Dedifferentiation and reprogramming are common
physiological responses to injury or cell ablation across various
tissues in a wide range of organisms (13–15). Therefore, dedifferentiation and
reprogramming may be a general characteristic of terminally
differentiated cells after injury. However, this event appears to
only occur after environmental perturbations around the cell but
not under conditions of homeostasis, tissue damage being one of the
main causes of these environmental disturbances. Previous studies
have shown that during the regeneration of Ambystoma
mexicanum tissues, including the jaw, lens, retina, large
region of heart, limbs and tail, cell identity changes occurred
within a 100 µm radius of the tissue resection site (16). In addition, the dedifferentiation
pattern of GATA6-positive cells during wound repair in the
epidermis suggests that dedifferentiation is more likely to occur
in close proximity to the wound (17). Therefore, tissue damage most
likely exposes cells to new stimuli that lead to dedifferentiation
or reprogramming, or cells are relieved of inhibitory signals that
suppress any phenotypic changes, ultimately promoting
dedifferentiation and reprogramming. These signals involving
cellular autonomic and non-autonomic factors altogether constitute
a regulatory mechanism for cell identity changes after tissue
injury.
Dedifferentiation or reprogramming of committed
cells occurs only after disturbances in the surrounding environment
of the cells; however, this does not occur under homeostatic
conditions (18,19). This suggests that committed cells
are inhibited by signals under homeostatic conditions, rendering
them unable to change phenotypically. This maintains the
equilibrium between stem cells and their differentiated
counterparts. This balance allows each cell type to have a clearly
defined function. Previous studies have shown that signals that
maintain intercellular balance may come from stem cells themselves
(15,20–22).
It should be noted that this mixed population of
cells appears to exist under a hierarchical structure. In this
structure, stem cells serve as ‘the leader’, where under their
‘rule’, cells form a strict hierarchy that is difficult to break.
However, once the leader is lost, new members will fill the vacancy
to stabilize the normal order of this hierarchy. The loss of stem
cells caused by tissue injury is one of the key factors that
releases the inhibition of committed cells and triggers
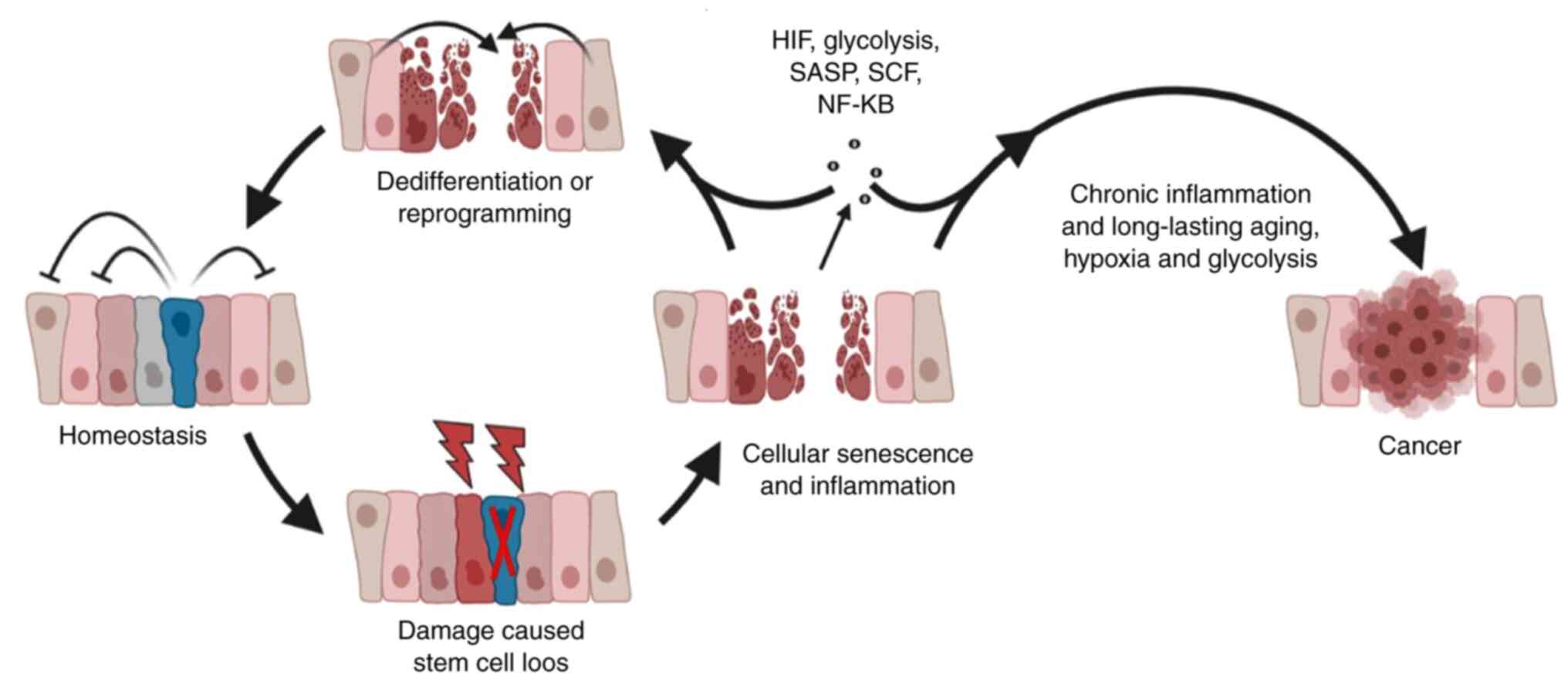
dedifferentiation or reprogramming (15,20–22) (Fig.
1).
After tissue damage, the loss of the epidermal
barrier can lead to a sudden influx of extracellular oxygen, which
is quickly consumed by metabolically active cells or converted into
reactive oxygen species (26). At
the same time, blood flow is interrupted due to vascular injury and
constriction, reducing oxygen delivery to the wound. At this time,
the wound site is in a state of local tissue hypoxia, where
hypoxia-inducible factor (HIF), which is normally degraded under
normoxic conditions, becomes stabilized in the hypoxic wound site
(27). Therefore, injury results
in the disturbance of the cell microenvironment at the wound site,
creating a hypoxic niche that embryonic stem cells (ESCs) and adult
stem cells rely on for self-renewal (28). Previous studies have shown that
hypoxic conditions can promote the self-renewal and maintenance of
pluripotency in embryonic and other types of stem cells (29–31). Under hypoxia, human ESCs control
HIF2α through glycolytic flux, thereby upregulating the expression
of C-terminal binding proteins 1 and 2 to sustain self-renewal
(29). In addition, HIF2α is
closely associated with the pluripotency regulatory network of
genes, such that HIF2α knockdown leads to the downregulation of
octamer-binding transcription factors 3 and 4 (OCT3/4), sex
determining region Y-box 2 (SOX2) and NANOG (30). In vitro reprogramming
experiments have also revealed that hypoxia can promote the
expression of pluripotent factors, such as OCT3, OCT4, SOX2, NANOG
and Krüppel-like factor 4 (KLF4), to increase the efficiency of
reprogramming, and can reduce the number of transcription factors
required for reprogramming (31).
Therefore, it is not surprising that hypoxia signaling serves a key
role in wound healing and tissue repair, largely by inducing cell
dedifferentiation or reprogramming at the wound site. In the
zebrafish model, myocardial hypoxia induced by ventricular
amputation has been shown to serve a positive role in myocardial
regeneration, whilst hyperoxic conditions or the overexpression of
HIF1α can strongly prevent the regeneration process after
ventricular amputation (32).
After culturing in vitro, it has been revealed that the
dedifferentiation of cardiomyocytes is significantly promoted under
hypoxic conditions compared with that under normoxia, whilst the
number of dedifferentiated cardiomyocytes is significantly
decreased following hyperoxic treatment (32). In addition, there is evidence that
hypoxia can induce the reprogramming of resident muscle cells
post-injury so that they exhibit the characteristics of pluripotent
cells known as injury-induced muscle-derived stem cells (33–35). A similar phenomenon has also been
observed in the brain tissues of patients with ischemic stroke
(36). It is noteworthy that
hypoxia has been found to induce dedifferentiation in a wide
variety of cell types, such as adipocytes, renal cells and
astrocytes (37–39).
A significant effect of hypoxia on cells is the
change to their metabolic state. Under hypoxia, the majority of
eukaryotic cells can switch their primary metabolic strategy from
mitochondrial respiration to glycolysis to maintain ATP levels
(40). Multiple previous studies
have shown that high levels of glycolysis can maintain the
self-renewal properties of stem cells (41–45). Mitochondrial function has been
found to be reduced in the inner cell mass (46), whereas ESCs obtained in
vitro also showed higher glycolytic rates (41). In addition, induced pluripotent
stem cells exhibit metabolic reprogramming from oxidative
phosphorylation to glycolysis (42). Similarly, in adult stem cells,
hematopoietic stem cells (HSCs) typically show more hypoxic states
with higher levels of HIF1α expression. These cells rely heavily on
anaerobic glycolysis and suppressed mitochondrial respiration,
which allows them to sustain their self-renewal characteristics
(43–45). Similar to HSCs, bone marrow stem
cells also rely on glycolysis for energy. When these cells are
transferred from hypoxic to normoxic conditions, their stem cell
properties become impaired and they differentiate (47,48). Metabolic conversion from oxidative
phosphorylation to aerobic glycolysis therefore likely serves a key
role in regeneration. Studies have previously shown that metabolic
transformation to glycolysis is an inevitable initial event during
blastema formation and tail regeneration in zebrafish after
amputation (49,50). During this process, cells undergo
epithelial-mesenchymal transition and dedifferentiation. Inhibiting
glycolysis leads to the failure of tail regeneration (49,50). In a zebrafish cardiac regeneration
model, glycolysis transfer has been found to be concentrated in the
vicinity of damaged tissues (51). Following treatment with
2-deoxy-D-glucose or when glycolysis is inhibited by affecting
pyruvate kinase M2 function, the dedifferentiation levels of
cardiomyocytes are decreased significantly (51). The importance of glycolytic
transfer on cell dedifferentiation after tissue damage is also
evident in mammals. Compared with normal mice, the Murphy Roths
Large (MRL) strain of mice exhibit higher glycolysis levels, which
enable MRL mice to retain the ability of forming blastema,
resulting in superior regenerative capabilities compared with
normal mice (52). A further
study has demonstrated that the HIF-1α pathway is key in
understanding the regenerative abilities of MRL mice. The
expression level of HIF-1α in MRL mice has been found to be
significantly increased after tissue injury, where the tissue
regeneration abilities of MRL mice are severely damaged after the
downregulation of HIF-1α expression (53). In normal mice, injection of HIF-1α
degradation inhibitors after ear perforation injury has been found
to promote perforation closure and healing, cartilage regeneration
and hair follicle formation (54). In addition, a number of studies
have shown that in vitro mammalian cell dedifferentiation
requires glycolysis transfer (42,55,56). Therefore, hypoxia and consequent
transformation into glycolysis have been shown to provide a
favorable condition at the site of the damaged tissue, allowing the
susceptible cells to re-enter pluripotency. However, there is also
a risk that if hypoxia and glycolysis are not switched off at the
early stages, specifically after the initiation of
dedifferentiation and during healing, then their persistence will
lead to chronic wound formation and the continued activation of
proinflammatory factor transcription (57–60). This accelerates cell senescence
and tissue damage. Such conditions, namely hypoxia, high glycolytic
rates, chronic inflammation and continuous aging of cells, provides
a favorable environment the promotion of carcinogenesis (61–64) (Fig.
2).
Cellular senescence, whether it is physiological or
pathological, is characterized by the secretion of inflammatory
cytokines and the inability to proliferate (65–68). It can be triggered by tissue
injury or increased with aging to prevent the unwarranted
proliferation of cells, through the induction of cell cycle arrest
(69–71). Accumulating evidence shows that
cell plasticity is closely associated with senescence (72–74). Recent studies have shown that
transient senescence can stimulate regeneration in the heart,
whilst the elimination of senescent cells can prevent regeneration
(75,76). Senescence can regulate
reprogramming and regeneration through a range of extracellular
mechanisms in vivo (72,73,77,78). Previous studies have shown that
when combined with injury, it can more effectively induce
reprogramming in the liver and pancreas in vivo (77,78). Similarly, in another previous
study, only after treatment with the DNA damaging agent bleomycin
can NANOG-positive cell clusters be observed in the lungs (73). During the regeneration process of
the skeletal muscle, reprogrammed cells can only be observed in the
injured area, where they appear in the vicinity of senescent cells
(72). Similarly, compared with
younger mice, older mice show a higher degree of reprogramming and
teratoma formation, as does a progeria mouse model that is
characterized by a premature aging phenotype (72,73). In addition, after senescent cells
are inoculated into the livers of mice, expression of
stemness-related genes can be detected (78).
Although they are no longer proliferative, senescent
cells remain metabolically and transcriptionally active and are
capable of a wide range of secretory activities. These secretory
proteins are capable of inducing cell cycle arrest and senescence
in a paracrine manner, in a process known as senescence-associated
secretory phenotype (SASP) (68,79,80). However, previous studies show that
the beneficial roles of senescence are mainly mediated through SASP
(79,80). SASP has been shown to promote the
regenerative response by inducing cell dedifferentiation in a
time-dependent manner. After being transiently exposed to SASP,
mouse keratinocytes can dedifferentiate into hair follicle stem
cells and regenerate the skin after transplantation (78). However, prolonged exposure to SASP
causes subsequent cell-intrinsic senescence that inhibits
continuous regenerative stimulation (78). Interleukin-6, which is the most
prominent cytokine released during SASP, has been identified to be
a critical mediator for creating a permissive tissue environment
for factor-mediated in vivo reprogramming (72,73). A similar event may occur under
physiological conditions, where tissue injury-induced senescence
can promote tissue repair by inducing cell dedifferentiation
(Fig. 1). Therefore,
understanding the beneficial paracrine effects of injury-induced
senescence would be instructive for the development of novel tissue
repair strategies.
Wound or tissue damage can trigger a series of
events in multicellular animals, such as acute inflammation and the
activation of local or systemic adaptive immunity, in response to
microorganism infection and the appearance of necrotic cells
(81). In addition to effectively
protecting the organism from foreign pathogens, another key
function of these early events involving inflammation and immunity
is to stimulate tissue repair in the damaged area, even if the
repair is typically defective and incomplete (31). During the process of response
following injury, cell reprogramming caused by tissue regeneration
is closely associated with inflammation and immunity (82–84). This involves a complex network of
associated growth factors, signal transduction pathways and
cytokines (85,86).
Stem cell factor (SCF) is one of the cytokines that
is accumulated during inflammation. Schmitt et al (87) has previously showed that a large
number of Leucine-rich repeat-containing G protein-coupled receptor
5 (Lgr5+) stem cells are lost and the expression of SCF is enhanced
after acute inflammation of the small intestine in mice. For
restoration, Paneth cells are induced to dedifferentiate and their
secretory phenotype is lost. This dedifferentiation process has
been revealed to be triggered by the SCF/c-Kit signaling pathway,
which eventually leads to glycogen synthase kinase 3β inhibition
and Wnt activation in Paneth cells.
Nuclear factor κB (NF-κB) is another key
transcription factor that is activated in the inflammatory and
immunity microenvironment. It has been shown to serve as a key link
between inflammation and cellular plasticity (65,72,88,89). In the brain, activation of the
NF-κB pathway by tumor necrosis factor has been shown to induce the
dedifferentiation of mature astrocytes into neural progenitor cell
phenotypes, which are capable of proliferating and differentiating
into neurons or astrocytes (90).
In the intestine, coactivation of Wnt/β-catenin and NF-κB signaling
can induce the dedifferentiation of villus cells (91). In the pancreas, previous
investigation has reported that NF-κB downstream of inflammation
can trigger the dedifferentiation of β-cells and acinar cells
(92). Accumulating evidence has
confirmed that activation of inflammatory signals cause global
changes in the expression and activity of several chromatin
modifying enzymes, such as the downregulation of histone
deacetylases and histone methyltransferases (with disruptor of
telomeric silencing 1-like being one of the examples) and the
upregulation of histone acetyltransferases, which can promote
epigenetic cell plasticity (82,83,93).
The notion that an inflammatory environment triggers
or promotes cell reprogramming remains controversial. However, an
appropriate level of inflammation appears to be critical for tissue
repair (94,95). A previous study into inflammatory
responses during amphibian limb regeneration has revealed that
potent, chronic inflammation induced by beryllium can inhibit limb
patterning by suppressing the expression of sonic hedgehog, T-box
transcription factor 3 and spalt-like transcription factor (Sall)
1; however, this had no effect on the expression of Sall4, a
genetic reprogramming marker (96). This suggests that under high
levels of inflammation, cellular reprogramming and
dedifferentiation can still occur locally; however, the
developmental mechanisms of limb regeneration cannot be replicated.
Although acute inflammation can trigger tissue repair or
regeneration, it will hinder the establishment of normal cell-cell
interactions and signaling gradients that promote limb patterning
if it is not released at appropriate times (96). Acute inflammatory responses
provide cells with a more plastic epigenetic state for repairing
tissue injury (97). However, if
the acute inflammatory response is not eliminated in time and
becomes a chronic inflammatory response, the cells then cannot be
effectively reprogrammed to initiate repair of the injury, thereby
hindering tissue regeneration (97). During chronic inflammatory
responses, dedifferentiation or reprogramming can still occur,
which may be one of the main causes of carcinogenesis (Fig. 1) (98–100).
Epigenetic modifications, such as DNA methylation,
histone acetylation and methylation and non-coding RNAs, are the
main methods of structural chromatin remodeling. Changes in the
chromatin structure affects DNA accessibility, leading to either
enhanced or decreased gene expression, or even gene silencing
(101). It is widely considered
that chromatin remodeling and epigenetic modification are critical
processes for controlling cell fate due to the unique epigenetic
features that exist for these two processes (102,103). The effect of epigenetic
modification on cell dedifferentiation or reprogramming after
tissue injury remain poorly understood. The present review
summarized the epigenetic mechanism underlying cell
dedifferentiation on a macroscopic level.
Dedifferentiation or reprogramming during damage
repair is a process of dramatic changes in cell identity that
involves a gradual but global remodeling of the epigenetic
signatures, resulting in generally more open chromatin. The newt
lens regeneration process involves the dedifferentiation of dorsal
pigment epithelial cells (PEC). Analysis of the global histone
modifications revealed that PEC dedifferentiation is accompanied by
an increase in trimethylated histone H3 lysine 4 (H3K4) and
acetylated histone H4 with a corresponding decrease in acetylated
histone H3 lysine 9 (H3K9) (104). Significant epigenetic
modifications were also observed during reprogramming triggered by
NF-κB and acute inflammation, including a decrease in H3K9
methylation and an increase in H3K4 methylation in the promoter
regions of endogenous pluripotency factors (83,93). In addition, activation of acute
inflammatory signals can cause changes in the expression of enzymes
involved in chromosomal modification, such as upregulation of
histone acetyltransferases, downregulation of histone deacetylases
and downregulation of histone methyltransferases, which enhance the
plasticity of the cells to facilitate dedifferentiation or
reprogramming (83,93). Successful tissue repair processes
should be accompanied by procedural dynamic changes in epigenetic
modification, such as early demethylation followed by de
novo methylation. Regeneration of the zebrafish retina provides
a good example (105). Müller
cell dedifferentiation into retinal progenitor cells occurs during
zebrafish retina regeneration (105). Studies into DNA methylation in
Müller cells have shown that methylation levels will typically
undergo an early reduction followed by a later increase (105). This coincides with the
dedifferentiation of Müller cells into retinal progenitor cells
during retinal regeneration and then redifferentiation into retinal
cells. This early reduction in DNA methylation facilitates the
dedifferentiation of Müller cells into retinal progenitor cells and
efficient progenitor cell proliferation. Subsequently, the increase
in DNA methylation during the later stages causes the retinal
progenitor cells to redifferentiate into retinal cells (105). This suggest that whilst
epigenetic modification can occur to complete tissue repair and
regeneration by regulating cell dedifferentiation, it can also
regulate subsequent redifferentiation.
The expression of stemness-related genes is one of
the most intuitive markers of cell dedifferentiation. Epigenetic
mechanisms regulate the effective expression and maintenance of
stemness-related genes, which determine the outcome of cell
dedifferentiation and subsequent regeneration (106). Compared with zebrafish, the
regenerative abilities of mammals after retinal injury are severely
limited (107,108). After
N-methyl-D-aspartate-induced retinal injury in mice, the expression
levels of pluripotent factors and progenitor cell-specific
transcription factors, such as OCT4, NANOG, KLF4 and paired box
protein (PAX) 6, increase rapidly during the early stages of
injury, suggesting that cells are undergoing dedifferentiation
(108). However, this change is
transient, where the expression of all the aforementioned genes are
greatly reduced or even become undetectable 18 to 24 h post-injury
(hpi) (108).
By contrast, the high expression of pluripotent
factors, such as OCT4, in damaged retinal cells can persist for up
to a week after injury in zebrafish (107). Detection of the expression of
methylation-related genes has revealed that the expression of
pluripotency genes is positively associated with growth arrest and
DNA damage inducible β whilst being negatively associated with DNA
methyltransferase (dnmt) 3β. Correspondingly, the methylation level
of OCT4 is decreased during the early stages of injury, which is
recovered 24 hpi (108).
Therefore, the rapid methylation of stemness-related genes after
tissue injury prevents successful Müller glia (MG)
dedifferentiation and their potential regeneration in mammals
(107,108).
In the intestinal epithelium, secretory cells can
dedifferentiate into Lgr5+ intestinal stem cells (ISCs) in response
to the ablation of native ISCs. Analysis of chromosomal status has
revealed the presence of thousands of transposase-accessible
genomic loci in the secretory cells controlling the expression of
lineage-restricted genes (109).
However, these sites are inaccessible in ISCs. When the secretory
cells dedifferentiate into ISCs after ISC ablation, the
accessibility signature dynamically converts into that of Lgr5+
ISCs (109). In the liver,
AT-rich interactive domain-containing protein 1A (Arid1a), a key
component of the switch/sucrose non-fermentable chromatin
remodeling complex, can regulate the expression of injury-induced
liver progenitor-like cell (LPLC)-related genes to promote liver
regeneration. Arid1a can establish an accessible state on
LPLC-enriched genes by regulating the accessibility of chromatin
(110). By contrast, Arid1a can
promote the Hippo-dependent transcriptional activation of
yes-associated protein/transcriptional enhanced associate domain at
the LPLC-enriched gene loci (110). Therefore, in response to tissue
damage, cells can turn on specific enhancers of stemness-related
genes whilst turning off specific enhancers of lineage restriction
genes by regulating chromatin accessibility to complete the
transformation of cell identity.
miRNA also serves a role in regulating the
expression of stemness and lineage-related genes (107,111). During the injury response in the
peripheral nervous system (PNS), a specific set of miRNAs have been
found to control myelination by silencing the positive and negative
regulators of Schwann cell dedifferentiation. Specifically,
miRNA-138 and miRNA-709 can directly target early growth response 2
(Egr2), c-Jun and SOX2, which are the major gene regulators of
dedifferentiation after PNS injury (111). During retinal regeneration,
lethal-7 miRNA is able to prevent MG dedifferentiation by
inhibiting the expression of regeneration-related genes, such as
achaete-scute homolog 1, heat shock protein family D member 1,
lin-28, OCT4, PAX6b and c-myc (107).
In addition to regulating the dedifferentiation
process of cells after tissue injury by directly regulating the
expression of stemness and lineage-related genes, the reversal of
dedifferentiation-associated signals through the expression of
tumor suppressor genes may provide another potential mechanism. The
tumor suppressor gene, especially the tumor protein p53 (TP53)
gene, is one of the major obstacles to the cellular
dedifferentiation process after tissue injury. As the blastema
forms after amputation, p53 expression decreases but is then absent
in completely dedifferentiated stem cells. However, p53 expression
is typically increased as the limb begins to regenerate (112). Studies into reprogramming in
mammalian cells in vitro also suggests that loss of p53
function leads to more efficient cell dedifferentiation (113).
In the liver, biliary epithelial cells (BECs)
dedifferentiate into bipotential progenitor cells (BPPCs) to
complete liver regeneration after injury. He et al (114) found that the significant
upregulation of the dnmt1 gene in BECs is able to methylate the
TP53 locus during this process, thereby inhibiting the expression
of TP53, which then reverses the inhibition of mammalian target of
rapamycin complex 1 signaling to activate the dedifferentiation of
BECs to BPPCs. By contrast, early DNA methylation inhibition using
5-azacytidine (5azaC) significantly reduces the methylation level
of the TP53 locus, where BEC dedifferentiation is dramatically
reduced and liver regeneration is blocked (114). In smooth muscle cells (SMCs),
the loss of phosphatase and tensin homolog (PTEN) is associated
with the dedifferentiation of SMCs (115), where a transcriptional blockade
of PTEN is observed during the dedifferentiation of SMCs induced by
platelet-derived growth factor (PDGF). However, 5azaC, an inhibitor
of dnmt1, is able to upregulate PTEN expression by decreasing the
methylation level whilst increasing the expression of genes
associated with the differentiated SMCs phenotype. Thus, blocking
PDGF-mediated SMC dedifferentiation (116).
After tissue injury, it can be hypothesized that the
effects of epigenetic modifications on cell dedifferentiation are
not achieved by a single pathway. In response to tissue injury,
genome-wide DNA methylation does not appear to affect gene
expression indiscriminately, such that other epigenetic mechanisms
will cooperate to maintain the silencing or activation of specific
DNA regions, showing compensatory effects among epigenetic
mechanisms (117,118). After deleting ubiquitin-like
with PHD and ring finger domains 1 expression, a protein that
maintains DNA methylation in the liver, Wang et al (117) found that this leads to
genome-wide DNA hypomethylation and the activation of regenerative
genes in hepatocytes, thereby promoting regeneration after partial
hepatectomy. However, genome-wide DNA hypomethylation does not
induce the expression of transposable elements, the re-mobilization
of which can lead to genome instability and cell death (118). Hypomethylated transposons are
repressed by the repositioning of the repressive histone marker,
trimethylated histone H3 lysine 27 (H3K27me3), whilst reduced
H3K27me3 expression can lead to activation of the regenerative
genes (117). After PNS injury,
miRNA-709 is able to form epigenetic silencing complexes together
with H3K27me3 and Agonaute-1 on the Egr2 promoter, a key regulator
of Schwann cell dedifferentiation, thereby regulating the process
of dedifferentiation and affecting myelination (111). Cell dedifferentiation after
tissue injury appears to be the result of a combinatorial action of
multiple epigenetic mechanisms.
Cellular plasticity may be the key to regeneration
after severe injury, which is the functional replacement of
tissue-specific stem cells lost due to injury by the transient
reprogramming of mature committed cells (119–121). However, elevated plasticity of
the tissue can also have potentially adverse consequences, such as
cancer (119–121). On a cellular level, as a
reaction to the signals released during tissue injury and
inflammation, the gene expression profile and chromatin structure
of the cells can undergo dramatic changes, which forms the basis of
cellular plasticity (83,93). These processes are normally
tightly controlled, because these same changes in the chromatin
structure and gene expression profile underlying cell plasticity
may also lead to oncogene expression or the inhibition of tumor
suppressors (112,114,115).
Indeed, dysregulation of plasticity has been
reported to be responsible for disrupting tissue stability and is a
key etiological factor in cancer. There is accumulating evidence
showing that several of the same signals that induce cell
reprogramming may also be carcinogenic, where cancer cells can
arise from somatic stem cells (90,91,122–126). NF-κB signaling has been reported
to induce the differentiation of mature astrocytes into neural
progenitor cells in the brain, whereas NF-κB signaling in the
intestines can induce non-stem cell dedifferentiation to promote
carcinogenesis (90,91). Similarly, the NOTCH signaling
pathway can induce hepatocyte transdifferentiation into biliary
cells, whereas activation of NOTCH in hepatocytes can lead to
hepatocellular carcinoma with biliary features or intrahepatic
cholangiocarcinomas (122,123). To explore the source of tumor
cells, Cobaleda et al (124) used the PAX5 conditional knockout
system to demonstrate that lymphoma can occur by the
dedifferentiation of mature B cells into a progenitor cell state. A
similar phenomenon of cancer caused by the dedifferentiation of
committed cells has also been found in gliomas (125,126). These findings suggest that
signals that cause committed cells to change their phenotype may
increase the risk of malignant transformation. This may be driven
by two factors. Firstly, directly by epigenetic alterations.
Genetic mutations are generally considered to be the main cause of
cancer and numerous types of cancer cells do have mutations in
multiple genes (127). However,
genetic mutations do not appear to be the only factor contributing
to cancer. Driver gene mutations are rarely found in childhood
tumors, including Wilms' tumor, medulloblastoma, neuroblastoma and
rhabdoid tumor. These tumors may be mainly due to epigenetic
disruption triggered by dedifferentiation (128–131). During in vivo
reprogramming in mice, it was also found that tumors distinct from
teratomas were detected in mice when Dox was withdrawn to induce
incomplete reprogramming (132).
DNA methylation patterns in these cancer cells exhibited partial
reprogramming and no mutations in cancer-related genes were
detected in these cells (132).
Thus, epigenetic alterations can directly drive the development of
cancer. Secondly, the synergistic drive of epigenetic alterations
on the basis of genetic mutation. Dedifferentiation exhibits strong
perturbations of epigenetic modifications and in some cases these
epigenetic alterations play a key boost to cancer development on
the basis of genetic variation. The findings in the Apc min/+ mouse
model indicated that mutations in the Apc gene, the driver gene of
intestinal neoplasms, were responsible for the initial colon
adenomas but were insufficient to trigger the overall tumor
progression (133–136). Thus, DNA methylation is a key
factor in the development of colon tumor (137–140). Understanding the idea that
epigenetic alterations drive cancer could allow us to develop
anti-cancer strategies that differ from genetic damage. For
example, epigenome reprogramming of human pancreatic ductal
adenocarcinoma can significantly reduce its tumorigenicity and
along this line, aberrant epigenetic modifications that drive
cancer may be new therapeutic targets (141). Increased risk of malignant
transformation during cell dedifferentiation meaning that cell
plasticity comes at a price that requires a trade-off between
maximizing tissue repair and minimizing the risk of malignant cell
transformation. This may explain why some tissues initiate a
process of dedifferentiation or reprogramming only after the stem
cell population has been completely ablated, whereas
differentiation or reprogramming is inhibited in the presence of
homeostasis or individual stem cells (15). Compared with dedifferentiation or
cell reprogramming, stem cell differentiation can control the
change of cell identity in a more accurate manner, phenotypically,
to reduce the frequency of identity changes. This serves to reduce
the risk of malignant transformation during tissue repair. It is
only when the stem cells are completely eliminated or signals that
allow normal differentiation are hindered, that dedifferentiation
and reprogramming can be used to maintain homeostasis or tissue
regeneration as the preferred mechanism.
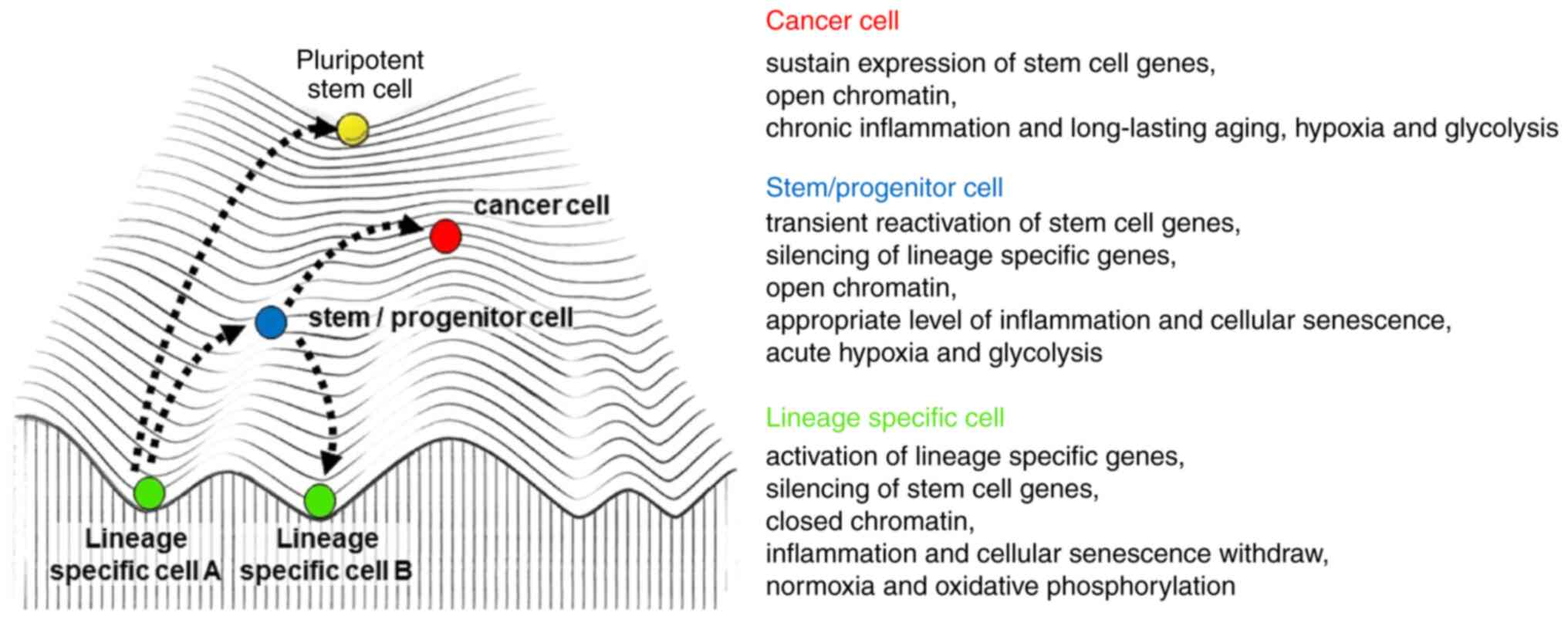
Similarly, maintaining a balance between maximizing
tissue repair and minimizing the risk of malignant cell
transformation also requires a moderate level of tissue
inflammation, cellular senescence and hypoxia after injury. Acute
inflammatory responses and transient cellular senescence can induce
dedifferentiation or reprogramming of cells by repairing damaged or
diseased tissues by providing the cells with a more plastic
epigenetic state (78,97). However, inflammation and cellular
senescence need to be maintained at an appropriate level and can be
withdrawn at appropriate times after the initiation of damaged or
diseased tissue repair, represented by decreased levels of NF-κB
and SASP-related factors. Dedifferentiated or reprogrammed cells
being redirected to differentiate and the plasticity of the cells
is then reduced to return to homeostasis. Once cells are
transformed into a chronic inflammatory and a long-lasting aging
status, represented by elevated levels and persistent expression of
NF-κB and SASP-associated factors, they will more likely exhibit a
persistently hyperplastic state in which dedifferentiated or
reprogrammed cells are unable to re-differentiate, resulting in
malignant transformation (Fig.
2). Although SASP can induce stemness-related gene expression
and promote cell dedifferentiation, long-term SASP exposure can
activate cell senescence arrest and lead to the formation of
papilloma in vivo (78).
This is why long-term use of non-steroidal anti-inflammatory drugs,
such as aspirin, can prevent the formation of tumors and
dramatically reduce the incidence of solid tumors, including
colorectal cancer (142–144).
The purpose of regenerative medicine is to restore
the physiological functions of organs and tissues that are
typically not easily repaired after injury, to promote changes in
cell characteristics and to enhance endogenous regeneration.
Dedifferentiation or reprogramming is a key method for tissue
repair after injury, where the local microenvironment serves a
crucial role. Hypoxia, inflammation and cellular senescence in
injury-induced local microenvironmental changes are key parameters
that can influence reprogramming. Hypoxia, inflammation and
senescence create a relaxed environment for cells that typically
promote reprogramming through a number of factors, such as HIF,
NF-κB and SASP (78,88,90).
There is increasing evidence that certain cell
types can be dedifferentiated by specific signals and therapeutic
stimuli. Injury-induced dedifferentiation of MG in the mammalian
retina can be promoted by treatment with insulin, epidermal growth
factor and Wnt3a (145–148). However, the majority of in
vivo reprogramming studies use viral delivery to allow key
transcription factors to be overexpressed in cells, where the
process of using transcription factor-induced reprogramming
typically activates endogenous pluripotency programs (149–151). This means that cells can
maintain their stem-state but also increase the risk of
tumorigenesis, which greatly limits the potential of clinical
applications. In addition, small molecules can be used to eliminate
epigenetic barriers and promote changes in cell characteristics.
For example, SGI-1027, a DNA-methyltransferase inhibitor, is used
to block DNA methylation and maintain OCT4 expression after retinal
injury (108).
5-aza-2′-deoxycytidine (5-aza-dC) or trichostatin A, the DNA
demethylation agent, have been used to increase stem cell numbers
at the amputation site and enhance digital regeneration (152). Therefore, establishment of small
molecule-mediated in vivo reprogramming has become a
promising new strategy (153–156). In addition, because they are
generally more cost-effective, more stable, less immunogenic and
can be more easily synthesized and standardized, small molecules
offer a more attractive alternative to transcription
factor-mediated in vivo reprogramming. Local
microenvironments that promote dedifferentiation or reprogramming
are generally specific for certain cell types, such that a specific
combination of small molecules can be designed to repair particular
tissue injury. Similarly, the local microenvironment that promotes
tissue repair can be maintained by adjusting the concentration of
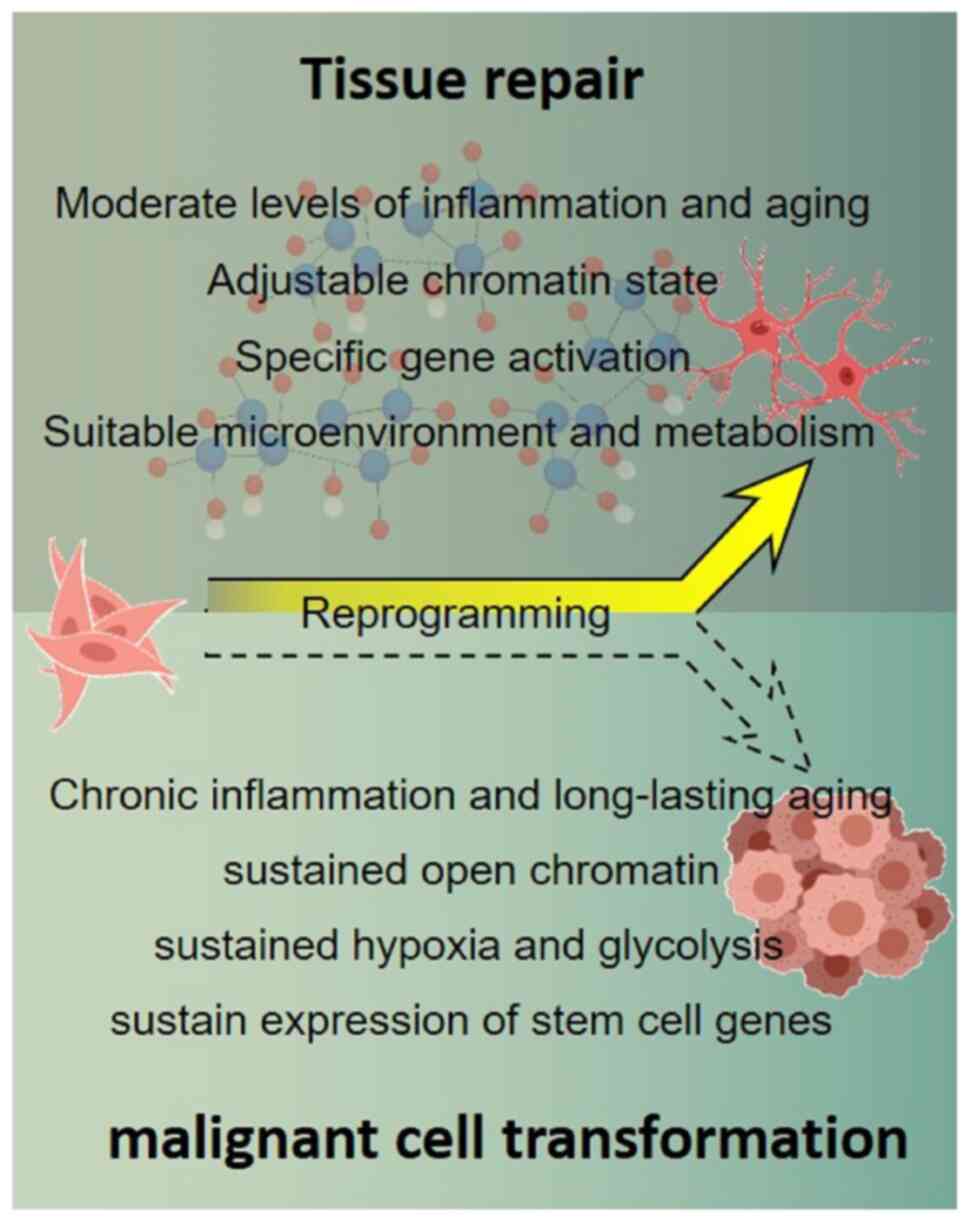
small molecules. The present study hypothesized that the cells
could maintain moderate levels of inflammation and senescence by
adjusting the combination and concentration of small molecules,
thus achieving the goal of maximizing tissue repair and minimizing
the risk of malignant cell transformation (Fig. 3).
Not applicable.
The present work was supported by the National Natural Science
Foundation of China (grant no. 31701121) and Project of Young
backbone Teachers in Henan Province (grant no. 2020GGJS196).
Not applicable.
YG, XY and XF conceptualized the study. YG, WW, XY
and XF wrote the manuscript. All authors read and approved the
final manuscript. Data authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Del Rio-Tsonis K and Tsonis PA: Eye
regeneration at the molecular age. Dev Dyn. 226:211–224. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gurdon JB: The developmental capacity of
nuclei taken from intestinal epithelium cells of feeding tadpoles.
J Embryol Exp Morphol. 10:622–640. 1962.PubMed/NCBI
|
|
3
|
Worley MI, Setiawan L and Hariharan IK:
Regeneration and transdetermination in Drosophila imaginal discs.
Annu Rev Genet. 46:289–310. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gurdon JB: Adult frogs derived from the
nuclei of single somatic cells. Dev Biol. 4:256–273. 1962.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Davis RL, Weintraub H and Lassar AB:
Expression of a single transfected cDNA converts fibroblasts to
myoblasts. Cell. 51:987–1000. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yamanaka S and Blau HM: Nuclear
reprogramming to a pluripotent state by three approaches. Nature.
465:704–712. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sisakhtnezhad S and Matin MM:
Transdifferentiation: A cell and molecular reprogramming process.
Cell Tissue Res. 348:379–396. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jopling C, Boue S and Izpisua Belmonte JC:
Dedifferentiation, transdifferentiation and reprogramming: Three
routes to regeneration. Nat Rev Mol Cell Biol. 12:79–89. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yao and Wang C: Dedifferentiation:
Inspiration for devising engineering strategies for regenerative
medicine. NPJ Regen Med. 5:142020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brawley C and Matunis E: Regeneration of
male germline stem cells by spermatogonial dedifferentiation in
vivo. Science. 304:1331–1334. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kai T and Spradling A: Differentiating
germ cells can revert into functional stem cells in Drosophila
melanogaster ovaries. Nature. 428:564–569. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kragl M, Knapp D, Nacu E, Khattak S, Maden
M, Epperlein HH and Tanaka EM: Cells keep a memory of their tissue
origin during axolotl limb regeneration. Nature. 460:60–65. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Blanpain C and Fuchs E: Stem cell
plasticity. Plasticity of epithelial stem cells in tissue
regeneration. Science. 344:12422812014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
van Es JH, Sato T, van de Wetering M,
Lyubimova A, Yee Nee AN, Gregorieff A, Sasaki N, Zeinstra L, van
den Born M, Korving J, et al: Dll1+ secretory progenitor cells
revert to stem cells upon crypt damage. Nat Cell Biol.
14:1099–1104. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tata PR, Mou H, Pardo-Saganta A, Zhao R,
Prabhu M, Law BM, Vinarsky V, Cho JL, Breton S, Sahay A, et al:
Dedifferentiation of committed epithelial cells into stem cells in
vivo. Nature. 503:218–223. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Brockes JP and Kumar A: Plasticity and
reprogramming of differentiated cells in amphibian regeneration.
Nat Rev Mol Cell Biol. 3:566–574. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Donati G, Rognoni E, Hiratsuka T,
Liakath-Ali K, Hoste E, Kar G, Kayikci M, Russell R, Kretzschmar K,
Mulder KW, et al: Wounding induces dedifferentiation of epidermal
Gata6+ cells and acquisition of stem cell properties.
Nat Cell Biol. 19:603–613. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Stange DE, Koo BK, Huch M, Sibbel G, Basak
O, Lyubimova A, Kujala P, Bartfeld S, Koster J, Geahlen JH, et al:
Differentiated Troy+ chief cells act as reserve stem cells to
generate all lineages of the stomach epithelium. Cell. 155:357–368.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Painter MW, Brosius Lutz A, Cheng YC,
Latremoliere A, Duong K, Miller CM, Posada S, Cobos EJ, Zhang AX,
Wagers AJ, et al: Diminished Schwann cell repair responses underlie
age-associated impaired axonal regeneration. Neuron. 83:331–343.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Buczacki SJ, Zecchini HI, Nicholson AM,
Russell R, Vermeulen L, Kemp R and Winton DJ: Intestinal
label-retaining cells are secretory precursors expressing Lgr5.
Nature. 495:65–69. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tian H, Biehs B, Warming S, Leong KG,
Rangell L, Klein OD and de Sauvage FJ: A reserve stem cell
population in small intestine renders Lgr5-positive cells
dispensable. Nature. 478:255–259. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Leushacke M, Tan SH, Wong A, Swathi Y,
Hajamohideen A, Tan LT, Goh J, Wong E, Denil SLIJ, Murakami K and
Barker N: Lgr5-expressing chief cells drive epithelial regeneration
and cancer in the oxyntic stomach. Nat Cell Biol. 19:774–786. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tulina N and Matunis E: Control of stem
cell self-renewal in Drosophila spermatogenesis by JAK-STAT
signaling. Science. 294:2546–2549. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kiger AA, Jones DL, Schulz C, Rogers MB
and Fuller MT: Stem cell self-renewal specified by JAK-STAT
activation in response to a support cell cue. Science.
294:2542–2545. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sheng XR, Brawley CM and Matunis EL:
Dedifferentiating spermatogonia outcompete somatic stem cells for
niche occupancy in the Drosophila testis. Cell Stem Cell.
5:191–203. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hameed LS, Berg DA, Belnoue L, Jensen LD,
Cao Y and Simon A: Environmental changes in oxygen tension reveal
ROS-dependent neurogenesis and regeneration in the adult newt
brain. Elife. 4:e084222015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
D'Ignazio L, Batie M and Rocha S: Hypoxia
and Inflammation in Cancer, Focus on HIF and NF-κB. Biomedicines.
5:212017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mohyeldin A, Garzon-Muvdi T and
Quinones-Hinojosa A: Oxygen in stem cell biology: A critical
component of the stem cell niche. Cell Stem Cell. 7:150–161. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Arthur SA, Blaydes JP and Houghton FD:
Glycolysis regulates human embryonic stem cell self-renewal under
hypoxia through HIF-2α and the glycolytic sensors CTBPs. Stem Cell
Reports. 12:728–742. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Forristal CE, Wright KL, Hanley NA, Oreffo
RO and Houghton FD: Hypoxia inducible factors regulate pluripotency
and proliferation in human embryonic stem cells cultured at reduced
oxygen tensions. Reproduction. 139:85–97. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yoshida Y, Takahashi K, Okita K, Ichisaka
T and Yamanaka S: Hypoxia enhances the generation of induced
pluripotent stem cells. Cell Stem Cell. 5:237–241. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jopling C, Sune G, Faucherre A, Fabregat C
and Izpisua Belmonte JC: Hypoxia induces myocardial regeneration in
zebrafish. Circulation. 126:3017–3027. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mu X, Xiang G, Rathbone CR, Pan H, Bellayr
IH, Walters TJ and Li Y: Slow-adhering stem cells derived from
injured skeletal muscle have improved regenerative capacity. Am J
Pathol. 179:931–941. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vojnits K, Pan H, Mu X and Li Y:
Characterization of an injury induced population of muscle-derived
stem cell-like cells. Sci Rep. 5:173552015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Vojnits K, Pan H, Dai X, Sun H, Tong Q,
Darabi R, Huard J and Li Y: Functional neuronal differentiation of
injury-induced muscle-derived stem cell-like cells with therapeutic
implications. Sci Rep. 7:11772017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tatebayashi K, Tanaka Y, Nakano-Doi A,
Sakuma R, Kamachi S, Shirakawa M, Uchida K, Kageyama H, Takagi T,
Yoshimura S, et al: Identification of multipotent stem cells in
human brain tissue following stroke. Stem Cells Dev. 26:787–797.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liao YJ, Gao JH, Jiang P and Lu F: Effect
of hypoxia on dedifferentiation of mature adipocytes: An
experimental study. Nan Fang Yi Ke Da Xue Xue Bao. 28:339–342.
2008.(In Chinese). PubMed/NCBI
|
|
38
|
Schmidt-Ott KM, Xu AD, Tuschick S,
Liefeldt L, Kresse W, Verkhratsky A, Kettenmann H and Paul M:
Hypoxia reverses dibutyryl-cAMP-induced stellation of cultured
astrocytes via activation of the endothelin system. FASEB J.
15:1227–1229. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sahai A, Mei C, Schrier RW and Tannen RL:
Mechanisms of chronic hypoxia-induced renal cell growth. Kidney
Int. 56:1277–1281. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kierans SJ and Taylor CT: Regulation of
glycolysis by the hypoxia-inducible factor (HIF): Implications for
cellular physiology. J Physiol. 599:23–37. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kondoh H, Lleonart ME, Nakashima Y, Yokode
M, Tanaka M, Bernard D, Gil J and Beach D: A high glycolytic flux
supports the proliferative potential of murine embryonic stem
cells. Antioxid Redox Signal. 9:293–299. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Folmes CD, Nelson TJ, Martinez-Fernandez
A, Arrell DK, Lindor JZ, Dzeja PP, Ikeda Y, Perez-Terzic C and
Terzic A: Somatic oxidative bioenergetics transitions into
pluripotency-dependent glycolysis to facilitate nuclear
reprogramming. Cell Metab. 14:264–271. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nombela-Arrieta C, Pivarnik G, Winkel B,
Canty KJ, Harley B, Mahoney JE, Park SY, Lu J, Protopopov A and
Silberstein LE: Quantitative imaging of haematopoietic stem and
progenitor cell localization and hypoxic status in the bone marrow
microenvironment. Nat Cell Biol. 15:533–543. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Takubo K, Nagamatsu G, Kobayashi CI,
Nakamura-Ishizu A, Kobayashi H, Ikeda E, Goda N, Rahimi Y, Johnson
RS, Soga T, et al: Regulation of glycolysis by Pdk functions as a
metabolic checkpoint for cell cycle quiescence in hematopoietic
stem cells. Cell Stem Cell. 12:49–61. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Warr MR and Passegue E: Metabolic makeover
for HSCs. Cell Stem Cell. 12:1–3. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lima A, Burgstaller J, Sanchez-Nieto JM
and Rodriguez TA: The mitochondria and the regulation of cell
fitness during early mammalian development. Curr Top Dev Biol.
128:339–363. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chen CT, Shih YR, Kuo TK, Lee OK and Wei
YH: Coordinated changes of mitochondrial biogenesis and antioxidant
enzymes during osteogenic differentiation of human mesenchymal stem
cells. Stem Cells. 26:960–968. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Pattappa G, Thorpe SD, Jegard NC, Heywood
HK, de Bruijn JD and Lee DA: Continuous and uninterrupted oxygen
tension influences the colony formation and oxidative metabolism of
human mesenchymal stem cells. Tissue Eng Part C Methods. 19:68–79.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Scott CA, Carney TJ and Amaya E: Aerobic
glycolysis is important for zebrafish larval wound closure and tail
regeneration. Wound Repair Regen. Sep 23–2022.(Epub ahead of
print). View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sinclair JW, Hoying DR, Bresciani E,
Nogare DD, Needle CD, Wu W, Bishop K, Elkahloun AG, Chitnis AB, Liu
PP, et al: A metabolic shift to glycolysis promotes zebrafish tail
regeneration through TGF-β dependent dedifferentiation of notochord
cells to form the blastema. bioRxiv. Mar 20–2020.(Epub ahead of
print). PubMed/NCBI
|
|
51
|
Fukuda R, Marin-Juez R, El-Sammak H,
Beisaw A, Ramadass R, Kuenne C, Guenther S, Konzer A, Bhagwat AM,
Graumann J and Stainier DY: Stimulation of glycolysis promotes
cardiomyocyte proliferation after injury in adult zebrafish. EMBO
Rep. 21:e497522020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Naviaux RK, Le TP, Bedelbaeva K,
Leferovich J, Gourevitch D, Sachadyn P, Zhang XM, Clark L and
Heber-Katz E: Retained features of embryonic metabolism in the
adult MRL mouse. Mol Genet Metab. 96:133–144. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sinha KM, Tseng C, Guo P, Lu A, Pan H, Gao
X, Andrews R, Eltzschig H and Huard J: Hypoxia-inducible factor 1α
(HIF-1α) is a major determinant in the enhanced function of
muscle-derived progenitors from MRL/MpJ mice. FASEB J.
33:8321–8334. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhang Y, Strehin I, Bedelbaeva K,
Gourevitch D, Clark L, Leferovich J, Messersmith PB and Heber-Katz
E: Drug-induced regeneration in adult mice. Sci Transl Med.
7:290ra2922015. View Article : Google Scholar
|
|
55
|
Pennock R, Bray E, Pryor P, James S,
McKeegan P, Sturmey R and Genever P: Human cell dedifferentiation
in mesenchymal condensates through controlled autophagy. Sci Rep.
5:131132015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Varum S, Rodrigues AS, Moura MB,
Momcilovic O, Easley CA IV, Ramalho-Santos J, Van Houten B and
Schatten G: Energy metabolism in human pluripotent stem cells and
their differentiated counterparts. PLoS One. 6:e209142011.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Schreml S, Szeimies RM, Prantl L, Karrer
S, Landthaler M and Babilas P: Oxygen in acute and chronic wound
healing. Br J Dermatol. 163:257–268. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Hong WX, Hu MS, Esquivel M, Liang GY,
Rennert RC, McArdle A, Paik KJ, Duscher D, Gurtner GC, Lorenz HP
and Longaker MT: The role of hypoxia-inducible factor in wound
healing. Adv Wound Care (New Rochelle). 3:390–399. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Baatar D, Jones MK, Tsugawa K, Pai R, Moon
WS, Koh GY, Kim I, Kitano S and Tarnawski AS: Esophageal ulceration
triggers expression of hypoxia-inducible factor-1 alpha and
activates vascular endothelial growth factor gene: Implications for
angiogenesis and ulcer healing. Am J Pathol. 161:1449–1457. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Elson DA, Ryan HE, Snow JW, Johnson R and
Arbeit JM: Coordinate up-regulation of hypoxia inducible factor
(HIF)-1alpha and HIF-1 target genes during multi-stage epidermal
carcinogenesis and wound healing. Cancer Res. 60:6189–6195.
2000.PubMed/NCBI
|
|
61
|
Coussens LM and Werb Z: Inflammation and
cancer. Nature. 420:860–867. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Muz B, de la Puente P, Azab F and Azab AK:
The role of hypoxia in cancer progression, angiogenesis,
metastasis, and resistance to therapy. Hypoxia (Auckl). 3:83–92.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Jiang B: Aerobic glycolysis and high level
of lactate in cancer metabolism and microenvironment. Genes Dis.
4:25–27. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Wyld L, Bellantuono I, Tchkonia T, Morgan
J, Turner O, Foss F, George J, Danson S and Kirkland JL: Senescence
and cancer: A review of clinical implications of senescence and
senotherapies. Cancers (Basel). 12:21342020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Lasry A and Ben-Neriah Y:
Senescence-associated inflammatory responses: Aging and cancer
perspectives. Trends Immunol. 36:217–228. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Munoz-Espin D and Serrano M: Cellular
senescence: From physiology to pathology. Nat Rev Mol Cell Biol.
15:482–496. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Tchkonia T, Zhu Y, van Deursen J, Campisi
J and Kirkland JL: Cellular senescence and the senescent secretory
phenotype: Therapeutic opportunities. J Clin Invest. 123:966–972.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Watanabe S, Kawamoto S, Ohtani N and Hara
E: Impact of senescence-associated secretory phenotype and its
potential as a therapeutic target for senescence-associated
diseases. Cancer Sci. 108:563–569. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Schmitt CA, Fridman JS, Yang M, Lee S,
Baranov E, Hoffman RM and Lowe SW: A senescence program controlled
by p53 and p16INK4a contributes to the outcome of cancer therapy.
Cell. 109:335–346. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Coppe JP, Patil CK, Rodier F, Sun Y, Muñoz
DP, Goldstein J, Nelson PS, Desprez PY and Campisi J:
Senescence-associated secretory phenotypes reveal
cell-nonautonomous functions of oncogenic RAS and the p53 tumor
suppressor. PLoS Biol. 6:2853–2868. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Campisi J: Aging, cellular senescence, and
cancer. Annu Rev Physiol. 75:685–705. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Chiche A, Le Roux I, von Joest M, Sakai H,
Aguín SB, Cazin C, Salam R, Fiette L, Alegria O, Flamant P, et al:
Injury-Induced senescence enables in vivo reprogramming in skeletal
muscle. Cell Stem Cell. 20:407–414. e42017. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Mosteiro L, Pantoja C, Alcazar N, Marión
RM, Chondronasiou D, Rovira M, Fernandez-Marcos PJ, Muñoz-Martin M,
Blanco-Aparicio C, Pastor J, et al: Tissue damage and senescence
provide critical signals for cellular reprogramming in vivo.
Science. 354:aaf44452016. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Taguchi J and Yamada Y: Unveiling the role
of senescence-induced cellular plasticity. Cell Stem Cell.
20:293–294. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Feng T, Meng J, Kou S, Jiang Z, Huang X,
Lu Z, Zhao H, Lau LF, Zhou B and Zhang H: CCN1-Induced cellular
senescence promotes heart regeneration. Circulation. 139:2495–2498.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Sarig R, Rimmer R, Bassat E, Zhang L,
Umansky KB, Lendengolts D, Perlmoter G, Yaniv K and Tzahor E:
Transient p53-mediated regenerative senescence in the injured
heart. Circulation. 139:2491–2494. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Heinrich C, Spagnoli FM and Berninger B:
In vivo reprogramming for tissue repair. Nat Cell Biol. 17:204–211.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Ritschka B, Storer M, Mas A, Heinzmann F,
Ortells MC, Morton JP, Sansom OJ, Zender L and Keyes WM: The
senescence-associated secretory phenotype induces cellular
plasticity and tissue regeneration. Genes Dev. 31:172–183. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Munoz-Espin D, Canamero M, Maraver A,
Gómez-López G, Contreras J, Murillo-Cuesta S, Rodríguez-Baeza A,
Varela-Nieto I, Ruberte J, Collado M and Serrano M: Programmed cell
senescence during mammalian embryonic development. Cell.
155:1104–1118. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Storer M, Mas A, Robert-Moreno A, Pecoraro
M, Ortells MC, Di Giacomo V, Yosef R, Pilpel N, Krizhanovsky V,
Sharpe J and Keyes WM: Senescence is a developmental mechanism that
contributes to embryonic growth and patterning. Cell.
155:1119–1130. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Hanna J, Guerra-Moreno A, Ang J and
Micoogullari Y: Protein degradation and the pathologic basis of
disease. Am J Pathol. 189:94–103. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Cooke JP, Sayed N, Lee J and Wong WT:
Innate immunity and epigenetic plasticity in cellular
reprogramming. Curr Opin Genet Dev. 28:89–91. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Lee J, Sayed N, Hunter A, Au KF, Wong WH,
Mocarski ES, Pera RR, Yakubov E and Cooke JP: Activation of innate
immunity is required for efficient nuclear reprogramming. Cell.
151:547–558. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
King MW, Neff AW and Mescher AL: The
developing Xenopus limb as a model for studies on the balance
between inflammation and regeneration. Anat Rec (Hoboken).
295:1552–1561. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Cavaillon JM: Pro-versus anti-inflammatory
cytokines: Myth or reality. Cell Mol Biol (Noisy-le-grand).
47:695–702. 2001.PubMed/NCBI
|
|
86
|
Lennartsson J and Ronnstrand L: Stem cell
factor receptor/c-Kit: From basic science to clinical implications.
Physiol Rev. 92:1619–1649. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Schmitt M, Schewe M, Sacchetti A, Feijtel
D, van de Geer WS, Teeuwssen M, Sleddens HF, Joosten R, van Royen
ME, van de Werken HJG, et al: Paneth cells respond to inflammation
and contribute to tissue regeneration by acquiring stem-like
features through SCF/c-Kit Signaling. Cell Rep. 24:2312–2328.
e72018. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Soria-Valles C, Osorio FG,
Gutierrez-Fernandez A, De Los Angeles A, Bueno C, Menéndez P,
Martín-Subero JI, Daley GQ, Freije JM and López-Otín C: NF-κB
activation impairs somatic cell reprogramming in ageing. Nat Cell
Biol. 17:1004–1013. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Soria-Valles C, Osorio FG and Lopez-Otin
C: Reprogramming aging through DOT1L inhibition. Cell Cycle.
14:3345–3346. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Gabel S, Koncina E, Dorban G, Heurtaux T,
Birck C, Glaab E, Michelucci A, Heuschling P and Grandbarbe L:
Inflammation promotes a conversion of astrocytes into neural
progenitor cells via NF-κB activation. Mol Neurobiol. 53:5041–5055.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Schwitalla S, Fingerle AA, Cammareri P,
Nebelsiek T, Göktuna SI, Ziegler PK, Canli O, Heijmans J, Huels DJ,
Moreaux G, et al: Intestinal tumorigenesis initiated by
dedifferentiation and acquisition of stem-cell-like properties.
Cell. 152:25–38. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Murtaugh LC and Keefe MD: Regeneration and
repair of the exocrine pancreas. Annu Rev Physiol. 77:229–249.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
O'Neill LA: ‘Transflammation’: When innate
immunity meets induced pluripotency. Cell. 151:471–473. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Jiang B and Liao R: The paradoxical role
of inflammation in cardiac repair and regeneration. J Cardiovasc
Transl Res. 3:410–416. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Cooke JP: Inflammation and its role in
regeneration and repair. Circ Res. 124:1166–1168. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Mescher AL, Neff AW and King MW: Changes
in the inflammatory response to injury and its resolution during
the loss of regenerative capacity in developing Xenopus limbs. PLoS
One. 8:e804772013. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Pietras EM, Mirantes-Barbeito C, Fong S,
Loeffler D, Kovtonyuk LV, Zhang S, Lakshminarasimhan R, Chin CP,
Techner JM, Will B, et al: Chronic interleukin-1 exposure drives
haematopoietic stem cells towards precocious myeloid
differentiation at the expense of self-renewal. Nat Cell Biol.
18:607–618. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Grivennikov SI, Greten FR and Karin M:
Immunity, inflammation, and cancer. Cell. 140:883–899. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Ben-Neriah Y and Karin M: Inflammation
meets cancer, with NF-κB as the matchmaker. Nat Immunol.
12:715–723. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Balkwill FR and Mantovani A:
Cancer-related inflammation: Common themes and therapeutic
opportunities. Semin Cancer Biol. 22:33–40. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Fedorova E and Zink D: Nuclear
architecture and gene regulation. Biochim Biophys Acta.
1783:2174–2184. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Boland MJ, Nazor KL and Loring JF:
Epigenetic regulation of pluripotency and differentiation. Circ
Res. 115:311–324. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Wu H and Sun YE: Epigenetic regulation of
stem cell differentiation. Pediatr Res. 59:21R–25R. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Nakamura K, Maki N, Trinh A, Trask HW, Gui
J, Tomlinson CR and Tsonis PA: miRNAs in newt lens regeneration:
Specific control of proliferation and evidence for miRNA
networking. PLoS One. 5:e120582010. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Powell C, Grant AR, Cornblath E and
Goldman D: Analysis of DNA methylation reveals a partial
reprogramming of the Muller glia genome during retina regeneration.
Proc Natl Acad Sci USA. 110:19814–19819. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Oliveri RS: Epigenetic dedifferentiation
of somatic cells into pluripotency: Cellular alchemy in the age of
regenerative medicine? Regen Med. 2:795–816. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Ramachandran R, Fausett BV and Goldman D:
Ascl1a regulates Muller glia dedifferentiation and retinal
regeneration through a Lin-28-dependent, let-7 microRNA signalling
pathway. Nat Cell Biol. 12:1101–1107. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Reyes-Aguirre LI and Lamas M: Oct4
Methylation-Mediated silencing as an epigenetic barrier preventing
muller glia dedifferentiation in a murine model of retinal injury.
Front Neurosci. 10:5232016. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Jadhav U, Saxena M, O'Neill NK, Saadatpour
A, Yuan GC, Herbert Z, Murata K and Shivdasani RA: Dynamic
reorganization of chromatin accessibility signatures during
dedifferentiation of secretory precursors into Lgr5+ Intestinal
stem cells. Cell Stem Cell. 21:65–77. e52017. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Li W, Yang L, He Q, Hu C, Zhu L, Ma X, Ma
X, Bao S, Li L, Chen Y, et al: A homeostatic arid1a-dependent
permissive chromatin state licenses hepatocyte responsiveness to
liver-injury-associated YAP signaling. Cell Stem Cell. 25:54–68.
e552019. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Adilakshmi T, Sudol I and Tapinos N:
Combinatorial action of miRNAs regulates transcriptional and
post-transcriptional gene silencing following in vivo PNS injury.
PLoS One. 7:e396742012. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Yun MH, Gates PB and Brockes JP:
Regulation of p53 is critical for vertebrate limb regeneration.
Proc Natl Acad Sci USA. 110:17392–17397. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Yi L, Lu C, Hu W, Sun Y and Levine AJ:
Multiple roles of p53-related pathways in somatic cell
reprogramming and stem cell differentiation. Cancer Res.
72:5635–5645. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
He J, Zhou Y, Qian C, Wang D, Yang Z,
Huang Z, Sun J, Ni R, Yang Q, Chen J and Luo L: DNA methylation
maintenance at the p53 locus initiates biliary-mediated liver
regeneration. NPJ Regen Med. 7:212022. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Nemenoff RA, Simpson PA, Furgeson SB,
Kaplan-Albuquerque N, Crossno J, Garl PJ, Cooper J and Weiser-Evans
MC: Targeted deletion of PTEN in smooth muscle cells results in
vascular remodeling and recruitment of progenitor cells through
induction of stromal cell-derived factor-1alpha. Circ Res.
102:1036–1045. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Strand KA, Lu S, Mutryn MF, Li L, Zhou Q,
Enyart BT, Jolly AJ, Dubner AM, Moulton KS, Nemenoff RA, et al:
High throughput screen identifies the DNMT1 (DNA
Methyltransferase-1) Inhibitor, 5-Azacytidine, as a potent inducer
of PTEN (Phosphatase and Tensin Homolog): Central role for PTEN in
5-Azacytidine protection against pathological vascular remodeling.
Arterioscler Thromb Vasc Biol. 40:1854–1869. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Wang S, Zhang C, Hasson D, Desai A,
SenBanerjee S, Magnani E, Ukomadu C, Lujambio A, Bernstein E and
Sadler KC: Epigenetic compensation promotes liver regeneration. Dev
Cell. 50:43–56. e62019. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Chuong EB, Elde NC and Feschotte C:
Regulatory activities of transposable elements: From conflicts to
benefits. Nat Rev Genet. 18:71–86. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Tosh D and Slack JM: How cells change
their phenotype. Nat Rev Mol Cell Biol. 3:187–194. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Corbett JL and Tosh D: Conversion of one
cell type into another: Implications for understanding organ
development, pathogenesis of cancer and generating cells for
therapy. Biochem Soc Trans. 42:609–616. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Abollo-Jimenez F, Jimenez R and Cobaleda
C: Physiological cellular reprogramming and cancer. Semin Cancer
Biol. 20:98–106. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Villanueva A, Alsinet C, Yanger K, Hoshida
Y, Zong Y, Toffanin S, Rodriguez-Carunchio L, Solé M, Thung S,
Stanger BZ and Llovet JM: Notch signaling is activated in human
hepatocellular carcinoma and induces tumor formation in mice.
Gastroenterology. 143:1660–1669. e72012. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Fan B, Malato Y, Calvisi DF, Naqvi S,
Razumilava N, Ribback S, Gores GJ, Dombrowski F, Evert M, Chen X
and Willenbring H: Cholangiocarcinomas can originate from
hepatocytes in mice. J Clin Invest. 122:2911–2915. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Cobaleda C, Jochum W and Busslinger M:
Conversion of mature B cells into T cells by dedifferentiation to
uncommitted progenitors. Nature. 449:473–477. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Liu C, Sage JC, Miller MR, Verhaak RG,
Hippenmeyer S, Vogel H, Foreman O, Bronson RT, Nishiyama A, Luo L
and Zong H: Mosaic analysis with double markers reveals tumor cell
of origin in glioma. Cell. 146:209–221. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Friedmann-Morvinski D, Bushong EA, Ke E,
Soda Y, Marumoto T, Singer O, Ellisman MH and Verma IM:
Dedifferentiation of neurons and astrocytes by oncogenes can induce
gliomas in mice. Science. 338:1080–1084. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Kandoth C, McLellan MD, Vandin F, Ye K,
Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al:
Mutational landscape and significance across 12 major cancer types.
Nature. 502:333–339. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Yamada Y, Haga H and Yamada Y: Concise
review: Dedifferentiation meets cancer development: Proof of
concept for epigenetic cancer. Stem Cells Transl Med. 3:1182–1187.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Rausch T, Jones DT, Zapatka M, Stütz AM,
Zichner T, Weischenfeldt J, Jäger N, Remke M, Shih D, Northcott PA,
et al: Genome sequencing of pediatric medulloblastoma links
catastrophic DNA rearrangements with TP53 mutations. Cell.
148:59–71. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Molenaar JJ, Koster J, Zwijnenburg DA, van
Sluis P, Valentijn LJ, van der Ploeg I, Hamdi M, van Nes J,
Westerman BA, van Arkel J, et al: Sequencing of neuroblastoma
identifies chromothripsis and defects in neuritogenesis genes.
Nature. 483:589–593. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Lee RS, Stewart C, Carter SL, Ambrogio L,
Cibulskis K, Sougnez C, Lawrence MS, Auclair D, Mora J, Golub TR,
et al: A remarkably simple genome underlies highly malignant
pediatric rhabdoid cancers. J Clin Invest. 122:2983–2988. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Yamada Y and Yamada Y: The causal
relationship between epigenetic abnormality and cancer development:
In vivo reprogramming and its future application. Proc Jpn Acad Ser
B Phys Biol Sci. 94:235–247. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Rao CV, Cooma I, Rodriguez JG, Simi B,
El-Bayoumy K and Reddy BS: Chemoprevention of familial adenomatous
polyposis development in the APC(min) mouse model by 1,4-phenylene
bis(methylene)selenocyanate. Carcinogenesis. 21:617–621. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Yamada Y, Hata K, Hirose Y, Hara A, Sugie
S, Kuno T, Yoshimi N, Tanaka T and Mori H: Microadenomatous lesions
involving loss of Apc heterozygosity in the colon of adult
Apc(Min/+) mice. Cancer Res. 62:6367–6370. 2002.PubMed/NCBI
|
|
135
|
Yamada Y and Mori H: Pre-cancerous lesions
for colorectal cancers in rodents: A new concept. Carcinogenesis.
24:1015–1019. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Moser AR, Pitot HC and Dove WF: A dominant
mutation that predisposes to multiple intestinal neoplasia in the
mouse. Science. 247:322–324. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Yamada Y, Jackson-Grusby L, Linhart H,
Meissner A, Eden A, Lin H and Jaenisch R: Opposing effects of DNA
hypomethylation on intestinal and liver carcinogenesis. Proc Natl
Acad Sci USA. 102:13580–13585. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Lin H, Yamada Y, Nguyen S, Linhart H,
Jackson-Grusby L, Meissner A, Meletis K, Lo G and Jaenisch R:
Suppression of intestinal neoplasia by deletion of Dnmt3b. Mol Cell
Biol. 26:2976–2983. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Linhart HG, Lin H, Yamada Y, Moran E,
Steine EJ, Gokhale S, Lo G, Cantu E, Ehrich M, He T, et al: Dnmt3b
promotes tumorigenesis in vivo by gene-specific de novo methylation
and transcriptional silencing. Genes Dev. 21:3110–3122. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Hatano Y, Semi K, Hashimoto K, Lee MS,
Hirata A, Tomita H, Kuno T, Takamatsu M, Aoki K, Taketo MM, et al:
Reducing DNA methylation suppresses colon carcinogenesis by
inducing tumor cell differentiation. Carcinogenesis. 36:719–729.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Khoshchehreh R, Totonchi M, Carlos Ramirez
J, Torres R, Baharvand H, Aicher A, Ebrahimi M and Heeschen C:
Epigenetic reprogramming of primary pancreatic cancer cells
counteracts their in vivo tumourigenicity. Oncogene. 38:6226–6239.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Rothwell PM, Fowkes FG, Belch JF, Ogawa H,
Warlow CP and Meade TW: Effect of daily aspirin on long-term risk
of death due to cancer: Analysis of individual patient data from
randomised trials. Lancet. 377:31–41. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Fraser DM, Sullivan FM, Thompson AM and
McCowan C: Aspirin use and survival after the diagnosis of breast
cancer: A population-based cohort study. Br J Cancer. 111:623–627.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Streicher SA, Yu H, Lu L, Kidd MS and
Risch HA: Case-control study of aspirin use and risk of pancreatic
cancer. Cancer Epidemiol Biomarkers Prev. 23:1254–1263. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Close JL, Liu J, Gumuscu B and Reh TA:
Epidermal growth factor receptor expression regulates proliferation
in the postnatal rat retina. Glia. 54:94–104. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Karl MO, Hayes S, Nelson BR, Tan K,
Buckingham B and Reh TA: Stimulation of neural regeneration in the
mouse retina. Proc Natl Acad Sci USA. 105:19508–19513. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Takeda M, Takamiya A, Jiao JW, Cho KS,
Trevino SG, Matsuda T and Chen DF: alpha-Aminoadipate induces
progenitor cell properties of Muller glia in adult mice. Invest
Ophthalmol Vis Sci. 49:1142–1150. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Osakada F, Ooto S, Akagi T, Mandai M,
Akaike A and Takahashi M: Wnt signaling promotes regeneration in
the retina of adult mammals. J Neurosci. 27:4210–4219. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Zhou Q, Brown J, Kanarek A, Rajagopal J
and Melton DA: In vivo reprogramming of adult pancreatic exocrine
cells to beta-cells. Nature. 455:627–632. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Qian L, Huang Y, Spencer CI, Foley A,
Vedantham V, Liu L, Conway SJ, Fu JD and Srivastava D: In vivo
reprogramming of murine cardiac fibroblasts into induced
cardiomyocytes. Nature. 485:593–598. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Kurita M, Araoka T, Hishida T, O'Keefe DD,
Takahashi Y, Sakamoto A, Sakurai M, Suzuki K, Wu J, Yamamoto M, et
al: In vivo reprogramming of wound-resident cells generates skin
epithelial tissue. Nature. 561:243–247. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Wang G, Badylak SF, Heber-Katz E, Braunhut
SJ and Gudas LJ: The effects of DNA methyltransferase inhibitors
and histone deacetylase inhibitors on digit regeneration in mice.
Regen Med. 5:201–220. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Ma X, Kong L and Zhu S: Reprogramming cell
fates by small molecules. Protein Cell. 8:328–348. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Tang Y and Cheng L: Cocktail of chemical
compounds robustly promoting cell reprogramming protects liver
against acute injury. Protein Cell. 8:273–283. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Niu W, Zang T, Zou Y, Fang S, Smith DK,
Bachoo R and Zhang CL: In vivo reprogramming of astrocytes to
neuroblasts in the adult brain. Nat Cell Biol. 15:1164–1175. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Guan J, Wang G, Wang J, Zhang J, Fu Y,
Cheng L, Meng G, Lyu Y, Zhu J, Li Y, et al: Chemical reprogramming
of human somatic cells to pluripotent stem cells. Nature.
605:325–331. 2022. View Article : Google Scholar : PubMed/NCBI
|