Introduction
Thalassemia is a hereditary blood disease common in
southern China, and the population carrier rate is 3–24%. It is
caused by a disorder in the synthesis of globin chains and can be
divided into two main subcategories, α-thalassemia and
β-thalassemia, and several rare categories (δ-, γ-, δβ- and
εγδβ-thalassemia) (1,2). Compound heterozygotes for
β-thalassemia usually have a severe transfusion-dependent phenotype
(3). Generally, some conditions
that decrease the production of α-globin (α-thalassemia) can reduce
the clinical severity (4). The
present study reports a case of a 15-year-old Chinese girl carrying
compound heterozygosity for Chinese
Gγ+(Aγδβ)°/βCD17-thalassemia
combined with an -SEA (Southeast Asian) deletion causing
a thalassemia intermedia phenotype.
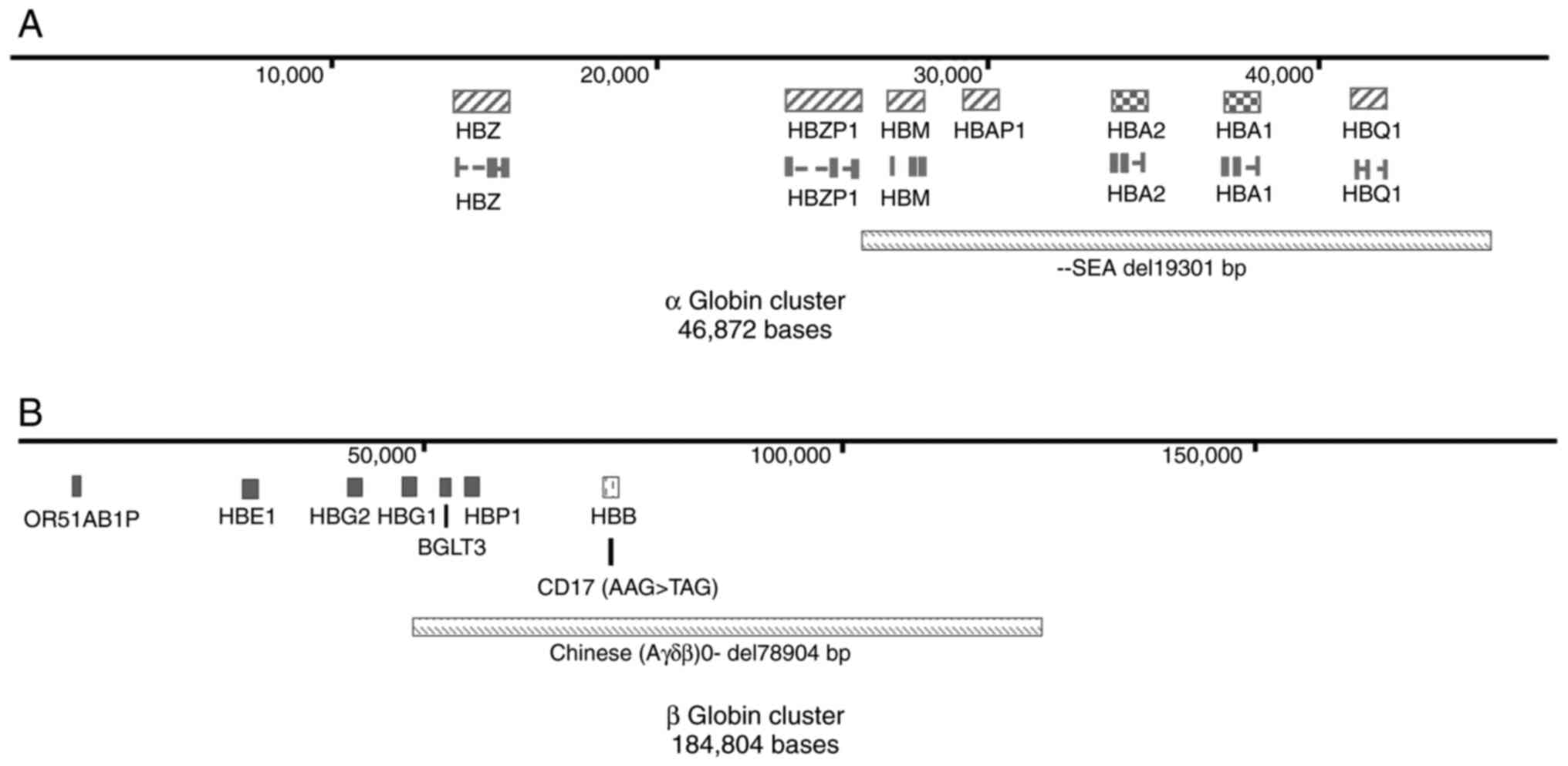
δβ-thalassemia is a rare variant that results from a
deletion in both the δ and β globin genes. The deletion causes the
fetal γ-globin gene to reopen and continue to be expressed into
adulthood, resulting in abnormal levels of fetal hemoglobin (HbF)
(5). Within the Chinese
population, Chinese type, Yunnan type and Guangzhou type δβ
thalassemia have been reported; these are all
Gγ+(Aγδβ)°-thalassemia, of which
Chinese Gγ+(Aγδβ)°-thalassemia is
the most common (6–9). The deletion range of Chinese
Gγ+(Aγδβ)°-thalassemia is ~78.9 kb
(Fig. 1B), which includes part of
the Aγ globin gene, all δ- and β-globin genes, and the
DNA sequences with regulatory function downstream of β-globin gene
(10). This thalassemia genotype
was first reported by Mann et al (11) in the 1970s. However, little
research has been focused on its clinical features. To date, only a
few cases of compound heterozygous mutations of Chinese
Gγ+(Aγδβ)°-thalassemia have been
reported, and these cases illustrated normal, moderate or major
anemia (12–15). Notably, whether the interaction
between Gγ+ (Aγδβ)°-thalassemia and other
common β-thalassemia mutations leads to moderate or major clinical
phenotypes mainly depends on the type of β-thalassemia allele
(β+ or β°).
The present study provides a detailed description of
clinical and molecular characteristics in a 15-year-old Chinese
girl carrying a compound heterozygosity for Chinese
Gγ+(Aγδβ)°/βCD17-thalassemia
combined with the-SEA deletion, which represents an
example of how combined types of thalassemia affect the clinical
severity of Chinese
Gγ+(Aγδβ)°-thalassemia.
Materials and methods
Ethical statement
The present study was approved by the ethics
committee of Shenzhen Second People's Hospital (Shenzhen, China;
approval no. 20210707004) and written informed consent were
obtained from the family.
Proband and parents
The proband was a 15-year-old girl from Yulin City,
Guangxi, China. 2 ml peripheral blood from the proband and her
parents (father, 44 years old; mother, 43 years old) were collected
using EDTA as anticoagulant.
Analysis of hematological parameters
and hemoglobin (Hb) components
Hematological parameters were analyzed using a
Sysmex XN-1,000 automated blood cell counter (Sysmex Corporation).
Hb components were tested using a V8 hemoglobin electrophoresis
instrument with the V8 Nexus Hemoglobin Ultrascreen kit (Helena
Laboratories Corporation).
DNA sample preparation
Genomic DNA was isolated from peripheral blood using
an automatic nucleic acid extractor (Xiamen Kaishuo Biological Co.,
Ltd.) according to the manufacturer's instructions.
Gene analysis
The α-thalassemia deletions (−α3.7,
-α4.2, -SEA, -THAI) were analyzed
using conventional gap-PCR with deletion type α-thalassemia gene
test kit (Shenzhen Yilifang Biological Products Co., Ltd.). The
non-deletion α-thalassemia mutations (Hb Constant Spring, Hb Quong
Sze and Hb Westmead) were analyzed using conventional PCR-reverse
dot blot hybridization (RDB) with non-deletion α-thalassemia gene
mutation test kit (Shenzhen Yilifang Biological Products Co.,
Ltd.). The 19 β-thalassemia mutations, including −28(A>G)(HBB:
c.-78A>G), −29 (A>G)(HBB: c.-79A>G), −30 (T>C)(HBB:
c.-80T>C), −32 (C>A)(HBB: c.-82C>A), −50(A>G)(HBB:
c.-100G>A), codons 14/15 (+G)(HBB: c.45_46insG),
codon17(A>T)(HBB: c.52A>T), codon26 (or Hb E)(G>A)
(HBB:c.79G>A), codons27/28 (+C)(HBB: c.84_85insC), codon31 (−C)
(HBB: c.94delC), codons 37 (G>A)(HBB: c.113G>A), codons 41/42
(−TTCT)(HBB:c.126_129delCTTT), codon43 (G>T)(HBB:
c.130G>T),codons71/72 (+A)(HBB: c.216_217insA), IVS-I-1
(G>T)(HBB: c.92 + 1G>T), IVS-I-5 (G>C)(HBB: c.92 +
5G>C), IVS-II-654 (C>T) (HBB: c.316-197C>T), CAP +1
(A>C)(HBB: c.-50A>C) and initiation codon (T>G) (HBB:
c.2T>G) were analyzed using conventional RDB with β-thalassemia
gene mutation test kit (Shenzhen Yilifang Biological Products Co.,
Ltd.). Chinese
Gγ+(Aγδβ)°-thalassemia was
analyzed by gap-PCR according to the manufacturer's instructions.
The gap-PCR primer sequences are provided in Table I. Multiplex ligation-dependent
probe amplification (MLPA) was performed according to the
manufacturer's protocol using a SALSA MLPA Probemix P102 HBB kit
(cat. no. P102; MRC-Holland) to further confirm the β-globin gene
cluster deletion identified by gap-PCR. PCR reaction mixtures
contained 0.1-1 µg of genomic DNA, 0.2 mmol/l each dNTP, 0.2 mmol/l
each primer, 20 mmol/l Tris-HCl (pH 8.4), 50 mmol/l KCl, 1.5 mmol/l
MgCl2 and 2.5 U of Platinum™ Taq polymerase
(Thermo Fisher Scientific, Inc.). After initial denaturation at
95°C for 5 min, a total of 35 PCR cycles were performed under the
following PCR conditions: 95°C for 45 sec, 60°C for 30 sec and 72°C
for 90 sec; and a final extension at 72°C for 5 min. After
amplification, 3 µl of product was run on a 1% agarose gel in 0.5X
Tris-Borate-EDTA buffer pre-stained with SYBR Safe (1:10,000;
Invitrogen; Thermo Fisher Scientific, Inc.) for 30 min at 5–6 V/cm.
After electrophoresis, the gel was visualized using an ultraviolet
transilluminator.
 | Table I.Primer sequences. |
Table I.
Primer sequences.
| Primer sequence
(5′-3′) | GenBank ID:
Nucleotides |
|---|
| F,
CAAGGGCACCTTTGCCCAGCT |
NC_000011.10:g.5249437 _5249417 |
| R,
CTAGAATTCTTCTGGTCCTCCCT |
NC_000011.10:g.5169215 _5169237 |
DNA sequencing
Two sets of primer pairs were designed and used to
amplify and sequence the hemoglobin subunit α genes (HBA1 and
HBA2): HBA1, forward 5′-CAAGCATAAACCCTGGCGCGC-3′, and reverse
5′-CCTGGCACGTTTGCTGAG-3′; HBA2, forward
5′-CAAGCATAAACCCTGGCGCGC-3′, and reverse
5′-ATTGTTGGCACATTCCGGGA-3′. The PCR mixture comprised: 50 ng of
genomic DNA; up to ddH2O, 30 µl; 10X LA buffer, 5.0 µl;
2.5 mmol/l dNTPs, 3.0 µl; 10 pmol/µl forward primer, 1.0 µl; 10
pmol/µl reverse primer, 1.0 µl; 5 mol/l betaine, 5.0 µl; DMSO, 2.5
µl; 2 U/µl Taq polymerase, 0.4 µl. The C1000 Thermal Cycler
(Bio-Rad Laboratories, Inc.) was used with the following
thermocycling conditions: Initial denaturation step of 95°C for 5
min, denaturation at 95°C for 40 sec, annealing at 66°C for 30 sec
and extension at 72°C for 70 sec, for a total of 33 cycles. DNA
sequencing was performed using the Sanger sequencing method, with
the reference sequence NC_000011.10. The PCR products were
sequenced using an ABI PRISM™ 3,130×l Genetic Analyzer (Applied
Biosystems; Thermo Fisher Scientific, Inc.). Sequencing results are
shown in Fig. S1, Fig. S2, Fig. S3, Fig. S4, Fig. S5.
Case report
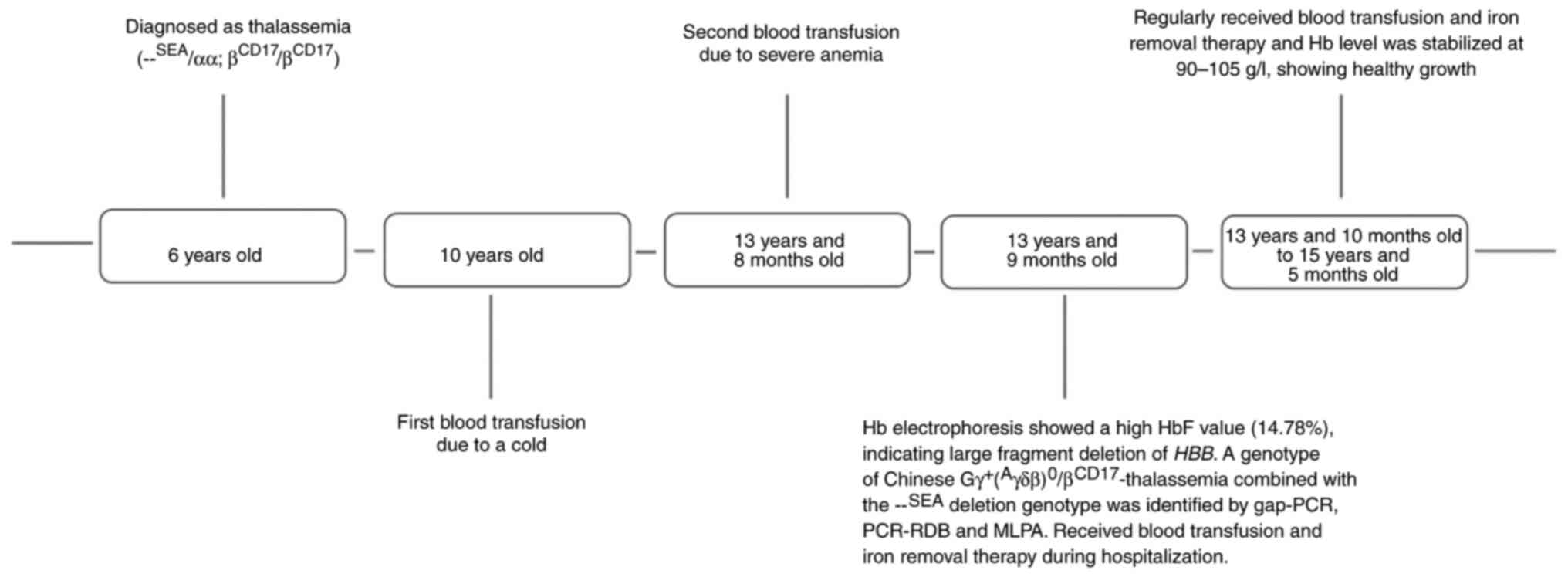
The proband, a 15-year-old girl from Yulin City,
Guangxi, China was born normally and without any observable
development delay. The proband was diagnosed with thalassemia
(-SEA/αα; βCD17/βCD17:homozygote)
in October 2002 at Yulin People's Hospital of Guangxi Province,
China at 6 years of age and did not initially receive regular blood
transfusion. By 10 years of age, she was admitted to a local
hospital due to cold symptoms and received a blood transfusion for
the first time. At 13 years and 8 months old, she had a Hb level of
55 g/l and received a transfusion of 4 units of leukocyte-poor red
blood cells (LPRC), her second blood transfusion, and was
subsequently discharged from the hospital. At 13 years and 9 months
old, she presented at Shenzhen Second People's Hospital (Shenzhen,
China), experiencing headaches and dizziness without obvious cause.
She had a Hb level of 51 g/l and was admitted to hospital. At the
time of admission, the proband weighed 33.9 kg, below the 3rd
percentile. She exhibited facial characteristics of thalassemia
(the skull was enlarged, the cheekbones prominent, the eye distance
widened and the bridge of the nose was low and flat.), marked
paleness and fatigue, but was not jaundiced. The proband's liver
was 4 cm below the midclavicular line of the right costal margin,
the spleen was 8 cm below the left costal margin and she had grade
III ejection systolic murmurs at the apex.
On the day of her admission, routine hematological
evaluation revealed a Hb level of 51 g/l, a mean corpuscular volume
(MCV) of 69 fl, a mean corpuscular Hb (MCH) of 21.3 pg, a white
blood cell (WBC) count of 8.83×109/l and a platelet
count of 143×109/l. On day 2, her Hb further dropped to
37 g/l and she subsequently received a transfusion of 2 units of
LPRC. Three days later, she remained fatigued and dizzy and
continued to exhibit grade III ejection systolic murmurs at the
apex. An echocardiogram showed that in the resting state, there was
no obvious abnormality in the intracardiac structure and blood
flow. As the proband's Hb level was 52 g/l, she received another
transfusion of 2 units of LPRC. Five days after admission, she
complained of dizziness in the morning with a Hb level of 66 g/l
and received a third transfusion of 2 units of LPRC. Six days after
admission, her Hb level recovered to 85 g/l and she received a
fourth transfusion of 2 units of LPRC. Blood tests for total
bilirubin, unconjugated bilirubin, aspartate aminotransferase,
alanine aminotransferase, serum ferritin (FER), human growth
hormone, triiodothyronine, total thyroid hormone, free
triiodothyronine, free thyroid hormone, serum thyroid stimulating
hormone, human chorionic gonadotropin, progesterone, estradiol and
follicle-stimulating hormone were performed (Table II). Seven days after admission,
T2*-weighted MRI analysis revealed moderate iron overload in the
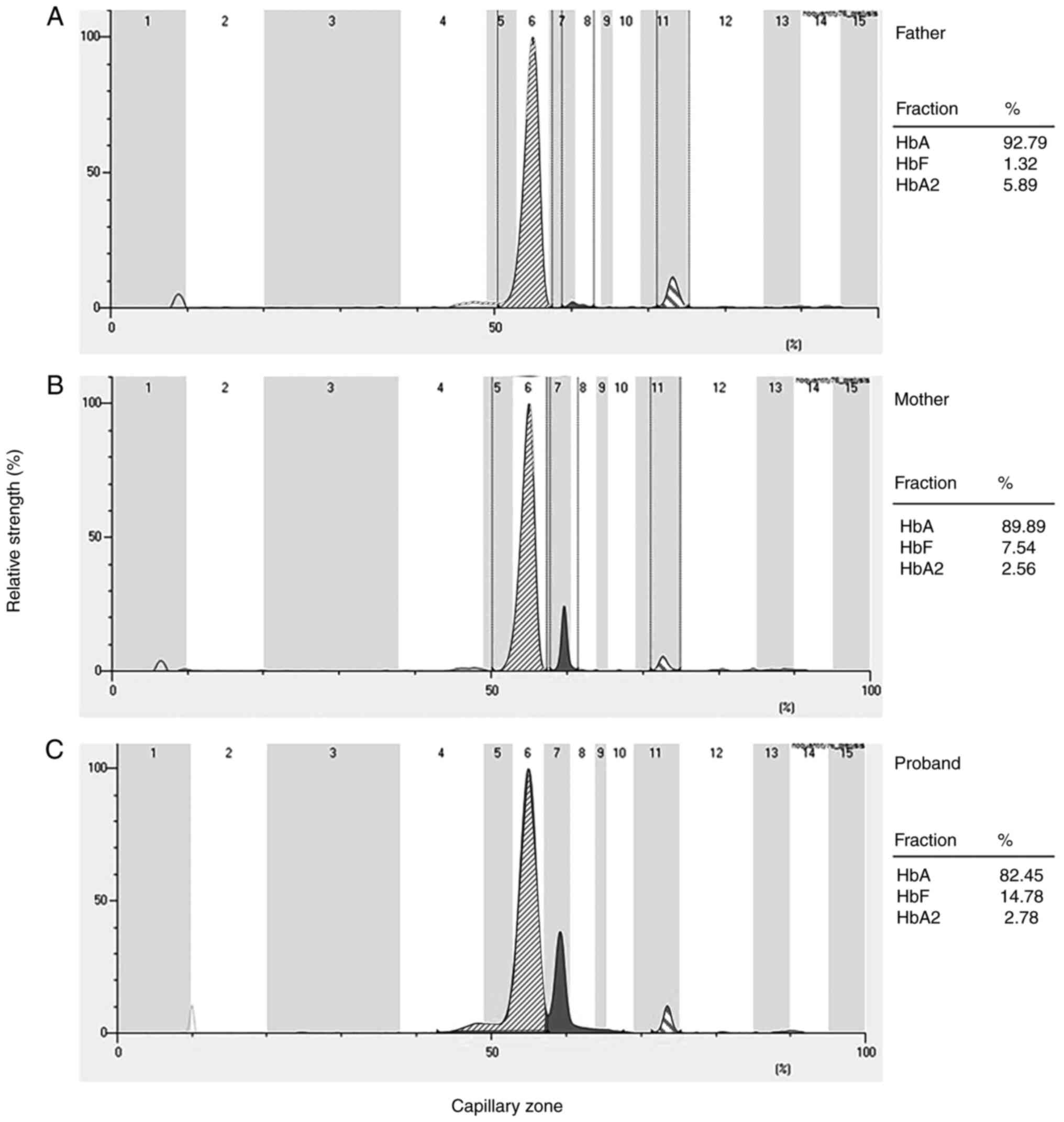
patients' liver. Notably, Hb electrophoresis showed changes of HbF
fraction (normal range, <2.3%) and HbA2 fraction (normal range,
2.5-3.5%). The father showed an increased HbA2 fraction, with HbA
(92.79%; lane 6), HbF (1.32%; lane 7) and HbA2 (5.89%; lane 11).
The probands mother showed increased HbF compared to the father
with HbA (89.89%; lane 6), HbF (7.54%; lane 7) and HbA2 (2.56 %;
lane 11), and the proband exhibited HbA (82.45%; lane 6), HbF
(14.78%; lane 7) and HbA2 (2.78%; lane 11) (Fig. 2). Hematological data of the family
showed changes of Hb (normal range, 110–150 g/l), MCV (normal
range, 80–100 fl) and MCH (normal range, 27–34 pg) The proband
showed data on fluctuations. The father and mother showed both MCV
and MCH decreased (Table
III).
| Table II.Relevant biochemical indicators
during hospitalization of proband. |
Table II.
Relevant biochemical indicators
during hospitalization of proband.
| Parameter | Results | Normal range (range
in different periods) |
|---|
| TBIL, µmol/l | 43.40 | 3.00-22.00 |
| UCB, µmol/l | 35.50 | 0.00-19.00 |
| AST, U/l | 95.00 | 14.00-36.00 |
| ALT, U/l | 51.00 | 9.00-52.00 |
| FER, ng/ml | 1,381.90 | 10.00-291.00 |
| hGH, ng/ml | 1.90 | <10.00 |
| TT3, ng/ml | 0.72 | 0.60-1.81 |
| TT4, ng/ml | 51.50 | 45.00-133.00 |
| FT3, pmol/l | 4.81 | 3.50-6.59 |
| FT4, pmol/l | 15.16 | 11.50-22.70 |
| TSH, mIU/l | 4.98 | 0.35-5.50 |
| HCG, mIU/ml | 2.00 | 0.00-10.00 |
| P4, ng/ml | 0.21 | 0.15-1.40
(follicular phase) |
| E2, pg/ml | 32.47 | 19.5-144.20
(follicular phase) |
| FSH, mIU/ml | 3.06 | 2.5-10.20
(follicular phase) |
| Table III.Hematological and genetic diagnosis
data of the proband and their mother and father. |
Table III.
Hematological and genetic diagnosis
data of the proband and their mother and father.
| Parameter | Proband | Mother | Father |
|---|
| Age, years | 15 | 43 | 44 |
| Hb, g/l |
3.20-12.00a | 11.80 | 11.50 |
| MCV, fl |
65.30-78.80a | 73.60 | 64.00 |
| MCH, pg |
20.80-27.70a | 25.20 | 20.10 |
| HbA, % | 82.45a | 89.89 | 92.79 |
| HbF, % | 14.78a | 7.54 | 1.32 |
| HA2, % | 2.78a | 2.56 | 5.89 |
| HBA
genotype |
-SEA/αα |
-SEA/αα | αα/αα |
| HBB
genotype |
Gγ+(Aγδβ)°/βCD17 |
Gγ+(Aγδβ)°/βCD17 |
βCD17/βN |
Since several studies have reported that Chinese
Gγ+(Aγδβ)°-thalassemia is
clinically manifested as no anemia or mild anemia, with increased
HbF (10–18%), and normal Hb A2 (2.5-3.5%) (16,17),
it was suspected that the proband may carry Chinese
Gγ+(Aγδβ)°-thalassemia, and
therefore the genotypes of the girl and her parents were analyzed
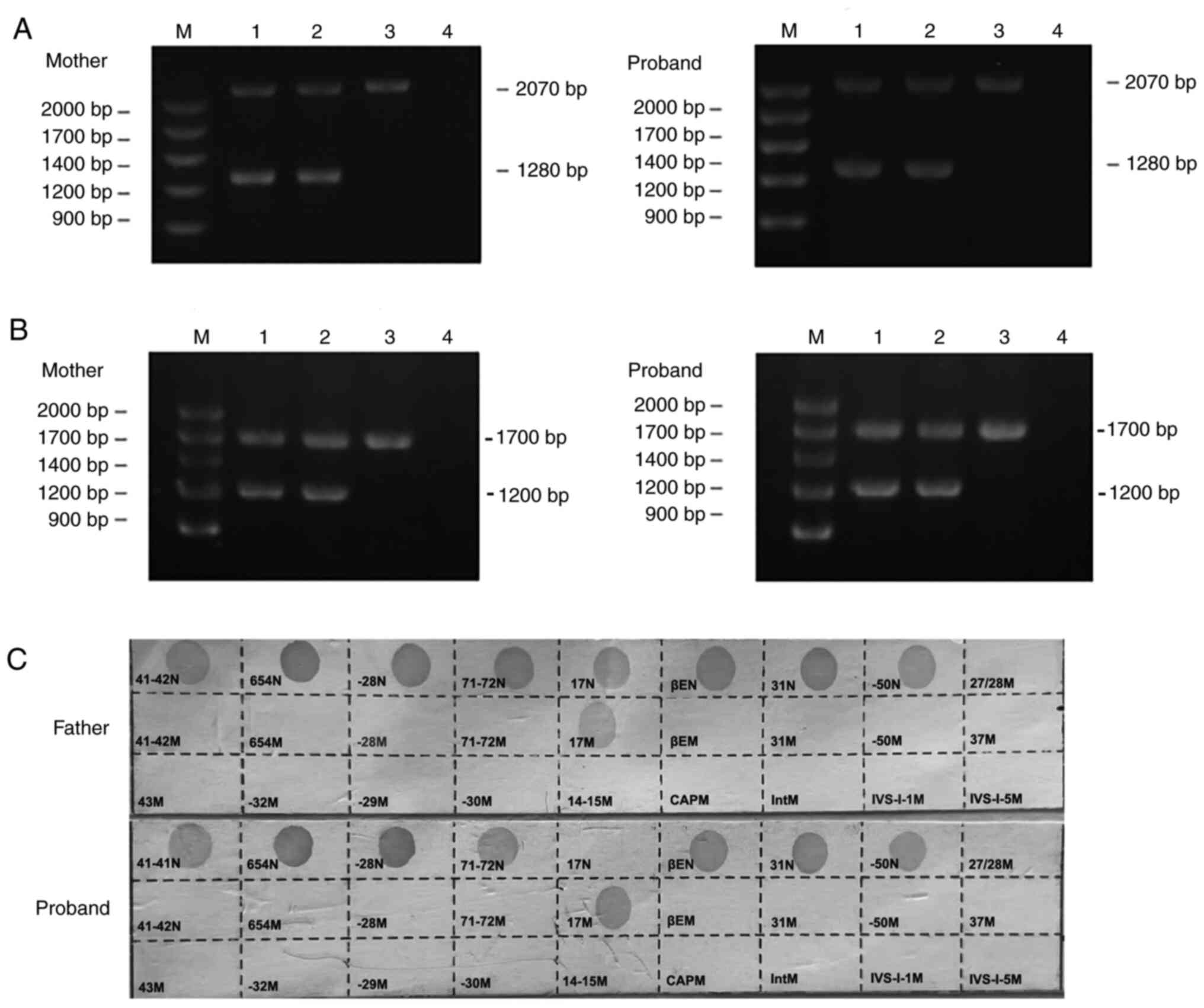
using gap-PCR, PCR-RDB and MLPA (Fig.
S6, Fig. S7). The
common-SEA deletion and Chinese
Gγ+(Aγδβ)°-thalassemia were
detected using gap-PCR and the codon17(A>T)(HBB: c.52A>T)
mutation was detected by PCR-RDB. The data showed that the father
was heterozygous for the codon17(A>T)(HBB: c.52A>T; Fig. 3C), the mother carried the Chinese
Gγ+(Aγδβ)°-thalassemia combined
with the-SEA deletion in a double heterozygous state and
no point mutations of HBB were detected by PCR-RDB (Fig. 3A and B, respectively; Fig. S8, and their daughter inherited
both mutations from her mother and father, thus carrying the
Chinese
Gγ+(Aγδβ)°/βCD17-thalassemia
combined with the-SEA deletion in a compound
heterozygous state (Fig. 3;
Table IV). Ten days later, the
proband had occasional dizziness. Her Hb level was 96 g/l and
hematological indexes were significantly improved. The proband
received a total of eight LPRC transfusions over 10 days, with Hb
fluctuating between 37–96 g/l. The patient's hematological indexes
are summarized in Table IV.
| Table IV.Changes in hematological data during
the proband's hospitalization. |
Table IV.
Changes in hematological data during
the proband's hospitalization.
| Date | Normal range | 11/06/2020 | 12/06/2020 | 14/06/2020 | 16/06/2020 | 17/06/2020 | 22/06/2020 |
|---|
| Transfusion,
units | n/a | n/a | n/a | 2 | 2 | 2 | 2 |
| WBC,
109/l | 5-15 | 8.38 | 5.09 | 5.20 | 6.12 | 5.49 | 7.91 |
| RBC,
1012/l | 3.5-5.5 | n/a | 1.78 | 2.35 | 2.78 | 3.43 | 3.67 |
| Hb, g/l | 110-150 | 51 | 37 | 52 | 66 | 85 | 96 |
| MCV, fl | 80-100 | 69 | 66.9 | 69.4 | 74.5 | 76.4 | 78.5 |
| MCH, pg | 27-34 | 21.3 | 20.8 | 22.1 | 23.7 | 24.8 | 26.2 |
| PLT,
109/l | 100-300 | 143 | 131 | 114 | 146 | 154 | 120 |
Since the diagnosis of the Chinese
Gγ+(Aγδβ)°/βCD17-thalassemia
combined with the-SEA deletion at 13 years and 10 months
old, the proband became transfusion dependent. She has since
received four units per month of LPRCs in view of her low Hb level
and persistent symptoms. Her Hb level remained a steady 90–105 g/l
throughout the following 18 months, prior to follow-up. FER levels
were regularly screened during follow-up and iron-chelating
therapy, desferrioxamine mesylate, was administered when FER was
>1,000 ng/ml. Liver and kidney function was monitored
periodically by clinical biochemical analysis. The proband, now 15
years and 3 months old is still under regular follow-up at the
Department of Hematology of Shenzhen Second People's Hospital
(Shenzhen, China), and is progressing well. Towards the start of
patient follow-up, the Hb level dropped to 62 g/l, and stabilized
between 90–105 g/l in the middle to late stages. WBCs were
consistently maintained within the biological reference interval,
whereas MCV and MCH were consistently below the biological
reference interval. RBC gradually rises towards the biological
reference range, indicates that the anemia symptoms are not further
aggravated. These results during follow-up are displayed in
Fig. 4. A timeline of the probands
disease progression is shown in Fig.
5.
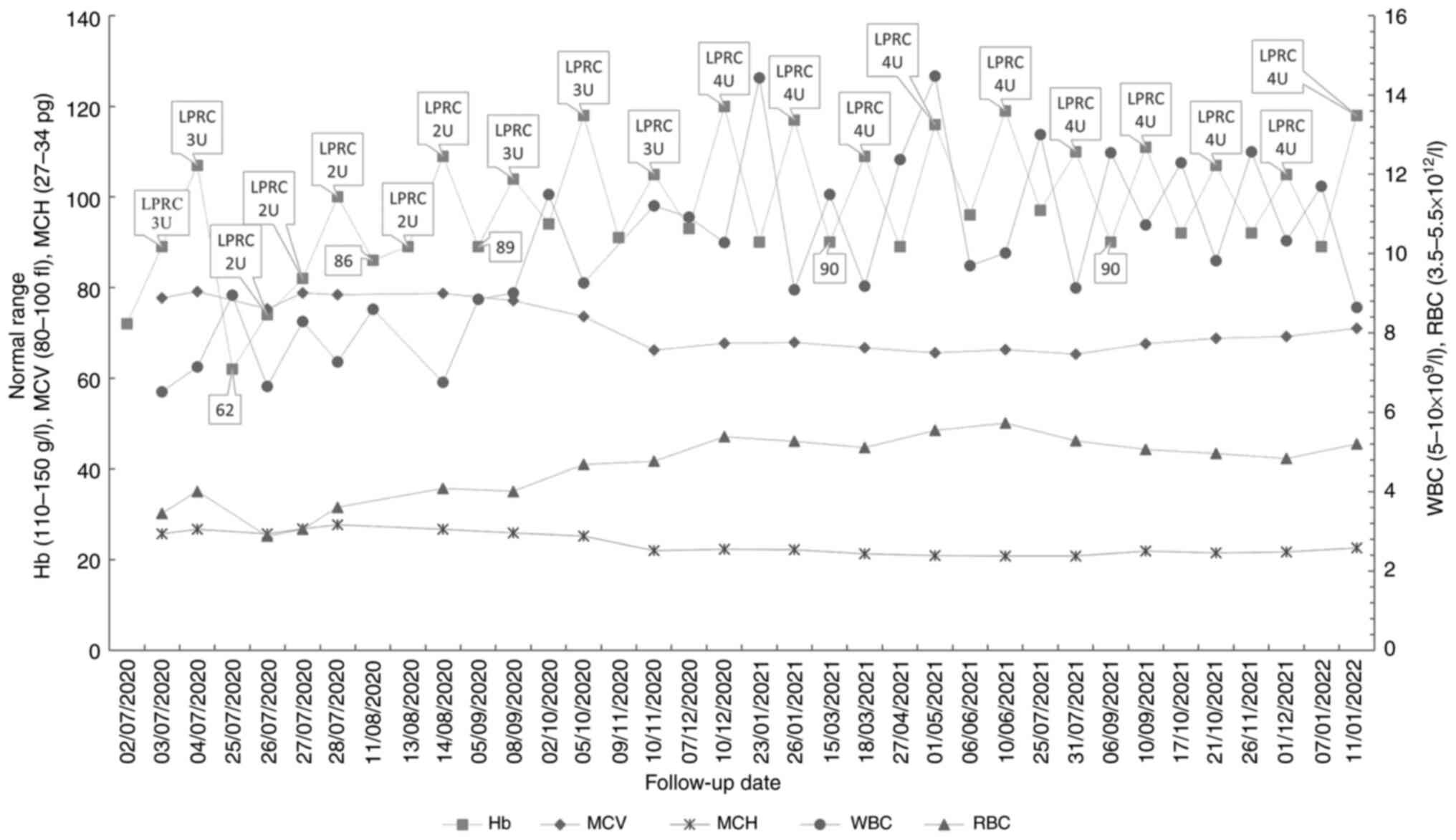 | Figure 4.Analysis of changes in Hb, MCV, MCH,
WBC and RBC during the proband's follow-up period. The label on the
Hb data shows dates of transfusion and dosage (e.g., 1 unit for
LPRC prepared from 200 ml whole blood) and intermittent Hb levels.
Hb, hemoglobin; LPRC, leukocyte-poor red blood cells; MCH, mean
corpuscular Hb; MCV, mean corpuscular volume; RBC, red blood cells;
U, unit; WBC, white blood cells. |
Discussion
Thalassemia is a hereditary hemolytic disease that
is prevalent in Southeast Asia, the Indian subcontinent, and
Africa. In China, there is a high incidence of α-thalassemia in
Guangdong is 8.53% and β-thalassemia is 2.54% (18). The large population base and high
carrying rate result in various types of thalassemia. In 1972,
Chinese Gγ+(Aγδβ)°-thalassemia was
first reported by Mann et al (11), the deletion range of which involves
Aγ, ψβ, δ, β-alleles and 3′-HS-1 regions of globin gene cluster,
covering ~100 kb (7). This
mutation is generated through non-homologous recombination
(5). Although a number of previous
studies have reported that the clinical manifestations of Chinese
Gγ+(Aγδβ)°-thalassemia
heterozygotes show significantly reduced MCV and MCH levels, it is
a typical characteristic of mild thalassemia with microcytic
hypochromic anemia (12–14,19,20).
However, when Chinese
Gγ+(Aγδβ)°-thalassemia coexists
with other common β-thalassemia gene mutations, the patients often
exhibit moderate or severe anemia with increased HbF levels
(12–14).
To date, there are few studies reporting a compound
heterozygous mutation of Chinese
Gγ+(Aγδβ)°/βN-thalassemia
combined with α-thalassemia (21).
In a previous study, the Chinese
Gγ+(Aγδβ)°/βCD41-42-thalassemia
combined with the-α3.7 deletions was reported to be
transfusion independent (13). In
the present report, the proband had no obvious disease symptoms
before 13 years of age in 2020. After a severe, unexplained
headache and dizziness, hepatosplenomegaly and marked paleness and
fatigue were discovered at 13 years and 9 months old and the
hematological results (Hb, 51 g/l; MCV, 69 fl; MCH, 21.3 pg) lead
to the diagnosis of β-thalassemia intermedia. As headache and
dizziness cannot be relieved by clinical treatment, Hb has been
reduced to 37 g/l, which can only be treated by regular blood
transfusion. The Hb level returned to normal, increased to 90–105
g/l and remained stable for ~18 months.
Intermediate thalassemia patients vary in degree of
anemia, the severity of onset is closely related to the amount of
β-chain synthesis, most symptoms in childhood, the clinical
manifestations of moderate anemia between mild and severe, mild or
moderate spleen enlargement, may have jaundice, varying degrees of
skeletal changes, sexual development delay. There is a wide range
of variation among individuals with different intermediate forms of
β-thalassemia: in mild cases, clinical symptoms are not
significant, and physical examination reveals small cell
hypothermic anemia and moderate hemoglobin reduction. In severe
cases, similar to severe beta-thalassemia, the liver and spleen are
enlarged and require irregular blood transfusion to maintain life
(21–23). The present study highlighted a case
of Chinese
Gγ+(Aγδβ)°/βCD17-thalassemia
combined with an -SEA deletion, an uncommon
β-thalassemia state (transitioning to severe β-thalassemia state
with age) caused by a rare β-globin genotype.
In the present report, analysis of the proband's
peripheral blood samples revealed βCD17 locus mutation
homozygous thalassemia combined with the-SEA deletion.
Her father presents with βCD17 locus mutation
thalassemia, and the mother presents with-SEA deletion
mutation thalassemia. As the proband's HbF value of 14.78% was
significantly elevated, she and her mother were subjected to
further genetic tests and were found to carry the Chinese
Gγ+(Aγδβ)°-thalassemia mutation.
The present findings suggested that when genetic tests of
thalassemia are found to be homozygous and further pedigree
analysis does not support this result, the possibility of the
presence of a Chinese
Gγ+(Aγδβ)°-thalassemia deletion
gene should be considered; that is, the impression of
‘homozygosity’ in the presence of a large segment of another
chromosome gene deletion results in wrong judgments. Within the
Chinese population,
Gγ+(Aγδβ)°-thalassemia and
South-East Asian-type hereditary persistent fetal hemoglobinemia
are the most common causes of significantly increased HbF levels
(≥10%) (16). Therefore, for cases
with increased HbF levels, the aforementioned deletions should be
analyzed first, and MLPA can be used to detect other types of
β-globin gene deletions when necessary. If the conventional
thalassemia gene tests reveal a homozygous state for the mutation
site and this result does not match clinical symptoms or
thalassemia screening results, deletion of the β-globin gene
cluster should be considered. In the present case, the proband was
accurately diagnosed with the rare Chinese
Gγ+(Aγδβ)°/βCD17 and
the-SEA deletion genotype. In clinical practice, the
diagnosis of a thalassemia gene is based on the diagnostic
principle of combining phenotype and genotype. When the hematologic
phenotype of the subject is inconsistent with the genotype of
thalassemia detected by routine tests, rare or undiscovered
mutations may exist, which should be further tested and analyzed
for confirmation. The results of this case also confirmed the
importance and practicability of this genetic diagnosis principle
for thalassemia.
The present case report is a textbook model for
β-thalassemia, outlining both the clinical and the genetical
heterogeneity of the disease, as well as the importance of
age-related complications. Complications of β-thalassemia
intermedia are associated with ineffective hematopoietic bone
marrow, chronic anemia and progressive iron overload. Symptoms
usually start between 6–10 years of age and increase in severity
with age. Timely blood transfusion and iron removal treatment can
delay and reduce the occurrence of childhood complications.
The molecular diagnosis of β-thalassemia in light of
the clinical progression of the disease should be carefully
discussed. The differences between β-thalassemia intermedia and
β-thalassemia major are usually difficult to distinguish; however,
according to the proband's blood transfusion requirements outlined
in the present report (average pre-transfusion Hb level <60 g/l)
the most reasonable diagnosis is severe thalassemia intermedia.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The work was supported by Shenzhen High-level Hospital
Construction Fund and by a grant from the Shenzhen Science and
Technology Innovation Commission (grant. no.
JCYJ20170817172241688).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HQ, WLL and WFL designed the present study. XHL,
XLZ, JX, WHZ and YW performed the experiments and analyzed the
data. HQ wrote the manuscript. HQ and WLL confirm the authenticity
of all the raw data. All authors read and approved the final
version of the manuscript.
Ethics approval and consent to
participate
The study protocol was conducted in line with The
Declaration of Helsinki, and was approved by the Ethics Committee
of Shenzhen Second People's Hospital (Shenzhen, China; approval no.
20210707004); written informed consent was obtained from the
parents for themselves and on behalf of the patient.
Patient consent for publication
Written informed consent was obtained from the
parents of the patient.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
MCH
|
mean corpuscular hemoglobin
|
|
MCV
|
mean corpuscular volume
|
|
MLPA
|
multiplex ligation-dependent probe
amplification
|
|
PLT
|
platelet count
|
|
PCR-RDB
|
PCR-reverse dot-blot hybridization
|
|
WBC
|
white blood cell
|
References
|
1
|
Martin A and Thompson AA: Thalassemias.
Pediatr Clin North Am. 60:1383–1391. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
He S, Li J, Li DM, Yi S, Lu X, Luo Y,
Liang Y, Feng C, Chen B, Zheng C and Qiu X: Molecular
characterization of α- and β-thalassemia in the Yulin region of
Southern China. Gene. 655:61–64. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cao A and Galanello R: Beta-thalassemia.
Genet Med. 12:61–76. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Galanello R, Sanna S, Perseu L, Sollaino
MC, Satta S, Lai ME, Barella S, Uda M, Usala G, Abecasis GR and Cao
A: Amelioration of Sardinian beta0 thalassemia by genetic
modifiers. Blood. 114:3935–3937. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Panyasai S, Fucharoen S, Surapot S,
Fucharoen G and Sanchaisuriya K: Molecular basis and hematologic
characterization of deltabeta-thalassemia and hereditary
persistence of fetal hemoglobin in Thailand. Haematologica.
89:777–781. 2004.PubMed/NCBI
|
|
6
|
Zeng YT, Huang SZ, Chen B, Liang YC, Chang
ZM, Harano T and Huisman TH: Hereditary persistence of fetal
hemoglobin or (delta beta)o-thalassemia: three types observed in
South-Chinese families. Blood. 66:1430–1435. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen W, Zhang X, Shang X, Cai R, Li L,
Zhou T, Sun M, Xiong F and Xu X: The molecular basis of
beta-thalassemia intermedia in southern China: Genotypic
heterogeneity and phenotypic diversity. BMC Med Genet. 11:312010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
So CC, So AC, Chan AY, Tsang ST, Ma ES and
Chan LC: Detection and characterisation of beta-globin gene cluster
deletions in Chinese using multiplex ligation-dependent probe
amplification. J Clin Pathol. 62:1107–1111. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang JW, Song WF, Zhao YJ, Wu GY, Qiu ZM,
Wang FN, Chen SS and Stamatoyannopoulos G: Molecular
characterization of a novel form of (A gamma delta beta)zero
thalassemia deletion in a Chinese family. Blood. 81:1624–1629.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhuang J, Jiang Y, Wang Y, Zheng Y, Zhuang
Q, Wang J and Zeng S: Molecular analysis of α-thalassemia and
β-thalassemia in Quanzhou region Southeast China. J Clin Pathol.
73:278–282. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mann JR, MacNeish AS, Bannister D, Clegg
JB, Wood WG and Weatherall DJ: δβ-thalassemia in a chinese famiIy.
Br J Haematol. 23:393–402. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Peng CT, Liu SC, Chiou SS, Kuo PL, Shih
MC, Chang JY and Chang JG: Molecular characterization of deletional
forms of beta-thalassemia in Taiwan. Ann Hematol. 82:33–36. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tan Jin Ai MA, Yap SF, Tan KL, Wong YC,
Wee YC and Kok JL: Mild beta-thalassemia intermedia caused by
compound heterozygosity for
(G)gamma((A)gammadeltabeta)(o)/beta-thalassemia and molecular
characterization of the defect in four Chinese families. Acta
Haematol. 109:169–175. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu Y, Yao Q, Zhong M, Wu J, Xie L, Su L
and Yu F: Genetic research and clinical analysis of deletional
Chinese Gγ+(Aγδβ)°-thalassemia and
Southeast Asian HPFH in South China. Ann Hematol. 99:2747–2753.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
He S, Wei Y, Lin L, Chen Q, Yi S, Zuo Y,
Wei H, Zheng C, Chen B and Qiu X: The prevalence and molecular
characterization of (δβ)° -thalassemia and hereditary persistence
of fetal hemoglobin in the Chinese Zhuang population. J Clin Lab
Anal. 32:e223042018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cai WJ, Li J, Xie XM and Li DZ: Screening
for common β-globin gene cluster deletions in Chinese individuals
with increased hemoglobin F. Int J Lab Hematol. 37:752–757. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Craig JE, Barnetson RA, Prior J, Raven JL
and Thein SL: Rapid detection of deletions causing delta beta
thalassemia and hereditary persistence of fetal hemoglobin by
enzymatic amplification. Blood. 83:1673–1682. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xu XM, Zhou YQ, Luo GX, Liao C, Zhou M,
Chen PY, Lu JP, Jia SQ, Xiao GF, Shen X, et al: The prevalence and
spectrum of alpha and beta thalassaemia in Guangdong Province:
Implications for the future health burden and population screening.
J Clin Pathol. 57:517–522. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Du L, QD, Wang JC, et al: Analysis and
prenatal diagnosis of 5 families with 813-thalassemia. J Mol Diagn
Ther. 7:27–32. 2015.
|
|
20
|
Chen M, Zhang M, Chen L, Lin N, Wang Y, Xu
L and Huang H: Genetic research and clinical analysis of β-globin
gene cluster deletions in the Chinese population of Fujian
province: A 14-year single-center experience. J Clin Lab Anal.
36:e241812022.PubMed/NCBI
|
|
21
|
Liang HF, Liang WM, Xie WG, Lin F, Liu LL,
Li LJ, Ge YY, Lu M, Liao YW, Zeng GK, et al: The gene spectrum of
thalassemia in Yangjiang of western Guangdong Province. Front
Genet. 14:11260992023. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Taher AT, Radwan A and Viprakasit V: When
to consider transfusion therapy for patients with
non-transfusion-dependent thalassaemia. Vox Sang. 108:1–10. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Weatherall DJ: The definition and
epidemiology of non-transfusion-dependent thalassemia. Blood Rev.
26 (Suppl 1):S3–S6. 2012. View Article : Google Scholar : PubMed/NCBI
|