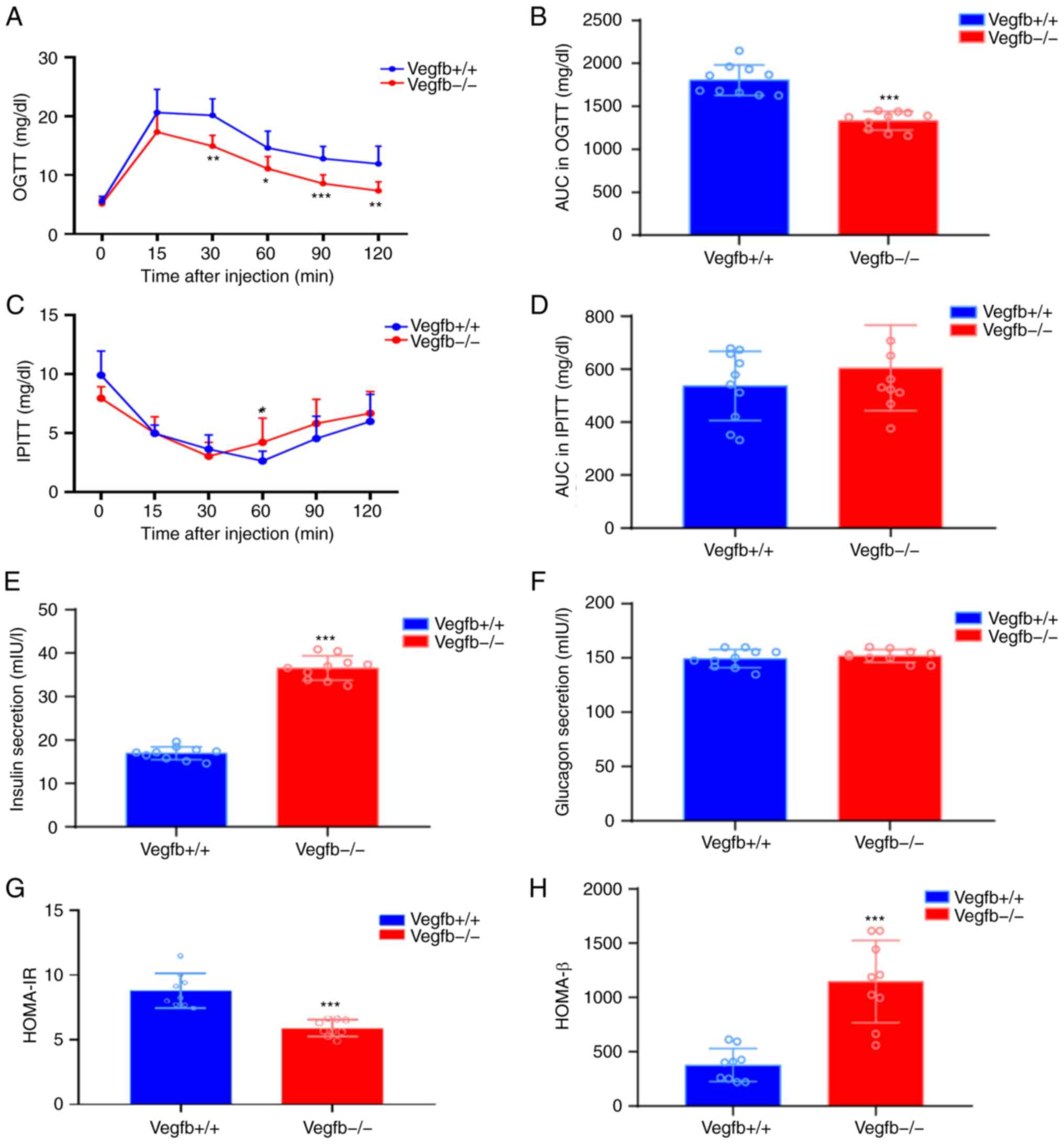
To determine whether loss of VEGFB affects glycemic
homeostasis, OGTT and IPITT tests were performed on
VEGFB+/+ and VEGFB−/− mice. The OGTT results
revealed that compared with that of VEGFB+/+ mice, the
blood glucose level of VEGFB−/− mice was significantly
reduced at 30, 60, 90 and 120 min (P<0.05; Fig. 4A), and the area under the curve
(AUC) was also significantly reduced (P<0.001; Fig. 4B). The IPITT results showed that
after intraperitoneal injection of insulin, the blood glucose level
of VEGFB−/− mice was significantly reduced at 0 min and
60 min (P<0.05; Fig. 4C). At
the other times, compared with VEGFB+/+ mice, there was
no significant change in blood glucose level (P>0.05; Fig. 4C), and there was no significant
difference in AUC (P>0.05; Fig.
4D). The ELISA results demonstrated that compared with that in
VEGFB+/+ mice, insulin secretion in VEGFB−/−
mice increased (P<0.001; Fig.
4E), while VEGFB had no effect on the secretion of glucagon in
mice (P>0.05; Fig. 4F).
Compared with that of VEGFB+/+ mice, HOMA-IR of
VEGFB−/− mice was significantly reduced (P<0.001;
Fig. 4G), while HOMA-β of
VEGFB−/− mice had significantly increased (P<0.001;
Fig. 4H).
Expression of VEGFR1 and NRP1 in the
pancreas and liver after VEGFB downregulation
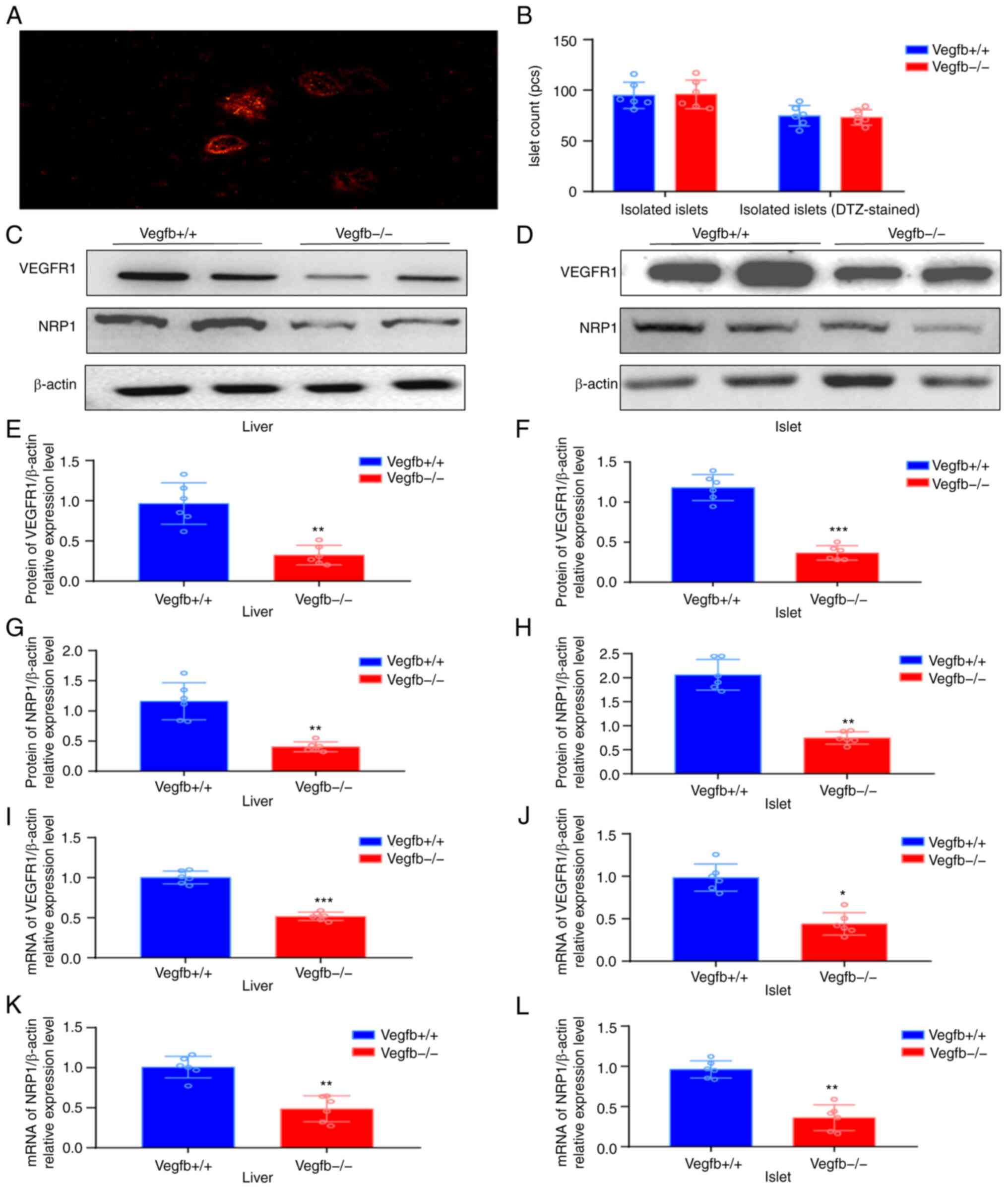
After the mouse islet tissue was stained with DTZ
solution, the isolated and purified intact or incomplete islet
cells were in the shape of red mass, and the peripheral pancreatic
exocrine cells were not stained (Fig.
5A). After islet culture and isolation, each mouse could
exchange 80–100 islet cell clusters (Fig. 5B). VEGFR1 and NRP1 protein
expression was also downregulated in islets and liver tissues of
VEGFB−/− mice (P<0.05; Fig. 5C-H). qPCR results revealed that
the expression of VEGFR1 and NRP1 in VEGFB−/− mice was
downregulated (P<0.05; Fig.
5I-L).
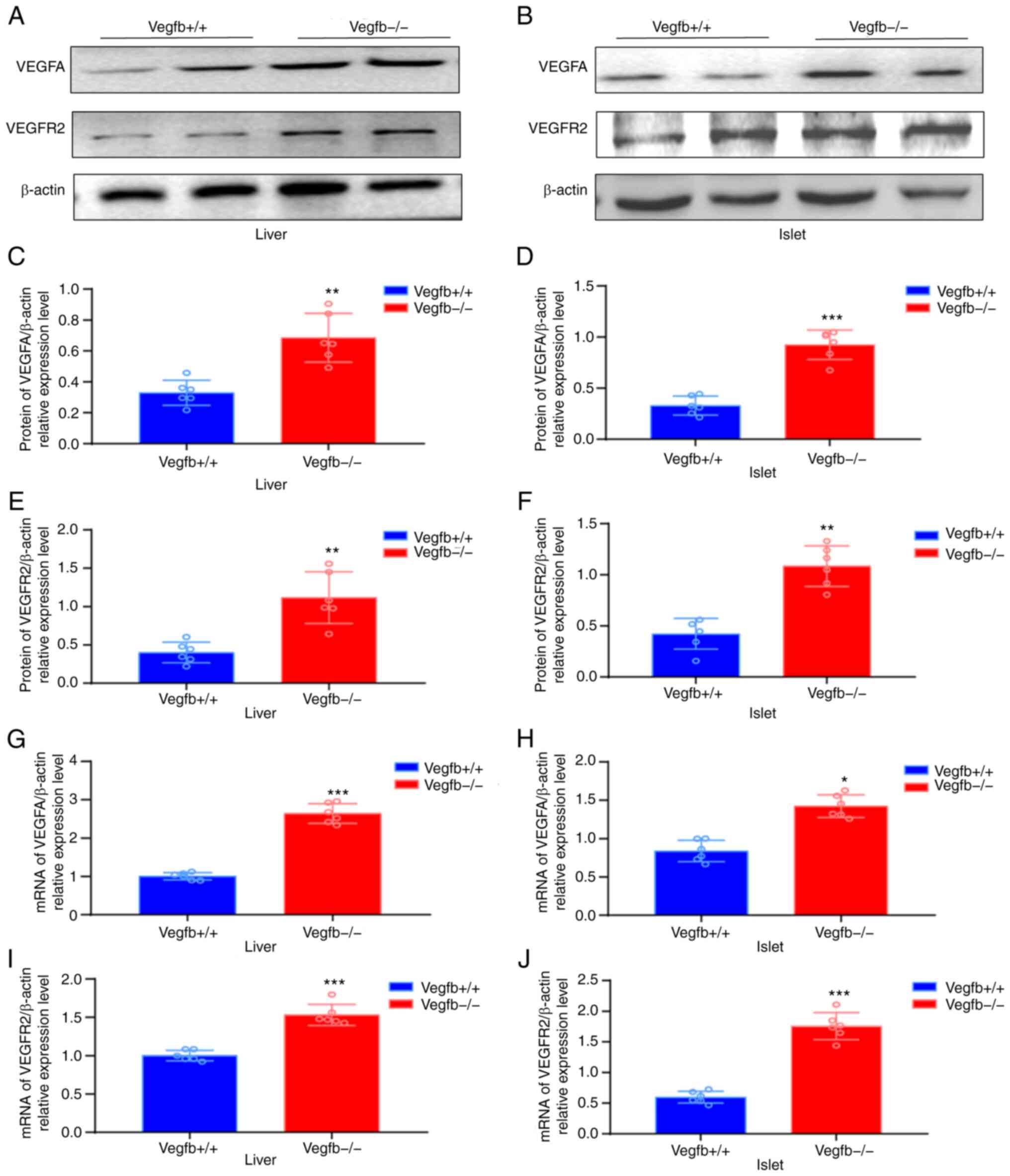
Expression of VEGFA and its receptor
VEGFR2 in the pancreas and liver after VEGFB downregulation
Western blot analysis showed that the expression
levels of VEGFA and VEGFR2 in VEGFB−/− mice were higher
than those in VEGFB+/+ mice (P<0.01; Fig. 6A-F). The qPCR results revealed
that the expression levels of VEGFA and VEGFR2 in
VEGFB−/− mice were higher than those in
VEGFB+/+ mice (P<0.01; Fig. 6G-J).
Discussion
VEGFB is a glycoprotein that is mainly present in
the heart, liver, skeletal muscle, brown fat and other tissues with
high metabolic activity (22,23). VEGF family is composed of 7
members: VEGFA, VEGFB, VEGFC, VEGFD, VEGFE, VEGFF and PIGF
(24,25). As a member of the VEGF family,
VEGFB not only maintains the development of vascular and promotes
neuroprotection and nutrition but also acts as a factor to maintain
homeostasis. In recent years, the role of VEGFB in regulating lipid
metabolism and blood glucose balance has attracted the attention of
numerous scholars.
Hagberg et al (10,12) reported on Nature that VEGFB can
regulate lipid uptake in endothelial cells and participate in lipid
metabolism. And in the study of db/db mice, VEGFB gene knockdown
prevented ectopic lipid deposition. Similar to the findings of
Hagberg et al (12), Falkevall et
al (14) also revealed that VEGFB
gene deletion can prevent pathological lipid deposition. Mehlem et
al (26) revealed that the
deletion of the VEGFB gene reduced lipid metabolism and also
prevented hyperglycaemia and hyperinsulinemia. In 2017, Wu et al
(27) observed that high plasma
VEGFB content causes abnormal glucose tolerance and elevates blood
glucose, aggravating the condition of patients with T2DM. By
contrast, Moessinger et al (15)
reported that reducing the VEGFB signal pathway can increase
cardiac glucose accumulation. Kivelä et al (28) revealed that in
cardiomyocyte-specific VEGFB transgenic rats, enhancing the effect
of VEGFB can alleviate glucose metabolism disorders. In 2021, Hu et
al (20) injected VEGFB into high
fat diet (HFD-induced obese mice for 10 weeks and observed that
both the blood glucose level and the blood glucose AUC in the VEGFB
treatment group were lower than that of the HFD group. Therefore,
VEGFB can improve glucose tolerance and increase insulin
sensitivity. An increasing number of scholars have discovered that
the VEGFB signaling pathway is involved in the development of
obesity and T2DM. It is predicted that VEGFB may become a key
factor in regulating blood glucose regulation and lipid metabolism
disorders. Altering the VEGFB signal pathway has certain
therapeutic potential for obesity and T2DM (10,12).
Abnormal blood glucose and lipid regulation are
closely related to T2DM, insulin resistance, non-alcoholic fatty
liver disease, hypertension, diabetic nephropathy and other
metabolic diseases. Abnormal deposition of lipids in non-fat
tissues such as the skeletal muscle and the myocardium can
interfere with the insulin signal pathway and indirectly affect key
factors associated with tissue glucose uptake, leading to insulin
resistance (29). Interfering
with lipid and glucose metabolism in early diabetes and diagnosed
patients with diabetes can help improve impaired insulin secretion
in the early stage. Anti-aliphatic deposition and improving glucose
intake may bring therapeutic benefits. However, drugs that improve
the abnormal deposition of adipose and glucose metabolism in
tissues are very rare at present. It has become one of the
important impediments to the treatment of metabolic diseases such
as obesity and diabetes. VEGFB has potential value as a target for
treatment, and may benefit from diseases such as lipid metabolism
disorders, diabetes, obesity and other diseases. Therefore,
elucidating the mechanism by which VEGFB regulates blood lipid
metabolism and blood glucose homeostasis has become the focus and
hot-spot of medical research.
In the present study, a systemic VEGFB knockdown
mouse model was constructed by CRISPR/cas9 genetic engineering to
investigate the effect of VEGFB gene deletion on lipid metabolism
and blood glucose homeostasis in mice and its mechanism. The
nutritional research results on lipid metabolism in mice after
VEGFB gene deletion are similar to those of Hagberg et al (10) and Robciuc et al (11). The research revealed that after
VEGFB gene deletion, the levels of serum TG, TC and LDL in mice
increased, suggesting that VEGFB gene deletion can lead to lipid
accumulation in mice. Through the morphological detection of liver
tissue, it was observed that following VEGFB gene deletion, mouse
hepatocytes have obvious steatosis, suggesting that VEGFB gene
deletion can accelerate the pathological progression of a mouse
fatty liver. In the present study, the glucose metabolism indexes
of mice with VEGFB gene deletion were determined. It was observed
that the contents of blood glucose, serum glucose and HbA1c
decreased after the VEGFB gene deletion. To further clarify the
effect of VEGFB on blood glucose, an OGTT and IPITT were performed.
It was observed that following the deletion of the VEGFB gene in
mice, the glucose tolerance increased, and the insulin resistance
index decreased significantly. This suggested that reducing the
VEGFB signal can improve glucose metabolism and insulin resistance
while aggravating the accumulation of lipids. By observing the
expression changes of VEGFR1, VEGFA and VEGFR2 in the liver and
islet tissues after the VEGFB gene deletion, it was concluded that
the VEGFB gene deletion may participate in the regulation of
glucose and lipid metabolism in mice by activating the VEGFA/VEGFR2
signal pathway.
Louzier et al revealed that the functional status of
mice lacking the VEGFB gene did not change significantly, and their
lifespan was similar to that of normal mice (30,31). In 2014, Sun et al (32) reported that there was no
significant difference between VEGFB−/− mice with
increased body weight and increased food intake and control group
mice. The weight of the constructed systemic VEGFB-knockdown mice
gradually increased as the age of weeks. The deletion of the VEGFB
gene did not affect the growth of mice, which was consistent with
the findings of a previous study by Sun et al (32) in a VEGFB-deficient mouse model. In
the present study, it was also found that although there was no
significant change in body weight compared with VEGFB+/+
mice, the organ ratio of VEGFB−/− mice liver increased
significantly, and there was steatosis in hepatocytes, suggesting
that VEGFB gene deletion affected liver lipid accumulation. In
2021, Hu et al (20) proposed
that VEGFB treatment can protect mice from liver lipid deposition
induced by an HFD, which is consistent with our results.
Furthermore, Shen et al revealed that VEGFB/IL22 fusion protein
therapy can reverse hepatic lipid accumulation induced by diabetes
mellitus (33).
In 2014, Sun et al (32) linked the VEGFB signal pathway with
human lipid metabolism, glucose metabolism and islet resistance
through genetic associations. Studies have shown that there was no
change in peripheral VEGFB levels between healthy individuals and
patients with diabetes, but there is a correlation between VEGFB
levels in patients with diabetes and the levels of C-reactive
protein, TC, TG and blood glucose levels (15,20,32). In 2015, Cheng et al (34) also observed an increase in serum
VEGFB levels in insulin-resistant subjects in clinical trials,
VEGFB was positively correlated with insulin resistance, and
metformin treatment could reduce plasma VEGFB levels. These studies
showed that there is a certain relationship between VEGFB levels
and obesity and insulin resistance. In 2016, Mehlem et al (26) examined model mice and revealed
that the TG diacylglycerol and PBG levels in the muscles of
VEGFB−/− mice were lower than those in normal mice, and
VEGFB−/− mice were not prone to hyperinsulinaemia. It
has been suggested that VEGFB gene deletion can prevent
hyperglycaemia and hyperinsulinaemia, reduce insulin resistance and
improve dyslipidaemia (26).
Robciuc et al (11) transfected
AAV-B186 into obese mice and found that VEGFB not only
significantly increased TG and cholesterol in mice but also
improved the response to abdominal glucose and insulin tolerance
tests and the metabolism of obese mice. The lipid metabolism
indexes of VEGFB−/− mice were examined, and the results
showed that after VEGFB gene deletion, the levels of TC, TG and LDL
were increased, while HDL decreased. This finding revealed that
VEGFB regulates lipid accumulation, which is consistent with the
results of Hagberg et al (10),
Robciuc et al (11) and other
previous studies (12,19).
Disorders of lipid metabolism in the body and
abnormal blood lipids can cause glucose metabolism disorders, which
results in impaired glucose regulation, diabetes and other
diseases. By detecting the glucose metabolism indexes of
VEGFB−/− mice, it was observed that after 32 weeks of
VEGFB knockdown, the FBG and PBG levels of mice decreased
significantly, along with the contents of serum glucose and HbA1c
when compared with that of VEGFB+/+ mice. According to
the analysis of the research results, the deletion of the VEGFB
gene can promote the glucose uptake of the body, which affects the
levels of serum glucose, HbA1c, FBG and PBG. Wu et al (27) revealed that the level of human
plasma VEGFB was positively correlated with FINS, HOMA-IR, blood
glucose and HbA1c, indicating that VEGFB was closely related to
insulin resistance and deterioration of glucose metabolism. In
2021, Hu et al (20) observed
that the level of blood glucose in the VEGFB treatment group was
lower than that in the HFD group and that the area under the curve
of the blood glucose curve was also significantly lower, indicating
that VEGFB could improve glucose tolerance and increase insulin
sensitivity. According to the results of the present study, it was
stated that glucose tolerance and level were improved after VEGFB
deletion which was similar to the result by Hu et al. Unlike the
aforementioned study, however, there was no significant effect on
insulin sensitivity. This was controversial with the present
findings.
Diabetes mellitus is the most common endocrine
disorder characterized by chronic hyperglycaemia owing to
relatively insufficient insulin secretion (35,36). Insulin or glucagon can impair
blood glucose homeostasis and cause diabetes and other diseases.
Currently, there have been no studies on the effect of VEGFB on the
secretion of glucagon and its mechanism. By detecting the fasting
serum insulin and glucagon levels of VEGFB−/− mice, it
was observed that insulin secretion increased after VEGFB gene
knockdown, while glucagon levels did not change significantly.
The detection of islet cell function is an important
part of the pathophysiology of abnormal glucose metabolism, and it
is important to clarify the β-cell functional status of abnormal
glucose metabolism. The amount of insulin secretion is closely
related to the number of vesicles in pancreatic islet cells
(37). There are ~9,000-13,000
dense-core secretory vesicles in each rodent (mouse/rat) β-cell
(38). In 2016, Pan et al
(39) concluded that the pattern
of insulin release corresponds to the number of cellular vesicles.
Insulin-secreting vesicles can be divided into two types, mature
and immature, according to their morphology. Mature vesicles are
formed by insulin, zinc and calcium crystals and contain dense core
particles. Immature vesicles are formed by the processing and
packaging of proinsulin in the reverse Golgi apparatus. They are
light grey and have low electron density (40). The maturation process of immature
vesicles requires acidification by an ATP-dependent proton pump,
and the coat protein must be removed for proinsulin to be converted
into insulin and C peptide through proteolysis (41). It was observed that the islet
vesicle density significantly increased in VEGFB−/− mice
compared with VEGFB+/+ mice, and the number of mature
and immature vesicles increased. The present study suggested that
deletion of VEGFB can cause increased serum insulin levels by
affecting the production of insulin secretory vesicles in islet
cells.
Insulin resistance is the main link in the
pathogenesis of T2DM. The effect of VEGFB gene deletion on insulin
resistance index and islet cell function index was examined and it
was found that VEGFB gene decreased insulin resistance index and
cell function index (HOMA-) increased after VEGFB gene deletion.
This finding suggested that the deletion of VEGFB gene can not only
affect insulin secretion, but also improved insulin resistance.
Robciuc et al (11) also found
similar results to the present study in obese and insulin-resistant
mice, suggesting that VEGFB can improve insulin secretion, insulin
supply and signal transduction. These findings predicted the
therapeutic potential of VEGFB in the treatment of disorders of
glucose and lipid metabolism.
VEGFB plays a biological role primarily by binding
to cell membrane receptors VEGFR1 and NRP1 (42). VEGFR1 was the first receptor in
the VEGF family that was identified. In addition to binding to
VEGFB, VEGFR1 has an increased affinity for VEGFA and placental
growth factors. NRP1, as a coreceptor of VEGFR1, performs
biological functions after binding to VEGFB (11,43,44). Falkevall et al (14) reported that VEGFB signal can
enhance the uptake and exocytosis of FAs by endothelial cells
through VEGFR1 and its coreceptor NRP1. In 2021, a study by Hu et
al (20) also revealed that the
effect of VEGFB on lipid metabolism depended on VEGFR1. In
addition, it has been observed that VEGFB gene knockout and VEGFR1
deletion in the db/db mouse model can maintain metabolic
homeostasis and restore lipid and glucose metabolism (45).
Anisimov et al (46) confirmed that VEGFB binding to
VEGFR1 does not induce VEGFR1 downstream signal. By contrast, VEGFB
is regarded to activate the VEGFA/VEGFR2 pathway by replacing
VEGFR1 with VEGFR2, thereby promoting the normal angiogenesis
pathway (47,48). In a study of the effect of VEGFB
on insulin secretion, Robciuc et al (11) reported that VEGFB and VEGFA
competitively bind VEGFR1. VEGFB and VEGFR1 binding promote the
binding of VEGFA to VEGFR2, thereby increasing vascular remodeling
and blood perfusion of adipose tissue and participating in
metabolic regulation, insulin secretion and signal transduction.
The expression of VEGFR1 and NRP1 in the liver and islet,
respectively, was verified. The results revealed that the
expression of VEGFR1 and NRP1 decreased significantly following
VEGFB gene deletion, suggesting that VEGFB gene deletion reduced
the expression of receptors VEGFR1 and NRP1 on the islet cell
membrane. The expression of VEGFA and VEGFR2 in the liver and islet
was evaluated and it was observed that their expression increased
significantly following VEGFB deletion. VEGFA is a vascular
permeability factor observed in tumor cells, which primarily plays
its biological role by connecting with the extracellular receptor
VEGFR2. The VEGFA/VEGFR2 signal pathway is involved in tumor
angiogenesis, inflammation and oxidative stress; however, it also
leads to endothelial cell dysfunction and lipid deposition
(49,50). Ghorbanzadeh et al (51) reported that the VEGFA and VEGFR2
signals are closely related to glucose. In 2017, Jin et al
(52) revealed that
overexpression of VEGFA protected against obesity caused by
high-fat foods and reduced insulin sensitivity.
VEGFA and VEGFB participate in multiple signal
pathways including cell proliferation and differentiation,
apoptosis and metabolic balance in vivo. In 2011, Osawa et al
(25) reported that VEGFB induced
signal transduction of pericytes and other cells by binding to
VEGFR1. Simultaneously, the binding of VEGFB to VEGFR1 increased
the binding of VEGFA to VEGFR2 and promoted tumor growth (25). Jin et al (52) reported that VEGFA overexpression
can resist obesity by increasing vascular density and browning of
white adipose tissue (WAT). In addition, it was observed that VEGFA
downregulation could upregulate the expression of VEGFB in WAT. It
was concluded that VEGFB and VEGFA have a certain relationship in
regulating adipose metabolism (52). Recent studies have reported that
VEGFB played its metabolic role by indirectly activating the
VEGFA/VEGFR2 pathway (11,28,47).
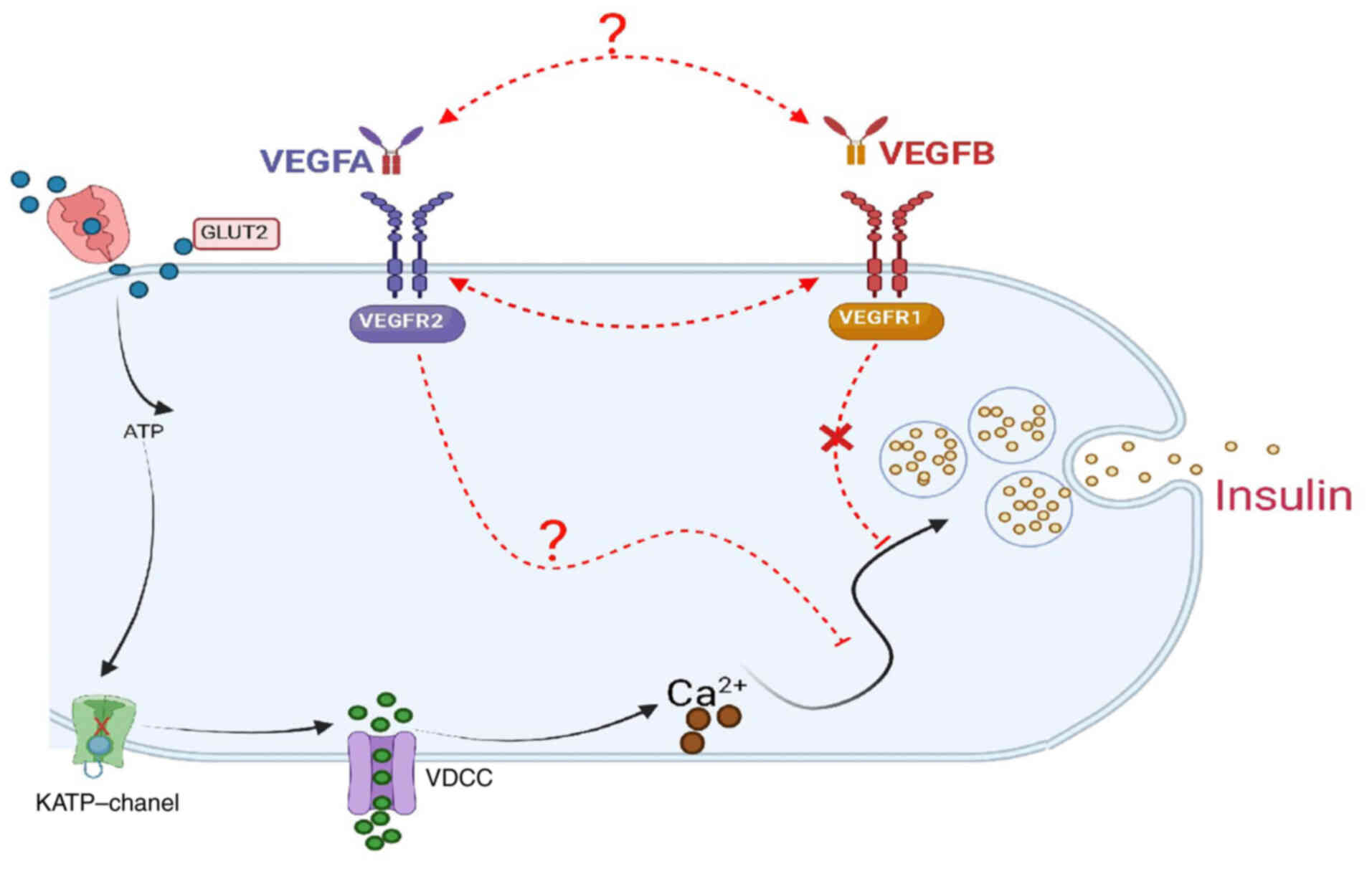
As a result of the downregulation of VEGFB and VEGFR1 expression
caused by the deletion of the VEGFB gene, it was concluded that
VEGFB may regulate the blood glucose homeostasis and lipid
metabolism in mice by activating the VEGFA/VEGFR2 signal pathway
(Fig. 7), which provided a novel
approach for VEGFB to participate in the pathogenesis of T2DM.
The present study analyzed the effect of VEGFB gene
knockdown on insulin secretion by observing the levels of glucose
and lipid metabolism in systemic VEGFB knockdown mice, and revealed
that VEGFB knockdown may participate in the regulation of glucose
and lipid metabolism in mice by activating the VEGFA/VEGFR2 signal
pathway. Since the mechanisms of glucose and lipid metabolism and
insulin secretion are not consistent under physiological and
pathological conditions, further exploration of the regulatory
mechanism of VEGFB on blood glucose, lipid metabolism and insulin
secretion is required via construction of animal models of obesity
and diabetes.
In conclusion, the present study identified that the
deletion of VEGFB gene could aggravate the lipid deposition in
mice, affect glucose metabolism, increase insulin secretion and
reduce blood glucose level. It was also found that the expression
of VEGFR1 decreased after downregulation of VEGFB, which may affect
the balance of glucose and lipid metabolism by activating
VEGFA/VEGFR2 pathway. The present findings provided theoretical and
experimental evidence for VEGFB in the diagnosis and treatment of
obesity and diabetes.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China Youth Project (grant no. 31702024), the
Shandong Province Higher Educational Science and Technology Plan
Project (grant no. J17KA258) and the Shandong University Student
Innovation Training Project (grant no. S202010440029).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YNL and WGJ designed the study, XL, RRL, YQL, HP and
HNY performed the experiments, wrote and revised the manuscript. XL
and YNL wrote the manuscript and analyzed the data. RRL searched
the literature. All authors read and approved the final manuscript.
YNL and XL confirm the authenticity of all the raw data. All
authors agree to the publication of the manuscript.
Ethics approval and consent to
participate
The present study was reviewed and approved by the
Institutional Review Board of Binzhou Medical University (Yantai,
China). All procedures involving animals were reviewed and approved
by the Institutional Animal Care and Use Committee of the Medical
Ethics Committee of Binzhou Medical University (IACUC protocol
number: 2021-300).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Stumvoll M, Goldstein BJ and van Haeften
TW: Type 2 diabetes: Principles of pathogenesis and therapy.
Lancet. 365:1333–1346. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Trayhurn P: Hypoxia and adipocyte
physiology: Implications for adipose tissue dysfunction in obesity.
Annu Rev Nutr. 34:207–236. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lysaght J, van der Stok EP, Allott EH,
Casey R, Donohoe CL, Howard JM, McGarrigle SA, Ravi N, Reynolds JV
and Pidgeon GP: Pro-inflammatory and tumour proliferative
properties of excess visceral adipose tissue. Cancer Lett.
312:62–72. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bai T, Li M, Liu Y, Qiao Z and Wang Z:
Inhibition of ferroptosis alleviates atherosclerosis through
attenuating lipid peroxidation and endothelial dysfunction in mouse
aortic endothelial cell. Free Radic Biol Med. 160:92–102. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sinha RA, Bruinstroop E, Singh BK and Yen
PM: Nonalcoholic fatty liver disease and hypercholesterolemia:
Roles of thyroid hormones, metabolites, and agonists. Thyroid.
29:1173–1191. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Santoleri D and Titchenell PM: Resolving
the paradox of hepatic insulin resistance. Cell Mol Gastroenterol
Hepatol. 7:447–456. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Laakso M and Kuusisto J: Insulin
resistance and hyperglycaemia in cardiovascular disease
development. Nat Rev Endocrinol. 10:293–302. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Khan RMM, Chua ZJY, Tan JC, Yang Y, Liao Z
and Zhao Y: From pre-diabetes to diabetes: Diagnosis, treatments
and translational research. Medicina (Kaunas). 55:5462019.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Aase K, Lymboussaki A, Kaipainen A,
Olofsson B, Alitalo K and Eriksson U: Localization of VEGF-B in the
mouse embryo suggests a paracrine role of the growth factor in the
developing vasculature. Dev Dyn. 215:12–25. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hagberg CE, Mehlem A, Falkevall A, Muhl L,
Fam BC, Ortsäter H, Scotney P, Nyqvist D, Samén E, Lu L, et al:
Targeting VEGF-B as a novel treatment for insulin resistance and
type 2 diabetes. Nature. 490:426–430. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Robciuc MR, Kivelä R, Williams IM, de Boer
JF, van Dijk TH, Elamaa H, Tigistu-Sahle F, Molotkov D, Leppänen
VM, Käkelä R, et al: VEGFB/VEGFR1-Induced expansion of adipose
vasculature counteracts obesity and related metabolic
complications. Cell Metab. 23:712–724. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hagberg CE, Falkevall A, Wang X, Larsson
E, Huusko J, Nilsson I, van Meeteren LA, Samen E, Lu L,
Vanwildemeersch M, et al: Vascular endothelial growth factor B
controls endothelial fatty acid uptake. Nature. 464:917–921. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wagenmakers AJM, Strauss JA, Shepherd SO,
Keske MA and Cocks M: Increased muscle blood supply and
transendothelial nutrient and insulin transport induced by food
intake and exercise: Effect of obesity and ageing. J Physiol.
594:2207–2222. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Falkevall A, Mehlem A, Palombo I, Heller
Sahlgren B, Ebarasi L, He L, Ytterberg AJ, Olauson H, Axelsson J,
Sundelin B, et al: Reducing VEGF-B signaling ameliorates renal
lipotoxicity and protects against diabetic kidney disease. Cell
Metab. 25:713–726. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Moessinger C, Nilsson I, Muhl L,
Zeitelhofer M, Heller Sahlgren B, Skogsberg J and Eriksson U:
VEGF-B signaling impairs endothelial glucose transcytosis by
decreasing membrane cholesterol content. EMBO Rep. 21:e493432020.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jensen N, Ning FC, Mi J, Lindström W,
Balan M, Muhl L, Eriksson U, Nilsson I and Nyqvist D: VEGF-B
ablation in pancreatic β-cells upregulates insulin expression
without affecting glucose homeostasis or islet lipid uptake. Sci
Rep. 10:9232020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shibuya M: VEGF-VEGFR system as a target
for suppressing inflammation and other diseases. Endocr Metab
Immune Disord Drug Targets. 15:135–144. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zachary I and Gliki G: Signaling
transduction mechanisms mediating biological actions of the
vascular endothelial growth factor family. Cardiovasc Res.
49:568–581. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shen Z, Zhang Z, Wang X and Yang K:
VEGFB-VEGFR1 ameliorates Ang II-induced cardiomyocyte hypertrophy
through Ca2+-mediated PKG I pathway. J Cell Biochem.
119:1511–1520. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hu L, Shan Z, Wang F, Gao X and Tong Y:
Vascular endothelial growth factor B exerts lipid-lowering effect
by activating AMPK via VEGFR1. Life Sci. 276:1194012021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Claesson-Welsh L: VEGF receptor signal
transduction-A brief update. Vascul Pharmacol. 86:14–17. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ferrara N, Gerber HP and LeCouter J: The
biology of VEGF and its receptors. Nat Med. 9:669–676. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bates DO: Vascular endothelial growth
factors and vascular permeability. Cardiovasc Res. 87:262–271.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Osawa T, Muramatsu M, Wang F, Tsuchida R,
Kodama T, Minami T and Shibuya M: Increased expression of histone
demethylase JHDM1D under nutrient starvation suppresses tumor
growth via down-regulating angiogenesis. Proc Natl Acad Sci USA.
108:20725–20729. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mehlem A, Palombo I, Wang X, Hagberg CE,
Eriksson U and Falkevall A: PGC-1α coordinates mitochondrial
respiratory capacity and muscular fatty acid uptake via regulation
of VEGF-B. Diabetes. 65:861–873. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu J, Wei H, Qu H, Feng Z, Long J, Ge Q
and Deng H: Plasma vascular endothelial growth factor B levels are
increased in patients with newly diagnosed type 2 diabetes mellitus
and associated with the first phase of glucose-stimulated insulin
secretion function of β-cell. J Endocrinol Invest. 40:1219–1226.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kivelä R, Bry M, Robciuc MR, Räsänen M,
Taavitsainen M, Silvola JM, Saraste A, Hulmi JJ, Anisimov A,
Mäyränpää MI, et al: VEGF-B-induced vascular growth leads to
metabolic reprogramming and ischemia resistance in the heart. EMBO
Mol Med. 6:307–321. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Samuel VT, Petersen KF and Shulman GI:
Lipid-induced insulin resistance: Unravelling the mechanism.
Lancet. 375:2267–2277. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Louzier V, Raffestin B, Leroux A,
Branellec D, Caillaud JM, Levame M, Eddahibi S and Adnot S: Role of
VEGF-B in the lung during development of chronic hypoxic pulmonary
hypertension. Am J Physiol Lung Cell Mol Physiol. 284:L926–L937.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Reichelt M, Shi S, Hayes M, Kay G, Batch
J, Gole GA and Browning J: Vascular endothelial growth factor-B and
retinal vascular development in the mouse. Clin Exp Ophthalmol.
31:61–65. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sun CY, Lee CC, Hsieh MF, Chen CH and Chou
KM: Clinical association of circulating VEGF-B levels with
hyperlipidemia and target organ damage in type 2 diabetic patients.
J Biol Regul Homeost Agents. 28:225–236. 2014.PubMed/NCBI
|
|
33
|
Shen Y, Chen W, Han L, Bian Q, Fan J, Cao
Z, Jin X, Ding T, Xian Z, Guo Z, et al: VEGF-B antibody and
interleukin-22 fusion protein ameliorates diabetic nephropathy
through inhibiting lipid accumulation and inflammatory responses.
Acta Pharm Sin B. 11:127–142. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cheng F, Zhao L, Wu Y, Huang T, Yang G,
Zhang Z, Wu Y, Jia F, Wu J, Chen C and Liu D: Serum vascular
endothelial growth factor B is elevated in women with polycystic
ovary syndrome and can be decreased with metformin treatment. Clin
Endocrinol (Oxf). 84:386–393. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Abdul Razak MK and Sultan AA: The
importance of measurement of plasma fibrinogen level among patients
with type-2 diabetes mellitus. Diabetes Metab Syndr. 13:1151–1158.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lam DW and LeRoith D: The worldwide
diabetes epidemic. Curr Opin Endocrinol Diabetes Obes. 19:93–96.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Clayton EL, Evans GJ and Cousin MA: Bulk
synaptic vesicle endocytosis is rapidly triggered during strong
stimulation. J Neurosci. 28:6627–6632. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhao A, Ohara-Imaizumi M, Brissova M,
Benninger RK, Xu Y, Hao Y, Abramowitz J, Boulay G, Powers AC,
Piston D, et al: Gαo represses insulin secretion by reducing
vesicular docking in pancreatic beta-cells. Diabetes. 59:2522–2529.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Pan JY, Yuan S, Yu T, Su CL, Liu XL, He J
and Li H: Regulation of L-type Ca2+ channel activity and
insulin secretion by huntingtin-associated protein 1. J Biol Chem.
291:26352–26363. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wollam J, Mahata S, Riopel M,
Hernandez-Carretero A, Biswas A, Bandyopadhyay GK, Chi NW, Eiden
LE, Mahapatra NR, Corti A, et al: Chromogranin A regulates vesicle
storage and mitochondrial dynamics to influence insulin secretion.
Cell Tissue Res. 368:487–501. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Vakilian M, Tahamtani Y and Ghaedi K: A
review on insulin trafficking and exocytosis. Gene. 706:52–61.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Nash AD, Baca M, Wright C and Scotney PD:
The biology of vascular endothelial growth factor-B (VEGF-B). Pulm
Pharmacol Ther. 19:61–69. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gilbert M, Jung SR, Reed BJ and Sweet IR:
Islet oxygen consumption and insulin secretion tightly coupled to
calcium derived from L-type calcium channels but not from the
endoplasmic reticulum. J Biol Chem. 283:24334–24342. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gauthier BR and Wollheim CB:
Synaptotagmins bind calcium to release insulin. Am J Physiol
Endocrinol Metab. 295:E1279–E1286. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Shang R, Lal N, Lee CS, Zhai Y, Puri K,
Seira O, Boushel RC, Sultan I, Räsänen M, Alitalo K, et al:
Cardiac-specific VEGFB overexpression reduces lipoprotein lipase
activity and improves insulin action in rat heart. Am J Physiol
Endocrinol Metab. 321:E753–E765. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Anisimov A, Leppänen VM, Tvorogov D,
Zarkada G, Jeltsch M, Holopainen T, Kaijalainen S and Alitalo K:
The basis for the distinct biological activities of vascular
endothelial growth factor receptor-1 ligands. Sci Signal.
6:ra522013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bry M, Kivelä R, Leppänen VM and Alitalo
K: Vascular endothelial growth factor-B in physiology and disease.
Physiol Rev. 94:779–794. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Rafii S and Carmeliet P: VEGF-B improves
metabolic health through vascular pruning of fat. Cell Metab.
23:571–573. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Feng S, Bowden N, Fragiadaki M, Souilhol
C, Hsiao S, Mahmoud M, Allen S, Pirri D, Ayllon BT, Akhtar S, et
al: Mechanical activation of hypoxia-inducible factor 1α drives
endothelial dysfunction at atheroprone sites. Arterioscler Thromb
Vasc Biol. 37:2087–2101. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Liu D, Lei L, Desir M, Huang Y, Cleman J,
Jiang W, Fernandez-Hernando C, Di Lorenzo A, Sessa WC and Giordano
FJ: Smooth muscle hypoxia-inducible factor 1α links intravascular
pressure and atherosclerosis-brief report. Arterioscler Thromb Vasc
Biol. 36:442–455. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ghorbanzadeh V, Mohammadi M, Dariushnejad
H, Chodari L and Mohaddes G: Effects of crocin and voluntary
exercise, alone or combined, on heart VEGF-A and HOMA-IR of HFD/STZ
induced type 2 diabetic rats. J Endocrinol Invest. 39:1179–1186.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Jin H, Li D, Wang X, Jia J, Chen Y, Yao Y,
Zhao C, Lu X, Zhang S, Togo J, et al: VEGF and VEGFB play balancing
roles in adipose differentiation, gene expression, and function.
Endocrinology. 159:2036–2049. 2018. View Article : Google Scholar : PubMed/NCBI
|