Introduction
Hepatocellular carcinoma (HCC) is the fourth leading
cause of global cancer-related mortality (1), and is characterized by a poor
prognosis due to its aggressive growth, propensity for metastasis
and intrinsic resistance to current therapeutic approaches
(2). Consequently, there is an
urgent need to develop effective therapeutic strategies for the
treatment of patients with HCC.
Triptolide (TPL), a structurally unique diterpene
triepoxide isolated from Tripterygium wilfordii Hook F, is a
traditional Chinese medicinal plant that has been used for
centuries to treat a wide range of diseases. Notably, TPL has
demonstrated potent anticancer activities, and exhibits cytotoxic
activity against cancer cells originating from diverse tissues,
including the colon, breast, stomach and liver (3–8).
Emerging evidence has demonstrated that TPL exerts potent anti-HCC
effects through multifaceted mechanisms. Studies have shown that
TPL suppresses the invasion and tumorigenesis of MHCC-97H HCC cells
by modulating the NF-κB signaling pathway (6). Furthermore, TPL can induce the
apoptosis of HCC cells independent of p53 status (7), while its targeted delivery in BALB/c
mice bearing HepG2 tumors effectively inhibits HCC progression
through suppression of de novo lipogenesis (8). Furthermore, synergistic therapeutic
outcomes have been observed when combining TPL with sorafenib for
HCC treatment (9,10). These findings indicate that TPL may
be a promising therapeutic candidate against HCC; however, its
clinical translation remains constrained by dose-dependent toxicity
(11).
Glutathione peroxidase 4 (GPx4), also known as
phospholipid hydroperoxide glutathione peroxidase, serves a pivotal
role in maintaining cellular redox homeostasis (12). Mechanistic studies have revealed
functional differences between the GPx4 isoforms: The 20-kDa
non-mitochondrial isoform suppresses ferroptosis through inhibition
of lipid peroxidation (13),
whereas the 23-kDa mitochondrial isoform prevents apoptosis by
neutralizing cardiolipin hydroperoxide-mediated cytochrome c
release (14,15). RAS-selective lethal 3 (RSL3)
induces ferroptosis through direct GPx4 inactivation in RAS-mutated
cancer cells, while sparing normal tissues (16). To potentiate the anti-HCC efficacy
of TPL by pharmacological inhibition of GPx4 activity, the present
study co-treated HCC cell lines in vitro with TPL and
RSL3.
Materials and methods
Cell culture
HCC cell lines Hep3B, PLC/PRF/5 (PLC) and Huh7 were
cultured in high-glucose Dulbecco's Modified Eagle Medium (cat. no.
C11995500BT; DMEM; Hyclone; Cytiva) supplemented with 10% fetal
bovine serum (cat. no. 35-015-CV; Corning, Inc.), streptomycin (100
µg/ml) and penicillin (100 U/ml) mix (cat. no. 15070063; Gibco;
Thermo Fisher Scientific, Inc). All cell cultures were maintained
in a humidified incubator with an atmosphere of 5% CO2
and a temperature of 37°C.
Cell viability assay
Hep3B/PLC/Huh7 cells were seeded at a density of
2,000 cells/well into 96-well plates and allowed to adhere for 24
h. Subsequently, TPL (cat. no. E-0316; Shanghai Tauto Biotech Co.
Ltd.) (0, 2.5, 5, 7.5, 10 and 12.5 ng/ml) or RSL3 (cat. no.
HY-100218A; MedChemExpress) (0, 0.125, 0.25, 0.5, 1, 2 and 4 µM)
was used for treating Hep3B/PLC cells for 48 h or Huh7 cells for 24
h at 37°C to determine the cytotoxicity of TPL or RSL3. To
investigate the cytotoxicity of TPL combined with RSL3, Hep3B or
PLC cells were incubated with TPL (5 and 7.5 ng/ml) plus RSL3 (0,
0.125, 0.25, 0.5 and 1 µM) for 48 h at 37°C, while Huh7 cells were
treated with TPL (10 ng/ml) and RSL3 (0.5 and 1 µM) for 24 or 48 h
at 37°C. In addition, Hep3B or PLC cells were treated with TPL (7.5
ng/ml) and RSL3 (1 µM) for 24 or 48 h with or without a 2-h
pretreatment with a reactive oxygen species (ROS) inhibitor
N-acetyl-cysteine (NAC) (12 mM) (cat. no. HY-B0215; MedChemExpress)
at 37°C to assess whether co-treatment with TPL and RSL3 increases
ROS levels. Likewise, Hep3B/PLC cell culture was subjected to a 2-h
pretreatment with a specific ferroptosis inhibitor Ferrostatin-1
(Fer-1) (5 µM) (cat. no. HY-100579; MedChemExpress) or a potent
free iron chelating agent Deferiprone (DEF) (100 µM) (cat. no.
HY-B0568; MedChemExpress) to evaluate if co-treatment with TPL and
RSL3 enhances ferroptosis. After that, Hep3B cells were treated
with TPL (10 ng/ml) plus RSL3 (2 µM) and PLC cells were treated
with TPL (7.5 ng/ml) plus RSL3 (1 or 2 µM) for 24 h at 37°C. Cell
viabilities were measured using the Cell Counting Kit 8 (CCK-8;
cat. no. HY-K0301; MedChemExpress) following the aforementioned
various cell treatments. CCK-8 solution (10 µl/well) was added to
the plate. The plates were then incubated for 2 h at 37°C.
Subsequently, the absorbance was measured at 450 nm using a
microplate reader (Biotek; Agilent Technologies, Inc.). All
experiments were performed independently at least three times.
Protein extraction and western blot
analysis
Hep3B/PLC/Huh7 cells were inoculated into 6-cm
dishes at a density of 1.7×105 cells/dish and cultured
for 24 h in the incubator. To investigate GPx4 expression, Hep3B or
PLC cells were treated with TPL at 0, 2.5, 5, 7.5, 10 and 12.5
ng/ml for 48 h or at 10 ng/ml for 0, 32, 48, 60 and 72 h. Huh7
cells were treated with TPL (0, 5, 10 and 15 ng/ml) for 24 or 48 h.
To analyze apoptosis, Hep3B or PLC cells were treated with TPL (7.5
ng/ml)/RSL3 (1 µM)/TPL (7.5 ng/ml) combined with RSL3 (1 µM) for 30
h. After that, total cell lysates were extracted using RIPA lysis
buffer containing PMSF (cat. no. C1055; Applygen Technologies,
Inc.). Protein concentration was determined with the BCA Protein
Assay Kit (cat. no. 23225; Thermo Fisher Scientific, Inc.).
Proteins (40 µg/lane) were separated on SurePAGE, Bis-Tris, 10×8 cm
gradient (4–20%) gels (cat. no. M00656; GenScript Biotech
Corporation) and transferred onto PVDF membranes (cat. no.
IPVH00010; Millipore; Merk Group), which were blocked in 5% BSA for
1 h at room temperature (RT). The membranes were then incubated
with specific primary antibodies (1:1,000) at 4°C overnight. GAPDH
(cat. no. BK7021-100 µl; Bioker Biotechnology Co.) was used as a
loading control. Antibodies against GPx4 (cat. no. 52455), both
cleaved and precursor proteins of PARP (cat. no. 9532), caspase-9
(cat. no. 9502) and caspase-3 (cat. no. 9662) were purchased from
Cell Signaling Technology, Inc. After being washed, the membranes
were incubated with HRP-conjugated Goat Anti-Rabbit-IgG (cat. no.
SA00001-2; Proteintech Group, Inc.) (1:10,000) at RT for 1 h and
detected using the FDbio-Femto ECL Kit (cat. no. FD8030; FDbio
Science Biotech Co. Ltd.). Protein bands were semi-quantified and
grayscale values were calculated using ImageJ 1.52a (National
Institutes of Health).
Real-time cellular analysis
(RTCA)
The inhibitory effect produced by TPL and RSL3 on
cell proliferation was evaluated by RTCA. Hep3B or PLC cells (2,000
cells/well) were seeded into E-Plate 16 (cat. no. 5469813001;
Agilent Technologies, Inc.) and cultured in medium containing TPL
(7.5 and 12.5 ng/ml) with or without RSL3 (1 µM) at 37°C for 80 h.
xCELLigence RTCA S16 Analyzer (ACEA Biosciences, Inc.) was used to
monitor cell proliferation in real-time. Cell proliferation signals
were transformed into cell indexes, which were displayed in RTCA
S16 software (version 1.0.1; ACEA Biosciences, Inc.).
Cell apoptosis assay
Hep3B or PLC cells were seeded into 6-cm dishes at
the density of 1.7×105 cells/dish, cultured for 24 h in
the incubator, and then treated with TPL (7.5 ng/ml) or RSL3 (1 µM)
or TPL (7.5 ng/ml) combined with RSL3 (1 µM) for 24 h. Thereafter,
cells were harvested, stained using BD Pharmingen™ FITC Annexin V
Apoptosis Detection Kit I (cat. no. 556547; BD Pharmingen; BD
Biosciences) according to the manufacturer's protocol and then
detected with flow cytometry (LSRFortessa SORP; BD Biosciences).
Data were recorded using BD FACSDiva™ software and analyzed with
FlowJo software (version V10; BD Biosciences). Annexin
V+/PI− stained cells represented apoptotic
cells.
Detection of ROS
Hep3B or PLC cells were seeded into 10-cm dishes at
a density of 5×105 cells/dish, allowed to adhere for 24
h in the incubator, then treated with TPL (7.5 and 10 ng/ml)/RSL3
(1 µM)/TPL (7.5, 10 ng/ml) combined with RSL3 (1 µM) for another 24
h at 37°C. Subsequently, intracellular ROS levels were determined
by detecting the fluorescence intensity of DCF as previously
described (17) with minor
modifications. That is, the cells were harvested and incubated with
DMEM containing 2.5 µM 2′-7′-dichlorodihydrofluorescein diacetate
(DCFH-DA) (cat. no. S0033S; Beyotime Institute of Biotechnology)
for 1 h at 37°C, with mixing performed every 5 min. The cells were
then washed three times with DMEM to remove DCFH-DA that had not
entered the cells. Finally, DCF emission was recorded on the FITC
channel of a flow cytometer (LSRFortessa SORP; BD Biosciences)
using the BD FACSDiva software. Data were analyzed with FlowJo
software (version V10; BD Biosciences).
Measurement of lipid peroxidation
levels
BODIPY® 581/591 C11 (cat. no. D3861;
Molecular Probes; Thermo Fisher Scientific, Inc.) is a fluorescent
probe used to determine lipid peroxidation in live cells. PLC cells
were seeded into 10-cm dishes at a density of 5×105
cells/dish, cultured for 24 h in the incubator, then treated with
TPL (7.5 ng/ml)/RSL3 (1 and 2 µM)/TPL (7.5 ng/ml) combined with
RSL3 (1 and 2 µM) for 24 h at 37°C. Cell pellets were obtained by
0.25% trypsin digestion and centrifugation at 200 × g for 3 min at
RT. After washing with PBS, the cells were resuspended in PBS
containing 10 µM BODIPY 581/591 C11 and incubated for 30 min at RT.
Subsequently, the excess dye was removed by rinsing with PBS and
centrifugation at 200 × g for 3 min at RT. Finally, the cells were
resuspended in PBS and detected by flow cytometry (LSRFortessa
SORP; BD Biosciences) and the fluorescence signals were recorded
using BD FACSDiva software. FlowJo software (version V10; BD
Biosciences) was used for data analysis.
Statistical analysis
SPSS 17.0 (SPSS, Inc.) was used for analyzing data.
All experiments were repeated three or more times. Quantitative
data from the experiments were analyzed and presented in graphs
using GraphPad Prism 8 (Dotmatics). Data are presented as the mean
± standard deviation. The statistical significance of differences
among three or more groups was determined by one-way ANOVA,
followed by Tukey's multiple comparisons test for post hoc
analysis. P<0.05 was considered to indicate a statistically
significant difference.
Results
TPL treatment induces accumulation of
GPx4
It has been reported that TPL inhibits the
proliferation of HCC cells in a concentration-dependent manner
(17). Consistent results were
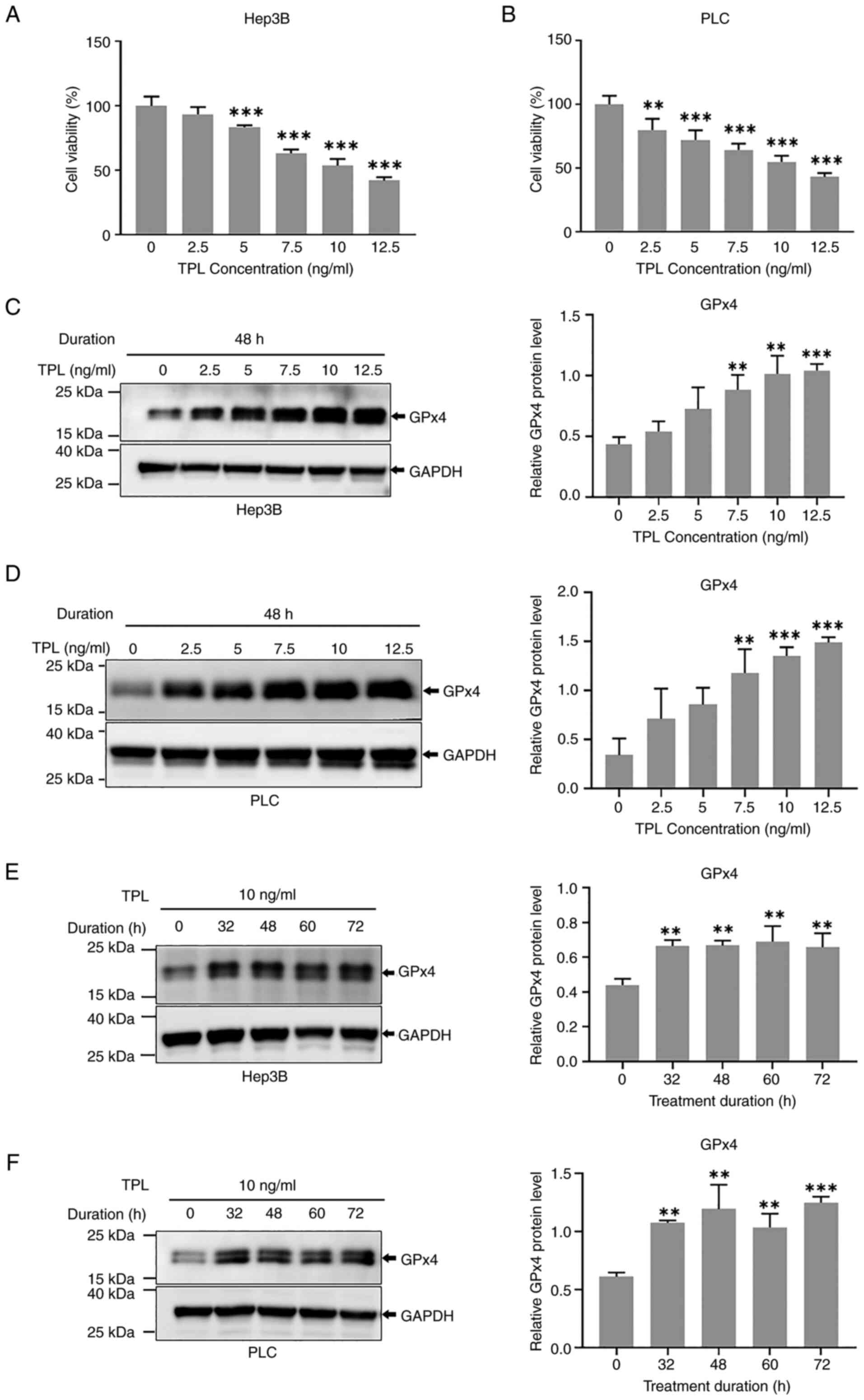
obtained in the present study. TPL significantly inhibited both
Hep3B and PLC cell viability in a concentration-dependent manner
(Fig. 1A and B). Furthermore, the
protein expression levels of GPx4 were elevated in Hep3B and PLC
cells 48 h after treatment with ≥7.5 ng/ml TPL (Fig. 1C and D). Furthermore, upregulation
of GPx4 was detected in cells 32, 48, 60 and 72 h after TPL (10
ng/ml) treatment (Fig. 1E and F).
GPx4 levels were also evidently increased in Huh7 cells after 48 h
of 10 or 15 ng/ml TPL treatment (Fig.
S1).
Co-treatment with TPL and RSL3
promotes the inhibition of cell viability
As an antioxidant enzyme, GPx4 protects cells from
oxidative stress-induced death (12); however, RSL3 can decrease the
activity of GPx4 (16). Therefore,
TPL in combination with RSL3 was used with the aim of improving the
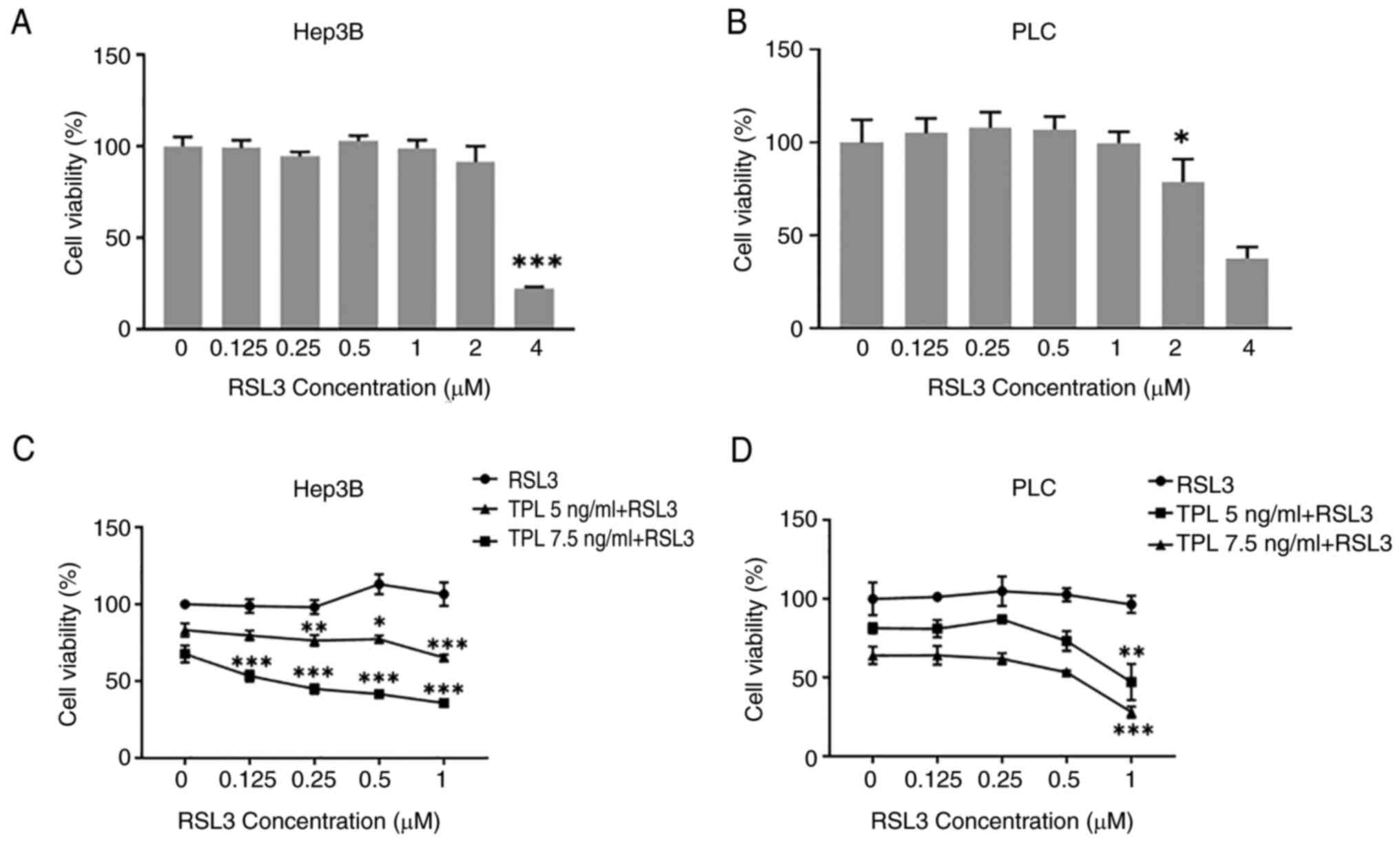
efficiency of TPL in inducing cell death. As shown in Fig. 2A and B, treatment of Hep3B or PLC
cells with RSL3 (≤1 µM) for 48 h did not result in reduced cell
viability. However, the inhibitory effect of TPL (7.5 ng/ml)
together with RSL3 (1 µM) on Hep3B or PLC cell viability was
greater than that produced by TPL (7.5 ng/ml) alone, and was
positively related to the concentration of RSL3 (Fig. 2C and D). In addition, the viability
of Huh7 cells was reduced by RSL3 (2 µM) (Fig. S2A), but not by TPL (≤10 ng/ml)
(Fig. S2B) after 24 h of
treatment duration. Furthermore, Huh7 cell viability was also
significantly reduced by co-treatment with TPL (10 ng/ml) and RSL3
(0.5 or 1 µM) for 24 or 48 h (Fig.
S2C). However, subsequent experiments used Hep3B and PLC cells,
and not Huh7 cells, considering that Huh7 cells were sensitive to
RSL3-induced cytotoxicity and might not necessitate co-treatment
with TPL and RSL3.
Additionally, cell indexes at the 80-h timepoint
monitored by RTCA indicated that TPL (7.5 ng/ml) together with RSL3
(1 µM) inhibited the proliferation of Hep3B and PLC cells more
effectively than TPL (12.5 ng/ml) alone (Table I). The mechanism underlying the
reduction in cell viability induced by the combination of TPL and
RSL3 was subsequently elucidated through analysis of Hep3B and PLC
cell death.
 | Table I.Effects of TPL and RSL3 on cell
proliferation. |
Table I.
Effects of TPL and RSL3 on cell
proliferation.
|
| Cell index at 80
h |
|---|
|
|
|
|---|
| Treatment | Hep3B cells | PLC cells |
|---|
| Control | 3.59±0.04 | 2.16±0.14 |
| RSL3 (1 µM) | 3.34±0.04 | 2.03±0.14 |
| TPL (7.5
ng/ml) | 2.17±0.00 | 2.88±0.02 |
| TPL (12.5
ng/ml) | 2.29±0.00 | 2.31±0.02 |
| TPL (7.5 ng/ml) +
RSL3 (1 µM) | 1.69±0.01 | 1.78±0.02 |
| TPL (12.5 ng/ml) +
RSL3 (1 µM) | 0.22±0.00 | 0.83±0.01 |
TPL combined with RSL3 increases
apoptosis
Since TPL induces mitochondrial pathway-mediated
apoptosis (18) and mitochondrial
GPx4 suppresses apoptosis mediated by the mitochondrial death
pathway (14), the present study
examined whether representative hallmarks of classic apoptosis were
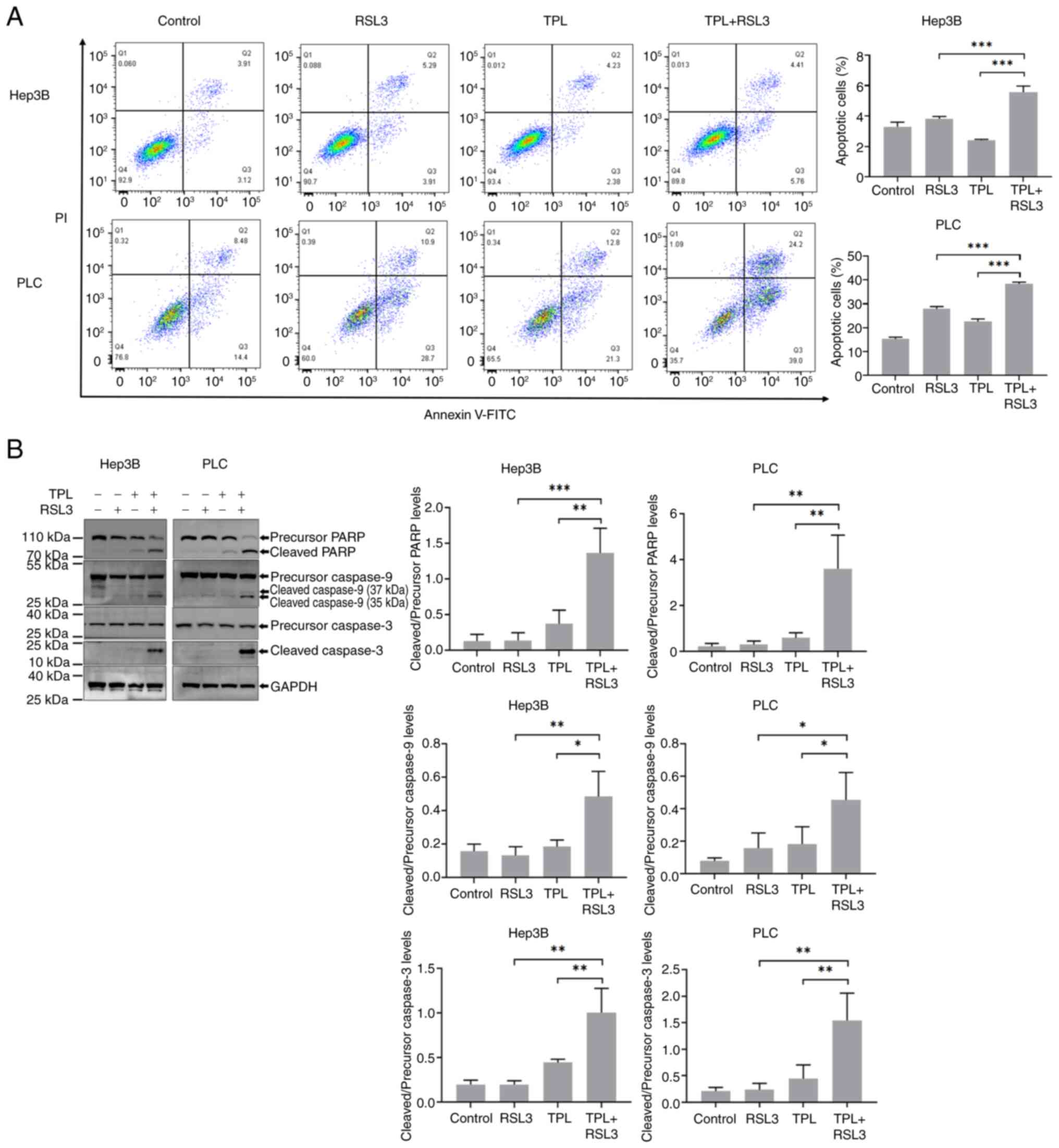
at higher levels in TPL and RSL3 co-treated Hep3B and PLC cells. As
shown in Fig. 3A, the proportion
of Annexin V+/PI− Hep3B or PLC cells was
higher in the group co-treated with TPL and RSL3 than that in the
group treated with TPL or RSL3 alone after 24 h. Furthermore, the
expression levels of cleaved PARP, cleaved caspase-9 and cleaved
caspase-3 were increased in Hep3B and PLC cells co-treated with TPL
and RSL3 compared with those treated with TPL or RSL3 alone
(Fig. 3B). Thus, an increase in
apoptosis was confirmed in HCC cells treated with TPL combined with
RSL3.
Co-treatment with TPL and RSL3
promotes the production of soluble ROS
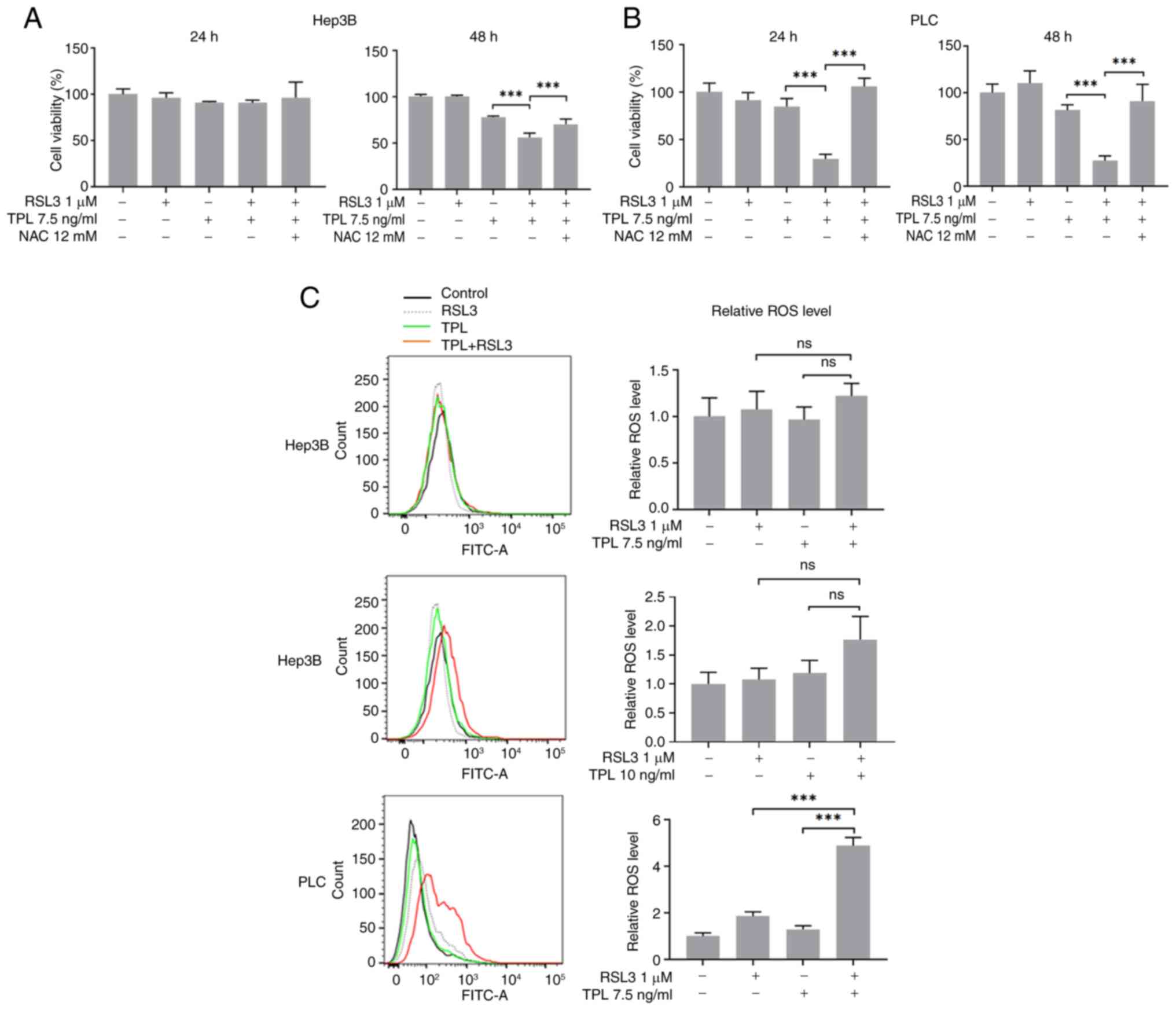
A comparison analysis of CCK-8 assay results
revealed that NAC inhibited the reduction in Hep3B and PLC cell
viability caused by TPL combined with RSL3, although this was not
significant in Hep3B cells at 24 h (Fig. 4A and B). To verify whether the
amount of soluble ROS was increased in TPL and RSL3 co-treated
cells, soluble ROS levels were measured by flow cytometry. As shown
in Fig. 4C, in the TPL (7.5 ng/ml)
and RSL3 (1 µM) co-treated PLC cells, though not the TPL (7.5 and
10 ng/ml) and RSL3 (1 µM) co-treated Hep3B cells, a significant
increase in soluble ROS levels was detected 24 h after
treatment.
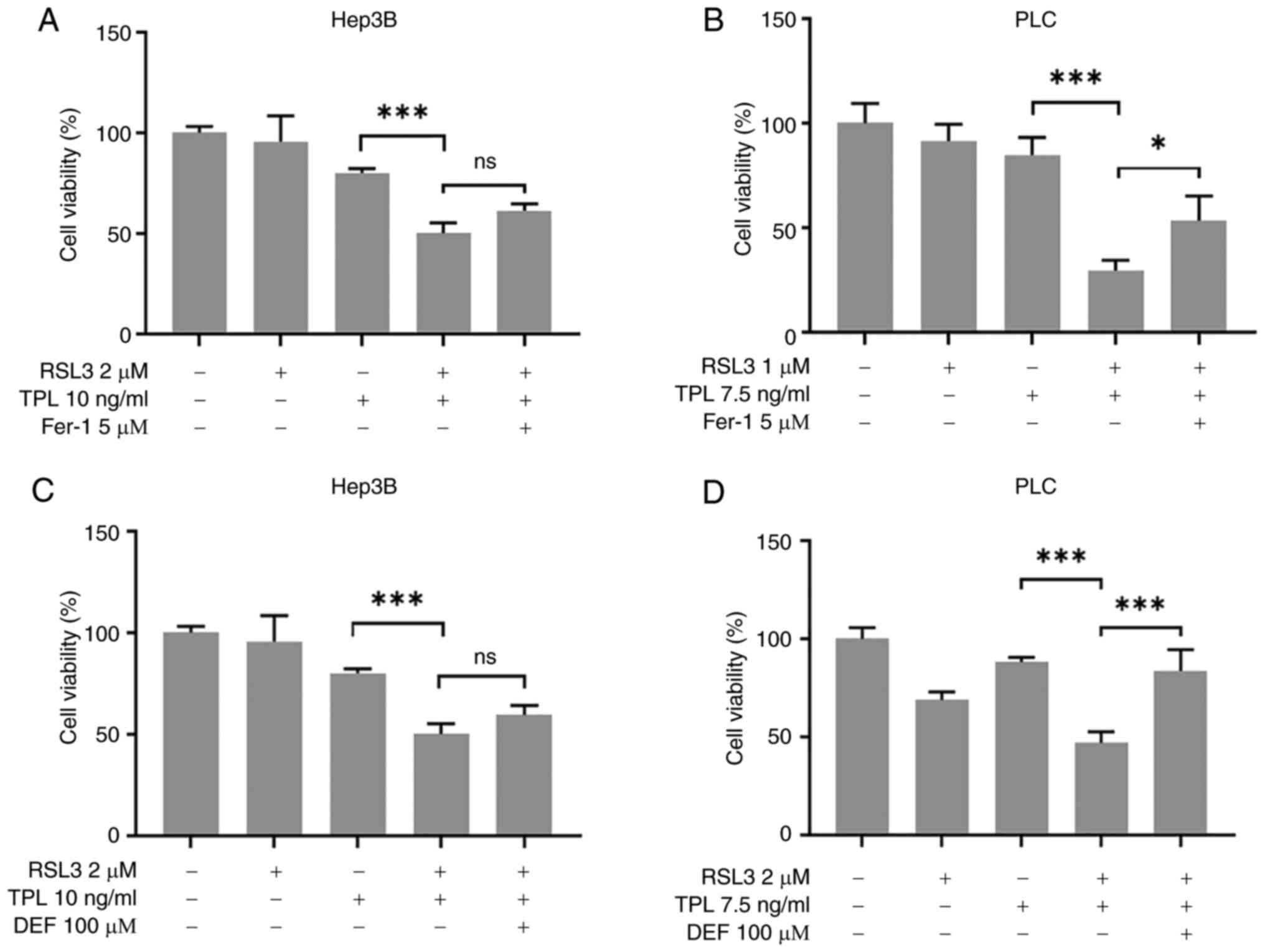
Co-treatment with TPL and RSL3 induces
ferroptosis
It has been reported that non-mitochondrial GPx4
inhibition induces ferroptosis (13). The present study investigated
whether ferroptosis of Hep3B and PLC cells was induced following
treatment with TPL in the presence of RSL3. Fer-1 was added to the
cell culture medium and cell viability was assessed 24 h after
co-treatment with TPL and RSL3. The results demonstrated that PLC
cell viability was enhanced by Fer-1, compared with the cell
viability in the TPL and RSL3 co-treatment group (Fig. 5B). In addition, the inhibition of
cell viability induced in PLC cells by TPL + RSL3 was reduced in
the presence of DEF when compared with that in TPL + RSL3 group
(Fig. 5D). In comparison to the
inhibition of Hep3B cell viability reduction by Fer-1 (P>0.05)
or DEF (P>0.05), the suppression of PLC cell viability reduction
by Fer-1 (P<0.05) or DEF (P<0.001) was significant.
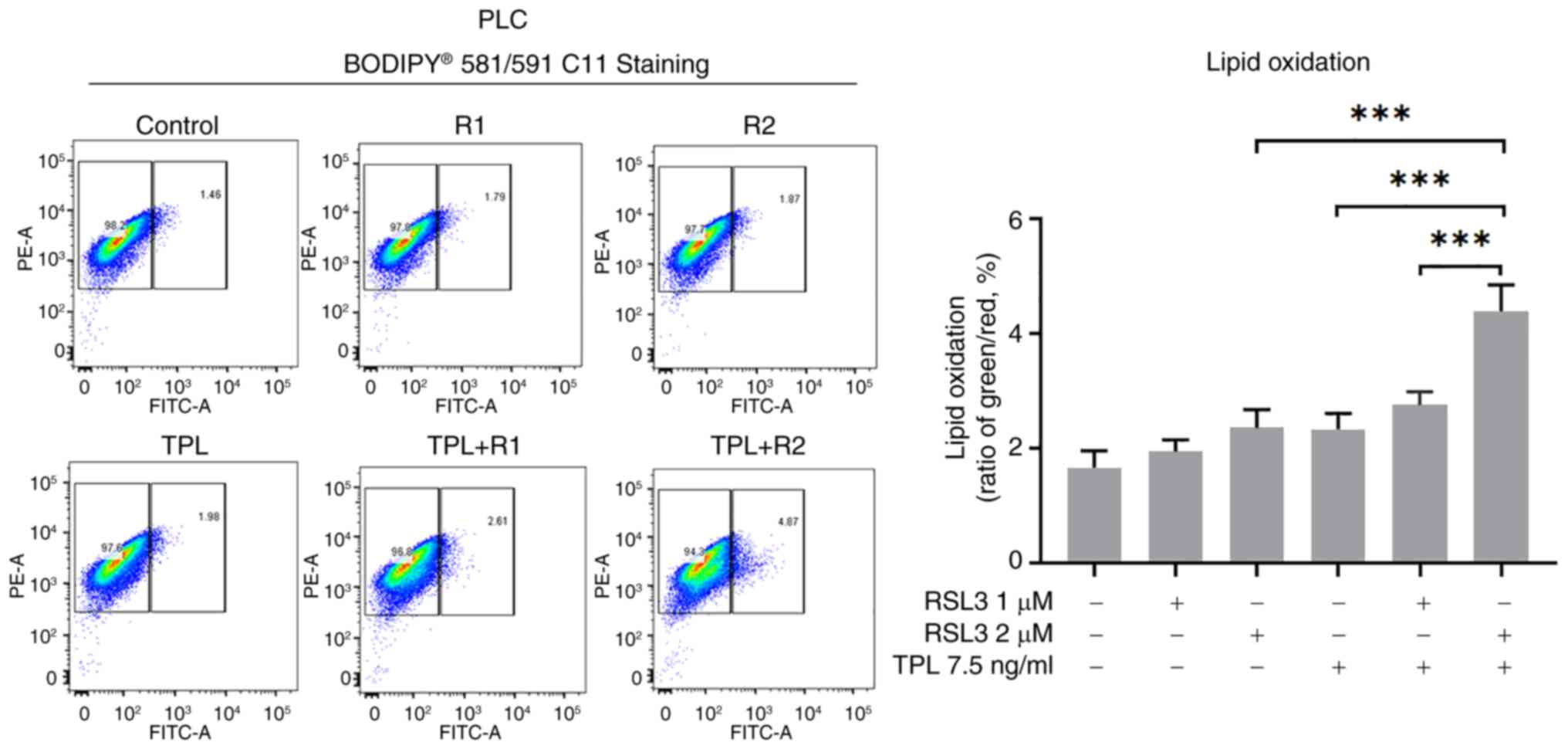
Therefore, PLC cells were selected to undergo lipid peroxidation
level evaluation after co-treatment with TPL and RSL3. Consistent
with our prediction, lipid peroxidation levels were significantly
increased in PLC cells after co-treatment with TPL (7.5 ng/ml) and
RSL3 (2 µM) for 24 h, compared with that in only TPL (7.5 ng/ml) or
RSL3 (2 µM) group or TPL (7.5 ng/ml) and RSL3 (1 µM) co-treatment
group (Fig. 6). These results
suggested that ferroptosis was induced by the combination of TPL
and RSL3 in HCC cells.
Discussion
Current therapeutic strategies for advanced HCC,
encompassing both surgical interventions and non-surgical
approaches, remain suboptimal regarding clinical efficacy.
Sorafenib, a multi-kinase inhibitor, is the first-line systemic
therapy for unresectable HCC, which received approval from the U.S.
Food and Drug Administration in 2007, and demonstrates survival
benefits in this patient population (19,20).
However, the clinical use of sorafenib is constrained by its modest
survival extension and the critical challenge of treatment
resistance (21). Emerging
evidence has indicated that both intrinsic and acquired resistance
mechanisms can undermine its therapeutic potential, necessitating
the urgent development of novel therapeutic approaches.
T. wilfordii Hook F (thunder god vine) is a
traditional medicinal plant with a long clinical history in the
treatment of autoimmune diseases. In recent years, the antitumor
effects of its active compound TPL have emerged as a new research
focus for this traditional therapy, demonstrating broad-spectrum
anticancer properties that make it a highly promising candidate as
a novel anticancer drug (22–25).
Notably, TPL demonstrates regulatory effects on lipid metabolism, a
mechanism that may antagonize HCC progression by altering cancer
cell metabolic dependencies (26,27).
However, the clinical application of TPL is limited by its narrow
therapeutic window and cumulative toxicity during prolonged use.
Researchers have explored various strategies for reducing toxicity
while enhancing efficacy, among which drug combination therapy has
shown particularly promising results (28). The present study investigated the
combination of TPL with RSL3, a compound known for its
ferroptosis-inducing activity, to observe their combined effects on
human HCC cell lines. The results revealed a cooperative effect
between the two agents, allowing dose reduction of both drugs while
maintaining inhibitory effects on cell viability, thereby
potentially mitigating their adverse effects.
Notably, the current study observed an elevation of
GPx4 protein levels in TPL-treated HCC cells. This finding
contrasts with the results of a recent report demonstrating
TPL-mediated GPx4 suppression in leukemia models, wherein GPx4
downregulation sensitized malignant cells to doxorubicin (29). The observed discrepancy suggests a
cell lineage-dependent dichotomy in the regulatory effects of TPL
on GPx4 expression, potentially reflecting tissue-specific redox
adaptation mechanisms across malignancies of distinct origins. This
tissue-specific regulation may originate from differential basal
Nrf2 activity between epithelial-derived HCC and hematopoietic
malignancies, as GPx4 expression is known to be transcriptionally
controlled by the Nrf2-Keap1-ARE pathway (30,31).
These findings highlight the necessity of context-specific
therapeutic strategies when targeting ferroptosis pathways in
different cancer types.
GPx4 exerts cytoprotective effects against oxidative
stress-mediated cell death. Based on this premise, it was
hypothesized that suppressing GPx4 upregulation with RSL3 in
combination with TPL would enhance cell death. As expected,
co-treatment with RSL3 (1 µM) and TPL (7.5 ng/ml) significantly
reduced HCC cell viability. However, RSL3 monotherapy (1 µM) for 48
h exhibited negligible cytotoxicity in both Hep3B and PLC cell
lines, a phenomenon consistent with the findings of a previous
study, which demonstrated that GPx4 ablation alone fails to induce
ferroptosis in glioblastoma models (32). This evidence suggested that GPx4
inhibition may require concomitant disruption of compensatory
antioxidant pathways to achieve therapeutic efficacy.
Co-treatment with TPL (7.5 ng/ml) and RSL3 (1 µM)
induced apoptosis in Hep3B and PLC cells in the present study.
Notably, the percentage of apoptotic cells in all groups was low in
Hep3B cells, with the maximum % being <10%. It is possible that
low treatment dose (7.5 ng/ml TPL and 1 µM RSL3) and short duration
(24 h) could not induce a high proportion of apoptosis in Hep3B
cells. In addition, Hep3B cells lack p53. They may rely on
alternative death mechanisms (e.g. necrosis), with the exception of
canonical apoptosis pathways. Soluble ROS levels were not markedly
increased in Hep3B cells after co-treatment with TPL (7.5 and 10
ng/ml) and RSL3 (1 µM) for 24 h, which may be reversed by
prolonging treatment duration, for NAC significantly increased
Hep3B cell viability after co-treatment with TPL (7.5 ng/ml) and
RSL3 (1 µM) for 48 h. Fer-1 is a synthetic antioxidant and prevents
damage to membrane lipids by a reductive mechanism and thereby
inhibits ferroptosis (33). Ample
iron ions engage in producing ROS by oxidizing lipid and thus
induce ferroptosis (34,35). DEF is a potent free iron chelating
agent and has antioxidant activity (36). In the present study, the reduction
in PLC but not Hep3B cell viability resulting from co-treatment
with TPL and RSL3 was inhibited by both Fer-1 and DEF, suggesting
that co-treatment with TPL and RSL3 at 1 or 2 µM induced
ferroptosis in PLC rather than Hep3B cells. This result might be
associated with the findings that RSL3 (2 µM) induced PLC (Fig. 2B) instead of Hep3B (Fig. 2A) cell death after 48 h of
treatment duration. Both results indicated that 2 µM of RSL3 might
be not high enough to induce Hep3B cell ferroptosis. Hence,
co-treatment with TPL and increased dose of RSL3 might induce Hep3B
cell ferroptosis.
In conclusion, building on the observation that TPL
upregulated GPx4 levels in HCC cells, the current study co-treated
HCC cells with TPL and RSL3 in vitro. The findings
demonstrated that these agents cooperated in inducing the apoptosis
and ferroptosis of HCC cells, thereby providing novel mechanistic
insights into TPL-mediated anti-HCC effects.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Dr Xue Liang (The
First Affiliated Hospital, Zhejiang University School of Medicine)
for maintaining cell lines.
Funding
This work was supported by the Independent Task of State Key
Laboratory for Diagnosis and Treatment of Infectious Diseases
(grant no. zz202224).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
WXL and ZC confirm the authenticity of all the raw
data. WXL designed and performed most of the experiments, analyzed
the data and wrote the manuscript. GDW and JW performed the flow
cytometry experiments. SSW participated in interpreting the flow
cytometry data. ZC contributed to the conception and secured
funding. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ganesan P and Kulik LM: Hepatocellular
carcinoma: New developments. Clin Liver Dis. 27:85–102. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Alawyia B and Constantina Constantinou:
Hepatocellular carcinoma: A narrative review on current knowledge
and future prospects. Curr Treat Options Oncol. 24:711–724. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu J, Shen M, Yue Z, Yang Z, Wang M, Li
C, Xin C, Wang Y, Mei Q and Wang Z: Triptolide inhibits
colon-rectal cancer cells proliferation by induction of G1 phase
arrest through upregulation of p21. Phytomedicine. 19:756–762.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Oliveira A, Beyer G, Chugh R, Skube SJ,
Majumder K, Banerjee S, Sangwan V, Li L, Dawra R, Subramanian S, et
al: Triptolide abrogates growth of colon cancer and induces cell
cycle arrest by inhibiting transcriptional activation of E2F. Lab
Invest. 95:648–659. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wei YS and Adachi I: Inhibitory effect of
triptolide on colony formation of breast and stomach cancer cell
lines. Zhongguo Yao Li Xue Bao. 12:406–410. 1991.PubMed/NCBI
|
|
6
|
Wang H, Ma D, Wang C, Zhao S and Liu C:
Triptolide inhibits invasion and tumorigenesis of hepatocellular
carcinoma MHCC-97H cells through NF-κB signaling. Med Sci Monit.
22:1827–1836. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li SG, Shi QW, Yuan LY, Qin LP, Wang Y,
Miao YQ, Chen Z, Ling CQ and Qin WX: C-Myc-dependent repression of
two oncogenic miRNA clusters contributes to triptolide-induced cell
death in hepatocellular carcinoma cells. J Exp Clin Cancer Res.
37:512018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yu L, Qian J, Xue X, Pang M, Wang X, Li X,
Tian M, Lu C, Xiao C and Liu Y: Application of galactosylated
albumin for targeted delivery of triptolide to suppress
hepatocellular carcinoma progression through inhibiting de novo
lipogenesis. Biomed Pharmacother. 179:1174322024. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Alsaied OA, Sangwan V, Banerjee S, Krosch
TC, Chugh R, Saluja A, Vickers SM and Jensen EH: Sorafenib and
triptolide as combination therapy for hepatocellular carcinoma.
Surgery. 156:270–279. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li Z, Yang G, Han L, Wang R, Gong C and
Yuan Y: Sorafenib and triptolide loaded cancer cell-platelet hybrid
membrane-camouflaged liquid crystalline lipid nanoparticles for the
treatment of hepatocellular carcinoma. J Nanobiotechnology.
19:3602021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xi C, Peng S, Wu Z, Zhou Q and Zhou J:
Toxicity of triptolide and the molecular mechanisms involved.
Biomed Pharmacother. 90:531–541. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Imai H and Nakagawa Y: Biological
significance of phospholipid hydroperoxide glutathione peroxidase
(PHGPx, GPx4) in mammalian cells. Free Radic Biol Med. 34:145–169.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang WS, SriRamaratnam R, Welsch ME,
Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji
AF, Clish CB, et al: Regulation of ferroptotic cancer cell death by
GPX4. Cell. 156:317–331. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nomura K, Imai H, Koumura T, Arai M and
Nakagawa Y: Mitochondrial phospholipid hydroperoxide glutathione
peroxidase suppresses apoptosis mediated by a mitochondrial death
pathway. J Biol Chem. 274:29294–29302. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nomura K, Imai H, Koumura T, Kobayashi T
and Nakagawa Y: Mitochondrial phospholipid hydroperoxide
glutathione peroxidase inhibits the release of cytochrome c from
mitochondria by suppressing the peroxidation of cardiolipin in
hypoglycaemia-induced apoptosis. Biochem J. 351:183–193. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang WS and Stockwell BR: Synthetic lethal
screening identifies compounds activating iron-dependent,
nonapoptotic cell death in oncogenic-RAS-harboring cancer cells.
Chem Biol. 15:234–245. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu W, Yang Y, Wang J, Wu S and Chen Z:
Triptolide-mediated downregulation of FLIP(S) in hepatoma cells
occurs at the post-transcriptional level independently of
proteasome-mediated pathways. Med Oncol. 40:72022. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yao J, Jiang Z, Duan W, Huang J, Zhang L,
Hu L, He L, Li F, Xiao Y, Shu B and Liu C: Involvement of
mitochondrial pathway in triptolide-induced cytotoxicity in human
normal liver L-02 cells. Biol Pharm Bull. 31:592–597. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Abou-Alfa GK, Schwartz L, Ricci S, Amadori
D, Santoro A, Figer A, De Greve J, Douillard JY, Lathia C, Schwartz
B, et al: Phase II study of sorafenib in patients with advanced
hepatocellular carcinoma. J Clin Oncol. 24:4293–4300. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Llovet JM, Ricci S, Mazzaferro V, Hilgard
P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A,
et al: Sorafenib in advanced hepatocellular carcinoma. N Engl J
Med. 359:378–390. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhai B and Sun XY: Mechanisms of
resistance to sorafenib and the corresponding strategies in
hepatocellular carcinoma. World J Hepatol. 5:345–352. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu Y, Xiao E, Yuan L and Li G: Triptolide
synergistically enhances antitumor activity of oxaliplatin in colon
carcinoma in vitro and in vivo. DNA Cell Biol. 33:418–425. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nakazato T, Sagawa M and Kizaki M:
Triptolide induces apoptotic cell death of multiple myeloma cells
via transcriptional repression of Mcl-1. Int J Oncol. 44:1131–1138.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chugh R, Sangwan V, Patil SP, Dudeja V,
Dawra RK, Banerjee S, Schumacher RJ, Blazar BR, Georg GI, Vickers
SM and Saluja AK: A preclinical evaluation of Minnelide as a
therapeutic agent against pancreatic cancer. Sci Transl Med.
4:156ra1392012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li J, Liu R, Yang Y, Huang Y, Li X, Liu R
and Shen X: Triptolide-induced in vitro and in vivo cytotoxicity in
human breast cancer stem cells and primary breast cancer cells.
Oncol Rep. 31:2181–2186. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang X, Xu M, Peng Y, Naren Q, Xu Y, Wang
X, Yang G, Shi X and Li X: Triptolide enhances lipolysis of
adipocytes by enhancing ATGL transcription via upregulation of p53.
Phytother Res. 34:3298–3310. 2020. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huang R, Guo F, Li Y, Liang Y, Li G, Fu P
and Ma L: Activation of AMPK by triptolide alleviates nonalcoholic
fatty liver disease by improving hepatic lipid metabolism,
inflammation and fibrosis. Phytomedicine. 92:1537392021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu Y, Chen F, Wang S, Guo X, Shi P, Wang
W and Xu B: Low-dose triptolide in combination with idarubicin
induces apoptosis in AML leukemic stem-like KG1a cell line by
modulation of the intrinsic and extrinsic factors. Cell Death Dis.
4:e9482013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wu X, Chen S, Huang K and Lin G:
Triptolide promotes ferroptosis by suppressing Nrf2 to overcome
leukemia cell resistance to doxorubicin. Mol Med Rep. 27:172023.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Deng Y, Chu X, Li Q, Zhu G, Hu J, Sun J,
Zeng H, Huang J and Ge G: Xanthohumol ameliorates drug-induced
hepatic ferroptosis via activating Nrf2/xCT/GPX4 signaling pathway.
Phytomedicine. 126:1554582024. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Luo L, Wang J, Zhao J, Yang B, Ma W and
Lin J: Dental pulp stem cells derived exosomes inhibit ferroptosis
via regulating the Nrf2-keap1/GPX4 signaling pathway to ameliorate
chronic kidney disease injury. Tissue Cell. 93:1026702025.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li S, He Y, Chen K, Sun J, Zhang L, He Y,
Yu H and Li Q: RSL3 drives ferroptosis through NF-κB pathway
activation and GPX4 depletion in glioblastoma. Oxid Med Cell
Longev. 2021:29150192021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Skouta R, Dixon SJ, Wang J, Dunn DE, Orman
M, Shimada K, Rosenberg PA, Lo DC, Weinberg JM, Linkermann A and
Stockwell BR: Ferrostatins inhibit oxidative lipid damage and cell
death in diverse disease models. J Am Chem Soc. 136:4551–4556.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fang X, Ardehali H, Min J and Wang F: The
molecular and metabolic landscape of iron and ferroptosis in
cardiovascular disease. Nat Rev Cardiol. 20:7–23. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shalev O, Repka T, Goldfarb A, Grinberg L,
Abrahamov A, Olivieri NF, Rachmilewitz EA and Hebbel RP:
Deferiprone (L1) chelates pathologic iron deposits from membranes
of intact thalassemic and sickle red blood cells both in vitro and
in vivo. Blood. 86:2008–2013. 1995. View Article : Google Scholar : PubMed/NCBI
|