Introduction
Acute lung injury (ALI), also known as acute
respiratory distress syndrome (ARDS), is a complex
pathophysiological process characterized by damage to alveolar
epithelial cells and vascular endothelial cells resulting from
various causes, such as infections, inhalation of toxic chemicals
and drowning. This leads to disruption of the pulmonary air-blood
barrier function, causing inflammatory cell exudation, pulmonary
edema and ultimately respiratory failure (1). The estimated incidence of ARDS can
reach 78.9/100000 person-years, and the incidence of hospital
admission ranges between 1.3 and 19% worldwide (2). In addition, the in-hospital mortality
rate for patients with severe ALI can reach 46.1% (3). Under physiological conditions,
endothelial and epithelial cells maintain an air-blood barrier that
constitutes the alveoli, preventing the accumulation of fluid or
the infiltration of inflammatory cells, such as neutrophils and
lymphocytes, into the alveoli, which is also fundamental for
maintaining effective gas exchange (4). In the presence of diverse
pathological states, such as hypoxia, mechanical strain and
bacterial infections, epithelial cells are damaged. This damage
results in the buildup of protein-rich fluid and inflammatory cells
within the alveolar spaces (1).
Consequently, there is an increase in oxidative stress and the
release of inflammatory cytokines, along with impaired gas
exchange; this ultimately results in the development of ALI
(3–5). Various mechanisms and pathways are
involved in the process of ALI, including apoptosis, autophagy,
senescence and ferroptosis (6),
implicating multiple genes; however, the complexity of this
pathophysiological process necessitates further research to
elucidate the exact mechanisms.
Previous studies on lung injury have focused on cell
and animal models (7–9); however, research at the cellular
level cannot reflect the influence of the microenvironment and
three-dimensional structure of alveoli (10). Furthermore, studies on animal
models typically require the collection of animal specimens for
research after modeling, making it impossible to visualize the
pathophysiological processes, and animal modeling is time-consuming
and expensive (11). Notably,
organ-on-a-chip, a new type of in vitro model, has attracted
attention in recent years; this model integrates tissue engineering
and microfluidic technology, which can partially simulate specific
tissue microstructures and facilitate the analysis and observation
(12). Organ-on-a-chip technology
first uses nano-processing methods to fabricate micro-scale chip
structures on different substrate materials [such as
polydimethylsiloxane (PDMS), polymethyl methacrylate,
polypropylene, glass, silicon and paper], and then implants
different types of cells or cell clusters wrapped in different
materials into the chip microstructure to construct models with
different functions. A lung-on-a-chip model approximating an
air-blood barrier can be constructed by co-culturing alveolar
epithelial cells and vascular endothelial cells on both sides of a
porous polymer membrane, which is a suitable model for research on
ALI (13). Some researchers have
used lung-on-a-chip models to study viral lung infections and drug
screening. Notably, there have been reports of simulating
rhinovirus, influenza or COVID-19 infections on lung-on-a-chip
models, and testing the effects of corresponding drugs; these
studies have also identified unique pathways and mechanisms
(14–16). However, these studies were all
conducted on chips based on regular cell lines, whereas
constructing lung-on-a-chip models using gene-edited cell lines and
investigating the function of specific genes in disease is a
promising direction.
Lung cancer metastasis-related protein 1
(LCMR1) is a gene that we previously reported to be highly
expressed in non-small cell lung cancer (NSCLC); notably, its
expression is highly associated with the stage of NSCLC (17). To further investigate the function
of LCMR1, a complete LCMR1 knockout mouse model was
generated utilizing CRISPR/Cas9 technology. Our findings indicated
that the complete knockout mouse model resulted in homozygous
lethality, suggesting that LCMR1 is indispensable in normal
embryonic growth and development (unpublished data). We then
established mice with conditional knockout of LCMR1 in type
II alveolar epithelial cells (AEC-II). The results revealed that
after AEC-II conditional knockout of the LCMR1 gene, the
mice gradually developed diffuse inflammatory cell infiltration in
lung tissue, alveolar hemorrhage, alveolar septal thickening and
alveolar edema, which eventually led to respiratory failure and
death (18). Previously, research
on LCMR1 mainly focused on the field of cancer (19–21);
however, based on our previous results, it was hypothesized that
the LCMR1 gene may contribute to the pathogenesis of ALI. To
the best of our knowledge, the present study is the first to
investigate the role of LCMR1 in the pathogenesis of ALI on
a mouse model of lipopolysaccharide (LPS)-induced ALI. In addition,
the underlying mechanism was explored using lung-on-a-chip
technology.
Materials and methods
Mouse models
The animal experiments in the present study were
approved by the Animal Ethics Committee of Chinese PLA General
Hospital (approval no. SQ2023672; Beijing, China) and were carried
out in accordance with the ARRIVE 2.0 guidelines (22). All mice were maintained in a
specific pathogen-free animal facility with free access to clean
food and water at the Laboratory Animal Center of the PLA General
Hospital. Mice status was observed every 12 h after LPS
administration and assessed according to a murine sepsis score
(23), and mice with a score
>21 were euthanized and all biological samples were collected
for subsequent analyses.
Wild-type male C57BL/6 mice [n=36; age, 8 weeks,
weight, 22–24 g, housed under a 12/12 h light/dark cycle in a room
with a controlled temperature (23°C) and humidity (50%)] provided
by the Laboratory Animal Center of the PLA General Hospital were
used to analyze LCMR1 expression after LPS challenge. The mice were
randomly assigned to the LPS group or control group. Anesthesia was
induced with 5% isoflurane inhalation and maintained with 2%
isoflurane inhalation. Following anesthesia, the mice in the LPS
group were intratracheally injected with LPS (10 mg/kg, dissolved
in saline, to reach a final concentration of 5 mg/ml LPS;
Sigma-Aldrich, cat. no. L2630) to generate an LPS-induced lung
injury model, whereas those in the control group were
intratracheally injected with saline. After injection, the mice
were placed on a vertical operating table and slowly shaken for 1
min to ensure uniform distribution of LPS or saline in the lungs,
after which the mice were returned to the animal facility. The lung
tissue samples of mice from both groups were collected at 0, 24,
48, 72 and 96 h after LPS/saline injection for histopathological
analysis, reverse transcription-quantitative PCR (RT-qPCR) and
western blot analysis (n=6 mice/group at 0, 24, 48, 72 and 96 h).
Notably, the mice were euthanized by cervical dislocation under
anesthesia as aforementioned prior to lung tissue harvest.
Male mice with LCMR1 conditional knockout in
AEC-II were generated based on our previous study (18). Briefly, mice with insertion of the
Cre recombinase genes at the pulmonary surfactant protein C
(Sftpc) locus (SftpcCreERT2 mice) based on
C57BL/6 mice (Purchased from Cyagen Biosciences, Inc.) and mice
carrying the floxed LCMR1 allele
(LCMR1flox/flox mice, Purchased from Cyagen
Biosciences, Inc.) were used. The SftpcCreERT2
mice have a tamoxifen-inducible Cre recombinase controlled by the
Sftpc promoter that specifically mediates the knockout of
target sequences located between LoxP sites in AEC-II.
SftpcCreERT2; LCMR1flox/flox
(LCMR1-CKO) mice were generated by crossing
SftpcCreERT2 mice with
LCMR1flox/flox mice, and the
LCMR1flox/flox littermates were used as controls
(LCMR1-C). A total of 6 SftpcCreERT2 and 6
LCMR1flox/flox mice (weight, 23–25 g) were use as
breeding pairs and housed under a 12/12 h light/dark cycle in a
room with a controlled temperature (23°C) and humidity (50%)
provided by the Laboratory Animal Center of the PLA General
Hospital.
To induce AEC-II specific deletion of LCMR1,
tamoxifen (50 mg/kg; Sigma-Aldrich) was dissolved in corn oil
(Shanghai Aladdin Biochemical Technology Co., Ltd.) and injected
intraperitoneally into mice for 5 consecutive days. Following
tamoxifen injection, both the LCMR1-C group and the
LCMR1-CKO group were intratracheally administered LPS (10
mg/kg) to generate an LPS-induced lung injury model, or with
sterile saline to serve as a control group. A total of 48 h after
LPS/saline administration, the mice (age, 8–10 weeks) were
anesthetized as aforementioned and subjected to pulmonary function
testing, after which the mice were euthanized by cervical
dislocation. The right lung of the mice was harvested and frozen in
liquid nitrogen, before being maintained at −80°C for further
biochemical measurements. The left lungs were harvested and stored
in 4% paraformaldehyde (PFA) for 24 h at 4°C, for subsequent
histological analysis. Each group contained 30 mice, among them, 6
were used for survival analysis after LPS stimulation, 12 were used
for pulmonary function testing followed with histological analysis,
lung wet/dry weight ratio analysis and transmission electron
microscopy (6 with LPS administration and 6 with saline
administration), and 12 were used for bronchoalveolar lavage fluid
(BALF) analysis (6 with LPS administration and 6 with saline
administration). All mice were housed in a specific pathogen-free
animal facility with free access to clean food and water at the
Laboratory Animal Center of the PLA General Hospital.
Survival analysis
The mice in the LCMR1-C and LCMR1-CKO
groups (n=6/group) were used to analyze the effect of LCMR1
knockout on the survival of the mice after LPS stimulation. The
number of deaths was counted every 24 h and the percentage of
survival was calculated. At 120 h, the remaining mice were
euthanized under anesthesia by cervical dislocation.
Genotyping
To genotype LCMR1-C and LCMR1-CKO
mice, the mouse tails were snipped between 9 and 14 days of age,
after which the mice were housed until 8–10 weeks of age for
subsequent experiments. The genomic DNA was extracted from the
tails of mice utilizing the Mouse Quick Genotyping Kit
(YangGuangBio, cat. no. C190801). The genotype was determined by
PCR employing the primers listed in Table SI. PCR amplification was carried
out using Premix Taq (Takara, cat. no. RR902A) on the T100™
thermocycler (Bio-Rad Laboratories, Inc.; Table SII. The resultant PCR products
were analyzed via agarose gel (1.5%) electrophoresis. For
visualization, the ChemiDoc XR imaging system (Bio-Rad
Laboratories, Inc.) and Image Lab version 3.0 (Bio-Rad) was
utilized.
Lung function testing
Lung function assessment was conducted utilizing the
AniRes2005 lung function testing apparatus (Best Lab International,
Inc.). Briefly, the process involved anesthetizing the mice,
performing a tracheotomy for cannulation and subsequently placing
them into a sealed plethysmograph chamber. The mice were then
linked to a ventilator through the tracheal cannula. The indices
used to evaluate the lung function of mice included forced vital
capacity (FVC), proportion of FVC expired in the first second,
static lung compliance and lung resistance (24,25).
Each mouse was measured five times and the mean value of these
indices was recorded.
Histopathological analysis
Lung tissues from mice were preserved in 4%
paraformaldehyde (PFA) for 24 h at 4°C for fixation. After
fixation, the tissue samples were dehydrated and embed in wax and
then sliced to a thickness of 4 µm. The tissue sections were
stained with hematoxylin solution for 3–5 min at room temperature
and then stained with Eosin dye for 15 sec at room temperature. The
stained tissue sections were analyzed using a NanoZoomer system
(Hamamatsu Photonics K.K.). The assessment of lung injury severity
was performed by two pathologists, who were unaware of the
specifics of the study, employing a previously established scoring
system (26).
Lung wet/dry weight ratio
The right lung of the mice was excised and weighed
to ascertain the wet weight. Subsequently, the samples underwent
desiccation in an oven maintained at 65°C for 120 h, after which
they were weighed again to acquire the dry weight data.
Subsequently, the wet/dry weight ratio of the lungs was
computed.
Bronchoalveolar lavage fluid (BALF)
analysis
Following anesthesia, the mice underwent intubation,
and the BALF was collected by three consecutive infusions and
withdrawal of 1-ml PBS into the tracheal cannula. The BALF was
centrifuged at 1,500 × g) for 10 min at 4°C. The resultant
supernatant was utilized to quantify total protein concentrations
employing the BCA Protein Assay kit (YangGuangBio), whereas
inflammatory cytokine levels were assessed utilizing the LEGENDplex
Mouse Inflammation Panel (cat. no. 740446; BioLegend, Inc.)
according to the manufacturer's protocol.
Transmission electron microscopy
Harvested mouse lung tissues were cut into tissue
blocks with a size of no more than 1 mm3. The tissue
blocks were then fixed in 2.5% glutaraldehyde at 4°C overnight. The
specimens were subsequently fixed in a 1% OsO4 solution
within 0.1 M PBS (pH 7.4) for 2 h at room temperature. Following
fixation, the samples underwent dehydration through a sequential
ethanol concentration and were ultimately embedded in epoxy resin
at room temperature for 3 h. Ultrathin sections, measuring 70 nm,
were then prepared utilizing an Ultracut E ultramicrotome (Leica
Microsystems GmbH) and were stained with a saturated alcoholic
solution of 2% uranium acetate for 8 min at room temperature. The
prepared sections were later analyzed using an HT7800 transmission
electron microscope (Hitachi, Ltd.).
Cell culture and chip fabrication
The human alveolar lung-on-a-chip device was kindly
provided by Professor Jianhua Qin (Division of Biotechnology,
Chinese Academy of Science Key Laboratory of separation science for
analytical chemistry, Dalian Institute of Chemical Physics, Chinese
Academy of Sciences, Dalian, China). This device mainly consists of
two channels made by casting a PDMS prepolymer on a mold fabricated
by a conventional soft lithography process (27). The device was sterilized in an
autoclave, and transferred to an ultraclean table under UV light
overnight. Both sides of the porous part of the device were covered
with 200 µg/ml type I rat tail collagen (diluted at a 1:100 ratio
with glacial acetic acid; Corning, Inc.) for 48 h at room
temperature before cell seeding.
The human AEC-II line (HPAEpiC; passage 4) and the
vascular endothelial cell line (Hulec-5A) derived from human
alveolar epithelial cells were also provided by Professor Jianhua
Qin. The HPAEpic cell line was maintained in RPMI 1640 medium (cat.
no. 12633020) supplemented with 10% FBS (cat. no. A5256701; both
Gibco, Thermo Fisher Scientific, Inc.). Hulec-5A cell line was
maintained in dedicated growth medium (cat. no. CM-0565; Procell
Life Science & Technology Co., Ltd.). All cells were cultured
at 37°C with 5% CO2. To construct the lung-on-a-chip
model, Hulec-5A and HPAEpiC cells (~1×105 cells) were
seeded on the bottom and upper channel of the aforementioned chip
device, respectively. After cell attachment, a constant flow of
medium (50 µl/h) was applied to both layers via a peristaltic pump.
The cells were cultured for 3 days until they reached 100%
confluence, and the chips were maintained in an incubator at 37°C
with 5% CO2.
The LCMR1-knockdown (LCMR1-KD) HPAEpic
cell lines were established by lentiviral vector transfection. The
vector was generated in cooperation with Vigen Biotechnology Co.,
Ltd. The short hairpin RNA (shRNA) sequences of the LCMR1 protein
and negative control (NC) were designed and synthesized by Vigen
Inc. The shRNA sequence and information regarding the lentiviral
vector are detailed in Table SIII
and Fig. S1. The lentivirus was
generated using 2nd generation lentivirus packaging plasmids by
co-transfection of psPAX2 (3 µg), pMD2G (1 µg) and pLKO.1 (Addgene
Inc.)-shRNA (4 µg) or pLKO.1-shNC (4 µg) into HEK-293T cells (Vigen
Biotechnology Co., Ltd, Zhenjiang, China). The 293T cells were
further cultured at 37°C for 48 h. Then, the supernatants of
HEK-293T cells were collected and concentrated by pEGit (Vigen
Biotechnology Co., Ltd, Zhenjiang, China). The
LCMR1-overexpressing (LCMR1-OE) HPAEpiC cells were
established by lentiviral vector transfection. Lentivirus was
generated by co-transfection of psPAX2 (3 µg), pMD2G (1 µg) and
pCDH-CMV-LCMR1(h)-EF1a-Puro (5 µg) or pCDH-MCS-EF1a-Puro (served as
NC) into HEK-293T cells (Vigen Biotechnology Co., Ltd, Zhenjiang,
China). The 293T cells were further cultured at 37°C for 48 h.
Then, the supernatants of HEK-293T cells were collected and
concentrated by pEGit (Vigen Biotechnology Co., Ltd.). The
information regarding the lentiviral vector was detailed in
Table SIV and Fig. S2.
To generate the LCMR1-KD and LCMR1-OE
HPAEpic cell lines, the HPAEpic cells were placed in 6-well plate,
cultivated until reaching a 70–80% density, and exposed to the
corresponding lentivirus with 8 µg/ml polybrene (Sigma-Aldrich)
with a multiplicity of infection (MOI) of 20 for 48 h at 37°C.
Stably transfected cell lines were selected using puromycin. The
concentration of selection was 2.5 µg/ml and maintenance
concentration were 0.4 µg/ml. After 10 days of puromycin selection,
the cells were used for subsequent experimentation. The
overexpression and knockdown efficiency of the target protein was
evaluated by western blotting, and cell proliferation rate after
transfection was detected using the Cell Counting Kit (CCK)8 assay
(cat. no. C0038; Beyotime Biotechnology) according to the
manufacturer's instruction.
To simulate ALI, LPS (1 µg/ml) was added for 72 h at
37°C. After which, the media in the alveolar channel were collected
from each chip. The concentrations of IL-6 and TNF-α were measured
using the LEGENDplex Human Inflammation Panel 1 (Biolegend, USA)
according to the manufacturer's instructions.
Immunofluorescence
For immunofluorescence imaging of the chips, the
cells were rinsed with PBS through the upper and lower channels,
and fixed with 4% PFA at room temperature for 20 min. The cells
were then infiltrated with 0.2% Triton X-100 in PBS (PBST) buffer
for 5 min and blocked with PBST buffer containing 5% goat serum
(cat. no. 16210064, Gibco, Thermo Fisher Scientific, Inc.) for 30
min at room temperature. They were subsequently stained with the
corresponding primary antibody overnight at 4°C and then with the
secondary antibody for 1 h at room temperature. After secondary
antibody staining, DAPI was used to stain the cell nucleus for 5
min at room temperature. Imaging was carried out using a confocal
fluorescence microscope system (LSM880; Carl Zeiss AG), and image
analysis was conducted employing Imaris software (Oxford
Instruments, version 10.2.0) along with ImageJ (National Institutes
of Health, version 1.54 g).
The primary antibodies used were as follows:
Anti-E-cadherin (cat. no. 60335-1-Ig; Proteintech Group, Inc.
1:100), anti-VE-cadherin (cat. no. 14-1449-82; Invitrogen; Thermo
Fisher Scientific, Inc. 1:100) and anti-pro-pulmonary surfactant C
precursor (proSP-C; cat. no. AB3786; MilliporeSigma 1:100). The
secondary antibodies were goat anti-mouse IgG (H+L) Cross-Adsorbed
Secondary Antibody, Alexa Fluor™ 647 (cat. no. AB_2535804,
Invitrogen; Thermo Fisher Scientific, Inc.; 1:1,000), goat
anti-mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa
Fluor™ 488 (cat. no. AB_2534069, Invitrogen; Thermo Fisher
Scientific, Inc. 1:1,000) and goat anti-Rabbit IgG (H+L) Highly
Cross-Adsorbed Secondary Antibody, Alexa Fluor™ Plus 647 (cat. no.
A32733, Invitrogen; Thermo Fisher Scientific, Inc. 1:1,000).
Permeability assay
Culture medium containing FITC-dextran (1 mg/ml;
MilliporeSigma) was added to the bottom channel of the
lung-on-a-chip model after LPS stimulation for 72 h. Subsequently,
media were collected from the bottom and upper channels at
different time points (0, 1 and 2 h). The fluorescence intensity of
the media collected from the upper and bottom channels was measured
with a microplate system (Spark®, Tecan Inc.), and the
permeability of the air-blood barrier of the chips was evaluated
based on their fluorescence intensity ratio.
RT-qPCR
Total RNA was extracted from cells or mouse tissue
samples using the Total RNA Kit (cat. no. DP451; Tiangen Biotech
Co., Ltd.) according to the manufacturer's instructions. The RNA
was then reverse-transcribed to cDNA using the PrimeScript RT
reagent Kit with a gDNA Eraser (cat. no. RR047A, Takara) according
to the manufacturer's instructions. qPCR was performed using KAPA
SYBR FAST Universal (cat. no. KK4601; KAPA Biosystems; Roche
Diagnostics). Primer sequences are detailed in Table SI. The thermocycling conditions
are listed in Table SV. The
standard 2−ΔΔCq assay was applied to calculate the mRNA
expression levels relative to β-actin (28).
Western blotting
Protein was extracted from mouse lung tissue or
cells using RIPA) lysis buffer supplemented with protease and
phosphatase inhibitor cocktails (cat. no. C1055, Applygen
Technologies Inc.). Protein concentration was determined with a
bicinchoninic acid protein assay kit and was then separated by gel
(12%) electrophoresis with 50 µg protein loaded per lane and
transferred to a 0.2-µm nitrocellulose membrane (Cytiva). The
membranes were blocked with Tris-buffered saline-0.05% Tween-20 and
5% bovine serum albumin (cat. no. A8020, Solarbio) for 2 h at room
temperature and then probed with primary antibodies overnight at
4°C. The membranes were then probed with a horseradish
peroxidase-labeled secondary antibody for 60 min at room
temperature. Protein bands were visualized using an automated
chemiluminescence imaging system (Tanon Science and Technology Co.,
Ltd.). The gray values of the bands were then semi-quantified using
ImageJ software (version 1.54g). The primary antibodies used were
as follows: Anti-tubulin (1:5,000; cat. no. 11224-1-AP; Proteintech
Group, Inc.) used as loading control, anti-LCMR1 (1:1,000; cat. no.
PA5-44383; Invitrogen; Thermo Fisher Scientific, Inc.), aquaporin 5
(AQP5; 1:1,000; cat. no. 20334-1-AP; Proteintech Group, Inc.),
anti-Bcl-2 (1:3,000; cat. no. 12789-1-AP; Proteintech Group, Inc.),
anti-Bax (1:3,000; cat. no. ab32503; Abcam) and cleaved caspase-3
(1:5,000; cat. no. ab214430; Abcam). The secondary antibody was
HRP-Goat Anti-Rabbit IgG (H&L; 1:3,000; cat. no. C081802;
YangGuangBio).
Statistical analysis
No specific statistical test was used to
predetermine the sample size. For group comparisons, GraphPad Prism
8 software (Dotmatics) was utilized. The data that conformed to a
normal distribution are presented as the mean ± SD. One-way
analysis of variance followed by Tukey's post hoc test, or two-way
analysis of variance followed by Sidak's post hoc test was applied
for statistical comparisons among three or more groups. Unpaired
Student's t-test was applied for statistical comparisons between
two groups. Data that did not conform to normal distribution are
presented as the median (range), and were compared using the
Kruskal-Wallis test followed by Dunn's post hoc test. Kaplan-Meier
analysis followed by Mantel-Cox test was used for survival
analysis. Two-sided P<0.05 was considered to indicate a
statistically significant difference.
Results
LCMR1 expression is downregulated in
wild-type mice with LPS-induced lung injury
To investigate whether LCMR1 was associated
with LPS-induced ALI in animal models, the protein and mRNA
expression levels of LCMR1 were assessed in lung tissues from LPS
(10 mg/kg)-treated mice at different time points. Briefly,
8-week-old male wild-type C57BL/6 mice were intratracheally
administered LPS (10 mg/kg) to generate an LPS-induced lung injury
model. The lung tissue samples were collected at 24, 48, 72 and 96
h after LPS injury for histopathological, RT-qPCR and western blot
analysis. The pathological results showed that there was obvious
fluid exudation, inflammatory cell infiltration, alveolar
hemorrhage and consolidation in the lungs within 48–96 h of LPS
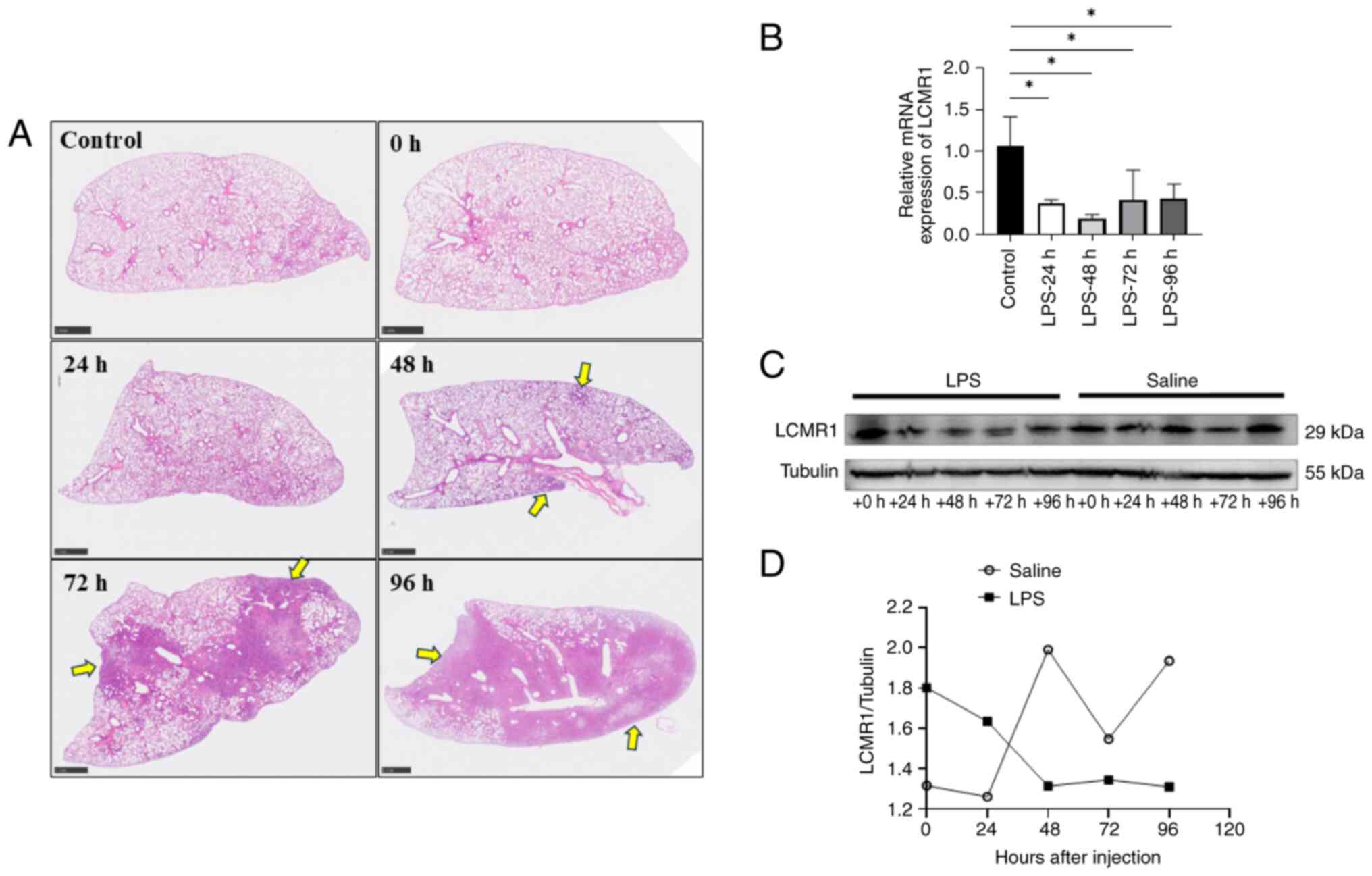
stimulation (Fig. 1A). Notably,
the mRNA expression levels of LCMR1 at 24–96 h after LPS
stimulation were downregulated compared with those in the control
group, and the protein expression levels of LCMR1 were
downregulated 48 h post-LPS stimulation (Fig. 1B-D). The expression of LCMR1
reached its lowest point at ~48 h after LPS challenge; therefore,
the intervention time of LPS was set to 48 h in subsequent
experiments.
LCMR1 conditional knockout in AEC-II
exacerbates LPS-induced ALI in mice
The method used to generate tamoxifen-induced
LCMR1 conditional knockout in AEC-II of mice was described
in our previous study (18). The
modeling pattern is shown in Fig.
2A. Subsequently, the LCMR1-CKO and LCMR1-C mice
were genotyped; LCMR1-CKO mice were carrying the
Sftpc-CreERT2 transgene (210bp; Fig. 2B. After 5 consecutive days of
intraperitoneal injection with tamoxifen to induce gene knockout in
LCMR1-CKO mice, western blot analysis revealed that LCMR1
protein expression was significantly lower in the LCMR1-CKO
group than that in the LCMR1-C group (Fig. 2D). These results indicated the
successful knockout of LCMR1 in AEC-II.
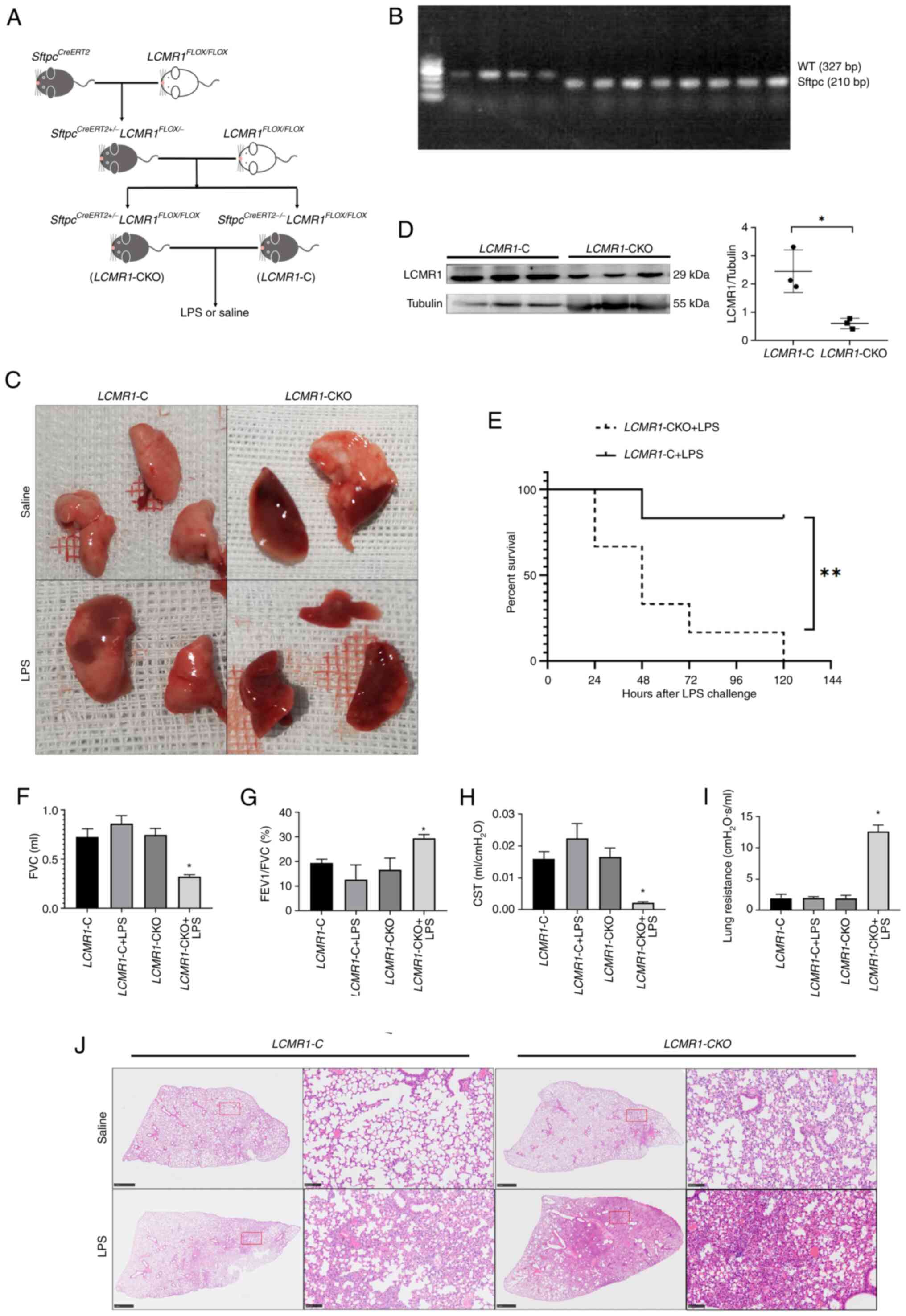 | Figure 2.LCMR1 deficiency in type II
alveolar epithelial cells exacerbates LPS-induced lung injury in
mice. (A) Schematic diagram of mouse model construction. (B)
Genotyping of mice by PCR for the presence of the
SftpcCreERT2 transgene. (C) Lung tissue harvested
48 h after LPS stimulation. (D) Western blot analysis of LCMR1
protein in lung tissue after tamoxifen administration (n=3/group)
(E) Kaplan-Meier survival curve of mice after LPS stimulation.
(F-I) Lung function test results, including (F) forced vital
capacity, (G) proportion of forced expiratory volume in 1 sec in
forced vital capacity, (H) static lung compliance and (I) lung
resistance (n=6/group). (J) Representative hematoxylin and eosin
staining of the left lung of LCMR1-C and LCMR1-CKO
mice. scale bar on the overall view: 1 mm, scale bar on the
magnified view: 100 µm. *P<0.05, **P<0.001. C, control; CKO,
conditional knockout; CST, static lung compliance; FEV1, forced
expiratory volume in 1 sec; FVC, forced vital capacity; LCMR1, lung
cancer metastasis-related protein 1; LPS, lipopolysaccharide; WT,
wild-type. |
After 5 consecutive days of intraperitoneal
injection with tamoxifen, the mice were administered LPS (10 mg/kg)
intratracheally. Survival analysis showed that the mortality rate
of LCMR1-CKO mice was significantly increased after LPS
stimulation compared with that in LCMR1-C mice (Fig. 2E). Lung function tests revealed
that LCMR1-CKO mice had decreased FVC, decreased lung
compliance and increased lung resistance after LPS-induced lung
injury compared with the LCMR1-C mice (Fig. 2F-I).
A total of 48 h after LPS stimulation, the mice
lungs were harvested. The LCMR1-CKO mice showed a more
severe lung injury after LPS challenge; macroscopically, the lung
of LCMR1-CKO mice showed obvious exudation and an overall
dark red color (Fig. 2C). The lung
pathology revealed that the LCMR1-CKO mice presented more
severe lung injury manifestations, such as hemorrhage and
exudation, than LCMR1-C mice after LPS stimulation, whereas
LCMR1-CKO mice administered with saline also developed
localized lesions similar to LCMR1-C mice challenged with
LPS (Fig. 2J). These findings were
confirmed by subsequent lung injury score analysis based on the
pathology images (Figs. 2J and
3B). Furthermore, the
LCMR1-CKO mice had increased wet/dry weight ratios (Fig. 3A) and increased levels of
inflammatory cytokines in the BALF (Fig. 3C-F) compared with LCMR1-C
mice following LPS stimulation. Among the inflammatory cytokines,
IL-1β, IL-6 and TNF-α were significantly increased in
LCMR1-CKO mice comparing to LCMR1-C mice, and the
protein content in the BALF of LCMR1-CKO mice was also
increased comparing to LCMR1-C mice. Western blot analysis
revealed that LCMR1 knockout significantly increased the
Bax/Bcl-2 protein ratio (Fig. 3G and
H) and cleaved caspase-3/caspase-3 protein ratio (Fig. 3I and J) comparing to LCMR1-C
mice, indicating enhanced apoptosis.
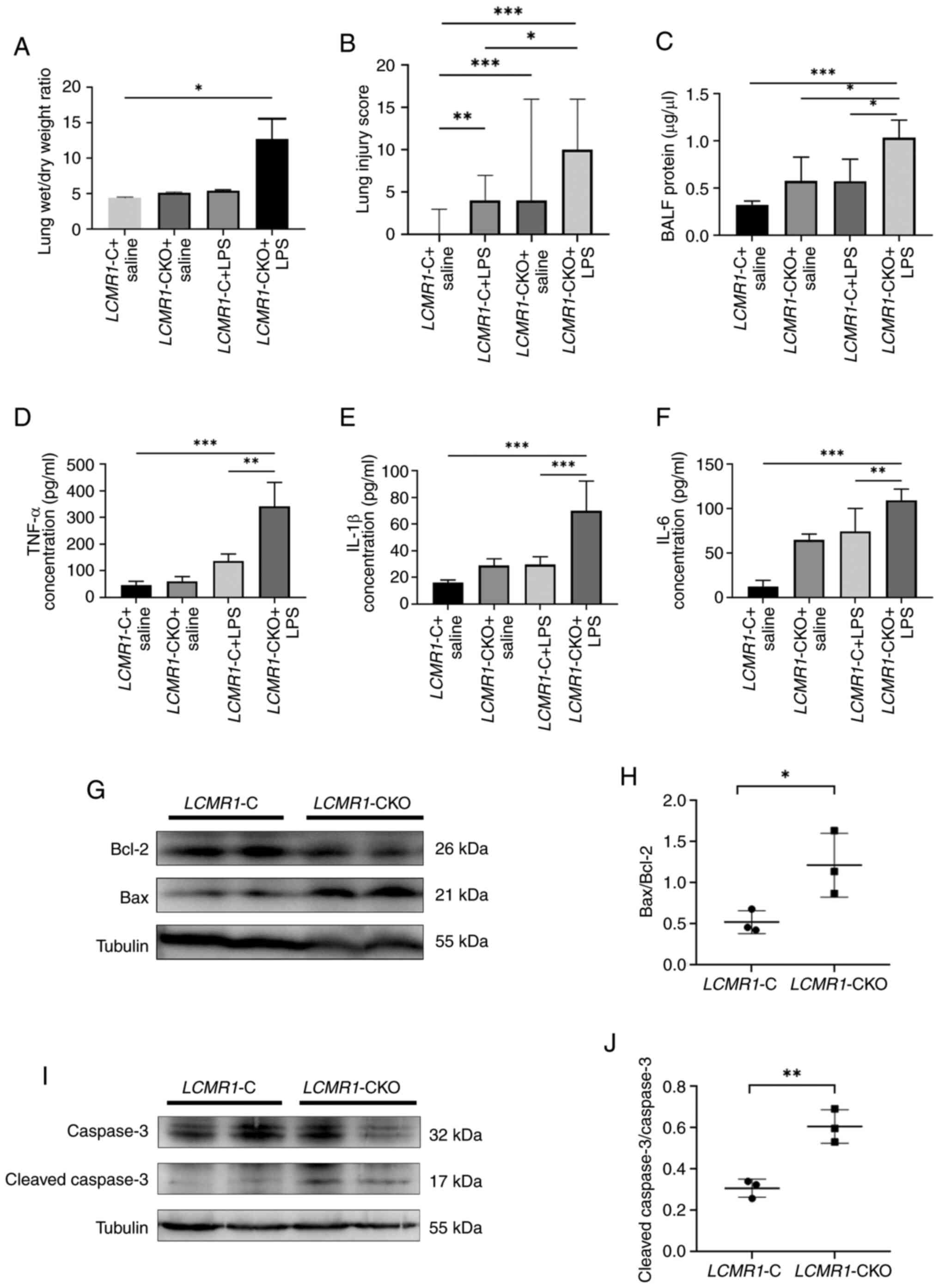 | Figure 3.LCMR1-CKO mice present with
more severe pathological damage, inflammatory response and enhanced
apoptosis after LPS-induced lung injury. (A) Mouse right lungs were
collected for measurement of wet/dry weight ratio. (B) Lung
pathological damage severity was accessed using lung injury scores.
BALF was collected and the concentration of (C) total protein, (D)
TNF-α, (E) IL-1β and (F) IL-6 in the BALF 48 h after LPS
administration were measured (n=6/group). (G) Western blot images
of Bax and Bcl-2 protein in murine lung tissues, and (H)
statistical results of Bax/Bcl-2 ratio (n=3/group). (I) Western
blot images of cleaved caspase-3 and caspase-3 protein in murine
lung tissues, and (J) statistical results of cleaved
caspase-3/caspase-3 ratio (n=6). *P<0.05, **P<0.001,
***P<0.0001. BALF, bronchoalveolar lavage fluid; C, control;
CKO, conditional knockout; LCMR1, lung cancer metastasis-related
protein 1; LPS, lipopolysaccharide. |
The present study further examined the alveolar
microstructure using transmission electron microscopy (Fig. 4). After LCMR1 conditional
knockout, the AEC-II cells of LCMR1-CKO mice were swollen
and the number of lamellar bodies was reduced. After LPS
stimulation, compared with the LCMR1-C mice, the AEC-II
cells of LCMR1-CKO mice shrank, and the lamellar bodies
disappeared indicating a lack of lung alveolar surfactant. The
air-blood barrier of was structurally disordered, and the barrier
becomes thinner after LPS-induced lung injury.
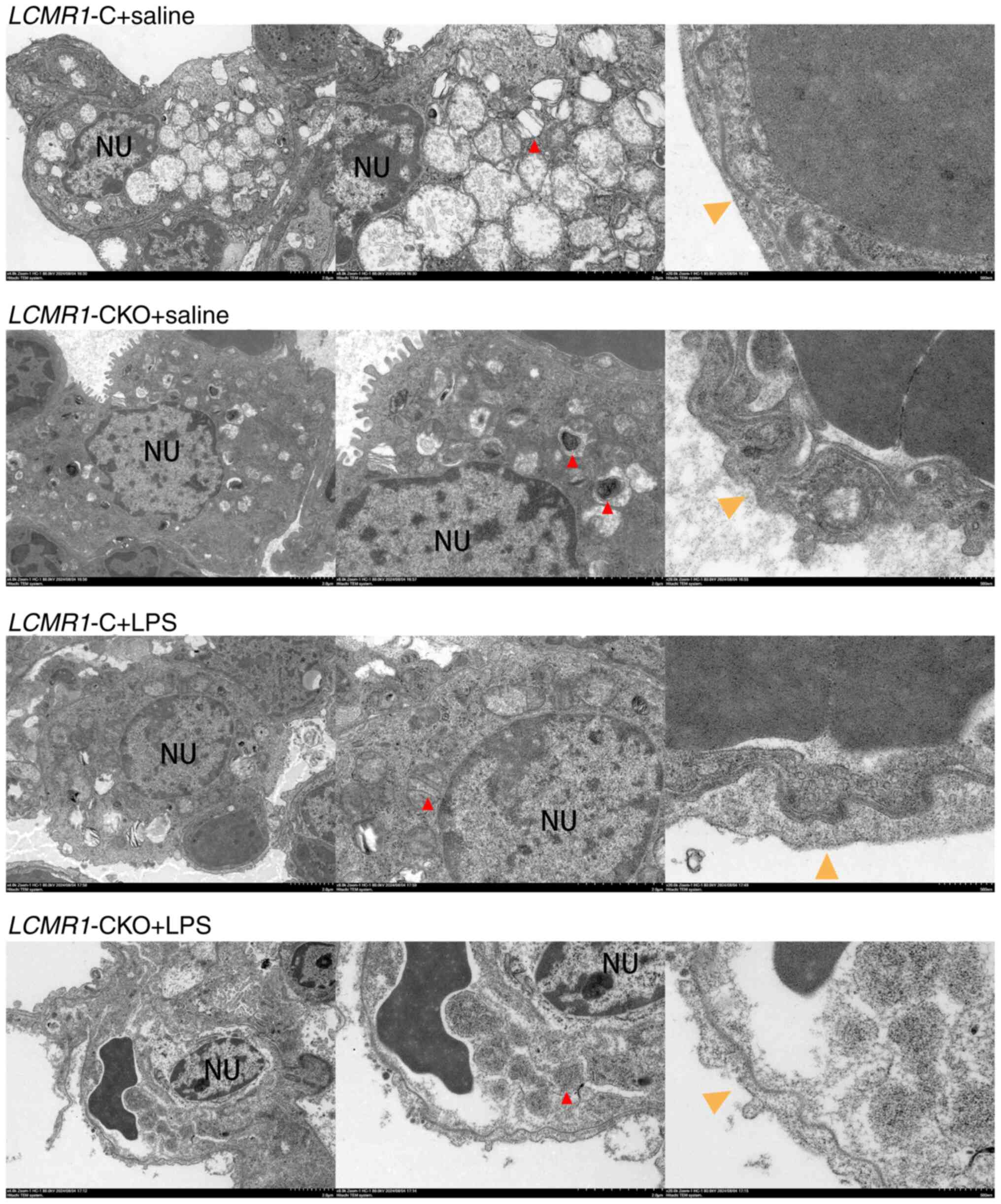 | Figure 4.Representative transmission electron
microscopy images of lung sections. Red arrows indicate lamellar
bodies in type II alveolar epithelial cells; yellow arrows indicate
the air-blood barrier. Scale bars: Left panels (2 µm, ×4,000),
right panels (500 nm, ×20,000), middle panels (2 µm, ×8,000). C,
control; CKO, conditional knockout; LCMR1, lung cancer
metastasis-related protein 1; LPS, lipopolysaccharide; NU,
nucleus. |
Generation of lung-on-a-chip
model
To further investigate the mechanism by which
LCMR1 deficiency aggravates lung injury, lung-on-a-chip
technology was used to simulate the alveolar environment under
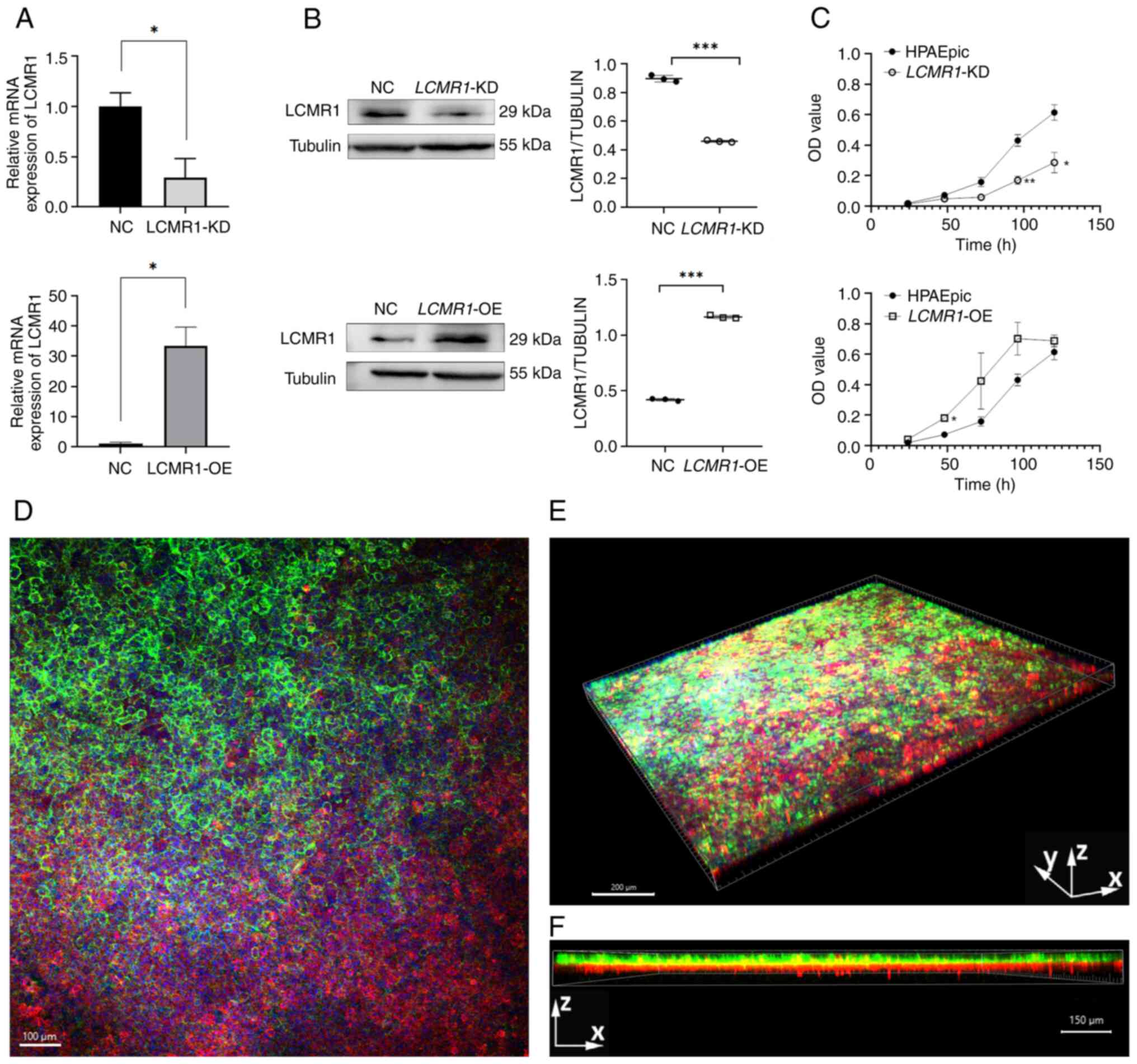
LPS-induced ALI. First, LCMR1-OE and LCMR1-KD cells,
based on the HPAEpiC cell line, were generated. The expression of
LCMR1 was upregulated in LCMR1-OE cells comparing to
negative controls and was downregulated in LCMR1-KD cells
comparing to negative controls, verified by RT-qPCR and western
blotting (Fig. 5A and B). In
addition, the proliferation rate of the gene-edited cells was
examined using the CCK8 assay, and the results revealed that the
proliferation rate of the LCMR1-OE cells increased, whereas
that of the LCMR1-KD cells decreased comparing to the
wild-type HPAEpic cells. (Fig.
5C).
The lung-on-a-chip model used in the present study
was constructed according to the method described by Zhang et
al (27). Immunofluorescence
demonstrated that epithelial cells were capable of establishing
adhesion junctions characterized by the presence of E-cadherin,
whereas endothelial cells were shown to form junctions identified
by VE-cadherin. These two types of cells successfully formed an
air-blood barrier on the chip device (Fig. 5D-F).
Knockdown of LCMR1 impairs the
structure and function of the air-blood barrier
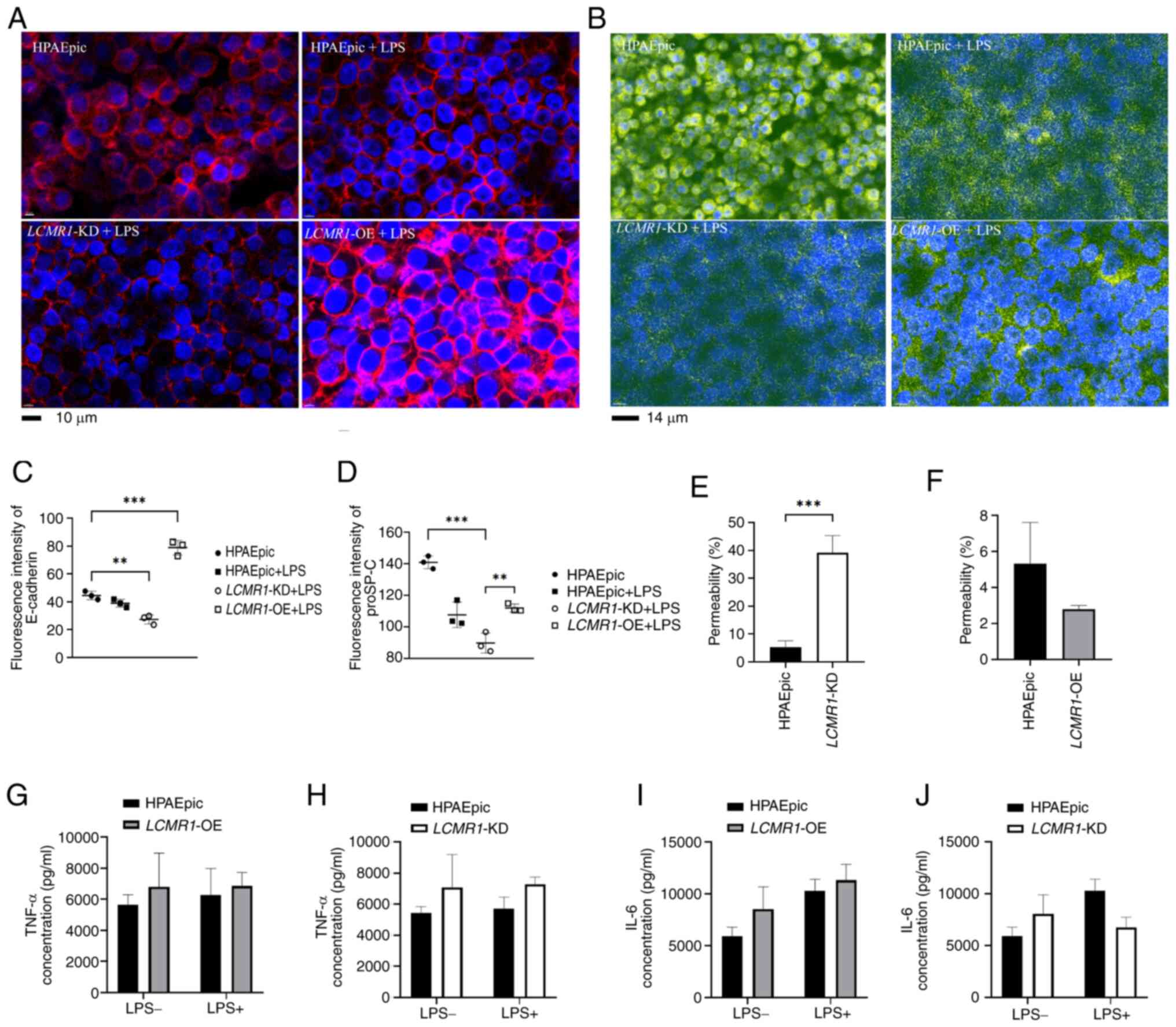
By adding LPS (1 µg/ml) to the flow medium, the
microenviroument of alveoli under LPS-induced lung injury was
mimicked, and the results revealed that E-cadherin expression was
attenuated in LCMR1-KD chips after 72 h compared with
HPAEpic chips; however, the E-cadherin expression of
LCMR1-OE group was significantly upregelated comparing to
the HPAEpic chips. (Fig. 6A and
C). In addition, proSP-C, an important synthetic substance of
AEC-II to maintain the homeostasis of the alveolar
microenvironment, was detected. The results showed that knockdown
of LCMR1 reduced proSP-C synthesis comparing to
LCMR1-OE group after LPS stimulation while the
LCMR1-OE group showed no significant difference in proSP-C
synthesis with the HPAEpic group after LPS stimulation (Fig. 6B and D). These findings suggested
that LCMR1 knockdown impaired the epithelial barrier
function. Therefore, FITC-glucan was administered to the bottom
channal of vascular cells to assess the integrity of the air-blood
barrier by comparing the fluorescence intensity of the two
channels. The results revealed increased permeability in the
LCMR1-KD comparing to the HPAEpic group, suggesting impaired
barrier integrity (Fig. 6E), while
the fluorescence intensity ratio of LCMR1-OE group showed no
significant change comparing to the HPAEpic group (Fig. 6E). The current study also examined
the inflammatory cytokines in the supernatant from the epithelial
channal of the chips, but the results showed no statistical
differences between the groups (Fig.
6G-J).
To further clarify the mechanism of air-blood
barrier dysfunction caused by LCMR1 deficiency, the possible
molecular mechanisms involved were examined. The protein expression
levels of apoptosis and differentiation-related molecules were
examined in samples extracted from lung-on-a-chip models. The
expression of AQP5, a marker of AEC-II differentiation into type I
alveolar epithelial cells (AEC-I), was analyzed. After LPS
stimulation, AQP5 expression was upregulated in LCMR1-OE
chips compared with in LPS-induced HPAEpic chips, suggesting
enhenced differentiation (Fig. 7A and
B). However, compared with that in the HPAEpic chips, the
protein expression levels of AQP5 in the LCMR1-KD chips were
decreased after LPS stimulation, suggesting dysfunctional
differentiation (Fig. 7A and C).
Markers of apoptosis were also examined, and increased Bax/Bcl-2
and cleaved caspase-3/caspse-3 protein ratios were detected in the
LCMR1-KD chips compared with HPAEpic chips, with or without
LPS, suggesting increased apoptosis. The LCMR1-OE chips,
however, showed no significant changes in Bax/Bcl-2 and cleaved
caspase-3/caspse-3 protein ratios comparing to the HPAEpic chips
(Fig. 7D-I).
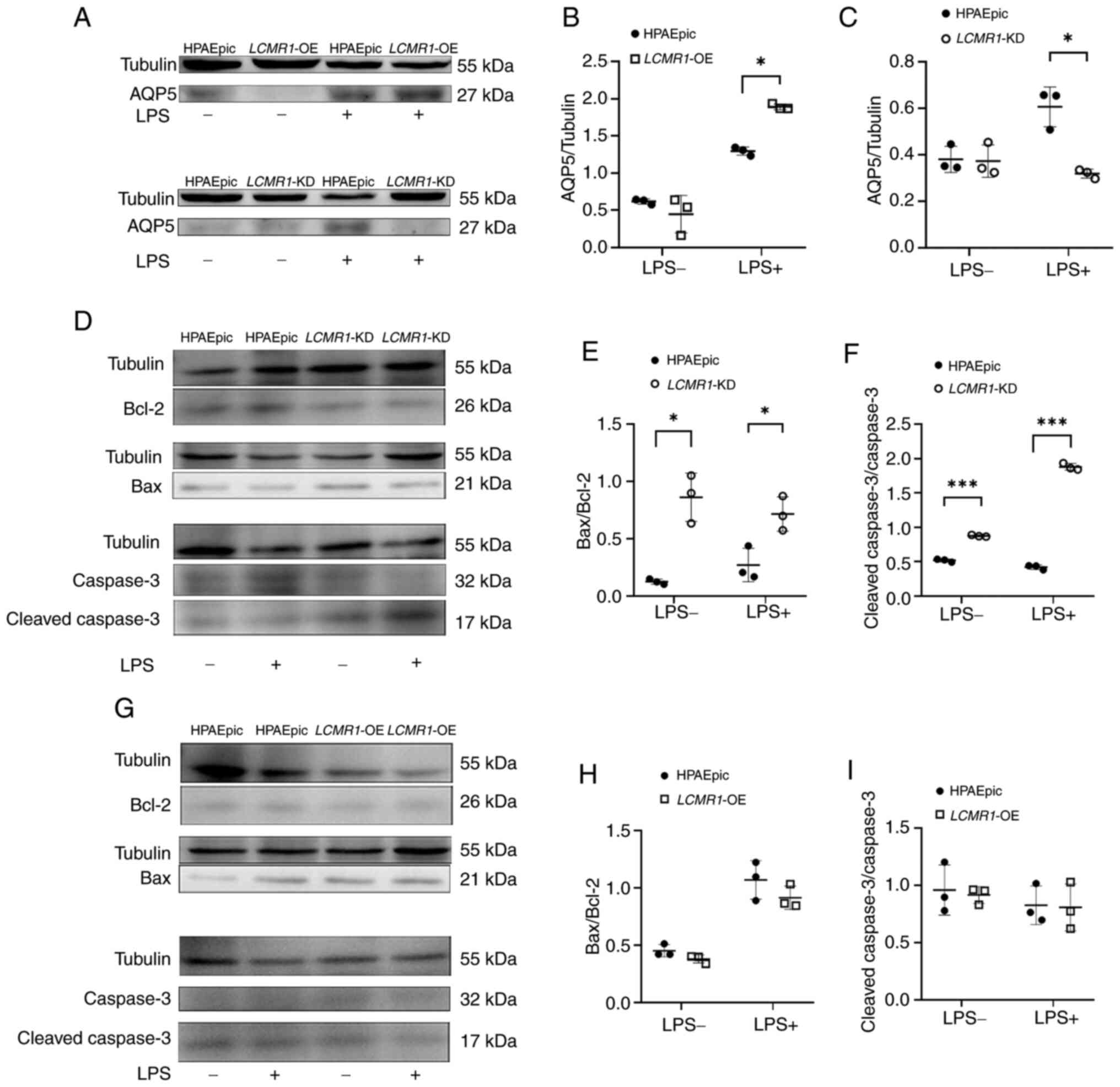 | Figure 7.Air-blood barrier impairment after
LCMR1 knockdown may be associated with impaired
differentiation and enhanced apoptosis. (A) Representative western
blot image of AQP5 protein in LCMR1-OE and LCMR1-KD
chips before and after LPS stimulation. (B) Gray value analysis of
AQP5 protein expression of the LCMR1-OE chips comparing to
the HPAEpic chips. (C) Gray value analysis of AQP5 protein
expression of the LCMR1-KD chips comparing to the HPAEpic
chips (n=3/group). *P<0.05, as determined by two-way analysis of
variance followed by Sidak's post hoc test. (D) Representative
western blot image of Bcl-2, Bax, cleaved caspase-3 and caspase-3
proteins in LCMR1-KD chips before and after LPS stimulation.
(E) Gray-scale analysis of Bax/Bcl-2 ratio in LCMR1-KD chips
(n=3). (F) Gray-scale analysis of cleaved caspase-3/caspase-3 ratio
in LCMR1-KD chips (n=3). (G) Representative western blot
image of Bcl-2, Bax, cleaved caspase-3 and caspase-3 protein in
LCMR1-OE chips before and after LPS stimulation. (H)
Gray-scale analysis of Bax/Bcl-2 ratio in LCMR1-OE chips
(n=3). (I) Gray-scale analysis of cleaved caspase-3/caspase-3 ratio
in LCMR1-OE chips (n=3). *P<0.05 and ***P<0.001, as
determined by two-way analysis of variance followed by Sidak's post
hoc test. AQP5, aquaporin 5; LCMR1, lung cancer
metastasis-related protein 1; LCMR1-KD,
LCMR1-knockdown; LCMR1-OE,
LCMR1-overexpressing; LPS, lipopolysaccharide. |
Discussion
Since the discovery of the LCMR1 gene in lung
cancer, research on LCMR1 has mainly focused on cancer;
notably, a number of reports have shown that LCMR1 serves an
important role not only in lung cancer, but also in a variety of
other types of cancer. Through large-scale RNA interference
screening, Agaësse et al (29) identified LCMR1 as a key
regulator of melanoma invasion, and Wang et al (20) reported that LCMR1 could
promote the metastasis of advanced metastatic prostate cancer.
However, the role of this gene in other diseases, particularly
inflammatory diseases, has yet to be investigated. The present
study, to the best of our knowledge, is the first to examine the
role of this gene in noncancerous diseases.
In experiments using wild-type mice to construct an
LPS-induced ALI model, 14) of mice died within 72 h after LPS
injection, and the results presented in the current study are from
mice that were still alive at each time point. Although these mice
survived for >72 h, it does not mean that the lungs of these
mice have returned to normal, and there may still be inflammatory
infiltration and edema; a previous study reported that the
inflammatory response peaked at ~2 days and returned to baseline at
~10 days after intratracheal LPS injection (30). The present study assessed the
expression of LCMR1 from 0 to 96 h after LPS injection to clarify
the change in LCMR1 expression after LPS-induced ALI. From the
findings of LPS-induced ALI in wild-type mice, it was revealed that
the expression of LCMR1 was significantly decreased during
the acute phase of LPS-induced lung injury (within 48 h after LPS
challenge). However, the expression of LCMR1 rebounded after
48 h (the acute phase). These results suggested that LCMR1
may be involved in the repair of lung injury and could serve a
protective role in the course of lung injury.
To further demonstrate the protective role of
LCMR1 in ALI, LCMR1-CKO mice were used to generate an
LPS-induced ALI model. The results revealed that mice with
conditional knockout of LCMR1 in AEC-II presented with more
severe lung function decline, lung inflammation and pathological
damage after LPS challenge than the LCMR1-C mice and
LCMR1-CKO mice without LPS administration. This finding is
consistent with our previous study, which showed that mice
developed lung injury at ~15 days after knockout (18). However, in the present study, LPS
injection was performed 24 h after tamoxifen induction, and lungs
were harvested 48 h after LPS administration (72 h after induced
knockout). The timepoint is earlier than the time of phenotypic
changes detected in mice in the previous study. At this timepoint,
the results showed that the LCMR1-CKO mice given saline did
not show significant pathological and functional changes, whereas
the LCMR1-CKO mice given LPS suffered significant lung
injury, which suggested that LCMR1 knockout aggravated the
course of LPS-induced lung injury.
Destruction of AEC-II and damage to the air-blood
barrier were observed by transmission electron microscopy,
suggesting LCMR1 may affect the homeostasis of the air-blood
barrier. In order to clarify the effect of LCMR1 on the
alveolar air-blood barrier, organ-on-a-chip technology was applied.
Organ-on-a-chip technology is an emerging method that combines
bioengineering and material technology, and can reconstruct the
microenvironment and microstructure of lung cells in vitro.
Some studies have used organ-on-a-chip models to mimic lung
diseases. Zhang et al used an alveolar-chip model to
simulate the infection process of SARS-CoV-2, which greatly reduced
the difficulty and cost of studying this pathogen (27). Dasgupta et al (31) constructed a lung-on-a-chip model of
radiation-induced ALI; compared with the preclinical animal model,
the radiation dose sensitivity observed on the chip was more
similar to that of the human lung, and the effect of therapeutic
drugs on radiation-induced ALI was intuitively evaluated using this
model. These findings indicated that lung-on-a-chip is an
intuitive, visual and easy-to-control model, which can be used to
simulate the microenvironment of various lung diseases. The current
study used this model to more intuitively and conveniently reflect
the effect of the LCMR1 gene on lung injury. The present
study revealed that knockdown of LCMR1 resulted in impaired
tight junctions in the alveolar epithelium and reduced expression
of E-cadherin, indicating impaired air-blood barrier function and
impaired barrier tightness as reflected by leakage of fluorescent
substances. In addition, knockdown of LCMR1 resulted in
decrease in proSP-C in epithelial cells after LPS stimulation
suggests a disorder of cellular synthetic function. As the proSP-C
is a crucial alveolar surfactant which maintain alveolar pressure
and preventing alveolar collapse (32), its reduction similarly affects
air-blood barrier function. Notably, in the current study, LPS
stimulation time on lung-on-a-chip model was 72 h, which is longer
than previous cell-based studies. In cell-based studies, the
duration of LPS stimulation has generally been reported to be
<24 h (33,34). This may be similar to the in
vivo environment, as the continuous flow of medium supplemented
with LPS mimicking the real in vivo injury stimulus.
The LCMR1 gene encodes a subunit of the
cellular transcription mediator complex and is also known as MED19
(35). Mediator complexes are
crucial in the proliferation and metastasis of malignant tumors as
they modulate various signal transduction pathways associated with
cell growth, differentiation, the cell cycle and apoptosis
(35). After the expression of the
LCMR1 gene was altered by lentiviral infection in HPAEpiC
cells, the proliferation rate of cells with overexpression of
LCMR1 accelerated, whereas the proliferation rate of cells
with knockdown of LCMR1 decreased, indicating that
LCMR1 may affect the proliferation of cells. This
observation is consistent with the clinical manifestation that
tumors with high expression of LCMR1 are more prone to metastasis
(36,37). By contrast, the present results
from the lung-on-a-chip model revealed that knockdown of the
oncogene LCMR1 increased the susceptibility of normal
alveolar epithelial cells to lung injury. The decreased expression
of E-cadherin suggested damage to epithelial tight junctions,
whereas the decrease in proSP-C suggested a decrease in cell
synthesis, all of which indicated that cell dysfunction may occur
after knockdown of LCMR1. Different protein isoforms of
LCMR1 have been reported to have different effects on gene
expression (38). It could be
hypothesized that LCMR1, which promotes metastasis in cancer, is a
different isoform than LCMR1 that maintains cell homeostasis, and
further studies are needed to clarify the specific mechanism of
LCMR1 isoforms.
The mechanism of LPS-induced ALI also includes an
increased inflammatory response. In the present study, mice with
LCMR1 conditional knockout in AEC-II exhibited a more severe
inflammatory response, manifested by increased protein and
inflammatory cytokine levels in the BALF. However, there was no
significant change in inflammatory cytokine levels in the
lung-on-a-chip model. It could be hypothesized that this may be due
to the absence of immune cells in the lung-on-a-chip model. In a
study of SARS-CoV-2 infection on a lung-on-a-chip model, more
severe epithelial damage and elevated inflammatory cytokines were
observed after the addition of circulating immune cells (27). A similar result has been observed
in a lung-on-a-chip model of radiation-induced ALI; the presence of
peripheral blood mononuclear cells at the time of radiation
exposure was shown to lead to a persistent and progressive
inflammatory response in the chips (31). The different results between the
in vivo and lung-on-a-chip models in the present study may
be related to this. The enhanced inflammatory response in
vivo may be a secondary result of the infiltration of
inflammatory cells after the impairment of the lung air-blood
barrier caused by LCMR1 knockdown.
The present study demonstrated that the knockdown
of LCMR1 could potentially disrupt cellular differentiation
processes. Following induction with LPS, the expression levels of
AQP5 in the LCMR1-KD chips were diminished in comparison
with the control and LCMR1-OE chips. AQP5 is a marker of
AEC-I. AEC-II serve a stem cell-like role in the lung and can
differentiate into AEC-I during lung injury to have a role in
repair (39). The present results
suggested that knockdown of LCMR1 may lead to impaired
differentiation and impaired epithelial repair, which in turn
aggravates injury. A similar phenomenon has been reported in a
P53 gene study; the p53 protein was shown to promote
alveolar regeneration following ALI by boosting the self-renewal of
AEC-II and facilitating their differentiation into mature AEC-I
(40). Taken together, these
results suggested the potential protective role of oncogenes in
inflammatory diseases such as ALI, and may provide new targets for
the prevention and treatment of these diseases.
Knockout of LCMR1 may also aggravate lung
injury through enhanced apoptosis. In the present study, both
lung-on-a-chip and animal models showed an increase in the
expression ratio of Bax/Bcl-2 after knockdown or knockout of
LCMR1, suggesting enhanced apoptosis. This is similar to the
results of some previous studies on tumors. Xu et al
(41) reported that the
interaction between LCMR1 and DEK can synergistically inhibit the
apoptosis of lung cancer cells, and this effect may be related to
the induction of the myeloid leukemia protein cell differentiation
protein 1 pathway. Zhang et al (42) reported that miroRNA-4778-3p can
specifically bind to LCMR1 and negatively regulate its expression,
which eventually leads to the downregulation of the expression of
the apoptosis-related molecules caspase-3, caspase-8 and caspase-9.
Notably, in the LCMR1-OE chip findings in the present study,
there was no significant difference in apoptotic proteins compared
with the control group. It could be hypothesized that
overexpression of LCMR1 inhibited apoptosis to a certain
extent, so that the apoptosis protein level was not significantly
increased even in the presence of LPS. Future studies should
investigate the relationship between LCMR1 and apoptosis,
particularly the relevant mechanism in the case of LCMR1
overexpression.
In conclusion, the present study indicated that
LCMR1 deficiency aggravated LPS-induced ALI in an animal
model and a lung-on-a-chip model. To the best of our knowledge, the
present study is the first regarding the function of this oncogene
in diseases other than cancer, which suggested that LCMR1
may serve a phylactic role in ALI by affecting the apoptosis and
differentiation of AEC-II. LCMR1 could contribute to the
process of inflammatory diseases such as ALI and may therefore be
considered a new therapeutic target. The present study also
demonstrated the possibility of using organ-on-chip technology for
gene editing research, which may make it more convenient, intuitive
and economical to study the function of specific genes in the
future.
The present study has some limitations. First, due
to the reduced lifespan of LCMR1-CKO mice, long-term
observation data were not acquired. Second, the specific pathways
and mechanism of LCMR1 have not been well studied. In future
studies, we aim to further clarify the in-depth mechanism based on
transcriptome sequencing data from LCMR1-OE and
LCMR1-KD lung-on-a-chip samples.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
The authors would like to thank Professor Jianhua
Qin (Division of Biotechnology, CAS Key Laboratory of SSAC, Dalian
Institute of Chemical Physics, Chinese Academy of Sciences) and
their team for technical assistance.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in this study are available on
request from the corresponding author.
Author's contributions
ZM, XM and LC conceived and designed the study, ZM
and XM conducted the experiments, and drafted and revised the
manuscript. CS, JL, JR, LL, YW and YL participated in cell and
animal experiments. CL and ZY analyzed data. All authors read and
approved the final version of the manuscript. ZM, XM and LC confirm
the authenticity of all the raw data.
Ethics approval and consent to
participate
The experimental protocol was approved by the
Animal Ethics Committee of Chinese PLA General Hospital (approval
no. SQ2023672; Beijing, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Long ME, Mallampalli RK and Horowitz JC:
Pathogenesis of pneumonia and acute lung injury. Clin Sci (Lond).
136:747–769. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wick KD, Ware LB and Matthay MA: Acute
respiratory distress syndrome. BMJ. 387:e0766122024. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bellani G, Laffey JG, Pham T, Fan E,
Brochard L, Esteban A, Gattinoni L, van Haren F, Larsson A, McAuley
DF, et al: Epidemiology, patterns of care, and mortality for
patients with acute respiratory distress syndrome in intensive care
units in 50 countries. JAMA. 315:788–800. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu H, Yu X, Yu S and Kou J: Molecular
mechanisms in lipopolysaccharide-induced pulmonary endothelial
barrier dysfunction. Int Immunopharmacol. 29:937–946. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bos LDJ and Ware LB: Acute respiratory
distress syndrome: Causes, pathophysiology, and phenotypes. Lancet.
400:1145–1156. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang Y, Wang L, Ma S, Cheng L and Yu G:
Repair and regeneration of the alveolar epithelium in lung injury.
FASEB J. 38:e236122024. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Woods SJ, Waite AA, O'Dea KP, Halford P,
Takata M and Wilson MR: Kinetic profiling of in vivo lung cellular
inflammatory responses to mechanical ventilation. Am J Physiol Lung
Cell Mol Physiol. 308:L912–L921. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li N, Liu B, Xiong R, Li G, Wang B and
Geng Q: HDAC3 deficiency protects against acute lung injury by
maintaining epithelial barrier integrity through preserving
mitochondrial quality control. Redox Biol. 63:1027462023.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen H, Lin X, Yi X, Liu X, Yu R, Fan W,
Ling Y, Liu Y and Xie W: SIRT1-mediated p53 deacetylation inhibits
ferroptosis and alleviates heat stress-induced lung epithelial
cells injury. Int J Hyperthermia. 39:977–986. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hiemstra PS, Tetley TD and Janes SM:
Airway and alveolar epithelial cells in culture. Eur Respir J.
54:19007422019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Perelson AS and Ribeiro RM: Introduction
to modeling viral infections and immunity. Immunol Rev. 285:5–8.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ma C, Peng Y, Li H and Chen W:
Organ-on-a-Chip: A new paradigm for drug development. Trends
Pharmacol Sci. 42:119–133. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tan J, Guo Q, Tian L, Pei Z, Li D, Wu M,
Zhang J and Gao X: Biomimetic lung-on-a-chip to model virus
infection and drug evaluation. Eur J Pharm Sci. 180:1063292023.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bai H, Si L, Jiang A, Belgur C, Zhai Y,
Plebani R, Oh CY, Rodas M, Patil A, Nurani A, et al: Mechanical
control of innate immune responses against viral infection revealed
in a human lung alveolus chip. Nat Commun. 13:19282022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nawroth JC, Lucchesi C, Cheng D, Shukla A,
Ngyuen J, Shroff T, Varone A, Karalis K, Lee HH, Alves S, et al: A
microengineered airway lung chip models key features of
Viral-induced exacerbation of asthma. Am J Respir Cell Mol Biol.
63:591–600. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang P, Jin L, Zhang M, Wu Y, Duan Z, Guo
Y, Wang C, Guo Y, Chen W, Liao Z, et al: Blood-brain barrier injury
and neuroinflammation induced by SARS-CoV-2 in a lung-brain
microphysiological system. Nat Biomed Eng. 8:1053–1068. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen L, Liang Z, Tian Q, Li C, Ma X, Zhang
Y, Yang Z, Wang P and Li Y: Overexpression of LCMR1 is
significantly associated with clinical stage in human NSCLC. J Exp
Clin Cancer Res. 30:182011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang Y, Li C, Wu Z, Dai Y, Liu L and Chen
L: Deletion of LCMR1 in alveolar type II cells induces lethal
impairment of lung structure and function in adult mice. J Thorac
Dis. 15:1445–1459. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bao X, Zhao L, Guan H and Li F: Inhibition
of LCMR1 and ATG12 by demethylation-activated miR-570-3p is
involved in the anti-metastasis effects of metformin on human
osteosarcoma. Cell Death Dis. 9:6112018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang K, Wang X, Fu X, Sun J, Zhao L, He H
and Fan Y: Lung cancer metastasis-related protein 1 promotes the
transferring from advanced metastatic prostate cancer to
castration-resistant prostate cancer by activating the
glucocorticoid receptor alpha signal pathway. Bioengineered.
13:5373–5385. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu L, Li C, Wu Z, Li Y, Yu H, Li T, Wang
Y, Zhao W and Chen L: LCMR1 promotes Large-cell lung cancer
proliferation and metastasis by downregulating HLA-Encoding genes.
Cancers (Basel). 15:54452023. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Percie du Sert N, Hurst V, Ahluwalia A,
Alam S, Avey MT, Baker M, Browne WJ, Clark A, Cuthill IC, Dirnagl
U, et al: The ARRIVE guidelines 2.0: Updated guidelines for
reporting animal research. PLoS Biol. 18:e30004102020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shrum B, Anantha RV, Xu SX, Donnelly M,
Haeryfar SM, McCormick JK and Mele T: A robust scoring system to
evaluate sepsis severity in an animal model. BMC Res Notes.
7:2332014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Stanojevic S, Kaminsky DA, Miller MR,
Thompson B, Aliverti A, Barjaktarevic I, Cooper BG, Culver B, Derom
E, Hall GL, et al: ERS/ATS technical standard on interpretive
strategies for routine lung function tests. Eur Respir J.
60:21014992022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Glaab T, Taube C, Braun A and Mitzner W:
Invasive and noninvasive methods for studying pulmonary function in
mice. Respir Res. 8:632007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fukumoto J, Fukumoto I, Parthasarathy PT,
Cox R, Huynh B, Ramanathan GK, Venugopal RB, Allen-Gipson DS,
Lockey RF and Kolliputi N: NLRP3 deletion protects from
hyperoxia-induced acute lung injury. Am J Physiol Cell Physiol.
305:C182–C189. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang M, Wang P, Luo R, Wang Y, Li Z, Guo
Y, Yao Y, Li M, Tao T, Chen W, et al: Biomimetic human disease
model of SARS-CoV-2-Induced lung injury and immune responses on
organ chip system. Adv Sci (Weinh). 8:20029282021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Agaesse G, Barbollat-Boutrand L, Sulpice
E, Bhajun R, El Kharbili M, Berthier-Vergnes O, Degoul F, de la
Fouchardière A, Berger E, Voeltzel T, et al: A large-scale RNAi
screen identifies LCMR1 as a critical regulator of Tspan8-mediated
melanoma invasion. Oncogene. 36:50842017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang L, Wang X, Tong L, Wang J, Dou M, Ji
S, Bi J, Chen C, Yang D, He H, et al: Recovery from acute lung
injury can be regulated via modulation of regulatory T cells and
Th17 cells. Scand J Immunol. 88:e127152018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dasgupta Q, Jiang A, Wen AM, Mannix RJ,
Man Y, Hall S, Javorsky E and Ingber DE: A human lung
alveolus-on-a-chip model of acute radiation-induced lung injury.
Nat Commun. 14:65062023. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sehlmeyer K, Ruwisch J, Roldan N and
Lopez-Rodriguez E: Alveolar dynamics and Beyond-The importance of
surfactant protein C and cholesterol in lung homeostasis and
fibrosis. Front Physiol. 11:3862020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xiao K, He W, Guan W, Hou F, Yan P, Xu J,
Zhou T, Liu Y and Xie L: Mesenchymal stem cells reverse EMT process
through blocking the activation of NF-κB and Hedgehog pathways in
LPS-induced acute lung injury. Cell Death Dis. 11:8632020.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li J, Deng SH, Li J, Li L, Zhang F, Zou Y,
Wu DM and Xu Y: Obacunone alleviates ferroptosis during
lipopolysaccharide-induced acute lung injury by upregulating
Nrf2-dependent antioxidant responses. Cell Mol Biol Lett.
27:292022. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang Y, Qin P, Tian L, Yan J and Zhou Y:
The role of mediator complex subunit 19 in human diseases. Exp Biol
Med (Maywood). 246:1681–1687. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang X, Fan Y, Liu B, Qi X, Guo Z and Li
L: Med19 promotes breast cancer cell proliferation by regulating
CBFA2T3/HEB expression. Breast Cancer. 24:433–441. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang X, Gao D, Fang K, Guo Z and Li L:
Med19 is targeted by miR-101-3p/miR-422a and promotes breast cancer
progression by regulating the EGFR/MEK/ERK signaling pathway.
Cancer Lett. 444:105–115. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ruoff R, Weber H, Wang Y, Huang H, Shapiro
E, Fenyo D and Garabedian MJ: MED19 encodes two unique protein
isoforms that confer prostate cancer growth under low androgen
through distinct gene expression programs. Sci Rep. 13:182272023.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu K, Meng X, Liu Z, Tang M, Lv Z, Huang
X, Jin H, Han X, Liu X, Pu W, et al: Tracing the origin of alveolar
stem cells in lung repair and regeneration. Cell.
187:2428–2445.e20. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kaiser AM, Gatto A, Hanson KJ, Zhao RL,
Raj N, Ozawa MG, Seoane JA, Bieging-Rolett KT, Wang M, Li I, et al:
p53 governs an AT1 differentiation programme in lung cancer
suppression. Nature. 619:851–859. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xu Y, Liang Z, Li C, Yang Z and Chen L:
LCMR1 interacts with DEK to suppress apoptosis in lung cancer
cells. Mol Med Rep. 16:4159–4164. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang Y, Li P, Hu J, Zhao LN, Li JP, Ma R,
Li WW, Shi M and Wei LC: Role and mechanism of miR-4778-3p and its
targets NR2C2 and Med19 in cervical cancer radioresistance. Biochem
Biophys Res Commun. 508:210–216. 2019. View Article : Google Scholar : PubMed/NCBI
|