Introduction
Sepsis is a potentially fatal condition
characterized by systemic organ damage and dysfunction induced by
an abnormal host response to infection (1). With the expansion of the ‘Save
Sepsis’ campaign, the international guidelines for sepsis and
infectious shock management in 2021 emphasized the role of
multi-organ dysfunction, early detection and sepsis treatment
(2). The heart is one of the most
sensitive organs to sepsis and sepsis-induced cardiomyopathy (SIC)
is frequently observed in the intensive care unit, with an
incidence of 10–70% in patients with sepsis in the United States
(3). The pathophysiology of SIC
includes dysregulation of inflammatory mediators, mitochondrial
dysfunction, oxidative stress, calcium regulatory abnormality,
dysregulation and endothelial dysfunction of the autonomic nervous
system, endoplasmic reticulum stress, autophagy and ferroptosis
(4,5).
Ferroptosis is a unique form of programmed cell
death dependent on reactive oxygen species (ROS) and iron and is
distinct from apoptosis, necrosis and autophagy (6). Ferroptosis is primarily caused by a
blockage of the cystine/glutamate reverse transporter system
xc− (system xc−),
oxidative stress, iron overload and lipid peroxidation
accumulation. System xc− comprises the
catalytic subunit solute carrier family 7 member 11 (SLC7A11) and
the chaperone subunit solute carrier family 3 member 2 (7); inhibition of system
xc− transport decreases cellular antioxidant
capacity and causes toxic lipid peroxide accumulation, eventually
leading to cellular ferroptosis (8). Beclin 1 (BECN1), an endogenous
SLC7A11-binding protein, is key for the regulation of ferroptosis
(9). Ferroptosis has been
associated with diabetic (10),
azithromycin-induced (11) and
iron overload cardiomyopathy (12) and lipopolysaccharide (LPS)-induced
acute lung (13) and acute kidney
injury (14). However, additional
research is required to determine the precise mechanism and
therapeutic targets of ferroptosis in LPS- and cecal ligation and
puncture (CLP)-induced cardiomyocyte injury in septic rats.
In mammals, H2S is regarded as the ‘third
novel medical signaling gas molecule’ and serves an important role
in protecting cells from oxidative stress and signal transduction
(15). H2S synthesis
is dependent on three enzymes: Cystathionine β-synthase,
cystathionine γ-lyase and 3-mercaptopyruvate sulfur-transferase
(16). H2S synthesis
is implicated in a range of pathological and physiological
processes, including inflammation (17), oxidative stress (18), apoptosis (19) and autophagy (20). NaHS has been reported to enhance
LPS-induced SIC via regulation of signaling pathways, such as the
toll-like receptor 4 pathway, and endoplasmic reticulum stress
(21). Although NaHS has been
widely investigated in the prevention and treatment of sepsis
(22,23), its regulatory mechanisms and
involvement in ferroptosis regulation in SIC require further
elucidation. In the present study, an in vitro model of
LPS-induced H9c2 cardiomyocyte injury and an in vivo model
of CLP-induced sepsis were employed to determine whether the
H2S donor NaHS alleviates septic myocardial injury by
decreasing oxidative stress and cardiomyocyte ferroptosis.
Materials and methods
Cell culture and processing
The rat cardiomyocyte H9c2 cell line (Procell Life
Science & Technology) was cultured in flasks containing 89%
DMEM (Gibco; Thermo Fisher Scientific, Inc.), 10% fetal bovine
serum (Gibco; Thermo Fisher Scientific, Inc.) and 1% 10,000 U/ml
penicillin-streptomycin (HyClone; Cytiva) at 37°C under 5%
CO2. The cells were pretreated with NaHS (Sigma-Aldrich;
Merck KGaA; 20, 50, 100, 150 and 200 µmol/l) for 1 h at 37°C prior
to cell stimulation with LPS (Sigma-Aldrich; Merck KGaA; 1, 3, 5,
10 and 20 µg/ml) for 24 h at 37°C. The control group received
double-distilled water to the same volume as the experimental
group.
Cell viability assay
Cell Counting Kit-8 (CCK-8) assay (cat. no. CK04;
Dojindo Laboratories, Inc.) was used to assess cell viability. H9c2
cells were seeded in 96-well plates, at a density of
2×104 cells/well and cultured for 24 h at 37°C. The
cells were pretreated with 20, 50, 100, 150 and 200 µM NaHS for 1 h
at 37°C prior to cell stimulation with 1, 3, 5, 10 and 20 µg/ml LPS
for 24 h at 37°C. Subsequently, 10 µl CCK-8 solution was added to
each well and the 96-well plates were incubated for 2 h at 37°C and
the cells were then incubated for 3 h. The optical density was
measured at 450 nm using a microplate reader (Thermo Fisher
Scientific, Inc.). The cell vitality was calculated as follows:
Cell viability (%)=(absorbance of treatment-absorbance of
blank)/(absorbance of control-absorbance of blank) ×100.
Determination of lactate dehydrogenase
(LDH), creatine kinase-myocardial band (CK-MB), glutathione (GSH),
malondialdehyde (MDA), Fe2+ and ROS levels and
mitochondrial membrane potential
H9c2 (2×104 cells/well) were pre-treated
with 50 µmol/l NaHS for 1 h prior to cell stimulation with 5 µg/ml
LPS for 24 h at 37°C. The cells were separated into three groups as
follows: i) Control, ii) LPS and iii) LPS + NaHS. LDH Activity
Assay (cat. no. BC0680; Beijing Solarbio Science and Technology
Co., Ltd.), CK-MB ELISA (cat. no. SEKM-0152; Beijing Solarbio
Science and Technology Co., Ltd.), Reduced GSH Content Assay (cat.
no. BC1170; Beijing Solarbio Science and Technology Co., Ltd.), MDA
Content Assay (cat. no. BC0020; Beijing Solarbio Science and
Technology Co., Ltd.) and Total Iron Content Colorimetric Assay
kits (cat. no. E1042; Applygen Technologies, Inc.) were used to
evaluate levels each marker in supernatant (4°C, 200 g, 5 min)
according to the manufacturers protocol. The ROS and mitochondrial
membrane potential changes in rat H9c2 cardiomyocytes were
quantified by measuring the fluorescence of DCFH-DA ROS (cat. no.
D6470; Beijing Solarbio Science and Technology Co., Ltd.) and JC-1
(cat. no. J8030; Beijing Solarbio Science and Technology Co., Ltd.)
fluorescent probes, respectively. The cells were incubated with
DCFH-DA ROS and JC-1 working solution at 37°C for 30 min.
Subsequently, 1X PBS was used to wash cells at least two times.
Images were captured under a fluorescence microscope
(magnification, ×200) and evaluated using ImageJ software (v146;
National Institutes of Health).
Western blotting
Total protein was extracted from H9c2 cells and
myocardial tissue using RIPA lysis buffer (cat. no. R0010; Beijing
Solarbio Science & Technology Co., Ltd.). Protein concentration
was evaluated using a Pierce BCA protein assay kit (cat. no.
PC0020; Beijing Solarbio Science & Technology Co., Ltd.). The
protein samples (10 µg/lane) were then separated using 10 and 12%
SDS-PAGE and electrophoresed. The proteins were electro-transferred
onto PVDF membranes. The membranes were blocked with 5% bovine
serum albumin (cat. no. A8020; Beijing Solarbio Science &
Technology Co., Ltd.) for 2 h at room temperature before overnight
incubation at 4°C with primary antibodies as follows: BECN1
(1:1,000; cat. no. ab210498; Abcam), phosphorylated (p)-BECN1
(1:1,000; cat. no. ab183313; Abcam), GPX4 (1:1,000; cat. no.
T56959; Abmart Pharmaceutical Technology Co., Ltd.), SLC7A11
(1:1,000; cat. no. T57046; Abmart Pharmaceutical Technology Co.,
Ltd.), ferritin (1:1,000; cat. no. T55648 Abmart Pharmaceutical
Technology Co., Ltd.) and β actin (1:1,000; cat. no. TA328071;
OriGene Technologies, Inc.). After washing the PVDF membranes, the
secondary antibodies were incubated for 1 h at room temperature.
Secondary antibodies were as follows: Peroxidase Conjugated
Affinity Purified Goat anti-Mouse IgG (H+L) (1:10,000; cat. no.
TA130003; OriGene Technologies, Inc.) and Goat Anti-Rabbit IgG
Secondary Antibody (1:10,000; cat. no. TA130015 OriGene
Technologies, Inc.). Gels were washed, incubated with
high-sensitivity ECL (cat. no. WBULS0100; Sigma-Aldrich; Merck
KGaA) according to manufacturer's protocol and imaged. Band
intensity was semi-quantified and evaluated using ImageJ software
(v146; National Institutes of Health).
Immunofluorescence staining
As aforementioned, H9c2 (2×104
cells/well) were pre-treated with 50 µmol/l NaHS for 1 h prior to
cell stimulation with 5 µg/ml LPS for 24 h at 37°C. Cells were
fixed using 4% paraformaldehyde for 24 h at room temperature,
blocked with PBS containing 1% BSA (cat. no. A8020; Beijing
Solarbio Science & Technology Co., Ltd.) for 1 h at room
temperature, followed by overnight incubation at 4°C with primary
antibodies BECN1 (1:1,000; cat. no. ab210498; Abcam) and SLC7A11
(1:1,000; cat. no. T57046; Abmart Pharmaceutical Technology Co.,
Ltd.). The next day, cultures were gently washed three times and
incubated for 50 min at room temperature with a Cy3 conjugated Goat
Anti-Rabbit IgG (H+L) (1:500; cat. no. GB21303; Wuhan Servicebio
Technology Co., Ltd.). The nuclei were counterstained with DAPI
(cat. no. G1012; Servicebio, Inc.) for 10 min at room temperature
and washed three times for 5 min each. The slides were gently
shaken before being sealed with Anti-Fluorescence Quenching Sealing
Reagent (cat. no. G1401; Servicebio, Inc.) for 10 min at room
temperature. Images were captured under a fluorescence microscope
(magnification, ×200) and evaluated using ImageJ software (v146;
National Institutes of Health). Finally, Pearson's correlation
coefficient and Mander's co localization coefficient were used to
evaluate the expression of fluorescence colocalization using Dunn
et al's scoring system (24).
BECN1 small interfering (si)RNA
transfection
BECN1 fragments and mutants were synthesized by
Shanghai GenePharma Co., Ltd. as follows: BECN1-siRNA,
5′-GCGGACAATTTGGCACGATCA-3′ and negative control (NC)-siRNA:
5′-TTCTCCGAACGTGTCACGT-3′. The Lentiviral plasmid
LV3(H1/GFP&Puro)-BECN1-Rat-1157 was obtained from Shanghai
GenePharma Co., Ltd. 293T cells (Shanghai GenePharma Co., Ltd.)
were co-transfected with the second-generation lentiviral plasmid
(10 µg) and packaging plasmids (pGag/Pol, pRev, PVSV-G), and mixed
according to the ratio of 2:1:1:1. Incubated at 37°C in 5%
CO2 incubator for 48 h. Lentivirus was produced and H9c2
cells were transduced for 48 h at 37°C with 5 µl/well BECN1-siRNA
(1 µg/µl)or NC-siRNA, the titer of BECN1-siRNA was determined to be
3×108 TU/ml, and the MOI for H9c2 infection was 20.
After lentiviral transduction and under the same conditions for 48
h. Then, 10,000 U/ml penicillin-streptomycin was used for
maintenance, 5 µg/ml puromycin was used to screen cells and protein
expression levels of BECN1 were assessed using western blotting.
Following stable transfection, H9c2 cells were treated with 50
µmol/l NaHS for 1 h at 37°C, then stimulated with 5 µg/ml LPS for
24 h at 37°C. H9c2 cells were divided into treatment groups as
follows: i) LPS; ii) BECN1-siRNA + LPS; iii) LPS + NaHS and iv)
BECN1-siRNA + LPS + NaHS.
Animal model
A total of 30 male Sprague-Dawley (SD) rats (age,
6–8 weeks; weight, 200±10 g) was purchased from the Animal
Experimentation Center of Xinjiang Medical University [animal
license no. SYXK (new) 2011-010101]. All SD rats were housed in the
Animal Experimentation Center of Shihezi University according to
the standards described by the National Institutes of Health
(25). All rats were housed under
standard conditions of 25°C, 60% relative humidity and 12/12-h
light/dark cycle with free access to standard laboratory food and
water. The present study was approved by the Institutional Ethics
Committee of The Medical Committee of The First Affiliated Hospital
of Shihezi University School of Medicine (approval no.
A2022-104-01).
Establishment of the SIC model and
model grouping
The septic myocardial injury model was established
using the previously reported CLP method (26). Briefly, rats were fasted for 12 h
before surgery without water restriction. Rats were anesthetized
using isoflurane inhalation (induction, 2.5%; maintenance, 1.0%).
The abdominal cavity was cut along the midline, the cecum was
ligated and the end of the cecum was perforated once with a
50-gauge needle; after squeezing out a small amount of feces, the
abdomen was closed. Following surgery, 1 ml/100 g saline was
administrated subcutaneously for resuscitation without diet
restriction. Rats in the sham group (n=10) underwent the same
surgical procedure without CLP. In the CLP + NaHS group (n=10), 8.9
µmol/kg NaHS was administered by intraperitoneal injection 1 h
after CLP (26). All rats in the
CLP group demonstrated symptoms such as increased respiratory and
heart rate (HR), listlessness, vertical hair, huddling, reduced
food intake and decreased activity following CLP modeling, which
was consistent with the previously reported rat sepsis model
(26,27). At 12 h after surgery, the cardiac
function of rats in each group was evaluated using
echocardiography. When rats were unable to move, slow to respond
and demonstrating symptoms such as diarrhea or urinary
incontinence, abdominal infection and suppuration or reached the
end of the experimental timeline, i.e. 12 h after the operation.
They were anesthetized with isoflurane (induction, 2.5%) and
euthanized by cervical dislocation. The death of rats was confirmed
by cardiac and respiratory arrest, muscle relaxation and lack of
reflex. The myocardial injury was assessed using hematoxylin and
eosin (H&E) staining. Changes in myocardial fiber
microstructure and mitochondria were evaluated using transmission
electron microscopy and myocardial tissue protein was extracted to
determine ferroptosis-associated protein expression using western
blotting.
Echocardiography
At 12 h after surgery, rats were anesthetized using
isoflurane (induction, 2.5%; maintenance, 1.0%) and the anterior
chest hair was shaved. Rates were placed in the left supine
position on the examination table, connected to an
electrocardiogram and GE Vivid E9 Ultrasound Machine with 12 sec
heart probe and probe frequency 12 Mhz was used to perform
echocardiography. HR and left ventricular end-diastolic diameter
(LVEDD) were measured under the M-mode ultrasound module and LV
ejection fraction (LVEF%) and shortening fraction (LVFS%) were
calculated. At least three cardiac cycles were recorded for each
rat to reflect the diastolic and systolic functions of the
heart.
Histopathological examination
Myocardial tissue specimens from sham, CLP and CLP +
NaHS rats were fixed in 4% paraformaldehyde for 24 h at room
temperature, paraffin-embedded and cut into 4 µm sections. The
tissue sections were stained with H&E for 24 h at room
temperature. Subsequently, the tissues were dehydrated in different
concentrations of ethanol, embedded in paraffin and sliced into
4-µm-thick sections. The tissue sections were then stained with
hematoxylin (0.4%) and eosin (0.1%) (H&E) solution at 37°C for
5 min and the pathological changes after myocardial injury were
observed using a light microscope. Finally, the extent of
Pathological score of myocardial injury was evaluated using the
Kishimoto scoring system (28).
Transmission electron microscopy
LV myocardium samples (2×2×2 mm) were collected and
fixed in 4% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M
phosphate buffer for 2 h at room temperature. Tissue samples were
embedded in 1% agarose, the samples were successively dehydrated
with 30–50–70–80–95–100–100% alcohol for 20 min each time, and 100%
acetone twice for 15 min each time at room temperature and cut into
ultrathin sections (70 nm) using an ultramicrotome. Sections were
stained using 2% uranyl acetate in pure ethanol for 15 min,
followed by 2.6% lead citrate for 15 min at room temperature. At
least five images were taken per sample. using a transmission
electron microscope (magnification ×10,000) at 80 kV. The extent of
mitochondrial damage was evaluated using the Flameng scoring system
(29).
Statistical analysis
SPSS 26.0 (IBM Corp.) statistical software was used
to analyze the data. Data are presented as the mean ± standard
deviation. Paired student's t test was used to compare the means of
two samples; ≥3 samples were compared using one-way ANOVA with
complete randomization and post hoc Bonferroni's correction.
P<0.05 was considered to indicate a statistically significant
difference. Each experiment was performed three times.
Results
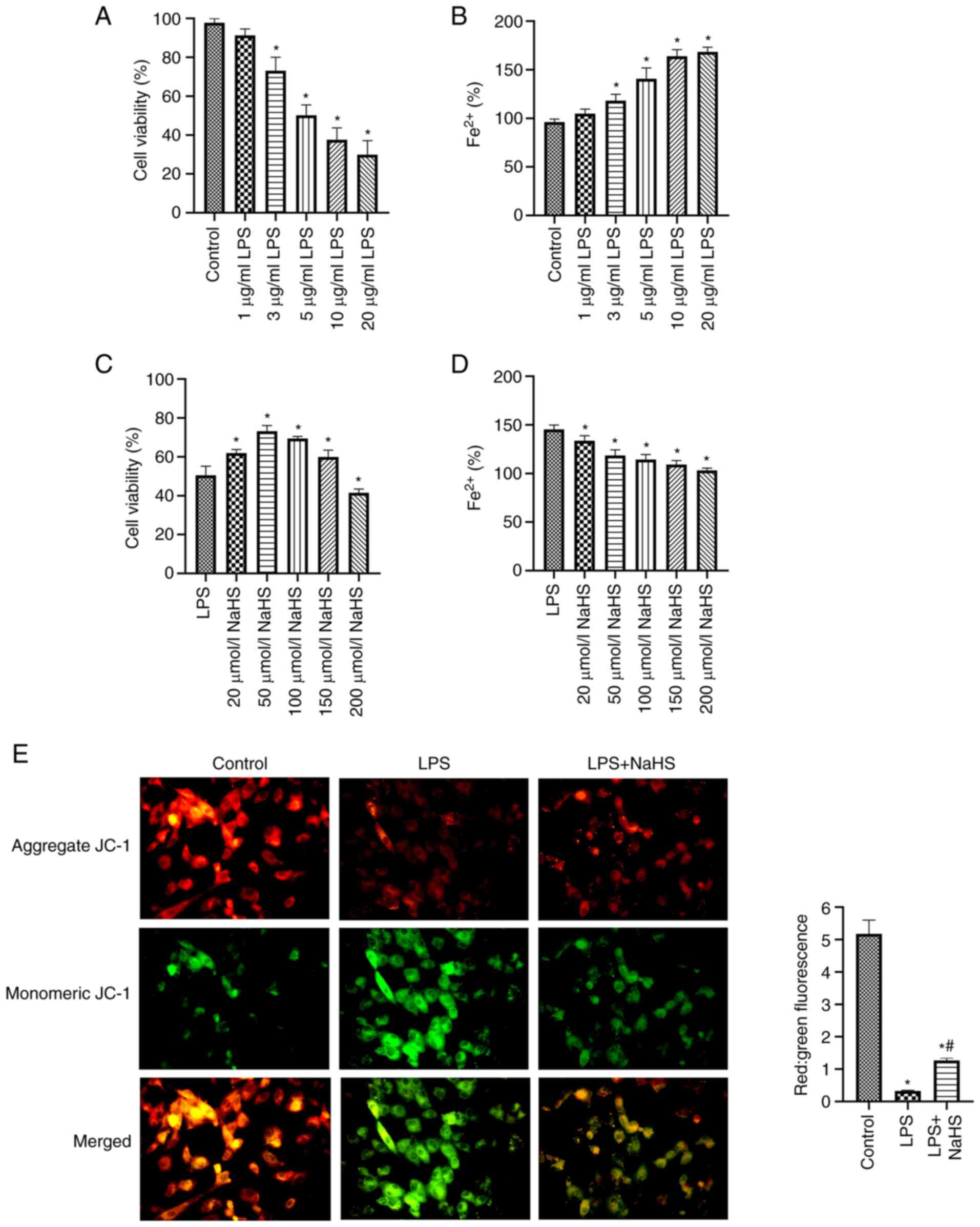
NaHS attenuates sepsis-induced
cardiomyocyte injury and impaired iron metabolism
The effect of LPS and the H2S donor NaHS
on cell viability were assessed to determine the appropriate dose.
When LPS concentration increased, H9c2 cell viability significantly
decreased and Fe2+ concentration significantly increased
compared with the control (both P<0.05; Fig. 1A and B). When treated with 5 µg/ml
LPS, the mean activity of the cell was 50.22% and the concentration
of Fe2+ released was 140.69% that of the control. These
results demonstrated that a SIC model was created using 5 µg/ml
LPS. In the LPS-induced myocardial injury model of sepsis in H9c2
cells, NaHS significantly improved the LPS-induced decrease in cell
viability for concentrations ≤150 µmol/ml and significantly
decreased intracellular Fe2+ concentration compared with
the LPS group (both P<0.05; Fig.
1C and D). These data demonstrated that NaHS treatment improved
cell viability, decreased cell injury and regulated impaired iron
metabolism. However, its effects were independent of concentration.
Cell viability was decreased when NaHS concentration was ≥100
µmol/l. NaHS at 50 µmol/l produced the greatest treatment effect,
therefore it was used in subsequent tests.
JC-1 staining demonstrated that mitochondrial JC-1
polymer (red fluorescence) decreased and JC-1 monomer (green
fluorescence) increased in the LPS group, with the red:green ratio
significantly decreased compared with the control (P<0.05;
Fig. 1E). This demonstrated that
the mitochondrial membrane potential was decreased, which suggested
increased cell damage. Treatment with NaHS significantly reversed
this phenomenon (P<0.05) compared with the LPS group and
demonstrated increased mitochondrial membrane potential level,
which suggested that cellular damage was alleviated. These data
indicate that NaHS improves cardiomyocyte viability and alleviates
LPS induced iron metabolism disorder and mitochondrial membrane
potential abnormality.
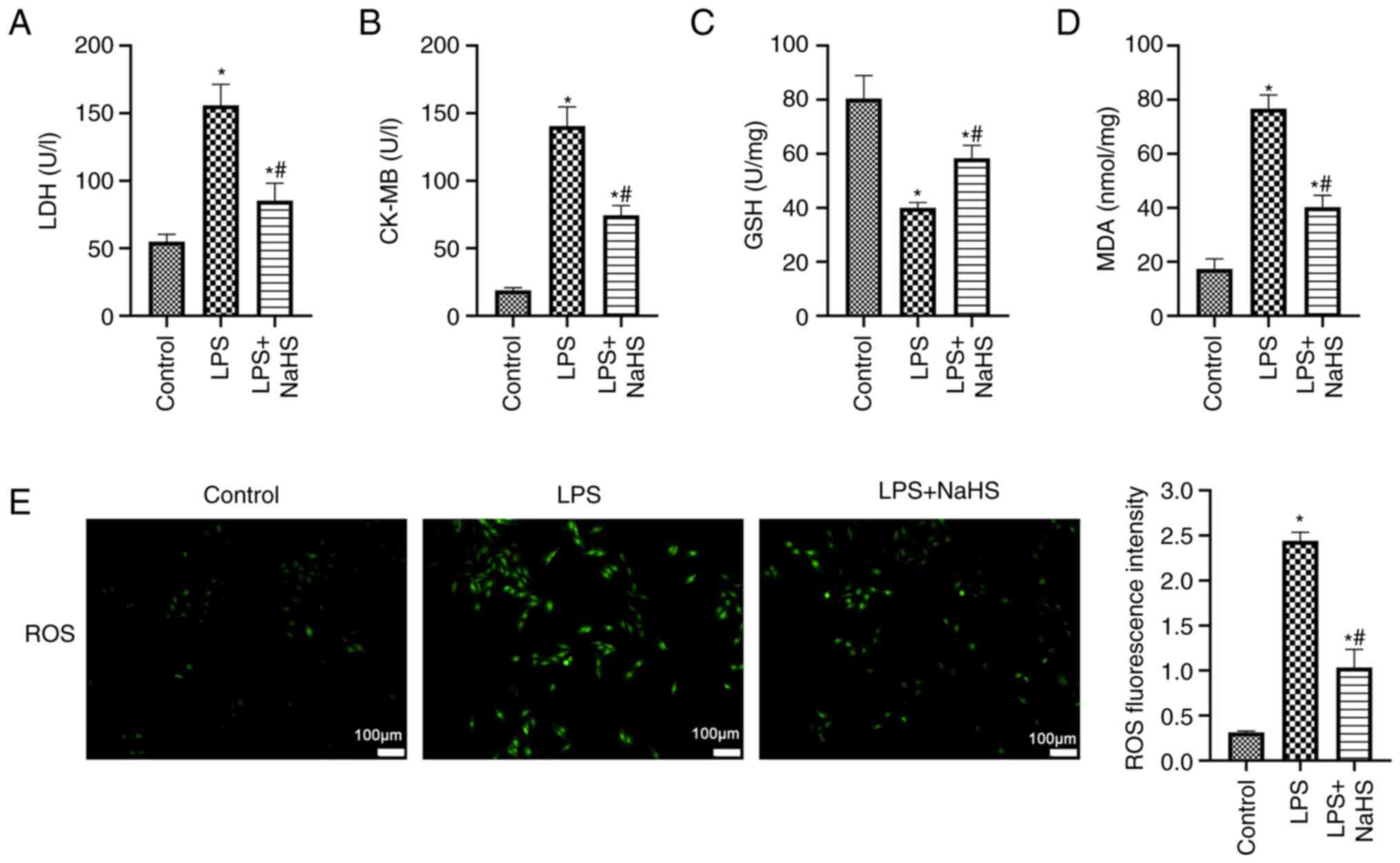
NaHS decreases sepsis-induced
oxidative stress and lipid peroxidation in cardiomyocytes
Significantly elevated release of cardiac enzymes
LDH and CK-MB from H9c2 cells was observed in the LPS group
compared with the control (P<0.05; Fig. 2A and B), which suggested increased
cardiomyocyte injury. NaHS intervention significantly decreased
levels of LDH and CK-MB compared with the LPS group (P<0.05),
which suggested NaHS attenuated cell injury. Following LPS
stimulation, the antioxidant GSH activity of H9c2 cells was
significantly decreased compared (Fig. 2C), whereas the lipid peroxide MDA
content significantly increased compared with the control (both
P<0.05; Fig. 2D). Furthermore,
green fluorescence (indicating ROS) was significantly enhanced in
the LPS group compared with the control (P<0.05; Fig. 2E), which suggested an imbalance
between oxidative and antioxidant effects in cardiomyocytes in
response to LPS stimulation and oxidative stress. Following NaHS
treatment, GSH activity significantly increased, MDA concentration
significantly decreased and ROS fluorescence intensity
significantly decreased compared with the LPS group (both
P<0.05; Fig. 2C, D and E).
These data suggested that NaHS enhanced intracellular antioxidant
capacity and mitigated oxidative stress and lipid peroxidation
caused by LPS in cardiomyocytes.
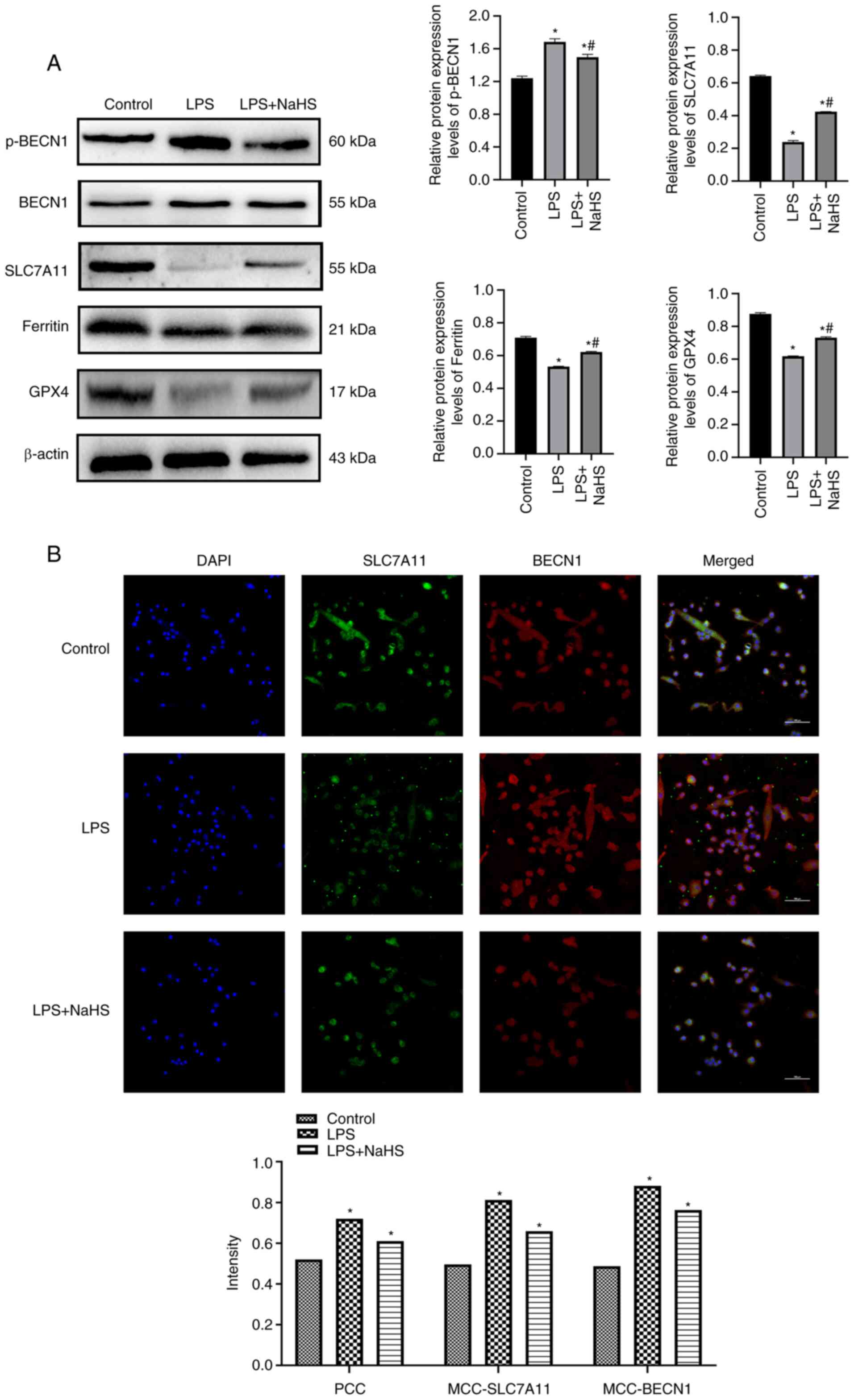
NaHS inhibits sepsis-induced
ferroptosis in cardiomyocytes
Expression levels of the ferroptosis regulatory
proteins SLC7A11 and GPX4 were significantly lower, whereas p-BECN1
expression was significantly higher, in the LPS group compared with
the control (P<0.05; Fig. 3A).
NaHS treatment significantly increased SLC7A11 and GPX4 protein
expression levels, whereas p-BECN1 protein expression levels were
significantly decreased compared with the LPS group (P<0.05).
Immunofluorescence demonstrated that co-localization
characterization parameters, such as Pearson's correlation
coefficient and Mander's co-localization coefficient, were
significantly elevated following LPS stimulation (P<0.05;
Fig. 3B). BECN1 co-localization
with SLC7A11, a key component of system xc−,
compared with the control group, the co-localization of BECN1 and
SLC7A11 was enhanced after LPS intervention. However, NaHS markedly
reversed this effect compared with the LPS group (P<0.05). These
results suggest that sodium thiohydride alleviates LPS induced
ferroptosis in cardiomyocytes by inhibiting the interaction of
BECN1 with SLC7A11.
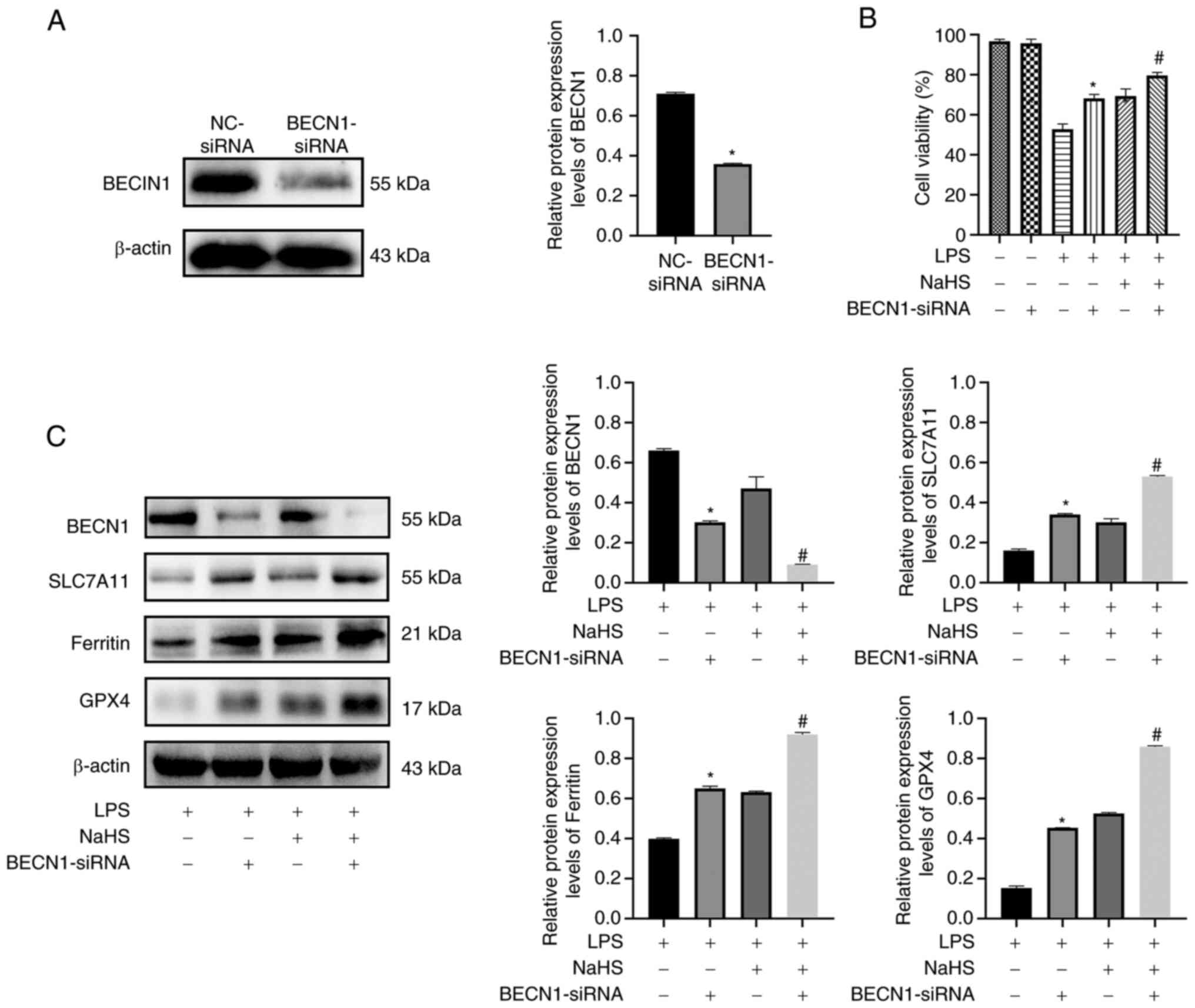
Inhibition of BECN1 expression by NaHS
attenuates ferroptosis in cardiomyocytes
The aforementioned data suggested that NaHS may
decrease LPS-induced ferroptosis in H9c2 cardiomyocytes via the
BECN1/SLC7A11 signaling pathway. To evaluate the association
between the BECN1 signaling pathway and ferroptosis, rat H9c2
cardiomyocytes were transfected with siRNA to silence BECN1. The
transfected cells were treated with 5 µg/ml LPS and 50 µmol/l NaHS
alone or in combination for 24 h. The protein expression levels of
BECN1 were significantly reduced in BECN-siRNA compared with
NC-siRNA (P<0.05; Fig. 4A).
Therefore, GPX4 protein expression reflects the occurrence of
ferroptosis. GPX4 and SLC7A11 protein expression levels were
significantly higher and BECN1 protein expression level was
decreased in the BECN1-siRNA + LPS group compared with the LPS
group (P<0.05; Fig. 4C).
Furthermore, ferritin, GPX4 and SLC7A11 protein expression levels
were significantly increased and BECN1 protein expression level was
significantly decreased in the BECN1-siRNA + LPS + NaHS group
compared with the LPS + NaHS group (P<0.05; Fig. 4C). These results suggested that
inhibiting BECN1 boosted cells antioxidant capacity and decreased
LPS-induced ferroptosis sensitivity in H9c2 cells, which indicated
BECN1 may serve as a regulator of system xc−.
NaHS may protect against LPS-induced ferroptosis in H9c2 cells by
disrupting the BECN1/SLC7A11 signaling pathway.
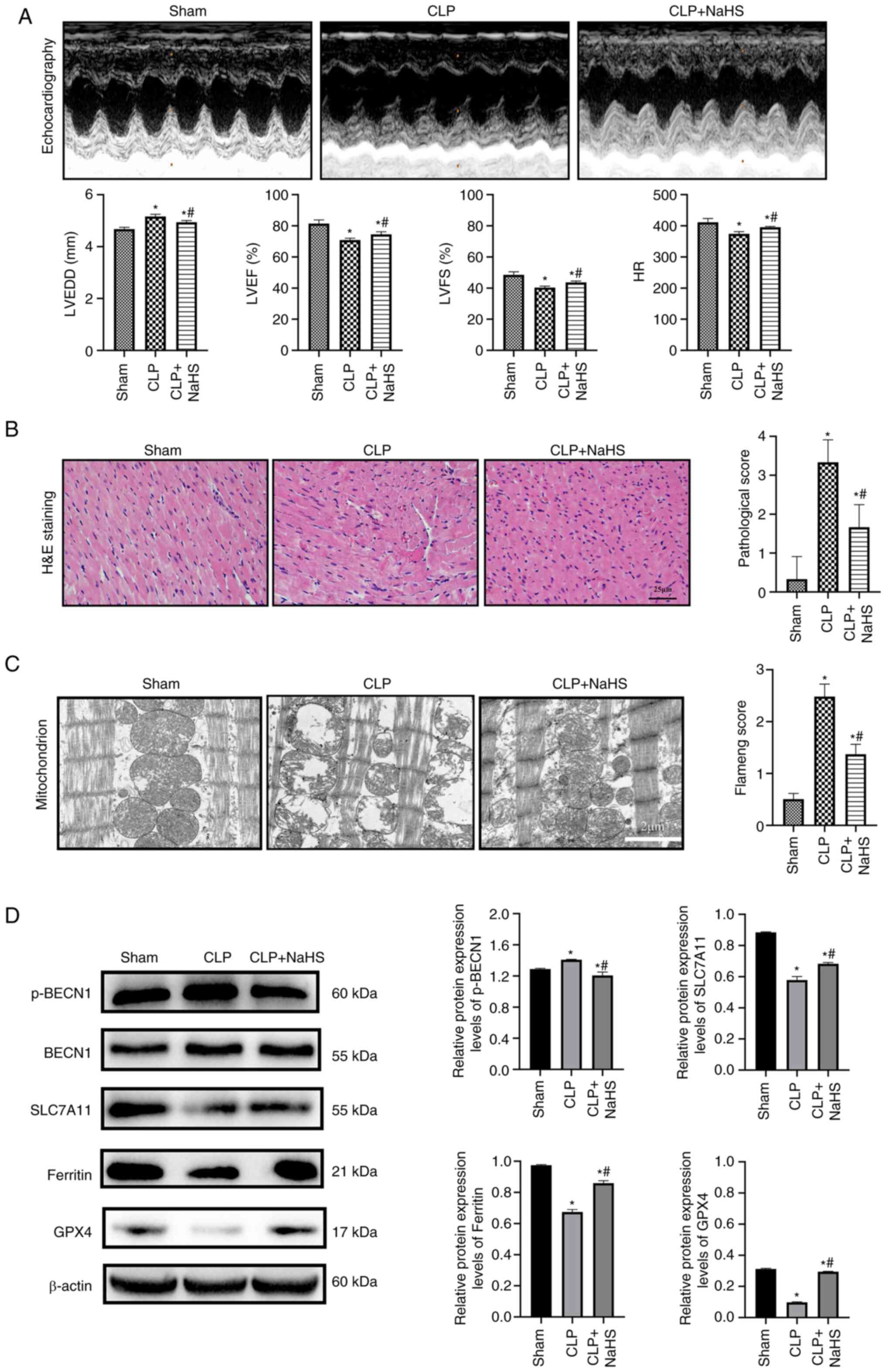
NaHS inhibits myocardial ferroptosis
and improves SIC
The effect of NaHS on ferroptosis in septic rat
heart tissue was assessed using a CLP-induced in vivo model
of septic myocardial injury. Compared with the sham group, the
LVEF%, LVFS% and HR significantly decreased and LVEED significantly
increased, and the systolic and diastolic functions of sepsis rats
were decreased in the CLP group (P<0.05; Fig. 5A). However, LVEF%, LVFS% and HR
significantly increased and LVEDD significantly decreased in the
CLP + NaHS group compared with the CLP group (P<0.05). There
were no pathological alterations visually apparent in the sham
group (Fig. 5B). However,
myocardial fibers in the CLP group were misaligned and partially
deteriorated compared with the sham group, demonstrating
inflammatory cell infiltration, interstitial edema, transverse
blurring and erythrocyte exudation, which resulted in significantly
higher pathological score (P=0.0017). Myocardial myofilaments in
the CLP + NaHS group were more structured than in the CLP group,
with no inflammatory infiltration or erythrocyte exudation,
Pathological score decreased (P=0.0285). The CLP group exhibited
disorganized mitochondrial arrangement, swelling, degeneration,
small size and increased density of mitochondria, decreased size
and breakage of cristae, partial dissolution and disappearance of
the outer membrane, which resulted in significantly increased
pathological score significantly higher (P=0.0009; Fig. 5C) when assessed using transmission
electron microscopy. The CLP + NaHS group demonstrated less
myocardial mitochondrial damage and more intact membrane compared
with the CLP group. However, mitochondrial cristae fractures and
swelling remained and the myocardial fibers were more precisely
aligned, Pathological score decreased (P=0.0023). Protein
expression levels of SLC7A11, GPX4, BECN1 and p-BECN1 were
semi-quantified using western blotting in cardiac tissue from rats
in each treatment group (Fig.
5D). SLC7A11, GPX4 and ferritin protein expression levels were
significantly lowered and p-BECN1 protein expression levels were
significantly elevated in the CLP group compared with the sham
(P<0.05), which demonstrated activation of ferroptosis. However,
NaHS significantly promoted GPX4, SLC7A11 and ferritin protein
expression levels and significantly decreased p-BECN1 protein
expression levels compared with the CLP group (P<0.05). These
results suggested that NaHS-mediated upregulation of SLC7A11 and
GPX4 and downregulation of p-BECN1 protected against ferroptosis
during SIC.
Discussion
Sepsis is a global health concern that continues to
be the leading cause of infection-associated death (30). A recent study reported that septic
myocardial injury is associated with increased risk of short- and
long-term death from septic shock and that this risk increases with
cardiac dysfunction (31). LPS is
an integral component of gram-negative bacteria outer membranes
that induces systemic inflammatory reactions, sepsis, infectious
shock and multi-organ failure by infiltrating the lymphatic and
circulatory systems (32).
Myocardial cell injury is the most commonly utilized model of SIC
(33). H2S has been
reported in substantial basic research to be involved in
controlling homeostasis, cardiac contraction, anti-inflammatory,
pro-apoptotic and other pathological functions (34–38).
NaHS is a conventional and stable H2S
donor with anti-inflammatory, anti-oxidative stress, anti-apoptotic
and autophagy-regulating properties (39). It has emerged as a target for
sepsis therapeutic research due to its reported regulation of
AMPK/mTOR and PI3K/Akt/mTOR pathways to decrease sepsis multi-organ
injury (27,40). To the best of our knowledge,
however, the role of NaHS in SIC and its mechanisms have not been
elucidated. To study the protective effect of NaHS on SIC, LPS was
used in the present study to induce rat H9c2 cardiomyocytes to
produce an in vitro model of sepsis myocardial injury; a
CLP-induced in vivo model of sepsis rat myocardial injury
was also used. The present study demonstrated that 50 µmol/l NaHS
decreased the decrease in cell viability caused by LPS stimulation
and significantly decreased release of cardiac enzymes compared,
thereby minimizing myocardial cell injury in vitro. In
vivo assessments demonstrated that NaHS pretreatment improved
cardiac systolic and diastolic dysfunction in septic rats and
decreased cardiac tissue degradation, edema and release of
inflammatory cells following CLP surgery, which significantly
improved myocardial histopathology scores.
Ferroptosis is a new type of cell death in which
iron-dependent lipid peroxidation co-exists with oxidative stress
(41). GSH, the primary
non-enzymatic antioxidant in cells, is a condensed form of
glutamate, cysteine and glycine with direct antioxidant effects and
serves as a synthetic substrate for GPX4 (42,43). ROS are created by normal cellular
physiological processes and are key for biological signaling and
cellular balance (44,45). MDA activity, a byproduct of lipid
peroxidation, represents the severity of oxygen radical damage to
body tissue and is also associated with ferroptosis (46). Oxidative stress and lipid
peroxidation are important in the pathophysiology of myocardial
injury and ferroptosis has recently been reported to be a key
element in SIC, with suppression of LPS-induced ferroptosis in
cardiac tissue alleviating SIC (41). H2S decreases
ferroptosis in acute lung injury by raising protein expression of
GPX4 and SLC7A11 in rat lung tissue following CLP surgery and
prevented autophagy activation in acute lung injury via blocking
the mTOR signaling pathway (47).
The involvement of NaHS in CLP-induced myocardial ferroptosis is
uncertain. The present study demonstrated that MDA levels rose
significantly following LPS stimulation compared with the control,
which suggested that lipid peroxidation was involved in this model
of cardiac cell injury. In combination with significantly decreased
GSH activity and GPX4 and SLC7A11 protein expression levels
following LPS treatment, these data suggested that
ferroptosis-induced oxidative stress and lipid peroxidation were
involved in the development of SIC. When treated with NaHS, the
levels of antioxidant marker GSH increased and the lipid
peroxidation product MDA was significantly decreased compared with
the LPS group. These data demonstrated that NaHS protected against
septic myocardial damage by decreasing oxidative stress and lipid
peroxidation in the heart.
Ferroptosis is an iron-dependent form of programmed
cell death characterized by lipid peroxidation and morphological
alteration (48). Ferritin, an
iron storage protein, is key in iron metabolism because it stores
and releases iron to maintain iron homeostasis in the body. Iron
homeostasis has been reported to be critical for normal heart
function (49). Iron shortage and
overload are linked to cardiomyopathy and heart failure via SLC7A11
pathway (50). When intracellular
Fe2+ levels rise, the body produces large amounts of
hydroxyl radicals and ROS, both of which are harmful to cells, via
the Fenton reaction, worsening the occurrence of ferroptosis
(51). LPS induces ferroptosis by
activating cardiomyocyte nuclear receptor coactivator 4 (NCOA4),
which increases its expression. NCOA4 interacts directly with
ferritin to degrade it, releasing large amounts of Fe2+
and inducing excessive mitochondrial ROS production (52). Aberrant changes in mitochondrial
membrane potential are not only key signs of mitochondrial damage
and ferroptosis initiation but also an early warning of the
occurrence of ferroptosis (53).
The present study demonstrated a significant increase in
Fe2+ content and significantly decreased mitochondrial
membrane potential and ferritin protein expression levels in cells
following LPS stimulation compared with the control. Following CLP,
transmission electron microscopy of cardiac tissue demonstrated
disrupted mitochondrial organization, swelling, degeneration of
mitochondria and decreased and fragmented cristae. The present
study demonstrated that NaHS administration significantly improved
iron metabolism in myocardial cells and tissue compared with the
LPS group, which suggested that NaHS decreased ferroptosis in
septic myocardial injury.
BECN1 is a key regulator of autophagy and has been
reported in the pathophysiology of diseases such as metabolic
disorders and tumors (54,55).
In sepsis, cardiac dysfunction is a key cause of multi-organ
dysfunction and BECN1-dependent autophagy has been reported to
protect the heart (56,57). However, the regulatory mechanisms
governing BECN1 and autophagy remain unclear. Song et al
(58) reported a potential
mechanism by which BECN1 may enhance ferroptosis in cancer cells
via modulation of system xc− activity,
thereby enhancing GSH depletion and lipid peroxidation. This
mechanism is dependent on formation of the BECN1-SLC7A11 complex
(58). BECN1 regulates
ferroptosis independently of autophagy, as reported by previous
studies (59,60). Liu et al (60) reported that BECN1 overexpression
aggravates isoflurane-induced cell damage via upregulation of
ferroptosis. This effect is significantly attenuated by silencing
of BECN1 in SH-SY5Y cells. Liu et al suggested that BECN1
may regulate ferroptosis via inhibition of the glutamate exchange
activity of system xc−, which is involved in
isoflurane-induced toxicity (60). These results are similar to the
present study, which demonstrated that LPS significantly increased
the phosphorylation of BECN1 while significantly decreasing the
protein expression levels of SLC7A11, a core component of system
xc− compared with the control. Furthermore,
immunofluorescence staining demonstrated significantly enhanced
co-localization of BECN1 and SLC7A11, which form a BECN1-SLC7A11
complex, decreasing system xc− activity and
thus regulating ferroptosis (58). BECN1 knockdown decreased
LPS-induced cell damage, which in turn prevented ferroptosis. When
cells were treated with NaHS, a significant decrease in p-BECN1 and
significant increase in SLC7A11 protein expression levels were
observed compared with the LPS group. Furthermore, inhibiting BECN1
expression markedly diminished the protective effect of NaHS on
H9c2 cells. It could therefore be hypothesized that BECN1 acts as a
key signaling component in the transition between autophagy and
ferroptosis in septic cardiac damage. However, there is a close
relationship between ferroptosis and autophagy, BECN1 knockout mice
were not used in animal experiments, the precise chemical mechanism
of this action requires further study. Evaluation of the use of
NaHS in sepsis multi-organ dysfunction based on autophagy and
ferroptosis requires further study.
In conclusion, the present study demonstrated that
NaHS alleviated SIC via modulation of the BECN1 signaling pathway,
decreased oxidative stress and lipid peroxidation levels and
inhibition of ferroptosis. Ferroptosis is predicted to be a novel
therapeutic target for SIC and NaHS may be an effective treatment
for SIC.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the National
Natural Science Foundation of China (grant no. 81860336) and the
Xinjiang Production and Construction Corps Science and Technology
Tackling and Achievement Transformation Program Project (grant no.
2016AD003).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
QC, GC and YCZ conceived and designed the present
study and wrote and revised the manuscript. GC, YCZ and YHZ wrote
the manuscript and analyzed the data. GC, YHZ, LL and XL performed
cell experiments and collected data. YCZ, LG and YXZ reviewed the
literature and performed animal experiments. GC, YCZ, LG, XL and
YXZ analyzed and interpreted the data and produced the figures. All
authors have read and approved the final manuscript. QC and GC
confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Ethics Committee of The Medical Committee of The First Affiliated
Hospital of Shihezi University School of Medicine (approval no.
A2022-104-01).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Shankar-Hari M, Phillips GS, Levy ML,
Seymour CW, Liu VX, Deutschman CS, Angus DC, Rubenfeld GD and
Singer M; Sepsis Definitions Task Force, : Developing a new
definition and assessing new clinical criteria for septic shock:
For the third international consensus definitions for sepsis and
septic shock (sepsis-3). JAMA. 315:775–787. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Evans L, Rhodes A, Alhazzani W, Antonelli
M, Coopersmith CM, French C, Machado FR, Mcintyre L, Ostermann M,
Prescott HC, et al: Surviving sepsis campaign: International
guidelines for management of sepsis and septic shock 2021.
Intensive Care Med. 47:1181–1247. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Beesley SJ, Weber G, Sarge T, Nikravan S,
Grissom CK, Lanspa MJ, Shahul S and Brown SM: Septic
cardiomyopathy. Crit Care Med. 46:625–634. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hollenberg SM and Singer M:
Pathophysiology of sepsis-induced cardiomyopathy. Nat Rev Cardiol.
18:424–434. 2021. View Article : Google Scholar
|
|
5
|
Fang X, Wang H, Han D, Xie E, Yang X, Wei
J, Gu S, Gao F, Zhu N, Yin X, et al: Ferroptosis as a target for
protection against cardiomyopathy. Proc Natl Acad Sci USA.
116:2672–2680. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2021. View Article : Google Scholar
|
|
7
|
Koppula P, Zhuang L and Gan B: Cystine
transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient
dependency, and cancer therapy. Protein Cell. 12:599–620. 2021.
View Article : Google Scholar
|
|
8
|
Zhang X, Zheng C, Gao Z, Chen H, Li K,
Wang L, Zheng Y, Li C, Zhang H, Gong M, et al: SLC7A11/xCT prevents
cardiac hypertrophy by inhibiting ferroptosis. Cardiovasc Drugs
Ther. 36:437–447. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zheng J and Conrad M: The metabolic
underpinnings of ferroptosis. Cell Metab. 32:920–937. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ge ZD, Lian Q, Mao X and Xia Z: Current
Status and challenges of NRF2 as a potential therapeutic target for
diabetic cardiomyopathy. Int Heart J. 60:512–520. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang H, Wang Z, Liu Z, Du K and Lu X:
Protective effects of dexazoxane on rat ferroptosis in
doxorubicin-induced cardiomyopathy through regulating HMGB1. Front
Cardiovasc Med. 8:6854342021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Khamseekaew J, Kumfu S, Wongjaikam S,
Kerdphoo S, Jaiwongkam T, Srichairatanakool S, Fucharoen S,
Chattipakorn SC and Chattipakorn N: Effects of iron overload, an
iron chelator and a T-type calcium channel blocker on cardiac
mitochondrial biogenesis and mitochondrial dynamics in thalassemic
mice. Eur J Pharmacol. 799:118–127. 2017. View Article : Google Scholar
|
|
13
|
Liu P, Feng Y, Li H, Chen X, Wang G, Xu S,
Li Y and Zhao L: Ferrostatin-1 alleviates
lipopolysaccharide-induced acute lung injury via inhibiting
ferroptosis. Cell Mol Biol Lett. 25:102020. View Article : Google Scholar
|
|
14
|
Belavgeni A, Meyer C, Stumpf J, Hugo C and
Linkermann A: Ferroptosis and necroptosis in the kidney. Cell Chem
Biol. 27:448–462. 2020. View Article : Google Scholar
|
|
15
|
Kimura H, Nagai Y, Umemura K and Kimura Y:
Physiological roles of hydrogen sulfide: Synaptic modulation,
neuroprotection, and smooth muscle relaxation. Antioxid Redox
Signal. 7:795–803. 2005. View Article : Google Scholar
|
|
16
|
Guo W, Cheng ZY and Zhu YZ: Hydrogen
sulfide and translational medicine. Acta Pharmacol Sin.
34:1284–1291. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bhatia M and Gaddam RR: Hydrogen sulfide
in inflammation: A novel mediator and therapeutic target. Antioxid
Redox Signal. 34:1368–1377. 2021. View Article : Google Scholar
|
|
18
|
Yang R, Teng X, Li H, Xue HM, Guo Q, Xiao
L and Wu YM: Hydrogen sulfide improves vascular calcification in
rats by inhibiting endoplasmic reticulum stress. Oxid Med Cell
Longev. 2016:90952422016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sen N, Paul BD, Gadalla MM, Mustafa AK,
Sen T, Xu R, Kim S and Snyder SH: Hydrogen sulfide-linked
sulfhydration of NF-κB mediates its antiapoptotic actions. Mol
Cell. 45:13–24. 2012. View Article : Google Scholar
|
|
20
|
Gotor C, García I, Crespo JL and Romero
LC: Sulfide as a signaling molecule in autophagy. Autophagy.
9:609–611. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen YH, Teng X, Hu ZJ, Tian DY, Jin S and
Wu YM: Hydrogen sulfide attenuated sepsis-induced myocardial
dysfunction through TLR4 pathway and endoplasmic reticulum stress.
Front Physiol. 12:6536012021. View Article : Google Scholar
|
|
22
|
Liang D and Huang A, Jin Y, Lin M, Xia X,
Chen X and Huang A: Protective effects of exogenous NaHS against
sepsis-induced myocardial mitochondrial injury by enhancing the
PGC-1α/NRF2 pathway and mitochondrial biosynthesis in mice. Am J
Transl Res. 10:1422–1430. 2018.PubMed/NCBI
|
|
23
|
Li T, Zhao J, Miao S, Chen Y, Xu Y and Liu
Y: Protective effect of H2S on LPS-induced AKI by
promoting autophagy. Mol Med Rep. 25:962022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dunn KW, Kamocka MM and McDonald JH: A
practical guide to evaluating colocalization in biological
microscopy. Am J Physiol Cell Physiol. 300:C723–C742. 2011.
View Article : Google Scholar
|
|
25
|
Hollands C: The animals (scientific
procedures) Act 1986. Lancet. 2:32–33. 1986. View Article : Google Scholar
|
|
26
|
Li X, Cheng Q, Li J, He Y, Tian P and Xu
C: Significance of hydrogen sulfide in sepsis-induced myocardial
injury in rats. Exp Ther Med. 14:2153–2161. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhao Y and Cheng Q: Exogenous
H2S protects against septic cardiomyopathy by inhibiting
autophagy through the AMPK/mTOR pathway. Contrast Media Mol
Imaging. 2022:84640822022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kishimoto C, Kawamata H, Sakai S,
Shinohara H and Ochiai H: Enhanced production of macrophage
inflammatory protein 2 (MIP-2) by in vitro and in vivo infections
with encephalomyocarditis virus and modulation of myocarditis with
an antibody against MIP-2. J Virol. 75:1294–1300. 2001. View Article : Google Scholar
|
|
29
|
Flameng W, Borgers M, Daenen W and
Stalpaert G: Ultrastructural and cytochemical correlates of
myocardial protection by cardiac hypothermia in man. J Thorac
Cardiovasc Surg. 79:413–424. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Baghela A, Pena OM, Lee AH, Baquir B,
Falsafi R, An A, Farmer SW, Hurlburt A, Mondragon-Cardona A, Rivera
JD, et al: Predicting sepsis severity at first clinical
presentation: The role of endotypes and mechanistic signatures.
EBioMedicine. 75:1037762022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Vallabhajosyula S, Shankar A, Vojjini R,
Cheungpasitporn W, Sundaragiri PR, DuBrock HM, Sekiguchi H, Frantz
RP, Cajigas HR, Kane GC and Oh JK: Impact of right ventricular
dysfunction on short-term and long-term mortality in sepsis: A
meta-analysis of 1,373 patients. Chest. 159:2254–2263. 2021.
View Article : Google Scholar
|
|
32
|
Dickson K and Lehmann C: Inflammatory
response to different toxins in experimental sepsis models. Int J
Mol Sci. 20:43412019. View Article : Google Scholar
|
|
33
|
Hao R and Su G, Sun X, Kong X, Zhu C and
Su G: Adiponectin attenuates lipopolysaccharide-induced cell injury
of H9c2 cells by regulating AMPK pathway. Acta Biochim Biophys Sin
(Shanghai). 51:168–177. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang Y, Wang S, Xin Y, Zhang J, Wang S,
Yang Z and Liu C: Hydrogen sulfide alleviates the anxiety-like and
depressive-like behaviors of type 1 diabetic mice via inhibiting
inflammation and ferroptosis. Life Sci. 278:1195512021. View Article : Google Scholar
|
|
35
|
Kabil O and Banerjee R: Enzymology of H2S
biogenesis, decay and signaling. Antioxid Redox Signal. 20:770–782.
2014. View Article : Google Scholar
|
|
36
|
Zhang Z, Jin S, Teng X, Duan X, Chen Y and
Wu Y: Hydrogen sulfide attenuates cardiac injury in takotsubo
cardiomyopathy by alleviating oxidative stress. Nitric Oxide.
67:10–25. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wallace JL, Blackler RW, Chan MV, Da Silva
GJ, Elsheikh W, Flannigan KL, Gamaniek I, Manko A, Wang L, Motta JP
and Buret AG: Anti-inflammatory and cytoprotective actions of
hydrogen sulfide: Translation to therapeutics. Antioxid Redox
Signal. 22:398–410. 2015. View Article : Google Scholar
|
|
38
|
Zhang S, Yang G, Guan W, Li B, Feng X and
Fan H: Autophagy plays a protective role in sodium
hydrosulfide-induced acute lung injury by attenuating oxidative
stress and inflammation in rats. Chem Res Toxicol. 34:857–864.
2021. View Article : Google Scholar
|
|
39
|
Chen L, Liu P, Feng X and Ma C:
Salidroside suppressing LPS-induced myocardial injury by inhibiting
ROS-mediated PI3K/Akt/mTOR pathway in vitro and in vivo. J Cell Mol
Med. 21:3178–3189. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang C, Yuan W, Hu A, Lin J, Xia Z, Yang
CF, Li Y and Zhang Z: Dexmedetomidine alleviated sepsis-induced
myocardial ferroptosis and septic heart injury. Mol Med Rep.
22:175–184. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ursini F and Maiorino M: Lipid
peroxidation and ferroptosis: The role of GSH and GPX4. Free Radic
Biol Med. 152:175–185. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sun L, Dong H, Zhang W, Wang N, Ni N, Bai
X and Liu N: Lipid peroxidation, GSH depletion, and SLC7A11
inhibition are common causes of EMT and ferroptosis in A549 cells,
but different in specific mechanisms. DNA Cell Biol. 40:172–183.
2021. View Article : Google Scholar
|
|
43
|
Park E and Chung SW: ROS-mediated
autophagy increases intracellular iron levels and ferroptosis by
ferritin and transferrin receptor regulation. Cell Death Dis.
10:8222019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Su LJ, Zhang JH, Gomez H, Murugan R, Hong
X, Xu D, Jiang F and Peng ZY: Reactive oxygen species-induced lipid
peroxidation in apoptosis, autophagy, and ferroptosis. Oxid Med
Cell Longev. 2019:50808432019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Imai H, Matsuoka M, Kumagai T, Sakamoto T
and Koumura T: Lipid peroxidation-dependent cell death regulated by
GPx4 and ferroptosis. Curr Top Microbiol Immunol. 403:143–170.
2017.
|
|
46
|
Li J, Li M, Li L, Ma J, Yao C and Yao S:
Hydrogen sulfide attenuates ferroptosis and stimulates autophagy by
blocking mTOR signaling in sepsis-induced acute lung injury. Mol
Immunol. 141:318–327. 2022. View Article : Google Scholar
|
|
47
|
Liang D, Minikes AM and Jiang X:
Ferroptosis at the intersection of lipid metabolism and cellular
signaling. Mol Cell. 82:2215–2227. 2022. View Article : Google Scholar
|
|
48
|
Gao M, Yi J, Zhu J, Yi J, Zhu J, Minikes
AM, Monian P, Thompson CB and Jiang X: Role of mitochondria in
ferroptosis. Mol Cell. 73:354–363.e3. 2019. View Article : Google Scholar
|
|
49
|
Fang X, Cai Z, Wang H, Han D, Cheng Q,
Zhang P, Gao F, Yu Y, Song Z, Wu Q, et al: Loss of cardiac ferritin
H facilitates cardiomyopathy via Slc7a11-mediated ferroptosis. Circ
Res. 127:486–501. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
He YJ, Liu XY, Xing L, Wan X, Chang X and
Jiang HL: Fenton reaction-independent ferroptosis therapy via
glutathione and iron redox couple sequentially triggered lipid
peroxide generator. Biomaterials. 241:1199112020. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Li N, Wang W, Zhou H, Wu Q, Duan M, Liu C,
Wu H, Deng W, Shen D and Tang Q: Ferritinophagy-mediated
ferroptosis is involved in sepsis-induced cardiac injury. Free
Radic Biol Med. 160:303–318. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Tang D, Chen X, Kang R and Kroemer G:
Ferroptosis: Molecular mechanisms and health implications. Cell
Res. 31:107–125. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kuramoto K and He C: The secretory
function of BECN1 in metabolic regulation. Autophagy. 17:3262–3263.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hu F, Li G, Huang C, Hou Z, Yang X, Luo X,
Feng Y, Wang G, Hu J and Cao Z: The autophagy-independent role of
BECN1 in colorectal cancer metastasis through regulating STAT3
signaling pathway activation. Cell Death Dis. 11:3042020.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lee S, Lee SJ, Coronata AA, Fredenburgh
LE, Chung SW, Perrella MA, Nakahira K, Ryter SW and Choi AM: Carbon
monoxide confers protection in sepsis by enhancing beclin
1-dependent autophagy and phagocytosis. Antioxid Redox Signal.
20:432–442. 2014. View Article : Google Scholar
|
|
56
|
Pi QZ, Wang XW, Jian ZL, Chen D, Zhang C
and Wu QC: Melatonin alleviates cardiac dysfunction via increasing
Sirt1-mediated beclin-1 deacetylation and autophagy during sepsis.
Inflammation. 44:1184–1193. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Song X, Zhu S, Chen P, Hou W, Wen Q, Liu
J, Xie Y, Liu J, Klionsky DJ, Kroemer G, et al: AMPK-mediated BECN1
phosphorylation promotes ferroptosis by directly blocking system
Xc− activity. Curr Biol. 28:2388–2399.e5.
2018. View Article : Google Scholar
|
|
58
|
Kang R, Zhu S, Zeh HJ, Klionsky DJ and
Tang D: BECN1 is a new driver of ferroptosis. Autophagy.
14:2173–2175. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Yin Z, Ding G, Chen X, Qin X, Xu H, Zeng
B, Ren J, Zheng Q and Wang S: Beclin1 haploinsufficiency rescues
low ambient temperature-induced cardiac remodeling and contractile
dysfunction through inhibition of ferroptosis and mitochondrial
injury. Metabolism. 113:1543972020. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Liu R, Li X and Zhao G: Beclin1-mediated
ferroptosis activation is associated with isoflurane-induced
toxicity in SH-SY5Y neuroblastoma cells. Acta Biochim Biophys Sin
(Shanghai). 51:1134–1141. 2019. View Article : Google Scholar : PubMed/NCBI
|