Introduction
Arsenic (As) is a natural element in earth crust
with nonmetallic and metallic properties (1,2),
arsenic combines with sulfur, chlorine and oxygen to form inorganic
compounds; while it will combine with hydrogen and carbon to form
organic compounds in animals and plants (1,2).
It is well known that organic arsenic is less toxic than inorganic
arsenic and pentavalent and zero-valent arsenic is less toxic than
trivalent arsenite (3,4). Arsenic trioxide (ATO), an ingredient
of Chinese medicine, has also been discovered upon its antitumor
potency first in patients with acute promyelocytic leukemia (APL)
(5). Several studies have
reported that ATO can induce apoptosis in tumor cells, including
APL (6), lung cancer (7) and multiple myeloma (8). Arsenic hexoxide, another inorganic
arsenic compound, is similarly found to possess anticancer effects
against MCF-7 human breast cancer cells (9). In addition, Darinaparsin
(S-dimethylarsino-glutathione), an organic arsenic compound, is
safer than other organic arsenic compounds in hematologic and solid
tumor treatment in preclinical models (10). Hence, not only inorganic arsenic
compounds but organic ones possess capability resisting in
vitro and in vivo tumor progression.
There are a number of mechanisms underlying cell
death, which occurs when cells sense environmental stresses or
intracellular signals (11). Cell
death is categorized into autophagy, apoptosis or necrosis
according to morphological changes (12). The first two types belong to
programmed cell death (13).
Defective apoptosis is considered as major relevant factor in
cancer development and that tumor cells avoid apoptosis serves an
important role in drug resistance to therapeutic regimens (14).
The features of apoptosis include cell shrinkage,
plasma membrane blebbing, DNA fragmentation and chromatin
condensation (15). Apoptosis can
be characterized into intrinsic and extrinsic pathways according to
the stimuli and the related aspartate-specific cysteine protease
(caspase) cascade (16). The
intrinsic pathway depends on the decrease of mitochondrial membrane
potential (MMP), which releases cytochrome c into the cytosol from
the mitochondrial intermembrane space when facing cellular stress,
such as DNA damage, UV exposure, or hypoxia (17). Then, cytochrome c will bind to
apoptotic protease activating factor 1 (Apaf-1) forming a complex
named ‘apoptosome’, which functions to recruit procaspase-9
(18). The active caspase-9 then
cleaves procaspase-3 and the cleaved caspase-3 enters cytosol to
stimulate the cleavage of poly ADP-ribose polymerase (PARP) for the
induction of apoptosis (17,19). By contrast, the extrinsic pathway
is initiated through death receptors, such as Fas/CD95, DR3, TNFR1,
DR4 and DR5, when these receptors are associated with certain
ligands (16). After binding,
activated receptor would recruit related signal molecules to
interact with death domains, leading to caspase-8 cleavage
(16,20). Activated caspase-8 will then
stimulate protease cascade to cleave caspase-3 and PARP within
cells, resulting in apoptosis (17).
MAPKs are essential signaling pathways maintaining
cell survival responding to various stresses (21). Conventional MAPKs include three
members: ERK, p38 and JNK (22),
which regulate cell mitosis, survival, gene expression,
proliferation, differentiation and apoptosis (23). According to different cell types
and stimuli, MAPKs can promote cells to survive and/or to undergo
cell death (20,21,24). In fact, ATO can induce p38 and JNK
phosphorylation, which result in human cervical cancer cell death
through mitochondrial apoptotic cascade death (25).
Head and neck squamous cell carcinoma (HNSCC)
develops from mucosa of upper aerodigestive tract (26) and usually occurs in males and it
is more often among individuals over aged >50 (27). It is strongly associated with
lifestyle and environmental risk causes, including betel chewing,
alcohol consumption, tobacco smoking, UV light exposure and
infection with human papillomavirus (28,29). HNSCC can be treated with
chemotherapy, radiation therapy, surgery, gene therapy and
immunotherapy (30). However,
these treatments are not good enough. In fact, oral cancer has been
in the top 10 commonest cancers for years in Taiwan, and the
age-standardized incidence rate of head and neck cancers has
increased by 5.4% per year among males and 3.1% among females from
1980–2014 (31,32). Thus, other alternative drugs with
improved efficacy and lower side effects for therapeutic remedy on
HNSCC will be needed. However, whether arsenic compounds could
serve as therapeutic agents for oral cavity cancers and the
relative regulating mechanisms remains to be elucidated.
Oral squamous cell carcinoma contributes 95% of
HNSCC, which is highly aggressive and malignant. Recently, studies
have shown the MAPK signaling pathway is stimulated in >50% of
human oral cancer cases (33).
Thus, an investigation on the MAPK signaling pathway with its
potential therapeutic mechanisms for oral squamous cell carcinoma
could be useful. To investigate anticancer effect of arsenic
compounds with underlying mechanisms associated with possible
therapeutic remedy treating oral cavity cancer, OC3 cells (a Taiwan
native oral cancer cell line with a long-term areca betel chewer
who did not smoke) (34) were
used in the present study. OC3 cells were treated with
NaAsO2 and DMA to examine if arsenic compounds would
influence cell viability, cell cycle and related signal pathways to
induce apoptosis with the anti-cancer capability. It was found that
both NaAsO2 and DMA could activate MAPK and caspase
pathways to cause cell cycle dysregulation, which lead to OC3 cell
apoptosis. These findings may provide therapeutic information for
oral cancer in Taiwan.
Materials and methods
Chemicals
Disodium hydrogen phosphate
(Na2HPO4), sodium bicarbonate
(NaHCO3) and potassium dihydrogen phosphate
(KH2PO4) were bought from Honeywell Specialty
Chemicals Seelze GmbH. FBS and Trypsin-EDTA were obtained from AG
Scientific. The Annexin V-FITC apoptosis detection kit was bought
from Strong Biotech Corporation. DMSO, hydrochloric acid, Tween-20,
SDS, sodium hydroxide DMA, NaAsO2, staurosporine,
high-glucose DMEM, PI, RNase A, MTT and penicillin-streptomycin
were purchased from MilliporeSigma. A Micro BCA protein assay kit
was purchased from Thermo Fisher Scientific, Inc. HEPES, potassium
chloride, sodium chloride and Tris base were procured from J.T.
Baker. Antibodies against β-actin (cat. no. 58169; 1:5,000), JNK
(cat. no. 9252), phosphorylated (p)-JNK (cat. no. 9251), ERK1/2
(cat. no. 9102), p-ERK1/2 (cat. no. 9101), p38 (cat. no. 9212),
p-p38 (cat. no. 9215), cleaved caspase-8 (cat. no. 9429), cleaved
PARP (cat. no. 9542), cleaved caspase-9 (cat. no. 9509) and cleaved
caspase-3 (cat. no. 9661) were attained from Cell Signaling
Technology, Inc. Donkey anti-rabbit IgG (cat. no. NEF81200-1EA)
conjugated to HRP was bought from PerkinElmer, Inc. ECL detection
kit was obtained from MilliporeSigma.
Cell culture
OC3 cell line, a Taiwan native oral cancer cell line
from a long-term areca betel chewer who did not smoke (34), was a gift from Professor Kuo-Wei
Chang (National Yang-Ming University). OC3 cells were kept in
DMEM/F12 and 2-fold volume of Keratinocyte-SFM medium (Thermo
Fisher Scientific, Inc.). All media were complemented with 25 mM
HEPES, 24 mM NaHCO3, 10,000 U streptomycin, 10,000 U
penicillin and 10% fetal bovine serum at pH 7.4. Cells were
incubated in a humidified atmosphere containing 95% air and 5%
CO2 at 37°C.
Morphology observation
OC3 cells (6×105) were cultured in 6 cm
Petri dish with 2 ml medium. After reaching 70–80% confluence,
cells were treated for 24 h with different dosages of sodium
arsenite (0, 0.1, 1, 10, 25, 50 and 100 µM) or dimethylarsenic acid
(0, 0.1, 1, 2, 5, 10, 25, 50 and 100 mM), respectively. The changes
of cell morphology were inspected with 10 random fields in each
treatment using light microscopy at 40× magnification (Olympus
CK40; Olympus Corporation) and images captured using an Olympus
DP20 digital camera (Olympus Corporation).
MTT viability assay
OC3 cells were cultured at 1×104 cells in
100 µl medium per well. After reaching 70–80% confluence, cells
were treated for 24 h with different dosages of sodium arsenite (0,
0.1, 1, 10, 25, 50 and 100 µM) or dimethylarsenic acid (0, 0.1, 1,
2, 5, 10, 25, 50 and 100 mM), respectively. MTT at 0.5 mg/ml final
concentration was then added and incubated at 37°C for another 4 h.
The medium was removed and 50 µl DMSO was put into all wells
dissolving the crystals through plate shaking for 20 min in the
dark. Absorbance values among treatments were then examined at
λ=570 nm using VersaMax ELISA reader (Molecular Devices, LLC.).
Cell cycle investigation
To test if sodium arsenite and dimethylarsenic acid
could stimulate apoptosis in OC3 cells, cell cycle redistribution
was studied using flow cytometry with propidium iodide staining.
OC3 cells (6×105) were cultured and, after 70–80%
confluence, treated with different dosages of sodium arsenite (0,
0.1, 1, 10, 25, 50 and 100 µM) and different dosages of
dimethylarsenic acid (0, 0.1, 1, 10, 25, 50 and 100 mM) for 24 h,
respectively. Cells were then collected with trypsin and
centrifuged at 400 × g and 4°C for 12 min, washed by isoton II and
fixed with 70% ethanol at −20°C for 2 h. After fixation, OC3 cells
were rinsed again by isoton II and harvested through centrifugation
at 400 × g for 12 min at 4°C. OC3 cell pellets were suspended again
by isoton II and then mixed with 40 µg/ml propidium iodide plus 100
µg/ml RNase for 30 min at 25°C. A flow cytometer (FACScan; BD
Biosciences) was used with excitation set at λ=488 nm to determine
stained cell distributions. DNA diploid will be observed in
G1 phase while DNA synthesis progress exists in
G2/M phase in normal cells. Less DNA content and
hypodiploid, is detected in subG1 phase cells,
considered as cell apoptosis (35).
Annexin V/PI double staining
assay
Treated OC3 cells were collected with trypsin and
then rinsed with 2 ml medium. After 160 × g centrifugation at 4°C
for 10 min, cell pellets were suspended again by cold isoton II and
centrifuged at 400 × g for 12 min at 4°C. Cell pellets were
subsequently mixed with staining solution (100 µl) and reacted for
15 min at 25°C. Stained OC3 cells were determined by FACScan flow
cytometer (BD Biosciences) with excitation at λ=488 nm using
>600 nm band pass filter for PI detection and 515 nm band pass
filter for FITC detection, respectively. Plots displayed four
quadrants with staining annexin V/PI double-positive (late
apoptosis) cells, PI-positive (necrosis) cells, annexin V-positive
(early apoptosis) cells and negative (viable) cells, respectively
(36). It should be noted that
control and staurosporine results in Fig. 5A and Fig. 6A are same. The reason is that
control, staurosporine, sodium arsenite and dimethylarsenic acid
treatments were conducted simultaneously in experiments. To clearly
show the results for sodium arsenite and dimethylarsenic acid
treatments, respectively, Figs. 5
and 6 were therefore created
using same control and staurosporine results for comparison.
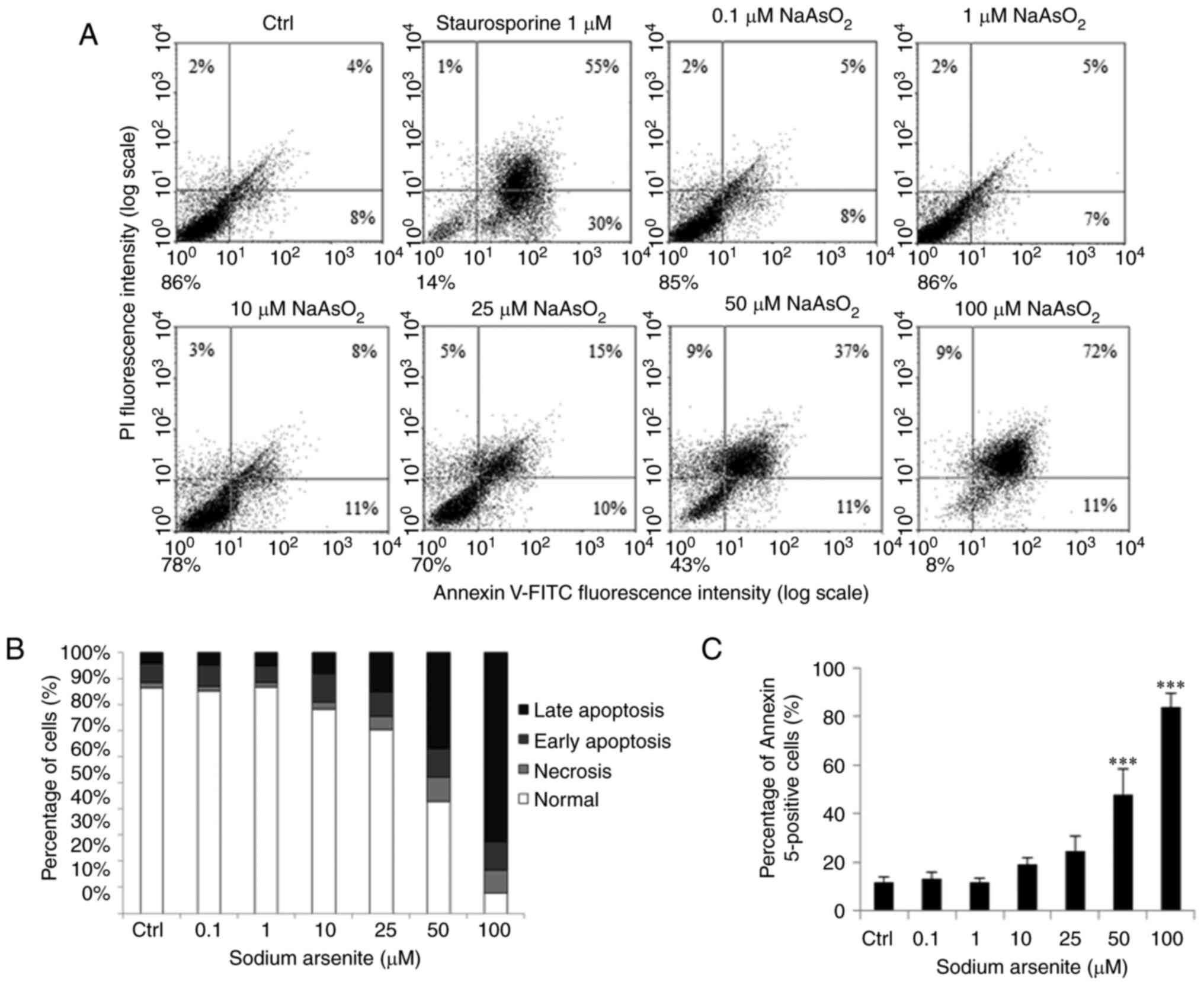 | Figure 5.Sodium arsenite induces cell
apoptosis in OC3 cells. OC3 cells were incubated with
NaAsO2 at 0, 0.1, 1, 10, 25, 5,0 and 100 µM for 24 h.
Staurosporine was used to treat cells for positive control. (A)
Annexin V/PI double staining assay was used to determine cell
apoptotic status. (B) Percentages of double-positive (late
apoptotic), annexin V single-positive (early apoptotic), PI
single-positive (necrotic) and double-negative (viable) cells. (C)
Double-positive (late apoptotic) and annexin V single-positive
(early apoptotic) cells were analyzed. Results are demonstrated as
mean ± standard error of the mean of three independent experiments.
***P<0.001 vs. control. Ctrl, control; PI, propidium iodide. |
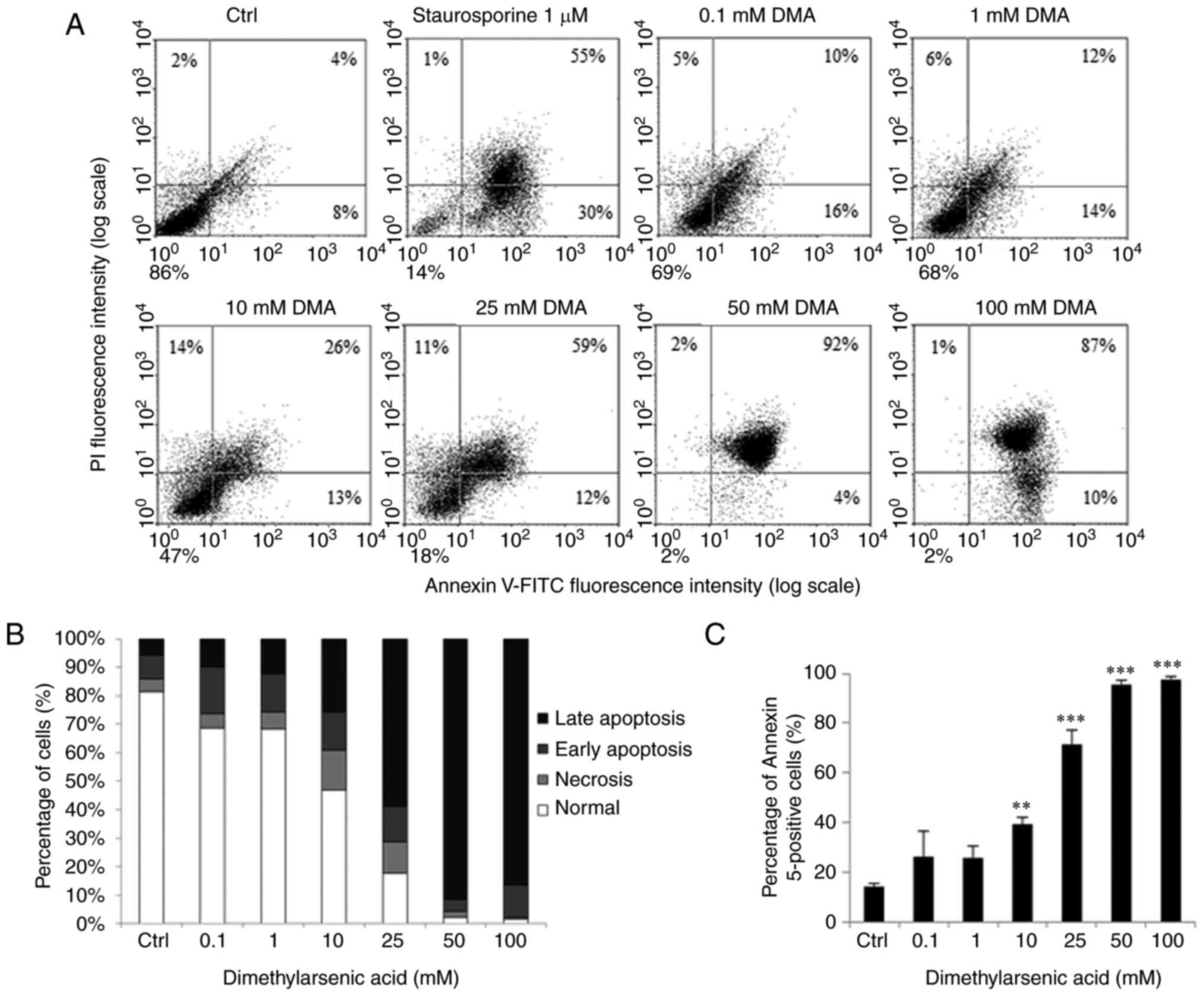 | Figure 6.Dimethylarsenic acid induces cell
apoptosis in OC3 cells. OC3 cells were incubated with DMA at 0,
0.1, 1, 10, 25, 50 and 100 mM for 24 h. Staurosporine was used to
treat cells for positive control. (A) Annexin V/PI double staining
assay was used to determine cell apoptotic status. (B) Percentages
of double-positive (late apoptotic), annexin V single-positive
(early apoptotic), PI single-positive (necrotic) and
double-negative (viable) cells. (C) Double-positive (late
apoptotic) and annexin V single-positive (early apoptotic) cells.
Results are demonstrated as mean ± standard error of the mean of
three independent experiments. Control and staurosporine results in
(A) are same as in Fig. 5, as
Control, staurosporine, sodium arsenite and dimethylarsenic acid
treatments were conducted simultaneously. **P<0.01 and
***P<0.001 vs. control. PI, propidium iodide; Ctrl, control. |
Western blotting
Cells at 6×105 were cultured with 60 mm
dish. After 70–80% confluence, different dosages of sodium arsenite
(0, 10 and 25 µM) or dimethylarsenic acid (0, 10 and 25 mM) were
added to OC3 cells for 3 to 24 h, respectively. Cell culture medium
was then moved to a 15 ml tube and collected by centrifugation
(1,500 × g; 10 min at 4°C). Cells were broken down by 100 µl lysis
solution plus proteinase inhibitor. Pellets were suspended again by
10 µl lysis solution to blend into cell lysates and conducted with
centrifugation (12,000 × g; 12 min at 4°C). Supernatants were then
collected and kept at −80°C. Protein concentrations of cell lysates
were determined by Bio-Rad protein assay dye reagent concentrate
(Bio-Rad Laboratories, Inc.) (19). Cell lysates (20–30 µg) were
resolved in SDS-PAGE gel (12%) with running buffer at 25°C and
transferred to PVDF membranes, electrophoretically, at 4°C.
Membranes was blocked in 2–4% skimmed milk at room temperature for
1 h and incubated with primary antibodies [cleaved caspase-3 (cat.
no. 9661; 1:1,000), cleaved caspase-9 (cat. no. 9509; 1:1,000),
cleaved PARP (cat. no. 9542; 1:1,000), cleaved caspase-8 (cat. no.
9429; 1:1,000), ERK1/2 (cat. no. 9102; 1:4,000), phospho-ERK1/2
(cat. no. 9101; 1:4,000), p38 (cat. no. 9212; 1:4,000), phospho-p38
(cat. no. 9215; 1:1,000), JNK (cat. no. 9252; 1:1,000), phospho-JNK
(cat. no. 9251; 1:4,000)] at 4°C overnight. After washing with TBS
Tween-20 and incubated with horseradish peroxidase-conjugated
secondary antibodies for 1 h at 25°C (Donkey anti-rabbit IgG; cat.
no. NEF81200-1EA; 1:2000), membranes were then visualized by
enhanced chemiluminescence detection kit using UVP EC3 (BioImaging
Systems). Quantification for each band was achieved using ImageJ
version 1.50 software (National Institutes of Health).
Statistical analysis
Data were expressed as mean ± SEM from ≥3 separate
experiments. Statistical significance between treatment and control
groups was examined by one-way ANOVA and then Tukey's test with
GraphPad Prism 9 software (GraphPad Software, Inc.). P<0.05 was
considered to indicate a statistically significant difference.
Results
Arsenic compounds induce morphological
changes in OC3 cells
OC3 cells were treated for 24 h in plain medium,
sodium arsenite (0, 0.1, 1, 10, 25, 50 and 100 µM; Fig. 1A-G), or dimethylarsenic acid (0,
0.1, 1, 10, 25, 50 and 100 mM; Fig.
1H-M), respectively. Cell morphological alterations were then
observed under light microscopy. In sodium arsenite experiment, OC3
cells showed spindle shape in control treatment (Fig. 1A). Cells appeared rounded and
floated in medium as the dosages increased and attached cells
significantly decreased from 25 to 100 µM groups in OC3 cells
(Fig. 1E to G), respectively. In
dimethylarsenic acid experiment, cells started to float in OC3
cells treated by 0.1 mM dimethylarsenic acid (Fig. 1H). As concentration of
dimethylarsenic acid increased, cells shrank with membrane
blebbing, implying that cells underwent apoptosis (Fig. 1I to M). Fig. 1N is a ×4 enlargement of cell
membrane blebbing in Fig. 1F,
which clearly demonstrates sodium arsenite could induce OC3 cell
apoptosis.
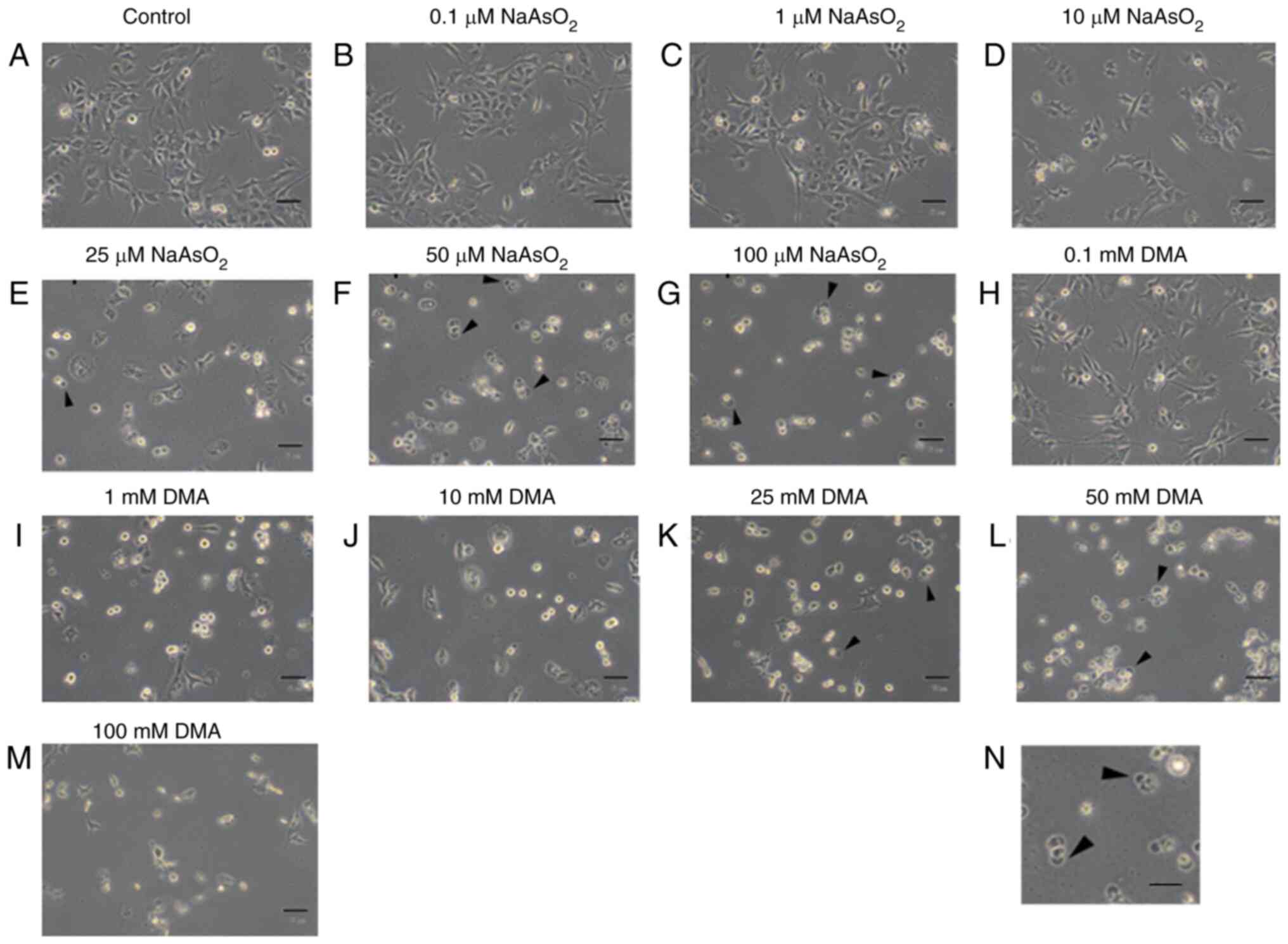 | Figure 1.Arsenic compounds induce
morphological changes in OC3 cells. OC3 cells were incubated with
(A) plain medium control, (B) 0.1 µM, (C) 1 µM, (D) 10 µM, (E) 25
µM, (F) 50 µM and (G) 100 µM sodium arsenite, or (H) 0.1 mM, (I) 1
mM, (J) 10 mM, (K) 25 mM, (L) 50 mM and (M) 100 mM dimethylarsenic
acid for 24 h, respectively, repeated three times. Changes of cell
morphology were observed under phase contrast microscopy (scale
bar, 50 µm). Cell membrane blebbings are arrowed. (N) the ×4
enlarged image of cell membrane blebbing in (F) (scale bar, 200
µm). DMA, dimethylarsenic acid. |
These data suggested both dimethylarsenic acid and
sodium arsenite could induce the abnormal morphological changes
associated with apoptotic cell death in OC3 oral cancer cells with
dose-dependent relationships.
Arsenic compounds reduces cell
viability in OC3 cells
Survival rates in OC3 cells following treatments of
arsenic compounds were further determined by MTT assay. OC3 cells
were treated for 24 h with sodium arsenite (0, 0.1, 1, 10, 25, 50
and 100 µM) or dimethylarsenic acid (0, 0.1, 1, 10, 25, 50 and 100
mM), respectively and results showed that both arsenic compounds
significantly reduced OC3 cell viability with dose-dependent
phenomena (P<0.05). The survival rate significantly decreased by
sodium arsenite from 10 to 100 µM (Fig. 2A) and by dimethylarsenic acid from
1 to 100 mM (Fig. 2B) in OC3
cells, respectively (P<0.05).
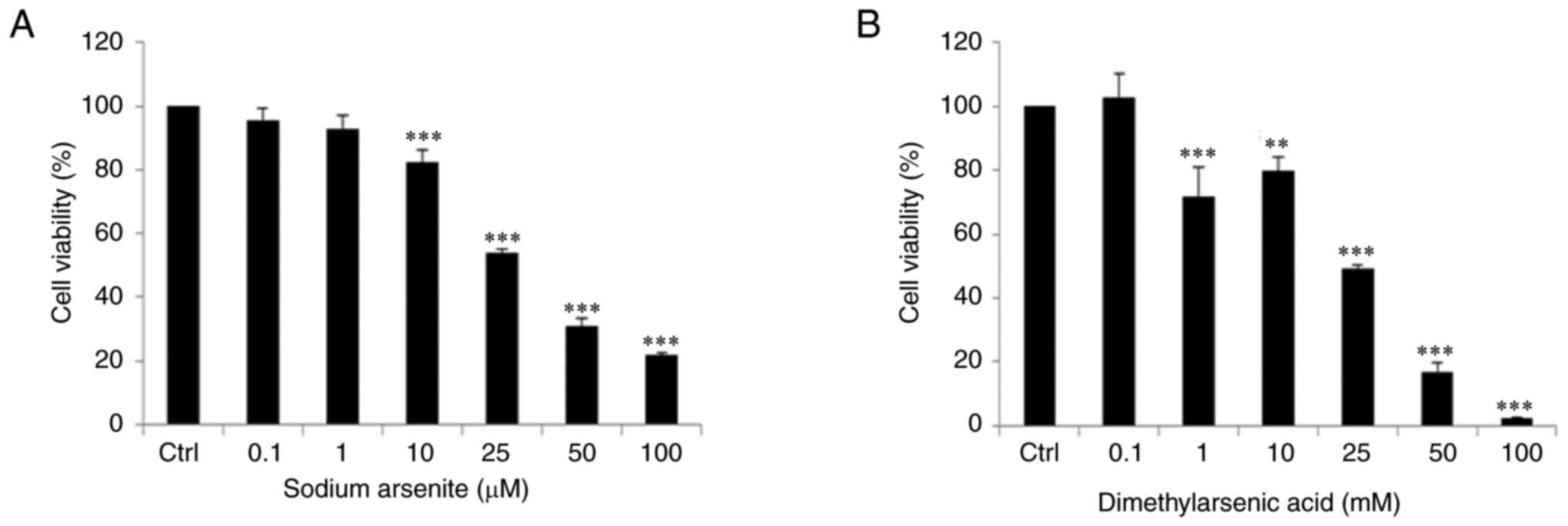 | Figure 2.Arsenic compounds reduce cell
viability in OC3 cells. Cells were incubated with (A) 0, 0.1, 1,
10, 25, 50 and 100 µM sodium arsenite and with (B) 0, 0.1, 1, 10,
25, 50 and 100 mM dimethylarsenic acid for 24 h, respectively. The
cell viability was determined through MTT assay. Data are
illustrated by percentages of cell proliferation rate compared with
control group. Results are mean ± standard error of the mean of
three independent experiments. **P<0.01 and ***P<0.001 vs.
control. Ctrl, control. |
The concentration of dimethylarsenic acid reducing
cell viabilities to 50% in OC3 cells was ~1,000 times higher
compared with the concentration of sodium arsenite. Therefore,
sodium arsenite showed stronger cytotoxicity than dimethylarsenic
acid in OC3 cells.
Arsenic compounds modulates
redistribution of cell cycle in OC3 cells
Studies have shown the subG1 phase is a
sign of DNA fragmentation for apoptosis and G2/M phase
arrest could lead cells to undergo apoptosis (20,37). To test whether sodium arsenite and
dimethylarsenic acid could induce cell death through apoptosis, OC3
cells were challenged with arsenic compounds and DNA contents were
detected through propidium iodide staining with flow cytometry
analysis. OC3 cells were treated with different concentrations of
sodium arsenite (0, 0.1, 1, 10, 25, 50 and 100 µM) and
dimethylarsenic acid (0, 0.1, 1, 10, 25, 50 and 100 mM) for 24 h to
examine possible impacts on cell cycle regulation (Figs. 3 and 4), respectively.
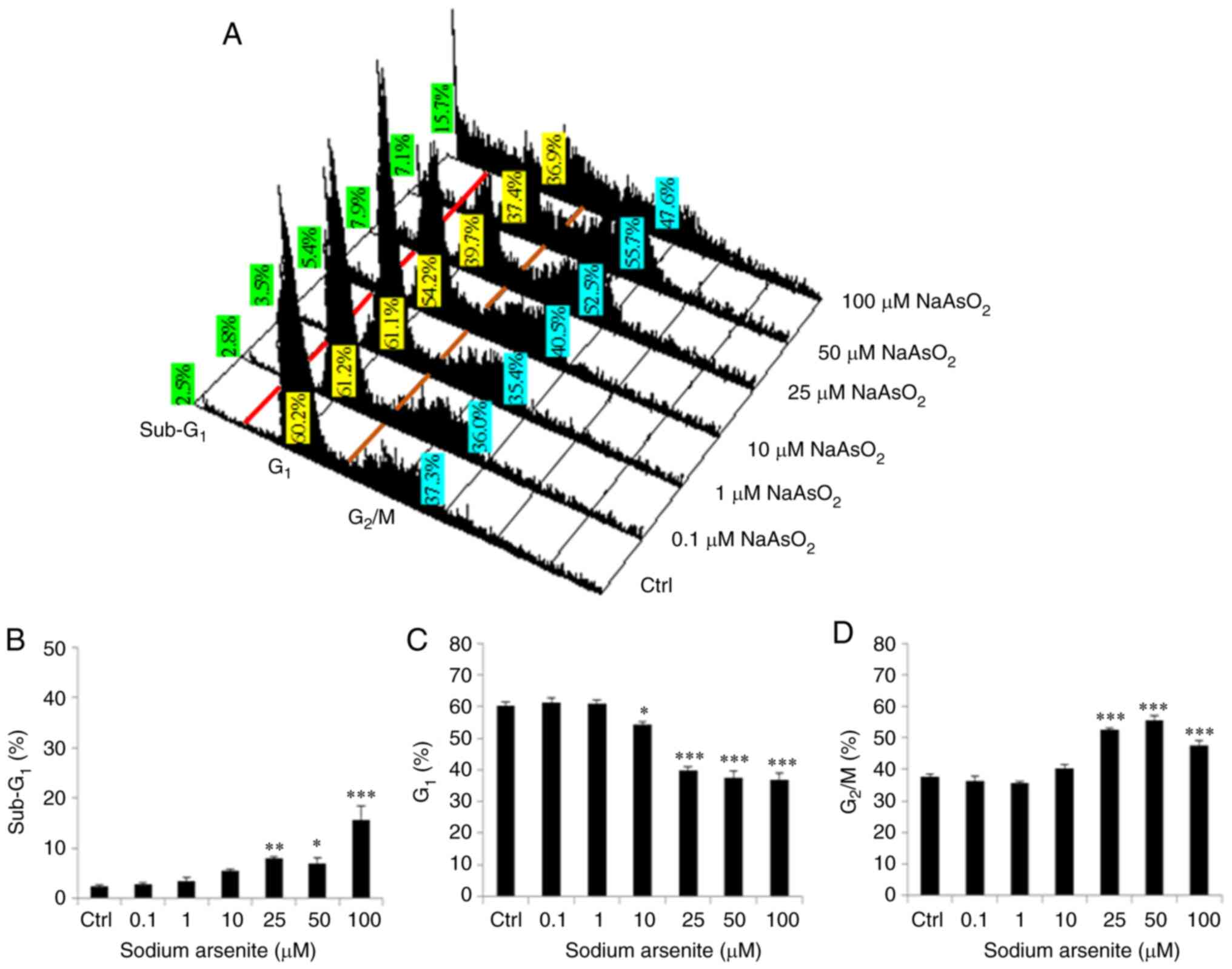 | Figure 3.Sodium arsenite modulates
redistribution of cell cycle in OC3 cells. OC3 cells were treated
with sodium arsenite at 0, 0.1, 1, 10, 25, 50 and 100 µM for 24 h
and cells were then fixed, stained with propidium iodide and
evaluated through flow cytometry assay. (A) Red and brown lines are
plotted to illustrate the changes of sub-G1 (left to red
line), G0/G1 (between red and brown lines) and
G2/M phases (right to brown line) in the different
treatment groups. Values with green background illustrate
percentages of sub G1 cells (left to red line) in
different treatments, which is also shown in (B) Values with yellow
background illustrate percentages of G0/G1 cells
(between red and brown lines) in different treatments, which is
also shown in (C) Values with blue background illustrate
percentages of G2/M cells (right to brown line) in
different treatments, which is also shown in (D) There is less DNA
content in sub-G1 phase cells than normal cells, which
indicates apoptosis. Results are demonstrated as mean ± standard
error of the mean of three independent experiments. *P<0.05,
**P<0.01 and ***P<0.001 vs. control. Ctrl, control. |
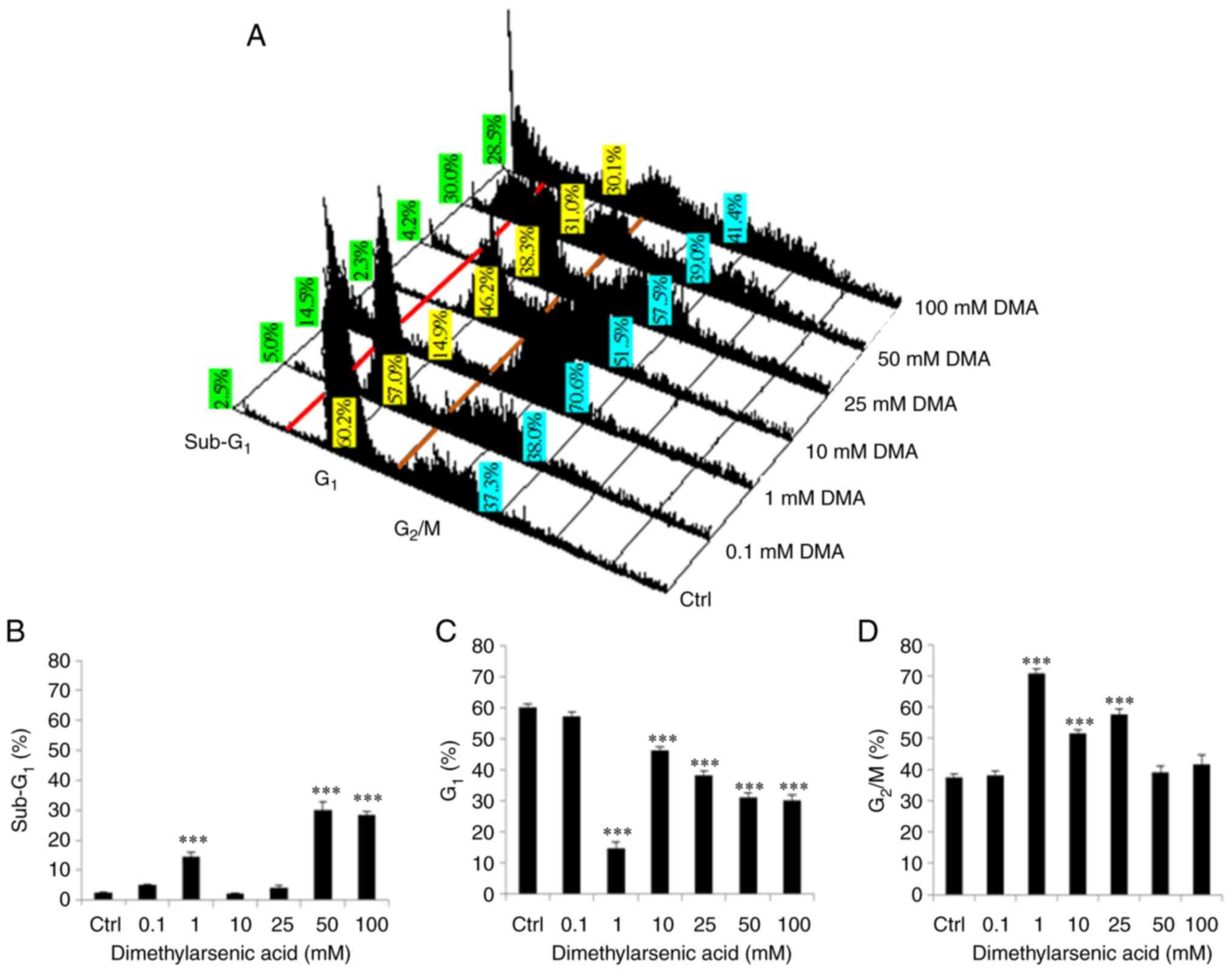 | Figure 4.Dimethylarsenic acid modulates
redistribution of cell cycle in OC3 cells. OC3 cells were treated
with dimethylarsenic acid at 0, 0.1, 1, 10, 25, 50 and 100 mM for
24 h and cells were then fixed, stained with propidium iodide and
evaluated through flow cytometry assay. (A) Red and brown lines are
plotted to illustrate the changes of sub-G1 (left to red
line), G0/G1 (between red and brown lines) and
G2/M phases (right to brown line) in the different
treatment groups. Values with green background illustrate
percentages of sub G1 cells (left to red line) in
different treatments, which is also shown in (B) Values with yellow
background illustrate percentages of G0/G1 cells
(between red and brown lines) in different treatments, which is
also shown in (C) Values with blue background illustrate
percentages of G2/M cells (right to brown line) in
different treatments, which is also shown in (D) There is less DNA
content in subG1 phase cells than normal cells, which
indicates apoptosis. Results are demonstrated as mean ± standard
error of the mean of three independent experiments. ***P<0.001
vs. control. Ctrl, control. |
In the present study, significant increase of
subG1 phase cell number was observed following 24 h
treatment of sodium arsenite at 25 to 100 µM in OC3 cells (Fig. 3A and B; P<0.05). The number of
G1 phase cells was decreased following 24 h treatment of
sodium arsenite from 10 to 100 µM in OC3 cells (Fig. 3A and C; P<0.05). In addition,
25 to 100 µM sodium arsenite significantly induced OC3 cell
G2/M phase arrest (Fig. 3A
and D; P<0.05).
In addition, dimethylarsenic acid at 1, 50 and 100
mM significantly increased subG1 phase cell numbers
(Fig. 4A and B; P<0.05); 1–100
mM significantly reduced G1 phase cell numbers (Fig. 4A and C; P<0.05); and 1 to 25 mM
significantly increased G2/M phase cell number,
respectively (Fig. 4A and D;
P<0.05).
It should be noted that dimethylarsenic acid at 1,
50 and 100 mM, but not 25 mM, could increase subG1 phase
cell number in OC3 cells (Fig. 4A and
B). Moreover, dimethylarsenic acid at high concentration could
increase G2/M phase cell number in OC3 cells (Fig. 4A and D). The data illustrated that
sodium arsenite and dimethylarsenic acid could modulate
subG1, G1 and G2/M phase numbers
in OC3 cells.
Arsenic compounds induce cell
apoptosis in OC3 cells
To confirm if apoptosis was induced with arsenic
compounds in OC3 cells, annexin V and propidium iodide double
staining assay with flow cytometry was used. Percentages of
double-positive (late apoptotic), annexin V single-positive (early
apoptotic), PI single-positive (necrotic) and double-negative
(viable) cells are shown in a four quadrants to determine cell
apoptosis (36). The results
showed that 24 h treatments with sodium arsenite (50 and 100 µM;
Fig. 5A-C) and dimethylarsenic
acid (10–100 mM; Fig. 6A-C) for
significantly induced OC3 cell apoptosis (P<0.05).
Arsenic compounds induces extrinsic
and intrinsic caspases pathways in OC3 cells
To further investigate whether arsenic compounds
could induce cell death involved in extrinsic (death receptor) or
intrinsic (mitochondrial) apoptotic pathways, expressions of
cleaved caspase-9 and caspase-8 with the downstream targets of
caspase-3 plus PARP cleavages were determined through western
blotting. The results showed that 24 h treatment with 25 µM sodium
arsenite could considerably stimulate cleaved caspase-8, −9, −3 and
PARP expressions in OC3 cells (Fig.
7A to E; P<0.05). Treatment for 24 h with 10 µM sodium
arsenite could also significantly stimulate the expressions of
cleaved caspase-9, −3 and PARP in OC3 cells (Fig. 7A to E; P<0.05). In addition,
treatment for 12 h with 25 µM sodium arsenite could significantly
induce cleaved caspase-3 expression (Fig. 7A and D; P<0.05). Treatments for
12 and 24 h with 1 mM dimethylarsenic acid significantly activated
expressions of cleaved caspase-8, −9 and −3, resulting in PARP
cleavage at 24 h treatment in OC3 cells (Fig. 8A to E; P<0.05).
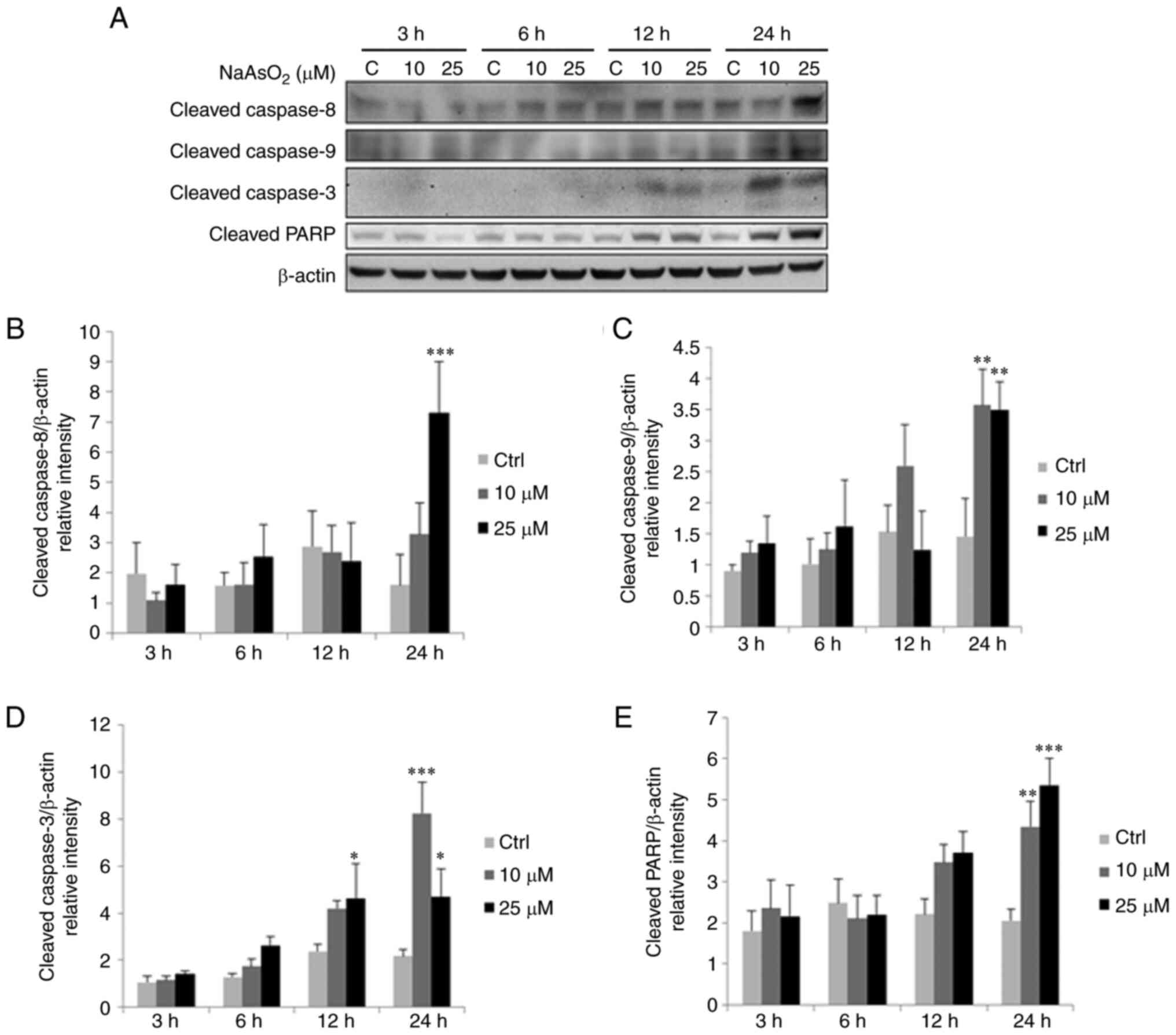 | Figure 7.Sodium arsenite upregulates cleaved
caspase-9, −8, −3 and PARP protein expressions in OC3 cells. OC3
cells were incubated with 0, 10 and 25 µM NaAsO2 for 3,
6, 12 and 24 h, respectively. (A) Expression of cleaved caspase-9
(35/37 kDa), −8 (43 kDa), −3 (17/19 kDa) and PARP (85~90 kDa) were
examine via western blotting. Integrated optical intensities of (B)
cleaved caspase-8, (C) −9, (D) −3 and (E) PARP were standardized by
β-actin (43 kDa) among lanes. Results are demonstrated as mean ±
standard error of the mean of three independent experiments.
*P<0.05, **P<0.01 and ***P<0.001 vs. control. Ctrl,
control. |
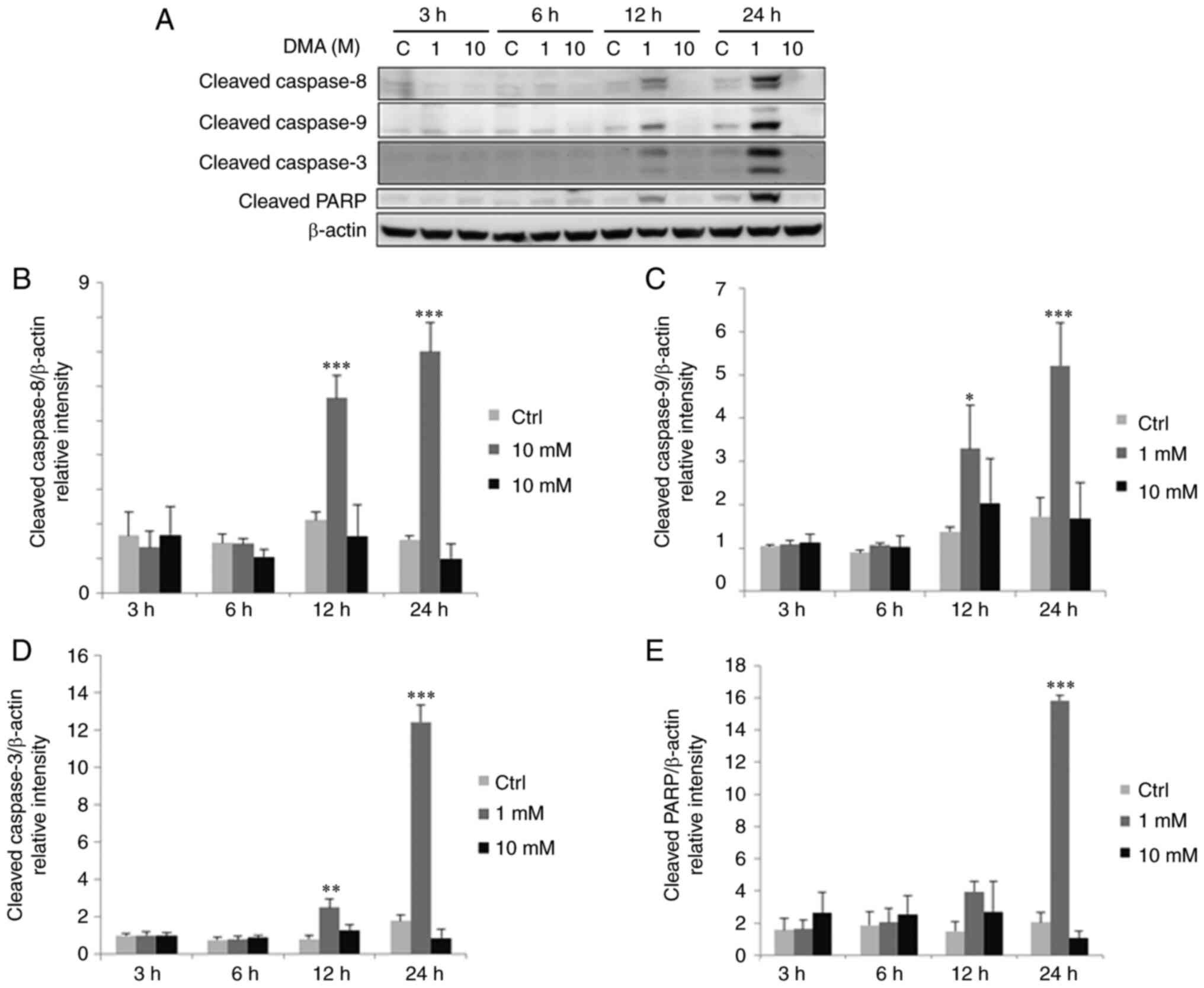 | Figure 8.Dimethylarsenic acid upregulates
cleaved caspase-9, −8, −3 and PARP protein expressions in OC3
cells. OC3 cells were incubated with 0, 1 and 10 mM DMA for 3, 6,
12 and 24 h. (A) Expression of cleaved caspase-9 (35/37 kDa), −8
(43 kDa), −3 (17/19 kDa) and PARP (85~90 kDa) were examine via
western blotting (A) Integrated optical intensities (B) cleaved
caspase-8, (C) −9, (D) −3 and (E) PARP were standardized by β-actin
(43 kDa) among lanes. Results are demonstrated as mean ± standard
error of the mean of three independent experiments. *P<0.05,
**P<0.01 and ***P<0.001 vs. control. DMA, dimethylarsenic
acid; C/Ctrl, control. |
Arsenic compounds induce MAPK pathways
in OC3 cells
Studies have shown MAPK signaling pathways are
involved to regulate cell survival, differentiation, gene
expression, proliferation, mitosis and/or apoptosis (21–24). To test if MAPK pathways were
associated with apoptosis induced by arsenic compounds in OC3
cells, JNK, ERK1/2 and p38 phosphorylation was analyzed with
western blotting. Results showed that treatments for 6 and 24 h
with 25 µM sodium arsenite significantly increased JNK
phosphorylation in OC3 cells (Fig. 9A
and B; P<0.05), while treatments for 3, 6, 12 and 24 h with
25 µM sodium arsenite significantly increased ERK1/2
phosphorylation in OC3 cells (Fig. 9A
and C; P<0.05). Phosphorylated p38 was significantly induced
after 24 h treatment of 25 µM sodium arsenite (Fig. 9A and D; P<0.05). Treatments for
3 and 6 h with 10 mM dimethylarsenic acid significantly increased
JNK phosphorylation (Fig. 10A and
B; P<0.05), while treatments for 3, 6, 12 and 24 h with 10
mM dimethylarsenic acid significantly increased ERK1/2
phosphorylation (Fig. 10A and C;
P<0.05). Phosphorylated p38 was significantly increased with 12
and 24 h treatments of 10 mM dimethylarsenic acid (Fig. 10A and D; P<0.05).
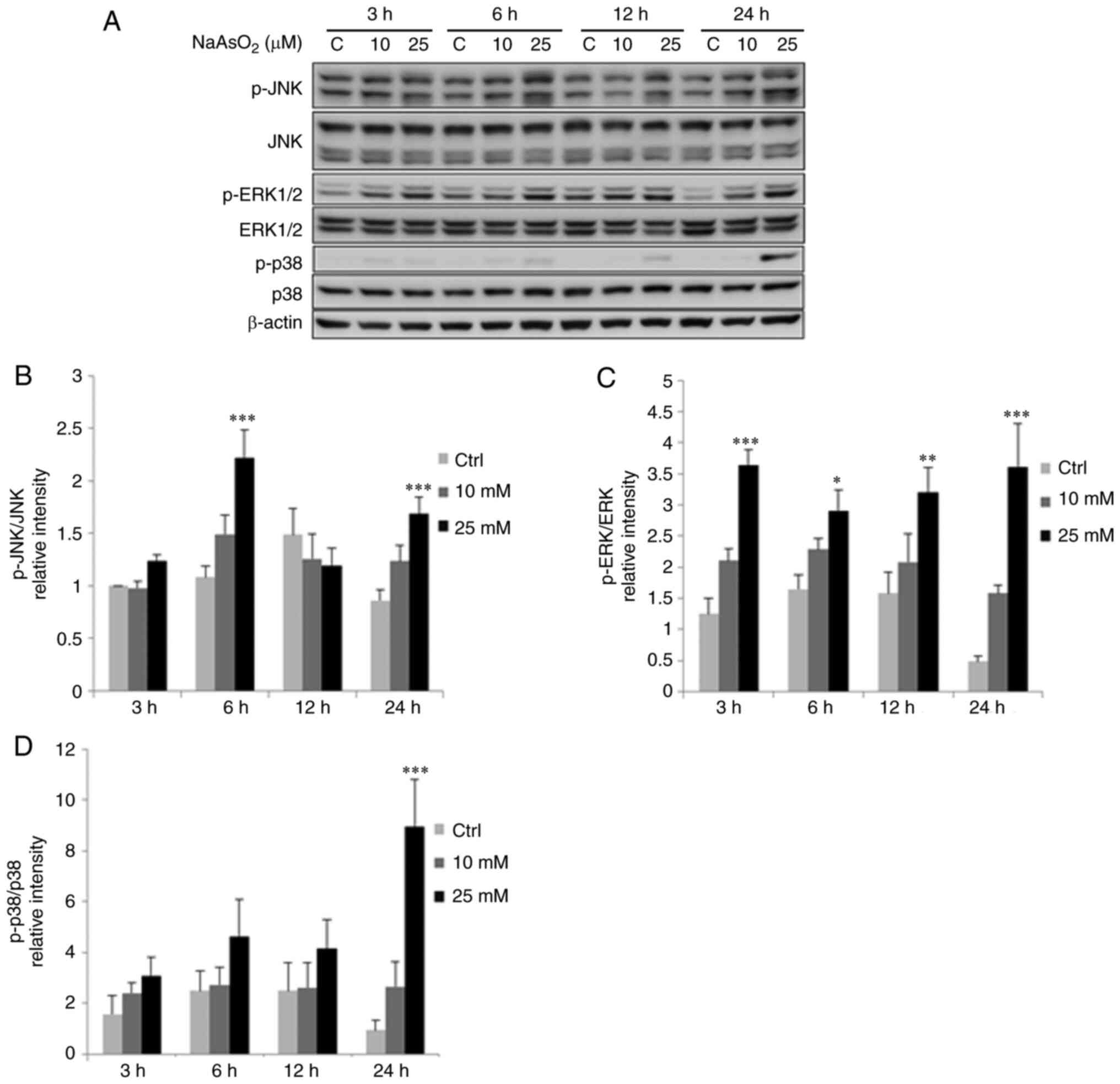 | Figure 9.Sodium arsenite modulates
phosphorylation of MAPK pathways in OC3 cells. OC3 cells were
incubated with 0, 10 and 25 µM NaAsO2 for 3, 6, 12 and
24 h, respectively. (A) JNK (46/54 kDa), p-JNK (46/54 kDa), ERK1/2
(42/44 kDa), p-ERK1/2 (42/44 kDa), p38 (43 kDa) plus p-p38 (43 kDa)
were examined via western blotting. Integrated optical intensities
of (B) p-JNK, (C) p-ERK and (D) p-p38 proteins were standardized
with their total forms in each lane, respectively. Results are
demonstrated as mean ± SEM of 3 independent experiments.
*P<0.05, **P<0.01 and ***P<0.001 vs. control. p-,
phosphorylated; C/Ctrl, control. |
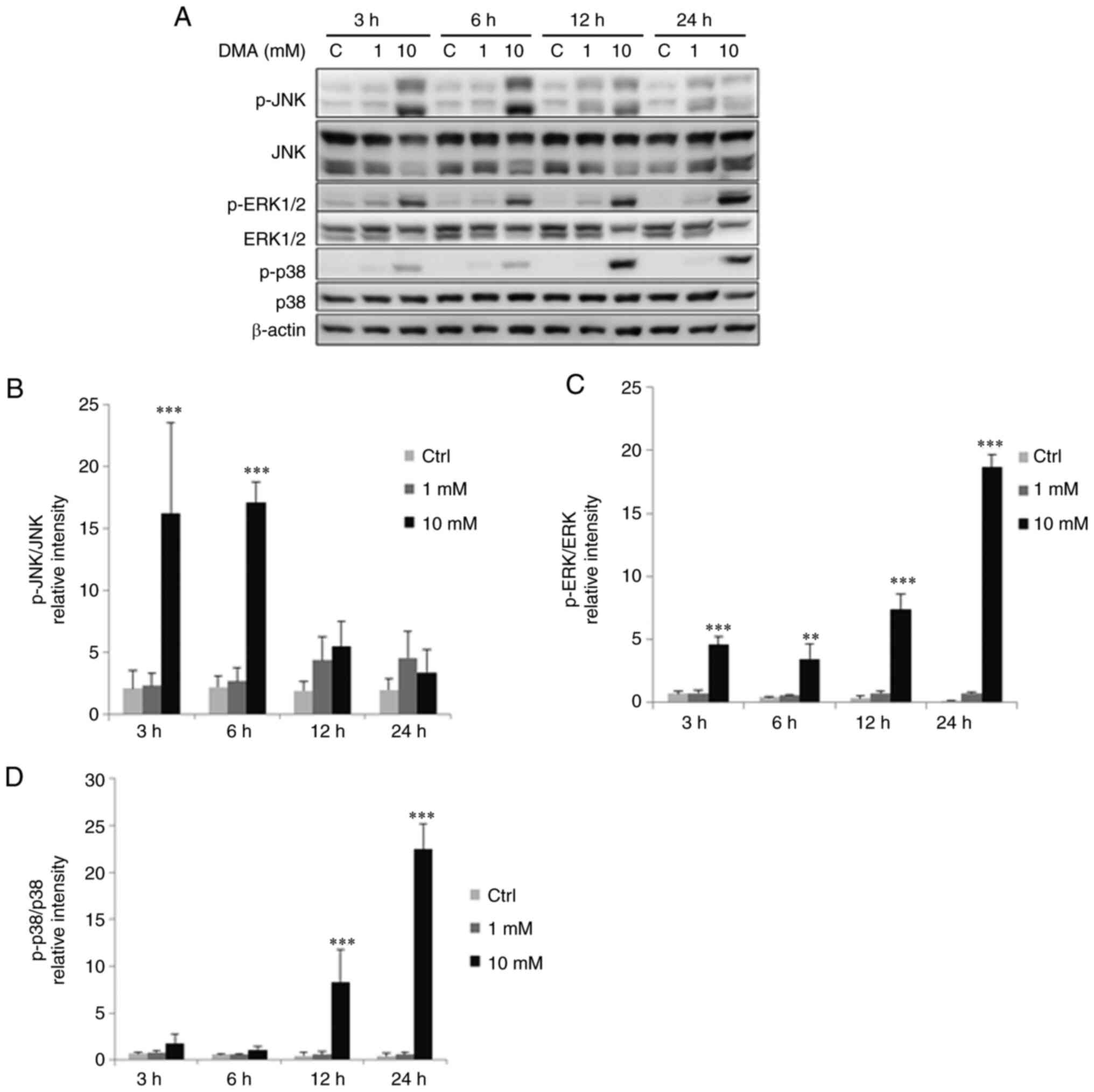 | Figure 10.DMA modulates phosphorylation of MAPK
pathways in OC3 cells. OC3 cells were incubated with 0, 1 and 10 mM
DMA for 3, 6, 12 and 24 h, respectively. (A) JNK (46/54 kDa), p-JNK
(46/54 kDa), ERK1/2 (42/44 kDa), p-ERK1/2 (42/44 kDa), p38 (43 kDa)
plus p-p38 (43 kDa) were examined via western blotting. Integrated
optical intensities of (B) p-JNK, (C) p-ERK and (D) p-p38 proteins
were standardized with their total forms in each lane,
respectively. Results are demonstrated as mean ± SEM of 3
independent experiments. **P<0.01 and ***P<0.001 vs. control.
DMA, dimethylarsenic acid; p-, phosphorylated; C/Ctrl, control. |
Discussion
ATO possesses anticancer ability in APL patients
(6). Studies have also shown that
arsenic compounds have been used to efficiently treat numerous
types of cancers, such as colon, breast, pancreatic, prostate and
neuroblastoma cancers, by inducing cell apoptosis (9,38–41). However, whether arsenic compounds
could serve as therapeutic agents for oral cavity cancers and the
relative regulating mechanisms remain to be elucidated. Oral
cancers have long been the refractory solid tumors, most of which
consist of squamous cell carcinoma (38–41). The incidence and mortality
increases year by year in Taiwan (31,32). Treatments remain poor and new
therapeutic strategies are required. In the present study, sodium
arsenite and dimethylarsenic acid showed significant effects to
induce apoptosis in OC3 cells.
In the present study, sodium arsenite and
dimethylarsenic acid decreased OC3 cell viability in dose-dependent
manners (Fig. 2), consistent with
the morphological change shown in Fig. 1. It should be noted that the
concentration of sodium arsenite was lower than that of
dimethylarsenic acid to perform similar effects; sodium arsenite at
25 µM for 24 h treatment significantly reduced cell viability to
53%, while dimethylarsenic acid 25 mM for 24 h treatment was needed
to reduce OC3 cell viability to 49% in MTT cell viability test. It
is known that inorganic arsenic compounds are more toxic than
organic arsenic compounds (3,4,42).
Inorganic arsenic compounds can be methylated to become less toxic
compounds processed by methyltransferases in liver, a
detoxification reaction, and the methylation occurs through
alternating reductive and oxidative methylation reactions (43). Thus, the findings of the present
study were not unprecedented.
Eukaryotic cell cycle proceeds to the next phase
when cellular upstream events accomplish checkpoint necessities
(44). After suffering cellular
injury, cell cycle arrest can occur at certain phase and cells can
proceed to programmed cell death as they cannot be properly
repaired (45). Arsenite can
stimulate microtubule network disruption, which can result in
spindle aberrations to induce cell apoptosis (46). Indeed, arsenic-induced cell cycle
arrest is a necessary step to activate apoptotic pathways among
various tumor cells (47). There
is a close relationship between apoptosis induction and
G2/M arrest in ovarian carcinoma responding to DNA
damage (48). In the present
study, sodium arsenite and dimethylarsenic acid significantly
stimulated subG1 phase cells and induced G2/M
phase arrest, which suggested that sodium arsenite and
dimethylarsenic acid stimulated apoptosis in OC3 cells. The present
study illustrated that arsenic-induced cell apoptosis was
associated with abnormal redistribution of cell cycles. Data
assessed by annexin V/PI double staining method also confirmed that
apoptosis induction with sodium arsenite and dimethylarsenic acid
was a dose-dependent phenomenon in OC3 oral cancer cells.
Apoptosis is a critical process for homeostasis in
individuals and apoptosis resistance is always hypothesized as a
central reason for cancer development (49). Apoptosis originates either by
extrinsic or intrinsic death signaling, which activates caspase
cascade (16). Arsenic trioxide
could stimulate apoptosis in laryngeal cancer through survivin mRNA
downregulation, which functions to inhibit caspases activation
(50). Arsenic trioxide can also
induce apoptosis among various types of cancer by activating
caspase-9, −8 and −3 (8). In the
present study, cleaved caspase-9, −8, −3 and PARP expressions were
significantly induced following treatments of sodium arsenite or
dimethylarsenic acid for 12 and/or 24 h in OC3 cells, respectively.
Consequently, the data suggested sodium arsenite and
dimethylarsenic acid could induce both extrinsic and intrinsic
apoptotic pathways to induce OC3 cell apoptosis.
Apoptotic cascades are regulated by a number of
cellular signal transduction pathways and the MAPK signaling
pathway is one associated with cellular stress (15,16,21–24). It has been shown that MAPK
signaling pathways can enhance cell survival or promote sensitivity
to apoptosis, which depends on different stimuli, cell types and
activation latency (23). JNK
contributes to cell survival by suppressing apoptosis through BAD
phosphorylation (51). By
contrast, the apoptosis induced by ATO in APL cells is through JNK
activation (52). ERK
demonstrates proapoptotic effect induced by various antitumor
compounds (53). A previous
report illustrates ATO can activate ERK1/2 and JNK1/2 pathways to
induce apoptosis in human mesothelioma cells (54). Furthermore, involvement of p38
associated with apoptosis is also varied and can suppress caspase-3
and caspase-8 activities in human neutrophils (55). In the present study, ERK1/2, p38
and JNK activities were stimulated by sodium arsenite and
dimethylarsenic acid in OC3 cells. Markedly, these results
demonstrated that activations of caspases plus PARP were detected
at 12 and 24 h treatments by arsenic compounds and the
phosphorylation of MAP kinases occurred at 3 to 6 h which is
earlier as compared with the caspase-induced pathway, demonstrating
both sodium arsenite and dimethylarsenic acid possibly stimulated
MAPK pathways before caspase pathways to stimulate OC3 cell
apoptosis (Table I). Therefore,
these results illustrated sodium arsenite and dimethylarsenic acid
activated MAPK pathways to stimulate apoptosis in OC3 oral cavity
cells.
 | Table I.Time course of activated apoptotic
and MAPK pathways by sodium arsenite and dimethylarsenic acid
treatments between FaDu (26),
OEC-M1 (44) and OC3 cells. Cells
were treated without or with NaAsO2 or DMA for 3, 6, 12
and 24 h. Apoptotic markers and MAPKs were detected by western
blotting. The arsenic-induced apoptotic and MAPK pathways were
time-dependent. |
Table I.
Time course of activated apoptotic
and MAPK pathways by sodium arsenite and dimethylarsenic acid
treatments between FaDu (26),
OEC-M1 (44) and OC3 cells. Cells
were treated without or with NaAsO2 or DMA for 3, 6, 12
and 24 h. Apoptotic markers and MAPKs were detected by western
blotting. The arsenic-induced apoptotic and MAPK pathways were
time-dependent.
| Cell line | As | Pathway | 3 h | 6 h | 12 h | 24 h |
|---|
| FaDu | 3+ | Apoptosis |
|
|
| V |
|
| 3+ | MAPK | V |
| V | V |
|
| 5+ | Apoptosis |
|
| V | V |
|
| 5+ | MAPK |
|
| V | V |
| OEC-M1 | 3+ | Apoptosis |
|
| V | V |
|
| 3+ | MAPK | V | V | V | V |
|
| 5+ | Apoptosis |
|
| V | V |
|
| 5+ | MAPK | V | V | V | V |
| OC3 | 3+ | Apoptosis |
|
| V | V |
|
| 3+ | MAPK | V | V | V | V |
|
| 3+ | Apoptosis |
|
| V | V |
|
| 5+ | MAPK | V | V | V | V |
Studies have revealed that sodium arsenite and
dimethylarsenic acid can modulate cell morphological changes
associated with cell apoptosis in dosage- and time-dependent
manners in FaDu and OEC-M1 cells, respectively (20,37). The present study showed that
sodium arsenite caused more shriveled and floating cells as
concentration increased in OC3 cells, compared with FaDu and OEC-M1
cells (20,37). Also, 50 and 100 mM dimethylarsenic
acid induced more floating OC3 cells with cell membrane blebbing.
In addition, the observations of the present study in OC3 cells is
similar to other finding that stimulations of MAPK and caspase
pathways by arsenic compound stimuli are similar as shown in OEC-M1
cells (Table I) (37). However, in FaDu cells, activation
of MAP kinases occurs at 3, 12 and 24 h, but caspase activation
occurs at 24 h under sodium arsenite treatment (Table I) (20). Notably, activations of MAP kinases
and caspase pathway occurred simultaneously at 12 and 24 h
treatments with dimethylarsenic acid in FaDu cells (Table I) (20). Hence, sodium arsenite and
dimethylarsenic acid activate MAPK and caspase pathways, however,
the dynamic phenomena are somewhat dissimilar between different
oral cavity cancer cells (Table
I). It would have been be more meaningful if non-cancerous oral
mucosal cells could be used to study and comparison for the
difference with the current data among tumor cell lines and this is
a limitation of the present study, which should be investigated in
the near future.
Taken together, sodium arsenite and dimethylarsenic
acid could stimulate apoptosis via intrinsic and extrinsic
apoptotic cascades, showing antitumor properties in OC3 cells.
Moreover, sodium arsenite and dimethylarsenic acid could activate
MAPKs to further regulate caspase cascade activation and then to
induce apoptosis in OC3 oral cancer cells.
Acknowledgements
Not applicable.
Funding
The present study was supported by Ministry of Science and
Technology of Taiwan, ROC (grant nos. MOST-107-2320-B-471-001 to
YL; MOST-110-2314-B-218-001 to HC; MOST 110-2320-6-B-025-MY3 to BH)
and Chi-Mei Medical Center; Liouying (grant nos. CLFHR11025 and
CLFHR11120 to SW and BH), Taiwan, ROC.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request, which adheres to the FAIR principles (https://www.go-fair.org/fair-principles/), including
the fundamental principles of Findability, Accessibility,
Interoperability and Reusability.
Authors' contributions
SW, YYL, CC and YPL designed and conducted the
experiments, interpreted results and wrote the manuscript. HC and
BH participated in the design and coordination of the present
study. SW, YYL, CC, HC and BH were involved in the statistical
analysis of results and revised manuscript for substantive and
methodological correctness. HC and BH confirm the authenticity of
all the raw data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mandal BK and Suzuki KT: Arsenic round the
world: A review. Talanta. 58:201–235. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Peng XX, Gai S, Cheng K and Yang F: Roles
of humic substances redox activity on environmental remediation. J
Hazard Mater. 435:1290702022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Calatayud M, Devesa V and Vélez D:
Differential toxicity and gene expression in Caco-2 cells exposed
to arsenic species. Toxicol Lett. 218:70–80. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Costa ASPN, Nascimento ALA, Botero WG,
Carvalho CM, Tonholo J, Santos JCC and Anunciação DS: Interaction
between humic substances and arsenic species simulating
environmental conditions. Sci Total Environ. 802:1497792022.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM,
Qiu QY, Zhu J, Tang W, Sun GL, Yang KQ, et al: Use of arsenic
trioxide (As2O3) in the treatment of acute
promyelocytic leukemia (APL): II. Clinical efficacy and
pharmacokinetics in relapsed patients. Blood. 89:3354–3360. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zamani-Moghaddam N, Mousavi FS, Esmaeili
S, Yousefi AM, Safaroghli-Azar A and Bashash D: Suppression of
proteasome induces apoptosis in APL cells and increases
chemo-sensitivity to arsenic trioxide: Proposing a perception in
APL treatment. Cancer Treat Res Commun. 26:1002842021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mandegary A, Torshabi M, Seyedabadi M,
Amirheidari B, Sharif E and Ghahremani MH: Indomethacin-enhanced
anticancer effect of arsenic trioxide in A549 cell line:
Involvement of apoptosis and phospho-ERK and p38 MAPK pathways.
Biomed Res Int. 2013:2375432013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu Q, Hilsenbeck S and Gazitt Y: Arsenic
trioxide-induced apoptosis in myeloma cells: p53-dependent
G1 or G2/M cell cycle arrest, activation of
caspase-8 or caspase-9, and synergy with APO2/TRAIL. Blood.
101:4078–4087. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim MJ, Jung JH, Lee WS, Yun JW, Lu JN, Yi
SM, Kim HJ, Chang SH, Kim GS, Hong SC and Ha WS: Arsenic hexoxide
enhances TNF-α-induced anticancer effects by inhibiting NF-κB
activity at a safe dose in MCF-7 human breast cancer cells. Oncol
Rep. 31:2305–2311. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mann KK, Wallner B, Lossos IS and Miller
WH Jr: Darinaparsin: A novel organic arsenical with promising
anticancer activity. Expert Opin Investig Drugs. 18:1727–1734.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kuwabara M, Asanuma T, Niwa K and Inanami
O: Regulation of cell survival and death signals induced by
oxidative stress. J Clin Biochem Nutr. 43:51–57. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
D'Arcy MS: Cell death: A review of the
major forms of apoptosis, necrosis and autophagy. Cell Biol Int.
43:582–592. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Danial NN and Korsmeyer SJ: Cell death:
Critical control points. Cell. 116:205–219. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wilson TR, Johnston PG and Longley DB:
Anti-apoptotic mechanisms of drug resistance in cancer. Curr Cancer
Drug Targets. 9:307–319. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kasibhatla S and Tseng B: Why target
apoptosis in cancer treatment? Mol Cancer Ther. 2:573–580.
2003.PubMed/NCBI
|
|
16
|
Westaby D, Jimenez-Vacas JM, Padilha A,
Varkaris A, Balk SP, de Bono JS and Sharp A: Targeting the
intrinsic apoptosis pathway: A window of opportunity for prostate
cancer. Cancers (Basel). 14:512021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Faramarzi F, Alimohammadi M, Rahimi A,
Alizadeh-Navaei R, Shakib RJ and Rafiei A: Naringenin induces
intrinsic and extrinsic apoptotic signaling pathways in cancer
cells: A systematic review and meta-analysis of in vitro and in
vivo data. Nutr Res. 105:33–52. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dorstyn L, Akey CW and Kumar S: New
insights into apoptosome structure and function. Cell Death Differ.
25:1194–1208. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lee YP, Huang WR, Wu WS, Wu YH, Ho SY,
Wang YJ and Huang BM: Cordycepin enhances radiosensitivity to
induce apoptosis through cell cycle arrest, caspase pathway and ER
stress in MA-10 mouse Leydig tumor cells. Am J Cancer Res.
12:3601–3624. 2022.PubMed/NCBI
|
|
20
|
Wu SZ, Lan YY, Chu CY, Wang YK, Lee YP,
Chang HY and Huang BM: Arsenic compounds induce apoptosis by
activating the MAPK and caspase pathways in FaDu oral squamous
carcinoma cells. Int J Oncol. 60:182022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Runchel C, Matsuzawa A and Ichijo H:
Mitogen-activated protein kinases in mammalian oxidative stress
responses. Antioxid Redox Signal. 15:205–218. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cargnello M and Roux PP: Activation and
function of the MAPKs and their substrates, the MAPK-activated
protein kinases. Microbiol Mol Biol Rev. 75:50–83. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wada T and Penninger JM: Mitogen-activated
protein kinases in apoptosis regulation. Oncogene. 23:2838–2849.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Juan WS, Mu YF, Wang CY, So EC, Lee YP,
Lin SC and Huang BM: Arsenic compounds activate MAPK and inhibit
Akt pathways to induce apoptosis in MA-10 mouse Leydig tumor cells.
Cancer Med. Aug 24–2022.(Epub ahead of print). View Article : Google Scholar
|
|
25
|
Kang YH and Lee SJ: The role of p38 MAPK
and JNK in Arsenic trioxide-induced mitochondrial cell death in
human cervical cancer cells. J Cell Physiol. 217:23–33. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
David MC, Randal SW and Stephen YL: Head
and neck cancer: An evolving treatment paradigm. Cancer. 113
(7Suppl):S1911–S1932. 2008. View Article : Google Scholar
|
|
27
|
Laraway DC, Lakshmiah R, Lowe D, Roe B and
Rogers SN: Quality of life in older people with oral cancer. Br J
Oral Maxillofac Surg. 50:715–720. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ko YC, Huang YL, Lee CH, Chen MJ, Lin LM
and Tsai CC: Betel quid chewing, cigarette smoking and alcohol
consumption related to oral cancer in Taiwan. J Oral Pathol Med.
24:450–453. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sudbø J: Human papillomavirus infection as
a risk factor for squamous-cell carcinoma of the head and neck. N
Engl J Med. 345:376–377. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bernier J and Cooper JS: Chemoradition
after surgery for high-risk head and neck cancer patients: How
strong is the evidence? Oncologist. 10:215–224. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen CJ, You SL, Lin LH, Hsu WL and Yang
YW: Cancer epidemiology and control in Taiwan: A brief review. Jpn
J Clin Oncol. 32 (suppl 1):S66–S81. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jhuang JR, Su SY, Chiang CJ, Yang YW, Lin
LJ, Hsu TH and Lee WC: Forecast of peak attainment and imminent
decline after 2017 of oral cancer incidence in men in Taiwan. Sci
Rep. 12:57262022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cheng Y, Chen J, Shi Y, Fang X and Tang Z:
MAPK signaling pathway in oral squamous cell carcinoma: Biological
function and targeted therapy. Cancers (Basel). 14:46252022.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lin SC, Liu CJ, Chiu CP, Chang SM, Lu SY
and Chen YJ: Establishment of OC3 oral carcinoma cell line and
identification of NF-kappa B activation responses to areca nut
extract. J Oral Pathol Med. 33:79–86. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mu YF, Chen YH, Chang MM, Chen YC and
Huang BM: Arsenic compounds induce apoptosis through caspase
pathway activation in MA-10 Leydig tumor cells. Oncol Lett.
18:944–954. 2019.PubMed/NCBI
|
|
36
|
van Engeland M, Ramaekers FC, Schutte B
and Reutelingsperger CP: A novel assay to measure loss of plasma
membrane asymmetry during apoptosis of adherent cells in culture.
Cytometry. 24:131–139. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Foo NP, Ko CL, Chu CY, Wang CY, So EC and
Huang BM: Arsenic compounds activate the MAPK and caspase pathways
to induce apoptosis in OEC-M1 gingival epidermal carcinoma. Oncol
Rep. 44:2701–2714. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nakagawa Y, Akao Y, Morikawa H, Hirata I,
Katsu K, Naoe T, Ohishi N and Yagi K: Arsenic trioxide-induced
apoptosis through oxidative stress in cells of colon cancer cell
lines. Life Sci. 70:2253–2269. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li X, Ding X and Adrian TE: Arsenic
trioxide induces apoptosis in pancreatic cancer cells via changes
in cell cycle, caspase activation, and GADD expression. Pancreas.
27:174–179. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Feng C, Wu Y, Chen Y, Xiong X, Li P, Peng
X, Li C, Weng W, Zhu Y, Zhou D and Li Y: Arsenic trioxide increases
apoptosis of SK-N-BE (2) cells partially by inducing GPX4-mediated
ferroptosis. Mol Biol Rep. 49:6573–6580. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mirzaei A, Jahanshahi F, Khatami F, Reis
LO and Aghamir SMK: Human prostate cancer cell
epithelial-to-mesenchymal transition as a novel target of arsenic
trioxide and curcumin therapeutic approach. Tissue Cell.
76:1018052022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Camacho J, de Conti A, Pogribny IP,
Sprando RL and Hunt PR: Assessment of the effects of organic vs.
inorganic arsenic and mercury in Caenorhabditis elegans. Curr Res
Toxicol. 3:1000712022. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Vahter M: Methylation of inorganic arsenic
in different mammalian species and population groups. Sci Prog.
82((Pt 1)): 69–88. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gordon EM, Ravicz JR, Liu S, Chawla SP and
Hall FL: Cell cycle checkpoint control: The cyclin G1/Mdm2/p53 axis
emerges as a strategic target for broad-spectrum cancer gene
therapy-A review of molecular mechanisms for oncologists. Mol Clin
Oncol. 9:115–134. 2018.PubMed/NCBI
|
|
45
|
Hartwell LH and Weinert TA: Checkpoints:
Controls that ensure the order of cell cycle events. Science.
246:629–634. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yih LH, Wu YC, Hsu NC and Kuo HH: Arsenic
trioxide induces abnormal mitotic spindles through a PIP4KIIγ/Rho
pathway. Toxicol Sci. 128:115–125. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ling YH, Jiang JD, Holland JF and
Perez-Soler R: Arsenic trioxide produces polymerization of
microtubules and mitotic arrest before apoptosis in human tumor
cell lines. Mol Pharmacol. 62:529–538. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Concin N, Stimpfl M, Zeillinger C, Wolff
U, Hefler L, Sedlak J, Leodolter S and Zeillinger R: Role of p53 in
G2/M cell cycle arrest and apoptosis in response to
gamma-irradiation in ovarian carcinoma cell lines. Int J Oncol.
22:51–57. 2003.PubMed/NCBI
|
|
49
|
Gullett JM, Tweedell RE and Kanneganti TD:
It's all in the PAN: Crosstalk, plasticity, redundancies, switches,
and interconnectedness encompassed by PANoptosis underlying the
totality of cell death-associated biological effects. Cells.
11:14952022. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Cheng B, Yang X, Han Z, An L and Liu S:
Arsenic trioxide induced the apoptosis of laryngeal cancer via
down-regulation of survivin mRNA. Auris Nasus Larynx. 35:95–101.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yu C, Minemoto Y, Zhang J, Liu J, Tang F,
Bui TN, Xiang J and Lin A: JNK suppresses apoptosis via
phosphorylation of the proapoptotic Bcl-2 family protein BAD. Mol
Cell. 3:329–340. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Davison K, Mann KK, Waxman S and Miller WH
Jr: JNK activation is a mediator of arsenic trioxide-induced
apoptosis in acute promyelocytic leukemia cells. Blood.
103:3496–3502. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Tewari R, Sharma V, Koul N and Sen E:
Involvement of miltefosine-mediated ERK activation in glioma cell
apoptosis through Fas regulation. J Neurochem. 107:616–627. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Eguchi R, Fujimori Y, Takeda H, Tabata C,
Ohta T, Kuribayashi K, Fukuoka K and Nakano T: Arsenic trioxide
induces apoptosis through JNK and ERK in human mesothelioma cells.
J Cell Physiol. 226:762–768. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Alvarado-Kristensson M, Melander F,
Leandersson K, Rönnstrand L, Wernstedt C and Andersson T: p38-MAPK
signals survival by phosphorylation of caspase-8 and caspase-3 in
human neutrophils. J Exp Med. 199:449–458. 2004. View Article : Google Scholar : PubMed/NCBI
|














