Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune
disorder that results in cartilage and bone damage, as well as
disability (1). It is estimated
that the prevalence of this disease is ~1% and women are at an
increased risk of developing RA compared with men (2). RA is primarily characterized by
warm, swollen and painful joints, predominantly the synovial joints
(3,4). Moreover, RA may impact other organs,
including the lungs, skin and eyes (5), which makes patients with RA more
prone to suffer from cancer, cardiovascular disease, infectious
disease, respiratory disease and osteoporosis (6). Increasing evidence supports the fact
that genetic and environmental factors are implicated in the
occurrence of RA, such as sex, tobacco smoking and dietary patterns
(7). It is widely recognized that
prompt and continuous treatment may ameliorate joint damage to
impede irreversible disability when patients are diagnosed in the
early stages (8,9). However, in spite of the
substantially improved clinical outcomes that have resulted from
the development of biological and targeted therapies in the past
two decades, the etiopathogenesis of RA remains elusive and the
therapeutic outcomes remain unsatisfactory (10,11).
Tripartite motif-containing (TRIM)22 is a member of
the TRIM family, an evolutionarily conserved gene family (12). The TRIM family participates in
numerous biological processes via E3 ubiquitin ligase activities
(13). There is also increasing
evidence that TRIM22 may serve a critical role in human diseases.
For instance, Liu et al (14) reported that TRIM22 contributes to
the proliferation and invasion of colon cancer cells. Furthermore,
Li et al (15)
demonstrated that TRIM22 knockdown inactivates the PI3K/Akt/mTOR
signaling pathway to prevent chronic myeloid leukemia. Moreover,
TRIM22 has also been reported to predict the poor prognosis of
non-small cell lung cancer and promote the epithelial-mesenchymal
transition process (16).
However, whether TRIM22 exerts regulatory functions in RA needs
further exploration.
Forkhead box C1 (FOXC1) is a member of the forkhead
family of transcription factors and is indispensable for tumor
development and metastasis (17).
Previous studies have highlighted the promoting role of FOXC1 in
the progression of non-small cell lung cancer (18), glioma (19), triple-negative breast cancer
(20) and pancreatic cancer
(21). Moreover, FOXC1 activates
the PI3K/AKT signaling pathway to increase fibroblast-like
synoviocyte (FLS) proliferation in RA (22). However, the interaction between
FOXC1 and TRIM22 in RA remains to be elucidated.
The NF-κB signaling pathway is composed of canonical
signaling pathways activated via various stimuli with a rapid but
transient transcriptional activity, as well as non-canonical
signaling pathways stimulated via TNF receptor superfamily members
(23). Previous studies have
verified the pivotal role of NF-κB signaling in tumors and diseases
(24,25). Furthermore, the NF-κB signaling
pathway promotes RA (26,27).
The aim of the present study was to test the
hypothesis that TRIM22 may be modulated via FOXC1 and influence the
FLS phenotype in RA via NF-κB signaling.
Materials and methods
Cell culture
The human rheumatoid FLS MH7A cell line (RA-FLS) was
purchased from the BeNa Culture Collection (Beijing Beina Chunglian
Institute of Biotechnology) and the immortalized normal FLS cell
line was purchased from the NTCC Type Culture Collection (BioVector
Science Lab, Inc.). The culture medium for both RA-FLSs and
normal-FLSs was DMEM (Hyclone; Cytiva) with 10% fetal bovine serum
(FBS; Thermo Fisher Scientific, Inc.). The cells were incubated at
37°C with 5% CO2.
Cell transfection
Small interfering RNAs (siRNAs/si) targeting TRIM22
(si-TRIM22#1/2; cat. no. siG000010346A-1-5/siG1072101848-1-5) were
constructed by Guangzhou RiboBio Co., Ltd., with an siRNA negative
control (NC; si-NC; non-targeting sequence; cat. no.
siB06525141922-1-5). The pcDNA3.1 overexpression vector containing
the FOXC1 gene (Ov-FOXC1) and NC overexpression vector (Ov-NC) were
purchased from Shanghai GenePharma Co., Ltd. For TRIM22 silencing,
RA-FLSs were transfected with 50 nM si-TRIM22#1/2 and si-NC. For
FOXC1 overexpression, RA-FLSs were transfected with 2 µg Ov-FOXC1
and Ov-NC. Untransfected cells were regarded as the Control group.
Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.) was used to transfect the plasmids into RA-FLSs,
which were seeded in 6-well plates at a density of 1×106
for 48 h at 37°C according to the manufacturer's instructions. The
cells were collected for subsequent experiments 48 h following
transfection.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from cells in 6-well plates
at a density of 2×105 cells/well using the
EasyPure® RNA Kit (TransGen Biotech, Co., Ltd.)
according to the manufacturer's instructions. Complementary DNA was
synthesized using a RevertAid First Strand cDNA Synthesis kit
(Fermentas; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. qPCR was performed using the SYBR Green
Premix PCR Master Mix [Roche Diagnostics (Shanghai) Co., Ltd.] and
the Applied Biosystems 7500 Real-Time PCR System (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The PCR conditions
were 95°C for 10 min for initial denaturation, 40 cycles of
denaturation 15 sec at 95°C, annealing 30 sec at 60°C, elongation
30 sec at 72°C, and final extension for 5 min at 72°C. The
2−ΔΔCq method (28)
was used to quantify relative mRNA expression levels, with GAPDH
used as the endogenous control. The qPCR primer sequences were as
follows: TRIM22 forward (F), 5′-GAGGTCAAGATGAGCCCACAG-3′ and
reverse (R), 5′-GCTTTTCCTGACATTCCTTGACC-3′; FOXC1 F,
5′-TAGCTACATCGCGCTCATCA-3′ and R, 5′-ACCTTGACGAAGCACTCGTT-3′; TNF-α
F, 5′-CTGGGCAGGTCTACTTTGGG-3′ and R, 5′-CTGGAGGCCCCAGTTTGAAT-3′;
IL-1β F, 5′-CCAAACCTCTTCGAGGCACA-3′ and R,
5′-AGCCATCATTTCACTGGCGA-3′; IL-6 F, 5′-GTCCAGTTGCCTTCTCCCTGG-3′ and
R, 5′-CCCATGCTACATTTGCCGAAG-3′ and GAPDH F,
5′-AATGGACAACTGGTCGTGGAC-3′ and R 5′-CCCTCCAGGGGATCTGTTTG-3′.
Cell counting kit-8 (CCK-8) assay
Transfected RA-FLSs were seeded into 96-well plates
(3×104 cells/well) and were then incubated for 12 h at
37°C. Cells were then maintained for 24, 48 and 72 h respectively
and 10 µl CCK-8 solution [Hangzhou Multi Sciences (Lianke) Biotech
Co., Ltd.] was added for additional 2 h. The absorbance at 450 nm
was assessed using a microplate reader (Perlong Medical Equipment
Co., Ltd.).
Flow cytometry
Cell apoptosis was assessed using the Annexin
V-FITC/PI Apoptosis kit (BD Biosciences). Following centrifugation
at 1,000 × g for 5 min at 4°C, transfected RA-FLSs in 6-well plates
(1×106 cells/well) were rinsed in cold PBS.
Subsequently, cells were re-suspended in 400 µl 1X binding buffer
after the supernatant was removed, followed by the addition of 4 µl
Annexin V-FITC at 4°C for 15 min and 4 µl PI for 30 min in the dark
at room temperature. Finally, the apoptotic rate of RA-FLSs was
detected using a BD FACSCalibur flow cytometer (BD Biosciences)
using BD Accuri C6 software (version no. 1.0.264.21; BD
Biosciences).
Transwell assays
Transwell assays were performed using Transwell
chambers (Corning, Inc.) with an 8.0 µm pore size. Briefly,
2×104 transfected RA-FLSs resuspended in 400 µl
serum-free DMEM medium were added to the upper chamber and 800 µl
DMEM medium containing 15% FBS was added to the lower chamber.
After 12 h at 37°C, the migrating cells were fixed using 100%
methanol for 10 min and were then stained with 0.1% crystal violet
for 15 min at room temperature. Images were captured using a light
microscope (magnification, ×100; Olympus Corporation) and analyzed
by ImageJ software (v1.8.0.112, National Institutes of Health). The
Transwell invasion assays followed the same procedure as the
migration assays, except that the Transwell chambers in the
invasion assays were precoated with Matrigel® (BD
Biosciences) for 1 h at 37°C.
ELISA
TNF-α (cat. no. ab181421), IL-1β (cat. no. ab214025)
and IL-6 (cat. no. ab178013) levels were detected using human TNF-α
ELISA kit, human IL-1β ELISA kit and human IL-6 ELISA kit purchased
from Abcam. Briefly, transfected RA-FLSs were seeded into 96-well
plates (5×103 cells/well). After centrifuging at 2,000 ×
g for 5 min at 4°C, cell supernatant was collected to examine the
secretion of inflammatory cytokines according to the instructions
of the ELISA kits. The absorbance was read at 450 nm using a
microplate reader (Thermo Fisher Scientific, Inc.).
Chromatin immunoprecipitation
(ChIP)
The EZ-ChIP kit (cat. no. 9005; Cell Signaling
Technology, Inc.) was used for the ChIP assays. RA-FLSs
(1×106) were crosslinked by 1% formaldehyde
(Sigma-Aldrich; Merck KGaA) by centrifugation at 300 × g for 3 min
at 25°C and washed in pre-cooled PBS for 10 min at 25°C. The
formaldehyde was quenched by the addition of glycine (Beijing
Solarbio Science & Technology Co., Ltd.). Then a total of 300
µl SDS lysis buffer (1% SDS, 10 mM EDTA and 50 mM Tris-HCl pH 8.0)
was then used to lyse the cells, which were subsequently sonicated
at 150 Hz and sheared with four sets of 10 sec pulses on wet ice
using a high intensity ultrasonic processor to obtain chromatin
fragments. An equal amount of chromatin (100 µl) was
immunoprecipitated at 4°C overnight, 5% of the supernatant was
collected as input DNA. Immunoprecipitated products were collected
after incubation with 100 ul Protein A/G magnetic beads coupled
with 2 µg anti-IgG (cat. no. ab6715; 1 µg/µl; Abcam) or 2 µg
anti-FOXC1 (cat. no. ab227977; 1 µg/µl; Abcam) for 90 min at 4°C.
The beads were washed using a magnetic separation rack and the
bound chromatin was eluted from the beads with SDS buffer and
subjected to RNase and proteinase K treatment (10 mg/ml) at 45°C.
Crosslinks were reversed by overnight incubation with 5 M NaCl at
65°C, and Chip DNA was purified using spin columns. The recovered
DNA fragments were subjected to PCR analysis using the SYBR Green
Premix PCR Master Mix [Roche Diagnostics (Shanghai) Co., Ltd.] and
the Applied Biosystems 7500 Real-Time PCR System (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The PCR conditions
were: 95°C 10 min for initial denaturation, 40 cycles of
denaturation 15 sec at 95°C, annealing 30 sec at 60°C, elongation
30 sec at 72°C, and final extension for 5 min at 72°C. PCR products
were separated by 1% gel electrophoresis using agarose gels
prestained with ethidium bromide. Bands were analyzed using ImageJ
v1.5.1 (National Institutes of Health, Bethesda). The primer
sequences used for ChIP were as follows: TRIM22 F,
5′-TGTGGTTGACACTCGGACTC-3′ and R, 5′-TCACGCCTGTCTTAGTCAAG-3′.
Dual-luciferase reporter assay
The wild-type (WT; TRIM22-WT) and mutant FOXC1
binding sites in the TRIM22 promoter were cloned into the pGL3
vector (E1761; Promega Corporation). These reporter plasmids were
co-transfected with 2 µg Ov-FOXC1 and Ov-NC into the cells seeded
at 5×104 cells per well in 24-well plates using
Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.) for 48 h at 37°C. After 48 h, luciferase activity
was assessed using the Dual Luciferase Reporter Gene Assay Kit
(Beyotime Institute of Biotechnology). Firefly luciferase activity
was normalized to Renilla luciferase activity.
Western blotting
A RIPA lysis buffer (Nanjing Jiancheng
Bioengineering Institute) was used to extract total protein from
cells (2×106) and protein concentrations were then
quantified using the BCA Protein Assay Kit (Pierce; Thermo Fisher
Scientific, Inc.). Subsequently, total protein (50 µg/lane) was
separated using SDS-PAGE on a 10% gel (Wuhan Boster Biological
Technology, Ltd.). Separated proteins were then transferred onto
PVDF membranes (Bio-Rad Laboratories, Inc.). The membranes were
blocked using 5% skimmed milk for 30 min at room temperature and
were then incubated with primary antibodies against the following
targets: TRIM22 (1:1,000; cat. no. ab68071; Abcam), Bcl-2 (1:1,000;
cat. no. 15071; Cell Signaling Technology, Inc.), Bax (1:1,000;
cat. no. ab216494; Abcam), cleaved caspase-3 (1:1,000; cat. no.
9661; Cell Signaling Technology, Inc.), caspase-3 (1:1,000; cat.
no. 9668; Cell Signaling Technology, Inc.), MMP2 (1:1,000; cat. no.
ab92536; Abcam), MMP9 (1:1,000; cat. no. ab283575; Abcam), FOXC1
(1:1,000; cat. no. ab223850; Abcam), phosphorylated (p)-IκBα
(1:1,000; cat. no. ab133462; Abcam), p-NF-κB (1:1,000; cat. no.
ab76302; Abcam), IκBα (1:1,000; cat. no. ab76429; Abcam), NF-κB
(1:1,000; cat. no. ab32536; Abcam) and GAPDH (1:1,000; cat. no.
ab8245; Abcam) at 4°C overnight. Following the primary antibody
incubation, the membranes were washed three times with TBS +0.05%
Tween-20 and were subsequently incubated with HRP-conjugated
anti-rabbit (1:2,000; cat. no. ab6721; Abcam) or HRP-conjugated
anti-mouse (1:2,000; cat. no. ab6789; Abcam) secondary antibodies
at room temperature for 2 h. A Pierce ECL Western Blotting
Substrate (Pierce; Thermo Fisher Scientific, Inc.) was applied to
visualize the protein bands and ImageJ (v1.8; National Institutes
of Health) was adopted for analysis.
Bioinformatics tools
TRIM22 expression levels in synovial membrane
samples from patients with RA and or healthy controls were
predicted using the Gene Expression Omnibus (GEO) database
(accession no. GSE12021; https//www.ncbi.nlm.nih.gov/geo/) (29) using GEO2R analysis of the GPL96
platform. The expression values of samples from patients with RA or
healthy controls were determined and GraphPad Prism 8.0 (GraphPad
Software, Inc.) was used to produce a scatter diagram. The
potential binding sites between FOXC1 and the TRIM22 promoter were
predicted using the JASPAR database (https://jaspar.genereg.net/).
Statistical analysis
All data were acquired from three independent
experiments and are presented as the mean ± SD. The results were
analyzed using SPSS 22.0 (IBM Corp.). The statistical differences
between two groups were determined using an unpaired Student's
t-test. The statistical differences between more than two groups
were compared using one-way ANOVA followed by Tukey's post hoc
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
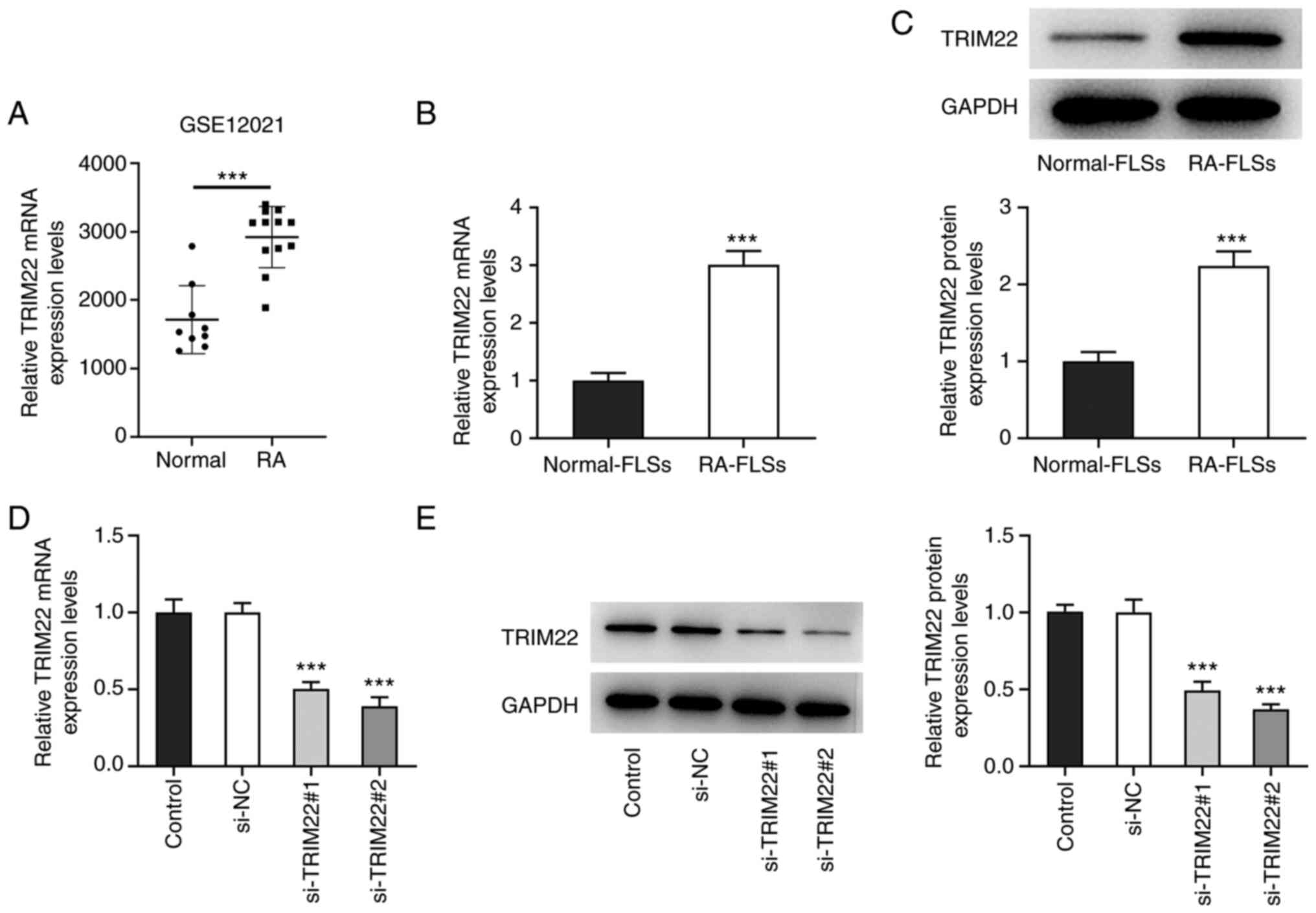
TRIM22 expression levels are increased
in RA-FLSs
To determine the role of TRIM22 in RA, TRIM22
expression levels were detected in synovial membrane samples from
patients with RA and healthy controls. The results demonstrated
that TRIM22 mRNA expression levels in the synovial membrane samples
of patients with RA was significantly higher compared with the
samples from the healthy individuals (Fig. 1A). Furthermore, RT-qPCR and
western blotting demonstrated that TRIM22 was also significantly
upregulated in RA-FLSs compared with normal FLSs (Fig. 1B and C). Subsequently, TRIM22
expression was knocked down using siRNA transfection. Compared with
the si-NC group, the mRNA and protein expression levels of TRIM22
were significantly reduced in the si-TRIM22#1 and #2 groups,
especially in the latter (Fig. 1D and
E). Therefore, si-TRIM22#2 was selected for the following
functional experiments. These results suggested that TRIM22 may be
highly expressed in RA-FLSs.
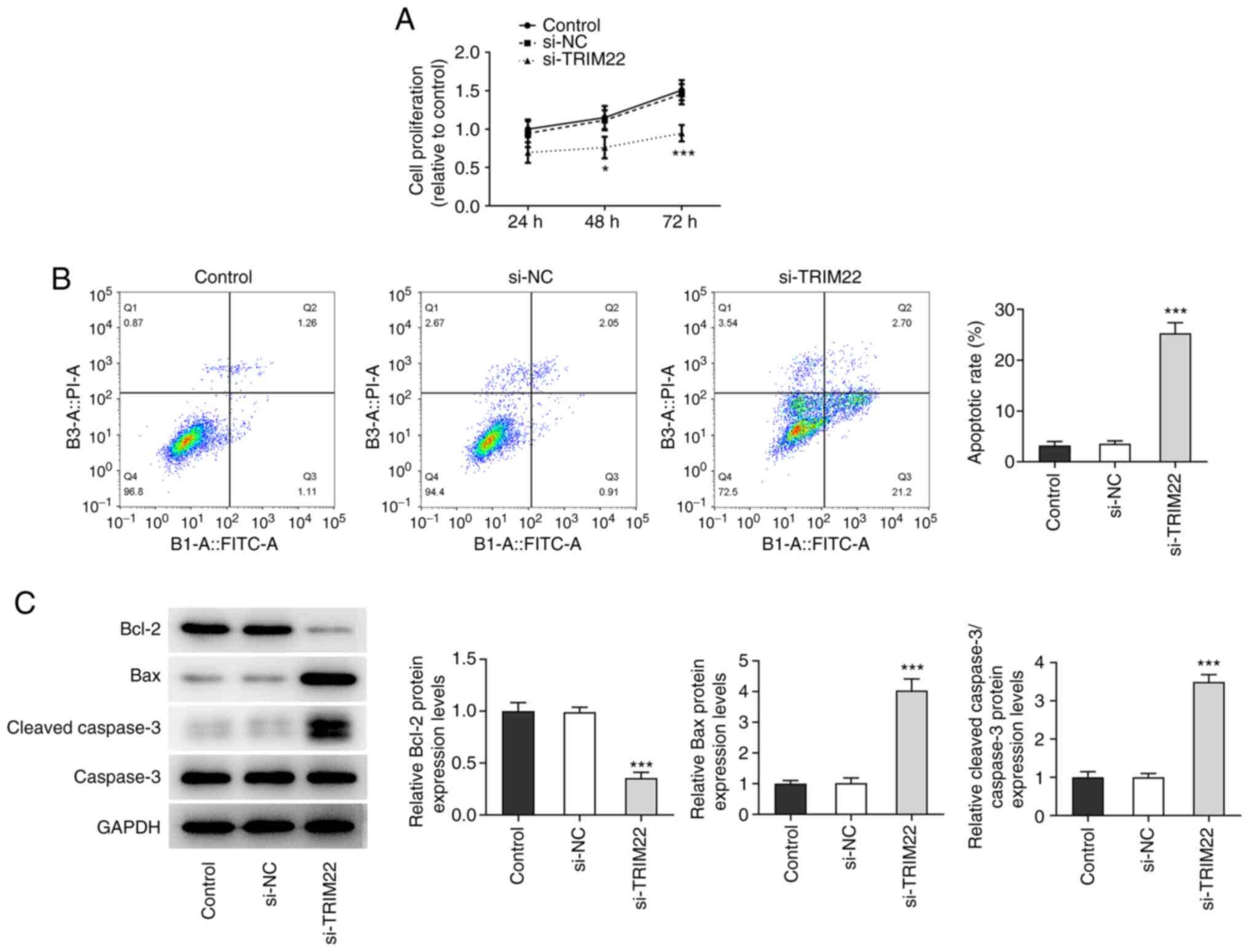
TRIM22 knockdown inhibits the
proliferation and promotes the apoptosis of RA-FLSs
CCK-8 assays were used to assess the effect of
TRIM22 knockdown on the proliferation of RA-FLSs. The results
demonstrated that the knockdown of TRIM22 significantly reduced
cell proliferation at 48 and 72 h compared with the si-NC group
(Fig. 2A). Furthermore, the
number of apoptotic cells in the si-TRIM22 group was significantly
increased compared with the si-NC group, which suggested that
TRIM22 knockdown potentially promoted the apoptosis of RA-FLSs
(Fig. 2B). Moreover, compared
with the si-NC group, western blotting indicated that TRIM22
knockdown significantly reduced the protein expression levels of
the antiapoptotic protein Bcl-2, whereas those of the proapoptotic
proteins Bax and cleaved caspase-3/caspase-3 were significantly
increased (Fig. 2C). These
results indicated that the knockdown of TRIM22 potentially
inhibited the proliferation and promoted the apoptosis of
RA-FLSs.
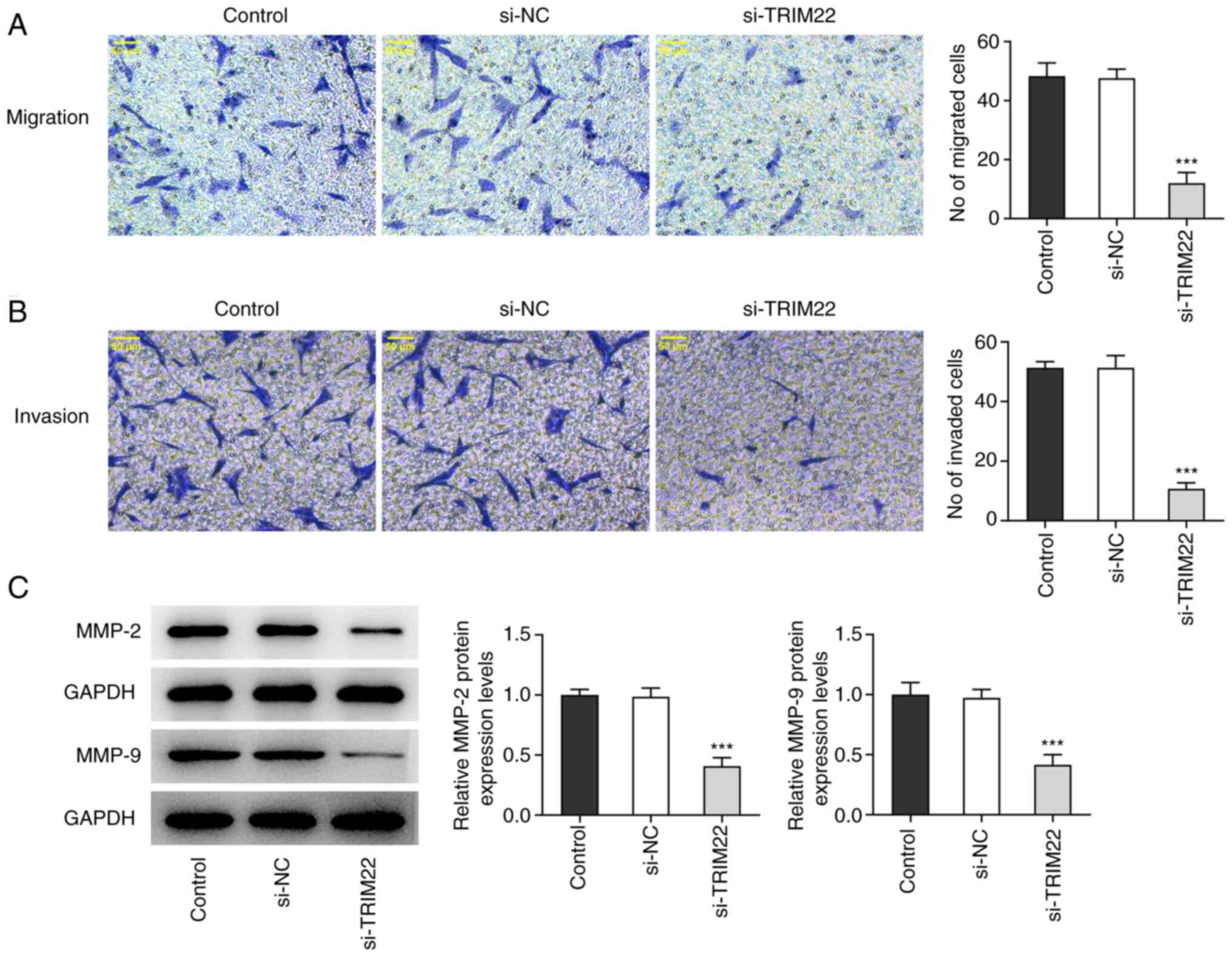
TRIM22 knockdown attenuates the
migratory and invasive capacities of RA-FLSs
Cell migration and invasion were assessed using
Transwell assays. The numbers of migrating cells and invading cells
were significantly reduced following TRIM22 knockdown compared with
the si-NC group (Fig. 3A and B).
Furthermore, as MMP2 and MMP9 are involved in cell migration and
invasion (30), western blotting
was used to analyze the expression levels of these proteins. MMP2
and MMP9 protein levels were both significantly downregulated
following TRIM22 knockdown compared with the si-NC group (Fig. 3C). Therefore, TRIM22 knockdown may
have potentially inhibited the migration and invasion of
RA-FLSs.
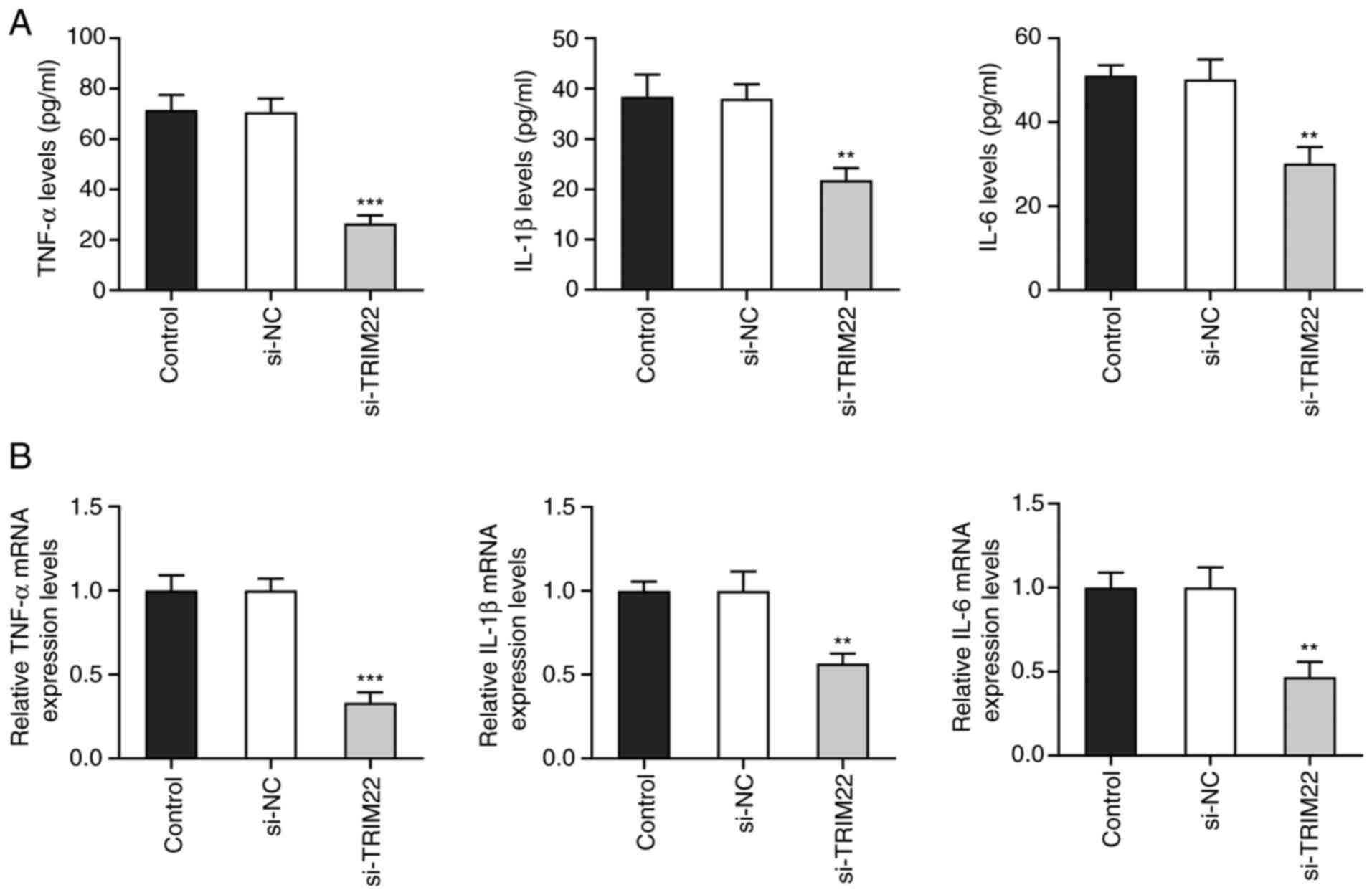
TRIM22 knockdown ameliorates
inflammatory responses in RA-FLSs
TNF-α, IL-1β and IL-6 are well-characterized
pro-inflammatory cytokines (31).
Using ELISA, it was observed that TNF-α, IL-1β and IL-6 levels were
significantly downregulated following TRIM22 knockdown compared
with the si-NC group (Fig. 4A).
Moreover, RT-qPCR further demonstrated that following TRIM22
knockdown, the mRNA expression levels of TNF-α, IL-1β and IL-6 were
also significantly downregulated compared with the si-NC group
(Fig. 4B). These results
indicated that TRIM22 knockdown potentially reduced the production
of pro-inflammatory cytokines by RA-FLSs.
FOXC1 induces the upregulation of
TRIM22 in RA-FLSs
FOXC1 is considered to be an oncogenic transcription
factor (15). The JASPAR database
was used to predict the potential binding site between FOXC1 and
the TRIM22 promoter (Fig. 5A).
Furthermore, RT-qPCR and western blotting demonstrated that FOXC1
mRNA and protein expression levels, respectively, were
significantly upregulated in RA-FLSs compared with normal-FLSs
(Fig. 5B and C). Subsequently,
FOXC1 mRNA and protein expression levels were significantly
increased following transfection with the Ov-FOXC1 plasmid,
compared with the Ov-NC group, and the overexpression efficiency
was assessed (Fig. 5D and E).
Moreover, the dual-luciferase reporter assay demonstrated that the
luciferase activity of TRIM22-WT significantly increased in the
Ov-FOXC1 group compared with the Ov-NC group while no obvious
changes were observed in the luciferase activity of TRIM22-MUT in
the Ov-FOXC1 group compared with the Ov-NC group (Fig. 5F). ChIP assays also demonstrated
that the TRIM22 promoter was co-precipitated with the FOXC1
antibody, which confirmed the binding between the TRIM22 promoter
and FOXC1 (Fig. 5G). Furthermore,
FOXC1 overexpression significantly upregulated the mRNA levels and
protein expression levels of TRIM22, which suggested that TRIM22
was potentially positively regulated via FOXC1 (Fig. 5H and I). Therefore, these results
indicated that FOXC1 may be a transcriptional activator of
TRIM22.
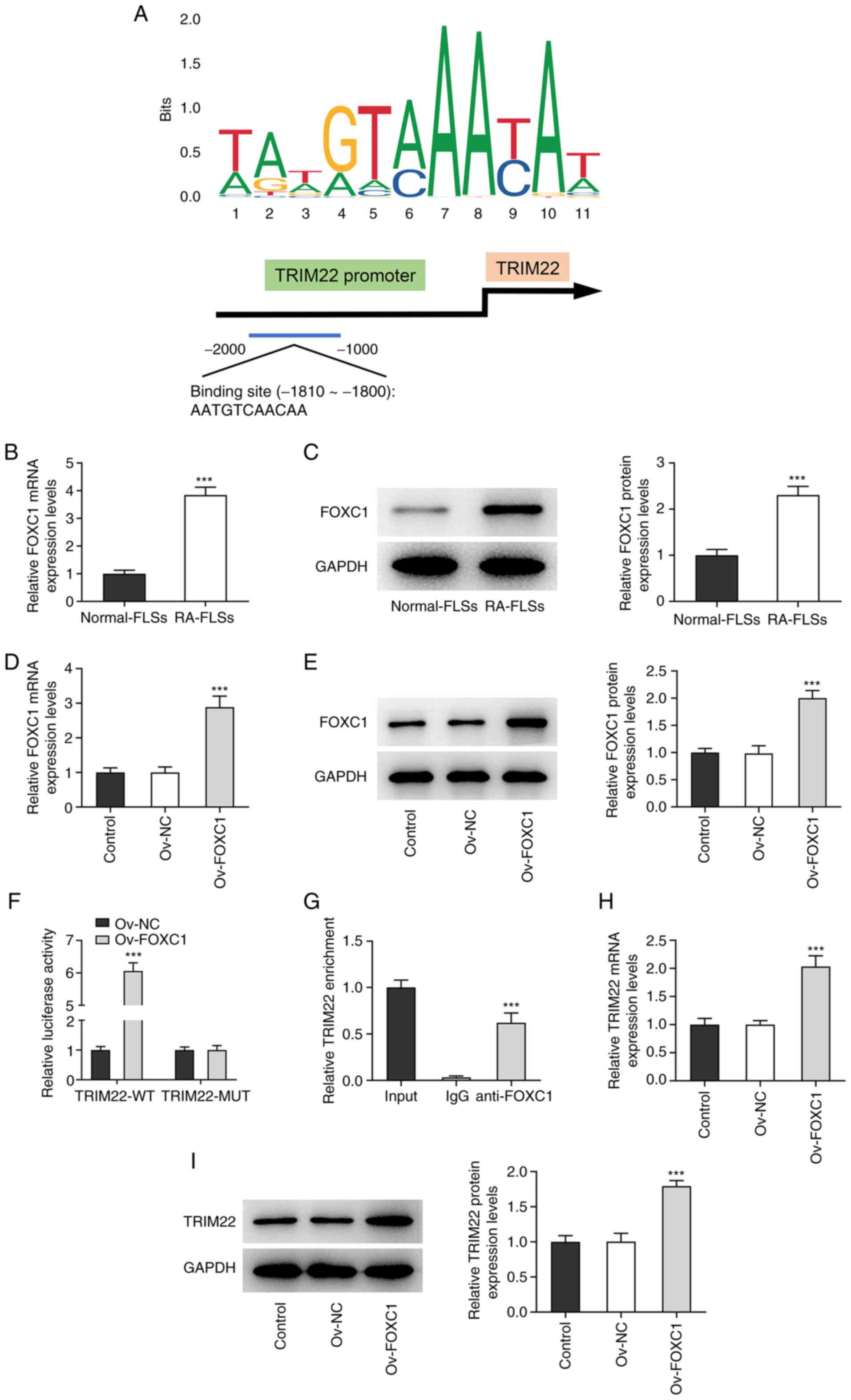 | Figure 5.FOXC1 induces the upregulation of
TRIM22 in RA-FLSs. (A) The possible binding site between FOXC1 and
the TRIM22 promoter was predicted using the JASPAR database. FOXC1
mRNA and protein expression levels in RA-FLSs and normal-FLSs was
determined using (B) RT-qPCR and (C) western blotting.
***P<0.001. Ov-FOXC1 transfection efficiency was analyzed using
(D) RT-qPCR and (E) western blotting. ***P<0.001 vs. Ov-NC. (F)
Dual-luciferase reporter assays were performed to assess the
luciferase activity of TRIM22-WT and TRIM22-MUT following
transfection with Ov-FOXC1. ***P<0.001 vs. Ov-NC. (G) Chromatin
immunoprecipitation confirmed the abundance of the TRIM22 promoter
in the FOXC1 antibody fraction. Input was the positive control.
***P<0.001 vs. IgG. TRIM22 mRNA and protein expression was
assessed using (H) RT-qPCR and (I) western blotting, respectively,
following FOXC1 overexpression. ***P<0.001 vs. Ov-NC. FOXC1,
Forkhead box C1; TRIM22, tripartite motif-containing 22; RA,
rheumatoid arthritis; FLS, fibroblast-like synoviocyte; RT-qPCR,
reverse transcription-quantitative PCR; Ov, overexpression; NC,
negative control; WT, wild-type; MUT, mutant. |
FOXC1 overexpression reverses the
effects of TRIM22 knockdown on the proliferation, apoptosis,
migration, invasion and inflammatory response of RA-FLSs
To confirm the interaction between FOXC1 and TRIM22
in RA-FLSs, rescue assays were performed. The mRNA and protein
expression levels of TRIM22 were significantly downregulated in
RA-FLSs transfected with si-TRIM22 compared with the control group.
However, this was significantly reversed by FOXC1 overexpression
compared with the si-TRIM22 + Ov-NC group (Fig. 6A and B). CCK-8 assays demonstrated
that the significant reduction in the proliferation of RA-FLSs
caused by TRIM22 knockdown could be significantly reversed via
FOXC1 overexpression compared with the si-TRIM22 + Ov-NC group
(Fig. 6C). Furthermore, TRIM22
silencing-induced cell apoptosis was significantly suppressed
following the overexpression of FOXC1 compared with the si-TRIM22 +
Ov-NC group (Fig. 6D). This
result was also confirmed by the significant downregulation of
Bcl-2 protein expression levels and the significant upregulation of
Bax and cleaved caspase-3/caspase-3 protein expression levels
following TRIM22 knockdown compared with the control. However,
these changes were significantly reversed via FOXC1 overexpression
compared with the si-TRIM22 + Ov-NC group (Fig. 6E). Furthermore, knockdown of
TRIM22 significantly reduced the migration and invasion of RA-FLSs
compared with the control; however, this effect was significantly
reversed by FOXC1 overexpression compared with the si-TRIM22 +
Ov-NC group (Fig. 6F and G).
Compared with the control the protein expression levels of MMP2 and
MMP9 in the si-TRIM22 group were significantly decreased. However,
they were significantly increased in the si-TRIM22 + Ov-FOXC1 group
compared with the si-TRIM22 + Ov-NC group (Fig. 6H). Moreover, ELISA and RT-qPCR
demonstrated that FOXC1 overexpression significantly reversed the
reduced inflammatory response in RA-FLSs transfected with si-TRIM22
compared with the si-TRIM22 + Ov-NC group (Fig. 6I and J). Collectively, these
results indicated that TRIM22 potentially contributes to the
progression of RA via FOXC1.
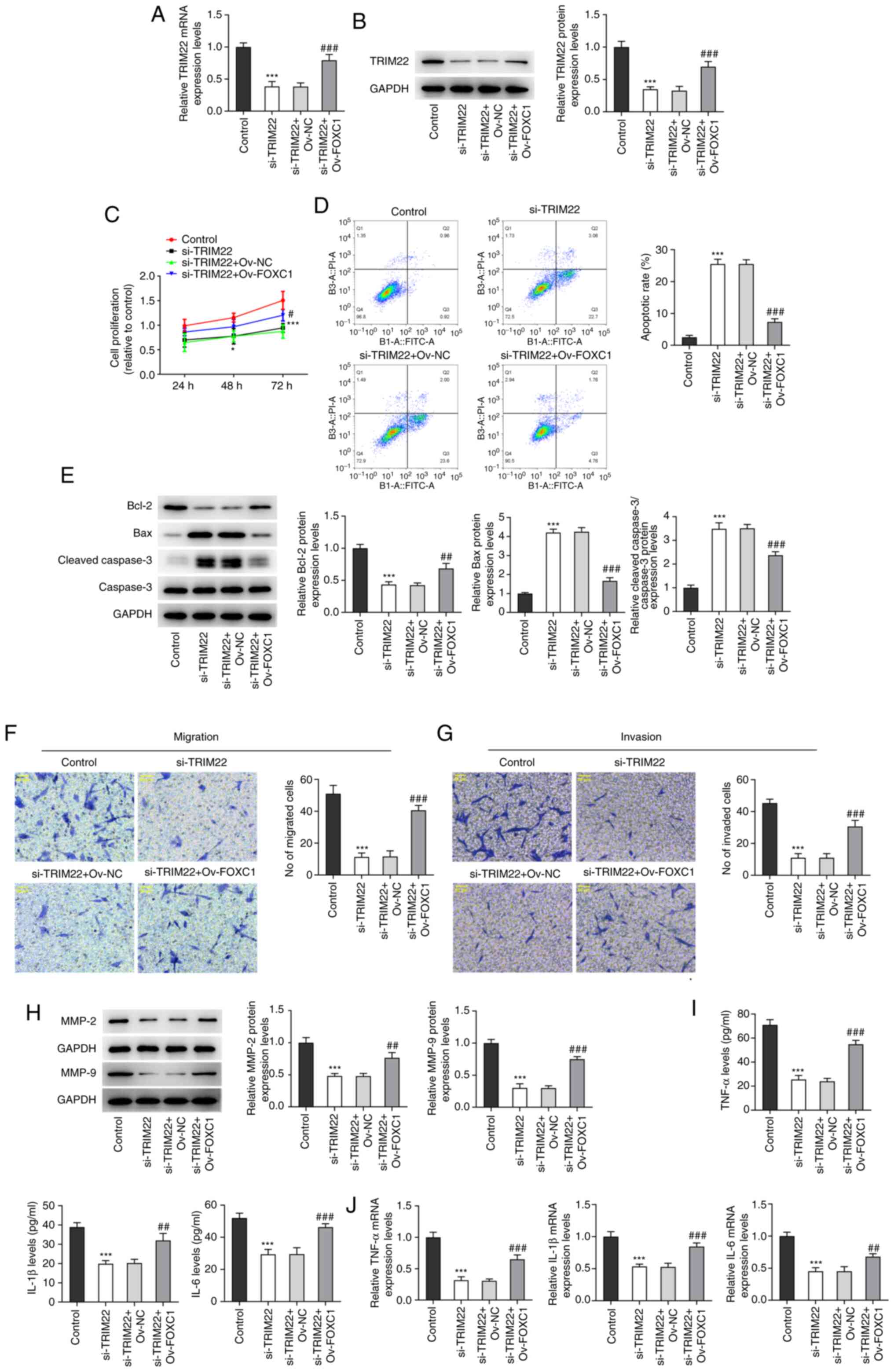 | Figure 6.FOXC1 overexpression counteracts the
impacts of TRIM22 reduction on the proliferation, apoptosis,
migration, invasion and inflammatory response of RA-FLSs. mRNA and
protein expression levels of TRIM22 in RA-FLSs transfected with
si-TRIM22 and Ov-FOXC1 were detected via (A) RT-qPCR and (B)
western blotting. (C) RA-FLS cell proliferation was detected using
the Cell Counting Kit-8 assay. (D) Cell apoptosis was assessed
using flow cytometry. (E) Western blotting was performed to analyze
the protein expression levels of apoptosis-related proteins. The
(F) migration and (G) invasion of RA-FLSs were assessed via
Transwell assays. Magnification, ×200. (H) Western blotting was
performed to analyze the protein expression levels of MMP2 and
MMP9. (I) Levels of TNF-α, IL-1β and IL-6 were assessed using
ELISA. (J) RT-qPCR determined the mRNA expression levels of TNF-α,
IL-1β and IL-6. *P<0.05 and***P<0.001 vs. si-NC; #P<0.05,
##P<0.01, ###P<0.001 vs. si-TRIM22 + Ov-NC. FOXC1, Forkhead
box C1; TRIM22, tripartite motif-containing 22; RA, rheumatoid
arthritis; FLS, fibroblast-like synoviocyte; si, small interfering
RNA; Ov, overexpression; RT-qPCR, reverse
transcription-quantitative PCR; NC, negative control. |
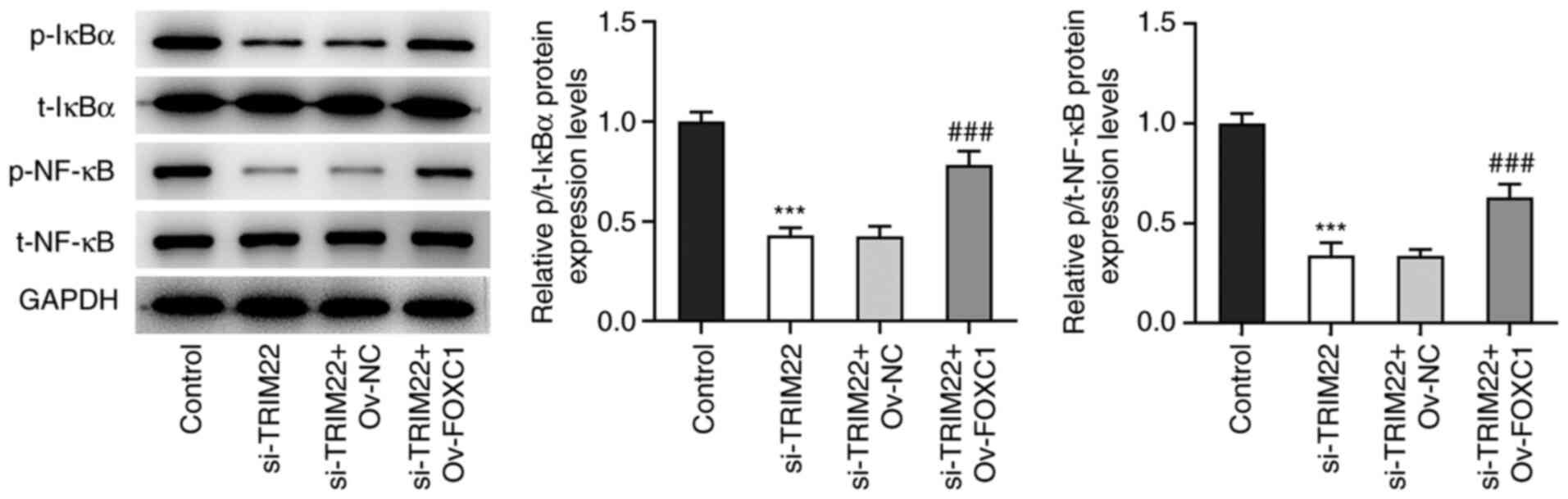
FOXC1-induced TRIM22 activates the
NF-κB signaling pathway
Western blotting demonstrated that TRIM22 knockdown
significantly reduced the protein expression levels of p/total
(t)-IκBα and p/t-NF-κB compared with the control. This effect was
significantly reversed following FOXC1 overexpression compared with
the si-TRIM22 + Ov-NC group. Therefore, these results indicated
that TRIM22 may be activated via FOXC1 to stimulate the NF-κB
signaling pathway (Fig. 7).
Discussion
RA is an autoimmune disease that is primarily
characterized by the development of aggressive FLS phenotypes
(32). FLSs are resident
joint-lining mesenchymal cells and aberrant hyperplasia of these
cells serves an important role in the pathogenesis of RA (32). Moreover, FLSs contribute to joint
inflammation via inducing the release of pro-inflammatory cytokines
(33). TRIM22 is an
evolutionarily ancient protein that is involved in cellular
differentiation and proliferation, which serves an integral role in
certain types of cancer and autoimmune diseases (12). Previous studies have suggested
that TRIM22 serves a role as a tumor suppressor in endometrial
cancer and gastric cancer (34,35). However, numerous studies have also
highlighted the oncogenic properties of TRIM22 in chronic myeloid
leukemia (15), glioblastoma
(36), non-small cell lung cancer
(14) and colon cancer (14). Moreover, Kang et al
(37) reported that TRIM22
knockdown exerts an anti-inflammatory effect over oxygen-glucose
deprivation/reoxygenation-stimulated HCN-2 cells. Similar to these
aforementioned studies, the present study highlighted the role of
TRIM22 in the inflammatory phenotypes of FLSs in RA. The results of
the present study demonstrated that TRIM22 was expressed at
significantly higher levels in the synovial membrane samples of
patients with RA and in RA-FLSs. TRIM22 knockdown significantly
inhibited the proliferation, migration and invasion of RA-FLSs and
promoted cell apoptosis.
TNF-α is a pro-inflammatory cytokine (38). It has previously been reported
that TNF-α is abundant in the serum and arthritic synovium of
patients with RA and that targeting TNF-α represents an efficacious
method for RA therapy (39,40). IL-1β and IL-6 are also central
regulators in the inflammatory response (31). In the present study, TRIM22
knockdown significantly reduced the secreted levels and the mRNA
expression levels of TNF-α, IL-1β and IL-6. These results therefore
suggested that TRIM22 may potentially drive the inflammatory
response of RA-FLSs.
The forkhead family of transcription factors has
been associated with diverse biological processes (41). For example, FOXC1 is an
indispensable member of the forkhead family and its upregulation
predicts poor survival outcomes in multiple types of carcinomas
(42). FOXC1 also promotes
aggressive tumor phenotypes in triple-negative breast cancer
(43), pancreatic cancer
(21), oral squamous cell
carcinoma (44) and prostate
cancer (45). Furthermore, Yu
et al (22) demonstrated
that FOXC1 exacerbates the proliferation of RA-FLSs. Moreover,
FOXC1 may elicit a potent activity in tumors via gene regulation.
For example, glutathione peroxidase 8 expression is induced via
FOXC1, which promotes the development of gastric cancer (46). Furthermore, lysyl oxidase is a
downstream effector of FOXC1 in non-small cell lung cancer
(47). Similarly, the present
study suggested that the FOXC1 transcription factor may have a
potential binding site on the TRIM22 promoter. The results
demonstrated that FOXC1 was significantly highly expressed at both
mRNA level and protein level in RA-FLSs and FOXC1 overexpression
significantly upregulated the mRNA expression and protein
expression of TRIM22 in RA-FLSs. The potent affinity of the TRIM22
promoter with FOXC1 was also verified using the dual-luciferase
reporter and ChIP assays. Furthermore, rescue assays demonstrated
that following FOXC1 overexpression the significantly reduced
proliferation, migration, invasion, inflammation and increased
apoptosis of RA-FLSs observed following TRIM22 silencing, were
significantly reversed.
NF-κB is a ubiquitous transcription factor involved
in fundamental cellular functions, including cell survival,
inflammation and immune responses (48). Increasing evidence has suggested
that the dysregulation of NF-κB signaling may influence the
development of RA (26,27,49). Furthermore, TRIM22 has been
regarded as an activator of the NF-κB signaling pathway (37,50). The role of FOXC1 in the
proliferation and invasion of cancer cells may also be attributed
to NF-κB modulation (51).
Consistent with these previous findings, the present study also
demonstrated that TRIM22 knockdown potentially inhibited the NF-κB
signaling pathway via the downregulation of the protein expression
levels of p/t-IκBα and p/t-NF-κB. However, this was significantly
reversed via the overexpression of FOXC1. A previous study reported
that FOXC1 activates the PI3K/AKT signaling pathway to increase FLS
proliferation in RA (22).
However, whether FOXC1 activates the PI3K/AKT signaling pathway to
aggravate FLS proliferation in RA via the NF-κB signaling pathway
should be studied further in the future.
In summary, the present study demonstrated that
TRIM22 overexpression potentially promoted RA via significantly
increasing the proliferation, migration, invasion, inflammation and
inhibiting the apoptosis of FLSs. Furthermore, the results
suggested that TRIM22 expression was potentially induced via the
FOXC1 transcription factor and activated the NF-κB signaling
pathway in RA. Overall, these findings demonstrated a potential
novel role of TRIM22 in RA and highlighted the underlying
regulatory mechanism. These results may have provided insight into
potential strategies for targeted RA therapeutics.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the Basic
Research Project of Shenzhen Science and Technology Innovation
Committee (grant no. JCYJ20210324120800001).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LD performed the experiments; YW and XH analyzed the
data; and LD, YM designed the experiments, interpreted the data and
wrote the manuscript. All authors read and approved the final
manuscript. LD and YW confirm the authenticity of all the raw
data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare they have no competing
interests.
References
|
1
|
Smolen JS, Aletaha D and McInnes IB:
Rheumatoid arthritis. Lancet. 388:2023–2038. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
van der Woude D and van der Helm-van Mil
AHM: Update on the epidemiology, risk factors, and disease outcomes
of rheumatoid arthritis. Best Pract Res Clin Rheumatol. 32:174–187.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lorenz HM: Rheumatoid arthritis:
Diagnostics and therapy 2012. Orthopade. 41:514–519. 2012.(In
German). View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Song X and Lin Q: Genomics,
transcriptomics and proteomics to elucidate the pathogenesis of
rheumatoid arthritis. Rheumatol Int. 37:1257–1265. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wasserman A: Rheumatoid arthritis: Common
questions about diagnosis and management. Am Fam Physician.
97:455–462. 2018.PubMed/NCBI
|
|
6
|
Sparks JA: Rheumatoid arthritis. Ann
Intern Med. 170:ITC1–ITC16. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Deane KD, Demoruelle MK, Kelmenson LB,
Kuhn KA, Norris JM and Holers VM: Genetic and environmental risk
factors for rheumatoid arthritis. Best Pract Res Clin Rheumatol.
31:3–18. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Burmester GR and Pope JE: Novel treatment
strategies in rheumatoid arthritis. Lancet. 389:2338–2348. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Aletaha D and Smolen JS: Diagnosis and
management of rheumatoid arthritis: A review. JAMA. 320:1360–1372.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
McInnes IB and Schett G: Pathogenetic
insights from the treatment of rheumatoid arthritis. Lancet.
389:2328–2337. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cheung TT and McInnes IB: Future
therapeutic targets in rheumatoid arthritis? Semin Immunopathol.
39:487–500. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hattlmann CJ, Kelly JN and Barr SD:
TRIM22: A diverse and dynamic antiviral protein. Mol Biol Int.
2012:1534152012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hatakeyama S: TRIM family proteins: Roles
in autophagy, immunity, and carcinogenesis. Trends Biochem Sci.
42:297–311. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu R, Zhao W, Wang H and Wang J: Long
noncoding RNA LINC01207 promotes colon cancer cell proliferation
and invasion by regulating miR-3125/TRIM22 axis. Biomed Res Int.
2020:12163252020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li L, Qi Y, Ma X, Xiong G, Wang L and Bao
C: TRIM22 knockdown suppresses chronic myeloid leukemia via
inhibiting PI3K/Akt/mTOR signaling pathway. Cell Biol Int.
42:1192–1199. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu L, Zhou XM, Yang FF, Miao Y, Yin Y, Hu
XJ, Hou G, Wang QY and Kang J: TRIM22 confers poor prognosis and
promotes epithelial-mesenchymal transition through regulation of
AKT/GSK3β/β-catenin signaling in non-small cell lung cancer.
Oncotarget. 8:62069–62080. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Han B, Bhowmick N, Qu Y, Chung S, Giuliano
AE and Cui X: FOXC1: An emerging marker and therapeutic target for
cancer. Oncogene. 36:3957–3963. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cao S, Wang Z, Gao X, He W, Cai Y, Chen H
and Xu R: FOXC1 induces cancer stem cell-like properties through
upregulation of beta-catenin in NSCLC. J Exp Clin Cancer Res.
37:2202018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cao Q, Wang X, Shi Y, Zhang M, Yang J,
Dong M, Mi Y, Zhang Z, Liu K, Jiang L, et al: FOXC1 silencing
inhibits the epithelial-to-mesenchymal transition of glioma cells:
Involvement of β-catenin signaling. Mol Med Rep. 19:251–261.
2019.PubMed/NCBI
|
|
20
|
Han B, Zhou B, Qu Y, Gao B, Xu Y, Chung S,
Tanaka H, Yang W, Giuliano AE and Cui X: FOXC1-induced
non-canonical WNT5A-MMP7 signaling regulates invasiveness in
triple-negative breast cancer. Oncogene. 37:1399–1408. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Subramani R, Camacho FA, Levin CI, Flores
K, Clift A, Galvez A, Terres M, Rivera S, Kolli SN, Dodderer J, et
al: FOXC1 plays a crucial role in the growth of pancreatic cancer.
Oncogenesis. 7:522018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yu Z, Xu H, Wang H and Wang Y: Foxc1
promotes the proliferation of fibroblast-like synoviocytes in
rheumatoid arthritis via PI3K/AKT signalling pathway. Tissue Cell.
53:15–22. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yu H, Lin L, Zhang Z, Zhang H and Hu H:
Targeting NF-κB pathway for the therapy of diseases: Mechanism and
clinical study. Signal Transduct Target Ther. 5:2092020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Poma P: NF-κB and disease. Int J Mol Sci.
21:91812020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hoesel B and Schmid JA: The complexity of
NF-κB signaling in inflammation and cancer. Mol Cancer. 12:862013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang HJ, Wei QF, Wang SJ, Zhang HJ, Zhang
XY, Geng Q, Cui YH and Wang XH: LncRNA HOTAIR alleviates rheumatoid
arthritis by targeting miR-138 and inactivating NF-κB pathway. Int
Immunopharmacol. 50:283–290. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shao L and Hou C: miR-138 activates NF-κB
signaling and PGRN to promote rheumatoid arthritis via regulating
HDAC4. Biochem Biophys Res Commun. 519:166–171. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Huber R, Hummert C, Gausmann U, Pohlers D,
Koczan D, Guthke R and Kinne RW: Identification of intra-group,
inter-individual, and gene-specific variances in mRNA expression
profiles in the rheumatoid arthritis synovial membrane. Arthritis
Res Ther. 10:R982008. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang JF, Wang P, Yan YJ, Li Y, Guan MW,
Yu JJ and Wang XD: IL-33 enhances glioma cell migration and
invasion by upregulation of MMP2 and MMP9 via the ST2-NF-κB
pathway. Oncol Rep. 38:2033–2042. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li QY, Xu HY and Yang HJ: Effect of
proinflammatory factors TNF-α,IL-1β, IL-6 on neuropathic pain.
Zhongguo Zhong Yao Za Zhi. 42:3709–3712. 2017.(In Chinese).
PubMed/NCBI
|
|
32
|
Wu Z, Ma D, Yang H, Gao J, Zhang G, Xu K
and Zhang L: Fibroblast-like synoviocytes in rheumatoid arthritis:
Surface markers and phenotypes. Int Immunopharmacol. 93:1073922021.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Han D, Fang Y, Tan X, Jiang H, Gong X,
Wang X, Hong W, Tu J and Wei W: The emerging role of
fibroblast-like synoviocytes-mediated synovitis in osteoarthritis:
An update. J Cell Mol Med. 24:9518–9532. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang L, Zhang B, Wei M, Xu Z, Kong W,
Deng K, Xu X, Zhang L, Ζhao X and Yan L: TRIM22 inhibits
endometrial cancer progression through the NOD2/NF-κB signaling
pathway and confers a favorable prognosis. Int J Oncol.
56:1225–1239. 2020.PubMed/NCBI
|
|
35
|
Zhou Z, Gao W, Yuan B, Zhang S, Wang K and
Du T: TRIM22 inhibits the proliferation of gastric cancer cells
through the Smad2 protein. Cell Death Discov. 7:2342021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ji J, Ding K, Luo T, Zhang X, Chen A,
Zhang D, Li G, Thorsen F, Huang B, Li X and Wang J: TRIM22
activates NF-κB signaling in glioblastoma by accelerating the
degradation of IκBα. Cell Death Differ. 28:367–381. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kang C, Lu Z, Zhu G, Chen Y and Wu Y:
Knockdown of TRIM22 relieves oxygen-glucose
deprivation/reoxygenation-induced apoptosis and inflammation
through inhibition of NF-κB/NLRP3 axis. Cell Mol Neurobiol.
41:341–351. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zelová H and Hošek J: TNF-α signalling and
inflammation: Interactions between old acquaintances. Inflamm Res.
62:641–651. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Radner H and Aletaha D: Anti-TNF in
rheumatoid arthritis: An overview. Wien Med Wochenschr. 165:3–9.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Moelants EA, Mortier A, Van Damme J and
Proost P: Regulation of TNF-α with a focus on rheumatoid arthritis.
Immunol Cell Biol. 91:393–401. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Golson ML and Kaestner KH: Fox
transcription factors: From development to disease. Development.
143:4558–4570. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sabapathi N, Sabarimurugan S, Madurantakam
Royam M, Kumarasamy C, Xu X, Xu G and Jayaraj R: Prognostic
significance of FOXC1 in various cancers: A systematic review and
meta-analysis. Mol Diagn Ther. 23:695–706. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li M, Lv H, Zhong S, Zhou S, Lu H and Yang
W: FOXC1. Arch Pathol Lab Med. Nov 16–2021.(Epub ahead of
print).
|
|
44
|
Liu Z, Xu S, Chu H, Lu Y, Yuan P and Zeng
X: Silencing FOXC1 inhibits growth and migration of human oral
squamous cell carcinoma cells. Exp Ther Med. 16:3369–3376.
2018.PubMed/NCBI
|
|
45
|
Zhang D, Liu X, Zhang Q and Chen X:
miR-138-5p inhibits the malignant progression of prostate cancer by
targeting FOXC1. Cancer Cell Int. 20:2972020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chen H, Xu L, Shan ZL, Chen S and Hu H:
GPX8 is transcriptionally regulated by FOXC1 and promotes the
growth of gastric cancer cells through activating the Wnt signaling
pathway. Cancer Cell Int. 20:5962020. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Gong R, Lin W, Gao A, Liu Y, Li J, Sun M,
Chen X, Han S, Men C, Sun Y and Liu J: Forkhead box C1 promotes
metastasis and invasion of non-small cell lung cancer by binding
directly to the lysyl oxidase promoter. Cancer Sci. 110:3663–3676.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Won M, Byun HS, Park KA and Hur GM:
Post-translational control of NF-κB signaling by ubiquitination.
Arch Pharm Res. 39:1075–1084. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wang Y, Xu N, Zhao S, Jiao T, Fu W, Yang L
and Zhang N: miR-410-3p suppresses cytokine release from
fibroblast-like synoviocytes by regulating NF-κB signaling in
rheumatoid arthritis. Inflammation. 42:331–341. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Tian H and He Z: miR-215 enhances HCV
replication by targeting TRIM22 and inactivating NF-κB signaling.
Yonsei Med J. 59:511–518. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wang J, Li W, Zheng X, Pang X and Du G:
Research progress on the forkhead box C1. Oncotarget.
9:12471–12478. 2017. View Article : Google Scholar : PubMed/NCBI
|