Introduction
Cysteinyl leukotriene receptor antagonists (LTRAs),
currently used for the treatment of asthma and allergic rhinitis,
demonstrate anticancer effects in in vitro and in
vivo models (1,2). Overexpression of 5-lipoxygenase, a
leukotriene synthesis enzyme, and leukotriene receptors are found
in numerous types of cancer, including breast cancer (BC) (3). A large population study in Taiwanese
patients with asthma using LTRAs identified a decreased risk of
breast, lung, colorectal and liver cancer compared with non-LTRA
users (4). In this study, a hazard
ratio of LTRA users was 0.31 and the increase of cumulative dose
progressively decreased cancer risk. Two subsequent large
retrospective cohort studies also reported a lower risk of cancers
in asthmatic patients taking LTRAs. Korean patients showed
significant lower risk of cancers after taking high dose of LTRAs
(adjusted HR=0.56) or 3 years or more of LTRA use (adjusted HR=0.68
after 3 years and 0.33 after 5 years) (5). In Japanese patients, the high
accumulative dose of LTRAs was associated with a decrease in risk
of cancer (adjusted HR=0.57) (6).
Cysteinyl leukotriene receptor 1 (CysLT1R), a receptor of
leukotriene D4, is highly expressed in breast tumor tissues
compared with in normal breast tissues, and its expression is
greater in higher grade tumors (3). MDA-MB-231, a triple-negative BC
(TNBC) cell line, expresses CysLT1R but not CysLT2R (3). Our previous study reported the
apoptotic and antiproliferative effects of two LTRAs, montelukast
and zafirlukast, on MDA-MB-231 cells (7). In addition, LTRAs have been reported
to induce the apoptosis of glioblastoma and lung cancer cells by
reducing ERK1/2 phosphorylation and Bcl-2 levels (8,9).
Zafirlukast has also been shown to induce
G0/G1 cell cycle arrest by upregulating p53
and p21 expression in both glioblastoma A172 and U-87 MG cells, and
p27 expression in U-87 MG cells (10). Similarly, only zafirlukast can
induce G0/G1 cell cycle arrest in MDA-MB-231
cells (7). However, the mechanisms
underlying the cytotoxic and antiproliferative effects of LTRAs on
MDA-MB-231 cells remain unknown. The present study aimed to compare
the effects of montelukast and zafirlukast on the molecular
mechanisms regulating proliferation and death in MDA-MB-231
cells.
Materials and methods
Cell line and culture
MDA-MB-231 cells (cat. no. HTB-26) were obtained
from American Type Culture Collection and cultured in DMEM (cat.
no. 12800-017) supplemented with 1% penicillin/streptomycin (cat.
no. 15140) and 10% fetal bovine serum (cat. no. 10270) (all Gibco;
Thermo Fisher Scientific, Inc.). Cells were maintained at 37°C in a
humidified atmosphere containing 5% CO2.
Chemicals
Montelukast (cat. no. SML0101) and zafirlukast (cat.
no. Z4152) were purchased from Sigma-Aldrich (Merck KGaA), and were
dissolved in DMSO (cat. no. D4540; Sigma-Aldrich; Merck KGaA).
GSK2606414 (cat. no. HY-18072) was purchased from MedChemExpress.
The final concentration of DMSO was 0.04% (v/v) in culture media,
as previously described (7).
Cell treatment
Cells were treated with DMSO, montelukast at 20 µM,
or zafirlukast at 20 µM at 37°C for 3 h in reverse
transcription-quantitative PCR analysis, 6, 12 or 48 h in western
blotting, and 6 or 12 h in flow cytometric analysis and apoptosis
assay. GSK2606414 was treated 1 h prior to the addition of
montelukast or zafirlukast.
Small interfering RNA (siRNA)
transfection
Cells were transfected with 50 nM ON-TARGETplus
siRNA Human DNA damage-inducible transcript 3 (DDIT3, also known as
C/EBP homologous protein or CHOP) SMARTpool (cat. no.
L-004819-00-0005) or ON-TARGETplus Non-targeting Control Pool (cat.
no. D-001810-10-05; both GE Healthcare Dharmacon, Inc.; Table I). All four siRNAs were mixed in
the same reaction. DharmaFECT 4 Transfection Reagent (cat. no.
T-2004-02; GE Healthcare Dharmacon, Inc.) was used according to the
manufacturer's instructions. Cells were transfected at 37°C for 48
h for gene expression studies and for 96 h before determining
protein expression. Cells were immediately treat with DMSO,
montelukast at 20 µM, or zafirlukast at 20 µM for 12 h.
 | Table I.siRNA target sequences. |
Table I.
siRNA target sequences.
| siRNA | Target sequence,
5′-3′ |
|---|
| ON-TARGET plus
Non-targeting Pool (control) |
UGGUUUACAUGUCGACUAA |
|
|
UGGUUUACAUGUUGUGUGA |
|
|
UGGUUUACAUGUUUUCUGA |
|
|
UGGUUUACAUGUUUUCCUA |
| ON-TARGET plus
Human DDIT3 |
GGUAUGAGGACCUGCAAGA |
|
|
CACCAAGCAUGAACAAUUG |
|
|
GGAAACAGAGUGGUCAUUC |
|
|
CAGCUGAGUCAUUGCCUUU |
Western blotting
Proteins were harvested from cells and detected as
previously described (10).
Protein concentrations were determined by Bradford Protein Assay
(cat. no. 5000006; Bio-Rad) and 25 µg protein were loaded into each
lane. Nitrocellulose membranes were blocked with 5% non-fat dry
milk (cat. no. 1706404; Bio-Rad) in 0.5% TBS-T at 4°C overnight and
probed with primary antibodies against phosphorylated (p)-ERK1/2
(rabbit; 1:4,000; cat. no. 4370), ERK1/2 (rabbit; 1:2,000; cat. no.
4695), Bcl-2 (rabbit; 1:600; cat. no. 4223), PARP-1 (rabbit;
1:2,000; cat. no. 9542), LC3-A/B (rabbit; 1:1,000; cat. no. 12741),
activating transcription factor 6 (ATF6; rabbit; 1:1,000; cat. no.
65880), CHOP (mouse; 1:1,000; cat. no. 2895), p21 (rabbit; 1:1,000;
cat. no. 2947), p27 (rabbit; 1:1,000; cat. no. 3686), CDK4 (rabbit;
1:1,000; cat. no. 12790), cyclin D1 (rabbit; 1:1,000; cat. no.
2978) (all Cell Signaling Technology, Inc.) and cyclin E (mouse;
1:1,000; cat. no. 05-363; Merck KGaA) for 2 h at room temperature.
β-actin (rabbit; 1:10,000; cat. no. 4970) and β-tubulin (mouse;
1:5,000; cat. no. 86298) (both Cell Signaling Technology, Inc.)
were incubated for 45 min at room temperature. After that,
membranes were incubated with anti-rabbit or anti-mouse conjugated
with horseradish peroxidase enzymes for 1 h at room temperature
(goat; 1:4,000; cat. no. 111-035-003 or 115-035-003; Jackson
ImmunoResearch Laboratories). Bands were visualized using Clarity
Western ECL substrate (cat. no. 1705061; Bio-Rad) and quantified
using ImageJ software (version 1.54; National Institutes of Health
and the Laboratory for Optical and Computational Instrumentation).
β-actin or β-tubulin was used as loading controls depending on the
molecular weight of proteins of interest. The band intensity was
normalized to the expression of the loading control.
Flow cytometric analysis
Cells were detached with 0.05% trypsin-EDTA (cat.
no. 25300054; Gibco; Thermo Fisher Scientific, Inc.) at 37°C
for 2 min and centrifuged at 320 × g for 10 min at room
temperature. The levels of monomeric JC-1, caspase 3/7 activity,
autophagic LC3, p-ATM and p-histone H2AX (H2AX) were measured using
Guava Mitochondrial Depolarization Kit, Guava® Caspase
3/7 FAM Kit (cat. no. 4500-0540; Merck Millipore; Merck KGaA),
FlowCellect Histone H2AX Phosphorylation Assay Kit (cat. no.
FCCS100182; Merck Millipore; Merck KGaA), phycoerythrin-conjugated
anti-p-ATM (Ser1981) (cat. no. FCMAB110P; Merck Millipore; Merck
KGaA) and FlowCellect™ Autophagy LC3 Antibody-based Assay Kit (cat.
no. FCCH100171; Merck Millipore; Merck KGaA), respectively. All
tests were performed according to the manufacturer's protocol. The
percentage of dead cells was determined by 7-aminoactinomycin D
(7-AAD) staining when assessing mitochondrial depolarization. Cells
were assessed using the Guava® easyCyte 5HT benchtop
flow cytometer with InCyte 3.1 (both Cytek Biosciences). A minimum
of 2,000 events was analyzed for each sample.
Reverse transcription-quantitative
(RT-q)PCR
Total RNA from cells was extracted using the Total
RNA Mini kit (cat. no. RB300; Geneaid Biotech Ltd.) according to
the manufacturer's protocol. cDNA synthesis and qPCR were performed
using superscript III reverse transcriptase (cat. no. 18080-044;
Invitrogen; Thermo Fisher Scientific, Inc.) and SensiFAST SYBR
Lo-ROX (cat. no. BIO-94005; Bioline; Meridian Bioscience) on an
Applied Biosystems Real-time PCR 7500 system (Applied Biosystems;
Thermo Fisher Scientific, Inc.) as previously described (11). All primers are listed in Table II. The thermocycling conditions
were as follows: Initial denaturation at 50°C for 2 min and 95°C
for 10 min, followed by 40 cycles at 95°C for 15 sec and 58°C for 1
min, with a melting curve stage of 95°C for 10 sec and 60°C for 1
min. The Cq value of each target gene was normalized to the
expression of GAPDH. Relative fold-change was calculated against
untreated cells based on the 2−ΔΔCq method (12).
| Table II.Reverse transcription-quantitative
PCR primers. |
Table II.
Reverse transcription-quantitative
PCR primers.
| Gene | Forward primer,
5′-3′ | Reverse primer,
5′-3′ |
|---|
| GAPDH |
AGCCTTCTCCATGGTGGTGAAGAC |
CGGAGTCAACGGATTTGGTCG |
| Ki-67 |
CTTTGGGTGCGACTTGACG |
GTCGACCCCGCTCCTTTT |
| PCNA |
GGCCGAAGATAACGCGGATAC |
GGCATATACGTGCAAATTCACCA |
| ATF4 |
CTTCCTGAGCAGCGAGGTG |
TCTCCAACATCCAATCTGTCC |
| ATF6 |
TCGGTCAGTGGACTCTTATT |
CCAGTGACAGGCTTATCTTC |
| CHOP |
AAGGCACTGAGCGTATCATGT |
TGAAGATACACTTCCTTCTTGAACA |
| GADD34 |
GAGGAGGCTGAAGACAGTGG |
AATTGACTTCCCTGCCCTCT |
| PERK |
GTCCGGAACCAGACGATGAG |
GGCTGGATGACACCAAGGAA |
| XBP-1s |
TCTGCTGAGTCCGCAGCAG |
GAAAAGGGAGGCTGGTAAGGAAC |
| CAT |
CCATTATAAGACTGACCAGGGC |
AGTCCAGGAGGGGTACTTTCC |
| GST |
GGATCTGCTGGAACTGCTTATCAT |
TGTCCGTGACCCCTTAAAATCTT |
| GR |
AACATCCCAACTGTGGTCTTCAGC |
TTGGTAACTGCGTGATACATCGGG |
| GPx1 |
TTCCCGTGCAACCAGTTTG |
TTCACCTCGCACTTCTCGAA |
| SOD |
CTGAAGGCCTGCATGGATTC |
CCAAGTCTCCAACATGCCTCTC |
Apoptosis assay
Floating cells and cells detached with 0.05%
trypsin-EDTA were collected and centrifuged at 320 × g for 10 min
at room temperature. Pellets were washed twice in Annexin V binding
buffer, and were then with Annexin V-FITC (cat. no. 556419) and
7-AAD (cat. no. 51-68981E) (both BD Biosciences). The percentage of
Annexin V positive cells representing early and late apoptotic
cells was determined using a BD Accuri C6 Plus with BD Accuri C6
Plus Software version 1.0.1 (both BD Biosciences). A minimum of
5,000 events/sample were collected.
Dichlorofluorescein (DCFH) assay
Cells were seeded in 96-well clear bottom black
plates and incubated with 20 µM DCFH diacetate (cat. no. D6883;
Sigma-Aldrich; Merck KGaA) in phenol red-free MEM (cat. no.
12800-017; Sigma-Aldrich; Merck KGaA) in the dark before being
treated with DMSO, montelukast at 20 µM, or zafirlukast at 20 µM
for 3 h. The DCF fluorescence intensity was detected using a
Varioskan™ Flash spectral scanning Multimode Reader (Thermo Fisher
Scientific, Inc.) with 490/535 nm excitation/emission wavelengths.
The DCF fluorescence intensity of each treatment was normalized to
fluorescence intensity of untreated cells.
Statistical analysis
Data are presented as the mean ± SEM (n≥3). Data
were analyzed by one- or two-way ANOVA followed by Tukey's post hoc
test using GraphPad Prism version 5.0 (Dotmatics). The level of
significance was defined as P<0.05.
Results
Zafirlukast induces p27 expression and
decreases cyclin D1 expression
Although both montelukast and zafirlukast at 20 µM
have been reported to inhibit MDA-MB-231 cell proliferation, only
20 µM zafirlukast can induce G0/G1 cell cycle
arrest at 48 h post-exposure, according to a previous study
(7). Therefore, the effects of
both drugs on the expression of two proliferating markers, Ki-67
and proliferating cell nuclear antigen (PCNA), were assessed.
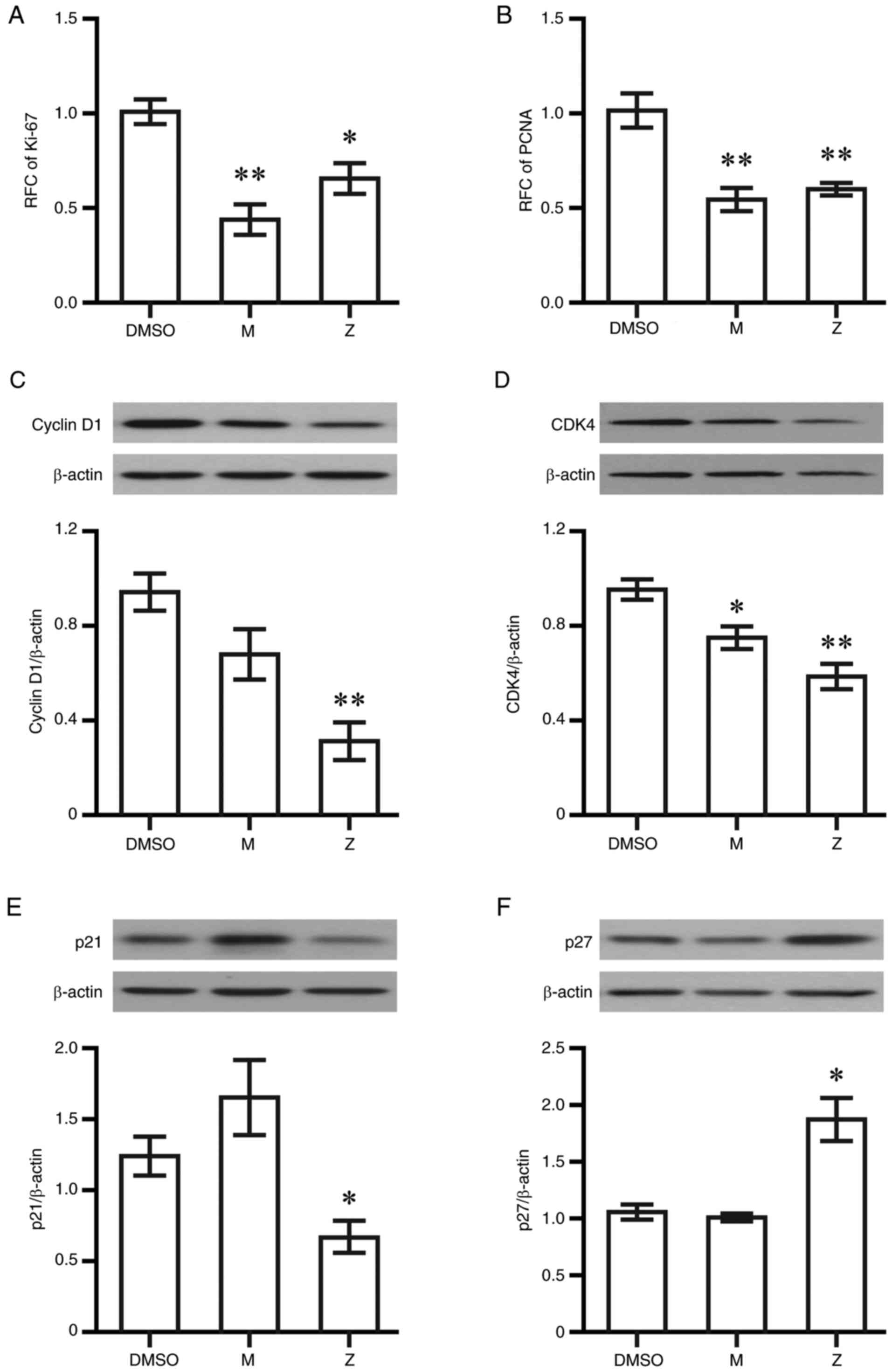
Montelukast and zafirlukast at 20 µM decreased Ki-67 and PCNA at 6
h post-exposure (Fig. 1A and B).
Cells were treated with 20 µM drugs for 48 h and the expression
levels of proteins regulating cell cycle progression were
determined using western blotting. Cyclin D1 and CDK4 levels were
decreased in cells that were treated with zafirlukast (Fig. 1C and D), while both drugs had no
effect on cyclin E levels (Fig.
S1). Zafirlukast significantly decreased p21 expression,
whereas the increase of p21 by montelukast was not significant
(Fig. 1E). Increased p27
expression was found only in cells treated with zafirlukast
(Fig. 1F).
LTRAs activate the intrinsic apoptosis
pathway in MDA-MB-231 TNBC cells
Montelukast at ≥10 µM and zafirlukast at 20 µM were
previously reported to induce the apoptosis of MDA-MB-231 cells at
24 h post-exposure (7); therefore,
20 µM was used to assess cell death in the present study. The
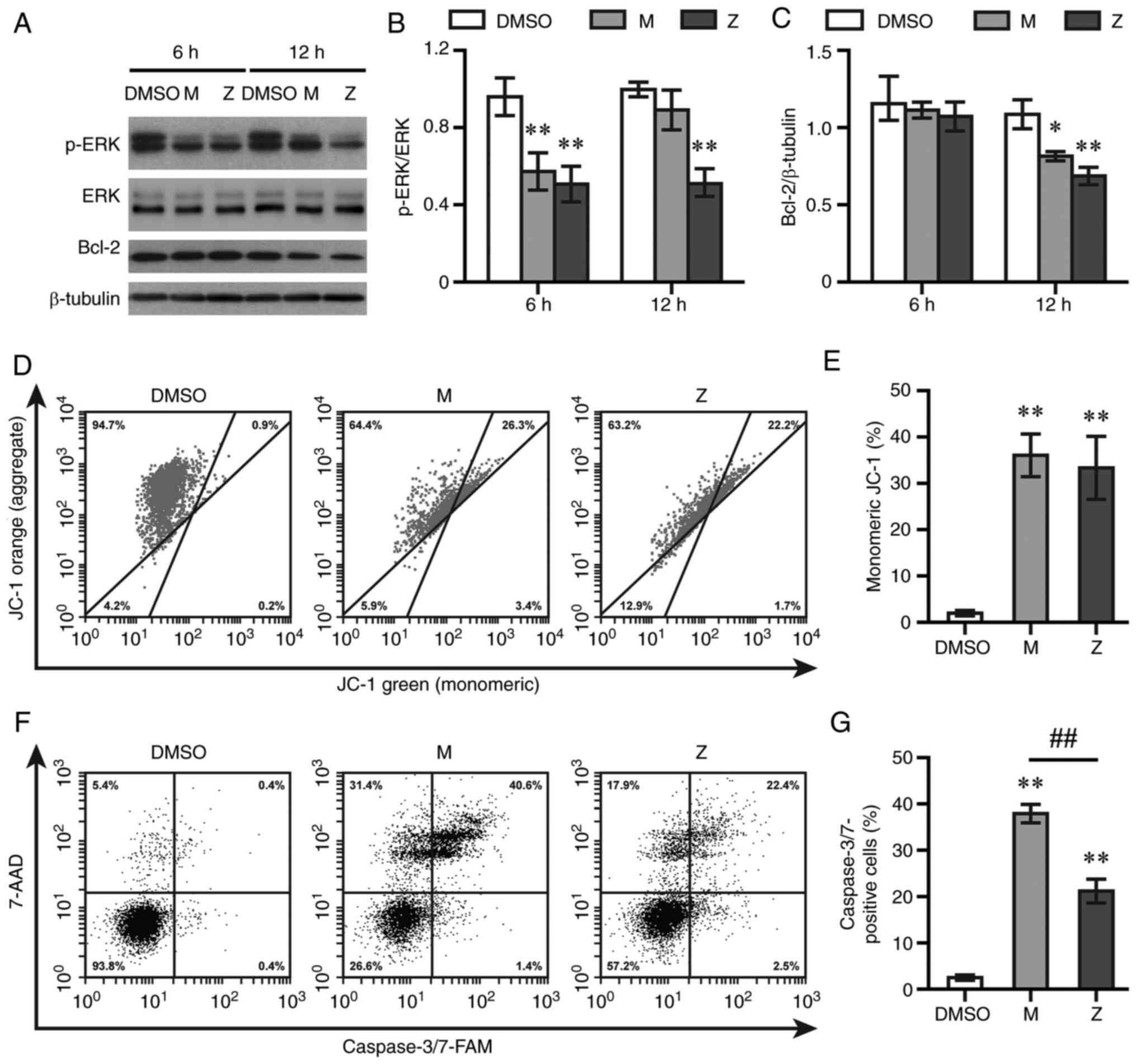
expression levels of p-ERK1/2 and Bcl-2 were detected using western
blotting. Montelukast and zafirlukast decreased the expression
levels of p-ERK1/2 at 6 h (Fig. 2A and
B) and the anti-apoptotic protein Bcl-2 at 12 h post-exposure
(Fig. 2A and C). Both LTRAs did
not alter levels of ERK1/2 (Fig. 2A
and B). Next, cells were stained with JC-1 and 7-AAD to assess
mitochondrial membrane potential and cell death, respectively. Both
LTRAs increased the levels of cells with green fluorescent signal,
indicative of monomeric JC-1, at 12 h post-exposure (Fig. 2D and E). The levels of
7-AAD-positive cells after being exposed to montelukast and
zafirlukast were 48.98±7.22 and 31.61±2.04%, respectively, compared
with 3.97±0.50% in DMSO-treated cells at 12 h (data not shown).
Caspase 3/7 activity was also measured by flow cytometry. Both
drugs increased caspase 3/7 activity at 12 h post-treatment, with a
greater effect observed in montelukast-treated cells (Fig. 2F and G)
LTRAs trigger autophagy in MDA-MB-231
TNBC cells
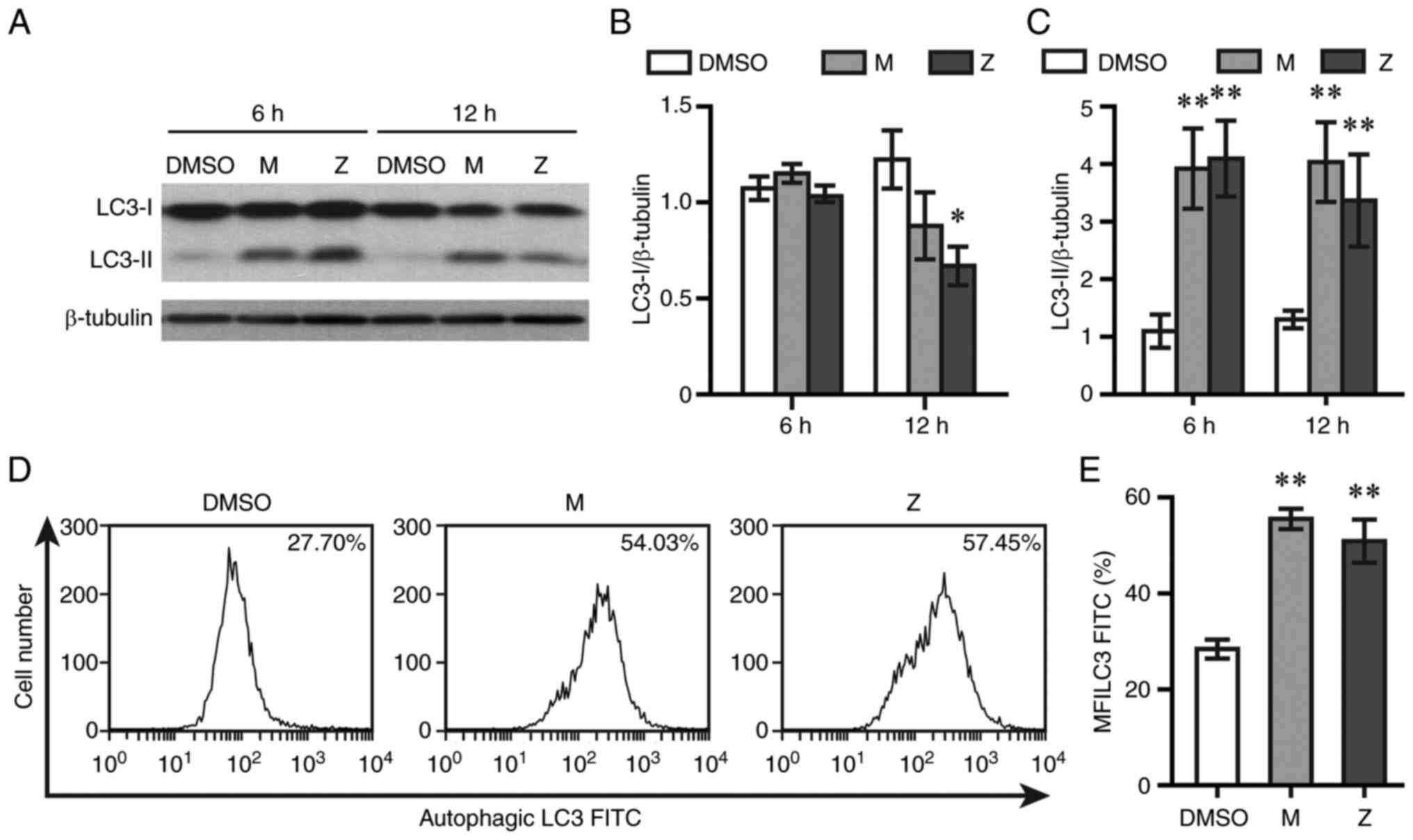
To test whether autophagy was caused by LTRAs, LC3
levels were measured by western blotting and flow cytometry.
Montelukast and zafirlukast did not alter LC3-I levels at 6 h
post-exposure, while zafirlukast decreased LC3-I levels at 12 h
post-exposure (Fig. 3A and B).
Both drugs elevated LC3-II levels at 6 and 12 h post-treatment
(Fig. 3A and C). The expression of
lipidated LC3 sequestered into autophagosomes was determined by
using flow cytometry. Both drugs caused a rightward shift of
autophagic LC3 fluorescence intensity and increased the mean
fluorescent intensity of autophagic LC3 at 12 h post-exposure
(Fig. 3D and E).
LTRAs induce DNA damage and
endoplasmic reticulum (ER) and oxidative stress in MDA-MB-231 TNBC
cells
Apoptosis and autophagy are triggered in response to
DNA damage (13). The levels of
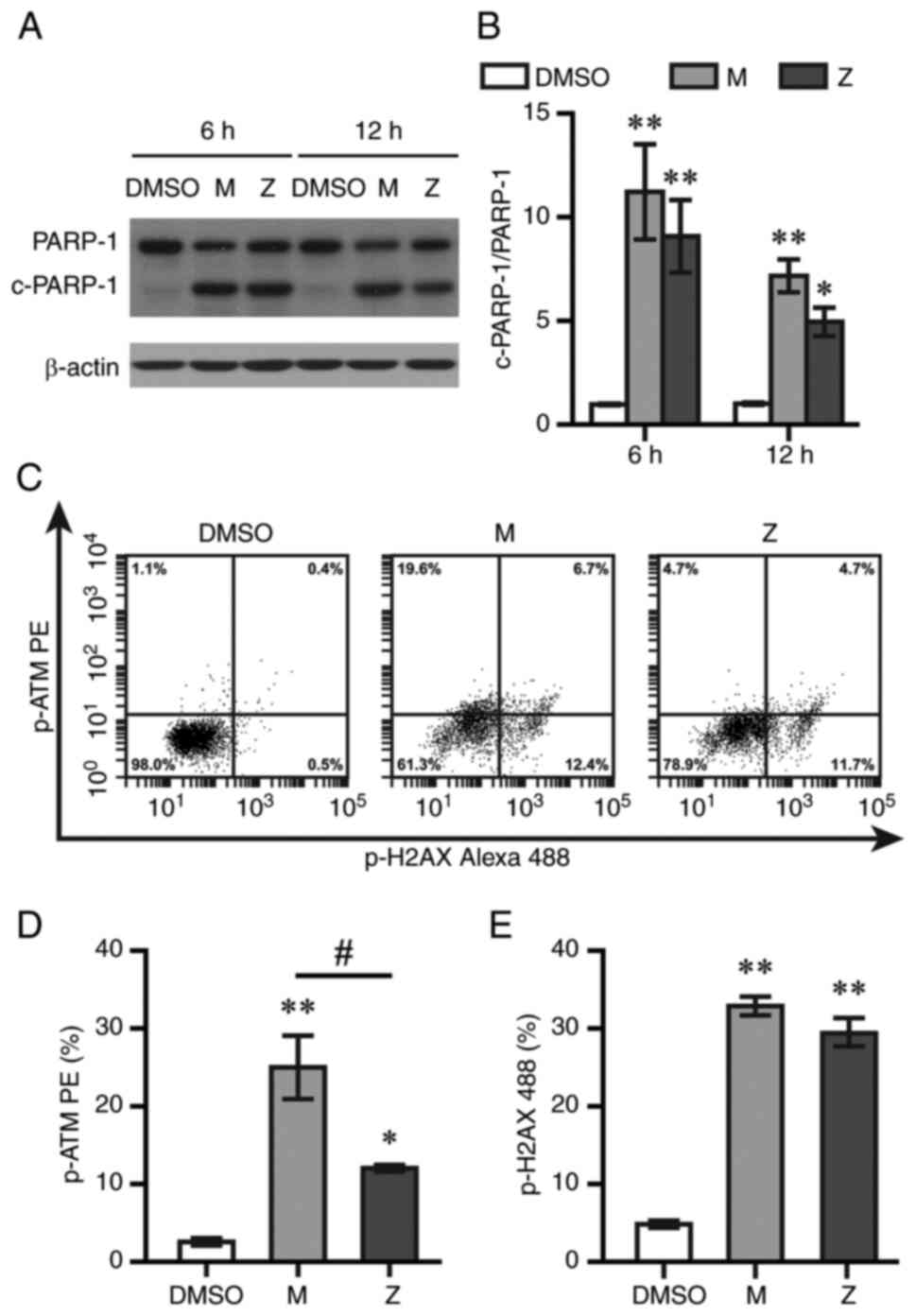
PARP-1, a key regulator of the DNA repair process, were measured by
western blotting in the present study. Montelukast and zafirlukast
increased levels of 89-kDa fragments of PARP-1 at 6 and 12 h after
treatment (Fig. 4A and B). Cells
with positive staining of p-ATM and p-H2AX, two DNA damage markers,
were counted by flow cytometry at 12 h post-exposure. Montelukast
and zafirlukast increased p-ATM and p-H2AX levels (Fig. 4C-E). Montelukast showed
significantly higher p-ATM levels than zafirlukast.
Activation of ER stress can lead to either autophagy
or apoptotic cell death (14,15).
In the present study, the expression of ER membrane-associated
sensors and signaling molecules was evaluated using RT-qPCR at 6 h
post-treatment. Montelukast, but not zafirlukast, upregulated the
expression levels of PERK and ATF4 (Fig. 5A and B). Both montelukast and
zafirlukast increased the mRNA expression levels of X-box binding
protein 1 splice variant (XBP-1s), CHOP and growth arrest and DNA
damage-inducible gene 34 (GADD34), but had no effect on ATF6 mRNA
levels (Fig. 5C-F). The protein
expression levels of CHOP in montelukast-treated cells were
significantly higher than those in zafirlukast-treated cells
(Fig. 5G and H). The activation of
ER stress triggers cleavage of ATF6 (16). Montelukast and zafirlukast
decreased the expression levels of total ATF6 protein (Fig. 5G), and the cleaved ATF6/ATF6 level
in zafirlukast-treated cells was greater than that in
montelukast-treated cells (Fig.
5I). The cleaved ATF6/ATF6 levels at 12 h post-zafirlukast
treatment were higher than those at 6 h.
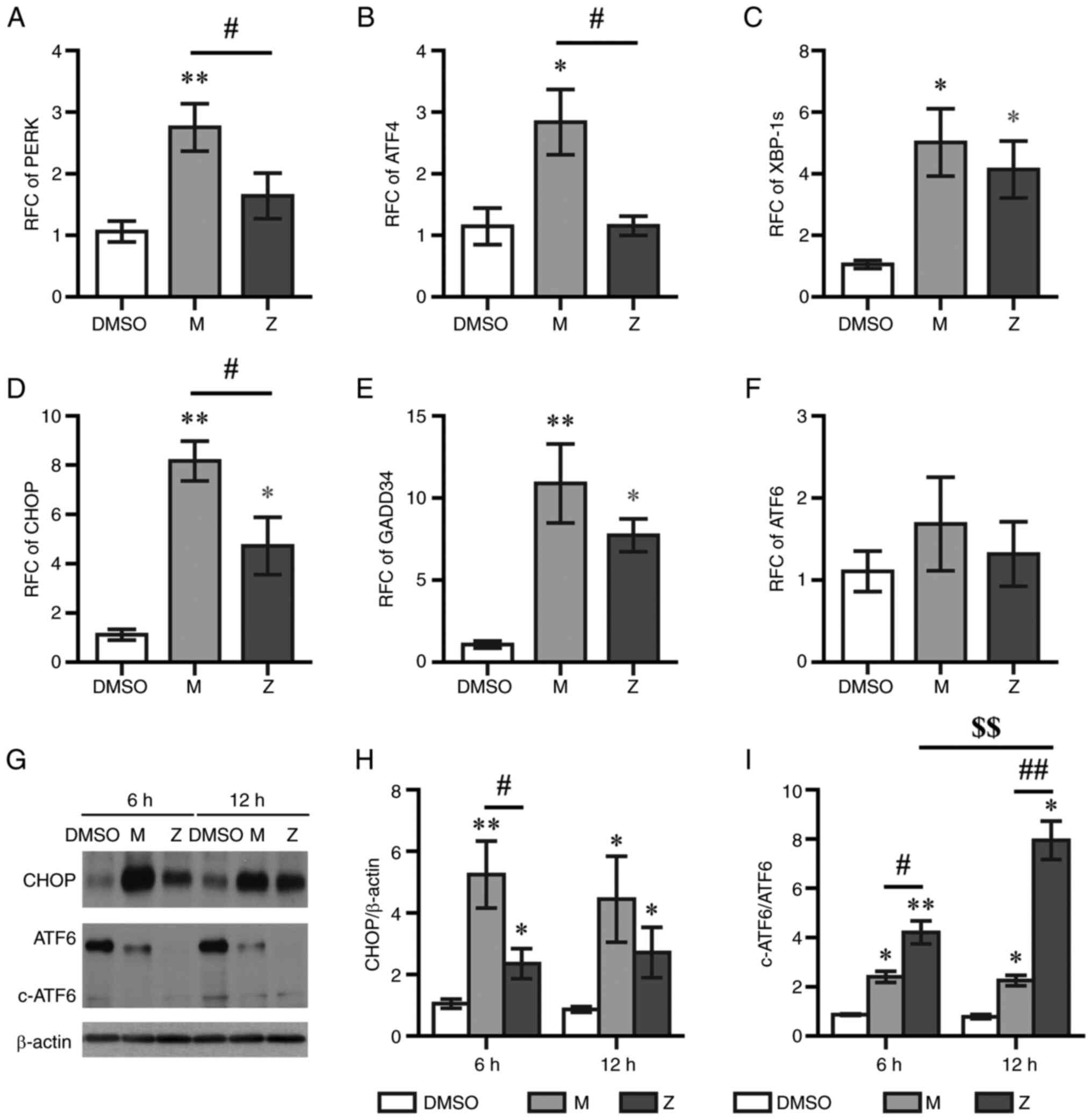 | Figure 5.LTRAs upregulate endoplasmic
reticulum stress markers. Cells treated with M exhibited increased
mRNA expression levels of (A) PERK, (B) ATF4, (C) XBP-1s, (D) CHOP
and (E) GADD34. Z triggered only the expression of XBP-1s, CHOP and
GADD34. (F) Both LTRAs did not alter mRNA expression of ATF6. (G)
Western blotting showed the increase of CHOP and the decrease of
ATF6 expression. (H) M significantly increased the levels of CHOP
protein compared with Z. (I) Z significantly increased the levels
of c-ATF6/ATF6 compared with M. Data are expressed as the mean ±
SEM of 3-5 independent experiments. *P<0.05 and **P<0.01 vs.
DMSO. #P<0.05 and ##P<0.01 vs.
zafirlukast. $$P<0.01 vs. zafirlukast at 12 h. LTRA,
leukotriene receptor antagonist; M, montelukast; Z, zafirlukast;
ATF, activating transcription factor; XBP-1s, X-box binding protein
1 splice variant; GADD34, growth arrest and DNA damage-inducible
gene 34; c-, cleaved; RFC, relative fold change. |
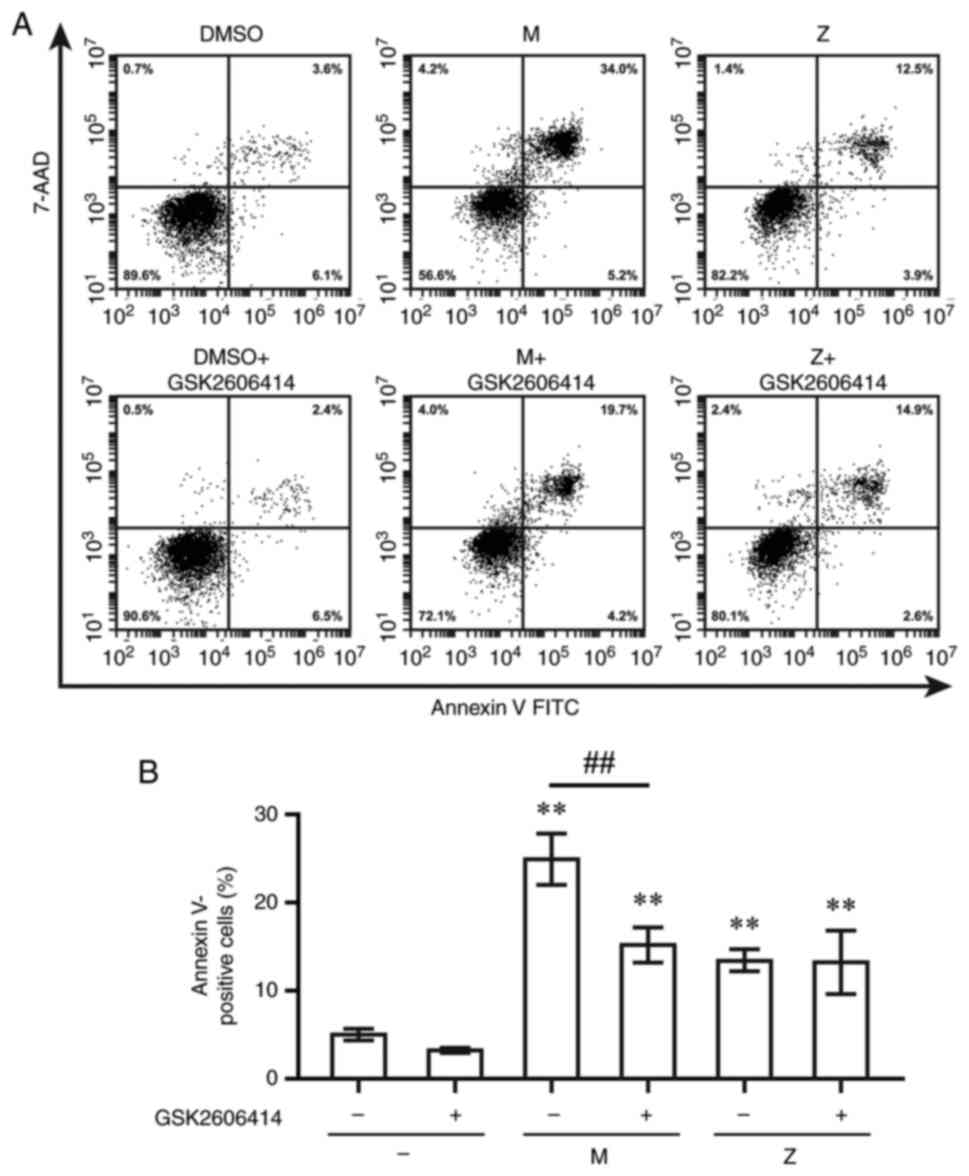
To determine the contribution of ER stress to
LTRA-induced apoptotic cell death, GSK2606414, a PERK inhibitor,
and siRNA targeting CHOP) were used. The percentage of cells with
Annexin V staining was measured at 12 h post-exposure to LTRAs.
Montelukast and zafirlukast increased the percentage of Annexin
V-positive cells; however, pre-treatment with GSK2606414 only
decreased the percentage of montelukast-induced Annexin V-positive
cells (Fig. 6). CHOP siRNA
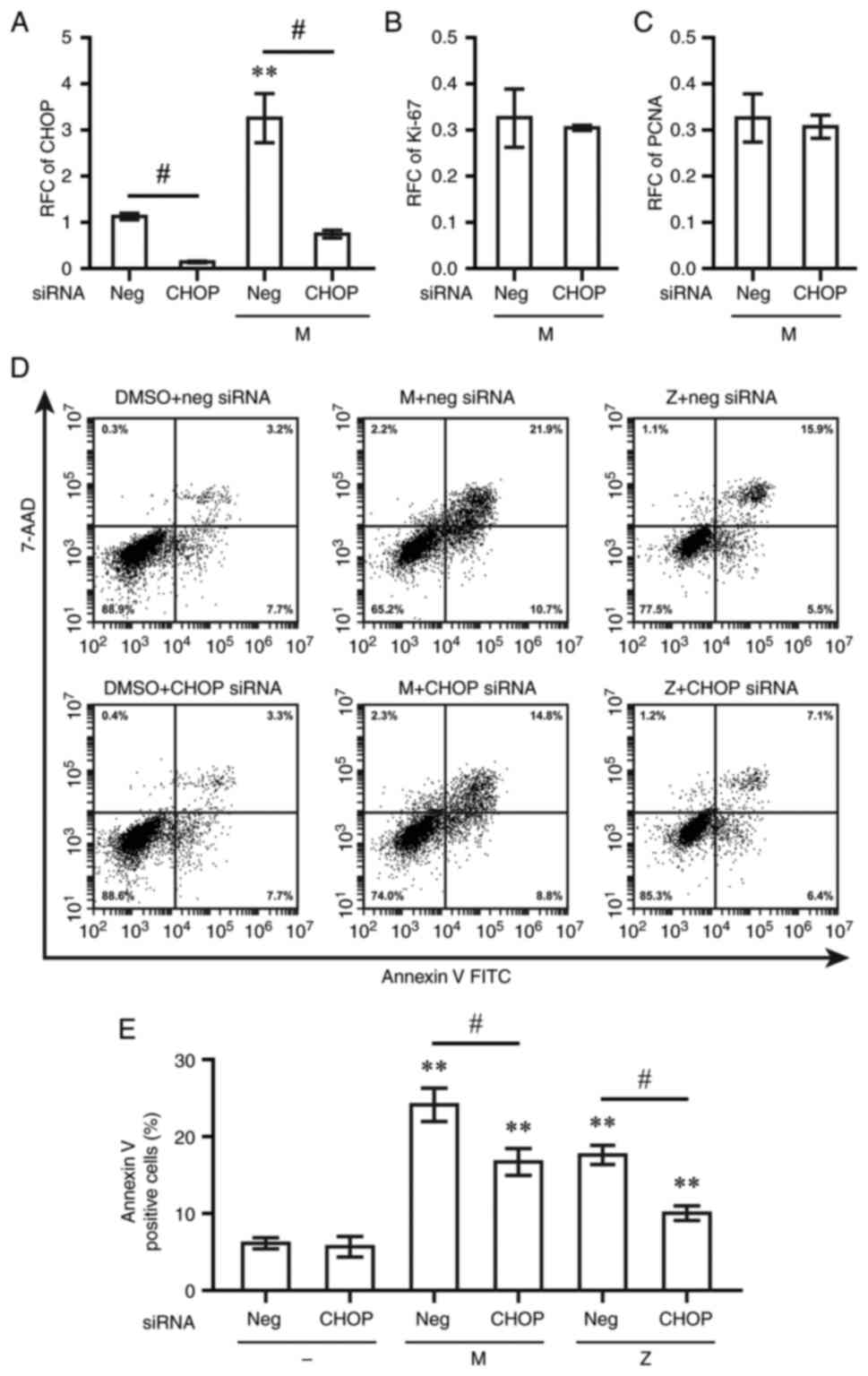
decreased the CHOP mRNA levels to 14% compared to non-targeting
control siRNA (Fig. 7A). The mRNA
expression levels of CHOP triggered by montelukast were decreased
to 23% at 48 h post-transfection with CHOP siRNA compared with
cells with non-targeting control siRNA (Fig. 7A). By contrast, the downregulation
of Ki-67 and PCNA by montelukast was not reversed by CHOP siRNA
(Fig. 7B and C). Transfection with
CHOP siRNA resulted in lower levels of Annexin V-positive cells
than in the group transfected with the non-targeting control siRNA
following exposure to montelukast or zafirlukast (Fig. 7D and E).
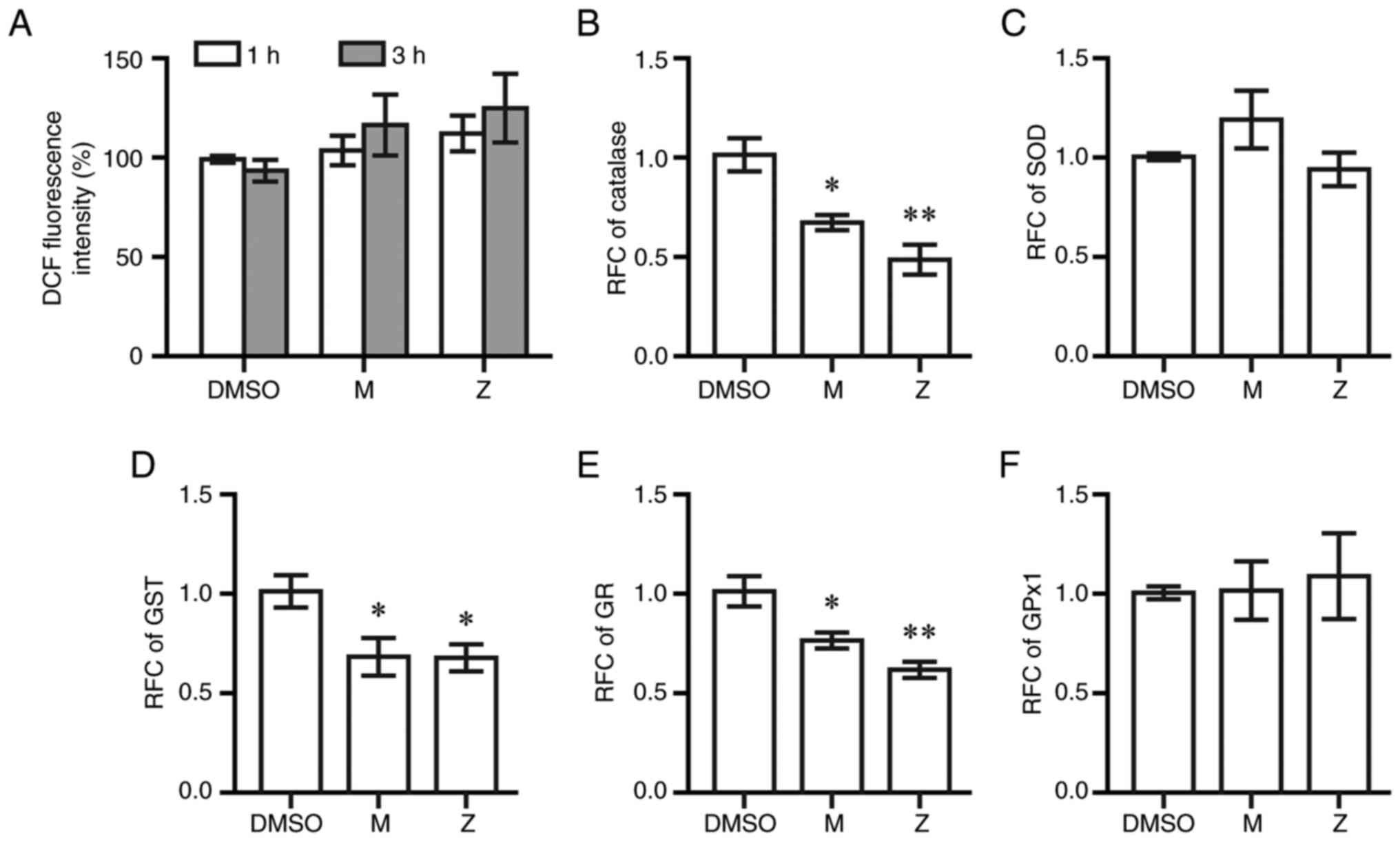
To examine whether LTRAs induced oxidative stress,
intracellular reactive oxygen species and the expression of
antioxidant enzymes were measured using DCF assays and RT-qPCR,
respectively. Montelukast and zafirlukast at 20 µM did not increase
DCF fluorescent intensity compared with DMSO at 1 and 3 h
post-treatment (Fig. 8A). Both
drugs decreased the expression of catalase, glutathione reductase
and glutathione S-transferase at 6 h post-exposure to montelukast
and zafirlukast; however, neither drug affected the expression of
superoxide dismutase and glutathione peroxidase (Fig. 8B-F).
Discussion
Our previous findings showed that LTRAs, including
montelukast and zafirlukast, have the potential for BC treatment by
inhibiting cancer progression and inducing apoptosis in the absence
of hormonal stimuli (7). The
apoptotic effects of montelukast are more potent than zafirlukast
in TNBC MDA-MB-231 cells. Zafirlukast induces cell cycle arrest at
G0/G1 phase, while montelukast treatment does
not induce cell cycle arrest at a specific phase and preferentially
induced cell death (7). The
present study demonstrated that montelukast and zafirlukast
differentially mediated CysLT1R signaling in MDA-MB-231 cells.
Montelukast and zafirlukast activates two ER stress sensors: ATF6
and IRE1; but only montelukast triggers PERK. The accumulation of
zafirlukast-treated cells in G0/G1 phase as
previously report (7) is mediated
by the upregulation of p27, and the decrease of CDK4 and cyclin D1.
Montelukast exhibited a greater effect than zafirlukast on the
upregulation of caspase 3/7, DNA damage and CHOP. Montelukast
activated all three pathways of ER stress: PERK, ATF6, and IRE1;
while zafirlukast only triggered the cleavage of ATF6 and XBP-1.
Both LTRAs also triggered autophagic response and decreased the
expression of antioxidant enzymes.
A key feature of cancer is the loss of cell cycle
regulation. Cell cycle arrest is the primary mechanism controlling
the proliferation of cancer cells and cell death (17). Ki-67 is a prognostic biomarker of
BC and was used in the recent American Joint Committee on Cancer
Staging System for BC (18,19).
In the present study, both LTRAs significantly decreased the
expression of two cell proliferating markers, Ki-67 and PCNA, in
MDA-MB-231 cells. The present results are consistent with a
previous study that showed inhibition of proliferation of non-tumor
and tumor-derived intestinal epithelial cells by LTRAs (20).
The upregulation of cyclin D1 promotes progression
of cells in the G1 phase while cyclin E is a key
regulator for cells to enter S phase. Cyclin D1 forms complexes
with CDK4 or CDK6 to trigger phosphorylation of retinoblastoma
protein (Rb), leading to the release of Rb from transcription
factor E2F. E2F then induces the synthesis of cyclin E and cyclin
A, which regulate the S phase (21). In the present study, zafirlukast
decreased the expression of cyclin D1 and CDK4 but showed no effect
on cyclin E expression. This suggested that the potential
G0/G1 cell cycle arrest was mediated by the
reduction of cyclin D1/CDK4 complexes. In addition to cyclin D1,
length of the G1 phase is regulated by the CDK inhibitor
p27. The inhibitory effect of p27 is key for entry into S phase
because p27 blocks the activity of cyclin E/CDK2, an initiator of
DNA synthesis. The inhibition of p27 expression decreases the
length of the G1 phase, and cyclin D1 inhibits the
activity of p27 (22). Therefore,
the G0/G1 cell cycle arrest induced by
zafirlukast in MDA-MB-231 cells in our previous study (7) may be mediated by the combination of
cyclin D1 downregulation and p27 upregulation. On the other hand,
decreased CDK4 expression without altered expression of cyclin D1,
cyclin E and p27 could explain the decrease of cell proliferation
without cell cycle arrest at specific phases induced by
montelukast. Our previous study showed that zafirlukast can
increase the levels of p53, p21 and p27 in U-87 MG glioblastoma
cells (10). In the present study,
zafirlukast decreased p21 expression. U-87 MG expresses wild-type
p53, while p53 in MDA-MB-231 is mutated (23). Although the most well-known
function of p21 is as a cell cycle inhibitor protein, there are
reports of p21 having oncogenic effects (24,25).
To date, three CDK 4/6 inhibitor agents
(palbociclib, ribociclib and abemaciclib) have been approved by the
Food and Drug Administration of United States and are available for
patients with BC. The major side effect of these CDK 4/6 inhibitors
is bone marrow suppression, resulting in anemia, neutropenia and
thrombocytopenia (26). LTRAs are
generally considered safe and well-tolerated drugs because of their
mild side effects (1,27). With the growing evidence of
anticancer effects and the inhibition of CDK4/6, LTRAs could be
repurposed as a chemopreventive agent or adjuvant in BC therapy. In
the present study, zafirlukast reduced the expression of both
cyclin D1 and CDK4; therefore, this drug may be a better candidate
than montelukast as a cyclin-CDK inhibitor. However, studies have
reported neuropsychiatric effects, such as depression,
hallucination and suicidal ideation, related to the use of
montelukast and zafirlukast (28,29).
The regulation of Bcl-2 by decreasing ERK1/2
activation following LTRA treatment has been reported in
glioblastoma (10), lung cancer
(9), leukemia (30) and colorectal cancer (31). In the present study, decreased
expression of p-ERK1/2 in the cells treated with LTRAs was observed
before a time-dependent reduction of Bcl-2 expression. Therefore,
LTRA-induced Bcl-2 downregulation in MDA-MB-231 cells is
potentially mediated by the inhibition of ERK1/2 phosphorylation.
The activation of the ERK1/2 pathway triggers the degradation of
p27 (22). Therefore, the
upregulation of p27 observed in the present study could be mediated
by inhibition of ERK1/2 phosphorylation. Bcl-2 expression maintains
the integrity of the mitochondrial outer membrane and prevents the
release of cytochrome c and caspase activation (32). In healthy cells, JC-1 dye enters
and forms aggregates with orange fluorescent signals. The decrease
of JC-1 aggregation by LTRAs indicated a loss of mitochondrial
membrane potential. Both LTRAs also increased levels of caspase
3/7-positive cells, indicating that LTRAs enhanced cell death via
the intrinsic mitochondrial apoptotic pathway in MDA-MB-231 cells.
The increase of caspase-3 activation by montelukast is consistent
with previous studies in colon cancer (33) and leukemia (30).
Caspase-mediated apoptotic cell death is
accomplished via cleavage of several key proteins, including
PARP-1. Cleavage of PARP-1 by caspases, a hallmark of apoptosis,
decreases DNA-binding capacity, which disrupts the routine repair
of DNA damage. Bcl-2 is an upstream regulator of PARP-1 activation
(34). Decreased Bcl-2 levels and
the upregulation of caspase 3/7 may lead to the increase of
cleaved-PARP-1 by LTRAs in MDA-MB-231 cells. This is consistent
with previous findings in chronic myeloid leukemia, which revealed
that montelukast triggers the mitochondrial apoptosis pathway by
increasing the activities of cytochrome c, caspase-3 and
cleavage of PARP-1 (20,30). The inhibition of PARP-1 can
increase ATM activation and promote H2AX phosphorylation (13). The elevation of cleaved-PARP-1,
p-ATM and p-H2AX by LTRAs in the present study indicated DNA damage
in MDA-MB-231 cells.
Autophagy serves a protective role in cancer cells
during chemotherapy, allowing cancer cells to alleviate cellular
stress, causing drug resistance. However, a number of cytotoxic
drugs promote cancer cell death via autophagy (35). The present study showed the
increased cleavage of LC3 (LC3-II), a marker of autophagy, and
decreased LC3-I in MDA-MB cells following LTRA treatment,
suggesting that the inhibition of leukotriene signaling could
induce autophagy. This observation aligns with the increase in
LC3-II induced by LTRAs in retinal pigment epithelial cells and a
rat model of hemorrhagic cystitis (36,37).
ER stress serves a dual role in cell survival and
death via apoptosis or autophagy in BC cells (14,15).
In the present study, the induction of key effectors involved in ER
stress and/or unfolded protein response (UPR) signaling by both
LTRAs was evidenced by an increase in ER stress markers, including
CHOP and XBP-1s, as well as the reduction of ATF6 due to
proteolytic cleavage. CHOP is a key link between the UPR and
pro-apoptotic activation (38).
The PERK/eukaryotic translation initiation factor 2α/ATF4 pathway
plays an important role in the upregulation of CHOP in cancer cells
(39). Activation of IRE1 induces
the splicing of XBP-1 mRNA. XBP-1s and cleaved-ATF6 also trigger
CHOP expression (38). In the
present study, the higher levels of CHOP induced by montelukast
compared with zafirlukast are likely caused by the activation of
all three ER stress sensors: PERK, ATF6, and IRE1. In contrast,
zafirlukast did not upregulate the PERK/ATF4 pathway. GADD34 is one
of the downstream transcriptional targets of CHOP. This protein
acts as a part of negative feedback loop for the response essential
for cell survival by prolonged CHOP expression (40). In the present study, both LTRAs
triggered the expression of CHOP and GADD34.
CHOP is a key protein responsible for ER
stress-induced cell death by downregulating Bcl-2 (41). CHOP activates caspase-3 and PARP-1
(42), and induces the expression
of autophagy-related genes (38).
In BC, the increased expression of CHOP indicates ER stress-induced
autophagic cell death (14,15).
In the present study, the downregulation of CHOP by siRNA decreased
LTRA-induced apoptosis. Montelukast upregulated PERK and the
inhibition of PERK using its specific inhibitor reduced
montelukast-induced apoptosis. These results suggested that the
upregulation of ER stress contributed to LTRA-induced cell death.
The expression of CHOP has been shown to be associated with a lower
risk of recurrence and prolonged disease-free survival in patients
with BC (43). Therefore, the
increase of CHOP expression by LTRAs could be an important
mechanism for BC treatment. Upregulation of CHOP could lead to cell
cycle arrest. Although the downregulation of CHOP via siRNA did not
alter the decrease the expression levels of the proliferating
markers Ki-67 and PCNA in MDA-MB-231 cells, further studies on CHOP
knockdown and cell proliferation are of interest in other types of
cancers to investigate the role of ER stress in the inhibition of
cell proliferation.
Excessive oxidative stress leads to cell death.
Antioxidants and olaparib, a PARP-1 inhibitor, have been shown to
decrease zafirlukast-induced cell death in renal cell carcinoma
(44). In the present study,
montelukast and zafirlukast decreased the expression of antioxidant
enzymes including catalase, glutathione S-transferase and
glutathione reductase. Montelukast exhibits protective effects
against oxidative and ER stress in non-cancerous models (45–48).
Montelukast can ameliorate acetaminophen-induced liver injury and
methotrexate-induced kidney damage by decreasing oxidative stress
(45,46). Montelukast has also been shown to
decrease the expression of ER stress markers, including CHOP,
GADD34, ATF4 and IRE1a, in models of hepatotoxicity and diabetes
(47,48). These differential effects require
further studies in other cancerous cells.
The toxic concentrations of LTRAs required for
anticancer effects are greater than plasma concentrations used in
asthma treatment. Pharmacokinetic studies of a single dose of 10 mg
montelukast and 20 mg zafirlukast detected 0.63 µM of montelukast
and 0.58 µM of zafirlukast in human plasma (49,50).
Novel localized drug delivery using polymeric wafers, nanofibrous
scaffolds and hydrogels could increase drug concentrations at
cancer sites (51). It is also
possible to use nanocarriers to deliver LTRAs via intraductal
delivery (52). Graphine oxide
nanoparticles of montelukast have been shown to increase apoptosis
of inflammatory cells in a mouse asthma model (53). The volume of distribution of
montelukast and zafirlukast is ~10 and 70 l, respectively (49,54).
The greater the volume of distribution, the greater the tissue
accumulation. Zafirlukast, with a higher volume of distribution and
effects on both cell cycle arrest and apoptosis, could be a better
candidate than montelukast. The anti-inflammatory effects of LTRAs
for capsular contracture after breast augmentation have suggested a
potential role for these drugs in BC treatment (55,56).
Zafirlukast has been shown to prevent the metastasis of MDA-MB-231
cells to the bone and lung by blocking CysLT1R on platelets,
subsequently inhibiting platelet aggregation and adhesion on cancer
cells (57). In addition,
Montelukast may suppress the epithelial-mesenchymal transition in
MDA-MB-231 cells (58). The
knockdown of CysLT1R using siRNA or CRISPR may confirm the role of
this receptor in cell death pathways.
In conclusion, LTRAs effectively inhibited
MDA-MB-231 cells via multiple downstream signaling pathways,
including induction of cell cycle arrest, apoptosis, autophagy, DNA
damage and ER stress. The use of one cell line is a limitation of
this study. The different effects on cell death and proliferation
induced by montelukast and zafirlukast should be further
investigated in other BC cells and animal models.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the Faculty of Medicine
Ramathibodi Hospital, Mahidol University (grant no. RF_64093).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
PV, NS and PW conceived the study. PV, TS and SP
analyzed data and wrote the manuscript. PV, KK and TS collected
data. PV and TS confirm the authenticity of all the raw data. PV
and TS edited the manuscript. All authors have read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Riccioni G, Bucciarelli T, Mancini B, Di
Ilio C and D'Orazio N: Antileukotriene drugs: clinical application,
effectiveness and safety. Curr Med Chem. 14:1966–1977. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tsai MJ, Chang WA, Chuang CH, Wu KL, Cheng
CH, Sheu CC, Hsu YL and Hung JY: Cysteinyl leukotriene pathway and
cancer. Int J Mol Sci. 23:1202021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Magnusson C, Liu J, Ehrnstrom R, Manjer J,
Jirström K, Andersson T and Sjölander A: Cysteinyl leukotriene
receptor expression pattern affects migration of breast cancer
cells and survival of breast cancer patients. Int J Cancer.
129:9–22. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tsai MJ, Wu PH, Sheu CC, Hsu YL, Chang WA,
Hung JY, Yang CJ, Yang YH, Kuo PL and Huang MS: cysteinyl
leukotriene receptor antagonists decrease cancer risk in asthma
patients. Sci Rep. 6:239792016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jang HY, Kim IW and Oh JM: Cysteinyl
leukotriene receptor antagonists associated with a decreased
incidence of cancer: A retrospective cohort study. Front Oncol.
12:8588552022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Maeda-Minami A, Hosokawa M, Ishikura Y,
Onoda A, Kawano Y, Negishi K, Shimada S, Ihara T, Sugamata M,
Takeda K and Mano Y: Relationship between leukotriene receptor
antagonists on cancer development in patients with bronchial
asthma: A retrospective analysis. Anticancer Res. 42:3717–3724.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Suknuntha K, Yubolphan R, Krueaprasertkul
K, Srihirun S, Sibmooh N and Vivithanaporn P: Leukotriene receptor
antagonists inhibit mitogenic activity in triple negative breast
cancer cells. Asian Pac J Cancer Prev. 19:833–837. 2018.PubMed/NCBI
|
|
8
|
Piromkraipak P, Sangpairoj K, Tirakotai W,
Chaithirayanon K, Unchern S, Supavilai P, Power C and Vivithanaporn
P: Cysteinyl leukotriene receptor antagonists inhibit migration,
invasion, and expression of MMP-2/9 in human glioblastoma. Cell Mol
Neurobiol. 38:559–573. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tsai MJ, Chang WA, Tsai PH, Wu CY, Ho YW,
Yen MC, Lin YS, Kuo PL and Hsu YL: Montelukast induces
apoptosis-inducing factor-mediated cell death of lung cancer cells.
Int J Mol Sci. 18:13532017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Piromkraipak P, Parakaw T, Phuagkhaopong
S, Srihirun S, Chongthammakun S, Chaithirayanon K and Vivithanaporn
P: Cysteinyl leukotriene receptor antagonists induce apoptosis and
inhibit proliferation of human glioblastoma cells by downregulating
B-cell lymphoma 2 and inducing cell cycle arrest. Can J Physiol
Pharmacol. 96:798–806. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Phuagkhaopong S, Ospondpant D, Kasemsuk T,
Sibmooh N, Soodvilai S, Power C and Vivithanaporn P:
Cadmium-induced IL-6 and IL-8 expression and release from
astrocytes are mediated by MAPK and NF-κB pathways.
Neurotoxicology. 60:82–91. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Aguilar-Quesada R, Munoz-Gamez JA,
Martin-Oliva D, Peralta A, Valenzuela MT, Matínez-Romero R,
Quiles-Pérez R, Menissier-de Murcia J, de Murcia G, Ruiz de
Almodóvar M and Oliver FJ: Interaction between ATM and PARP-1 in
response to DNA damage and sensitization of ATM deficient cells
through PARP inhibition. BMC Mol Biol. 8:292007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Clarke R, Cook KL, Hu R, Facey CO,
Tavassoly I, Schwartz JL, Baumann WT, Tyson JJ, Xuan J, Wang Y, et
al: Endoplasmic reticulum stress, the unfolded protein response,
autophagy, and the integrated regulation of breast cancer cell
fate. Cancer Res. 72:1321–1331. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sisinni L, Pietrafesa M, Lepore S,
Maddalena F, Condelli V, Esposito F and Landriscina M: Endoplasmic
reticulum stress and unfolded protein response in breast cancer:
The balance between apoptosis and autophagy and its role in drug
resistance. Int J Mol Sci. 20:8572019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ye J, Rawson RB, Komuro R, Chen X, Davé
UP, Prywes R, Brown MS and Goldstein JL: ER stress induces cleavage
of membrane-bound ATF6 by the same proteases that process SREBPs.
Mol Cell. 6:1355–1364. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vermeulen K, Berneman ZN and Van
Bockstaele DR: Cell cycle and apoptosis. Cell Prolif. 36:165–175.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Finkelman BS, Zhang H, Hicks DG and Turner
BM: The Evolution of Ki-67 and Breast Carcinoma: Past observations,
present directions, and future considerations. Cancers (Basel).
15:8082023. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhu H and Dogan BE: American Joint
Committee on Cancer's Staging System for Breast Cancer, Eighth
Edition: Summary for Clinicians. Eur J Breast Health. 17:234–238.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Paruchuri S, Mezhybovska M, Juhas M and
Sjolander A: Endogenous production of leukotriene D4 mediates
autocrine survival and proliferation via CysLT1 receptor signalling
in intestinal epithelial cells. Oncogene. 25:6660–6665. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vermeulen K, Van Bockstaele DR and
Berneman ZN: The cell cycle: A review of regulation, deregulation
and therapeutic targets in cancer. Cell Prolif. 36:131–149. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Stacey DW: Three observations that have
changed our understanding of cyclin D1 and p27 in cell cycle
control. Genes Cancer. 1:1189–1199. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hui L, Zheng Y, Yan Y, Bargonetti J and
Foster DA: Mutant p53 in MDA-MB-231 breast cancer cells is
stabilized by elevated phospholipase D activity and contributes to
survival signals generated by phospholipase D. Oncogene.
25:7305–7310. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Georgakilas AG, Martin OA and Bonner WM:
p21: A Two-Faced Genome Guardian. Trends Mol Med. 23:310–319. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Warfel NA and El-Deiry WS: p21WAF1 and
tumourigenesis: 20 years after. Curr Opin Oncol. 25:52–58. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Braal CL, Jongbloed EM, Wilting SM,
Mathijssen RHJ, Koolen SLW and Jager A: Inhibiting CDK4/6 in breast
cancer with palbociclib, ribociclib, and abemaciclib: similarities
and differences. Drugs. 81:317–331. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Matsuse H and Kohno S: Leukotriene
receptor antagonists pranlukast and montelukast for treating
asthma. Expert Opin Pharmacother. 15:353–363. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Law SWY, Wong AYS, Anand S, Wong ICK and
Chan EW: Neuropsychiatric events associated with
leukotriene-modifying agents: A systematic review. Drug Saf.
41:253–265. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Marques CF, Marques MM and Justino GC:
Leukotrienes vs. Montelukast-Activity, Metabolism, and Toxicity
Hints for Repurposing. Pharmaceuticals (Basel). 15:10392022.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zovko A, Yektaei-Karin E, Salamon D,
Nilsson A, Wallvik J and Stenke L: Montelukast, a cysteinyl
leukotriene receptor antagonist, inhibits the growth of chronic
myeloid leukemia cells through apoptosis. Oncol Rep. 40:902–908.
2018.PubMed/NCBI
|
|
31
|
Burke L, Butler CT, Murphy A, Moran B,
Gallagher WM, O'Sullivan J and Kennedy BN: Evaluation of cysteinyl
leukotriene signaling as a therapeutic target for colorectal
cancer. Front Cell Dev Biol. 4:1032016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sharpe JC, Arnoult D and Youle RJ: Control
of mitochondrial permeability by Bcl-2 family members. Biochim
Biophys Acta. 1644:107–113. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Savari S, Liu M, Zhang Y, Sime W and
Sjolander A: CysLT(1)R antagonists inhibit tumor growth in a
xenograft model of colon cancer. PLoS One. 8:e734662013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Konopleva M, Zhao S, Xie Z, Segall H,
Younes A, Claxton DF, Estrov Z, Kornblau SM and Andreeff M:
Apoptosis. Molecules and mechanisms. Adv Exp Med Biol. 457:217–236.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li X, He S and Ma B: Autophagy and
autophagy-related proteins in cancer. Mol Cancer. 19:122020.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Elrashidy RA and Hasan RA: Modulation of
autophagy and transient receptor potential vanilloid 4 channels by
montelukast in a rat model of hemorrhagic cystitis. Life Sci.
278:1195072021. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Koller A, Bruckner D, Aigner L, Reitsamer
H and Trost A: Cysteinyl leukotriene receptor 1 modulates
autophagic activity in retinal pigment epithelial cells. Sci Rep.
10:176592020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hu H, Tian M, Ding C and Yu S: The C/EBP
Homologous Protein (CHOP) transcription factor functions in
endoplasmic reticulum stress-induced apoptosis and microbial
infection. Front Immunol. 9:30832018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Rozpedek W, Pytel D, Mucha B, Leszczynska
H, Diehl JA and Majsterek I: The Role of the PERK/eIF2α/ATF4/CHOP
signaling pathway in tumor progression during endoplasmic reticulum
stress. Curr Mol Med. 16:533–544. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Brush MH, Weiser DC and Shenolikar S:
Growth arrest and DNA damage-inducible protein GADD34 targets
protein phosphatase 1 alpha to the endoplasmic reticulum and
promotes dephosphorylation of the alpha subunit of eukaryotic
translation initiation factor 2. Mol Cell Biol. 23:1292–1303. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Iurlaro R and Munoz-Pinedo C: Cell death
induced by endoplasmic reticulum stress. FEBS J. 283:2640–2652.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hsu HY, Lin TY, Hu CH, Shu DTF and Lu MK:
Fucoidan upregulates TLR4/CHOP-mediated caspase-3 and PARP
activation to enhance cisplatin-induced cytotoxicity in human lung
cancer cells. Cancer Lett. 432:112–120. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zheng YZ, Cao ZG, Hu X and Shao ZM: The
endoplasmic reticulum stress markers GRP78 and CHOP predict
disease-free survival and responsiveness to chemotherapy in breast
cancer. Breast Cancer Res Treat. 145:349–358. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wolf C, Smith S and van Wijk SJL:
Zafirlukast Induces VHL- and HIF-2alpha-dependent oxidative cell
death in 786-O clear cell renal carcinoma cells. Int J Mol Sci.
23:35672022. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Abdel-Raheem IT and Khedr NF:
Renoprotective effects of montelukast, a cysteinyl leukotriene
receptor antagonist, against methotrexate-induced kidney damage in
rats. Naunyn Schmiedebergs Arch Pharmacol. 387:341–353. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Pu S, Liu Q, Li Y, Li R, Wu T, Zhang Z,
Huang C, Yang X and He J: Montelukast prevents mice against
acetaminophen-induced liver injury. Front Pharmacol. 10:10702019.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Fei Z, Zhang L, Wang L, Jiang H and Peng
A: Montelukast ameliorated pemetrexed-induced cytotoxicity in
hepatocytes by mitigating endoplasmic reticulum (ER) stress and
nucleotide oligomerization domain-like receptor protein 3 (NLRP3)
activation. Bioengineered. 13:7894–7903. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Fleifel AM, Soubh AA, Abdallah DM, Ahmed
KA and El-Abhar HS: Preferential effect of Montelukast on
Dapagliflozin: Modulation of IRS-1/AKT/GLUT4 and ER stress response
elements improves insulin sensitivity in soleus muscle of a type-2
diabetic rat model. Life Sci. 307:1208652022. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cheng H, Leff JA, Amin R, Gertz BJ, De
Smet M, Noonan N, Rogers JD, Malbecq W, Meisner D and Somers G:
Pharmacokinetics, bioavailability, and safety of montelukast sodium
(MK-0476) in healthy males and females. Pharm Res. 13:445–448.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Dekhuijzen PN and Koopmans PP:
Pharmacokinetic profile of zafirlukast. Clin Pharmacokinet.
41:105–114. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Woodring RN, Gurysh EG, Bachelder EM and
Ainslie KM: Drug delivery systems for localized cancer combination
therapy. ACS Appl Bio Mater. 6:934–950. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Pandey M, Wen PX, Ning GM, Xing GJ, Wei
LM, Kumar D, Mayuren J, Candasamy M, Gorain B, Jain N, et al:
Intraductal delivery of nanocarriers for ductal carcinoma in situ
treatment: A strategy to enhance localized delivery. Nanomedicine
(Lond). 17:1871–1889. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Du C, Zhang Q, Wang L, Wang M, Li J and
Zhao Q: Effect of montelukast sodium and graphene oxide
nanomaterials on mouse asthma model. J Nanosci Nanotechnol.
21:1161–1168. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Accolate (zafirlukast). AstraZeneca
Pharmaceuticals LP; Wilmington, DE: 2009
|
|
55
|
Huang CK and Handel N: Effects of
Singulair (montelukast) treatment for capsular contracture. Aesthet
Surg J. 30:404–408. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Scuderi N, Mazzocchi M, Fioramonti P and
Bistoni G: The effects of zafirlukast on capsular contracture:
Preliminary report. Aesthetic Plast Surg. 30:513–520. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Saier L, Ribeiro J, Daunizeau T, Houssin
A, Ichim G, Barette C, Bouazza L and Peyruchaud O: Blockade of
Platelet CysLT1R receptor with zafirlukast counteracts platelet
protumoral action and prevents breast cancer metastasis to bone and
lung. Int J Mol Sci. 23:122212022. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
El-Ashmawy NE, Khedr EG, Khedr NF and
El-Adawy SA: Suppression of epithelial-mesenchymal transition and
SIRT1/AKT signaling pathway in breast cancer by montelukast. Int
Immunopharmacol. 119:1101482023. View Article : Google Scholar : PubMed/NCBI
|