Introduction
Homocystinuria (HCU) is a rare genetic metabolic
disorder that results from the deficiency of the enzyme
cystathionine β-synthase (CBS), which is a key player in the
metabolism of the amino acid methionine. As it is an autosomal
recessive genetic disease, a child must inherit two defective
copies of the gene from the parents to be affected. Other enzymes
involved in homocysteine metabolism include
methylenetetrahydrofolate reductase (MTHFR) and methionine synthase
(MS), especially in vitamin B12 or folate deficiency. This CBS
enzymatic deficiency leads to the accumulation of homocysteine and
methionine in the blood and urine of affected patients (1), eventually leading to a wide range of
notable and irreversible multi-organ complications.
HCU imposes a notable and multifaceted healthcare
burden, particularly when misdiagnosed in early childhood or when
inadequate metabolic control is achieved. HCU requires lifelong
management, strict dietary restrictions, multivitamin
supplementation and frequent biochemical monitoring, all of which
place financial and logistical burdens on patients, families and
healthcare systems. This burden is escalated by the required
management and hospitalization of patients due to disease-related
complications with often life-threatening consequences. Recent
population-based analysis further quantified this burden,
documenting substantial rates of thromboembolism, ophthalmic
morbidity and healthcare utilization in classical HCU (2).
Foundational syntheses, such as that reported by
Schneede et al (3), have
established the enzymatic basis of HCU, emphasized homocysteine as
a pathogenic vascular risk factor and summarized the then-standard
treatments: i) Dietary methionine restriction; ii) supplementation
with high-dose pyridoxine, folate or B12; and iii) betaine.
Current therapies for HCU such as dietary methionine
restriction and supplementation with vitamins B6, B12 and folate
often fail to prevent complications, especially in patients who are
non-responsive to treatment. With the understanding of disease
pathogenesis, patient-focused novel interventions that correct
fundamental metabolic imbalances, such as enzyme replacement, gene
therapy and RNA-based treatments, have become important (2). The present review revisits the
foundational enzymology of HCU in the context of modern
genotype-phenotype insights and incorporates novel epidemiological
data quantifying the healthcare burden of classical HCU. The
present review also integrates developments through early 2025,
including the discontinuation of the pegtarviliase program and the
rebranding of CDX-6512 as SYNT-202, with corresponding updates from
clinical trial data, regulatory announcements and pipeline
disclosures.
Epidemiology
Consanguinity and gene mutations are the predominant
risk factors for HCU, which explains the high incidence of HCU in
some populations, such as Irish and Middle Eastern populations
(4,5).
Although HCU is a rare genetic enzymatic disorder,
with global prevalence estimates of 1 in 300,000 individuals, its
prevalence rates vary considerably across different regions due to
factors such as genetic mutations and consanguinity (4). Qatar has the highest known incidence
at ~1 in 1,800 births, most likely due to the high rates of
consanguineous marriages (4),
while Kuwait and the eastern area of Saudi Arabia have reported
comparably lower rates of 1 in 43,000 (6,7).
In the United States, the estimated reported
incidence is 1 in 100,000 (8);
however, a previous analysis has suggested that the actual
prevalence may be ≥10 times this estimate, most likely because of
underdiagnosis (8). In Norway, a
study reported a prevalence of 1 in 6,400 (9), whereas surveys in Ireland and Germany
have reported incidences of ~1 in 65,000 and 1 in 17,800 births,
respectively (5,10).
Clinical presentation and complications
The clinical presentation of HCU spans ocular,
skeletal, neurologic and thromboembolic complications (11–15)
and often differs between pediatric and adult patients, influenced
by the severity of enzyme deficiency and responsiveness to therapy
(16,17).
Thromboembolism is a life-threatening complication
of homocysteine accumulation. Hyperhomocysteinemia damages the
vascular endothelium, causing oxidative stress, inflammation and
endothelial dysfunction, which leads to platelet activation and
coagulation cascade dysregulation, markedly increasing the risk of
arterial and venous thrombosis. Furthermore, homocysteine impairs
nitric oxide production, reducing vasodilation and contributing to
vascular stiffness, leaving patients at an increased risk of
developing deep vein thrombosis (DVT), pulmonary embolism (PE),
myocardial infarction and stroke, even at a young age (11,12).
An ocular complication of hyperhomocysteinemia is
ectopia lentis (eye lens dislocation). Elevated homocysteine levels
interfere with collagen cross-linking, thus disrupting the normal
connective tissue integrity of the zonular fibers, which are
fibrillin-rich structures that anchor the lens within the eye,
leading to ectopia lentis. The displaced lens impairs vision and
predisposes the patient to secondary complications, such as myopia,
glaucoma, retinal detachment and eventual permanent vision loss.
Ectopia lentis typically develops in early childhood and is often
bilateral (13).
In the same context, impaired collagen cross-linking
and connective tissue integrity lead to impaired bone matrix
formation that resembles the features of Marfanoid body habitus,
such as long limbs, scoliosis and pectus deformities, as well as
osteopenia or osteoporosis, and increases the risk of fractures.
These skeletal manifestations are typically more evident during
growth spurts in childhood and adolescence (14).
Neurological complications occur because HCU induces
oxidative stress, impairs methylation processes that are important
for neurotransmitter synthesis and damages the vascular
endothelium, thereby increasing the risk of cerebral microvascular
injury and stroke. This can result in developmental delays, mental
disability, seizures and motor dysfunction during childhood. In
adolescents and adults, untreated disease may manifest as
behavioral disorders, psychiatric symptoms such as depression,
anxiety and psychosis, or cognitive decline (15).
In pediatric patients, developmental delay, failure
to thrive and hypotonia are the earliest signs. Intellectual
disability is commonly detected in untreated children in
combination with behavioral disorders. Ectopia lentis appears
within the first 10 years of life and is usually associated with
severe myopia or glaucoma, while musculoskeletal anomalies can
emerge during growth. Children may also experience seizures or
exhibit a clumsy gait due to neurological disorders (16).
In adults, especially those with delayed treatment
during childhood, thromboembolic events such as DVT, PE and stroke
frequently predominate, even in the absence of known risk factors.
Psychiatric symptoms such as depression, anxiety and psychosis can
also show delayed manifestations in adults with low treatment
responsiveness, who may have impaired intellectual or physical
signs (17).
Molecular background of HCU
Transsulfuration pathway
The transsulfuration pathway is a hepatic pathway
that facilitates the conversion of homocysteine into cysteine to
detoxify excess homocysteine, and contributes to the synthesis of
glutathione, taurine and coenzyme A, which are important as
antioxidants, for bile acid metabolism and for energy production
(18). The transsulfuration
pathway starts with the CBS enzyme, which catalyzes the conversion
of homocysteine and serine into cystathionine, using pyridoxal
5′-phosphate (PLP), the active form of pyridoxine, as a cofactor.
Afterwards, cystathionine γ-lyase (CGL) breaks down cystathionine
into cysteine, α-ketobutyrate and ammonia. Absolute deficiency or
malfunction of CBS disrupts this pathway, leading to elevated blood
and urine levels of homocysteine and methionine, and reduced
cysteine levels (18).
CBS mutations show genotype-phenotype variability,
influencing the severity of the disease and the responsiveness to
pyridoxine therapy. High-dose B6 supplementation may be beneficial
in the case of partial CBS activity to enhance residual enzyme
function, while total enzyme deficiency requires a
methionine-restricted diet, cysteine-supplemented diets and
betaine, folate and B12 supplementation to promote alternate
remethylation pathways (19,20).
Remethylation pathway
The remethylation pathway works in parallel with the
transsulfuration pathway to normalize plasma homocysteine levels,
especially in the brain where the transsulfuration pathway is
inactive. In this reaction, homocysteine is remethylated to
methionine, which is necessary for protein synthesis, through
catalysis by MS, which requires B12 as a cofactor and employs
5-methyltetrahydrofolate produced by MTHFR as the methyl group
donor (21).
Another hepatic remethylation route involves the
enzyme betaine-homocysteine methyltransferase (BHMT), which uses
betaine as a methyl donor to remethylate homocysteine into
methionine. The BHMT pathway provides an alternative route for
homocysteine metabolism in cases of folate or B12 deficiency or
genetic disorders that affect the primary route (22).
These interrelated transsulfuration and
remethylation pathways are summarized in Fig. 1, which illustrates the metabolic
conversion of homocysteine and the key cofactors involved.
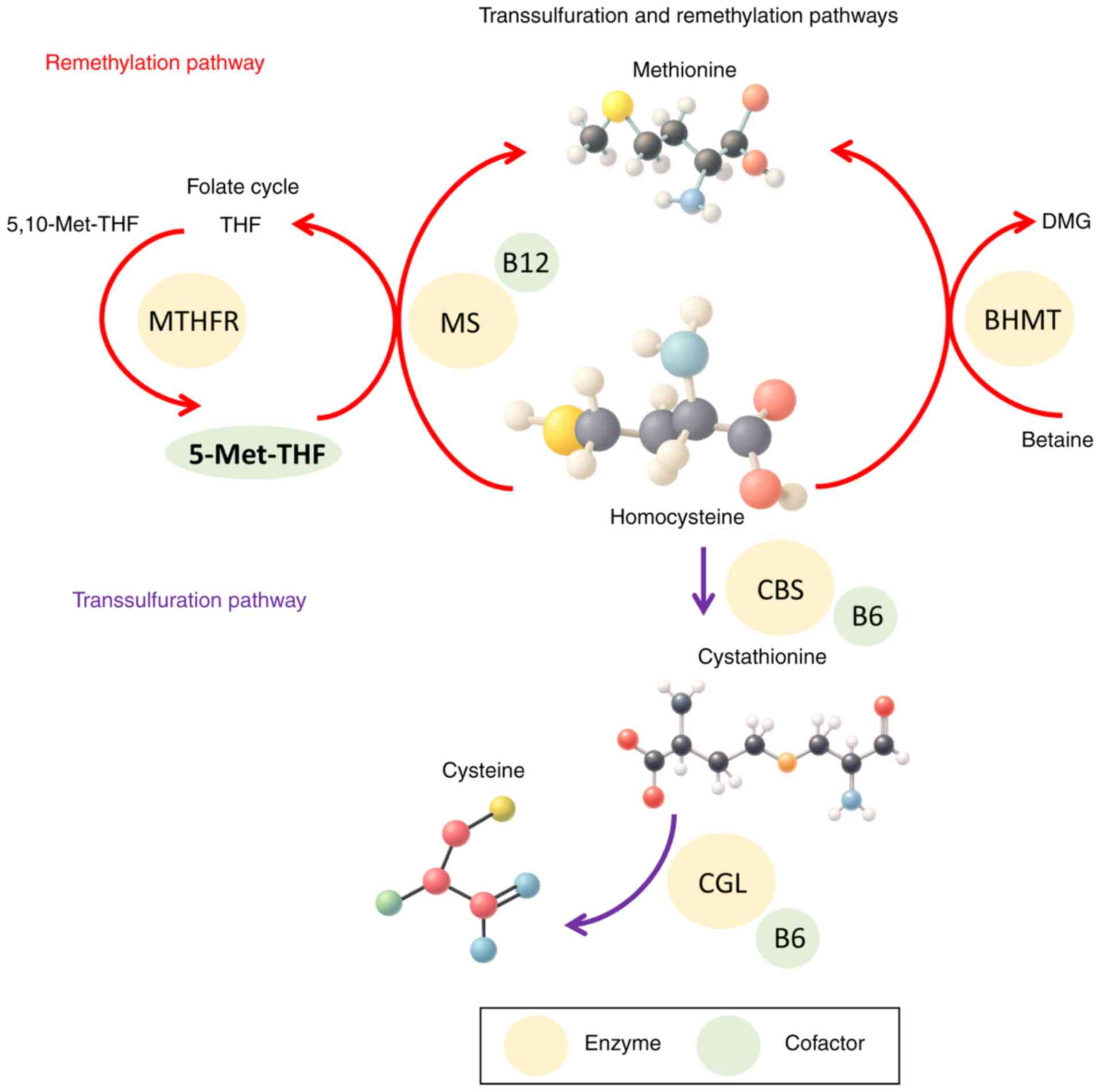 | Figure 1.Molecular pathways of homocysteine
metabolism. The figure illustrates the normal transsulfuration and
remethylation pathways of homocysteine metabolism, as well as the
folate cycle. Key enzymes include CBS, MS, MTHFR and BHMT.
Important cofactors such as pyridoxal phosphate (vitamin B6),
folate (5-Met-THF) and methylcobalamin (vitamin B12) are indicated.
Substrates and intermediates shown include homocysteine,
methionine, cystathionine, cysteine, 5,10-Met-THF and DMG. THF,
tetrahydrofolate; MTHFR, methylenetetrahydrofolate reductase; MS,
methionine synthase; DMG, dimethylglycine; BHMT,
betaine-homocysteine methyltransferase; CBS, cystathionine
β-synthase; CGL, cystathionine γ-lyase; 5-Met-THF,
5-methyltetrahydrofolate; 5,10-Met-THF,
5,10-methylenetetrahydrofolate. |
Genetic variant associations
Genetic variants of HCU serve important roles in
determining case severity, response to treatment and clinical
outcomes (23,24). The CBS gene is located on
chromosome 21q22.3 (23). More
than 200 pathogenic mutations have been identified in CBS,
resulting in varying degrees of residual enzyme activity, which
markedly influence both phenotype and clinical expression (24).
The I278T mutation, which is widely prevalent
in Western and Far Eastern populations, has been extensively
studied. Patients homozygous for I278T often retain residual
enzyme activity and respond to B6 therapy, whereas the G307S
mutation, common in Irish and Australian populations, is associated
with a severe phenotype and non-responsiveness to B6 (24–26).
Individuals with partial enzymatic activity tend to
show delayed manifestations, milder laboratory abnormalities and
more favorable clinical outcomes. This genotype-phenotype
association is an important consideration in guiding initial
vitamin B6 therapy and in predicting disease prognosis (25).
Variants of other genes involved in homocysteine
metabolism, such as MTHFR,
5-methyltetrahydrofolate-homocysteine methyltransferase
(MTR), MTR reductase (MTRR) and metabolism of
cobalamin associated C, also contribute to homocysteine toxicity.
The MTHFR C677T polymorphism produces a thermolabile enzyme with
reduced activity, leading to mild-to-moderate hyperhomocysteinemia,
especially in the context of low folate levels (27–29).
While not typically causing classical HCU, these variants can
worsen biochemical imbalances in genetically predisposed
individuals (27–29).
Genetic workups, even in asymptomatic individuals,
are valuable for early diagnosis and for predicting vitamin B6
responsiveness (27–29).
Table I summarizes
the key genetic variants associated with classical HCU,
highlighting their impact on enzymatic activity, B6 responsiveness
and clinical severity.
 | Table I.Summary of key genetic variants of
CBS and related genes, their geographic prevalence, enzyme
activity, vitamin B6 responsiveness and associated clinical
severity of HCU. |
Table I.
Summary of key genetic variants of
CBS and related genes, their geographic prevalence, enzyme
activity, vitamin B6 responsiveness and associated clinical
severity of HCU.
| Mutation | Geographic
prevalence | B6
responsiveness | Residual CBS
activity | Clinical severity
(phenotypic severity of homocystinuria) |
|---|
| I278T | Western (Europe and
North America) and Far Eastern (China) | Responsive | Partial | Mild to
moderate |
| G307S | Ireland and
Australia | Non-responsive | Minimal to
none | Severe, early
onset |
| Other CBS
variants | Global | Variable | Variable | Variable |
| (p.R125Q,
p.R266K, p.R336C and c.1224-2A>C) |
|
|
|
|
| MTHFR
C677T | Global (especially
Europe and Asia) | No (not classical
HCU) | Thermolabile
MTHFR | Mild to moderate
hyperhomocysteinemia |
Diagnosis of HCU
The diagnosis of HCU involves a combination of
clinical evaluation, biochemical testing and molecular genetic
analysis. Early and accurate diagnosis is important because prompt
treatment can improve the prognosis of HCU and prevent irreversible
complications (16,17).
Symptoms and phenotypic features
HCU usually presents with a wide range of clinical
symptoms that affect multiple organs. Ocular complications,
including ectopia lentis, myopia, astigmatism, glaucoma and
blindness, are the earliest and most common manifestations,
affecting 90% of poorly managed individuals (13–17).
In the early childhood growth phase, skeletal
deformities are common, including marfanoid body habitus, long
limbs, scoliosis, pectus deformities, genu valgum and severe
osteoporosis, which predispose patients to recurrent bone fractures
(14). Neurological deficits and
developmental delays commonly occur in the early years of life, are
noticed as intellectual disabilities and may progress to repeated
seizures (17).
Thromboembolic events, such as DVT, PE and stroke,
are the most serious complications that may affect both adolescents
and adults. The severity of clinical presentation is markedly
dependent on the degree of enzyme deficiency and therapeutic
responsiveness (11,12).
Biochemical diagnosis
Plasma homocysteine level
Plasma total homocysteine (tHcy) is a sensitive
diagnostic biomarker for the primary diagnosis and therapeutic
monitoring of HCU. Impaired metabolism of homocysteine to
cystathionine results in elevated tHcy blood levels of >100
µmol/l compared with <15 µmol/l in healthy individuals (30).
Plasma tHcy levels combined with elevated methionine
levels can distinguish CBS deficiency from other
homocysteine-related disorders, such as remethylation defects,
which usually present with high tHcy levels but low methionine
levels. Furthermore, tHcy levels also guide clinical decisions
during diagnostic B6 challenge tests, in which a notable drop in
homocysteine confirms B6-responsive HCU (30).
Plasma methionine levels
Decreased homocysteine metabolism leads to a buildup
of methionine, with levels ranging from 60 to 100 µmol/l. This
concurrent elevation of tHcy and methionine can distinguish
classical HCU from hyperhomocysteinemia caused by other
remethylation pathway disorders, such as MTHFR deficiency or
cobalamin metabolism malfunction, which lead to elevated tHcy
levels while maintaining normal methionine levels. In patients on
methionine-restricted diets, the normalization of plasma methionine
and tHcy levels indicates effective metabolic control. The B6 trial
also identifies B6-responsive individuals who show partial
improvement in methionine levels (17).
Urinary homocysteine
Measurement of urinary homocysteine levels provides
evidence for the diagnosis of classical HCU, particularly in
limited-resource settings where plasma tHcy testing is not
available. In CBS deficiency, elevated plasma homocysteine levels
exceed the renal reabsorption capacity, leading to increased renal
excretion of both free and tHcy. Although the urinary homocysteine
level is less precise than the plasma tHcy test, it is used as an
initial screening test, especially in symptomatic children.
However, elevated urinary homocysteine levels can also be detected
in other homocysteine metabolism disorders, and may vary with
hydration status and renal function (31).
Vitamin B12, folate and methylmalonic
acid (MMA)
Being cofactors in the remethylation pathway that
converts homocysteine to methionine, deficiencies in vitamin B12 or
folate can lead to elevated plasma tHcy levels but are not
indicative of CBS deficiency. Thus, measuring serum B12 and folate
levels rules out nutritional or acquired causes of
hyperhomocysteinemia (32,33).
Elevated MMA levels suggest functional B12
deficiency or disorders of cobalamin metabolism, which may be
associated with elevated tHcy levels. However, in classical HCU,
MMA levels are typically normal, which helps to exclude
remethylation disorders from the potential diagnosis and indicates
that the elevated homocysteine level is due to transsulfuration
pathway impairment rather than defective remethylation (34).
Molecular genetic testing
Genetic testing using next-generation sequencing
that utilizes the CBS gene, or whole-exome sequencing in
asymptomatic cases, is a decisive tool for confirming the diagnosis
of classical HCU, uncovering the underlying CBS gene
mutations responsible for CBS deficiency, as well as predicting B6
responsiveness and guiding treatment decisions (35,36).
Over 190 CBS gene mutations with clinically
significant genotype-phenotype associations have been isolated. The
common p.I278T mutation is often associated with partial or
full responsiveness to B6 therapy, whereas p.R125Q, p.G307S
and p.R266K mutations are associated with non-responsiveness
and severe symptoms (37).
Enzyme activity assays
CBS activity assays performed on cultured skin
fibroblasts or fresh liver biopsy samples evaluate the functional
capacity of CBS, which is typically absent or markedly reduced in
HCU. Enzymatic activity testing assists in distinguishing between
the B6-responsive and non-responsive forms of CBS, as residual
activity may be enhanced by the addition of PLP in vitro.
The use of enzyme assays has lost its applicability in clinical
practice as it requires an invasive tissue sampling technique, a
long turnaround time and challenging interpretation in cases with
residual activity (36,38).
Treatment
Conventional therapies
Pyridoxine (vitamin B6) supplementation
High-dose B6 therapy is the first line of treatment
for patients with residual CBS enzyme activity. B6 is converted
into PLP, an important cofactor for the enzymatic activity of CBS.
Genotypes such as I278T typically retain fractional enzyme
functions and respond satisfactorily to B6 therapy (39,40).
Notably, a study involving European and sub-Saharan populations has
identified multiple haplotypes of the CBS gene, suggesting
that an ongoing mutational process may contribute to the diminished
long-term effectiveness of pyridoxine therapy in sustaining optimal
clinical outcomes (39). By
contrast, null mutations in G307S result in no functional
CBS enzymes and are usually pyridoxine-non-responsive; the CBS
c.1224-2A>C mutation is a null, splice-site variant
associated with vitamin B6 non-responsiveness (41). Other mutations such as
p.R336C alter the molecular properties of CBS, leading to a
severe HCU phenotype unresponsive to B6 therapy (42).
Comprehensive genotyping and in vitro enzyme
activity assays are not available in numerous clinical settings.
Therefore, clinicians rely on empirical pyridoxine trials without
accurately predicting the outcomes. A pyridoxine trial is performed
in all newly diagnosed cases, typically starting at 100–500 mg/day,
followed by therapeutic monitoring of plasma tHcy and methionine
levels. Patients able to achieve tHcy levels <50 µmol/l with B6
therapy alone are classified as responsive and usually exhibit
milder clinical presentations (17).
B6 therapy faces some challenges, as not all
pyridoxine-responsive patients demonstrate clinical improvement
with B6 supplementation. This may be due to additional confounders
such as epigenetics, coexisting polymorphisms and nutrient status.
Furthermore, some patients exhibit only a partial response and may
continue to develop further complications. These patients often
require combination therapy, and their classifications can be
ambiguous (17). Additionally,
long-term adherence to B6 therapy may be challenging because of
rising concerns regarding peripheral neuropathy caused by chronic
high-dose use (43).
Dietary methionine restriction
A lifelong methionine-restricted diet remains the
basic strategy for managing HCU due to CBS gene mutations.
The diet aims to reduce methionine accumulation while ensuring
adequate nutrition using methionine-free nutritional formulas. This
approach requires careful follow-up of plasma amino acid levels to
maintain metabolic control, prevent protein malnutrition and ensure
the fulfillment of demands for normal growth, especially in
pediatric patients (44).
Although a methionine-restricted diet is effective
in lowering plasma tHcy levels, it poses a long-term compliance
challenge because of the poor palatability of dietary formulas
(45). In addition, dietary
restrictions alone may not be sufficient to prevent disease
complications and may require adjunctive therapies (44,45).
Numerous preclinical trials in animal models have
attempted to overcome the challenges related to dietary
restrictions. Using a mouse model, CDX-6512, an engineered
orally-stable methionine γ-lyase (MGL), was administered following
a high-protein meal and led to a dose-dependent ability to locally
degrade methionine in the gastrointestinal tract (GIT), suppressing
plasma methionine and homocysteine (46). Another experimental model by
Perreault et al (47) used
a bolus dose of an engineered probiotic E. coli Nissle
strain to degrade methionine in the GIT. The resulting SYNB1353
strain metabolized methionine in mice, non-human primates and
humans, resulting in lower plasma methionine levels.
Betaine supplementation
Betaine, also known as trimethylglycine, is a
commonly used adjunctive therapy for B6-unresponsive patients with
HCU that acts as a methyl donor in the hepatic BHMT remethylation
pathway (48). In a study
enrolling patients with B6-unresponsive HCU, betaine administration
resulted in individual mean reductions in plasma tHcy levels
ranging from 47.4 to 105.0 µmol/l (49). Another study involving healthy
subjects showed that betaine supplementation of 6 g/day for 3 weeks
significantly reduced homocysteine levels (P=0.030) (50). A study by Lu et al (51) reported a 10% reduction in the
plasma homocysteine concentration when using a combination of
betaine and low-dose B vitamins: 400 µg folic acid, 8 mg vitamin
B6, 6.4 µg vitamin B12 and 1 g betaine.
However, prolonged betaine therapy of >100
mg/kg/day in patients with inadequate dietary methionine
restriction led to notable hypermethioninemia-related adverse
events (52), while another study
reported marked elevations in total cholesterol levels with betaine
supplementation, necessitating close monitoring in patients at risk
of cardiovascular disease (53).
Folate and B12 supplementation
Folate and B12 are important drugs in HCU management
protocols, particularly in patients with MTHFR, MTR and
MTRR mutations. These vitamins serve important roles in the
remethylation of homocysteine to methionine (54). The active form of folate,
5-methyltetrahydrofolate, donates a methyl group to homocysteine,
converting it to methionine. This reaction is catalyzed by MS with
methylcobalamin, the active form of vitamin B12, as a cofactor.
Insufficient folate and B12 supplies impair this pathway, leading
to elevated tHcy levels (55). A
randomized clinical trial by Kok et al (56) demonstrated marked reductions in
tHcy levels in elderly participants consuming 400 g folic acid and
500 g vitamin B12 daily over 2 years.
Limitations and challenges of
conventional therapies
Despite being the basis of HCU management,
conventional therapies have non-negligible limitations, including
variability in patient responsiveness to B6. Adherence to
low-methionine diets is often difficult, affecting metabolic
control, even with strict diet control and vitamin supplements.
Numerous patients fail to meet the desired tHcy levels, leaving
them at risk of complications. Furthermore, the long-term outcomes
of current interventions are inconsistent and there is limited
capacity to reverse established complications (17,43–45).
Novel treatment approaches
Mechanistic and structural studies on CBS have
provided the basis for emerging therapeutic concepts (57–62).
Building on these and on prior reviews, such as that by Majtan
et al (63), this section
emphasizes treatment developments since 2023 and practical
translational considerations across enzyme replacement, gene- and
proteostasis-directed strategies.
Fig. 2 provides an
integrated overview of these molecular pathways with annotated
therapeutic targets and intervention points.
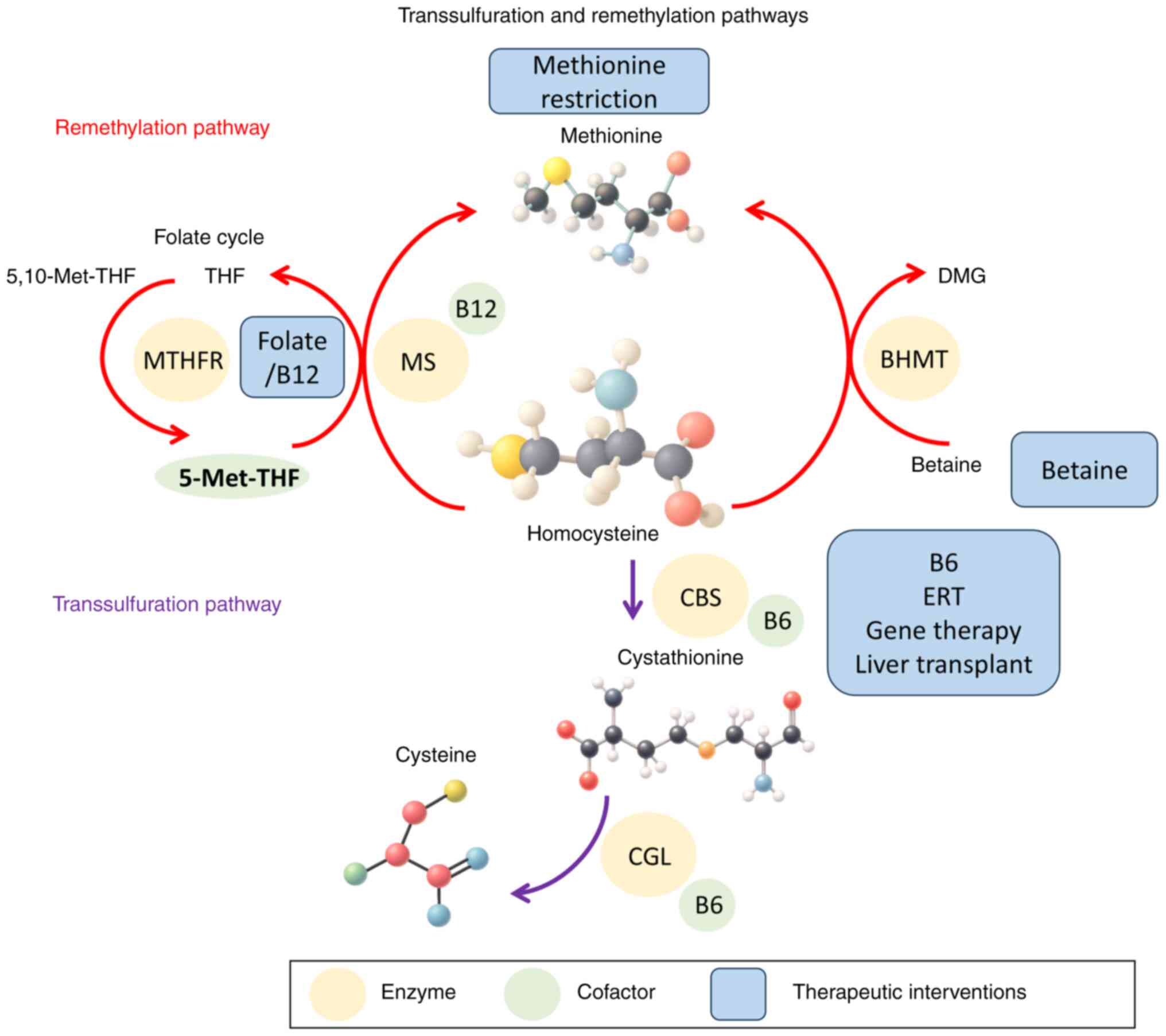 | Figure 2.Therapeutic interventions and
intervention points in homocysteine metabolism. The figure presents
the molecular pathway of homocysteine metabolism with annotated
sites of action for conventional and novel therapies, including
dietary modification, vitamin supplementation, betaine therapy,
ERT, gene therapy and orthotopic liver transplantation. THF,
tetrahydrofolate; MTHFR, methylenetetrahydrofolate reductase; MS,
methionine synthase; DMG, dimethylglycine; BHMT,
betaine-homocysteine methyltransferase; CBS, cystathionine
β-synthase; ERT, enzyme replacement therapy; CGL, cystathionine
γ-lyase; 5-Met-THF, 5-methyltetrahydrofolate; 5,10-Met-THF,
5,10-methylenetetrahydrofolate. |
Enzyme replacement therapy (ERT)
Human CBS is a complex multidomain enzyme that
requires PLP for its activity and binds to a heme group that serves
a regulatory role in its enzyme activity. The CBS protein is
composed of four identical subunits comprising 551 amino acids.
Each subunit consists of three domains, with the N-terminal domain
containing the heme-binding site. In this region, the heme is
linked by cysteine at position 52 and histidine at position 65
(57).
ERT is being developed as a mutation-agnostic
approach for CBS deficiency that delivers functional enzymes
directly, bypassing reliance on residual endogenous activity. While
ERT can normalize biochemistry quickly, the need for chronic
administration and the typical costs and immunogenicity risks of
biologics remain practical challenges for widespread adoption
(58).
i) Pegtibatinase. Pegtibatinase, also known as OT-58
or TVT-058, is a recombinant human CBS catalytic core (amino acids
1–413) that has been modified with the addition of polyethylene
glycol (PEG) and lacks the CBS autoinhibitory regulatory domain,
yielding a constitutively active, predominantly dimeric enzyme.
This modification stimulates the catalytic activity of the enzyme,
rendering it predominantly in a dimeric state (58). To boost pegtibatinase stability and
minimize unwanted protein aggregation, a cysteine-to-serine
substitution at position 15 is introduced to prevent the formation
of interdimer disulfide bonds. The enzyme is also chemically
modified by PEGylation, in which five PEG chains of 20 kDa each are
attached to each CBS subunit. This extends the circulation
half-life by 10-fold, making the PEGylated form more suitable for
therapeutic use in HCU than the unmodified enzyme (59).
In the phase 1/2 COMPOSE study, pegtibatinase
demonstrated promising tolerability and a safety profile with only
two incidents of mild injection site urticaria that resolved upon
temporary dose interruption and subsequent dose titration. Using a
subcutaneous dose of 1.5 mg/kg twice weekly of pegtibatinase
achieved a 67.1% mean relative reduction in tHcy levels from
baseline (97 to 32 µM) at follow-up intervals of 6, 8, 10 and 12
weeks, maintaining tHcy levels below the clinically meaningful
threshold of ≥100 µM (60). A
preclinical study in CBS-deficient animal models has demonstrated
that pegtibatinase ameliorated related complications such as ocular
and skeletal manifestations, decreased facial alopecia, and
enhanced liver metabolism of glucose and lipids (61).
ii) Pegtarviliase. Pegtarviliase, also known as
AGLE-177, is an engineered CGL variant (CGL-ILMDRGVS) with markedly
increased affinity for homocysteine compared with wild-type CGL. It
contains eight missense mutations, referred to as the CGL-ILMDRGVS
construct, and exhibits a 60-fold increased affinity for
homocysteine compared with the wild-type human CGL enzyme (62). Furthermore, pegtarviliase shows an
enhanced capability for degrading multiple forms of homocysteine
compared with wild-type CGL, along with improved stability and
circulation time, potentially resulting in a more comprehensive
reduction in tHcy levels (62).
A preclinical study of pegtarviliase in mice using
subcutaneous doses of 1, 3 and 10 mg/kg twice weekly between
postnatal days 10 and 70 reported a marked improvement in 30-day
survival rates, at >75 vs. <20% in the treated and control
groups, respectively. The treated mice also showed resolution of
liver steatosis and alopecia. Furthermore, a dose of 10 mg/kg led
to an ~43% reduction in plasma tHcy levels and an 87% reduction in
brain tHcy levels (63).
In a phase 1/2 dose-escalation trial aiming to
assess the safety, tolerability, pharmacokinetics and efficacy of
pegtarviliase in patients with classical HCU, three cohorts of
participants received weekly subcutaneous injections of
pegtarviliase at doses of 0.15, 0.45 and 1.35 mg/kg, respectively
over the course of 4 weeks. A 3-day post-treatment dose-dependent
reduction in tHcy levels of 26.3 and 33.0% was observed in cohorts
1 and 2, respectively. However, some participants in cohort 3
experienced injection site reactions and increased tHcy levels,
which can be explained by the development of anti-drug antibodies.
The study highlighted the promising tolerability and efficacy of
pegtarviliase at low doses, with a clear need for modified clinical
strategies to resolve the immunogenicity issues (64). As of 2023, Aeglea BioTherapeutics,
Inc., announced the exploration of strategic alternatives for the
pegtarviliase program following interim phase 1/2 results (65), which ultimately led to the
discontinuation of its clinical development in 2024.
iii) CDX-6512. CDX-6512 is an investigational
modified MGL enzyme that is specifically engineered to maintain
stability and activity in the GIT. In 2022, the U.S. Food and Drug
Administration granted CDX-6512 the ‘orphan drug designation’ for
the treatment of HCU. Using artificial intelligence and machine
learning, 12 iterative rounds of enzyme evolution were performed,
and >27,000 variants were screened for activity under simulated
gastric and intestinal conditions. The resulting enzyme exhibited
high resistance to deactivation by gastric pH and other enzymes
(46).
In a preclinical study using the Tg-I278T
CBS−/−mouse model of HCU, an oral dose of 148
mg/kg CDX-6512 administered after a high-protein meal led to an
almost 50% reduction in plasma tHcy levels after 4 h. Plasma
methionine levels also declined in a non-significant dose-dependent
manner, with up to a one-third decrease at the highest dose
(66).
A study in healthy non-human primates that received
a high-protein meal followed by an oral dose of 370 mg/kg CDX-6512
treatment led to a statistically significant, dose-dependent
reduction in plasma methionine levels, demonstrating the
effectiveness of the enzyme in breaking down dietary methionine in
a physiologically-similar model to humans (46). In the same preclinical program
using Tg-I278T CBS−/− mice, daily oral
administration of CDX-6512 with a high-protein meal over 2 weeks
effectively maintained baseline plasma tHcy levels. By contrast,
untreated controls showed a 39% increase in homocysteine levels,
suggesting that homocysteine levels can be sustained with long-term
CDX-6512 treatment (46). In 2024,
the program was acquired and rebranded as SYNT-202 by Syntis Bio,
with continued preclinical optimization focused on gut-restricted
methionine degradation; no human data under the new designation
have been disclosed at present (67).
Limitations and challenges of ERT
Although ERT offers mutation-independent metabolic
control, injectable agents such as pegtibatinase face lifetime
adherence and immunogenicity hurdles, and oral agents such as
SYNT-202 still require durable efficacy and safety data;
cost-of-goods and access considerations further suggest that ERT
will likely complement, rather than fully replace, diet and
vitamin-based care (58–61).
Gene therapy
Gene therapy offers a potential one-time treatment
for CBS deficiency by restoring intrinsic CBS function at the
genetic level. The strategy delivers a functional CBS gene to the
liver, via viral or non-viral vectors, to achieve sustained
metabolic correction.
Adeno-associated virus serotype rh.10
(AAVrh.10)-CBS-based gene therapy
AAVrh.10-CBS uses an AAVrh.10 vector to deliver a
functional human CBS gene under a cytomegalovirus early
enhancer/chicken β-actin promoter, targeting hepatocytes involved
in homocysteine metabolism. In the CBS-deficient mouse model I278T,
a single intravenous dose reduced plasma tHcy levels by ~97% within
1 week and sustained an ~81% reduction at 1 year from
administration, with observed reversal of alopecia, skeletal
defects and abnormal fat distribution (68). Pre-existing neutralizing antibodies
to AAVrh.10 appear to be less prevalent than those to AAV2 or AAV8,
supporting the suitability of AAVrh.10 for liver-directed
applications (69). Furthermore,
the durability of liver-directed AAV expression observed in a human
hemophilia B gene therapy trial (70) suggests the potential for long-term
transgene persistence in clinical applications.
Minicircle DNA-CBS gene therapy
Minicircular DNA vectors, which lack bacterial
backbone sequences, can more efficiently enhance transgene
expression and reduce innate immune activation compared with
plasmids. Foundational work on vector design and dosing has also
highlighted safety principles to minimize hepatic genotoxicity in
gene-delivery programs (71).
In CBS-deficient mice, a single liver-targeted
injection of the minicircle CBS construct MC.P3-hCBS reduced plasma
homocysteine levels by ~50% within 1 week, restored hepatic CBS
activity ~34-fold and sustained metabolic correction for >6
months (72).
Advantages, limitations and challenges
of gene therapy
Gene therapy directly addresses the genetic cause of
HCU, offering the possibility of durable metabolic correction.
However, immune responses to viral vectors, the dilution of
treatment effects with hepatic growth in children and complex
manufacturing remain key challenges (68–72).
Pharmacological chaperones
Pharmacological chaperones are small molecules that
bind to misfolded CBS proteins, stabilizing their structure and
enhancing catalytic activity. Examples include glycerol,
trimethylamine-N-oxide and dimethyl sulfoxide, which have been
shown to restore activity in certain CBS mutants in cell and animal
models (73,74). Yeast expression systems for human
CBS mutants have also demonstrated that chaperones can promote
proper assembly and tetramerization, supporting mutation-specific
therapeutic potential (75).
S-adenosylmethionine (SAM)
SAM, an allosteric activator of CBS, binds to the
regulatory domain of the enzyme to boost activity (76,77).
A study by Mendes et al (77) demonstrated that ~50% of tested
patients with HCU showed defective SAM activation, often linked to
specific CBS mutations.
Heme arginate
CBS is a heme-dependent enzyme; some CBS gene
mutations impair heme binding. Heme arginate can stabilize these
variants and restore heme-binding activity in cell models (78). A study by Melenovská et al
(78) showed that administration
of heme arginate restored heme binding, improved tetramer formation
and enhanced catalytic activity of the CBS enzyme in several
B6-resistant mutants.
Advantages and limitations of
pharmacological chaperones
Chaperones may complement diet and gene therapy, but
most remain unvalidated in clinical trials. Mutation-specific
responsiveness suggests that genotype-guided therapy may be
required (73–78).
Proteasome inhibitors (PIs)
Proteasome inhibition can stabilize partially active
but misfolded CBS proteins. Bortezomib, an oncology drug, increased
hepatic CBS activity >20-fold and reduced plasma tHcy levels by
97% in a p.R266K mouse model (79–81).
Carfilzomib has shown similar biochemical benefits in preclinical
HCU models, although its long-term safety remains yet to be fully
elucidated (82).
Advantages and limitations of PIs
Potential risks of PIs include systemic proteasome
suppression. Liver-targeted delivery strategies and dose
minimization are being explored to mitigate toxicity (80–82).
Probiotic treatment (SYNB1353)
SYNB1353 is an engineered E. coli Nissle
strain expressing methionine γ-lyase to degrade dietary methionine.
In preclinical models, it reduces plasma homocysteine and
methionine levels without adverse events (83). A phase 1 trial in healthy
volunteers showed promising tolerability and dose-dependent
methionine reduction (84). A
phase 2 study in classical HCU (ClinicalTrials.gov identifier,
NCT05651054) is ongoing, with efficacy data pending.
Orthotopic liver transplantation
(OLT)
OLT has emerged as the only definitive treatment for
severe pyridoxine-non-responsive HCU. Multiple case reports have
demonstrated complete metabolic normalization following OLT. A case
report by Lin et al (85)
described a 24-year-old patient with HCU with notable
complications, including cerebellar infarctions and hypertension,
who underwent a liver transplant, after which their metabolic
control normalized without dietary restrictions. Another case
report described a patient with HCU who demonstrated
post-transplant normalized homocysteine and methionine levels at
the age of 4 years and remained stable for 6 years, indicating the
resolution of HCU. The patient had no complications before the
transplantation, highlighting the potential of early liver
transplantation as a curative strategy for HCU (86).
A retrospective genetic analysis uncovered 18 CBS
variants in 13 patients diagnosed with classic HCU between 10 days
of age and 14 years of age, all of whom exhibited elevated
methionine and tHcy levels. Three B6 non-responders underwent liver
transplantation at the ages of 3, 8 and 8 years, and achieved
normalized methionine and homocysteine levels within a week
post-transplant (87).
Although liver transplantation offers a curative
approach for B6 non-respondents, it carries notable challenges,
including surgical risks, the need for prolonged immunosuppression
and limited donor availability. Additionally, OLT does not reverse
existing complications, and early diagnosis is important to
optimize outcomes (85–87).
Personalized medicine and biomarkers in HCU
management
The integration of genetic and metabolic profiling
allows physicians to tailor treatment according to the molecular
profile of a patient (25,40). Patients with p.I278T mutations may
exhibit variable responses to B6, whereas those with p.R125Q or
p.R266K mutation variants require different therapeutic approaches
(25,40,81).
Early identification of these mutations through genetic workups not
only supports accurate diagnosis but also allows for prompt and
tailored treatments, as summarized in Table II, which compares treatment
modalities, their mechanisms, genotype associations, effectiveness
and limitations (17,38).
| Table II.Comparison of treatment modalities in
homocystinuria. |
Table II.
Comparison of treatment modalities in
homocystinuria.
| Treatment
modality | Mechanism of
action | Genotype
association | Effectiveness |
Limitations/challenges |
|---|
| Pyridoxine (vitamin
B6) | Cofactor for CBS
enzyme; enhances residual CBS activity. | Effective primarily
in CBS mutations with residual enzymatic activity such as
p.Ile278Thr. | Highly effective in
pyridoxine-responsive genotypes; reduces homocysteine and improves
prognosis. | Not effective in
pyridoxine-non-responsive genotypes; responsiveness must be
tested. |
| Betaine | Remethylates
homocysteine to methionine via betaine-homocysteine
methyltransferase, bypassing the CBS pathway. | Effective in both
responsive and non-responsive CBS genotypes. | Reduces plasma
homocysteine levels in non-responsive patients. | Can increase
methionine to toxic levels; requires careful monitoring. |
|
Methionine-restricted diet | Limits precursor
amino acid (methionine) to reduce homocysteine production | Universally
applied, regardless of genotype. | Reduces total
homocysteine load; supportive in both responsive and non-responsive
cases. | Difficult
compliance, especially in older children and adults; risk of
nutritional deficiency. |
| Folic acid and
B12 | Cofactors in
remethylation of homocysteine to methionine. | Useful in
remethylation defects and cases with mild elevation in
homocysteine. | Beneficial in
methylenetetrahy-drofolate reductase and cobalamin defects; used
adjunctively in patients with CBS mutations. | Not curative for
CBS deficiency; requires a combined approach |
| Experimental enzyme
replacement therapy | Supplements
defective or absent CBS enzyme. | Under investigation
for CBS-deficient genotypes. | Potential for
correcting enzymatic deficiency directly. | Not yet clinically
available; challenges in enzyme delivery and immunogenicity. |
| Experimental gene
therapy | Introduces
functional CBS gene into host cells. | Targeted at
CBS-null or severe mutation genotypes. | Preclinical studies
show promise; long-term correction potential. | Experimental;
delivery vectors and sustained expression are hurdles. |
| Liver
transplantation | Provides normal CBS
activity from the liver of the donor. | Effective in
CBS-deficient patients with severe mutations. | Can normalize
homocysteine metabolism; curative in some cases. | Invasive; limited
to severe, refractory cases; lifelong immunosuppression
required. |
Metabolic biomarkers are important tools for
therapeutic monitoring and follow-up of disease progression
(17,30). The plasma tHcy level is the primary
biochemical marker for assessing metabolic control in HCU (17,30).
However, novel biomarkers such as cystathionine, methionine and SAM
are being investigated for their potential to provide a more
accurate view of disease status (30,55).
Stratifying patients according to their residual CBS activity or
proteostatic disparities may help guide the use of emerging
therapies (73,74,81,82).
Limitations of the present review
The present narrative review synthesizes
peer-reviewed literature, clinical trial data, and select pipeline
updates on metabolic and molecular therapies for HCU. As a
narrative rather than systematic review, study selection and
emphasis in the present review may reflect some degree of author
judgment. Furthermore, regional differences in prevalence, genotype
distribution and patterns of consanguinity, together with
heterogeneity in CBS mutations and pyridoxine responsiveness, may
limit the generalizability of certain therapeutic approaches or
outcome expectations across diverse patient populations. Several
discussed interventions are supported mainly by early-phase or
preclinical data, limiting conclusions on long-term safety and
efficacy. Additionally, some pipeline updates are based on press
releases or conference reports, which may change as further
evidence emerges. Despite these constraints, the present review
provides a timely, integrated synthesis of current and emerging
therapeutic strategies, contextualized with the latest clinical and
translational developments through early 2025.
Conclusion
HCU is an underrecognized metabolic disorder
associated with severe multi-organ complications. Timely diagnosis
and individualized therapy are important to prevent long-term
morbidity and improve patient outcomes. Advances in molecular
genetics have deepened the current understanding of disease
pathophysiology and provided the basis for innovative therapeutic
strategies.
By integrating updates from therapeutic pipelines
with clinical considerations and emphasizing how regional and
genetic variability shape the applicability of emerging treatments,
the present review offers a novel and clinically-oriented
synthesis. It underscores both the implications for current
practice and the priorities for future research aimed at
transforming care for individuals with HCU.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
AAA was solely responsible for the conception,
design, literature review, writing and revision of the manuscript.
Data authentication is not applicable. The author has read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The author declares that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
HCU
|
homocystinuria
|
|
CBS
|
cystathionine β-synthase
|
|
MTHFR
|
methylenetetrahydrofolate
reductase
|
|
DVT
|
deep vein thrombosis
|
|
PE
|
pulmonary embolism
|
|
PLP
|
pyridoxal 5′-phosphate
|
|
CGL
|
cystathionine γ-lyase
|
|
MS
|
methionine synthase
|
|
BHMT
|
betaine-homocysteine
methyltransferase
|
|
tHcy
|
total homocysteine
|
|
MMA
|
methylmalonic acid
|
|
MGL
|
methionine γ-lyase
|
|
ERT
|
enzyme replacement therapy
|
|
SAM
|
S-adenosylmethionine
|
|
AAVrh.10
|
adeno-associated virus serotype
Rh.10
|
|
PI
|
proteasome inhibitor
|
|
OLT
|
orthotopic liver transplantation
|
References
|
1
|
Carson NA, Dent C, Field CM and Gaull GE:
Homocystinuria: Clinical and pathological review of ten cases. J
Pediatr. 66:565–583. 1965. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jain M, Shah M, Thakker KM, Rava A,
Pelts-Block A, Ndiba-Markey C and Pinto L: High clinical burden of
classical homocystinuria in the United States: A retrospective
analysis. Orphanet J Rare Dis. 20:372025. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schneede J, Refsum H and Ueland PM:
Biological and environmental determinants of plasma homocysteine.
Semin Thromb Hemost. 26:263–279. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Al-Sadeq DW and Nasrallah GK: The spectrum
of mutations of homocystinuria in the MENA region. Genes (Basel).
11:3302020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Naughten ER, Yap S and Mayne PD: Newborn
screening for homocystinuria: Irish and world experience. Eur J
Pediatr. 157:S84–S87. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Alsharhan H, Ahmed AA, Ali NM, Alahmad A,
Albash B, Elshafie RM, Alkanderi S, Elkazzaz UM, Cyril PX,
Abdelrahman RM, et al: Early diagnosis of classic homocystinuria in
Kuwait through newborn screening: A 6-year experience. Int J
Neonatal Screen. 7:562021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Moammar H, Cheriyan G, Mathew R and
Al-Sannaa N: Incidence and patterns of inborn errors of metabolism
in the Eastern Province of Saudi Arabia, 1983–2008. Ann Saudi Med.
30:271–277. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sellos-Moura M, Glavin F, Lapidus D, Evans
K, Lew CR and Irwin DE: Prevalencecharacteristics, costs of
diagnosed homocystinuria, elevated homocysteine, phenylketonuria in
the United States: A retrospective claims-based comparison. BMC
Health Serv Res. 20:1832020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Refsum H, Fredriksen Å, Meyer K, Ueland PM
and Kase BF: Birth prevalence of homocystinuria. J Pediatr.
144:830–832. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Schnabel E, Kölker S, Gleich F, Feyh P,
Hörster F, Haas D, Fang-Hoffmann J, Morath M, Gramer G, Röschinger
W, et al: Combined newborn screening allows comprehensive
identification also of attenuated phenotypes for methylmalonic
acidurias and homocystinuria. Nutrients. 15:33552023. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yap S, Naughten E, Wilcken B, Wilcken D
and Boers GH: Vascular complications of severe hyperhomocysteinemia
in patients with homocystinuria due to cystathionine β-synthase
deficiency: Effects of homocysteine-lowering therapy. Semin Thromb
Hemost. 2000:335–340. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kalil MAB, Donis KC, de Oliveira Poswar F,
Dos Santos BB, Santos ÂBS and Schwartz IVD: Cardiovascular findings
in classic homocystinuria. Mol Genet Metab Rep.
25:1006932020.PubMed/NCBI
|
|
13
|
Rahman M, Sharma M, Aggarwal P, Singla S
and Jain N: Homocystinuria and ocular complications-A review.
Indian J Ophthalmol. 70:2272–2278. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Weber DR, Coughlin C, Brodsky JL,
Lindstrom K, Ficicioglu C, Kaplan P, Freehauf CL and Levine MA: Low
bone mineral density is a common finding in patients with
homocystinuria. Mol Genet Metab. 117:351–354. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cordaro M, Siracusa R, Fusco R, Cuzzocrea
S, Di Paola R and Impellizzeri DJM: Involvements of
hyperhomocysteinemia in neurological disorders. Metabolites.
11:372021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yap S and Naughten E: Homocystinuria due
to cystathionine β-synthase deficiency in Ireland: 25 years'
experience of a newborn screened and treated population with
reference to clinical outcome and biochemical control. J Inherit
Metab Dis. 21:738–747. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gerrard A and Dawson C: Homocystinuria
diagnosis, management: It is not all classical. J Clin Pathol.
75:744–750. 2022. View Article : Google Scholar
|
|
18
|
Kožich V, Ditrói T, Sokolová J, Křížková
M, Krijt J, Ješina P and Nagy P: Metabolism of sulfur compounds in
homocystinurias. Br J Pharmacol. 176:594–606. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Horowitz JH, Rypins EB, Henderson JM,
Heymsfield SB, Moffitt SD, Bain RP, Chawla RK, Bleier JC and Rudman
D: Evidence for impairment of transsulfuration pathway in
cirrhosis. Gastroenterology. 81:668–675. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sacharow SJ, Picker JD and Levy HL:
Homocystinuria caused by cystathionine beta-synthase deficiency.
2017.
|
|
21
|
Richard E, Desviat LR, Ugarte M and Pérez
B: Oxidative stress and apoptosis in homocystinuria patients with
genetic remethylation defects. J Cell Biochem. 114:183–191. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Obeid R: The metabolic burden of methyl
donor deficiency with focus on the betaine homocysteine
methyltransferase pathway. Nutrients. 5:3481–3495. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Münke M, Kraus JP, Ohura T and Francke U:
The gene for cystathionine beta-synthase (CBS) maps to the
subtelomeric region on human chromosome 21q and to proximal mouse
chromosome 17. Am J Hum Genet. 42:5501988.PubMed/NCBI
|
|
24
|
Liu X, Liu X, Liu J, Guo J, Nie D and Wang
J: Identification and functional analysis of cystathionine
beta-synthase gene mutations in chinese families with classical
homocystinuria. Biomedicines. 13:9192025. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Poloni S, Sperb-Ludwig F, Borsatto T, Hoss
GW, Doriqui MJR, Embiruçu EK, Boa-Sorte N, Marques C, Kim CA, de
Souza CFM, et al: CBS mutations are good predictors for
B6-responsiveness: A study based on the analysis of 35 Brazilian
Classical Homocystinuria patients. Mol Genet Genomic Med.
6:160–170. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li DX, Li XY, Dong H, Liu YP, Ding Y, Song
JQ, Jin Y, Zhang Y, Wang Q and Yang YL: Eight novel mutations of
CBS gene in nine Chinese patients with classical homocystinuria.
World J Pediatr. 14:197–203. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sibani S, Christensen B, O'Ferrall E,
Saadi I, Hiou-Tim F, Rosenblatt DS and Rozen R: Characterization of
six novel mutations in the methylenetetrahydrofolate reductase
(MTHFR) gene in patients with homocystinuria. Hum Mutat.
15:280–287. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li WX, Dai SX, Zheng JJ, Liu JQ and Huang
JFJN: Homocysteine metabolism gene polymorphisms (MTHFR C677T,
MTHFR A1298C, MTR A2756G and MTRR A66G) jointly elevate the risk of
folate deficiency. Nutrients. 7:6670–6687. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li WX, Cheng F, Zhang AJ, Dai SX, Li GH,
Lv WW, Zhou T, Zhang Q, Zhang H, Zhang T, et al: Folate deficiency
and gene polymorphisms of MTHFR, MTR and MTRR elevate the
hyperhomocysteinemia risk. Clin Lab. 63:523–533. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zaric BL, Obradovic M, Bajic V, Haidara
MA, Jovanovic M and Isenovic ER: Homocysteine and
hyperhomocysteinaemia. Curr Med Chem. 26:2948–2961. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sellos-Moura M, Glavin F, Lapidus D, Evans
KA, Palmer L and Irwin DE: Estimated prevalence of moderate to
severely elevated total homocysteine levels in the United States: A
missed opportunity for diagnosis of homocystinuria? Mol Genet
Metab. 130:36–40. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Levy HL, Mudd SH, Schulman JD, Dreyfus PM
and Abeles RH: A derangement in B12 metabolism associated with
homocystinemia, cystathioninemia, hypomethioninemia and
methylmalonic aciduria. Am J Med. 48:390–397. 1970. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Carey MC, Fennelly JJ and FitzGerald O:
Homocystinuria: II. Subnormal serum folate levels, increased folate
clearance and effects of folic acid therapy. Am J Med. 45:26–31.
1968. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bartholomew DW, Batshaw ML, Allen RH, Roe
CR, Rosenblatt D, Valle DL and Francomano CA: Therapeutic
approaches to cobalamin-C methylmalonic acidemia and
homocystinuria. J Pediatr. 112:32–39. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kraus JP: Molecular basis of phenotype
expression in homocystinuria. J Inherit Metab Dis. 17:383–390.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tsai MY, Garg U, Key NS, Hanson NQ, Suh A
and Schwichtenberg K: Molecular and biochemical approaches in the
identification of heterozygotes for homocystinuria.
Atherosclerosis. 122:69–77. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mohamed AS, AlAnzi T, Alhashem A, Alrukban
H, Al Harbi F and Mohamed S: Clinicalbiochemical molecular
characteristics of classic homocystinuria in Saudi Arabia, the
impact of newborn screening on prevention of the complications: A
tertiary center experience. JIMD Rep. 66:e124542025.PubMed/NCBI
|
|
38
|
Alcaide P, Krijt J, Ruiz-Sala P, Ješina P,
Ugarte M, Kožich V and Merinero B: Enzymatic diagnosis of
homocystinuria by determination of cystathionine-ß-synthase
activity in plasma using LC-MS/MS. Clin Chim Acta. 438:261–265.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Vyletal P, Sokolová J, Cooper DN, Kraus
JP, Krawczak M, Pepe G, Rickards O, Koch HG, Linnebank M,
Kluijtmans LA, et al: Diversity of cystathionine β-synthase
haplotypes bearing the most common homocystinuria mutation c.
833T> C: A possible role for gene conversion. Hum Mutat.
28:255–264. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kruger WD, Wang L, Jhee KH, Singh RH and
Elsas II LJ: Cystathionine β-synthase deficiency in Georgia (USA):
Correlation of clinical and biochemical phenotype with genotype.
Hum Mutat. 22:434–441. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Linnebank M, Janosik M, Kozich V, Pronicka
E, Kubalska J, Sokolova J, Linnebank A, Schmidt E, Leyendecker C,
Klockgether T, et al: The cystathionine β-synthase (CBS) mutation
c.1224-2A>C in Central Europe: Vitamin B6 nonresponsiveness and
a common ancestral haplotype. Hum Mutat. 24:352–353. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Al-Sadeq DW, Thanassoulas A, Islam Z,
Kolatkar P, Al-Dewik N, Safieh-Garabedian B, Nasrallah GK and
Nomikos M: Pyridoxine non-responsive p. R336C mutation alters the
molecular properties of cystathionine beta-synthase leading to
severe homocystinuria phenotype. Biochim Biophys Acta Gen Subj.
1866:1301482022. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gdynia N, Gratzer A and Gdynia HJ:
Polyneuropathy due to vitamin B6 hypervitaminosis: A case series
and call for more education. J Neurosci Rural Pract. 16:99–103.
2025. View Article : Google Scholar
|
|
44
|
Liu Y, Guo J, Cheng H, Wang J, Tan Y,
Zhang J, Tao H, Liu H, Xiao J, Qu D, et al: Methionine restriction
diets: Unravelling biological mechanisms and enhancing brain
health. Trends Food Sci Technol. 149:1045322024. View Article : Google Scholar
|
|
45
|
Pokrzywinski R, Bartke D, Clucas C,
Machuzak K and Pinto L: ‘My dream is to not have to be on a diet’:
A qualitative study on burdens of classical homocystinuria (HCU)
from the patient perspective. Orphanet J Rare Dis. 20:1–11. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Skvorak K, Mitchell V, Teadt L, Franklin
KA, Lee HO, Kruse N, Huitt-Roehl C, Hang J, Du F, Galanie S, et al:
An orally administered enzyme therapeutic for homocystinuria that
suppresses homocysteine by metabolizing methionine in the
gastrointestinal tract. Mol Genet Metab. 139:1076532023. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Perreault M, Means J, Gerson E, James M,
Cotton S, Bergeron CG, Simon M, Carlin DA, Schmidt N, Moore TC, et
al: The live biotherapeutic SYNB1353 decreases plasma methionine
via directed degradation in animal models and healthy volunteers.
Cell Host Microbe. 32:382–395. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Truitt C, Hoff WD and Deole R: Health
functionalities of betaine in patients with homocystinuria. Front
Nutr. 8:6903592021. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Singh RH, Kruger WD, Wang L, Pasquali M
and Elsas LJ II: Cystathionine β-synthase deficiency: Effects of
betaine supplementation after methionine restriction in
B6-nonresponsive homocystinuria. Genet Med. 6:90–95. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Schwab U, Törrönen A, Toppinen L, Alfthan
G, Saarinen M, Aro A and Uusitupa M: Betaine supplementation
decreases plasma homocysteine concentrations but does not affect
body weight, body composition, or resting energy expenditure in
human subjects. Am J Clin Nutr. 76:961–967. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Lu XT, Wang YN, Mo QW, Huang BX, Wang YF,
Huang ZH, Luo Y, Maierhaba W, He TT, Li SY, et al: Effects of
low-dose B vitamins plus betaine supplementation on lowering
homocysteine concentrations among Chinese adults with
hyperhomocysteinemia: A randomized, double-blind, controlled
preliminary clinical trial. Eur J Nutr. 62:1599–1610. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Imbard A, Toumazi A, Magréault S,
Garcia-Segarra N, Schlemmer D, Kaguelidou F, Perronneau I, Haignere
J, de Baulny HO, Kuster A, et al: Efficacy pharmacokinetics of
betaine in CBS, cblC deficiencies: A cross-over randomized
controlled trial. Orphanet J Rare Dis. 17:4172022. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zawieja EE, Bogna Z and Chmurzynska A:
Betaine supplementation moderately increases total cholesterol
levels: A systematic review and meta-analysis. J Diet Suppl.
18:105–117. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Marroncini G, Martinelli S, Menchetti S,
Bombardiere F and Martelli FS: Hyperhomocysteinemia and disease-is
10 µmol/l a suitable new threshold limit? Int J Mol Sci.
25:122952024. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
McCaddon A and Miller JW: Homocysteine-a
retrospective and prospective appraisal. Front Nutr.
10:11798072023. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kok DEG, Dhonukshe-Rutten RAM, Lute C,
Heil SG, Uitterlinden AG, van der Velde N, van Meurs JB, van Schoor
NM, Hooiveld GJ, de Groot LC, et al: The effects of long-term daily
folic acid and vitamin B12 supplementation on genome-wide DNA
methylation in elderly subjects. Clin Epigenetics. 7:1212015.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Meier M, Janosik M, Kery V, Kraus JP and
Burkhard P: Structure of human cystathionine beta-synthase: A
unique pyridoxal 5′-phosphate-dependent heme protein. EMBO J.
20:3910–3916. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Bublil EM and Majtan T: Classical
homocystinuria: From cystathionine beta-synthase deficiency to
novel enzyme therapies. Biochimie. 173:48–56. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Bublil EM, Majtan T, Park I, Carrillo RS,
Hůlková H, Krijt J, Kožich V and Kraus JP: Enzyme replacement with
PEGylated cystathionine β-synthase ameliorates homocystinuria in
murine model. J Clin Invest. 126:2372–2384. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Ficicioglu C, Ganesh J, Smith W, Lah M,
Kudrow D, Güner J, McDermott S, Vaidya S, Wilkening L, Thomas J and
Levy H: P012: Pegtibatinase, an investigational enzyme replacement
therapy for the treatment of classical homocystinuria: Latest
findings from the COMPOSE phase 1/2 trial. Genetics in Medicine
Open. 2:1008892024. View Article : Google Scholar
|
|
61
|
Majtan T, Jones W Jr, Krijt J, Park I,
Kruger WD, Kožich V, Bassnett S, Bublil EM and Kraus JP: Enzyme
replacement therapy ameliorates multiple symptoms of murine
homocystinuria. Mol Ther. 26:834–844. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Cramer SL, Saha A, Liu J, Tadi S, Tiziani
S, Yan W, Triplett K, Lamb C, Alters SE, Rowlinson S, et al:
Systemic depletion of L-cyst (e) ine with cyst (e) inase increases
reactive oxygen species and suppresses tumor growth. Nat Med.
23:120–127. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Majtan T, Kožich V and Kruger WD: Recent
therapeutic approaches to cystathionine beta-synthase-deficient
homocystinuria. Br J Pharmacol. 180:264–278. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
A Phase 1/2 Multiple Ascending-Dose Study
in Subjects With Homocystinuria Due to Cystathionine β-Synthase
(CBS) Deficiency to Investigate the Safety, Pharmacokinetics, and
Pharmacodynamics of ACN00177. https://clinicaltrials.gov/study/NCT05154890August
11–2025
|
|
65
|
Aeglea BioTherapeutics: Aeglea
BioTherapeutics Announces Interim Results from Ongoing Phase 1/2
Clinical Trial of Pegtarviliase for the Treatment of Classical
Homocystinuria and Begins Process to Explore Strategic
Alternatives. https://ir.aeglea.com/press-releases/news-details/2023/Aeglea-BioTherapeutics-Announces-Interim-Results-from-Ongoing-Phase-12-Clinical-Trial-of-Pegtarviliase-for-the-Treatment-of-Classical-Homocystinuria-and-Begins-Process-to-Explore-Strategic-Alternatives/default.aspxAugust
11–2025
|
|
66
|
Wang L, Chen X, Tang B, Hua X,
Klein-Szanto A and Kruger WD: Expression of mutant human
cystathionine β-synthase rescues neonatal lethality but not
homocystinuria in a mouse model. Hum Mol Genet. 14:2201–2208. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Syntis Bio, . Syntis Bio Launches to
Revolutionize Oral Therapy for Obesity, Diabetes and Rare Diseases
by Unlocking the Full Therapeutic Potential of the Small Intestine.
https://syntis.bio/syntis-bio-launches-to-revolutionize-oral-therapy-for-obesity-diabetes-and-rare-diseases-by-unlocking-the-full-therapeutic-potential-of-the-small-intestine/August
11–2025
|
|
68
|
Lee HO, Salami CO, Sondhi D, Kaminsky SM,
Crystal RG and Kruger WD: Long-term functional correction of
cystathionine β-synthase deficiency in mice by adeno-associated
viral gene therapy. J Inherit Metab Dis. 44:1382–1392. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Calcedo R, Vandenberghe LH, Gao G, Lin J
and Wilson JM: Worldwide epidemiology of neutralizing antibodies to
adeno-associated viruses. J Infect Dis. 199:381–390. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
George LA, Sullivan SK, Giermasz A, Rasko
JEJ, Samelson-Jones BJ, Ducore J, Cuker A, Sullivan LM, Majumdar S,
Teitel J, et al: Hemophilia B gene therapy with a
high-specific-activity factor IX variant. N Engl J Med.
377:2215–2227. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Chandler RJ, LaFave MC, Varshney GK,
Trivedi NS, Carrillo-Carrasco N, Senac JS, Wu W, Hoffmann V,
Elkahloun AG, Burgess SM and Venditti CP: Vector design influences
hepatic genotoxicity after adeno-associated virus gene therapy. J
Clin Invest. 125:870–880. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Lee HO, Gallego-Villar L, Grisch-Chan HM,
Häberle J, Thöny B and Kruger WD: Treatment of cystathionine
β-synthase deficiency in mice using a minicircle-based naked DNA
vector. Hum Gene Ther. 30:1093–1100. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Collard R and Majtan T: Genetic and
pharmacological modulation of cellular proteostasis leads to
partial functional rescue of homocystinuria-causing
cystathionine-beta synthase variants. Mol Cell Biol. 43:664–674.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Majtan T, Pey AL, Gimenez-Mascarell P,
Martínez-Cruz LA, Szabo C, Kožich V and Kraus JP: Potential
pharmacological chaperones for cystathionine
beta-synthase-deficient homocystinuria. Handb Exp Pharmacol.
245:345–383. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Kopecká J, Krijt J, Raková K and Kožich V:
Restoring assembly and activity of cystathionine β-synthase mutants
by ligands and chemical chaperones. J Inherit Metab Dis. 34:39–48.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Kluijtmans LA, Boers GH, Stevens EM,
Renier WO, Kraus JP, Trijbels FJ, van den Heuvel LP and Blom HJ:
Defective cystathionine beta-synthase regulation by
S-adenosylmethionine in a partially pyridoxine responsive
homocystinuria patient. J Clin Invest. 98:285–289. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Mendes MI, Colaço HG, Smith DE, Ramos RJ,
Pop A, van Dooren SJ, de Almeida IT, Kluijtmans LA, Janssen MC,
Rivera I, et al: Reduced response of Cystathionine Beta-Synthase
(CBS) to S-Adenosylmethionine (SAM): Identification and functional
analysis of CBS gene mutations in Homocystinuria patients. J
Inherit Metab Dis. 37:245–254. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Melenovská P, Kopecká J, Krijt J, Hnízda
A, Raková K, Janošík M, Wilcken B and Kožich V: Chaperone therapy
for homocystinuria: The rescue of CBS mutations by heme arginate. J
Inherit Metab Dis. 38:287–294. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Kim D and Li GC: Proteasome inhibitors
lactacystin and MG132 inhibit the dephosphorylation of HSF1 after
heat shock and suppress thermal induction of heat shock gene
expression. Biochem Biophys Res Commun. 264:352–358. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Gupta S, Wang L, Anderl J, Slifker MJ,
Kirk C and Kruger WD: Correction of cystathionine β-synthase
deficiency in mice by treatment with proteasome inhibitors. Hum
Mutat. 34:1085–1093. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Gupta S, Wang L and Kruger WD: The c.797
G>A (p.R266K) cystathionine β-synthase mutation causes
homocystinuria by affecting protein stability. Hum Mutat.
38:863–869. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Gupta S, Lee HO, Wang L and Kruger WD:
Examination of two different proteasome inhibitors in reactivating
mutant human cystathionine β-synthase in mice. PLoS One.
18:e02865502023. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Isabella VM, Ha BN, Castillo MJ, Lubkowicz
DJ, Rowe SE, Millet YA, Anderson CL, Li N, Fisher AB, West KA, et
al: Development of a synthetic live bacterial therapeutic for the
human metabolic disease phenylketonuria. Nat Biotechnol.
36:857–864. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Puurunen MK, Vockley J, Searle SL,
Sacharow SJ, Phillips JA III, Denney WS, Goodlett BD, Wagner DA,
Blankstein L, Castillo MJ, et al: Safety pharmacodynamics of an
engineered E. coli Nissle for the treatment of phenylketonuria: A
first-in-human phase 1/2a study. Nat Metab. 3:1125–1132. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Lin NC, Niu DM, Loong CC, Hsia CY, Tsai
HL, Yeh YC, Tsou MY and Liu CS: Liver transplantation for a patient
with homocystinuria. Pediatr Transplant. 16:E311–E314. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Kerkvliet SP, Rheault MN and Berry SA:
Liver transplant as a curative treatment in a pediatric patient
with classic homocystinuria: A case report. Am J Med Genet A.
185:1247–1250. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Li DX, Chen ZH, Jin Y, Song JQ, Li MQ, Liu
YP, Li XY, Chen YX, Zhang YN, Lyu GY, et al: Clinical
characteristics and CBS gene analysis of 13 cases with classic
homocystinuria. Zhonghua Er Ke Za Zhi. 60:533–538. 2022.(In
Chinese). PubMed/NCBI
|














