Introduction
Hereditary spastic paraplegia (HSP) is a group of
clinically and genetically heterogeneous neurodegenerative diseases
with a global incidence of 4.3-9.8 per 100,000 individuals
(1). Four inheritance patterns
have been identified: i) Autosomal-dominant (AD), ii)
autosomal-recessive, iii) X-linked recessive and iv) mitochondrial;
additionally, de novo mutations have been found in a number
of patients with HSP (1). Thus
far, >70 related pathogenic genes have been identified in
patients with HSP (1). According
to the clinical phenotype, HSP can be divided into pure and complex
forms. The pure form is characterized by progressive spasticity and
weakness of the lower limbs, with occasional sensory impairment or
bladder dysfunction; the complex form includes the pure phenotype
with additional abnormalities, including mental and cognitive
changes, optic atrophy, muscular atrophy, ataxia, deafness,
ichthyosis and/or peripheral neuropathy (2).
Spastic paraplegia type 4 (SPG4) is the most common
subtype of AD-SPG; it is present in ~40% of patients with AD-SPG.
SPG4 is caused by mutations in the spastin (SPAST) gene and
usually manifests as pure HSP (3).
Mutations in SPAST are the most common causes of familial
and sporadic HSP. In China, the frequency of SPAST mutations
is 40% in patients with familial HSP and 33.33% in patients with
sporadic HSP (2). The SPAST
gene is located on chromosome 2p22.3 and contains 17 exons; it
encodes the Spastin protein, a member of the ATPase associated with
diverse cellular activities (AAA) protein family. Spastin comprises
four functional domains: i) Hydrophobic domain (HD; amino acid
residues 1–86); ii) microtubule interacting and trafficking domain
(MIT; amino acid residues 116–194), which is involved in
cytokinesis and endosomal-tubule recycling (4); iii) microtubule-binding domain (MTBD;
amino acid residues 270–328), which enables Spastin to bind to
microtubules; and iv) AAA domain (amino acid residues 342–599),
which is required for Spastin hexamerization and
microtubule-severing activity (5).
There are two translation initiation codons in SPAST, which
encode the full-length isoform M1 (expressed in the spinal cord)
and the slightly shorter isoform M87 (expressed in the spinal cord
and cerebral cortex) (5). HD is
present only in the M1 Spastin isoform; MIT, MTBD and AAA are
present in both Spastin isoforms (4).
Spastin is an ATPase that severs microtubules.
During the severing process, the six Spastin subunits assemble into
a ring-shaped hexamer that is attached to the microtubule and
energy from adenosine triphosphate (ATP) hydrolysis is used to
sever the microtubule by pulling the negatively charged C-terminus
of tubulin through the central pore of the hexamer (4). In neurons, this process can convert
long microtubules into short microtubules that move rapidly and
harmoniously within axons, thereby enabling efficient microtubule
transport that is crucial to axonal growth and axon branch
formation; microtubule transport also maintains neurite complexity
(5). Mutations in SPAST are
presumed to cause partial loss of Spastin microtubule-severing
activity or the production of a neurotoxic mutant protein; these
changes contribute to the onset of SPG4 (5). Additionally, SPG4 penetrance may be
influenced by biological sex and age (6).
In the present study, the clinical characteristics
of affected individuals and sequencing analysis of a mutation that
caused SPG4 only among male members of a family are reported.
Materials and methods
Clinical characteristics
The family was recruited in November 2021 from the
Affiliated Hospital of Jining Medical University (Jining, China).
Familial history and clinical data of the patients were collected.
All patients provided written informed consent to participate in
the study.
Whole-exome sequencing (WES)
Genetic screening was performed to determine the
genetic etiology of the disease. Peripheral blood (5 ml) was
collected from the proband's father (II-4) and a portion (2 ml) was
sent to Beijing Kangxu Medical Laboratory Co., Ltd. for WES. A
total of 2 ml of peripheral blood were collected from the proband
(III-6), his uncle (II-3), third aunt (II-6), brother (III-5) and
sister (III-7). The proband's other two aunts (II-1 and II-2)
declined to be tested Genomic DNA was extracted from blood samples
(2 ml) using a FlexiGene DNA Kit (Qiagen). The degree of DNA
degradation, the presence of RNA, and protein contamination were
analyzed by agarose gel electrophoresis (data not shown). The DNA
concentration was accurately quantified using a Qubit 2.0
fluorometer (Thermo Fisher Scientific, Inc.). Genomic DNA was
fragmented using a Covaris bath sonicator (duty cycle, 10%;
intensity, 5; cycles per burst, 200; 3 min for 25°C) into 180–280
bp fragments to construct a DNA library. Adaptors from
TransNGS® Index Primers (384) Kit for
Illumina® (TransGen Biotech Co., Ltd.), were ligated to
both ends of each DNA fragment, and cohesive ends of the DNA
fragments were trimmed. Next, the DNA library was amplified by
polymerase chain reaction under the following thermocycling
conditions according to the TransNGS® Index
Primers (384) Kit manufacturer's protocol: Initial denaturation at
98°C for 3 min; followed by 5 cycles of 30 sec at 98°C, 35 sec at
60°C and 30 sec at 72°C; with a final extension at 72°C for 3 min.
The following adaptor-specific primers were used to amplify DNA
library: Forward,
5′-AATGATACGGCGACCACCGAGATCTACACTAGCTGCCACACTCTTTCCCTACACGACCTCTTCCGATC-s-T-3′
and reverse,
5′-CAAGCAGAAGACGGCATACGAGATTCCGCGAAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATC-s-T-3′;
where -s- represents a phosphorothioate bond. The PCR products were
then purified using MagicPure® Size Selection DNA
Beads (TransGen Biotech Co., Ltd.) according to the manufacturer's
protocol. The subsequent DNA fragments were hybridized in liquid
phase using up to 500,000 biotin-labeled Agilent SureSelect Human
All Exon V6 probes (Agilent Technologies, Inc.), which were then
captured using streptomycin magnetic beads and amplified using the
SureSelect Target Enrichment System (Agilent Technologies, Inc.)
under the following thermocycling conditions: Initial denaturation
at 98°C for 2 min; followed by 15 cycles of 30 sec at 98°C, 30 sec
at 62°C and 1 min at 72°C; with a final extension at 72°C for 10
min. The following adaptor-specific primers were used to amplify:
Forward, 5′-AATGATACGGCGACCACCGA-3′ and reverse primer,
5′-CAAGCAGAAGACGGCATACGA-3′. The products were subsequently
purified using MagicPure® Size Selection DNA
Beads as aforementioned, and the Qubit 2.0 fluorometer and an
Agilent Technologies 2200 TapeStation qPCR (7500 Fast Dx Real-Time
PCR Instrument, Thermo Fisher Scientific, Inc.) were used to
accurately quantify the effective concentration (3 nM) of the
library, which was then subjected to single-read sequencing using a
NextSeq500 (Illumina, Inc.).
Sequencing reads were aligned to the human reference
genome (version hg19) using Burrows-Wheeler Alignment (version
0.7.15). Quality control was conducted by analyzing global
alignment depth, interval alignment depth, interval coverage, site
alignment quality and other indicators, such as data volume and
duplication rate. The alignment results were translated into BAM
format; GATK UnifiedGenotyper (v3.6) (https://www.broadinstitute.org) was used to detect
single-nucleotide variants and small insertion or deletion
variants. Possible copy number variations were analyzed using CODEX
(v1.14.1), XHMM (v1.0) and KSCNV (developed by Beijing Kangxu
Medical Laboratory Co., Ltd.) (7,8).
ANNOVAR (v2016-02-01) was used to annotate the locations of
variants in genes and transcripts. Subsequently, gene-related
annotations were conducted, involving databases such as RefSeq
(version of the reference genome, GRCH37/Hg19; http://www.ncbi.nlm.nih.gov/refseq), Ensembl
(April 2021 update; http://www.ensembl.org/index.html) and UCSC (version
of the reference genome, GRCH37/Hg19; http://genome.ucsc.edu). The frequencies of annotated
variants in the population were investigated using 1000G (2015
update; http://www.1000genomes.org), dbSNP
(v150; http://www.ncbi.nlm.nih.gov/SNP), ESP6500 (2014
update) (https://evs.gs.washington.edu/EVS) and ExAC (v0.3;
ExAC is now in gnomAD; (www.gnomad-sg.org) databases. SIFT (version 2;
http://sift.bii.a-star.edu.sg),
PolyPhen2 (version 2; http://genetics.bwh.harvard.edu/pph2) and
MutationTaster (NCBI 37/Ensembl 69; http://www.mutationtaster.org) were used to
investigate the impacts of mutations on Spastin protein function
(9,10). Disease-related annotations were
performed using Online Mendelian Inheritance in Man, The Human Gene
Mutation Database and ClinVar databases. Next, annotation results
were filtered according to mutation location, mutation type,
mutation frequency and mutation site characteristics to retain
mutations potentially associated with disease. MutationTaster were
used to assess sequence conservation across species. Variants
(pathogenic, likely pathogenic, benign, likely benign and variants
of uncertain significance) were classified according to the
American College of Medical Genetics and Genomics (ACMG) Variation
Interpretation guidelines (11).
Finally, clinical analyses, such as clinical symptom matching and
genetic pattern matching, were conducted for each sample according
to ACMG (11), family history and
protein damage prediction of gene locus according to the results of
SIFT, Polyphen2 and MutationTaster and the clinical
presentation.
Sanger sequencing
Peripheral blood (2 ml) was collected from the
proband (III-6), proband's father (II-4), his uncle (II-3), third
aunt (II-6), brother (III-5) and sister (III-7). The proband's
other two aunts (II-1 and II-2) declined to be tested. The blood
samples were sent to Beijing Kangxu Medical Laboratory Co., Ltd.
for Sanger sequencing. Genomic DNA was extracted from the blood
samples using a FlexiGene DNA Kit (Qiagen). According to the
results of WES of the proband's father, the c.1785C>A mutation
in SPAST gene was selected for further validation. Primers
were designed using the website Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast)
(12) and synthesized by Tianyi
Huiyuan Biotech Co., Ltd (101.43.170.175/tyhy). The primer
sequences for SPAST were designed using the gene sequence
from GenBank (accession no. NM_014946), as are as follows: Forward,
5′-TCCATCATTTCGTTAACCACCA-3′; reverse, 5′-GCCGATGACGTTCATTGAAGA-3′.
The c.1785C>A mutation in SPAST was amplified by PCR
using a EasyTaq PCR SuperMix (Beijing TransGen Biotech Co., Ltd.),
under the following thermocycling conditions: Initial denaturation
at 95°C for 10 min; followed by 35 cycles of 30 sec at 95°C, 30 sec
at 60°C and 45 sec at 72°C; with a final extension at 72°C for 5
min. The amplicons were then sequenced by Sanger sequencing using
an ABI 3730×l DNA analyzer (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The resulting sequences were aligned with the
results of WES, and false-positive sites obtained by
next-generation sequencing were excluded.
Whole-exome sequences accession
numbers in ClinVar
Whole-exome sequences of the family members in this
study have been deposited in ClinVar under the following accession
number: SCV002761959.
Bioinformatics analysis
Bioinformatics analysis was used to predict the
possible effects of gene mutations on relevant mRNAs and proteins.
The RNAfold web server (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi)
was used to predict the effects of new mutations on mRNA structure.
PSIPRED (V4.0) (http://bioinf.cs.ucl.ac.uk/psipred) and RaptorX
(http://raptorx.uchicago.edu) were used
to predict the secondary and tertiary structures of mutated and
wild-type Spastin proteins, respectively.
Summary of various pathogenic
mutations
ClinVar and gnomAD databases were searched for
pathogenic variant data regarding the SPAST gene, using the
search term ‘SPAST’. We did not include mutations that were
considered ‘benign’ or ‘likely pathogenic.’
Results
The proband was a 30-year-old male who was admitted
in November 2021 to The Affiliated Hospital of Jining Medical
University with a >10-year history of unstable gait; the patient
first began to experience difficulty walking because of tremors in
the lower limbs. Subsequently, weakness and tremors in the lower
limbs during walking gradually worsened, and the patient could not
bend his knees; however, he could stand and walk slowly unaided at
the time of admission. His urinary and bowel functions were normal.
Neurological examination revealed increased muscle tone in both
lower limbs, muscle strength grade 3–4 (normal muscle strength is
grade 5), tendon hyperreflexia and bilateral Babinski sign
positive; lower limb sensation was normal. Examination of cranial
nerves and upper limbs revealed no abnormalities. General physical
examination, routine laboratory tests and magnetic resonance
imaging of the brain, cervical spine and thoracic spine did not
reveal any obvious pathognomonic alterations (Fig. 1). The patient had been born through
full-term vaginal delivery, and he had exhibited good health from
birth until the onset of the present symptoms.
The proband's deceased paternal grandfather had
exhibited difficulty walking (Fig.
2); his age at death was not provided by the family. The
proband's 66-year-old uncle (II-3) had begun to experience
difficulty walking at the age of 36 and he began walking with
crutches at the age of 56 (Video
SI). The proband's 61-year-old father (II-4) had begun to
experience difficulty walking at the age of 41 and is currently
confined to a wheelchair. The proband's youngest uncle (II-5) died
of diabetes at the age of 54; he could not walk before death,
although his age at the onset of walking difficulty was unknown.
The proband's 33-year-old brother (III-5) exhibited muscle weakness
at the age of 15 and has been confined to a wheelchair since the
age of 29. The urinary and bowel functions of these living
relatives were normal. Moreover, neurological examinations of these
relatives revealed increased muscle tone, reduced muscle strength,
tendon hyperreflexia, positive bilateral Babinski sign and sensory
deficits in both lower limbs. Muscle strength was normal in the
upper limbs; examinations of the cranial nerves and upper limbs
revealed no abnormalities. General physical examinations of these
relatives also did not reveal any abnormalities. The proband's
72-year-old aunt (II-1), 70-year-old aunt (II-2), 51-year-old aunt
(II-6) and 27-year-old sister (III-7) did not exhibit signs of
difficulty walking or muscle weakness; general physical and
neurological examinations revealed no abnormalities in these
relatives. Because no equipment for MRI was available at the local
hospital, magnetic resonance imaging scans of family members other
than the proband could not be obtained. Patient information and
clinical features of affected individuals in this family are
summarized in Table I.
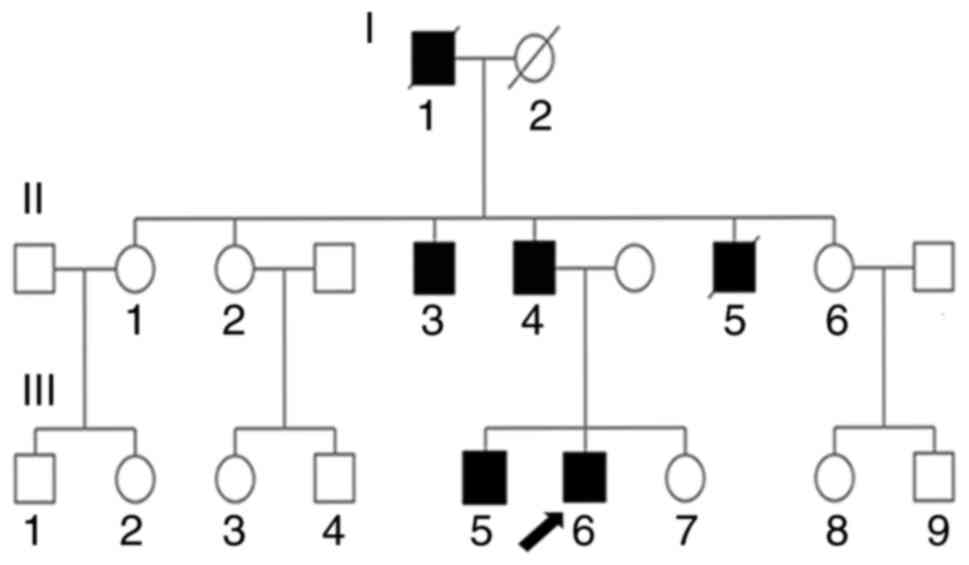 | Figure 2.SPG4 family pedigree of 17 members.
I-1, grandfather, deceased, exhibited difficulty walking. I-2,
grandmother, deceased, who did not exhibit muscle weakness. II-1,
oldest aunt who has no muscle weakness. II-2, second aunt who has
no muscle weakness. II-3, oldest uncle, 66 years old, who has SPG4.
II-4, father, 61 years old, who has SPG4. II-5, youngest uncle,
deceased, who had SPG4. II-6, third aunt who has no muscle
weakness. III-1 and III-2, children of older aunt who have no
muscle weakness. III-3 and III-4, children of second aunt who have
no muscle weakness. III-5, older brother, 33 years old, who has
SPG4. III-6, proband (arrow), 30 years old, who has SPG4. III-7,
sister who has no muscle weakness. III-8 and III-9, third aunt's
children who have no muscle weakness. /, deceased family member;
SPG4, spastic paraplegia type 4. |
 | Table I.Clinicopathological features of
affected individuals within the family. |
Table I.
Clinicopathological features of
affected individuals within the family.
| Individual ID | I-1 | II-3 | II-4 | II-5 | III-5 | III-6 |
|---|
| Sex | M | M | M | M | M | M |
| Age at onset,
years | – | 36 | 41 | – | 15 | 20 |
| Age at examination,
years | – | 66 | 61 | – | 33 | 30 |
| Disease duration,
years | – | 30 | 20 | – | 18 | >10 |
| Lower limb
hyperreflexia | – | + | + | – | + | + |
| Lower limb
spasticity | – | + | + | – | + | + |
| Lower limb muscle
strengtha | – | 2-3 | 1-2 | – | 1-2 | 3-4 |
| Reflex | – | + | + | – | + | + |
| Babinski sign | – | + | + | – | + | + |
| Upper limb
hyperreflexia | – | _ | _ | – | _ | _ |
| Upper limb
spasticity | – | _ | _ | – | _ | _ |
|
SPATAX-EUROSPAb disability score | 6 | 5 | 6 | – | 6 | 3 |
| Neurogenic bladder
dysfunction | _ | _ | _ | _ | _ | _ |
| Sensory
deficits | – | + | + | – | + | _ |
| Mental
retardation | – | _ | _ | – | _ | _ |
| Concomitant
diseases | – | _ | _ | Diabetes | _ | _ |
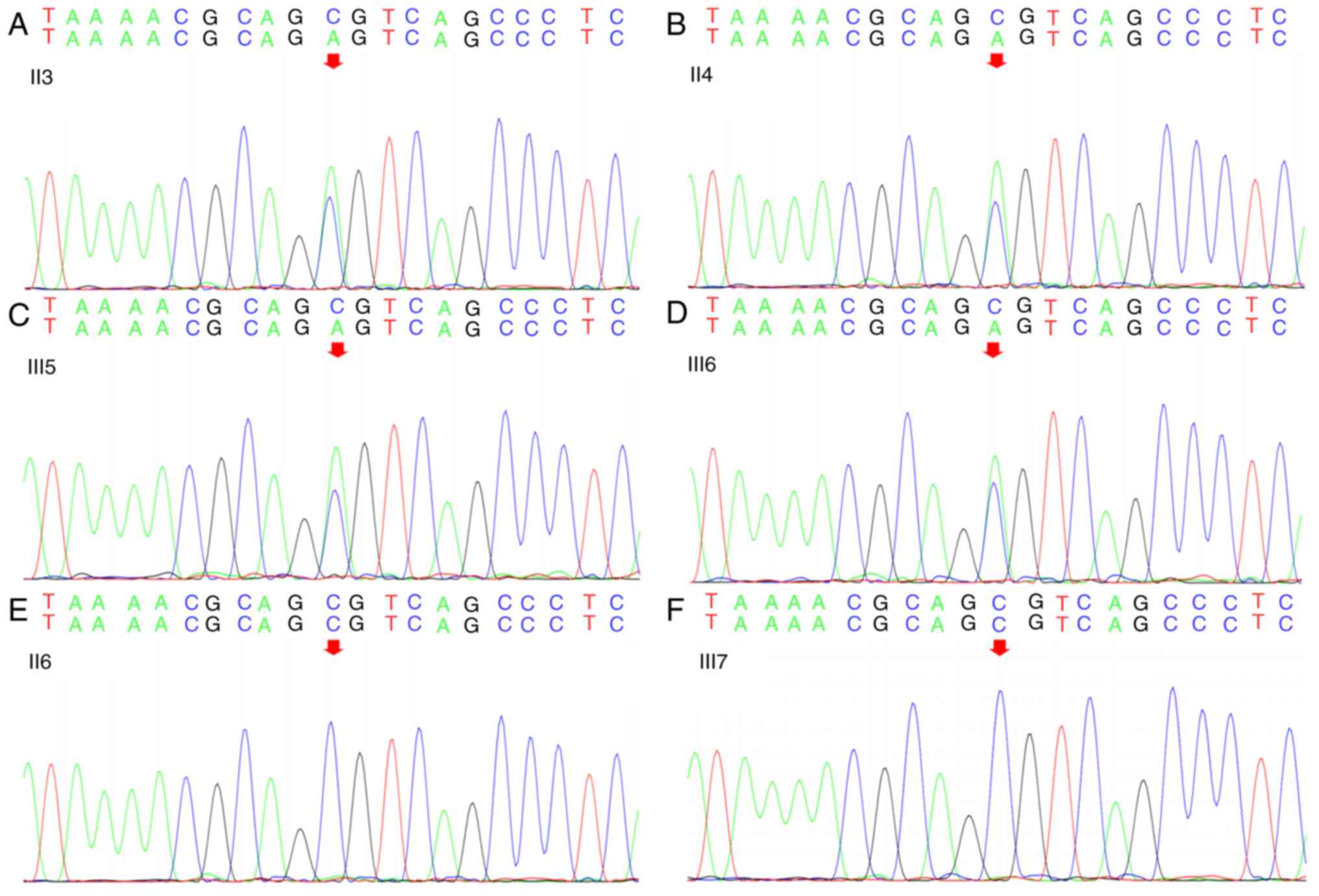
WES identified the missense mutation in the
SPAST gene: c.1785C>A as a likely pathogenic variant
according to ACMG guidelines (11). Sanger sequencing analysis of the
proband, his father (II-4), uncle (II-3), third aunt (II-6),
brother (III-5) and sister (III-7) revealed that all tested male
family members carried the same heterozygous missense mutation in
the SPAST gene: c.1785C>A in exon 17; genomic
coordinates, Chr2:32379499 (Fig.
3). This mutation resulted in a serine to arginine substitution
at position 595 (Ser595Arg) in the AAA ATPase cassette. However,
the missense mutation was not present in the proband's aunt and
sister.
According to American College of Medical Genetics
and Genomics criteria, this variant was considered likely
pathogenic: PM1 (located in mutation hotspots and/or in critical
functional areas where no benign variation is known), PM2 (no
variation or very low frequency in recessive mode was found in
normal control populations), PP3 (variants that would have a
deleterious effect on the function of the gene or gene product) and
PP4 (the clinical phenotype or family history of the mutation
carrier is highly consistent with the characteristics of a certain
monogenic genetic disease). SIFT and PolyPhen2 analyses indicated
that the Ser595Arg mutation is likely to be ‘deleterious’ and
‘probably damaging,’ respectively. MutationTaster software
identified the Ser595Arg mutation as a ‘disease-causing’
mutation.
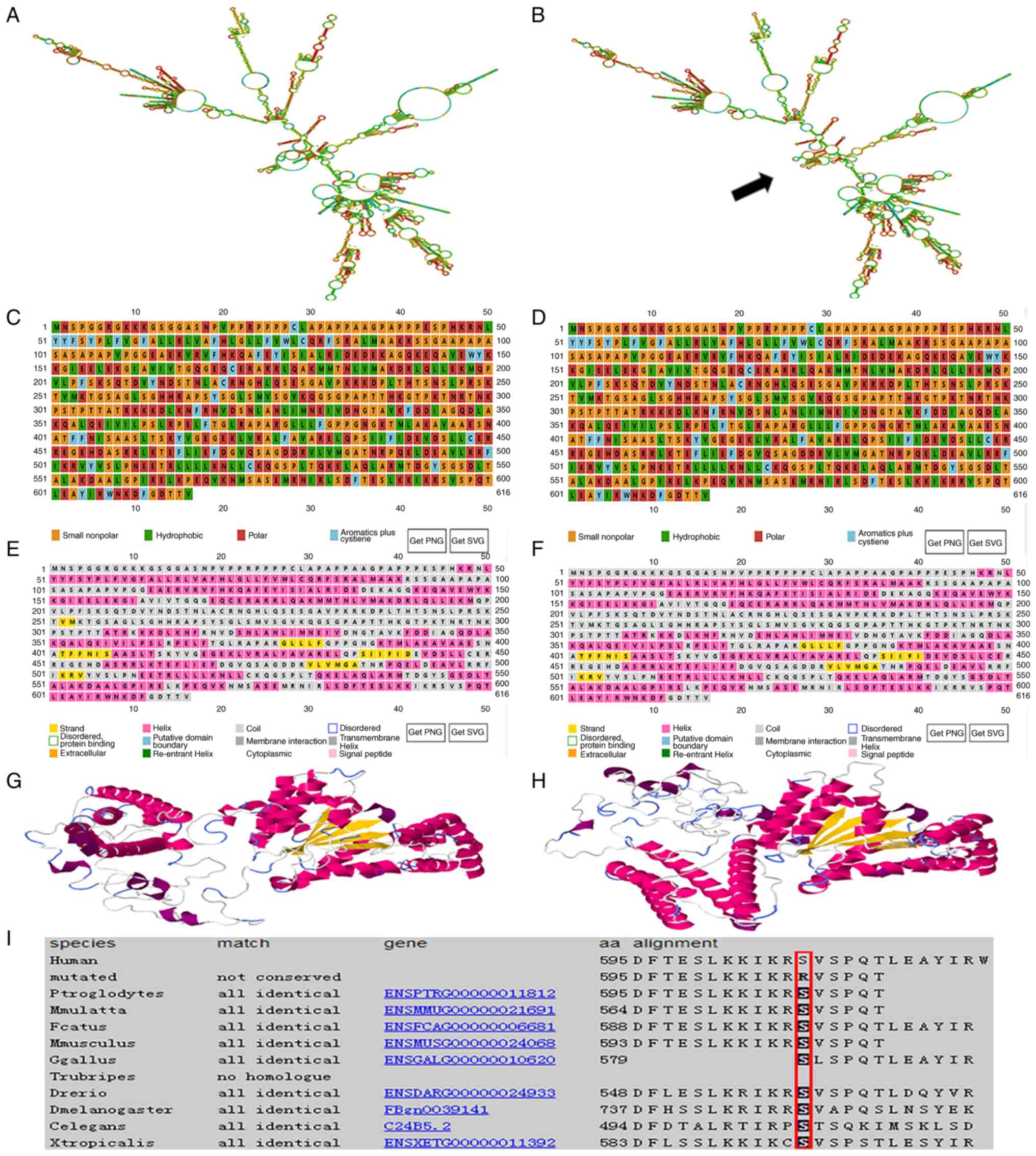
The RNAfold web server was used to predict the
effects of the SPAST mutation on mRNA structure; the results
revealed that it could lead to changes in mRNA structure, as shown
by the increase of hairpin structure at 1564–1904 ribonucleic acid
site (Fig. 4A and B). PSIPRED
(V4.0) was used to predict and compare the mutant and wild-type
Spastin secondary protein structure; the c.1785C>A missense
mutation was predicted to cause changes in Spastin amino acid
polarity, from neutral to positively charged (Fig. 4C and D), and in the secondary
structure, which was manifested as the strand at amino acids
252–253 changed to coil, the helix shortened at amino acids
331–337, the helix shortened at amino acids 436–441, and the helix
lengthened at amino acids 443–447 (Fig. 4E and F). RaptorX was used to
predict the mutated Spastin tertiary structure, which revealed the
spatial structure of the HD, MIT and MTBD domains was reversed
compared with wild-type Spastin (Fig.
4G and H). Protein sequence alignment revealed that the Spastin
amino acid sequence is highly conserved across species at amino
acid 595 (red frame in Fig. 4I).
Therefore, we hypothesized that the mutation causes a change in
tertiary structure that affects protein function.
A summary of all mutations occurring in the four
domains of spastin and the various pathogenic mutations (according
to the clinical significance of mutations shown in the ClinVar and
gnomAD databases) affecting the four domains of Spastin is provided
in Table II. Among these
pathogenic mutations, frameshift mutations were most common,
followed by missense mutations, nonsense mutations were least
common. Approximately 84% (78/93) of the frameshift mutations were
pathogenic; most occurred in the AAA domain (47/78), followed by
the MIT domain (13/78) and MTBD (13/78), and then the HD (5/78).
Among all missense mutations, ~30% (65/208) were considered
pathogenic; most missense mutations occurred in the AAA domain
(63/65), and two occurred in the MTBD, whereas no pathogenic
missense mutations occurred in the MIT domain or the HD. Similar to
frameshift mutations, ~65% (32/49) nonsense mutations were
pathogenic, and most occurred in the AAA domain.
| Table II.Summary of the numbers of mutations
affecting the Spastin domains. |
Table II.
Summary of the numbers of mutations
affecting the Spastin domains.
|
|
|
Pathogenica |
|---|
|
|
|
|
|---|
| Gene mutation | Total | HD | MIT | MTBD | AAA | Total |
|---|
| Frameshift | 93 | 5 | 13 | 13 | 47 | 78 |
| Missense | 208 | 0 | 0 | 2 | 63 | 65 |
| Nonsense | 49 | 7 | 11 | 6 | 18 | 32 |
Pathogenic mutations in exons that encode AAA
functional structure and splicing mutations in introns that may
affect AAA were identified as shown in Table III. Further analysis revealed
that point mutations (specifically missense mutations) were most
common overall (Table III). Base
deletions were also common. Analysis of exons 7–17 encoding the AAA
domain did not reveal obvious differences in the distribution of
pathogenic mutation types among exons or differences in the
locations of splice site mutations.
| Table III.Summary of all pathogenic mutations
affecting the AAA domain. |
Table III.
Summary of all pathogenic mutations
affecting the AAA domain.
| Region and type of
mutation |
|
| Affected exon
region |
|---|
| Intron (splice
mutations) |
|
| Exon8:
c.1098+1G>A; c.1098+2T>A; c.1099-4371_1245+1010del;
c.1099-1062_1246-1342del; c.1099-956_1246-1672del and
c.1099-1G>A |
|
|
|
| Exon9:
c.1173+1G>A; c.1173+185_1245del; c.1174-270_1246-1724dup;
c.1174-2A>T; c.1174-1G>C and c.1174-1G>A |
|
|
|
| Exon10:
c.1245+1G>A; c.1246-2897_1493+523dup and
c.1246-2896_1493+523dup |
|
|
|
| Exon11:
c.1321+1G>A; c.1321+2T>G; c.1322-30_1322-2del;
c.1322-2A>C; c.1322-2A>G; c.1322-1G>T and
c.1322-1G>A |
|
|
|
| Exon12:
c.1413+1_1413+2del; c.1413+3_1413+6del; c.1413+2dup;
c.1413+1G>T; c.1414-2A>T and c.1414-1G>C |
|
|
|
| Exon13:
c.1493+2_1493+5del; c.1493+1G>T; c.1493+1G>A; c.1493+2T>A;
c.1493+2T>C; c.1494-1393_1688-466dup; c.1494-2A>G;
c.1494-2A>C and c.1494-1G>A |
|
|
|
| Exon14:
c.1536+1G>T and c.1537-1G>A |
|
|
|
| Exon15:
c.1617-15_1624dup; c.1617-2A>G and c.1617-1G>A |
|
|
|
| Exon16:
c.1688-378_1728+1541del; c.1688-2A>G; c.1688-1G>C and
c.1688-1G>A |
|
|
|
| Exon17:
c.1728+1G>T; c.1728+1G>A; c.1728+1G>C; c.1728+2T>G;
c.1729-3331_*1641del; c.1729-884_*1715del; c.1729-1G>C and
c.1729-1G>A |
| Exon | Single
nucleotide | Missense | Exon7:
c.1031T>A; c.1047G>C; c.1066G>A; c.1067A>G;
c.1082C>T; c.1085C>T and c.1085C>G |
|
|
|
| c.1103T>C;
c.1104C>G; c.1105A>C; c.1112T>G; c.1121C>T and
c.1133T>A |
|
|
|
| Exon8:
c.1148C>G; c.1151C>T; c.1157A>G; c.1164G>T;
c.1165A>G; c.1168A>G and c.1169T>A |
|
|
|
| Exon9:
c.1181C>A; c.1196C>T; c.1216A>G; c.1219A>C;
c.1225G>A; c.1226C>T and c.1238C>T |
|
|
|
| Exon10:
c.1250G>T; c.1250G>A; c.1253A>G; c.1266G>C;
c.1271G>C; c.1276C>G; c.1306T>C; c.1307C>A and
c.1307C>T |
|
|
|
| Exon11:
c.1322A>G; c.1331A>G; c.1335C>A; c.1343G>A;
c.1360G>A; c.1375A>G; c.1378C>T; c.1379G>A;
c.1382T>C; c.1384A>C; c.1396C>G; c.1405T>G and
c.1409A |
|
|
|
| Exon12:
c.1462A>G; c.1472A>G and c.1484C>T |
|
|
|
| Exon13:
c.1507C>T |
|
|
|
| Exon14:
c.1540A>G; c.1550T>C and c.1601T>C |
|
|
|
| Exon15:
c.1637G>A; c.1667C>T; c.1684C>G and c.1685G>A |
|
|
|
| Exon16:
c.1715T>C |
|
|
|
| Exon17:
c.1742G>C; c.1775T>A and c.1785C>A |
|
|
| Nonsense | Exon7:
c.1039C>T; c.1054C>T and c.1063C>T |
|
|
|
| Exon9:
c.1238C>A |
|
|
|
| Exon10:
c.1252G>T and c.1291C>T |
|
|
|
| Exon11:
c.1344T>A and c.1390G>T |
|
|
|
| Exon12:
c.1417C>T and c.1468C>T |
|
|
|
| Exon14:
c.1573C>T; c.1591C>T and c.1606C>T |
|
|
|
| Exon15:
c.1634C>G and c.1684C>T |
|
|
|
| Exon17:
c.1741C>T; c.1774del and c.1245del |
| Deletion |
|
| Exon7: c.1053del;
c.1056del; c.1069del and c.1091_1092del |
|
|
|
| Exon8: c.1118del
and c.1167_1168del |
|
|
|
| Exon9: c.1180del;
c.1204del; c.1209_1212del and c.1215_1219del |
|
|
|
| Exon10: c.1245del;
c.1253_1255del; c.1262_1263del and c.1263del |
|
|
|
| Exon11:
c.1339_1340del; c.1350_1351del; c.1375del; c.1392del; c.1395del and
c.1407del |
|
|
|
| Exon12:
c.1437_1438del; c.1454_1463del and c.1469_1470del |
|
|
|
| Exon13: c.1496del;
c.1506del and c.1514_1515del |
|
|
|
| Exon14:
c.1539_1540del; c.1561_1564del; c.1577_1580del; c.1583del and
c.1601del |
|
|
|
| Exon16:
c.1702_1712del |
|
|
|
| Exon17: c.1740del;
c.1767del; c.1774del and c.1781del |
| Duplication |
|
| Exon7:
c.1027_1031dup |
|
|
|
| Exon9:
c.1224dup |
|
|
|
| Exon11:
c.1359_1360dup; c.1361dup and c.1368dup |
|
|
|
| Exon12:
c.1458_1459dup |
|
|
|
| Exon14:
c.1560_1561insTT and c.1562dup |
|
|
|
| Exon17:
c.1774dup |
Discussion
All affected family members in this report were
<40 years old at the time of onset and presented with clinical
manifestations of spastic paraplegia, including lower limb
spasticity and weakness, increased muscle tone in both lower limbs,
varying degrees of reduction in muscle strength, tendon
hyperreflexia and bilateral Babinski sign positive and had a family
history suggesting autosomal dominant inheritance; furthermore, all
affected family members carried the c.1785C>A missense mutation
in the SPAST gene, met the diagnostic criteria of HSP-SPG4
(13). Although all affected
family members showed progressive spasticity and weakness of the
lower limbs, they did not exhibit mental, cognitive or other
abnormalities that have been associated with complex HSP (2). The clinical manifestations of the
patients in the present belong to simple HSP. However, this
diagnosis is limited by the lack of MRI results for other family
members. There is no cure for the disease, and only symptomatic
treatment is available, such as intrathecal baclofen
administration, botulinum toxin injections and functional
electrical stimulation (13).
Medication and surgical treatment were not provided to the proband
or his affected family members.
The c.1785C>A missense mutation occurs in the AAA
domain, which is involved in Spastin hexamerization,
microtubule-severing activity and ATP hydrolysis (4). This mutation leads to changes in mRNA
structure, causes a p.Ser595Arg amino acid change in the AAA ATPase
cassette that may alter amino acid polarity and modify both the
secondary and tertiary structures of the Spastin protein. The
mutation is suspected to influence normal protein function,
resulting in reduced microtubule-severing activity, which affects
axon growth and development, ultimately leading to disease.
Additionally, some missense mutations in the M1 isoform can reduce
the activity of wild-type Spastin in a dominant-negative manner,
thereby exacerbating the disease (4). The results of recent studies have
suggested that the neurotoxic effects of mutant Spastin (mainly the
M1 isoform) contribute to disease onset (14–16).
Therefore, the neurotoxicity of this mutant protein is presumed to
serve a role in the pathogenesis experienced by the proband's
affected family members.
SPG4 is an autosomal dominant disease (1); in the present study, it was noted
that all male members of this family carried the pathogenic gene
and exhibited varying degrees of disease presentation. Among the
female family members who provided samples for sequencing, none
carried the SPAST mutation. There is a need to investigate
whether the high heritability of the pathogenic gene among men in
this family is a chance event. Moreover, the family members
differed in terms of age at onset, disease progression and symptom
severity, indicating substantial intrafamilial variability.
The present study determined that the proportion of
pathogenic mutations was greatest in the AAA domain, probably
because the long sequence of nucleotides encoding the AAA domain
carries a greater risk of mutation. The high rates of pathogenicity
for frameshift and nonsense mutations may be related to the early
termination of translation caused by frameshift and nonsense
mutations, which seriously affect protein structure and function,
thus leading to the onset of disease. Although missense mutations
occur most frequently, pathogenic missense mutations comprise ~30%
of all missense mutations. These findings suggested that the
reduced microtubule-severing activity or loss of AAA domain
function is a critical component of SPG4 pathogenesis. No
significant difference was identified in the distribution of
pathogenic mutation types among exons or differences in the
locations of splice site mutations. However, few pathogenic
mutations occurred in exon 16, although this finding requires
confirmation in future studies.
Approximately 75% of HSP-SPAST cases are inherited,
and the remaining 25% of cases involve de novo mutations;
patients with SPG4 mainly receive symptomatic treatment because no
cure is available (17).
Therefore, genetic counseling is essential for affected families.
When progressive walking difficulties, lower limb spasticity and
other symptoms, such as tendon hyperreflexia, occur in patients
without a family history, HSP should also be suspected and genetic
screening should be conducted. In the present report, the clinical
characteristics and sequencing analysis results of a family with
SPG4 were reported. The identification of a novel SPAST
mutation expands the spectrum of pathogenic mutations that cause
SPG4; it also provides information for use in genetic counseling.
Furthermore, specific pathogenic mutations in the SPAST gene
were analyzed and the findings suggested that the loss of AAA
domain function is a crucial component of SPG4 pathogenesis.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
The authors would like to thank Beijing Kangxu
Medical Laboratory Co., Ltd. (Beijing, China) for conducting the
whole exon sequencing.
Funding
The present study was supported by Natural Science Foundation of
Shandong Province (grant no. ZR2019MH060) and The Health Science
and Technology Development Program of Shandong Province (grant no.
202006010928).
Availability of data and materials
The datasets generated and analyzed in this study
are available in the ClinVar (accession no. SCV002761959;
http://www.ncbi.nlm.nih.gov/clinvar).
Other datasets used and/or analyzed in this study are available
from the corresponding author upon reasonable request.
Authors' contributions
XCW, RHL and QXK designed the study. YLW, DDC, YJ,
XYW and TSH collected the data. XCW, RHL and TW contributed to data
analysis and interpretation. XCW and RHL drafted the manuscript;
QXK and RHL contributed to the revision. XCW and RHL confirm the
authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
This study was approved by The Ethics Committee of
The Affiliated Hospital of Jining Medical University (Jining,
China). The proband, his father (II-4), his uncle (II-3), third
aunt (II-6), brother (III-5) and sister (III-7) provided written
informed consent to participate.
Patient consent for publication
The proband, his father (II-4), his uncle (II-3),
third aunt (II-6), brother (III-5) and sister (III-7) provided
written informed consent for the publication of any associated
data, as well as accompanying images and videos. The two medical
staff in the Supplementary data video also provided consent for
publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Shen T, Zhang W, Li L, Zuo RX, Wang ZJ,
Xiao T and Zheng KW: A novel variant of SPAST in a pedigree with
pure hereditary spastic paraplegia in Yunnan Province. Ann Transl
Med. 10:672022. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wei QQ, Chen Y, Zheng ZZ, Chen X, Huang R,
Yang Y, Burgunder J and Shang HF: Spastin mutation screening in
Chinese patients with pure hereditary spastic paraplegia.
Parkinsonism Relat Disord. 20:845–849. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhu Z, Zhang C, Zhao G, Liu Q, Zhong P,
Zhang M, Tang W, Zhan F, Tian W, Wang Y, et al: Novel mutations in
the SPAST gene cause hereditary spastic paraplegia. Parkinsonism
Relat Disord. 69:125–133. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Solowska JM and Baas PW: Hereditary
spastic paraplegia SPG4: What is known and not known about the
disease. Brain. 138((Pt 9)): 2471–2484. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Parodi L, Fenu S, Barbier M, Banneau G,
Duyckaerts C, Tezenas du Montcel S, Monin ML, Ait Said S, Guegan J,
Tallaksen CME, et al: Spastic paraplegia due to SPAST mutations is
modified by the underlying mutation and sex. Brain. 141:3331–3342.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Finsterer J, Löscher W, Quasthoff S,
Wanschitz J, Auer-Grumbach M and Stevanin G: Hereditary spastic
paraplegias with autosomal dominant, recessive, X-linked, or
maternal trait of inheritance. J Neurol Sci. 318:1–18. 2012.
View Article : Google Scholar
|
|
7
|
Fromer M, Moran JL, Chambert K, Banks E,
Bergen SE, Ruderfer DM, Handsaker RE, McCarroll SA, O'Donovan MC,
Owen MJ, et al: Discovery and statistical genotyping of copy-number
variation from whole-exome sequencing depth. Am J Hum Genet.
91:597–607. 2012. View Article : Google Scholar
|
|
8
|
Jiang Y, Oldridge DA, Diskin SJ and Zhang
NR: CODEX: A normalization and copy number variation detection
method for whole exome sequencing. Nucleic Acids Res. 43:e392015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Schwarz JM, Cooper DN, Schuelke M and
Seelow D: MutationTaster2: Mutation prediction for the
deep-sequencing age. Nat Methods. 11:361–362. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tsai MF, Lin YJ, Cheng YC, Lee KH, Huang
CC, Chen YT and Yao A: PrimerZ: Streamlined primer design for
promoters, exons and human SNPs. Nucleic Acids Res. 35((Web Server
Issue)): W63–W65. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shribman S, Reid E, Crosby AH, Houlden H
and Warner TT: Hereditary spastic paraplegia: From diagnosis to
emerging therapeutic approaches. Lancet Neurol. 18:1136–1146. 2019.
View Article : Google Scholar
|
|
14
|
Qiang L, Piermarini E, Muralidharan H, Yu
W, Leo L, Hennessy L, Fernandes S, Connors T, Yates PL, Swift M, et
al: Hereditary spastic paraplegia: Gain-of-function mechanisms
revealed by new transgenic mouse. Hum Mol Genet. 28:1136–1152.
2019. View Article : Google Scholar
|
|
15
|
Solowska JM, Rao AN and Baas PW:
Truncating mutations of SPAST associated with hereditary spastic
paraplegia indicate greater accumulation and toxicity of the M1
isoform of spastin. Mol Biol Cell. 28:1728–1737. 2017. View Article : Google Scholar
|
|
16
|
Chen R, Du S, Yao Y, Zhang L, Luo J, Shen
Y, Xu Z, Zeng X, Zhang L, Liu M, et al: A Novel SPAST mutation
results in spastin accumulation and defects in microtubule
dynamics. Mov Disord. 37:598–607. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mo A, Saffari A, Kellner M, Döbler-Neumann
M, Jordan C, Srivastava S, Zhang B, Sahin M, Fink JK, Smith L, et
al: Early-Onset and severe complex hereditary spastic paraplegia
caused by de novo variants in SPAST. Mov Disord. 37:2440–2446.
2022. View Article : Google Scholar : PubMed/NCBI
|