Introduction
Disorders of sex development (DSDs) represent a
broad and heterogeneous group of congenital conditions,
characterized by discordance between chromosomal, gonadal and
anatomical sex (1). The clinical
presentation of DSDs is highly variable, encompassing numerous
conditions, such as hypospadias, ambiguous genitalia and complete
sex reversal in 46, XX or 46, XY individuals (2). In 2006, the Chicago Consensus
redefined DSD classifications into three major categories based on
karyotype; namely, sex chromosome DSDs, 46, XY DSDs and 46, XX DSDs
(3). Among these, 46, XY DSDs
exhibit the greatest level of complexity, often involving atypical
female genitalia, incomplete intrauterine masculinization and the
absence of Müllerian structures (4,5).
Androgen receptor dysfunction remains the most prevalent etiology
in these cases. The psychological, physical and reproductive
consequences of DSDs are profound, with patients facing an elevated
risk of sex cord-stromal tumors, such as gonadoblastoma and
experiencing considerable social and medical burdens (6).
The clinical heterogeneity of DSDs complicates the
accuracy of diagnosis based solely on phenotypic assessments
(6). Genetic factors underlying
DSD pathogenesis remain to be elucidated, necessitating molecular
diagnostics to complement clinical evaluations. The
mitogen-activated protein 3 kinase 1 (MAP3K1) gene plays a crucial
role in the genetic network associated with gonadal development
(7). MAP3K1 mediates sex
differentiation through modulating the balance between the
pro-testicular SOX9/FGF9 pathway and the pro-ovarian WNT/β-catenin
pathway (4,8,9).
Variants in MAP3K1 have been identified in large families
with 46, XY DSD, exhibiting an autosomal dominant, sex-limited mode
of inheritance (7). In addition,
MAP3K1 variants have been detected in 18% of sporadic cases
of 46, XY gonadal dysgenesis, with phenotypic manifestations
ranging from complete gonadal dysgenesis to milder presentations,
such as hypospadias, micropenis and cryptorchidism (4,5).
Results of a previous study demonstrated that gain-of-function
variants in MAP3K1 enhance the phosphorylation of downstream
targets, leading to reduced SOX9 expression levels and
increased β-catenin levels (7).
The shift in signaling pathways results in the disruption of normal
testicular development, leading to various degrees of gonadal
dysgenesis.
Gain-of-function variants in MAP3K1 include
p.L189P, p.L189R and p.K246E and splice-site variants include
c.634-8T>A and c.2180-2A>G. Notably, the aforementioned
variants may enhance WNT/β-catenin signaling and suppress the
testis-promoting pathway driven by SOX9 (7,8,10).
These variants also increase phosphorylation events that skew the
signaling cascade towards ovarian development in 46, XY
individuals, leading to gonadal dysgenesis with partial or complete
female characteristics. In addition, the existence of novel
variants, such as c.3020A>G and c.2117T>G, highlight the role
of MAP3K1 variants in human sex differentiation (5,11).
Results of previous studies reveal that
Map3k1 null mutant mouse embryonic stem (ES) cells exhibit
increased apoptosis under hyperosmotic stress, low-temperature
shock and microtubule disruption (12,13).
Moreover, cardiac myocytes derived from Map3k1-mutant ES
cells display heightened susceptibility to oxidative stress-induced
apoptosis (14), underscoring the
critical role of Map3k1 in protecting mammalian cells from
cell death. However, whether MAP3K1 variants contribute to
apoptosis in patients with DSD remains unclear. Previous studies
have reported increased DNA damage in individuals with DSD
(15) and elevated DNA damage and
chronic inflammation in male patients with idiopathic germ cell
aplasia (16). Collectively, these
results highlight the potential association between DNA damage and
gonadal disorders. Notably, excessive DNA damage triggers apoptosis
in various cell types, including germ cells (17). Thus, the present study hypothesized
that increased DNA damage leads to abnormal apoptosis, ultimately
contributing to the clinical phenotype observed in patients with
DSD.
In the present study, a 3-year-old male presenting
with an abnormal urethral opening was found to have a recurved
penis with the urethral opening located at the penile-scrotal
junction. Family history suggested a potential familial pattern of
inheritance, and cytogenetic analysis revealed a 46, XY karyotype.
Genetic analysis was conducted to identify potential pathogenic
variants, followed by in vitro experiments to explore the
underlying regulatory mechanisms. These findings contribute to a
deeper understanding of the pathogenesis of 46, XY DSD.
Materials and methods
Patients
A family with 46, XY DSD was recruited from the
Prenatal Diagnosis Center of Guizhou Medical University in December
2020. A total of five family members were enrolled in this study,
including four males and one female: the proband, his brother,
father, mother and cousin. The affected individuals were between 3
and 5 years of age. Ethics approval was obtained from the Ethics
Committee of the Affiliated Hospital of Guizhou Medical University
(approval no. 2020-325) and all individuals provided written
informed consent.
Karyotype analysis and sex hormone
examination
Peripheral blood samples were collected from the
proband and the younger sibling and parents of the proband for
karyotype analysis and C-banding. Peripheral blood chromosomes were
analyzed as previously described (18). Briefly, venous blood anticoagulated
with sodium heparin was inoculated into a medium containing
phytohemagglutinin and incubated at 37°C for 66–72 h. Subsequently,
40 µg/ml colchicine was added to arrest cells at metaphase and
cells were incubated for 1 h at 37°C. Chromosome harvesting was
performed using an automated chromosome harvester. Cells were
resuspended and dispersed at 25°C with 50% humidity. In total, 1–2
drops of each sample were deposited onto a slide and incubated at
80°C for 3 h.
For G-banding, slides were digested with 0.025%
trypsin at 37°C for 30 sec, rinsed twice in saline, stained with
Giemsa at 37°C for 5 min and rinsed with water. Images of
chromosomes were obtained using the GSL-120 automated chromosome
scanner and subsequently analyzed due to the clinical significance
of abnormal sexual development.
For C-banding, slides were treated with a 5%
Ba(OH)2 solution at 60°C for 10–20 min. Following
treatment, slides were rinsed and incubated in 2X SSC solution at
60°C for 90 min. For the observation of chromosomal heterochromatin
regions, slides were stained with Giemsa at 37°C for 50% less of
the Ba(OH)2 exposure time. An additional 5 ml of venous
blood was collected and serum was used for sex hormone profile
analysis.
Gene sequencing and bioinformatics
analysis
Genomic DNA was extracted from peripheral blood
samples obtained from the proband and the younger sibling and
parents of the proband using the Qiagen DNA Blood Mini kit (cat.
no. 51104; Qiagen GmbH) following the manufacturer's protocol. The
genomic DNA of the proband underwent whole exome sequencing (WES)
and copy number variation sequencing (CNV-Seq), following the
manufacturer's protocols (Tiangen Biotech Co., Ltd.) (19). Sequencing was performed using the
Illumina NextSeq 2000 platform (Illumina, Inc.; Berry Genomics Co
Ltd. with the reference genome, GRCh37/hg19. Variant filtering
utilized multiple databases, including the 1,000 Genomes Project
(https://www.internationalgenome.org/), GnomAD
(https://gnomad.broadinstitute.org),
ESP 6500 (https://esp.gs.washington.edu/drupal/) and ExAC
(http://exac.broadinstitute.org/). Sanger
sequencing (Sangon Biotech Co., Ltd.; http://www.sangon.com/) was used to validate
identified variants in family members, including the proband, his
brother, father and mother. Interpretation and pathogenicity were
assessed following the American College of Medical Genetics and
Genomics (ACMG) guidelines (20).
For variants of uncertain significance, databases were used to
predict pathogenicity, conservation and protein structural effect
of missense variants (https://www.internationalgenome.org/; https:/gnomad broadinstitute.org/; http://evs.gs.washington.edu/EVS/; http://exac.broadinstitute.org/). Data analysis
was conducted using the Verita Trekker® variant
detection system and the Enliven® variant annotation and
interpretation system (Berry Genomics; http://www.berrygenomics.com/). The pathogenicity
analysis was conducted using PolyPhen (version 2, http://genetics.bwh.harvard.edu/pph2/index.shtml).
The genetic conservation of the variant site was assessed using the
MEGA tool (version 7.0.14, http://www.megasoftware.net/). SWISS-MODEL (https://swissmodel.expasy.org/) was used to
predict potential structural changes in the protein due to the
amino acid substitution and PyMOL software (version 3.1, http://www.pymol.org/) was used for visualization of
the alterations. The 1,000 Genomes Project database (https://www.internationalgenome.org/)
was used to assess the novelty of the variant.
Cell culturing
293T cells were obtained from The Cell Bank of Type
Culture Collection of The Chinese Academy of Sciences and cultured
in high-glucose Dulbecco's Modified Eagle's Medium (DMEM; cat. no.
PM150210; Pricella; Elabscience Bionovation Inc.) with 10% fetal
bovine serum (FBS; cat. no. 164210-50; Pricella; Elabscience
Bionovation Inc.) at 37°C in a 5% CO2 incubator
(21).
Construction of the heterozygous
variant cell line using CRISPR/Cas9
To investigate the pathogenicity of the variant, a
heterozygous variant cell line with MAP3K1 gene c.4445G>A
was established in 293T cells through electroporation (22). The site of guide RNA (gRNA) was
designed using CRISPOR (https://crispor.gi.ucsc.edu/). The target sequence,
5′-AGAGCCACATCTCGTAAACC-3′, is located within exon 20 of the
MAP3K1 gene. Genome editing at this site affects a region
corresponding to the protein kinase domain. Cas9 protein
(NLS-Cas9-NLS Nuclease, cat. no. Z03469, GenScript, Inc.,
http://www.genscript.com/) was incubated
with gRNA and subsequently co-electroporated into cells using
oligonucleotides for monoclonal culture. A point mutation,
c.4445G>A, was introduced, resulting in an amino acid
substitution from arginine to glutamine at position 1482
(p.Arg1482Gln) of the MAP3K1 protein, which may alter the function
of the kinase domain. Reverse transcription-quantitative (RT-q) PCR
and gel electrophoresis were used to confirm cell genotypes. The
MAP3K1 c.4445 G>A heterozygous variant cell line was obtained
from Cyagen Biosciences, Inc.
Cell counting kit (CCK)-8 assay
The CCK-8 assay (cat. no. HY-K0301; MedChemExpress)
was used to assess cell viability (23). Following culturing at 37°C for 48
h, cells were terminated with 2 ml of standard medium and
resuspended in 2 ml of medium without antibiotics. Cells were
incubated at 37°C for an additional 24 h and analyzed using an
inverted microscope. CCK-8 reagent was added to cells and incubated
for 2 h and the absorbance was measured at 450 nm using an enzyme
marker.
Flow cytometry
Following culturing at 37°C for 48 h, cells were
digested, resuspended and stained with 5 µl Annexin V-FITC and
propidium iodide (PI) in the dark at room temperature for 15 min.
Apoptosis was analyzed using a flow cytometer (Navios; Beckman
Coulter, Inc.) within 1 h. For cell cycle analysis, cells were
cultured at 37°C for 48 h and 1.5×106 cells were
digested and fixed with 75% ethanol at 4°C for 24 h. Subsequently,
cells were treated with 100 µl of RNase A solution and incubated at
37°C for 30 min. In total, 400 µl of PI dye was added and cells
were incubated for 30 min at 4°C in the dark. Samples were filtered
through a 400-mesh cell strainer and the cell cycle was assessed
via flow cytometry. FlowJo (version 10.10; FlowJo LLC) was used to
analyze the data.
Western blot analysis
Total protein was extracted from cells on ice using
a protein extraction kit (Beijing Solarbio Science and Technology
Co., Ltd.). Total protein was quantified using a bicinchoninic acid
assay and samples were separated by SDS-PAGE on a 10% gel (25
µg/lane). The separated proteins were transferred onto 0.45-µm
thick PVDF membranes via wet transfer. Membranes were blocked with
5% skimmed milk for 2 h at room temperature, followed by overnight
incubation at 4°C with primary antibodies, including anti-GAPDH
(1:5,000; cat. no. HRP-6004; Proteintech Group, Inc.), anti-Bax
(1:2,000; cat. no. 50599-2-Ig; Proteintech Group, Inc.), anti-Bcl-2
(1:2,000; cat. no. 12789-1-AP; Proteintech Group, Inc.),
anti-Caspase 3 (1:1,000; cat. no. WL02117; Wanleibio Group, Inc.),
anti-cleaved Caspase 3 (1:1,000; cat. no. WL01992; Wanleibio Co.,
Ltd.), anti-SOX9 (1:5,000; cat. no. 67439-1-Ig; Proteintech Group,
Inc.), anti-FOXL2 (1:5,000; cat. no. 84144-1-RR; Proteintech Group,
Inc.), anti-ERK (1:2,000; cat. no. 16443-1-AP; Proteintech Group,
Inc.), anti-phosphorylated (p)-ERK (1:2,000; cat. no. 28733-1-AP;
Proteintech Group, Inc.), anti-p38 (1:2,000; cat. no. 14064-1-AP;
Proteintech Group, Inc.) and anti-p-p38 (1:2,000; cat. no.
28796-1-AP; Proteintech Group, Inc.). Following primary incubation,
membranes were washed three times with TBS-0.1% Tween-20 and
incubated with HRP-conjugated goat anti-rabbit IgG (1:20,000; cat.
no. BS22357; Bioworld Technology, Inc.) or HRP-conjugated goat
anti-mouse IgG (1:20,000; cat. no. BS22356; Bioworld Technology,
Inc.) for 2 h at room temperature. Protein bands were visualized
using ECL (cat. no. WBKLS0050, MilliporeSigma). Images were
obtained using an exposure meter (Universal Hood II; Bio-Rad
Laboratories, Inc.) and protein expression was quantified using
ImageJ software (version 1.6.0; National Institutes of Health).
RT-qPCR
Following culturing for 48 h, total RNA was
extracted from cells (1×106 cells) using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.), according to the manufacturer's protocol. Total RNA was
reverse-transcribed into cDNA using the SYBR Fluorescence
Quantification kit (Takara Bio, Inc.), according to the
manufacturer's protocol. Thermocycling conditions: Pre-denaturation
at 95°C for 3 min, followed by 40 cycles of denaturation at 95°C
for 30 sec, annealing at 60°C for 35 sec and extension at 72°C for
10 sec. The expression levels were determined using the
2−ΔΔCq method (24).
The following primer pairs were used for qPCR: SOX9 forward,
5′-GAGGAAGTCGGTGAAGAACGG-3′ and reverse, 5′-CCCTCTCGCTTCAGGTCAG-3′;
FOXL2 forward, 5′-GAGAAGAGGCTCACGCTGTC-3′ and reverse,
5′-CTCGTTGAGGCTGAGGTTGT-3′; and GAPDH forward,
5′-GGTCTCCTCTGACTTCAACA-3′ and reverse, 5′-GTGAGGGTCTCTCTCTTCCT-3′.
All experiments were repeated at least three times.
Statistical analysis
Data are presented as the mean ± standard deviation
using at least three independent experiments. Analyses were
performed using SPSS statistical software (version 19.0; IBM
Corp.). Differences between groups were analyzed using independent
Student's t-tests (unpaired). P<0.05 was considered to indicate
a statistically significant difference.
Results
Clinical evaluation of a family with
the DSD phenotype
The proband investigated in the present study was a
3-year-old male who presented with abnormal urethral opening that
had been observed since birth. Physical examination revealed a
recurved penis with the urethral opening located at the
penile-scrotal junction. A review of the family history indicated
that the proband's brother presented with the same clinical
features and the son of a maternal aunt also exhibited comparable
manifestations of hypospadias, suggesting a potential familial
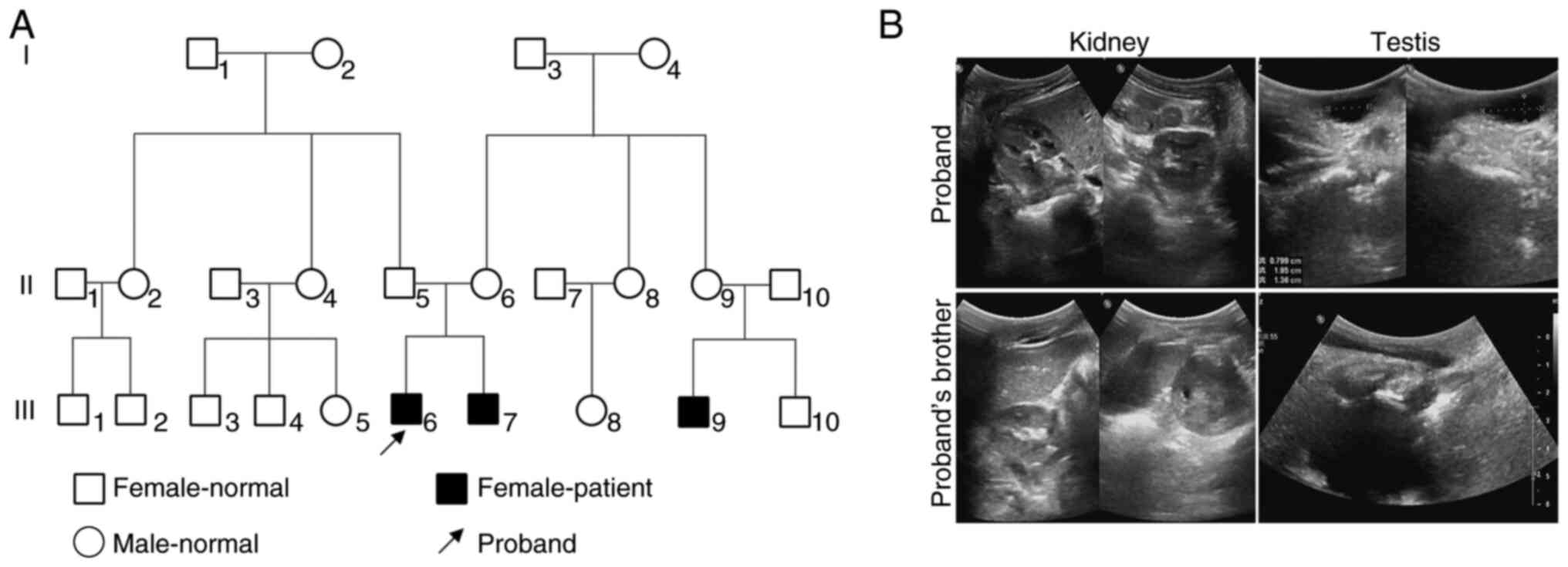
pattern. A pedigree chart illustrating the family lineage was
constructed based on the genetic data (Fig. 1A). Pelvic and scrotal ultrasound
examinations were conducted for both the proband and the brother of
the proband. Imaging revealed bilateral kidneys and testes in both
individuals, with no obvious solid masses detected posterior to the
bladder (Fig. 1B). The proband
underwent ultrasound examination and the results indicated right
testicular hydrocele with no evidence of female reproductive
structures, such as a uterus or ovaries (Fig. 1B). Notably, these results were
indicative of the DSD phenotype. To rule out hormonal causes of
this case, venous blood samples were collected from both the
proband and the brother of the proband for sex hormone analysis.
Results of the present study demonstrated that levels of
luteinizing hormone, estradiol and testosterone were decreased,
while levels of anti-Müllerian hormone and inhibin B remained
unaltered (Table I), indicating
that the phenotype may be not a result of hormones. Collectively,
these results demonstrated that the pathogenesis of the patient may
be attributed to a genetic variant.
 | Table I.Levels of sex hormones. |
Table I.
Levels of sex hormones.
| Hormone | Proband | Proband's
brother | Reference value
range |
|---|
| Luteinizing
hormone | 0.18 ↓ | 0.22 ↓ | 1.70–8.60 IU/l |
|
Follicle-stimulating hormone | 1.59 | 2.40 | 1.50–12.40
IU/l |
| Prolactin | 410.00 | 190.20 | 81.80–483.00
mlU/l |
| Estradiol (E2) | <18.35 ↓ | <18.35 ↓ | 41.40–159.00
pmol/l |
| Progesterone
(PRGE) | 0.25 | <0.16 | 0.00–0.47
nmol/l |
| Testosterone
(T) | <0.09 ↓ | <0.09 ↓ | 8.64–29.00
nmol/l |
|
Deoxycorticosterone | <80 | <80 | ≤350 pg/ml |
|
17-Hydroxyprogesterone | 239.7 | <100 | <1100 pg/ml |
|
11-Deoxycortisol | 310.0 | 209.7 | <3440 pg/ml |
| Cortisol |
0.83×105 |
0.96×105 |
(0.3–2.5)x105 pg/ml |
|
Dehydroepiandrosterone | 537.3 | <500 | <2300 pg/ml |
|
Androstenedione | <50.0 | <50.0 | <510 pg/ml |
| anti-Müllerian
hormone | >18 | >18 | 2.04–19.22
ng/ml |
| Inhibin B | 120.53 | 204.38 | 21–166 pg/ml |
Identification of 46, XY DSD
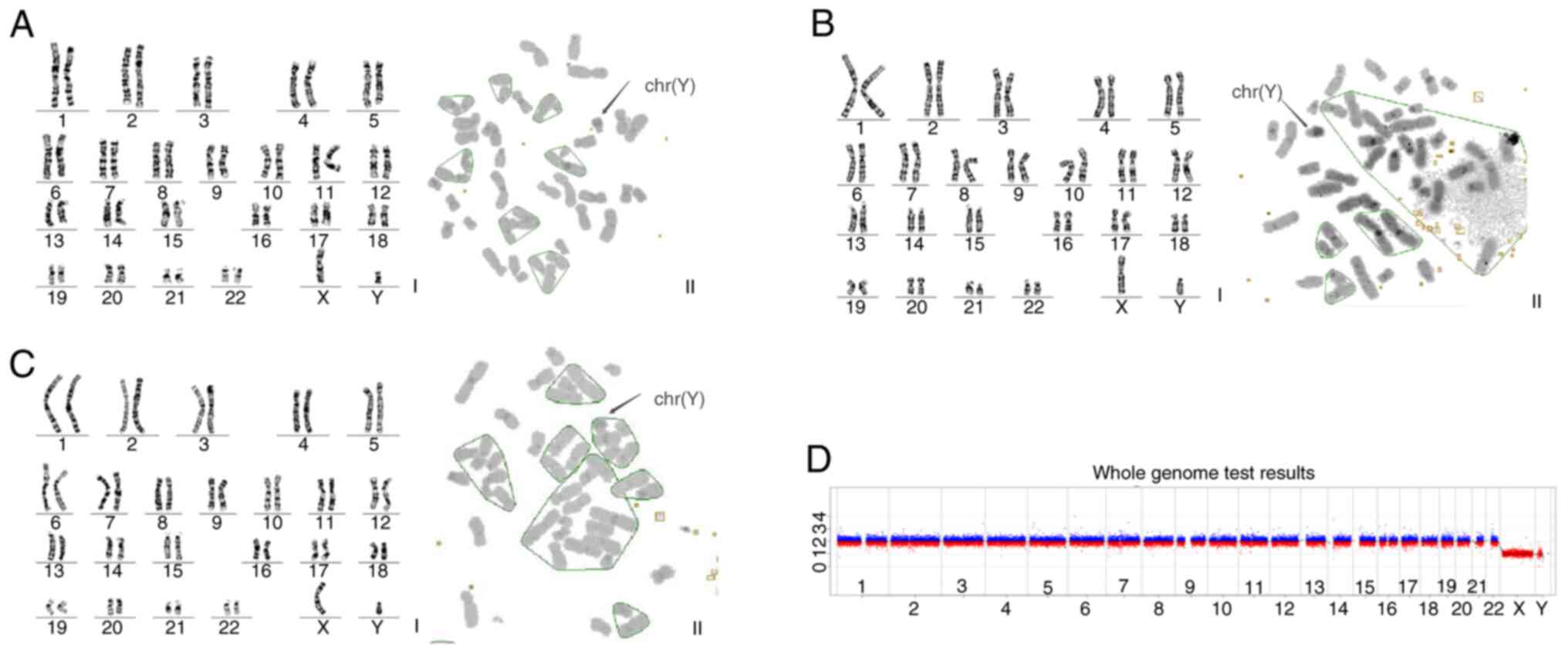
Karyotype analysis of peripheral blood obtained from
the proband and two other affected family members revealed a 46, XY
chromosome pattern. In addition, C-band analysis identified a
prominent heterochromatin region on the long arm of chromosome Y
(q12) in all three individuals (Fig.
2A-C). CNV-Seq of peripheral blood obtained from the proband
revealed no abnormalities (Fig.
2D). Collectively, these findings suggested that the disease
affecting this family was consistent with 46, XY DSD.
Identification and bioinformatics
analysis of the mutated gene
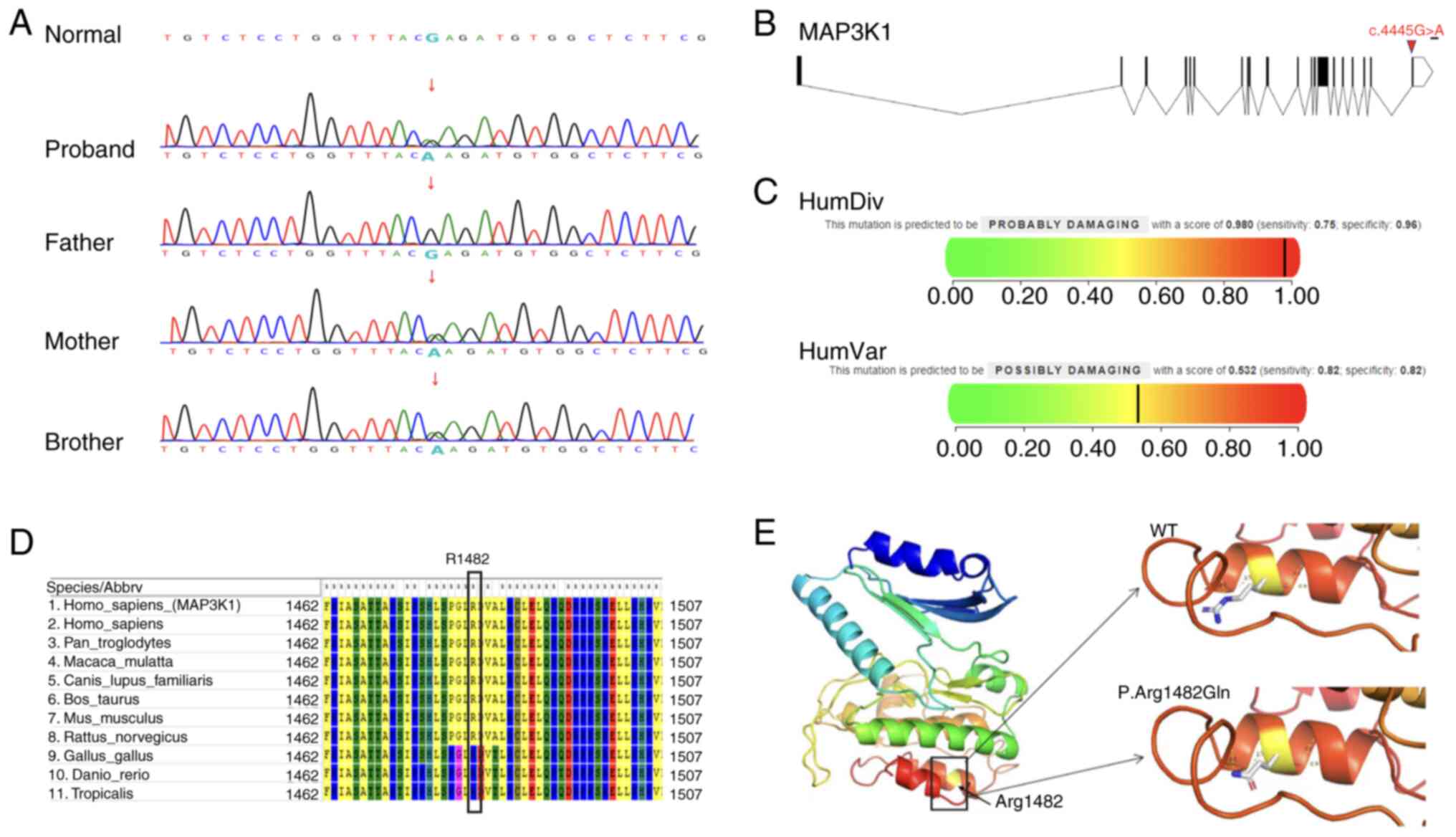
WES of peripheral blood obtained from the proband
identified a heterozygous variant at the MAP3K1 gene locus
c.4445G>A, associated with autosomal dominant 46, XY DSD type 6.
Sanger sequencing was subsequently performed on the peripheral
blood obtained from the brother and parents of the proband
(Fig. 3A) and the results
confirmed that the variant was inherited from the mother. Notably,
these results were consistent with an autosomal dominant
inheritance pattern. The MAP3K1 c.4445G>A variant is a
missense variant, resulting in the substitution of arginine with
glutamine at position 1482. This variant was not present in the
1,000 Genomes Project database and the frequencies observed in the
ExAC and gnomAD databases were 8.30×106 and
2.90×105, respectively. Based on the ACMG guidelines
(20), this variant was classified
as a variant of unknown clinical significance (PM1 + PM2) and a
search of existing databases did not identify relevant studies or
case reports (Fig. 3B). Further
pathogenicity analysis was conducted using PolyPhen and the results
revealed a HumanDiv score of 0.98, indicative of disruption
(Fig. 3C). Although the HumanVar
score of 0.532 indicated a moderate likelihood of pathogenicity
(Fig. 3C), it remained within the
range of potentially damaging variants. Considering the
complementary nature of these models, the results supported the
classification of this variant as potentially disruptive. The MEGA
tool was used to assess the genetic conservation of this variant
site and the results revealed that the variant is highly conserved
across species (Fig. 3D). In
addition, SWISS-MODEL was used to predict potential structural
changes in the protein due to the amino acid substitution and PyMOL
software was used for visualization of the alterations. Results of
the analysis revealed notable modifications in the tertiary
structure of the protein (Fig.
3E). Collectively, these results suggested that the novel point
variant c.4445G>A in the MAP3K1 gene may lead to protein
dysfunction; thus, triggering the 46, XY DSD phenotype.
The heterozygous c.4445G>A variant
in MAP3K1 gene reduces cell viability in vitro
To validate whether the MAP3K1 c.4445G>A
variant was the causative factor of disease in the family
investigated in the present study, the heterozygous variant was
established in 293T cells using the CRISPR/Cas9-mediated gene
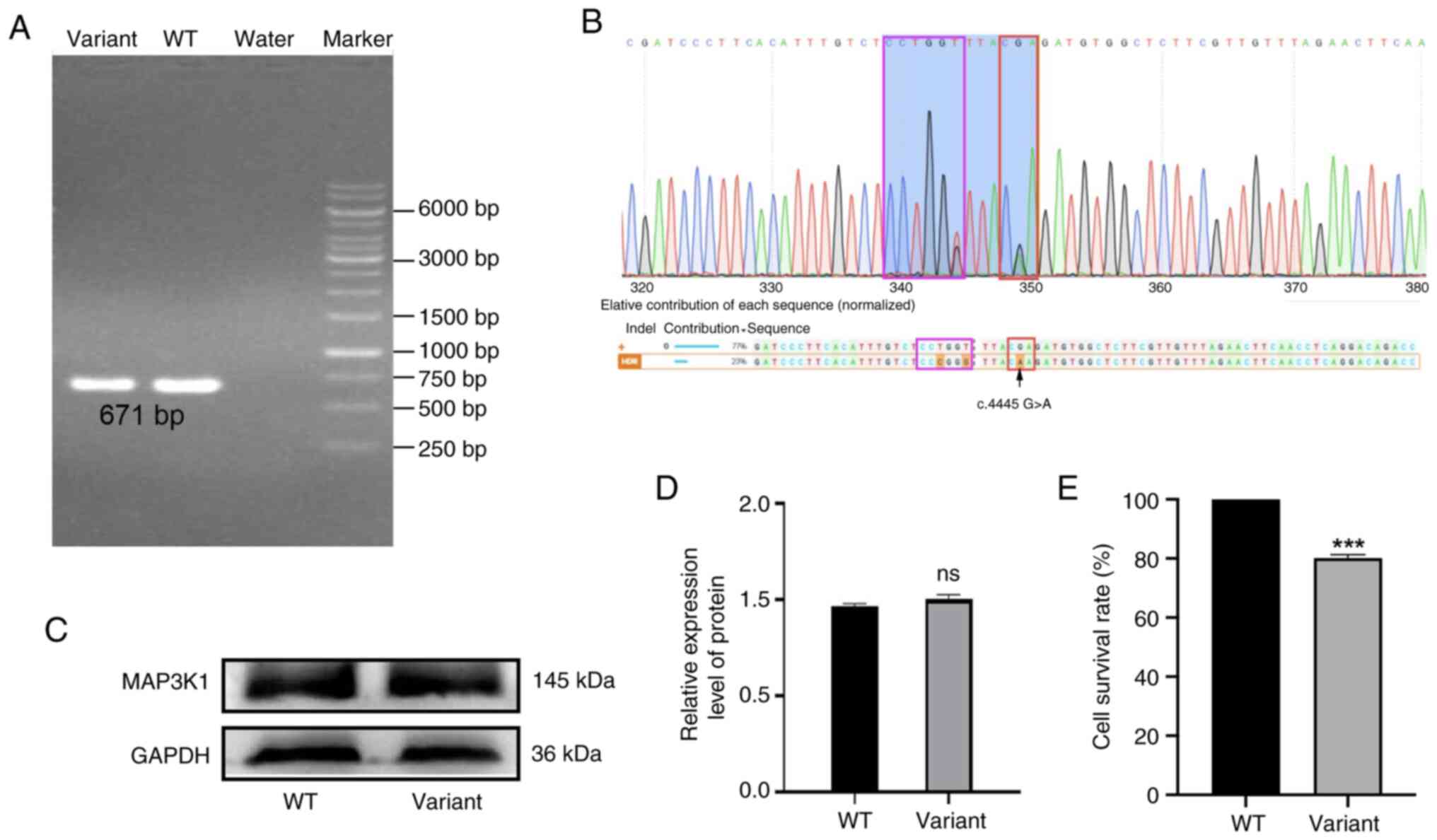
editing system. Single clones were selected following
electro-transformation and verified using RT-qPCR and sequencing.
Results of the present study revealed that heterozygous
MAP3K1 variant cells were successfully generated (Fig. 4A and B). To determine whether the
variant affected the translation or stability of the MAP3K1
protein, MAP3K1 expression levels were assessed. Results of the
western blot analysis revealed that MAP3K1 protein expression was
not affected by the presence of the variant (Fig. 4C and D). However, the viability of
variant cells was markedly reduced, with levels at 80.15% of that
of wild-type cells (Fig. 4E).
Collectively, these results indicated that the variant site may be
required for MAP3K1 function; however, it is not essential for
protein expression.
The heterozygous variant activates
pathways associated with apoptosis and induces cell cycle arrest in
vitro
To investigate the potential effects of the
MAP3K1 c.4445G>A point variant on cell proliferation and
apoptosis, the expression of proteins associated with apoptosis was
investigated. Results of the western blot analysis revealed that
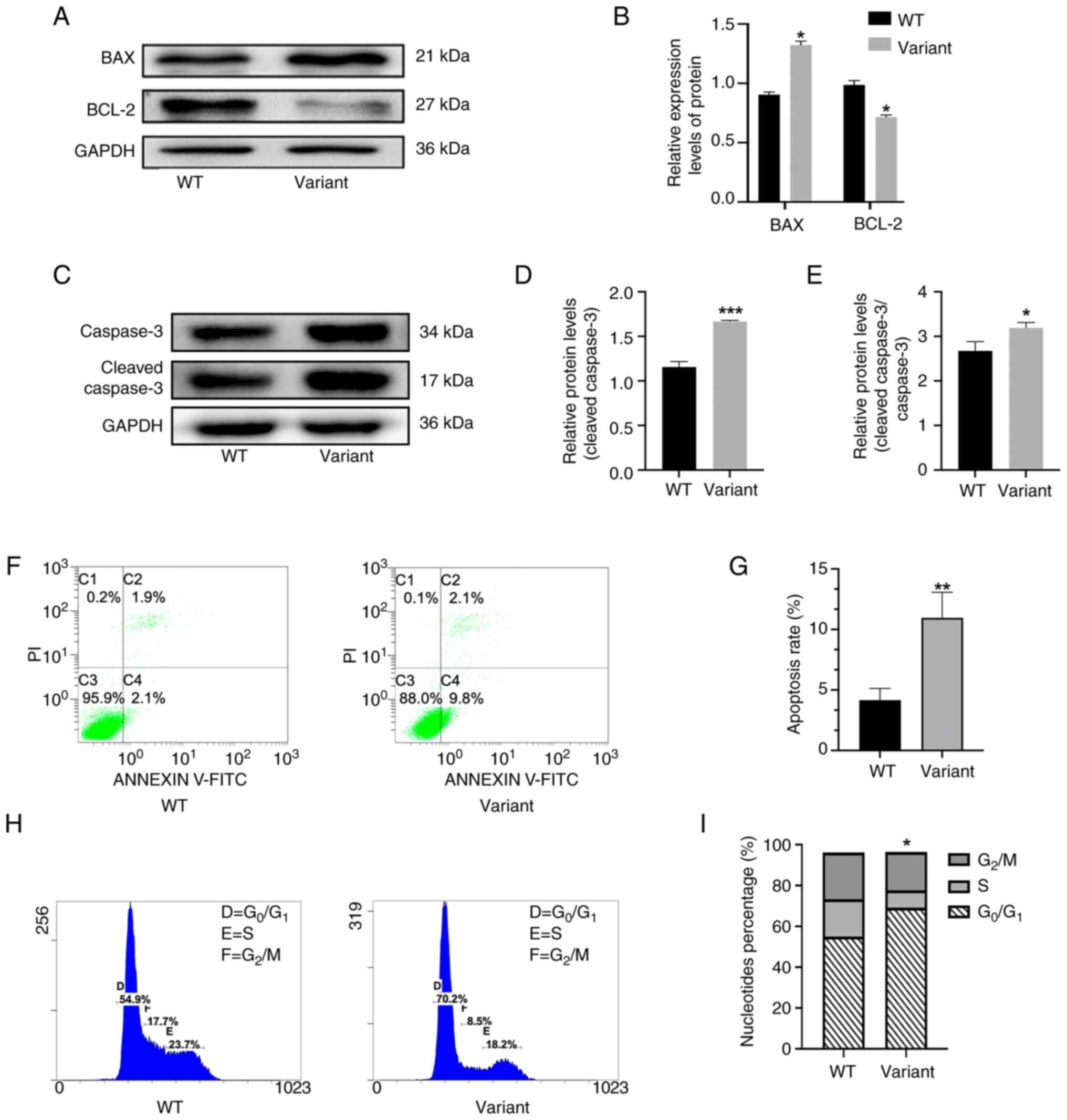
expression levels of pro-apoptotic proteins in the variant group;
namely, Bax and cleaved Caspase 3, were markedly increased
following 48 h incubation, while expression levels of the
anti-apoptotic protein, Bcl-2, were markedly reduced, compared with
the control group (Fig. 5A-E).
These results indicated that apoptosis may be associated with the
MAP3K1 c.4445G>A point variant. Results of flow cytometry
also revealed a significant increase in the rate of variant cell
apoptosis (Fig. 5F and G) and
results of the cell cycle analysis revealed a significant reduction
in the number of cells in S phase following 48 h incubation. These
results highlighted that the variant cells were predominantly
arrested in the G0/G1 phase (Fig. 5H and I). Collectively, these
findings suggested that the MAP3K1 c.4445G>A variant may
induce abnormal activation of apoptosis and cell cycle arrest in
the G0/G1 phase.
MAP3K1 c.4445 G>A point variant
disrupts the expression of sexual developmental factors
To investigate the underlying mechanism by which the
MAP3K1 c.4445G>A variant contributes to 46, XY DSD, the
expression levels of sexual developmental factors were
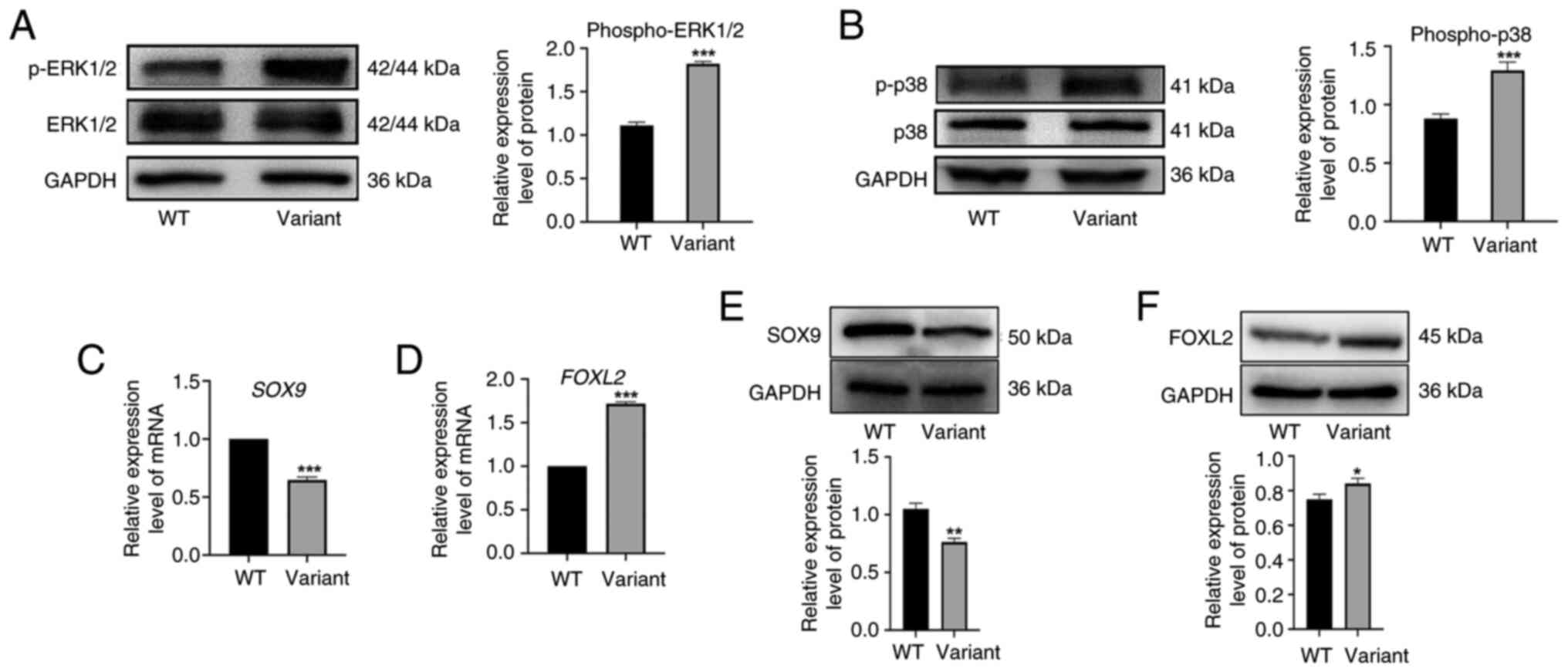
investigated. Results of the western blot analysis revealed no
significant difference in the protein expression of total ERK1/2
and p38 between the wild-type and variant groups. However, results
of the present study demonstrated that the expression levels of
p-ERK1/2 and p-p38 were markedly elevated in the variant group
(Fig. 6A and B). These results
highlighted that the MAP3K1 c.4445G>A variant may
activate downstream signaling pathways, leading to a
gain-of-function phenotype. To assess the impact of the variant on
the expression levels of sexual developmental factors, RT-qPCR was
performed in the present study. Compared with the wild-type group,
the relative mRNA and protein expression levels of SOX9 were
markedly reduced in the variant group (Fig. 6C and E), while the expression
levels of FOXL2 were notably increased (Fig. 6D and F). These results suggested
that the MAP3K1 c.4445G>A variant may activate the MAPK
pathway, leading to ERK1/2 and p38 hyperphosphorylation. This may
in turn modulate the expression of key sex-determining genes;
namely, SOX9 and FOXL2, ultimately contributing to
the pathogenesis of 46, XY DSD.
Discussion
The present study investigated a family with 46, XY
DSD following identification of a novel MAP3K1 gene variant
(c.4445G>A) in a 3-year-old male proband who presented with
clinical features of hypospadias and gonadal dysgenesis. This
variant resulted in a shift from testis to ovarian differentiation
and was associated with apoptotic dysfunction, cell cycle arrest
and hyperphosphorylation of key signaling molecules, such as ERK
and p38. Notably, these molecules are crucial for sex determination
and gonadal development. Results of the present study revealed that
the mother of the proband also carried the MAP3K1 variant;
however, this individual presented with no abnormalities in
phenotype. Therefore, it was hypothesized that the unaffected 46,
XX carrier may have transmitted a pathogenic variant in an
autosomal gene to the affected 46, XY proband. In addition, both
the proband and the sibling of the proband presented with the DSD
phenotype, supporting the notion that MAP3K1-associated 46, XY DSD
follows a sex-limited, autosomal dominant inheritance pattern.
These results were comparable with those of a previous study
(25).
Gain-of-function variants in the MAP3K1 gene
have been increasingly recognized as key contributors to the
development of 46, XY DSD (25).
These variants, such as p.L189P and p.P153L, lead to abnormal
activation of downstream signaling pathways, such as the
WNT/β-catenin and MAPK pathways, which are crucial for sex
determination (8). In healthy
individuals, MAP3K1 acts as a regulator of cell signaling through
modulating the balance between testis-promoting SOX9/FGF9 signaling
and ovary-promoting WNT/β-catenin signaling (26,27).
However, gain-of-function variants in MAP3K1 may result in
enhanced activation of the WNT/β-catenin pathway, leading to a
shift towards ovarian differentiation and suppression of testis
formation. These variants cause an imbalance in the expression of
critical sex-determining genes, such as SOX9 and
FOXL2, contributing to gonadal dysgenesis in 46, XY
individuals (6,26,27).
Notably, the MAP3K1 c.4445G>A variant is a representative
example of how hyperactivation of MAP3K1 may disrupt healthy
testicular development, through increased phosphorylation of
downstream kinases, such as ERK1/2 and p38. Results of the present
study are comparable with those of previous studies, which
demonstrated that gain-of-function variants in MAP3K1 not
only alter gonadal development, but also contribute to the
phenotypic heterogeneity observed in patients with 46, XY DSD
(8,10). Collectively, these findings
highlight the critical role of MAP3K1 in regulating sex
differentiation and emphasize the importance of understanding
MAP3K1 variants for the clinical management of DSD.
To the best of the authors' knowledge, the present
study was the first to demonstrate that apoptosis may play a key
role in the pathogenesis of 46, XY DSD. Previous studies assessed
the expression of the downstream effector in vitro; however,
cell viability was not investigated. Notably, the presence of
apoptosis in heterozygous MAP3K1 variant cells remained to
be fully elucidated (8,10). Results of the present study also
revealed elevated levels of apoptosis and alterations in
apoptosis-associated protein expression in cells harboring the
MAP3K1 c.4445G>A variant. In addition, variant cell
viability was markedly reduced, indicating that the MAP3K1
c.4445G>A variant may differ from variants previously described.
Collectively, results of the present study highlighted that
MAP3K1 variant-induced apoptosis and the associated cell
cycle arrest may disrupt healthy testicular differentiation,
promoting ovarian pathways in 46, XY individuals. Further
investigations are required to determine whether apoptosis plays a
role in cell lines with alternative genetic variants.
The c.4445G>A variant identified in present study
is a novel MAP3K1 variant implicated in 46, XY DSD.
Gain-of-function variants were described in a previous study
(8) and these were comparable with
the c.4445G>A variant, which resulted in hyperphosphorylation of
downstream targets, such as ERK and p38. High levels of
phosphorylation may promote ovarian differentiation through
increased FOXL2 expression and reduced SOX9
expression. Notably, splice-site variants disrupt healthy splicing
and promote aberrant protein function. By contrast, the missense
variant observed in the present study may affect protein
conformation independent of protein expression, leading to
potential alterations in the interaction of MAP3K1 with key
cofactors. These results highlighted that various variant types
within MAP3K1 may exert comparable downstream effects; however,
they may involve different molecular pathways and mechanisms.
The most notable clinical phenotype of patients with
46, XY DSD is abnormal external genitalia. The development of
external genitalia occurs in three phases; namely, genital tubercle
outgrowth, cloacal septation and urethral tubularization. In males,
the external genitalia differentiate into the penis, with the
urethral tube extending along its entire length. Incomplete
urethral tubularization leads to hypospadias, characterized by an
abnormally positioned urethral opening on the ventral side of the
penis (28). Previous studies have
reported increased apoptosis in the peri-cloacal, peri-urethral and
urorectal septum mesenchyme of Pdgfra-cKO mutants,
accompanied by p53 induction and Caspase 3 activation. Dysregulated
Pdgfra signaling may be associated with apoptosis-mediated
urorectal malformations, including anorectal defects and
hypospadias (28). Moreover,
excessive apoptosis and impaired mesenchymal growth in the
peri-urethral region may disrupt urethral fold fusion, directly
contributing to hypospadias (29).
Failure of urethral fold fusion at the midline prevents the
formation of a urethral groove, leading to urethral defects
associated with hypospadias (30).
Consistent with these findings, results of the present study
demonstrated that the MAP3K1 variant observed in patients
with 46, XY DSD may promote elevated levels of apoptosis, further
supporting the role of apoptosis in hypospadias development.
Collectively, these findings suggested that dysregulated apoptosis
may act as a key mechanism in the pathogenesis of hypospadias in
patients with 46, XY DSD.
Abnormalities in sex development are associated with
an increased risk of tumor development, particularly during puberty
(31). Malignancy of these tumors
is associated with negative consequences, including the requirement
for organ removal and complete loss of fertility (32). Gonadoblastoma typically arises from
either embryonic or hypoplastic gonads and >90% of affected
individuals carry a Y chromosome, with the 46, XY karyotype being
the most common (33). Results of
the in vitro analysis in the present study suggested that
the c.4445G>A heterozygous variant in MAP3K1 may affect
the cell cycle and cell viability and promote abnormal apoptosis.
However, these results are not indicative of this variant being the
cause of gonadoblastoma. Notably, the in vivo
microenvironment is more complex and apoptosis in early phase may
trigger apoptosis resistance, a feature of tumor formation
(34). Thus, further
investigations are required to explore whether this variant
contributes to tumor development.
While the present study provides novel insights into
the role of the MAP3K1 c.4445 G>A variant in 46, XY DSD,
certain limitations should be acknowledged. For example, the
experiments were conducted using 293T cells, which, while commonly
used for studying gene function, do not fully replicate the
physiological environment. Future studies utilizing more
physiologically relevant cell models (for example, human granulosa
KGN cells and PSC-derived human PGC-like cells) are required to
further validate the findings of the present study. Moreover,
results of the present study revealed that apoptosis may exhibit
potential in the pathogenesis of the disease; however, the present
study did not investigate whether similar apoptotic effects occur
in other reported MAP3K1 variants. Examination of additional
variants may provide a broader understanding of the role of MAP3K1
in 46, XY DSD. In addition, results of the present study revealed
that reduced cell viability may be associated with the variant;
however, the molecular pathways leading to this reduction, such as
the potential involvement of mitochondrial dysfunction or intrinsic
apoptosis pathways, were not examined. Therefore, future
investigations should explore the aforementioned mechanisms to
further elucidate the functional consequences of the variant.
In conclusion, the present study identified a novel
MAP3K1 variant (c.4445G>A) associated with 46, XY DSD and
demonstrated the associated impact on apoptosis, cell cycle
regulation and disruption of key signaling pathways. These findings
expand the current understanding of how gain-of-function variants
in MAP3K1 contribute to gonadal dysgenesis, through shifting
the balance between testis and ovary differentiation pathways. The
present study provides novel insights for improved management and
personalized treatment of patients with DSD.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Science and Technology
Foundation of Guizhou Provincial Health and Construction Commission
(grant no. gzwkj2021-300).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author. The data generated in the
present study may be found in the Sequence Read Archive under
accession number (BioProject no. PRJNA1250182) or at the following
URL: https://www.ncbi.nlm.nih.gov/sra/PRJNA1250182.
Authors' contributions
ZW designed the study. YL and SW participated in all
experiments. YX and CH analyzed the experimental data and drafted
the manuscript. SW and JZ carried out the cell experiments. WP
guided the operation of the flow cytometer. ZW, YL and SW confirmed
the authenticity of all the raw data. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Guizhou Medical University [approval no. 2020 Ethics
(No 325)]. Written informed consent was obtained from all
participants.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lucas-Herald AK, Ali SR, McMillan C, Rodie
ME, McMillan M, Bryce J and Ahmed SF: I-DSD: The first 10 years.
Horm Res Paediatr. 96:238–246. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Grinspon RP, Bergadá I and Rey RA: Male
Hypogonadism and disorders of sex development. Front Endocrinol
(Lausanne). 11:2112020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hughes IA, Houk C, Ahmed SF and Lee PA;
Lawson Wilkins Pediatric Endocrine Society/European Society for
Paediatric Endocrinology Consensus Group, : Consensus statement on
management of intersex disorders. J Pediatr Urol. 2:148–162. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jiali C, Huifang P, Yuqing J, Xiantao Z
and Hongwei J: Worldwide cohort study of 46, XY
differences/disorders of sex development genetic diagnoses:
Geographic and ethnic differences in variants. Front Genet.
15:13875982024. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xue M, Wang X, Li C, Zhao M, He F and Li
X: Novel pathogenic mutations in disorders of sex development
associated genes cause 46,XY complete gonadal dysgenesis. Gene.
718:1440722019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Reyes AP, León NY, Frost ER and Harley VR:
Genetic control of typical and atypical sex development. Nat Rev
Urol. 20:434–451. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Granados A, Alaniz VI, Mohnach L,
Barseghyan H, Vilain E, Ostrer H, Quint EH, Chen M and Keegan CE:
MAP3K1-related gonadal dysgenesis: Six new cases and review of the
literature. Am J Med Genet C Semin Med Genet. 175:253–259. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Loke J, Pearlman A, Radi O, Zuffardi O,
Giussani U, Pallotta R, Camerino G and Ostrer H: Mutations in
MAP3K1 tilt the balance from SOX9/FGF9 to WNT/β-catenin signaling.
Hum Mol Genet. 23:1073–1083. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Stewart MK, Bernard P, Ang CS, Mattiske DM
and Pask AJ: Oestrogen activates the MAP3K1 cascade and β-catenin
to promote granulosa-like cell fate in a human testis-derived cell
line. Int J Mol Sci. 22:100462021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pearlman A, Loke J, Le Caignec C, White S,
Chin L, Friedman A, Warr N, Willan J, Brauer D, Farmer C, et al:
Mutations in MAP3K1 cause 46,XY disorders of sex development and
implicate a common signal transduction pathway in human testis
determination. Am J Hum Genet. 87:898–904. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cheng Y, Xu C, Yang J, Zhou X and Chen N:
Identification of a novel MAP3K1 variant in a family with 46, XY
DSD and partial growth hormone deficiency. Mol Med Rep. 26:3382022.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yujiri T, Fanger GR, Garrington TP,
Schlesinger TK, Gibson S and Johnson GL: MEK kinase 1 (MEKK1)
transduces c-Jun NH2-terminal kinase activation in response to
changes in the microtubule cytoskeleton. J Biol Chem.
274:12605–12610. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yujiri T, Sather S, Fanger GR and Johnson
GL: Role of MEKK1 in cell survival and activation of JNK and ERK
pathways defined by targeted gene disruption. Science.
282:1911–1914. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Minamino T, Yujiri T, Papst PJ, Chan ED,
Johnson GL and Terada N: MEKK1 suppresses oxidative stress-induced
apoptosis of embryonic stem cell-derived cardiac myocytes. Proc
Natl Acad Sci USA. 96:15127–15132. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Krivega M, Zimmer J, Slezko A,
Frank-Herrmann P, Rehnitz J, Hohenfellner M, Bettendorf M,
Luzarowski M and Strowitzki T: Genomic instability in individuals
with sex determination defects and germ cell cancer. Cell Death
Discov. 9:1732023. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Alfano M, Tascini AS, Pederzoli F,
Locatelli I, Nebuloni M, Giannese F, Garcia-Manteiga JM, Tonon G,
Amodio G, Gregori S, et al: Aging, inflammation and DNA damage in
the somatic testicular niche with idiopathic germ cell aplasia. Nat
Commun. 12:52052021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bailly A and Gartner A: Germ cell
apoptosis and DNA damage responses. Adv Exp Med Biol. 757:249–276.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang LF, Wang KY, Tu HJ, Lin K and Lin H:
Clinical investigation of chromosome karyotype analysis with
amniotic fluids cell and parental peripheral blood. Clin Lab.
68:2022. View Article : Google Scholar
|
|
19
|
Yi T, Sun H, Fu Y, Hao X, Sun L, Zhang Y,
Han J, Gu X, Liu X, Guo Y, et al: Genetic and clinical features of
heterotaxy in a prenatal cohort. Front Genet. 13:8182412022.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Castro S, Brunello FG, Sansó G, Izquierdo
A, Zaiat J, Urrutia M, Martí M, Rey RA, Tellechea ML and Grinspon
RP: Clinical presentation of congenital hypogonadotropic
hypogonadism in males with delayed puberty according to genetic
etiology: A systematic review and meta-analysis after
reclassification of gene variants. Hum Reprod. 40:904–918. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cheng Y, Chen J, Zhou X, Yang J, Ji Y and
Xu C: Characteristics and possible mechanisms of 46, XY differences
in sex development caused by novel compound variants in NR5A1 and
MAP3K1. Orphanet J Rare Dis. 16:2682021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li T, Yang Y, Qi H, Cui W, Zhang L, Fu X,
He X, Liu M, Li PF and Yu T: CRISPR/Cas9 therapeutics: progress and
prospects. Signal Transduct Target Ther. 8:362023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liao S, Wu J, Liu R, Wang S, Luo J, Yang
Y, Qin Y, Li T, Zheng X, Song J, et al: A novel compound DBZ
ameliorates neuroinflammation in LPS-stimulated microglia and
ischemic stroke rats: Role of Akt(Ser473)/GSK3β (Ser9)-mediated
Nrf2 activation. Redox Biol. 36:1016442020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ostrer H: Pathogenic variants in MAP3K1
cause 46,XY gonadal dysgenesis: A review. Sex Dev. 16:92–97. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Elzaiat M, McElreavey K and Bashamboo A:
Genetics of 46,XY gonadal dysgenesis. Best Pract Res Clin
Endocrinol Metab. 36:1016332022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chamberlin A, Huether R, Machado AZ,
Groden M, Liu HM, Upadhyay K, O V, Gomes NL, Lerario AM, Nishi MY,
et al: Mutations in MAP3K1 that cause 46,XY disorders of sex
development disrupt distinct structural domains in the protein. Hum
Mol Genet. 28:1620–1628. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Qian C, Wu Z, Ng RC, Garcia-Barceló MM,
Yuan ZW, Wong KKY, Tam PKH and Lui VCH: Conditional deletion of
platelet derived growth factor receptor alpha (Pdgfra) in urorectal
mesenchyme causes mesenchyme apoptosis and urorectal developmental
anomalies in mice. Cell Death Differ. 26:1396–1410. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yamada G, Satoh Y, Baskin LS and Cunha GR:
Cellular and molecular mechanisms of development of the external
genitalia. Differentiation. 71:445–460. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Glenister TW: The origin and fate of the
urethral plate in man. J Anat. 88:413–425. 1954.PubMed/NCBI
|
|
31
|
Lee PA, Nordenström A, Houk CP, Ahmed SF,
Auchus R, Baratz A, Baratz Dalke K, Liao LM, Lin-Su K, Looijenga LH
III, et al: Global disorders of sex development update since 2006:
Perceptions, approach and care. Horm Res Paediatr. 85:158–180.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rosenfield RL: Normal and premature
adrenarche. Endocr Rev. 42:783–814. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Arya S, Kumar S, Lila AR, Sarathi V, Memon
SS, Barnabas R, Thakkar H, Patil VA, Shah NS and Bandgar TR: Exonic
WT1 pathogenic variants in 46,XY DSD associated with
gonadoblastoma. Endocr Connect. 10:1522–1530. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dehghan N, Mousavikia SN, Qasempour Y and
Azimian H: Radiation-induced senescence in glioblastoma: An
overview of the mechanisms and eradication strategies. Life Sci.
359:1232182024. View Article : Google Scholar : PubMed/NCBI
|