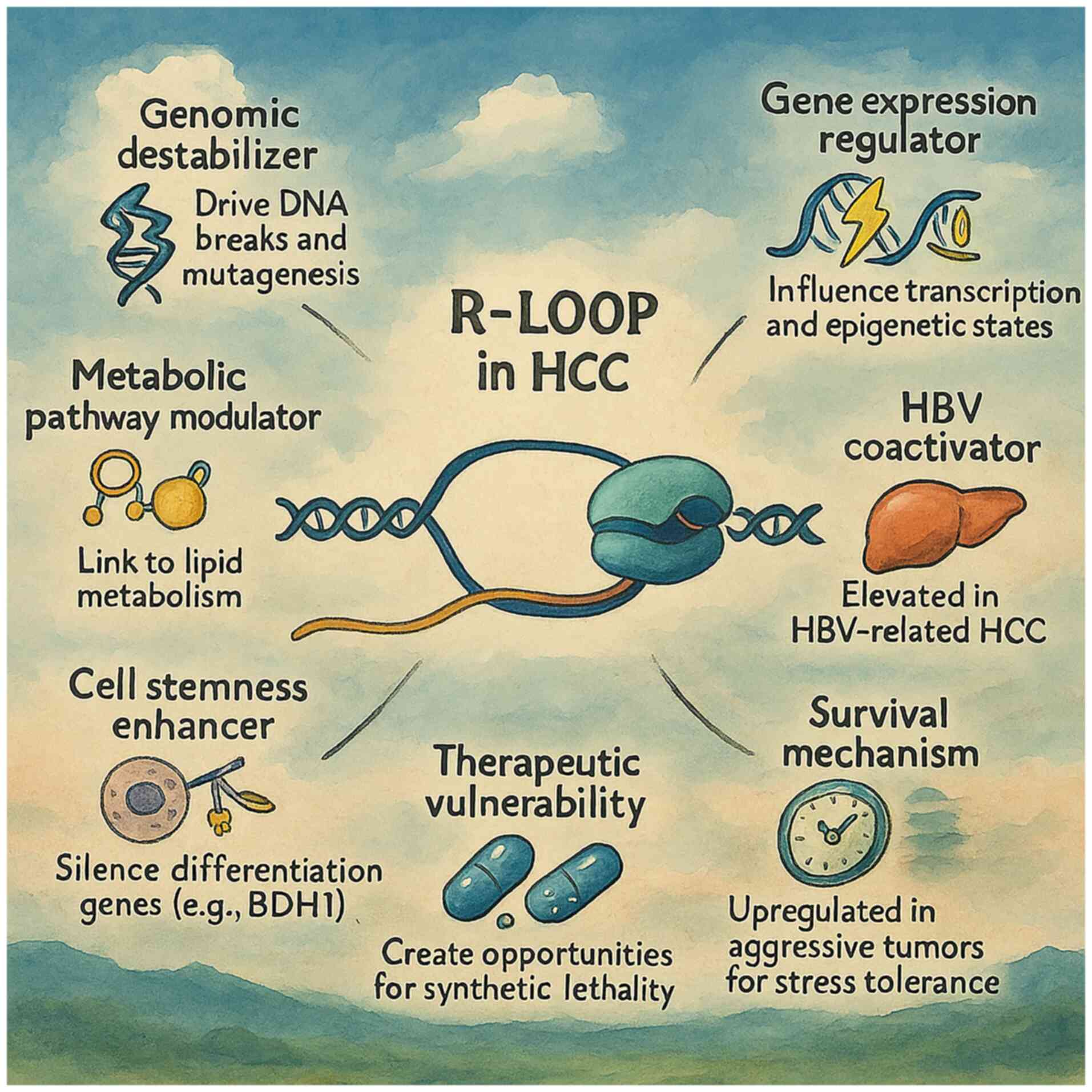
Introduction
R-loops are triple-stranded nucleic acid structures
comprising an RNA:DNA hybrid and displaced single-stranded DNA,
typically forming co-transcriptionally when nascent RNA reanneals
to its template DNA strand (1).
Once considered by-products of transcription, R-loops have been
recognized as important regulators of gene expression, DNA
replication and genome maintenance (1). Under physiological conditions,
controlled R-loop formation plays a role in normal transcriptional
termination, RNA processing, epigenetic regulation and
mitochondrial DNA replication (1,2).
However, unscheduled or persistent R-loops can become pathological
and pose a threat to genomic integrity (1). Excess R-loops can stall replication
forks, induce DNA breaks and promote mutagenesis, thereby
contributing to genomic instability, which is a well-known hallmark
of cancer (1–3). Indeed, aberrant accumulation of
R-loops has been observed in breast cancer, colorectal cancer and
leukemia and has been linked to dysregulated proliferation and
oncogene activation (2,4,5).
Hepatocellular carcinoma (HCC), the most common
primary liver cancer, is a disease characterized by pronounced
genomic instability (1,6). HCC typically arises in cases of
chronic liver injury, including hepatitis B virus (HBV) or HCV
infection, alcohol abuse or fatty liver disease, and is
characterized by ongoing DNA damage and chromosomal aberrations
(1,6). Such chronic stress may predispose the
liver cells to aberrant R-loop accumulation. Recent evidence
suggests that R-loops play a role in HCC pathophysiology; improper
regulation of R-loop homeostasis can drive DNA breakage and
replication stress, fueling the genomic instability that propels
hepatocarcinogenesis (1,6).
Furthermore, HCC cells may co-opt mechanisms to
tolerate or exploit R-loops. HCCs generally have defence mechanisms
to minimize R-loop-induced damage (6) and aggressive tumors often upregulate
R-loop-resolving factors to maintain survival despite high
transcriptional output (2). HCC
tumors frequently exhibit widespread transcriptional dysregulation
due to inflammation and oncogene activation, which can alter
signaling pathways, epigenetic marks and transcription factor
activity, leading to aberrant gene expression, replication stress
and increased R-loop formation. In addition, HBV infection, a major
etiological factor of HCC, is associated with changes in the
expression of R-loop regulatory genes (7). An integrative single-cell study found
that HCC cells from HBV-infected livers tend to have higher ‘R-loop
scores’ linking viral oncogenesis with R-loop dysregulation
(1).
In summary, R-loops represent a notable interface
between transcriptional regulation and genomic stability in cancer
cells. In HCC, a cancer characterized by extensive genomic
instability and complex molecular drivers, understanding R-loop
biology is particularly relevant. The present review provided an
overview of R-loop formation and functions and delves into their
mechanistic impact on transcriptional regulation and genome
stability in HCC. The key findings on the role of the R-loop in HCC
are summarized in Table I
(1,2,6,8,9).
Selected R-loop-associated genes and their functions in HCC are
presented in Table II (1,2,10–12).
The R-loop-related signaling pathways in HCC are shown in Table III (1,8,9,13).
Therapeutic strategies targeting R-loops in HCC are presented in
Table IV (1,11,12,14).
The present review also discussed how HCC cells manage or mismanage
R-loops and how this knowledge has contributed to novel therapeutic
strategies.
 | Table I.Key findings on the roles of R-loops
in HCC. |
Table I.
Key findings on the roles of R-loops
in HCC.
| Key finding | Description and
significance | (Refs.) |
|---|
| R-loops contribute
to genomic instability in HCC | Persistent R-loops
induce DNA breaks and replication stress, fueling HCC genomic
instability | (1,6) |
| HCC cells modulate
R-loop levels for survival | Cancer cells
upregulate defense mechanisms to limit R-loop-associated
damage | (2,6) |
| High R-loops linked
to HBV-related HCC | HBV-infected HCCs
show elevated R-loop regulator-gene expression and higher R-loop
scores | (1) |
| R-loops impact HCC
tumor microenvironment | R-loops activate
innate immune pathways, influencing tumor-immune interactions | (1) |
| R-loop
dysregulation alters metabolism in HCC | Aberrant R-loops
are linked to metabolic reprogramming affecting tumor
progression | (1) |
| R-loop-associated
factors affect chemosensitivity | THO complex 1
knockdown increases R-loops and DNA damage, sensitizing cells to
cisplatin | (8) |
| Oncogenic drivers
exploit R-loops |
Metastasis-associated protein 2 enhances
R-loop formation to silence differentiation genes such as
β-hydroxybutyrate dehydrogenase 1 | (9) |
| Table II.Selected R-loop-associated genes and
proteins and their roles in HCC. |
Table II.
Selected R-loop-associated genes and
proteins and their roles in HCC.
| Gene/factor | Role in R-loop
regulation | Relevance in
HCC | (Refs.) |
|---|
| RNASEH1 | Degrades RNA in
hybrids | RNASEH1-antisense 1
long non-coding RNA is upregulated in advanced HCC | (2,10) |
| RNASEH2
(A/B/C) | Removes RNA from
hybrids | RNASEH2 deficiency
creates synthetic lethality when combined with ATR inhibition | (11) |
| Senataxin | Unwinds R-loops
during transcription | Important for
resolving R-loops at highly transcribed HCC genes | (12) |
| Fanconi anemia
group G protein (Fanconi anemia complex) | Binds and resolves
R-loops at forks | Part of the 8-gene
prognostic signature; affects HCC outcomes | (1,2) |
| Breast cancer
susceptibility gene 1/2 | Recruits factors to
suppress R-loops | Suppresses
telomeric R-loops that cause DNA damage | (1,2) |
| TOP1 | Prevents excessive
R-loop formation | TOP1 inhibitors
induce DNA damage partly via R-loop accumulation | (12) |
| ATR/checkpoint
kinase 1 pathway | Responds to
R-loop-stalled forks | High R-loop HCCs
show ATR dependency; this shows potential for ATR inhibition in
therapeutics | (2,12) |
|
Metastasis-associated protein 2/histone
deacetylase 2 (nucleosome remodeling and deacetylase) | Promotes R-loops at
target genes | Enhances stemness
via R-loop-mediated silencing of differentiation genes | (1) |
| Table III.R-loop-related signaling pathways in
HCC. |
Table III.
R-loop-related signaling pathways in
HCC.
| Pathway |
Trigger/context | Function in
HCC | (Refs.) |
|---|
| DNA damage response
(ATR/ATM-checkpoint kinase 1) |
Transcription-replication conflicts cause
R-loop accumulation, stalling replication forks and causing DNA
double-strand breaks. For example, the loss of the RNA export
factor THO complex 1 in HCC cells increases R-loops and activates
DNA damage signaling | Activates ATR/ATM
and p53 pathways, leading to genomic instability or growth arrest.
In HCC models, R-loop-induced DNA damage reduces tumor cell
proliferation and sensitizes cells to chemotherapy | (8) |
| Innate immune
(cGAS-STING) pathway | Unresolved nuclear
R-loops can be processed into cytosolic RNA:DNA hybrids, such as in
breast cancer susceptibility gene 1 or Senataxin-deficient
contexts. These hybrids and R-loop-derived DNA fragments are sensed
by cGAS and other pattern recognition receptors | Triggers
STING-interferon regulatory factor 3 signaling and inflammatory
cytokine production. In HCC, excessive R-loops aberrantly activate
innate immunity, which can induce tumor cell apoptosis and reshape
the tumor microenvironment | (13) |
| Metabolic
reprogramming (lipid metabolism) | Upregulation of
certain R-loop regulator genes, such as CLTC, in HCC promotes
persistent R-loop formation at metabolic gene loci, for example at
fatty acid synthase regulators | Enhances lipid
biosynthesis and storage to fuel tumor growth. High R-loop levels
in HCC cells are linked to increased fatty acid metabolism and a
more aggressive tumor phenotype. For instance, CLTC-driven R-loops
upregulate lipogenic pathways, supporting rapid proliferation | (1) |
| Epigenetic
regulation (histone modification) | R-loops can be
exploited by chromatin-modifying complexes to silence genes. In
HCC, the chromatin remodeler metastasis-associated protein 2,
acting with histone deacetylase 2/chromodomain-helicase-DNA-
binding protein 4, induces R-loop formation at the BDH1
locus, which is a metabolic gene | Transcriptional
repression of BDH1 leads to altered β-hydroxybutyrate levels
and increased histone β-hydroxybutyrylation, an epigenetic mark
that promotes cancer stem cell traits. This R-loop-dependent
epigenetic silencing enhances HCC stemness and tumor
propagation | (9) |
| Table IV.Therapeutic strategies impacting
R-loops in HCC. |
Table IV.
Therapeutic strategies impacting
R-loops in HCC.
| Therapeutic
agent/class | Mechanism (R-loop
context) | Potential
application in HCC | (Refs.) |
|---|
| Topoisomerase I
inhibitors | Increase R-loop
formation and associated DNA breaks | Target HCCs with
high transcriptional output; combine with DNA damage response
inhibitors | (12) |
| ATR inhibitors | Block responses to
R-loop-induced fork collapses | Target HCCs with
R-loop resolution defects or high stress | (11,14) |
| Poly(ADP-ribose)
polymerase inhibitors | Block repair of
R-loop-induced DNA breaks | Effective in HCCs
with homologous recombination deficiencies or R-loop-mediated
breast cancer susceptibility gene sequestration | (12) |
| Splicing
modulators | Induce R-loop
overload via unspliced transcripts | Create synthetic
lethality when combined with ATR inhibitors | (14) |
| Guanine-quadruplex
stabilizers | Prevent R-loop
resolution by locking Guanine-quadruplex structures | Target R-loop-prone
regions in HCC, such as recombinant DNA and telomeres | (12) |
| Histone deacetylase
inhibitors | Alter chromatin
states affecting R-loop dynamics | Reverses
metastasis-associated protein 2-induced R-loop silencing of
differentiation genes | (1,12) |
Mechanisms of R-loop formation and function
in HCC
R-loop formation and homeostasis
Formation
R-loops commonly form during transcription when
nascent RNA threads back and hybridizes with the template DNA
strand, displacing the non-template strand (1). Certain DNA sequences and contexts
promote R-loop formation; for example, GC-rich regions, especially
those capable of forming G-quadruplexes, and repetitive sequences
increase the stability of RNA:DNA hybrids. Negative supercoiling
behind a moving RNA polymerase can also facilitate re-annealing of
RNA strands to DNA (12). By
contrast, positive supercoiling before the polymerase can impede
hybrid formation. This topological balance is managed by
topoisomerases (TOPs) such as TOP1, TOP2 and TOP3B, which relax the
supercoils and prevent excessive R-loop formation (12).
Regulation and resolution
The cell maintains R-loop homeostasis through
dedicated enzymes that remove R-loops once formed. Most of these
are ribonuclease H (RNASEH)1 and 2, which specifically recognize
RNA:DNA hybrids and cleave RNA strands (2). RNASEH1 is active throughout the cell
cycle and serves as the first line of defence against R-loops,
degrading RNA components and dismantling the hybrid (15). RNASEH2 operates especially during
the S/G2 phases, not only to resolve R-loops but also to
excise ribonucleotides embedded in DNA (16).
Along with RNASEH, several DNA and RNA helicases
resolve R-loops by unwinding the RNA:DNA hybrid. Senataxin (SETX)
is a key helicase that resolves R-loops during transcription
termination, preventing persistent hybrids at gene 3′ ends
(17). The Asp-Glu-Ala-Asp box
(DEAD-box) RNA helicases DEAD-box helicase (DDX)19 and DDX21 are
also important; DDX21 unwinds R-loops at ribosomal RNA (rRNA) genes
and collaborates with NAD-dependent protein deacetylase sirtuin-7
to suppress R-loops during replication stress (18,19).
Other factors involved in R-loop suppression include
the transcription-export (THO) complex/3′ repair exonuclease
complex, which couples transcription to mRNA export and various
RNA-processing factors, such as splicing factors. Comprehensive
mRNA splicing and 3′ end processing are important to prevent
R-loops; if transcription outpaces processing or if splicing is
defective, the unprocessed nascent RNA is more prone to hybridize
with DNA. This increase in hybridization due to insufficient mRNA
processing explains why mutations in spliceosome genes, such as
splicing factor 3b subunit 1 and U2 small nuclear RNA
auxiliary factor 1, can induce R-loop accumulation and activate
DNA damage responses (DDRs) (12,14).
HCC context
Homeostatic mechanisms may be altered in HCC. Tumor
cells often have markedly elevated transcriptional activity, which
can favor R-loop formation beyond the capacity of normal regulatory
mechanisms. There is evidence that HCC cells upregulate certain
R-loop-resolving factors, presumably to cope with the increased
R-loop load (1,2). In HCC, dysregulated R-loops
contribute to genomic instability by inducing replication stress
and DNA double-stranded breaks, thereby activating the ataxia
telangiectasia and Rad3-related protein (ATR)-checkpoint kinase
(CHK)1 and ataxia telangiectasia mutated (ATM)-CHK2 DNA damage
response pathways (2,6). Oncogenic drivers such as
metastasis-associated protein 2 (MTA2) promote R-loop formation at
the promoters of differentiation genes such as β-hydroxybutyrate
dehydrogenase 1 (BDH1), leading to epigenetic silencing
and enhanced stemness in HCC cells (9). Furthermore, unresolved R-loops can
initiate inflammatory responses through cytosolic DNA-sensing
pathways, such as cyclic GMP-AMP synthase and stimulator of
interferon genes (cGAS-STING), contributing to immune modulation
within the tumor microenvironment (TME) (1). For instance, RNASEH1 expression is
elevated in breast cancer, colorectal cancer, leukemia, and HCC and
associated with poor prognosis (2). One study noted that a long non-coding
RNA (lncRNA) antisense to RNASEH1 (RNASEH1-AS1) is frequently
upregulated in HCC and associated with worse survival prognosis
(10). Additionally, certain
oncogenic pathways in HCC actively induce R-loops to alter gene
expression, such as the chromatin modifier MTA2, which increases
R-loops at specific gene promoters (9). Thus, the balance between R-loop
formation and resolution is maintained in HCC cells.
R-loops in transcriptional regulation
and epigenetics
In addition to their impact on DNA stability,
R-loops can influence gene expression and the chromatin state.
Physiological R-loops often occur in the promoter or terminator
regions of genes and have regulatory functions (2). For example, R-loops formed over
5′-cytosine-phosphate-guanine-3′ island promoters maintain an open
chromatin configuration by displacing nucleosomes, thereby
facilitating transcription initiation of these genes (19). R-loops at the gene termini have
been implicated in pausing RNA polymerase II and promoting
transcription termination (20,21).
These co-transcriptional R-loops can recruit or
occlude specific factors. One study showed that promoter-proximal
R-loops can attract DNA demethylases, leading to local DNA
hypomethylation and robust gene expression (2). Conversely, stable R-loops on gene
bodies or regulatory elements may interfere with transcriptional
elongation, effectively repressing these loci. Furthermore, R-loops
interact with chromatin-modifying complexes and alter histone
marks, modulate chromatin accessibility and either promote or
repress gene transcription (5).
For example, R-loop structures are recognized by the polycomb
repressive complex and DNA methyltransferases, linking hybrid
formation to heritable epigenetic changes (2).
These R-loops may be co-opted or dysregulated in
cancer cells, including HCC cells. A notable HCC-related example is
the MTA2 pathway; MTA2, a component of the nucleosome remodeling
and deacetylase chromatin-remodeling complex, increases R-loop
formation at the promoter of BDH1, which is a gene that
influences both differentiation and metabolism (9,22).
The R-loop recruits histone deacetylase
(HDAC)2/chromodomain-helicase-DNA-binding protein 4 (CHD4)
specifically to the promoter region of BDH1, leading to the
epigenetic silencing of BDH1 and stemness in HCC cells
(9,23). Thus, an oncogenic factor exploits
R-loop formation as part of a transcriptional repression mechanism
that favors tumor cell dedifferentiation and self-renewal.
Another example involves the THO complex (THOC).
THOC1 knockdown impairs the THO complex's role in
co-transcriptional RNA processing and export, causing RNA-DNA
hybrids to accumulate, which increases R-loop formation and
consequent DNA damage, which paradoxically leads to reduced tumor
cell proliferation, indicating that THOC1 aids HCC cells by
preventing lethal levels of R-loop accumulation (8). This suggests that HCC cells require
proper mRNA processing to avoid transcription-associated damage.
Therefore, R-loops serve as both outputs and regulators of
transcription. With widespread transcriptional reprogramming, HCC
cells are likely to exhibit an altered landscape of R-loops across
the genome. The influence of R-loops on oncogenic gene expression
is bidirectional: Certain R-loops promote tumor progression by
activating oncogenes or enhancing stemness, while others suppress
tumor progression by inducing transcriptional repression or DNA
damage-mediated checkpoints (1,6). The
interplay between R-loops and epigenetic machinery is a frontier
area; for instance, how R-loop formation might interface with the
frequent epigenetic alterations seen in HCC.
R-loops and genome instability
One of the most important aspects of R-loops is
their ability to induce DNA damage, if not properly mitigated.
During the S phase, the coexistence of transcription and
replication machinery on the DNA template can lead to
transcription-replication conflicts. R-loops are a prominent source
of such conflicts. An R-loop can impede the replication fork,
especially when it collides with the transcription complex
(2). Conflicts are particularly
deleterious in a head-on orientation, when transcription and
replication move toward each other on opposite strands, as the
replication fork encounters the R-loop and stalled RNA polymerase
headlong (23). Head-on
transcription-replication conflicts markedly increase the
likelihood of fork stalling or collapse, resulting in DNA
double-strand breaks (DSBs) and chromosome rearrangements (2,24).
ATR and ATM activation
ATR kinase is a central responder to replication
stress caused by R-loops. Stalled replication forks with stretches
of single-stranded DNA coated with replication protein A activate
ATR, which, in turn, phosphorylates CHK1 and coordinates fork
stabilization (25). ATR signaling
is associated with head-on collisions. Notably, ATR not only halts
the cell cycle to provide time for repair, but also actively
promotes R-loop resolution (3), as
ATR signaling recruits SETX to transcription-replication clash
sites to help unwind R-loops (2).
Similar to ATR, the ATM kinase is activated when
R-loop-associated DSBs are formed. ATM can promote end resection
and homologous recombination (HR) or facilitate non-homologous end
joining, depending on the cell cycle stage and local chromatin
environment (2). ATM-CHK2 and
ATR-CHK1 are both engaged in response to R-loops; however, R-loops
that result in DSBs preferentially trigger ATM, whereas those that
cause stalled forks without immediate breakage primarily trigger
ATR (26).
Notably, the ATR and ATM signaling pathways suppress
R-loop-driven instability. R-loop accumulation is notably toxic to
cells lacking these kinases or their downstream factors. For
example, deficiencies in the Fanconi anemia (FA) pathway lead to
hypersensitivity to R-loop-induced DNA damage (27). FA proteins such as FA group D2
protein bind R-loops and stalled forks, stabilizing them and
recruiting RNASEH2 to remove RNA:DNA hybrids. Breast cancer
susceptibility gene (BRCA)1, apart from its role in HR repair,
directly contributes to resolving R-loops by recruiting SETX to
stalled transcription sites (28).
Similarly, BRCA2 knockdown leads to abnormal R-loop accumulation,
DNA breaks and chromosomal aberrations (29).
Genomic instability in HCC
The relevance of these mechanisms in HCC is
supported by several observations. HCC tumors often harbor
mutations or dysregulation of DNA repair genes; for example,
tumor protein P53 is commonly mutated, whilst BRCA2
or ATM mutations are less frequent but occur in subsets.
Even without specific DDR mutations, chronic replication stress in
HCC likely indicates that HCC cells rely heavily on ATR-CHK1
signaling to survive (30,31).
If R-loops are prevalent in HCC cells, ATR would be
expected to remain continuously engaged in managing
replication-transcription conflicts. Recent research has shown that
HCC cells with high R-loop scores exhibit an activated defined set
of genes involved in the DDR pathway and are more reliant on stress
response pathways (1). When R-loop
levels become excessive, cells may enter senescence or apoptosis
because of irreparable DNA damage (1,6).
This could act as a barrier to tumor proliferation, which HCC cells
must overcome by enhancing R-loop tolerance. The aforementioned
THOC1 example illustrates that reducing the ability of HCC cells to
resolve R-loops leads to notable DNA damage and slow proliferation
in HCC cells (8).
Furthermore, etiological factors such as HBV can
exacerbate genomic instability in HCC via R-loops. HBV genome
integration and HBV X protein (HBx) expression interfere with host
DNA repair. Although not yet supported by evidence, it is
conceivable that HBV infection facilitates R-loop accumulation, for
instance, by causing replication stress or by HBx-mediated
downregulation of the RNASEH or ATR pathways (32).
In summary, R-loop-induced DNA damage is likely
associated with myriad sources of genomic instability in HCC. The
ability of HCC cells to survive and proliferate may depend on how
effectively they activate ATR/ATM and associated repair factors to
manage R-loop conflicts.
Signaling pathway and function of
R-loop in HCC
Aberrant R-loop accumulation can provoke DDRs,
contributing to genomic instability in liver cancer (6). THOC1 knockdown triggers R-loop
accumulation and DNA damage and curbs tumor proliferation (8). Excess R-loops can also initiate
innate immune signaling; cytosolic RNA:DNA hybrids excised from
nuclear R-loops activate the cGAS-STING pathway and related
receptors, leading to interferon regulatory factor 3-driven
inflammatory cascades and tumor cell apoptosis (13). Furthermore, the dynamics of R-loops
intersect with cancer metabolism. High R-loop levels are associated
with metabolic reprogramming, for instance, the HCC R-loop score
correlates with elevated lipid synthesis and an immunosuppressive
microenvironment (1). Elevated
lipid synthesis provides energy and membrane components to support
rapid HCC cell proliferation, while an immunosuppressive
microenvironment inhibits anti-tumor immune responses, together
promoting tumor growth and progression. Upregulation or increased
activity of specific R-loop regulators such as clathrin heavy chain
1 (CLTC) enhances fatty acid metabolism, providing energy and
biosynthetic substrates that support tumor growth and survival
(1). Finally, R-loops interact
with epigenetic mechanisms and result in both tumor promotion and
suppression; the chromatin modifier MTA2 creates R-loops to repress
BDH1. The increase in histone β-hydroxybutyrylation is a
direct consequence of BDH1 repression. BDH1 normally metabolizes
β-hydroxybutyrate (BHB); when BDH1 is repressed, BHB accumulates,
serving as a substrate for histone β-hydroxybutyrylation, which
promotes pro-tumorigenic gene expression and stemness (9).
Similarly, RNA-binding proteins such as G patch
domain-containing protein 4 (GPATCH4) guard ribosomal DNA loci by
unwinding nucleolar R-loops. This activity of GPATCH4 primarily
drives HCC progression. By unwinding nucleolar R-loops, it
maintains rRNA transcription and protein synthesis, supporting
rapid proliferation and tumor growth. However, GPATCH4 could also
represent a therapeutic vulnerability, as its inhibition may
increase R-loop accumulation, impair ribosome biogenesis, and
selectively stress HCC cells (33). Collectively, these findings
indicate that R-loops engage multiple signaling pathways, including
DNA damage repair, immune surveillance, metabolic adaptation and
epigenetic regulation, all of which can drive HCC progression or
present therapeutic vulnerabilities (34).
R-loop dysregulation and its dual role
in HCC
Dysregulation of R-loops in HCC is associated with
context-dependent pro- and antitumor effects (6). Abnormal R-loop accumulation causes
genomic instability and DNA breaks that, beyond repair thresholds,
trigger replicative senescence or cell death, thereby suppressing
cancer proliferation (6). For
example, unresolved R-loops arising from THOC1 depletion or GPATCH4
loss in HCC cells can lead to DNA damage, impaired proliferation
and heightened chemosensitivity (8). Thus, cancer cells often upregulate
R-loop-resolving factors such as THOC1, GPATCH4 and
tonicity-responsive enhancer-binding protein to avoid lethal R-loop
stress.
By contrast, dysregulation of R-loop formation can
drive HCC progression. R-loop formation can activate oncogenic
pathways or silence tumor suppressors (1,6). As
aforementioned, MTA2 induces R-loops at the BDH1 locus,
repressing differentiation-linked metabolism and sustaining cancer
stemness (9). Similarly, elevated
R-loop levels (via CLTC or other R-loop regulators) reprogram lipid
metabolism and correlate with an immunosuppressive
microenvironment, ultimately promoting HCC proliferation (1). Excessive R-loops activate cGAS-STING
signaling, inducing type I interferons and cytokines that recruit
cytotoxic immune cells, which directly kill tumor cells and enhance
immune surveillance, thereby constraining HCC growth and
progression (1).
R-loops and the TME
R-loop dysregulation contributes to the malignant
behavior of HCC cells and is closely associated with changes in the
TME. As aforementioned, MTA2 promote R-loop accumulation at the
BDH1 locus, silencing this metabolic gene via the
recruitment of HDAC2/CHD4 and enhancing cancer stemness and
invasiveness (9). Additionally,
elevated R-loop levels in HCC have been shown to upregulate lipid
metabolic pathways via regulators such as CLTC, promoting tumor
cell proliferation and tumor aggressiveness (1). These metabolic shifts reshape the TME
by increasing immunosuppressive signaling and nutrient competition
with paracancerous cells. Furthermore, unresolved R-loops generate
cytosolic DNA fragments that activate cGAS-STING signaling, drive
type I interferon responses and alter immune cell infiltration
(6,35). Hypoxic or inflammatory TMEs may
also exacerbate R-loop formation by impairing RNA processing or TOP
activity, further aggravating replication stress (3). Thus, R-loops and HCC TMEs exist in a
bidirectional relationship that fuels tumor progression.
Therapeutic strategies targeting R-loops in
HCC
Given their notable roles in genome stability and
gene regulation, R-loops and their regulatory machinery are
promising targets for any other cancer therapy methods (2). In HCC, where standard treatments
often achieve a relatively low rate of curing HCC in advanced
stages, exploiting R-loop vulnerabilities may offer novel
approaches. Therapeutic strategies can broadly aim to either reduce
pathological R-loops or increase the R-loop burden beyond a
tolerable threshold. The latter approach leverages the concept of
synthetic lethality, targeting the ‘Achilles heel’ of R-loop
processing in cancer cells.
Small-molecule approaches
TOP inhibitors
TOPs prevent excessive R-loop formation; therefore,
drugs that inhibit them can increase R-loop levels, leading to
cytotoxic effects (12). TOP1
inhibitors such as camptothecin and its derivatives trap TOP1 on
DNA, preventing it from relieving supercoils (36,37).
This increases negative supercoiling behind transcription complexes
and promotes R-loop formation (12). Trapped TOP1 also leads to transient
DNA breaks that are converted into lethal collisions in the
presence of R-loops (36). In HCC,
TOP inhibitors have shown some therapeutic efficacy in preclinical
experimental models, including cell lines and mouse xenograft
models, by inducing DNA damage; their ability to exacerbate R-loop
formation is a notable contributor to this damage (12).
ATR and CHK1 inhibitors
As described, ATR kinase is important for cancer
cells to survive R-loop-driven replication stress. Therefore,
inhibition of ATR can experimentally reveal the lethal consequences
of R-loops on tumor cells. Preclinical studies have shown strong
synergy between ATR inhibitors and R-loop accumulation (24,38).
For example, cancer cells with RNASEH2 dysfunction accumulate
R-loops and rely on ATR to mitigate the harmful effects of
accumulated R-loops; treating these cells with an ATR inhibitor
causes notable DNA breaks and apoptosis (1). The same study found that cells
harboring spliceosome mutations accumulate R-loops and show
selective sensitivity to ATR inhibitors (1). Translating this to HCC, ATR
inhibitors may be effective therapeutic agents in tumors with a
high R-loop load or impaired R-loop resolution. ATR inhibitors are
currently in clinical trials for solid tumors and hold potential
for use in combination with an agent that elevates R-loops for the
treatment of HCC (28).
Poly(ADP-ribose) polymerase (PARP)
inhibitors
PARP1 inhibitors are the preferred treatment option
for tumors with HR deficiencies. There is an emerging connection
between R-loop incidence and sensitivity to PARP inhibition
(39). R-loops can cause
replication fork collapse and one-ended DSBs, which normally
require HR for repair (40,41).
If HR is compromised, cells become markedly reliant on
PARP-mediated repair of replication-associated breaks, making them
vulnerable to PARP inhibition; in cancer and HCC, PARP inhibitors
exacerbate DNA damage, promote R-loop-induced genomic instability
and trigger apoptosis, selectively suppressing tumor proliferation
(39). Furthermore, R-loops can
sequester BRCA1 and prevent it from localizing to DNA damage sites,
impairing homologous recombination repair (12). Although BRCA1 is wild-type, its
functional unavailability creates an HR defect, increasing genomic
instability (12). In Ewing
sarcoma cells, transcription of the EWS RNA binding protein
1-friend leukemia integration 1 transcription factor fusion
oncogene leads to R-loops that sequester BRCA1, rendering the cells
sensitive to PARP inhibitors, similar to BRCA1 mutants (12). Similarly, conditions in HCC that
compromise HR repair can potentially be exploited using PARP
inhibitors.
Splicing/transcription modulators
Compounds that perturb RNA processing can induce
R-loops by leaving nascent transcripts unprocessed. For example,
spliceosome inhibitors such as sudemycin and H3B-8800 cause
widespread splicing disruption, leading to R-loop accumulation and
DNA damage in cancer cells (14).
These inhibitors are proposed for splice-mutant cancers because
such cells already have impaired RNA splicing, which increases
baseline R-loop formation. Inhibiting relevant pathways exacerbates
R-loop accumulation beyond a tolerable threshold, overwhelming DNA
repair and promoting cancer cell death (14). In HCC, direct spliceosome mutations
are rare; however, the use of a splicing modulator can artificially
create an R-loop overload.
Gene editing and molecular
strategies
In addition to conventional drugs, gene editing and
molecular biology techniques offer ways to directly manipulate
R-loops and their regulators in cancer cells. One innovative
approach involves the use of engineered RNASEH to eliminate R-loops
at specific genomic sites. Researchers have developed a
CRISPR/dCas9-RNASEH1 fusion protein, which can be guided by single
guide RNAs to genomic loci and locally resolve R-loops (42,43).
This tool has been used experimentally to target tumorigenic
R-loops; for instance, site-specific R-loop removal improved
cellular reprogramming efficiency in cultured cells (42,43).
In principle, a similar strategy could be used to
target a pathogenic R-loop in HCC; for example, if a particular
R-loop silences a tumor suppressor gene, a targeted RNASEH1 might
reactivate that gene. Conversely, gene editing can be used to
create synthetic vulnerabilities in tumor cells. The CRISPR
knockout of R-loop in HCC cells removes backup mechanisms that
normally compensate for each other, exacerbating DNA damage
(11). This heightened stress
increases sensitivity to DNA-damaging agents, such as TOP
inhibitors, ATR inhibitors and PARP inhibitors, which exploit
defects in replication and repair pathways. Another gene-focused
strategy targets non-coding RNAs that modulate R-loops.
RNASEH1-AS1, an antisense lncRNA upregulated in HCC, may attenuate
RNASEH1 function. Using antisense oligonucleotides to knockdown
RNASEH1-AS1 could free RNASEH1 to better resolve R-loops,
potentially reducing genomic instability in tumor cells.
Synthetic lethality and combination
strategies
The concept of synthetic lethality, in which two
non-lethal perturbations kill the cell when combined, is notably
relevant to R-loops (11). The
inherent DNA damage in cancer cells, arising from defects in HR
repair or RNASEH function, activates compensatory pathways like ATR
signaling. ATR responds to replication stress and R-loop-induced
DNA breaks, stabilizing replication forks and coordinating repair
to maintain cell survival. The present review has already discussed
some synthetic lethal pairs, such as RNASEH2 loss and ATR
inhibition (11) or splicing
mutation and ATR inhibition (14).
One combined strategy for therapeutic application to
HCC could be coupling ATR inhibitors with agents that increase
R-loop formation, as aforementioned. Another notable possibility is
the combination of R-loop targeting and immunotherapy. HCC
treatment has included immune checkpoint inhibitors (ICIs) in
recent years (44–46). There is evidence that tumors with
increased DNA damage can be more immunogenic due to the activation
of cGAS-STING and type I interferon responses (35). Excess R-loops can activate the
cGAS-STING pathway, as displaced single-stranded DNA or
R-loop-driven DNA breaks generate ligands for this pathway
(1).
A transient burst of R-loop-induced damage may
acutely increase the immunostimulatory signals via the activated
cGAS-STING and type I interferon pathways. Therefore, a short
course of R-loop-inducing therapy can enhance tumor immunogenicity
by increasing DNA damage and immune signaling, making individual
tumors, including HCC, more susceptible to immune checkpoint
inhibitor-mediated targeting (13,47).
Another scenario using synthetic lethality could
involve targeting metabolic pathways that become lethal when the
number of R-loops is high. Single-cell analysis by Chen et
al (1) highlighted a link
between R-loops and lipid metabolism in HCC. The study identified
an 8-gene signature of fatty acid metabolism-related R-loop
regulator genes; inhibiting CLTC reduces fatty acid metabolism,
decreasing lipid synthesis and accumulation in HCC cells. This
metabolic disruption impairs tumor cell proliferation and overall
tumor growth (1).
Challenges in targeting R-loops
Although targeting R-loop biology holds promise for
HCC therapy, notable challenges must be addressed for such
strategies to be effective and safe.
Detection and measurement of
R-loops
The accurate detection of R-loops in cells and
tissues is important. The primary method, DNA-RNA
immunoprecipitation (DRIP) using the S9.6 monoclonal antibody, has
known limitations in terms of specificity (48). Thus, a key challenge is the
identification of patients with HCC who have high R-loop levels or
dysfunctional R-loop regulation. Without reliable diagnostic
assays, patient stratification for R-loop-targeted therapy is
difficult.
Specificity and off-target
effects
Targeting R-loops can affect normal cells because
they are not unique to cancer cells. All rapidly dividing cells
generate R-loops (49). Therapies
such as ATR inhibitors or TOP poisons also affect healthy
proliferating tissues, causing side effects, such as bone marrow
suppression (anemia, neutropenia), gastrointestinal toxicity
(nausea, diarrhea) and hair loss (12). However, cancer selectivity remains
a major challenge. Cancer selectivity is observed in cells with
~2–3 times higher R-loop levels than normal tissues, providing a
therapeutic window for targeting R-loop-dependent
vulnerabilities.
Resistance mechanisms
As with any cancer therapy, tumors may develop
resistance to R-loop-targeting strategies. There are several
plausible resistance routes: i) A tumor under ATR inhibitor
pressure may upregulate compensatory pathways such as ATM or
DNA-dependent protein kinase catalytic subunit (50); ii) a HCC cell may slow its
proliferation or transcription rate to manage R-loop stress
(51); iii) if a small molecule
such as an ATR inhibitor is used, classical drug-specific
resistance mechanisms can occur (52,53);
and iv) the liver TME might shield HCC cells from treatments.
Therapeutic index and safety
A number of R-loop-targeting approaches run the risk
of causing global DNA damage if not controlled. It is important to
avoid therapies that could induce notable genomic instability in
healthy tissues, resulting in secondary malignancies or organ
failure.
Limitations in current knowledge
The mechanisms of R-loops in HCC remain yet to be
fully elucidated. Most mechanistic insights have been obtained from
cell-line studies or other cancer types such as breast cancer,
colorectal cancer and leukemia. Care must be taken when suggesting
that HCC has unique aspects that could influence R-loop dynamics in
ways that have not yet been fully understood.
Future directions
The intersection of R-loop biology and cancer
research is a rapidly advancing field (2,3).
Several promising avenues of investigation are emerging:
Integrative multi-omics profiling of
R-loops in HCC
A more comprehensive understanding of R-loop
dynamics in HCC may be achieved by integrating multiple layers of
data: Genomics, transcriptomics, epigenomics and proteomics. At
single-cell resolution, it is possible to profile transcription and
chromatin states, while R-loop mapping remains largely bulk;
emerging methods like scDRIP-seq combined with single-cell ATAC-seq
or RNA-seq enable integrated analyses. These investigative methods
are capable of refining the current understanding of where R-loops
form in HCC genomes, as well as their causes and consequences.
Identifying new therapeutic targets in
the R-loop pathway
The present study has focused on known molecules
that interact with R-loops, such as ATR and RNASEH; however, future
studies may unveil HCC-specific interactions. The list of R-loop
regulatory genes used in recent HCC studies (1) provides a starting point for
identifying novel HCC-specific interactions that includes
unexpected genes that are not previously linked to canonical R-loop
regulation or cancer progression pathways.
R-loop biomarkers for patient
stratification
As therapies that exploit R-loops are introduced to
clinical settings, the parallel development of biomarkers is
needed. Potential biomarkers could be the expression of an R-loop
resolver, mutations in an R-loop-related gene or the direct
measurement of RNA:DNA hybrids in tumor samples, if feasible.
Overcoming challenges: Improved
delivery and combination regimens
Future research should aim to overcome the
challenges outlined in the present review. Nanoparticle- or
virus-based delivery of gene therapies may be refined for drug
delivery. With small-molecule approaches, prodrugs that are
activated in the TME can localize the R-loop-targeting effect
(52,53). This refers to a subset of drugs,
specifically those designed to target R-loops or exploit
R-loop-associated vulnerabilities.
Exploring the R-loop-immune
interface
The relationship between R-loops and the immune
system is an area of study with direct implications for HCC
(1,13). Future studies could clarify this by
manipulating R-loops in vivo and observing immune
responses.
Clinical translation and trials
Finally, the culmination of these future directions
of study will be clinical trials that test R-loop-targeted
therapies in patients with HCC. For instance, a trial of an ATR
inhibitor in combination with an approved HCC therapy could be
designed for patients whose tumors exhibit a predefined R-loop
signature.
Study limitations
The present review had several limitations that
warrant consideration. First, most mechanistic insights into
R-loops in HCC have been derived from in vitro studies or
non-HCC cancer models (6).
Although previous data from single-cell and bulk transcriptomic
analyses have suggested altered R-loop dynamics in HCC, these
findings require validation using in vivo models and human
tissue samples. Second, current methods for R-loop detection, such
as DRIP-seq using the S9.6 antibody, suffer from limited
specificity and potential cross-reactivity with double-stranded
RNA, which may lead to the misinterpretation of hybrid prevalence
and localization. Furthermore, the heterogeneity of HCC, driven by
diverse etiologies such as HBV infection, metabolic syndrome and
alcohol-related liver disease, poses challenges in generalizing
R-loop-associated mechanisms across all patient subsets. Finally,
although the concept of targeting R-loops therapeutically, such as
via ATR or PARP inhibition, is compelling, clinical evidence in HCC
remains lacking of evidence of the efficacy of these inhibitors
in vivo in HCC, and potential off-target effects or
resistance mechanisms remain to be fully elucidated. Future studies
should focus on refining detection techniques, validating key
pathways in patient-derived models and integrating R-loop profiling
into prospective therapeutic trials.
Conclusion
R-loops represent a notable interface between
transcriptional regulation and genomic stability in cancer.
Understanding R-loop biology is particularly relevant in HCC, a
cancer characterized by genomic instability and complex molecular
drivers. The present review provided an overview of R-loop
formation and function (Fig. 1)
and discussed the mechanistic impact of R-loops on transcriptional
regulation and genome stability in HCC. The present review has
discussed how HCC cells manage or mismanage R-loops and how this
knowledge may provide the foundation for novel therapeutic
strategies.
By integrating advanced genomic tools, identifying
novel targets and biomarkers and addressing current challenges,
therapies can be developed that either exploit the toxic potential
of R-loops to destroy cancer cells or correct the underlying
dysregulation of R-loops that contributes to HCC development.
Ultimately, these efforts aim to improve the outcomes of patients
with HCC by composing novel strategies of cancer therapy that
target the fundamental intersection of transcription and genome
stability.
Acknowledgements
Not applicable.
Funding
The present research was funded by Chang Gung Memorial Hospital
(grant nos. CMRPG8Q0181 and CORPG8N0241).
Availability of data and materials
Not applicable.
Authors' contributions
CHH was responsible for writing the original draft,
conceptualization and funding acquisition. YWL revised the
manuscript. PCC was responsible for editing the manuscript. Data
authentication is not applicable. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ATM
|
ataxia telangiectasia mutated
|
|
ATR
|
ataxia telangiectasia and
Rad3-related protein
|
|
BDH1
|
β-hydroxybutyrate dehydrogenase 1
|
|
BRCA
|
breast cancer susceptibility gene
|
|
CHK1
|
checkpoint kinase 1
|
|
DDR
|
DNA damage response
|
|
DRIP
|
DNA-RNA immunoprecipitation
|
|
DSB
|
double-strand break
|
|
FA
|
Fanconi anemia
|
|
HBV
|
hepatitis B virus
|
|
HCC
|
hepatocellular carcinoma
|
|
HDAC
|
histone deacetylase
|
|
HR
|
homologous recombination
|
|
lncRNA
|
long non-coding RNA
|
|
MTA2
|
metastasis-associated protein 2
|
|
PARP
|
poly(ADP-ribose) polymerase
|
|
RNASEH
|
ribonuclease H
|
|
SETX
|
Senataxin
|
|
THOC
|
THO complex
|
|
TOP1
|
topoisomerase I
|
References
|
1
|
Chen L, Yang H, Wei X, Liu J, Han X, Zhang
C, Liu Y, Zhang Y, Xu Y, Li Y, et al: Integrated single-cell and
bulk transcriptome analysis of R-loop score-based signature with
regard to immune microenvironment, lipid metabolism and prognosis
in HCC. Front Immunol. 15:14873722025. View Article : Google Scholar
|
|
2
|
Li F, Zafar A, Luo L, Denning AM, Gu J,
Bennett A, Yuan F and Zhang Y: R-Loops in genome instability and
cancer. Cancers (Basel). 15:49862023. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rinaldi C, Pizzul P, Longhese MP and
Bonetti D: Sensing R-Loop-Associated DNA damage to safeguard genome
stability. Front Cell Dev Biol. 8:6181572021. View Article : Google Scholar
|
|
4
|
Li D, Shao F, Li X, Yu Q, Wu R, Wang J,
Wang Z, Wusiman D, Ye L, Guo Y, et al: Advancements and challenges
of R-loops in cancers: Biological insights and future directions.
Cancer Lett. 610:2173592025. View Article : Google Scholar
|
|
5
|
Qiu Y, Man C, Zhu L, Zhang S, Wang X, Gong
D and Fan Y: R-loops' m6A modification and its roles in cancers.
Mol Cancer. 23:2322024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Baek H, Park SU and Kim J: Emerging role
for R-loop formation in hepatocellular carcinoma. Genes Genomics.
45:543–551. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Peng TW, Ma QF, Li J, Wang X, Zhang CH, Ma
J, Li JY, Wang W, Zhu CL and Liu XH: HBV promotes its replication
by up-regulating RAD51C gene expression. Sci Rep. 14:26072024.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cai S, Bai Y, Wang H, Zhao Z, Ding X,
Zhang H, Zhang X, Liu Y, Jia Y, Li Y, et al: Knockdown of THOC1
reduces the proliferation of hepatocellular carcinoma and increases
the sensitivity to cisplatin. J Exp Clin Cancer Res. 39:1352020.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang H, Chang Z, Qin LN, Liang B, Han JX,
Qiao KL, Yang C, Liu YR, Zhou HG and Sun T: MTA2 triggered R-loop
trans-regulates BDH1-mediated β-hydroxybutyrylation and potentiates
propagation of hepatocellular carcinoma stem cells. Signal
Transduct Target Ther. 6:1352021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sun J, Li Y, Tian H, Chen H, Li J and Li
Z: Comprehensive analysis identifies long non-coding RNA
RNASEH1-AS1 as a potential prognostic biomarker and oncogenic
target in hepatocellular carcinoma. Am J Cancer Res. 14:996–1014.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang C, Wang G, Feng X, Shepherd P, Zhang
J, Tang M, Chen Z, Srivastava M, McLaughlin ME, Navone NM, et al:
Genome-wide CRISPR screens reveal synthetic lethality of RNASEH2
deficiency and ATR inhibition. Oncogene. 38:2451–2463. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Saha S and Pommier Y: R-loops, type I
topoisomerases and cancer. NAR Cancer. 5:zcad0132023. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Crossley MP, Song C, Bocek MJ, Choi JH,
Kousouros JN, Sathirachinda A, Lin C, Brickner JR, Bai G, Lans H,
et al: R-loop-derived cytoplasmic RNA-DNA hybrids activate an
immune response. Nature. 613:187–194. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Flach J, Jann JC, Knaflic A, Riabov V,
Streuer A, Altrock E, Xu Q, Schmitt N, Obländer J, Nowak V, et al:
Replication stress signaling is a therapeutic target in
myelodysplastic syndromes with splicing factor mutations.
Haematologica. 106:2906–2917. 2021. View Article : Google Scholar
|
|
15
|
Yang S, Winstone L, Mondal S and Wu Y:
Helicases in R-loop formation and resolution. J Biol Chem.
299:1053072023. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kellner V and Luke B: Molecular and
physiological consequences of faulty eukaryotic ribonucleotide
excision repair. EMBO J. 39:e1023092020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hasanova Z, Klapstova V, Porrua O, Stefl R
and Sebesta M: Human senataxin is a bona fide R-loop resolving
enzyme and transcription termination factor. Nucleic Acids Res.
51:2818–2837. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Song C, Hotz-Wagenblatt A, Voit R and
Grummt I: SIRT7 and the DEAD-box helicase DDX21 cooperate to
resolve genomic R loops and safeguard genome stability. Genes Dev.
31:1370–1381. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hodroj D, Serhal K and Maiorano D: Ddx19
links mRNA nuclear export with progression of transcription and
replication and suppresses genomic instability upon DNA damage in
proliferating cells. Nucleus. 8:489–495. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mérida-Cerro JA, Maraver-Cárdenas P,
Rondón AG and Aguilera A: Rat1 promotes premature transcription
termination at R-loops. Nucleic Acids Res. 52:3623–3635. 2024.
View Article : Google Scholar
|
|
21
|
Xu C, Li C, Chen J, Xiong Y, Qiao Z, Fan
P, Li C, Ma S, Liu J, Song A, et al: R-loop-dependent
promoter-proximal termination ensures genome stability. Nature.
621:610–619. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang Y, Qiu R, Zhao S, Shen L, Tang B,
Weng Q, Xu Z, Zheng L, Chen W, Shu G, et al: SMYD3 associates with
the NuRD (MTA1/2) complex to regulate transcription and promote
proliferation and invasiveness in hepatocellular carcinoma cells.
BMC Biol. 20:2942022. View Article : Google Scholar
|
|
23
|
Zhou T, Cheng X, He Y, Xie Y, Xu F, Xu Y
and Huang W: Function and mechanism of histone
β-hydroxybutyrylation in health and disease. Front Immunol.
13:9812852022. View Article : Google Scholar
|
|
24
|
Liu J, Li F, Cao Y, Lv Y, Lei K, Tu Z,
Gong C, Wang H, Liu F and Huang K: Mechanisms underlining R-loop
biology and implications for human disease. Front Cell Dev Biol.
13:15377312025. View Article : Google Scholar
|
|
25
|
Matos DA, Zhang JM, Ouyang J, Nguyen HD,
Genois MM and Zou L: ATR protects the genome against R Loops
through a MUS81-triggered feedback loop. Mol Cell. 77:514–527.e4.
2020. View Article : Google Scholar
|
|
26
|
González-Marín B, Calderón-Segura ME and
Sekelsky J: ATM/Chk2 and ATR/Chk1 pathways respond to DNA damage
induced by Movento® 240SC and Envidor® 240SC
Keto-enol insecticides in the germarium of drosophila melanogaster.
Toxics. 11:7542023. View Article : Google Scholar
|
|
27
|
Okamoto Y, Hejna J and Takata M:
Regulation of R-loops and genome instability in Fanconi anemia. J
Biochem. 165:465–470. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sharma AB, Ramlee MK, Kosmin J, Higgs MR,
Wolstenholme A, Ronson GE, Jones D, Ebner D, Shamkhi N, Sims D, et
al: C16orf72/HAPSTR1/TAPR1 functions with BRCA1/Senataxin to
modulate replication-associated R-loops and confer resistance to
PARP disruption. Nat Commun. 14:50032023. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang Y, Ma B, Liu X, Gao G, Che Z, Fan M,
Meng S, Zhao X, Sugimura R, Cao H, et al: ZFP281-BRCA2 prevents
R-loop accumulation during DNA replication. Nat Commun.
13:34932022. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Smith MA, Van Alsten SC, Walens A,
Damrauer JS, Maduekwe UN, Broaddus RR, Love MI, Troester MA and
Hoadley KA: DNA damage repair classifier defines distinct groups in
hepatocellular carcinoma. Cancers (Basel). 14:42822022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wu X, Xu S, Wang P, Wang ZQ, Chen H, Xu X
and Peng B: ASPM promotes ATR-CHK1 activation and stabilizes
stalled replication forks in response to replication stress. Proc
Natl Acad Sci USA. 119:e22037831192022. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li D, Hamadalnil Y and Tu T: Hepatitis B
viral protein HBx: Roles in viral replication and
hepatocarcinogenesis. Viruses. 16:13612024. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhao YM, Jiang Y, Wang JZ, Cao S, Zhu H,
Wang WK, Yu J, Liu J and Hui J: GPATCH4 functions as a regulator of
nucleolar R-loops in hepatocellular carcinoma cells. Nucleic Acids
Res. 53:gkaf4382025. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tan SLW, Chadha S, Liu Y, Gabasova E,
Perera D, Ahmed K, Constantinou S, Renaudin X, Lee M, Aebersold R
and Venkitaraman AR: A class of environmental and endogenous toxins
induces BRCA2 haploinsufficiency and genome instability. Cell.
169:1105–1118.e15. 2017. View Article : Google Scholar
|
|
35
|
Du H, Xu T and Cui M: cGAS-STING signaling
in cancer immunity and immunotherapy. Biomed Pharmacother.
133:1109722021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Promonet A, Padioleau I, Liu Y, Sanz L,
Biernacka A, Schmitz AL, Skrzypczak M, Sarrazin A, Mettling C,
Rowicka M, et al: Topoisomerase 1 prevents replication stress at
R-loop-enriched transcription termination sites. Nat Commun.
11:39402020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Duardo RC, Marinello J, Russo M, Morelli
S, Pepe S, Guerra F, Gómez-González B, Aguilera A and Capranico G:
Human DNA topoisomerase I poisoning causes R loop-mediated genome
instability attenuated by transcription factor IIS. Sci Adv.
10:eadm81962024. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Huang TT, Chiang CY, Nair JR, Wilson KM,
Cheng K and Lee JM: AKT1 interacts with DHX9 to Mitigate R
Loop-Induced Replication Stress in Ovarian Cancer. Cancer Res.
84:887–904. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Petropoulos M, Karamichali A, Rossetti GG,
Freudenmann A, Iacovino LG, Dionellis VS, Sotiriou SK and
Halazonetis TD: Transcription-replication conflicts underlie
sensitivity to PARP inhibitors. Nature. 628:433–441. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Pavani R, Tripathi V, Vrtis KB, Zong D,
Chari R, Callen E, Pankajam AV, Zhen G, Matos-Rodrigues G, Yang J,
et al: Structure and repair of replication-coupled DNA breaks.
Science. 385:eado38672024. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Nickoloff JA, Sharma N, Taylor L, Allen SJ
and Hromas R: The safe path at the fork: Ensuring
replication-associated DNA double-strand breaks are repaired by
homologous recombination. Front Genet. 12:7480332021. View Article : Google Scholar
|
|
42
|
Khosraviani N, Abraham K, Chan J and
Mekhail K: Locus-associated R-loop repression in cis. bioRxiv.
2021.DOI: 10.1101/2021.05.25.445644. View Article : Google Scholar
|
|
43
|
Li Y, Song Y, Xu W, Li Q, Wang X, Li K,
Wang J, Liu Z, Velychko S, Ye R, et al: R-loops coordinate with
SOX2 in regulating reprogramming to pluripotency. Sci Adv.
6:eaba07772020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Boilève J, Guimas V, David A, Bailly C and
Touchefeu Y: Combining immune checkpoint inhibitors with
loco-regional treatments in hepatocellular carcinoma: Ready for
prime time? Curr Oncol. 31:3199–3211. 2024. View Article : Google Scholar
|
|
45
|
Childs A, Aidoo-Micah G, Maini MK and
Meyer T: Immunotherapy for hepatocellular carcinoma. JHEP Rep.
6:1011302024. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hao L, Li S, Ye F, Wang H, Zhong Y, Zhang
X, Hu X and Huang X: The current status and future of
targeted-immune combination for hepatocellular carcinoma. Front
Immunol. 15:14189652024. View Article : Google Scholar
|
|
47
|
Maxwell MB, Hom-Tedla MS, Yi J, Li S,
Rivera SA, Yu J, Burns MJ, McRae HM, Stevenson BT, Coakley KE, et
al: ARID1A suppresses R-loop-mediated STING-type I interferon
pathway activation of anti-tumor immunity. Cell. 187:3390–3408.e19.
2024. View Article : Google Scholar
|
|
48
|
Bou-Nader C, Bothra A, Garboczi DN, Leppla
SH and Zhang J: Structural basis of R-loop recognition by the S9.6
monoclonal antibody. Nat Commun. 13:16412022. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Jazinaki MS, Bahari H, Rashidmayvan M,
Arabi SM, Rahnama I and Malekahmadi M: The effects of raspberry
consumption on lipid profile and blood pressure in adults: A
systematic review and meta-analysis. Food Sci Nutr. 12:2259–2278.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Turchick A, Zimmermann A, Chiu LY, Dahmen
H, Elenbaas B, Zenke FT, Blaukat A and Vassilev LT: Selective
inhibition of ATM-dependent double-strand break repair and
checkpoint control synergistically enhances the efficacy of ATR
inhibitors. Mol Cancer Ther. 22:859–872. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Lam FC, Kong YW, Huang Q, Vu Han TL, Maffa
AD, Kasper EM and Yaffe MB: BRD4 prevents the accumulation of
R-loops and protects against transcription-replication collision
events and DNA damage. Nat Commun. 11:40832020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Baxter JS, Zatreanu D, Pettitt SJ and Lord
CJ: Resistance to DNA repair inhibitors in cancer. Mol Oncol.
16:3811–3827. 2022. View Article : Google Scholar
|
|
53
|
O'Leary PC, Chen H, Doruk YU, Williamson
T, Polacco B, McNeal AS, Shenoy T, Kale N, Carnevale J, Stevenson
E, et al: Resistance to ATR inhibitors is mediated by loss of the
nonsense-mediated decay factor UPF2. Cancer Res. 82:3950–3961.
2022. View Article : Google Scholar
|