Introduction
Intestinal ischemia-reperfusion (I/R) injury
represents a life-threatening condition precipitated by a transient
interruption of blood flow to the intestines, followed by the
abrupt restoration of circulation (1). This sequence of events triggers a
cascade of detrimental effects, including oxidative stress,
inflammation and apoptosis, all of which culminate in substantial
tissue damage due to the sudden influx of oxygenated blood
(2). I/R injury is frequently
encountered in clinical scenarios such as organ transplantation or
intestinal obstruction, where the restoration of blood supply,
while intended to rescue tissue viability, often exacerbates
cellular injury (3). Extensive
investigations into the molecular mechanisms underlying I/R injury
have revealed the involvement of multiple genes and signaling
pathways in these pathological processes. Specifically,
ferroptosis-induced intestinal tissue damage during I/R can be
mitigated by inhibiting acyl-CoA synthetase long-chain family
member 4 before reperfusion, highlighting the therapeutic potential
of targeting ferroptosis-related genes (4). Additionally, it has been demonstrated
that the activation of JUN and FOS transcription
factors during I/R injury orchestrates a dual role, promoting both
apoptosis and compensatory cell proliferation, thereby underscoring
the complexity of cellular responses to I/R stress (5). Furthermore, heme oxygenase-1 has
emerged as a critical regulator of the oxidative stress response
during I/R injury, with its induction serving as a protective
mechanism against oxidative damage (6). Despite these advances, the complex
interplay between signaling pathways implicated in I/R injury
remains incompletely understood. Oxidative stress, a cornerstone of
I/R-induced tissue damage, is orchestrated by a multitude of
molecular factors, the interactions and cross-talk of which dictate
the extent of cellular injury and recovery (7). Thus, further research is required to
determine the complex regulatory networks governing I/R injury,
with the aim of identifying novel therapeutic targets to mitigate
tissue damage and improve clinical outcomes.
The lysosomal cysteine protease cathepsin B
(CTSB) serves a role in various digestive system disorders.
Studies have demonstrated that CTSB deficiency effectively
mitigates symptoms linked to hypoxia-ischemia, inflammation and
pain (8,9). For example, Xihuang pills have been
shown to alleviate colitis by modulating CTSB levels, which
in turn improve mucosal barrier integrity and suppress inflammation
(10). Furthermore, Dong et
al (11) revealed that
administering mannose to treat inflammatory bowel disease (IBD)
supports lysosomal stability and curtails CTSB release,
thereby safeguarding against mitochondrial dysfunction during
intestinal epithelial injury. Notably, CTSB serves as a
mediator in the activation of NLR family pyrin domain-containing 3
(NLRP3) inflammasomes; it binds to NLRP3, thereby amplifying the
activity of the NLRP3/caspase-1 inflammasome pathway (12). Research has indicated that colon
inflammation induced by deoxycholic acid can be substantially
diminished by inhibiting the NLRP3 inflammasome through the use of
S1PR2 inhibitors and CTSB antagonists (13). Despite the progress made in
understanding the involvement of CTSB in intestinal
diseases, research regarding its function and underlying mechanisms
in I/R-induced intestinal damage remains relatively limited. Th
present study aimed to address this knowledge gap by exploring the
role of CTSB in oxidative stress and disruption of the
intestinal barrier during I/R injury.
In the setting of I/R injury, an overabundance of
oxidative stress and inflammation represents a driving force behind
epithelial barrier dysfunction, thereby intensifying cellular
damage (14). Prior research has
identified the pivotal role of CTSB in fostering
inflammation and cell death in pathological states (15). Nevertheless, its precise
contribution to I/R-triggered intestinal injury has been relatively
under-investigated. The present study aimed to assess the effects
of CTSB knockdown on alleviating oxidative stress,
mitigating intestinal barrier impairment and curbing NLRP3
inflammasome activation in Caco-2 cells subjected to oxygen-glucose
deprivation/reoxygenation (OGD/R). The results of this
investigation may provide novel insights into the therapeutic
promise of targeting CTSB for the management of I/R
injury.
Materials and methods
Screening of datasets and
identification of differentially expressed genes (DEGs)
For in-depth analysis, the GSE37013 dataset was
accessed from the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) repository,
originally reported by Kip et al (16). This dataset comprises 28 samples,
including seven non-ischemic surgical control jejunal samples
collected intraoperatively and 21 I/R case samples. Differential
expression analysis on the GSE37013 dataset was conducted using the
limma package (version 3.48.3; http://bioconductor.org/packages/limma) implemented in
R (version 4.1.2; R Foundation for Statistical Computing;
http://www.r-project.org/). Genes
exhibiting a fold change (FC) of >1.3 were categorized as
upregulated, whereas those with an FC of <0.77 were deemed
downregulated; P<0.05 was employed to denote a statistically
significant difference. To visualize the DEGs, the ggplot2 package
(https://cran.r-project.org/package=ggplot2) was
utilized in R.
Expression validation of overlapping
genes in protein-protein interaction (PPI) networks
To elucidate the interactions among DEGs, a PPI
network analysis was conducted using the Search Tool for the
Retrieval of Interacting Genes (https://string-db.org/). The top 15 genes were
identified using the Cytohubba plugin within Cytoscape (version
3.9.1; http://cytoscape.org), employing
algorithms such as the maximum neighborhood component (MNC),
maximal clique centrality (MCC) and degree. Subsequently, an
intersection analysis was performed on the top 15 genes derived
from these three network modules using the Bioinformatics and
Evolutionary Genomics online tool (http://bioinformatics.psb.ugent.be/webtools/Venn/).
The gene expression levels of the intersection genes in both the
control and case groups of the GSE37013 dataset were then analyzed
using the SangerBox platform (http://sangerbox.com/). P<0.05 was deemed
statistically significant for the scores obtained in this
analysis.
Cell culture
Caco-2 cells, from a human colorectal
adenocarcinoma, were sourced from National Collection of
Authenticated Cell Cultures. These cells were cultured in DMEM
(Procell Life Science & Technology Co., Ltd.), supplemented
with 10% fetal bovine serum (Procell Life Science & Technology
Co., Ltd.) and 1% penicillin-streptomycin solution (Procell Life
Science & Technology Co., Ltd.). Cell culture was performed at
37°C within a humidified incubator maintained at 5%
CO2.
I/R and inflammation models
An OGD/R model was established to simulate I/R
injury. The cells were subjected to OGD/R by being incubated in
glucose-free DMEM at 37°C within a microaerobic environment
(comprising 1% O2, 94% N2, 5% CO2)
for varying durations of 0, 2, 4 and 6 h. Subsequently, the cells
underwent reoxygenation in complete DMEM under normoxia at 37°C
(21% O2, 5% CO2, 74% N2) for 24 h.
Control cells were maintained in complete DMEM (with glucose and
10% FBS) at 37°C under normoxia (21% O2, 5%
CO2, balance N2) and underwent the same
medium-change schedule as the OGD/R groups. Additionally, to induce
inflammation, Caco-2 cells (which had been treated with OGD for 4 h
followed by reoxygenation for 24 h) were exposed to 1 µg/ml
lipopolysaccharide (LPS; Beijing Solarbio Science & Technology
Co., Ltd.) for 6 h at 37°C. Finally, adenosine triphosphate (ATP;
Beyotime Institute of Biotechnology) was introduced at a
concentration of 5 mM and the cells were incubated for 30 min at
37°C.
Cell transfection
Transfection was performed after OGD/R. After
seeding at a density of 2×105 cells/well in 6-well
plates, Caco-2 cells were cultured until they reached 60–70%
confluence. To achieve knockdown of CTSB, the cells were
transfected with specific small interfering RNA (siRNA; final
concentration, 50 nM) designed to target CTSB, adhering
strictly to the manufacturer's protocol. The transfection was
carried out using 10 µl Lipofectamine™ 2000 reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) at 37°C for 6 h. As a negative
control (NC), a non-targeting siRNA was employed. Following
transfection, the cells were incubated for an additional 48 h
before being harvested for use in further experiments. The siRNA
sequences utilized for transfection were as follows:
si-CTSB, sense 5′-CCUGUCGGAUGAGCUGGUCAACU-3′, antisense
5′-AUAGUUGACCAGCUCAUCGACAG-3′; si-NC, sense
5′-CCUAGCGAGUGGUCAACUGUAU-3′, antisense
5′-AUACAGAGUUGACCACUCGCCUAG-3′.
Assessment of Caco-2 cell
viability
The Cell Counting Kit-8 (CCK-8) assay (Beyotime
Institute of Biotechnology) was utilized to evaluate cell
viability, according to the manufacturer's instructions. Initially,
96-well plates were seeded with 5×103 Caco-2 cells/well
in 100 µl complete growth medium. The plates were then incubated
overnight at 37°C in a humidified incubator maintained at 5%
CO2, allowing the cells to adhere and achieve a stable
growth state. Following this incubation period, each well was
supplemented with 10 µl CCK-8 solution, and the plates were further
incubated for 2 h at 37°C. Subsequently, cell viability was
assessed by measuring the absorbance at 450 nm using a microplate
reader (Bio-Rad Laboratories, Inc.). To ensure the robustness and
reliability of the findings, data from a minimum of three
independent experiments were subjected to statistical analysis.
Detection of lactate dehydrogenase
(LDH) levels
Caco-2 cells were exposed to OGD for 2, 4 and 6 h,
followed by 24 h of reoxygenation to evaluate the resultant impact
on LDH levels. According to the manufacturer's guidelines, culture
supernatants were carefully collected, and LDH levels were measured
using an LDH cytotoxicity assay kit (Beyotime Institute of
Biotechnology). To ensure the reliability of the data, the assay
was conducted in triplicate for each time point. LDH activity was
quantified by recording the absorbance at 490 nm using a microplate
reader. Data obtained from a minimum of three independent
experiments were subjected to rigorous statistical analysis to
discern the significance of the differences in LDH levels across
the various OGD/R treatment durations.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from Caco-2 cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's detailed instructions, after
the aforementioned OGD/R and/or LPS/ATP treatments. Subsequently, 1
µg isolated total RNA was reverse-transcribed into cDNA utilizing
the PrimeScript RT reagent kit (Takara Biotechnology, Ltd.)
according to the manufacturer's protocol. qPCR was performed on a
Bio-Rad CFX96 Real-Time PCR Detection System (Bio-Rad Laboratories,
Inc.), employing the SYBR Green kit (Takara Biotechnology, Ltd.).
The expression levels of the target genes, as well as the internal
control GAPDH, were quantified using gene-specific primers
(Table I). The thermocycling
conditions were as follows: 95°C for 30 sec, followed by 40 cycles
at 95°C for 5 sec and 60°C for 30 sec; a melt-curve analysis was
then performed from 65–95°C with 0.5°C increments. The relative
expression of each candidate gene was determined using the
2−ΔΔCq method (17). To
ensure the robustness of the findings, data from a minimum of three
independent experiments were subjected to statistical analysis.
 | Table I.Primer sequences for reverse
transcription-quantitative PCR. |
Table I.
Primer sequences for reverse
transcription-quantitative PCR.
| Target | Sequence,
5′-3′ |
|---|
| CTSB | F:
GTACACCTGCGAGTGTTCCA |
|
| R:
CAAGTGCACGCTCTGGTAGA |
| ZO-1 | F:
CTCAAGAGGAAGCTGTGGGT |
|
| R:
CCATTGCTGTGCTAGTGAGC |
| CLDN1 | F:
ATGACCCCAGTCAATGCCAG |
|
| R:
GCTGGAAGGTGCAGGTTTTG |
| TNF-α | F:
CACAGTGAAGTGCTGGCAAC |
|
| R:
AGGAAGGCCTAAGGTCCACT |
| IL-6 | F:
TTCGGTCCAGTTGCCTTCTC |
|
| R:
CTGAGATGCCGTCGAGGATG |
| IL-1β | F:
AGCCATGGCAGAAGTACCTG |
|
| R:
GAAGCCCTTGCTGTAGTGGT |
| GAPDH | F:
CATGTTGCAACCGGGAAGGA |
|
| R:
ATCACCCGGAGGAGAAATCG |
Western blot (WB) analysis
To lyse Caco-2 cells, RIPA lysis buffer (Beyotime
Institute of Biotechnology) was prepared with the addition of
protease and phosphatase inhibitors. The total protein content in
the lysates was then quantified using a BCA protein assay kit
(Beyotime Institute of Biotechnology). Equal amounts of protein (20
µg) were separated by SDS-PAGE on 10% polyacrylamide gels and were
subsequently transferred onto PVDF membranes (Beyotime Institute of
Biotechnology). To prevent non-specific binding, the membranes were
blocked for 1 h at room temperature with 5% skim milk dissolved in
Tris-buffered saline containing 0.1% Tween-20 (TBST). To detect the
target proteins, the antibodies for each target protein and the
internal control GAPDH antibody were mixed separately, and the PVDF
membranes were incubated with the primary antibody solutions at 4°C
overnight. The following primary antibodies were used: CTSB (cat.
no. 12216-1-AP; 1:1,000; Wuhan Sanying Biotechnology), active CTSB
(cat. no. 12216-1-AP; 1:1,000; Wuhan Sanying Biotechnology), ZO-1
(cat. no. ab276131; 1:1,000; Abcam), claudin-1 (cat. no. ab211737;
1:2,000; Abcam), NLRP3 (cat. no. 30109-1-AP; 1:2,000; Wuhan Sanying
Biotechnology), gasdermin D (GSDMD; cat. no. ab210070; 1:1,000;
Abcam), cleaved N-terminal GSDMD (GSDMD-N; cat. no. ab215203;
1:2,000; Abcam), ASC (cat. no. 10500-1-AP; 1:5,000; Wuhan Sanying
Biotechnology), cleaved caspase-1 (p20; cat no. AF4005; 1:2,000;
Affinity Biosciences), caspase-1 (cat. no. ab207802; 1:1,000;
Abcam), IL-1β (cat. no. ab283818; 1:1,000; Abcam), IL-18 (cat. no.
82696-15-RR; 1:2,000; Wuhan Sanying Biotechnology) and GAPDH (cat.
no. 80570-1-RR; 1:2,500; Wuhan Sanying Biotechnology). After
washing the membranes three times with TBST, they were incubated
for 1 h at room temperature with HRP-conjugated goat anti-rabbit
IgG (cat. no. A0208; 1:1,000; Beyotime Institute of Biotechnology).
Protein bands were detected using an enhanced chemiluminescence
detection system (Bio-Rad Laboratories, Inc.). Band intensities
were semi-quantified using ImageJ software (version 2.0.0; National
Institutes of Health). To ensure accurate normalization, the bands
of both the proteins of interest and GAPDH were developed from the
same membrane, subjected to the same blocking, incubation and
washing steps, and visualized under identical conditions.
Co-immunoprecipitation (Co-IP)
To investigate the interaction between CTSB and
NLRP3, co-IP was employed. Cells were harvested and lysed in RIPA
buffer (Beyotime Institute of Biotechnology) supplemented with a
protease-inhibitor cocktail. Lysates were clarified at 12,000 × g
for 10 min at 4°C, and protein concentrations were determined by
BCA assay. The resulting cell lysates were then mixed with 30 µl
protein A/G agarose bead slurry (Santa Cruz Biotechnology, Inc.)
per 1 mg total protein for 1 h at 4°C on a rotator to remove
non-specific binding. Following centrifugation (2,500 × g, 3 min,
4°C), the agarose beads were discarded, and the supernatant,
containing the pre-cleared lysates, was collected. Primary
antibodies specific for either CTSB (cat. no. ab30443; Abcam) or
NLRP3 (cat. no. 30109-1-AP; Wuhan Sanying Biotechnology), or an
equal amount of species-matched normal IgG (negative control), were
added at 2 µg per IP and mixtures were incubated for 6 h at 4°C on
a rotator to allow antigen-antibody binding. Subsequently, an
additional 30 µl protein A/G agarose beads was added, and the
suspensions were rotated overnight at 4°C to capture the
antigen-antibody complexes. Beads were collected by centrifugation
(2,500 × g, 3 min, 4°C) and washed four times with ice-cold RIPA
lysis buffer (1 ml/wash) to remove any unbound proteins. The
antigen-antibody complexes were eluted from the beads using 2X
Laemmli SDS sample buffer at 95°C for 5 min. The eluate was boiled
and denatured to prepare it for subsequent WB analysis.
Enzyme-linked immunosorbent assay
(ELISA)
ELISA was carried out to quantify CTSB, TNF-α, IL-6
and IL-1β using the following commercially available kits: Human
CTSB ELISA Kit (cat. no. ab119584; Abcam), Human TNF-αELISA Kit
(cat. no. BMS2034; Invitrogen; Thermo Fisher Scientific, Inc.),
Human IL-6 ELISA Kit (cat. no. EH2IL6; Invitrogen; Thermo Fisher
Scientific, Inc.), and Human IL-1β Instant ELISA Kit (cat. no.
BMS224INST; Invitrogen; Thermo Fisher Scientific, Inc.) according
to the manufacturer's instructions. For each assay, 100 µl Caco-2
cell culture supernatant/well was carefully transferred to
pre-coated 96-well plates, and the plates were incubated according
to the manufacturer's guidelines. The enzymatic reaction was
developed using the tetramethylbenzidine substrate, and the
absorbance at 450 nm was measured using a microplate reader.
Protein concentrations were calculated based on standard curves
generated with kit-supplied standards. To ensure the robustness and
reliability of the results, each experiment was replicated at least
three times. Data from these independent experiments were subjected
to statistical analysis to evaluate the significance of differences
in protein levels between experimental groups.
Immunofluorescence analysis
After OGD for 4 h followed by 24 h reoxygenation,
cells were fixed for 15 min at room temperature in 4%
paraformaldehyde solution. Subsequently, the cells were
permeabilized for 10 min with 0.1% Triton X-100 to facilitate
antibody penetration and then blocked for 1 h with 5% bovine serum
albumin (BSA; Beyotime Institute of Biotechnology) dissolved in
phosphate-buffered saline (PBS) to mitigate non-specific binding.
Primary antibodies targeting ZO-1 (cat. no. ab276131; 1:500) and
claudin-1 (cat. no. ab211737; 1:1,000) (both from Abcam) were
diluted in 1% BSA and incubated with the cells overnight at 4°C to
allow for specific antigen-antibody interactions. The following
day, the cells were incubated with Alexa Fluor®
488-labeled goat anti-rabbit IgG secondary antibodies (cat. no.
A-11008; 1:1,000; Invitrogen; Thermo Fisher Scientific, Inc.) for 1
h at room temperature in the dark to preserve the fluorescence
signal. The nuclei were counterstained DAPI (Beyotime Institute of
Biotechnology) for 5 min at room temperature after washing with
PBS. Fluorescence microscopy images were captured using a
fluorescence microscope (Nikon Eclipse Ti-U; Nikon Corporation). To
ensure the reproducibility and reliability of our findings, the
entire immunofluorescence staining protocol was replicated at least
three times, with three technical replicates per condition in each
experiment.
Oxidative stress markers assay
After 4 h of OGD followed by 24 h of reoxygenation,
cells were lysed on ice using the kit-supplied sample lysis buffer
(Beyotime Institute of Biotechnology). The lysates were
subsequently analyzed using commercially available detection kits
for reactive oxygen species (ROS; cat. no. S0033S), malondialdehyde
(MDA; cat. no. S0131S), superoxide dismutase (SOD; cat. no. S101S)
and glutathione peroxidase (GSH-Px; cat. no. S0059S) (all from
Beyotime Institute of Biotechnology). These assays were conducted
according to the manufacturer's instructions. Absorbance values
were recorded using a microplate reader at the following specific
wavelengths: 488 nm (excitation) and 525 nm (emission) for ROS, 412
nm for GSH-Px, 450 nm for SOD and 532 nm for MDA. To ensure the
reliability and reproducibility of the data, each test was
performed in triplicate.
Statistical analysis
All results are presented as the mean ± standard
deviation. To determine statistical significance, one-way analysis
of variance was utilized, followed by Tukey's post hoc test for
multiple comparisons between groups. For pairwise comparisons
between two specific groups, a two-tailed unpaired Student's t-test
was employed. P<0.05 was considered to indicate a statistically
significant difference. All statistical analyses were conducted
using the R programming language. Additionally, graphs were
generated exclusively for visualization purposes using GraphPad
Prism software (version 8.0; Dotmatics) to facilitate
interpretation of the results.
Results
Differential expression and PPI
network analysis to obtain key intersection genes
First, a differential expression analysis was
conducted using the GSE37013 dataset, comparing I/R jejunal samples
(n=21) with non-ischemic surgical controls (n=7), which yielded 181
downregulated and 224 upregulated DEGs (Fig. 1A). Subsequently, a PPI network
analysis was performed on the DEGs. This analysis revealed the top
15 genes as identified by three different algorithms: Degree (15
nodes and 44 edges; Fig. 1B), MCC
(15 nodes and 50 edges; Fig. 1C)
and MNC (15 nodes and 52 edges; Fig.
1D). Following this, a cross-analysis of the top 15 genes from
each of the three algorithms was conducted, resulting in the
identification of 13 key intersection genes (Fig. 1E). Expression analysis was then
performed to examine the expression levels of these genes in both
the control and case samples from the GSE37013 dataset (Fig. 1F). The results demonstrated that in
the case samples, BIRC3, CTSB, EZH2, GSK3B, LAMP1, PTPRC and
TLR3 were significantly upregulated, whereas EGFR, NCAM1,
PLCG1, SQSTM1, STAT5B and ZAP70 were significantly
downregulated.
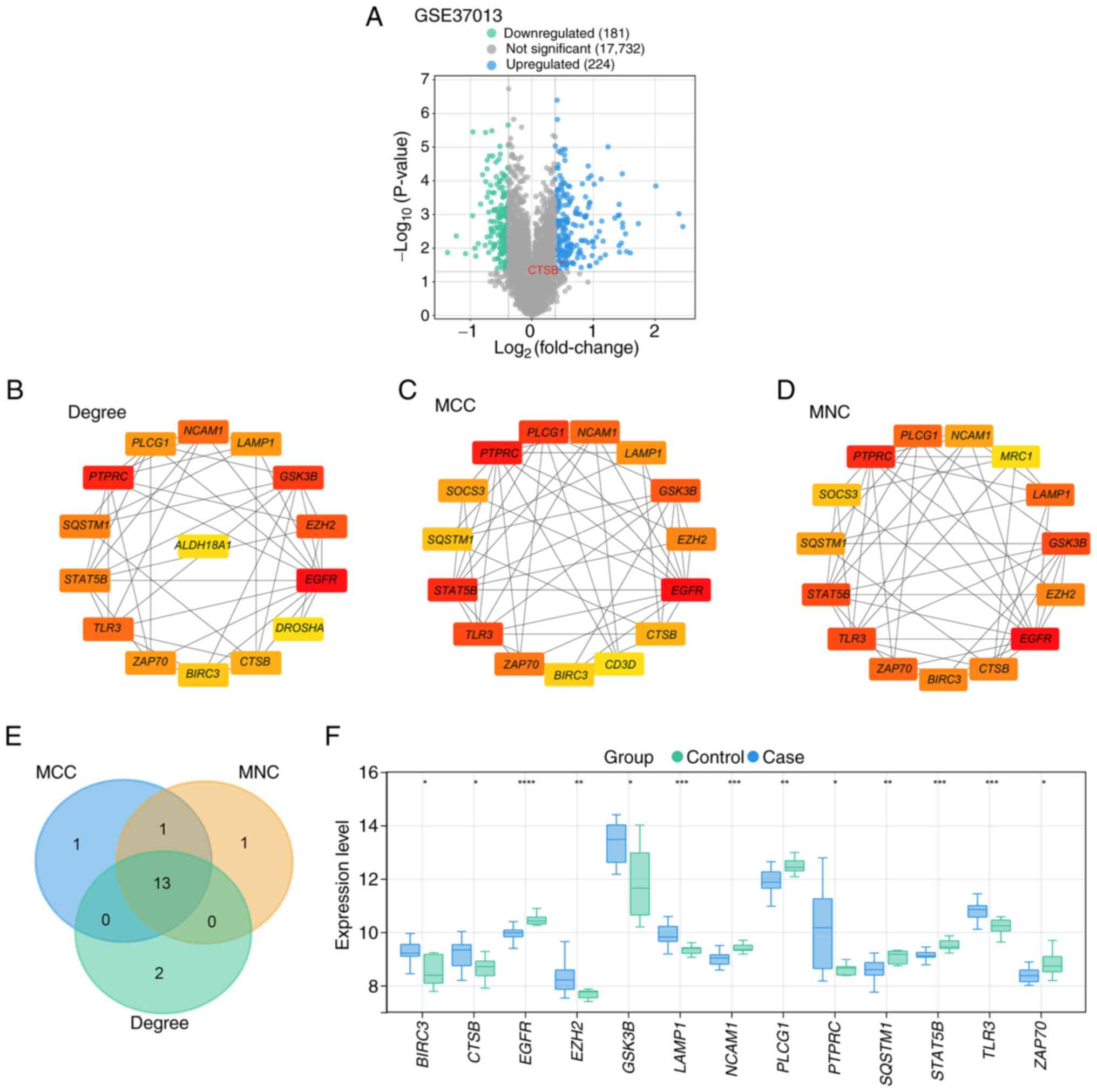 | Figure 1.Screening of DEGs, PPI network
construction and intersection analysis based on the GSE37013
dataset to identify hub genes. (A) Volcano plot of DEGs in the
GSE37013 dataset. Green represents 181 downregulated DEGs and blue
represents 224 upregulated DEGs. PPI network analysis of
upregulated DEGs using (B) degree, (C) MCC and (D) MNC algorithms,
and visualization of the top 15 genes in each network. (E) Venn
diagram of the intersection analysis of the top 15 genes of the
degree, MCC and MNC algorithms, where the overlapping genes
represent the intersection genes. (F) Box plot of the expression
levels of 13 intersection genes in control and case samples from
the GSE37013 dataset. *P<0.05, **P<0.01, ***P<0.001,
****P<0.0001. CTSB, cathepsin B; DEG, differentially
expressed gene; MCC, maximal clique centrality; MNC, maximum
neighborhood component; PPI, protein-protein interaction. |
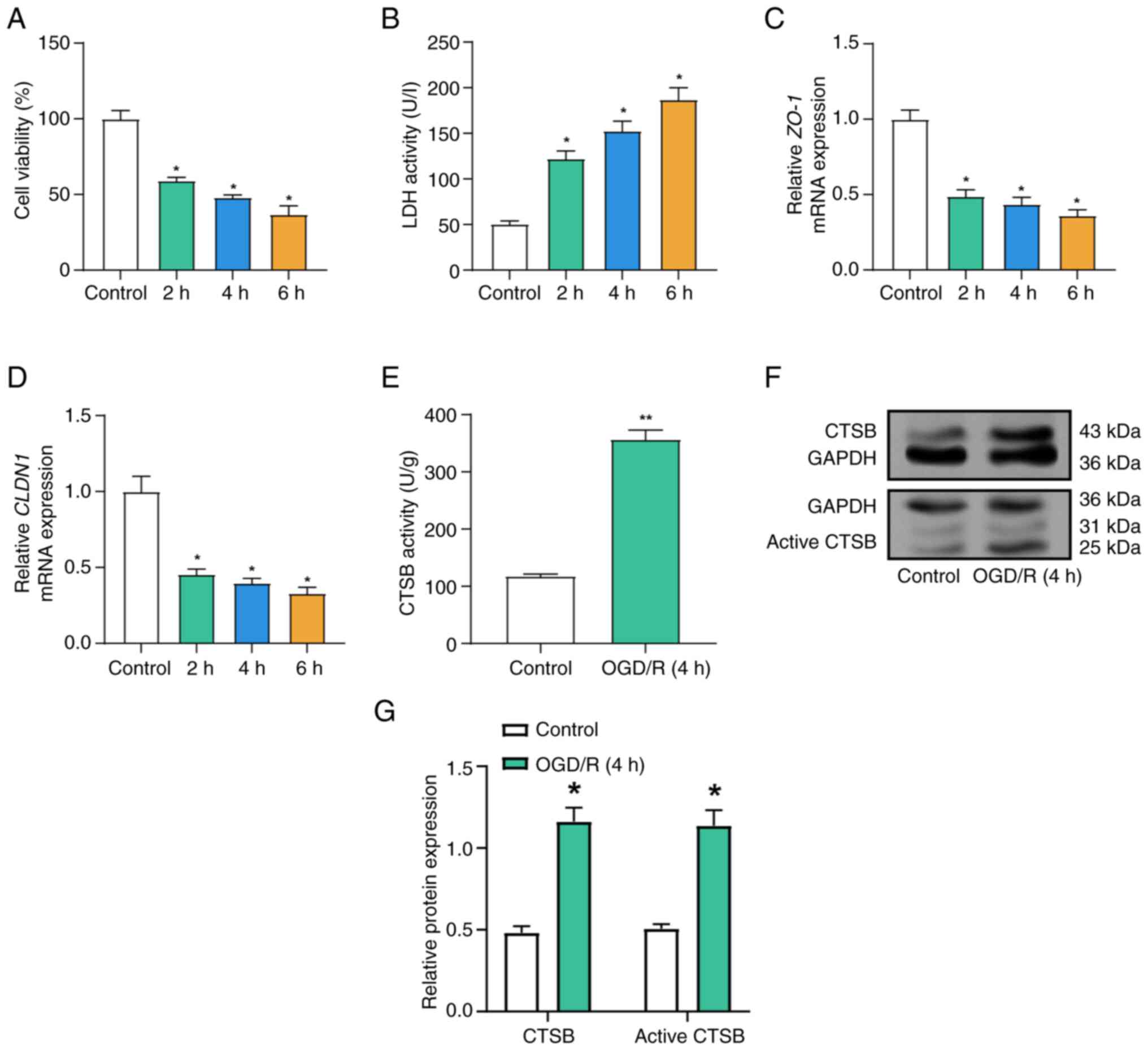
OGD/R decreases cell viability,
disrupts tight junction protein expression and increases CTSB
activity
The current study assessed the effects of OGD/R on
the expression of tight junction proteins and CTSB, LDH release and
cell survival in Caco-2 cells. Compared with in the control group,
as the duration of OGD/R treatment was extended, the results from
the CCK-8 assay revealed a progressive decline in cell viability
(Fig. 2A). Concurrently, a
significant increase in LDH levels was observed with prolonged
OGD/R exposure (Fig. 2B).
Furthermore, following OGD/R treatment, there was a gradual
decrease in the mRNA expression levels of ZO-1 and
CLDN1, with their levels declining progressively as the
duration of OGD/R exposure increased (Fig. 2C and D). Based on preliminary
experiments (Fig. 2A and B), which
demonstrated significant but sub-lethal injury at the 4-h OGD mark,
consistent with established in vitro I/R models (18), subsequent experiments used the same
4-h OGD treatment to further examine its effect on CTSB activity.
Subsequently, an ELISA was employed to examine CTSB activity in
Caco-2 cells. In the OGD/R model, CTSB activity was markedly
elevated compared with that in the control group (Fig. 2E). WB analysis further confirmed
that both total CTSB protein and active CTSB protein levels were
increased following OGD/R treatment (Fig. 2F and G).
CTSB knockdown alleviates
OGD/R-induced Caco-2 cell injury
RT-qPCR and WB analysis confirmed the efficient
knockdown of CTSB, resulting in a significant reduction in
CTSB expression following transfection (Figs. S1 and 3A-C). To evaluate the impact of
CTSB knockdown on the viability of Caco-2 cells exposed to
OGD/R, the CCK-8 assay was employed. OGD/R resulted in a marked
reduction in cell viability compared with that in the control
group, whereas CTSB knockdown effectively reversed this
reduction, thereby preserving cell viability (Fig. 3D). Furthermore, LDH levels, which
serve as an indicator of cell damage, were markedly elevated in the
OGD/R-treated cells; however, CTSB knockdown notably reduced
LDH release, suggesting a decrease in cellular injury (Fig. 3E). WB analysis results further
revealed that the protein levels of ZO-1 and claudin-1, both of
which were reduced in the OGD/R model, were restored following
CTSB knockdown (Fig. 3F and
G). Immunofluorescence staining provided additional
confirmation, demonstrating the recovery of ZO-1 and claudin-1
expression after CTSB knockdown (Fig. 3H and I). Collectively, these
findings suggested that CTSB knockdown may mitigate
I/R-induced cellular injury and preserve epithelial barrier
integrity in Caco-2 cells.
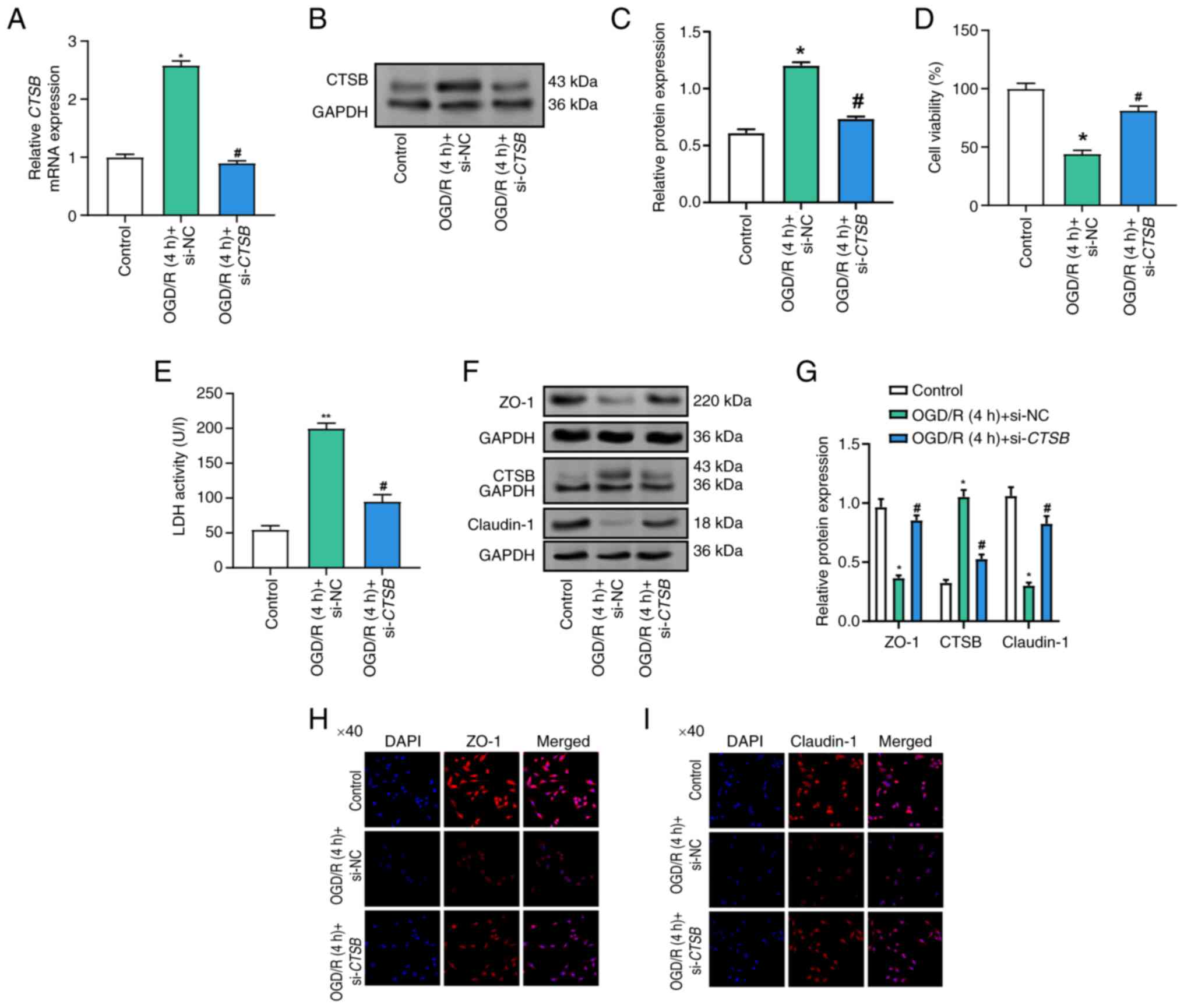 | Figure 3.Effects of CTSB knockdown on
cell viability, LDH activity and tight junction protein expression
in OGD/R-treated Caco-2 cells. (A) Reverse
transcription-quantitative PCR analysis of the relative expression
levels of CTSB mRNA in the control group, the OGD/R + si-NC
group and the OGD/R + si-CTSB group. (B) Representative
western blot of CTSB protein in the Control, OGD/R + si-NC and
OGD/R + si-CTSB groups. GAPDH was used as the loading
control. (C) Densitometric semi-quantification of CTSB protein
expression (normalized to GAPDH). (D) Cell Counting Kit-8 detected
the viability of Caco-2 cells in the control, OGD/R (4 h) + si-NC
and OGD/R (4 h) + si-CTSB groups. (E) LDH activity of Caco-2
cells was detected in the control, OGD/R (4 h) + si-NC and OGD/R (4
h) + si-CTSB groups. (F) WB analysis of the protein
expression of CTSB, ZO-1 and claudin-1 in control, OGD/R (4 h) +
si-NC and OGD/R (4 h) + si-CTSB Caco-2 cell groups, and (G)
semi-quantitative analysis. Immunofluorescence analysis of (H) ZO-1
and (I) claudin-1 in Caco-2 cells in the control, OGD/R (4 h) +
si-NC, and OGD/R (4 h) + si-CTSB groups. Magnification, ×40.
All data were obtained from at least three independent biological
experiments (n=3). *P<0.05, **P<0.01 vs. control;
#P<0.05 vs. OGD/R (4 h) + si-NC. CTSB, cathepsin B;
LDH, lactate dehydrogenase; NC, negative control; OGD/R,
oxygen-glucose deprivation/reoxygenation; si, small interfering;
WB, western blot. |
CTSB knockdown alleviates
OGD/R-induced oxidative stress and inflammatory response
To elucidate the role of CTSB in oxidative
stress and inflammatory responses within I/R-injured Caco-2 cells,
a series of experiments were conducted. The levels of ROS in the
OGD/R model, as assessed using an assay kit, were significantly
higher than those in the control group, indicating an increase in
oxidative stress (Fig. 4A).
Notably, CTSB knockdown attenuated this elevation in ROS
levels, suggesting that CTSB serves an important role in
regulating oxidative stress. Furthermore, MDA levels, evaluated
using an MDA detection kit, were markedly elevated in the OGD/R
model group compared with those in the control group, whereas
CTSB knockdown mitigated this increase (Fig. 4B), indicating a reduction in lipid
peroxidation. SOD activity, which was markedly decreased in the
OGD/R model group, was partially restored following CTSB
knockdown (Fig. 4C), suggesting an
enhancement in antioxidative capacity. Similarly, GSH-Px activity,
which was significantly decreased in the OGD/R model group, was
reversed by CTSB knockdown (Fig. 4D), further supporting its role in
oxidative stress regulation. The results of ELISA revealed that the
OGD/R model group exhibited higher levels of pro-inflammatory
cytokines, including IL-6, IL-1β and TNF-α; by contrast,
CTSB knockdown significantly reduced the levels of these
inflammatory markers (Fig. 4E-G).
Collectively, these findings suggested that CTSB knockdown
may alleviate oxidative stress and inflammatory responses in Caco-2
cells following I/R injury, thereby highlighting the potential
therapeutic benefits of targeting CTSB in this context.
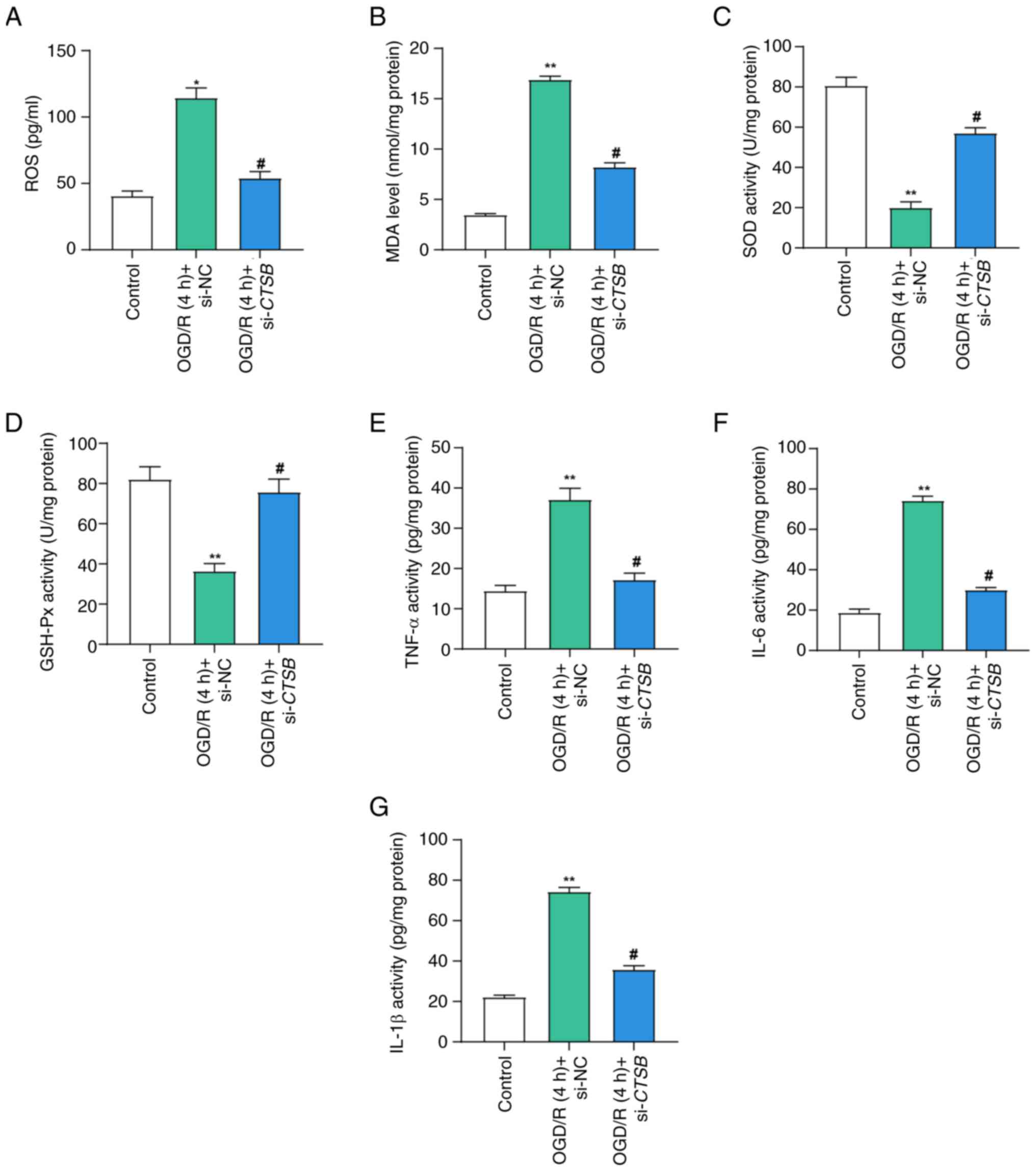 | Figure 4.CTSB knockdown mitigates
ischemia-reperfusion-induced oxidative stress and inflammation.
Detection of (A) ROS levels, (B) MDA levels, (C) SOD activity and
(D) GSH-Px activity in Caco-2 cells in the control, OGD/R (4 h) +
si-NC and OGD/R (4 h) + si-CTSB groups. Enzyme-linked
immunosorbent assay detected the activities of the cytokines (E)
TNF-α, (F) IL-6 and (G) IL-1β in Caco-2 cells in the control, OGD/R
(4 h) + si-NC and OGD/R (4 h) + si-CTSB groups. All data
were obtained from at least three independent biological
experiments (n=3). **P<0.01 vs. control; #P<0.05
vs. OGD/R (4 h) + si-NC. CTSB, cathepsin B; GSH-Px,
glutathione peroxidase; MDA, malondialdehyde; NC, negative control;
OGD/R, oxygen-glucose deprivation/reoxygenation; ROS, reactive
oxygen species; si, small interfering; SOD, superoxide
dismutase. |
CTSB knockdown attenuates
OGD/R-induced NLRP3 inflammasome activation and proinflammatory
cytokine expression
To investigate the role of CTSB in the
inflammatory responses induced by OGD/R in Caco-2 cells, the
expression of key inflammasome components and pro-inflammatory
cytokines was examined. WB analysis revealed that OGD/R treatment
significantly elevated the protein expression levels of GSDMD-N,
NLRP3, ASC, cleaved caspase-1, IL-1β and IL-18 compared with those
in the control group (Fig. 5A-D).
However, CTSB knockdown effectively reduced the protein
levels of these inflammatory mediators. Given the pivotal role of
CTSB in the inflammatory process, the current study aimed to
explore whether CTSB directly interacts with NLRP3, a key component
of the inflammasome. Notably, a previous study has suggested an
interaction exists between CTSB and NLRP3 (11). To further investigate this, co-IP
experiments were conducted. The results confirmed that CTSB
interacts with NLRP3 in the intestinal I/R model (Fig. 5E and F). When CTSB was
immunoprecipitated (Fig. 5E),
NLRP3 was detected only in the OGD/R + si-NC group. This is
consistent with the strong induction of NLRP3 by OGD/R and its
marked reduction after CTSB silencing, rendering the
co-precipitated NLRP3 in the control and OGD/R + si-CTSB
groups below the detection limit. Conversely, when NLRP3 was
immunoprecipitated (Fig. 5F), CTSB
was recovered in all groups because CTSB is constitutively
expressed, with the signal highest in the OGD/R + si-NC groups and
decreased after CTSB knockdown. These reciprocal co-IP
findings support a specific CTSB-NLRP3 interaction in this model.
Furthermore, OGD/R treatment significantly elevated the mRNA
expression levels of pro-inflammatory cytokines (Fig. 5G-5I), whereas CTSB knockdown
markedly decreased the expression levels of these cytokines.
Notably, when cells were further treated with LPS and ATP following
CTSB knockdown, cytokine expression was partially restored,
suggesting a CTSB-independent pathway of inflammatory
activation.
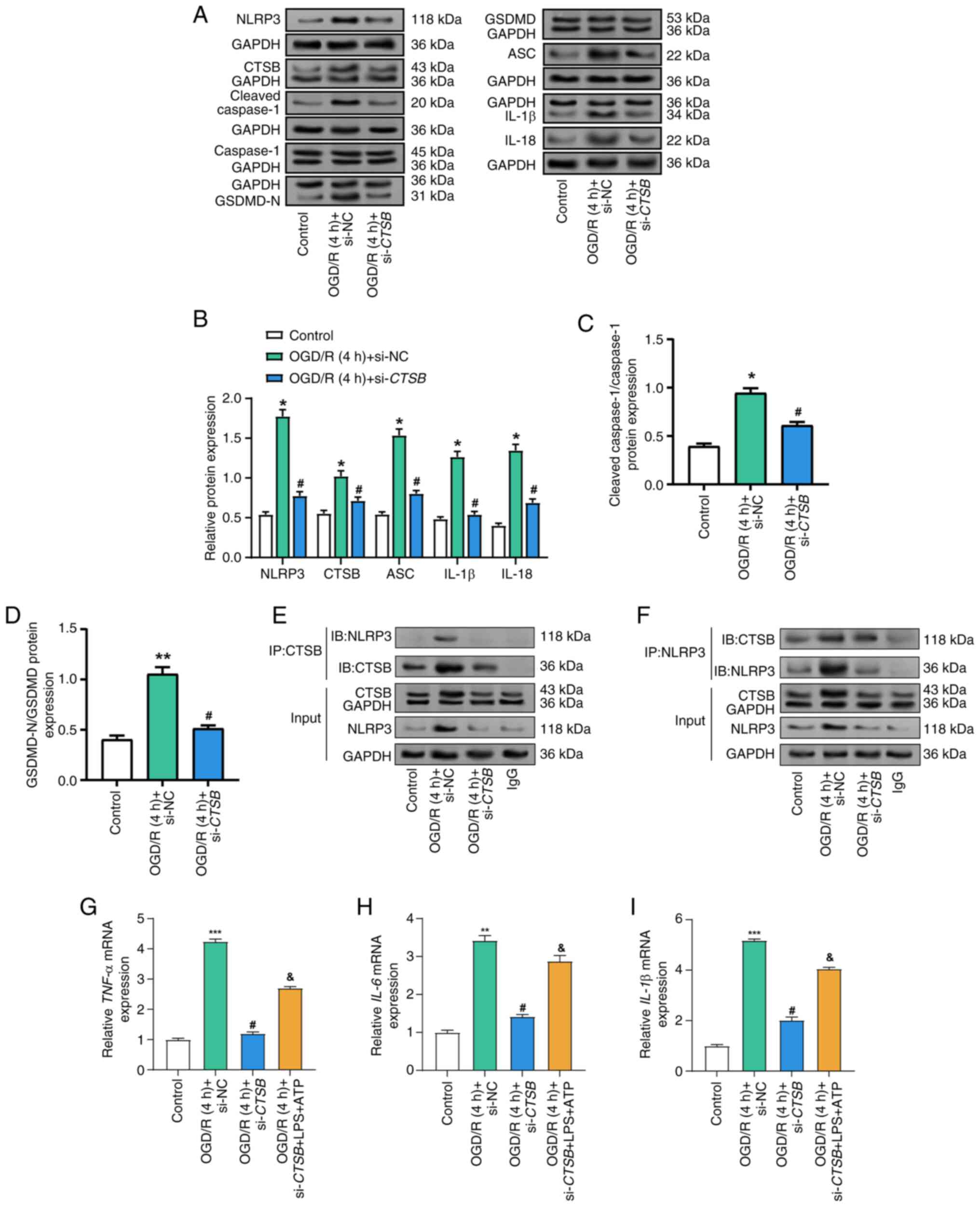 | Figure 5.Knockdown of CTSB inhibits
activation of the NLRP3 inflammasome in Caco-2 cells injured by
OGD/R. (A) Representative WB analysis of inflammasome-related
proteins in Caco-2 cells under control conditions, OGD/R (4 h) +
si-NC and OGD/R (4 h) + si-CTSB. Each GAPDH blot shown
corresponds to the target protein immediately above it.
Semi-quantitative analysis of protein expression levels: (B) NLRP3,
CTSB, ASC, IL-1β and IL-18; (C) cleaved caspase-1/caspase-1 ratio;
and (D) GSDMD-N/GSDMD ratio. (E) Co-IP of CTSB and NLRP3 (IP: CTSB;
IB: NLRP3 and CTSB). Inputs and IgG controls are shown. (F)
Reciprocal co-IP (IP: NLRP3; IB: CTSB and NLRP3) confirming the
interaction between CTSB and NLRP3 under OGD/R conditions. Reverse
transcription-quantitative PCR detection of the relative mRNA
expression levels of the cytokines (G) TNF-α, (H)
IL-6 and (I) IL-1β in Caco-2 cells in the control,
OGD/R (4 h) + si-NC, OGD/R (4 h) + si-CTSB and OGD/R (4 h) +
si-CTSB + LPS + ATP groups. All data were obtained from at
least three independent biological experiments (n=3). *P<0.05,
**P<0.01, ***P<0.001 vs. control; #P<0.05 vs.
OGD/R (4 h) + si-NC; &P<0.05 vs. OGD/R (4 h) +
si-CTSB. ATP, adenosine triphosphate; CTSB, cathepsin B;
GSDMD, gasdermin D; GSDMD-N, cleaved N-terminal GSDMD; IB,
immunoblot; IP, immunoprecipitation; LPS, lipopolysaccharide; NC,
negative control; NLRP3, NLR family pyrin domain-containing 3;
OGD/R, oxygen-glucose deprivation/reoxygenation; si, small
interfering; WB, western blot. |
CTSB knockdown alleviates
OGD/R-induced oxidative stress and impaired intestinal barrier
function
Compared with those in the control group, WB
analysis demonstrated that OGD/R treatment significantly reduced
the expression levels of tight junction proteins, specifically ZO-1
and claudin-1 (Fig. 6A and B).
However, CTSB knockdown significantly restored these protein
levels. Notably, further treatment with LPS and ATP following
CTSB knockdown partially reversed this recovery, suggesting
a regulatory role of CTSB in maintaining tight junction integrity
under inflammatory conditions. This suggests that LPS and ATP may
counteract the benefits of CTSB knockdown. The results of
the cell viability experiment indicated a substantial decrease in
cell viability following OGD/R treatment compared with that in the
control group (Fig. 6C). By
contrast, CTSB knockdown significantly enhanced cell
viability under OGD/R conditions, indicating its protective effect;
however, the subsequent addition of LPS and ATP attenuated this
protective effect, suggesting that LPS and ATP may counteract the
benefits of CTSB knockdown. Subsequently, it was revealed
that MDA levels in OGD/R-treated cells were significantly elevated,
indicating an increase in oxidative stress (Fig. 6D). By contrast, CTSB
knockdown significantly reduced MDA levels, demonstrating its
antioxidant potential; however, treatment with LPS and ATP
partially reversed this decrease, suggesting that LPS and ATP may
exacerbate oxidative stress despite CTSB knockdown.
Furthermore, OGD/R-treated cells exhibited a substantial reduction
in SOD activity, which was restored by CTSB knockdown
(Fig. 6E). Notably, the protective
impact of CTSB knockdown on SOD activity was, however,
diminished by subsequent LPS and ATP treatment, further emphasizing
the complex interplay between CTSB, inflammation and
oxidative stress.
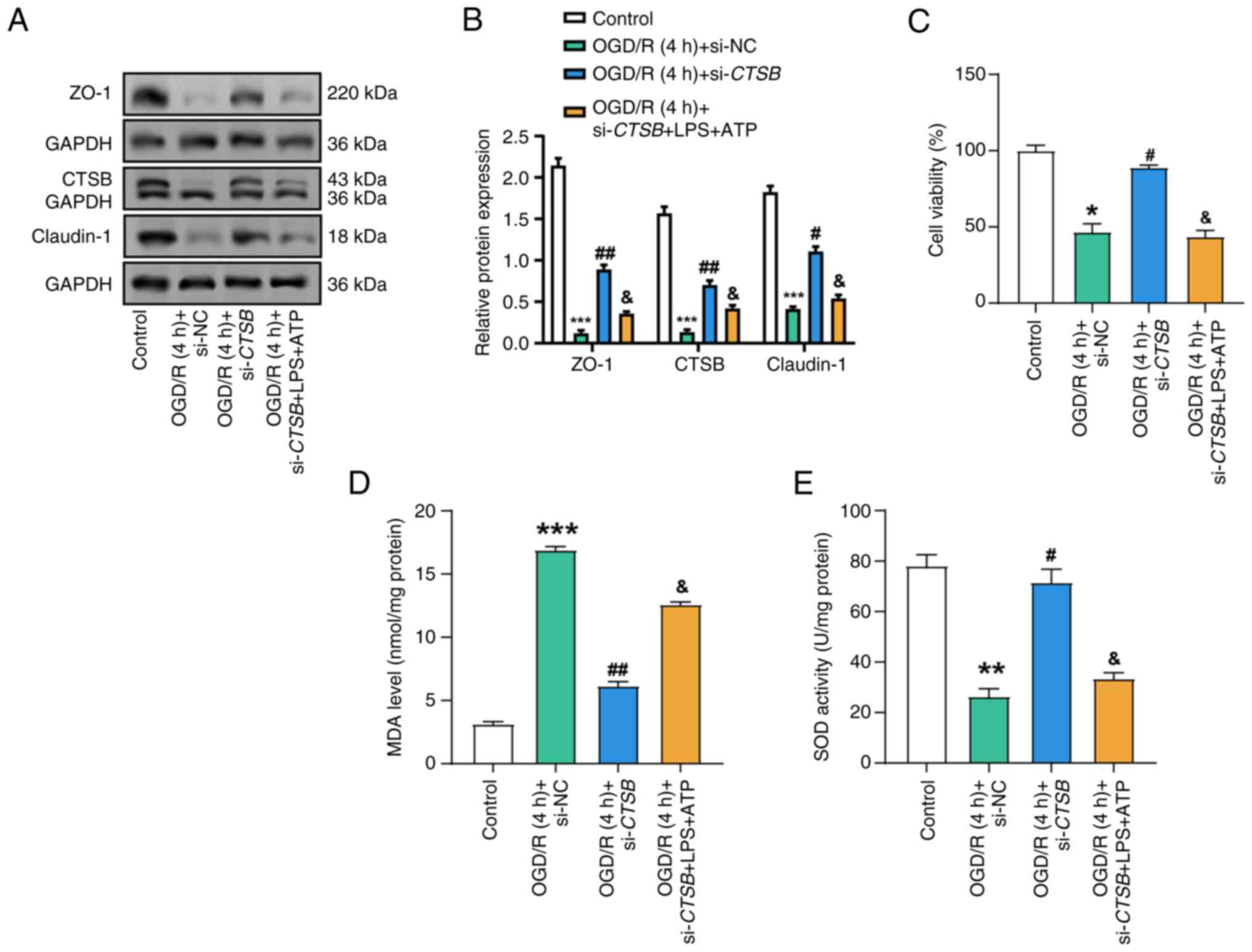 | Figure 6.Effects of CTSB knockdown and
LPS + ATP treatment on cell viability, oxidative stress and tight
junction protein expression in OGD/R-treated Caco-2 cells. (A)
Western blot analysis of the protein expression of CTSB, ZO-1 and
claudin-1 in Caco-2 cells in the control, OGD/R (4 h) + si-NC,
OGD/R (4 h) + si-CTSB and OGD/R (4 h) + si-CTSB + LPS
+ ATP groups, and (B) semi-quantitative analysis. (C) Cell Counting
Kit-8 detected the viability of Caco-2 cells in the control, OGD/R
(4 h) + si-NC, OGD/R (4 h) + si-CTSB and OGD/R (4 h) +
si-CTSB + LPS + ATP groups. Detection of (D) MDA levels and
(E) SOD activity in Caco-2 cells in the control, OGD/R (4 h) +
si-NC, OGD/R (4 h) + si-CTSB and OGD/R (4 h) +
si-CTSB + LPS + ATP groups. All data were obtained from at
least three independent biological experiments (n=3). *P<0.05,
**P<0.01, ***P<0.001 vs. Control. #P<0.05,
##P<0.01 vs. OGD/R (4 h) + si-NC.
&P<0.05 vs. OGD/R (4 h) + si-CTSB + LPS +
ATP. ATP, adenosine triphosphate; CTSB, cathepsin B; LPS,
lipopolysaccharide; MDA, malondialdehyde; NC, negative control;
OGD/R, oxygen-glucose deprivation/reoxygenation; si, small
interfering; SOD, superoxide dismutase. |
Discussion
I/R injury represents a complex pathogenic process
that arises from the transient interruption of blood flow, followed
by the reintroduction of oxygen to tissues; this sequence of events
precipitates cellular damage and inflammation (19). Previous studies have identified the
critical molecular mediators in I/R injury, elucidating their roles
in amplifying tissue destruction and inflammatory responses. For
example, research has demonstrated the involvement of microRNAs
(miRs) in I/R. Specifically, the overexpression of miR-146a-5p has
been shown to target TXNIP, modulating the PRKAA/mTOR
pathway, which in turn inhibits autophagy and mitigates intestinal
barrier damage (20). Through
rigorous bioinformatics analysis, the present study identified
several genes that exhibit significant differential expression in
the context of I/R injury. Some of these genes have already been
validated in prior studies for their association with I/R. For
example, PTPRC has emerged as a potential focal point for
pharmacotherapy targeting I/R damage (21). Furthermore, in an I/R-induced mouse
model, TLR3 expression has been reported to be elevated in
the intestinal mucosa, and its knockdown was found to reduce
apoptosis of intestinal epithelial cells and lower levels of
inflammatory cytokines post-I/R (22). Additionally, TLR4 has been
implicated in exacerbating I/R injury by augmenting the NF-κB
signaling pathway (23). Although
CTSB has been recognized as a pivotal contributor to I/R
injury in various organs, exacerbating renal I/R injury through
apoptosis, inflammation and dysregulation of autophagy (24), and amplifying myocardial I/R
inflammatory injury via the NLRP3 inflammasome (12), dedicated studies on its specific
role in I/R injury remain sparse (25,26).
Consequently, knowledge gaps persist regarding the regulatory
mechanisms of CTSB, the breadth of its impact across diverse
tissues and its potential as a therapeutic target. By conducting a
comprehensive bioinformatics analysis, CTSB has been
identified as a hub gene that is upregulated during I/R injury.
Given the lack of research on CTSB in the context of I/R
injury and its demonstrated capacity to mediate cellular responses
during this process, CTSB was chosen as the focus of the
current study. The objective of the study was to systematically and
precisely elucidate its regulatory mechanisms, and to assess its
influence on the pathophysiology of I/R injury, thereby addressing
a crucial void in current research.
The lysosomal enzyme CTSB serves a pivotal role in
regulating oxidative stress, inflammation and cell death (24). For example, in cells infected with
a high dosage of Legionella pneumophila, knockdown of
CTSB has been shown to effectively suppress cell death and
inflammation (27). Prior research
has also indicated that inhibiting CTSB can mitigate
intestinal epithelial cell death and safeguard barrier function
(10). Elevated CTSB levels
have been reported to be associated with heightened inflammation
and cellular damage in IBD (28).
The in vivo process of I/R injury is frequently simulated
using the OGD/R approach, which facilitates comprehension of the
complex mechanisms underlying I/R damage, including oxidative
stress, inflammation and apoptosis (29). The current study successfully
replicated I/R-induced inflammation and intestinal barrier
disruption in Caco-2 cells by employing the well-established OGD/R
in vitro model. Caco-2 cells subjected to OGD/R exhibited
decreased cell viability, disruption of tight junction proteins,
increased LDH release, and elevated CTSB protein levels and
activity. Research has shown that IL-28A can protect the expression
of occludin, ZO-1 and claudin-1, thereby enhancing the function of
the epithelial barrier and reducing damage caused by intestinal I/R
(30). A reduction in the
expression of tight junction proteins can exacerbate inflammatory
responses, augment vascular permeability and compromise tissue
barrier function (31). These
adverse effects were alleviated by CTSB knockdown, which
also enhanced cell survival, diminished LDH release and restored
tight junction protein levels. These findings suggest that
CTSB knockdown may shield Caco-2 cells from damage induced
by OGD/R. CTSB could thus serve as a key mediator of
I/R-induced damage by diminishing cell viability and disrupting
barrier proteins.
I/R injury is closely associated with oxidative
stress, a pathological state characterized by an imbalance between
the antioxidant defense mechanisms of the body and the generation
of ROS (32). Within the framework
of I/R, oxidative stress has been extensively studied, with
compelling evidence indicating its pivotal role in tissue damage.
For example, by promoting sirtuin 1-mediated suppression of
epithelial ROS generation and apoptosis, the knockdown of
miR-34a-5p has been shown to mitigate I/R-induced harm (33). Furthermore, oxidative stress
indicators, such as MDA, SOD and GSH-Px, are commonly utilized to
assess antioxidant capability and oxidative damage (34). Research has demonstrated that in
I/R-injured cells, dexmedetomidine can alleviate oxidative stress
by decreasing MDA levels and increasing SOD levels (35). Additionally, elevated GSH-Px
activity in I/R injury can reduce oxidative stress, as it is often
associated with enhanced antioxidant defense (36). Caco-2 cells originate from human
colorectal adenocarcinoma but differentiate into enterocyte-like
cells that form tight junctions and exhibit absorptive and barrier
properties similar to intestinal epithelial cells (37,38).
Therefore, in the current study, Caco-2 cells were selected as an
in vitro model to investigate intestinal I/R injury. The
present study explored how CTSB regulates oxidative stress
in Caco-2 cells subjected to I/R injury. The OGD/R model revealed a
significant elevation in cellular oxidative stress levels; however,
this increase was attenuated by CTSB knockdown. These
findings suggest the potential of CTSB as a therapeutic
target to mitigate oxidative damage, implying its contribution to
oxidative stress in I/R injury.
I/R injury exacerbates tissue damage and impedes the
healing process by triggering a robust inflammatory response
(1,39). The NLRP3 inflammasome, a
multiprotein complex that is instrumental in activating
inflammatory cytokines, serves as a crucial mediator of this
inflammatory cascade (40).
Numerous studies have identified the role of the NLRP3 inflammasome
in I/R injury, with its activation leading to heightened
inflammation and tissue damage (18,41).
For example, VX-765 has been shown to mitigate I/R-induced
harm and alleviate oxidative stress by diminishing NLRP3
inflammasome activation (42).
Additional research has highlighted the strong association between
CTSB and the NLRP3 inflammasome (43). The activation of the NLRP3
inflammasome, which is induced by CTSB, along with the
maturation of pro-inflammatory cytokines IL-1β and IL-18, can be
significantly inhibited by Ilex saponin I (ISI) (25). Concurrently, by disrupting the
CTSB/NLRP3 complex, ISI curtails the inflammatory response through
inactivation of the NLRP3 inflammasome. According to Li et
al (44), hollow cerium-oxide
(CeO2) nanospheres coated with non-stoichiometric copper
oxide (Cu5.4O) (abbreviated as
hCeO2@Cu5.4O nanoparticles) can
reduce inflammation by modifying the CTSB-NLRP3 signaling pathway.
The experimental findings of the current study demonstrated that
the OGD/R model exhibited elevated levels of pro-inflammatory
cytokines, and that CTSB knockdown markedly reduced these
levels while obstructing NLRP3 inflammasome activation. These
results underscore the pivotal role of CTSB in modulating
NLRP3 inflammasome activation, offering promising therapeutic
implications for alleviating I/R-induced intestinal inflammation.
Notably, a direct interaction exists between CTSB and NLRP3, and it
has been shown that CTSB can activate NLRP3 inflammatory vesicles
through direct engagement with NLRP3 (45). This interaction may involve direct
modification of NLRP3 by CTSB or may indirectly influence NLRP3
activity by altering the intracellular milieu. Additionally, the
release of CTSB may activate the NLRP3 inflammasome via lysosomal
rupture. In the current study, through co-IP experiments, the
presence of a direct interaction between CTSB and NLRP3 was
confirmed in an intestinal I/R model. This finding provides
molecular-level evidence that CTSB may regulate its activity by
directly interacting with NLRP3.
In the current study, it was observed that in
CTSB-knockdown Caco-2 cells, the levels of inflammatory
factors, such as TNF-α, IL-6 and IL-1β, were partially restored
following LPS and ATP treatment. This observation suggests that
while CTSB has a pivotal role in the inflammatory response
induced by I/R injury, other regulators or pathways may circumvent
the inhibitory effects of CTSB knockdown, thereby partially
reinstating the inflammatory response. CTSB is crucial in
I/R injury through its activation of the NLRP3 inflammasome, a
process essential for the maturation of inflammatory factors, and
CTSB can directly bind to NLRP3 and enhance its activity (11). In the present study, CTSB
knockdown significantly suppressed OGD/R-induced NLRP3 inflammasome
activation and inflammatory factor expression, further
corroborating the key regulatory role of CTSB in I/R injury.
LPS and ATP are well-established activators of the NLRP3
inflammasome, functioning through distinct signaling pathways. LPS
activates NF-κB via the TLR4 signaling pathway, promoting the
transcription of inflammatory factors (20). Conversely, ATP enters the cell
through the P2X7 receptor and pannexin-1 channel, thereby
activating the NLRP3 inflammasome (33). The partial recovery of inflammatory
factors following LPS + ATP treatment in CTSB-knockdown
cells may indicate that these alternative pathways can bypass the
inhibitory effects of CTSB knockdown and reactivate the
NLRP3 inflammasome. Although CTSB has a critical role in the
inflammatory response, the recovery of inflammatory factors after
LPS + ATP treatment suggests that CTSB may not be the sole
regulator. The activation of the NLRP3 inflammasome involves
multiple signaling pathways, including ROS generation,
mitochondrial damage and lysosomal rupture (33). In the context of CTSB
knockdown, other signaling pathways may be activated by LPS and
ATP, thereby partially restoring the levels of inflammatory
factors. For example, research has shown that ISI can inhibit the
inflammatory response by disrupting the CTSB/NLRP3 complex;
however, this inhibition is not absolute, implying the existence of
other compensatory mechanisms (40). This phenomenon does not undermine
the significance of CTSB as a potential therapeutic target.
Instead, it underscores the complexity of the inflammatory response
and suggests that co-targeting multiple key molecules may be
necessary to effectively inhibit the inflammatory response in
clinical settings. For example, combined inhibition of CTSB
and the TLR4 signaling pathway may prove more effective than
inhibiting CTSB alone.
The present study was confined to in vitro
experiments utilizing a Caco-2 cell model to simulate I/R injury.
While in vitro models can offer valuable insights into the
mechanism of action of CTSB in I/R injury, these findings
necessitate further validation in in vivo models. For
example, future investigations could employ mouse I/R models to
assess the effects of CTSB knockdown or inhibition on
intestinal injury, inflammatory response and oxidative stress. Such
in vivo validation would aid in determining the clinical
potential of CTSB as a therapeutic target. In the present
study, despite CTSB knockdown significantly reducing the
levels of inflammatory factors, these factors remained slightly
elevated following LPS + ATP stimulation. This suggests that, in
the absence of CTSB, other inflammasomes or alternative
pathways may be activated, thus promoting the inflammatory
response. Future studies are warranted to further explore these
potential mechanisms, such as conducting a detailed analysis of the
signaling pathways activated by LPS and ATP using specific
inflammasome inhibitors or by knocking out other inflammasome
components. Additionally, investigating the interaction between
CTSB and other lysosomal enzymes or proteases may provide clues to
understanding the compensatory mechanisms at play. It was
hypothesized that the interaction between CTSB and NLRP3 may occur
through multiple molecular mechanisms. CTSB may directly bind to
NLRP3 and enhance the activity of the NLRP3 inflammasome, thereby
promoting the maturation and release of inflammatory factors.
Furthermore, CTSB may indirectly activate the NLRP3 inflammasome by
affecting the integrity and function of lysosomes. However, a
limitation of the current study is the lack of further verification
regarding the specific details of CTSB activating NLRP3 through
lysosomal rupture. Future studies may further verify this using
advanced tools such as co-IP, mass spectrometry and lysosomal
functional imaging techniques. Although the present study provides
evidence for the critical role of CTSB in I/R injury,
further research is needed to translate these findings into
clinical applications. Future work should include validating the
therapeutic effects of CTSB in in vivo models, and
developing small-molecule inhibitors or gene therapy strategies
targeting CTSB. Additionally, studying the role of
CTSB in different tissues and cell types may help to devise
more specific treatment options to reduce inflammation and tissue
damage in I/R-related diseases.
In conclusion, the present study identified
CTSB as a pivotal regulator of I/R-induced damage in Caco-2
cells, underscoring its critical role in intestinal barrier
dysfunction, inflammation and oxidative stress. Notably,
CTSB knockdown effectively mitigated cellular damage by
reducing oxidative stress, restoring tight junction proteins and
modulating NLRP3 inflammasome activation. Conversely, CTSB
expression was significantly upregulated under OGD/R conditions.
These findings elucidate how CTSB orchestrates inflammatory
signals and influences intestinal barrier function during I/R
injury.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was funded by the Key Specialty Construction Project
of the Pudong Health Commission of Shanghai (grant no.
PWZzk2022-09) and the Qihang Project of Shanghai Pudong New Area
People's Hospital (grant no. PRYQH202502).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
SH, LW, ZW, HX and YW conceived and designed the
research. SH, LW and ZW acquired the data. SH, LW, ZW, SY, QZ, YX
and YJ analyzed and interpreted the data. SY, QZ, YX and YJ
performed statistical analysis. SH, LW and ZW drafted the
manuscript. HX and YW revised the manuscript for important
intellectual content. SH and HX confirm the authenticity of all the
raw data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wang J, Zhang W and Wu G: Intestinal
ischemic reperfusion injury: Recommended rats model and
comprehensive review for protective strategies. Biomed
Pharmacother. 138:1114822021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li G, Wang S and Fan Z: Oxidative stress
in intestinal ischemia-reperfusion. Front Med (Lausanne).
8:7507312022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang J, Liu Z, Liu Y, Shi Y, Chen F and
Leng Y: Role of non-coding RNA in the pathogenesis of intestinal
ischemia-reperfusion injury. Curr Med Chem. 30:4130–4148. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li Y, Feng D, Wang Z, Zhao Y, Sun R, Tian
D, Liu D, Zhang F, Ning S, Yao J and Tian X: Ischemia-induced ACSL4
activation contributes to ferroptosis-mediated tissue injury in
intestinal ischemia/reperfusion. Cell Death Differ. 26:2284–2299.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Meng Q, Ye C and Lu Y: miR-181c regulates
ischemia/reperfusion injury-induced neuronal cell death by
regulating c-Fos signaling. Pharmazie. 75:90–93. 2020.PubMed/NCBI
|
|
6
|
Zu G, Zhou T, Che N and Zhang X:
Salvianolic acid A protects against oxidative stress and apoptosis
induced by intestinal ischemia-reperfusion injury through
activation of Nrf2/HO-1 pathways. Cell Physiol Biochem.
49:2320–2332. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wu L, Xiong X, Wu X, Ye Y, Jian Z, Zhi Z
and Gu L: Targeting oxidative stress and inflammation to prevent
ischemia-reperfusion injury. Front Mol Neurosci. 13:282020.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hook G, Reinheckel T, Ni J, Wu Z, Kindy M,
Peters C and Hook V: Cathepsin B gene knockout improves behavioral
deficits and reduces pathology in models of neurologic disorders.
Pharmacol Rev. 74:600–629. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
De Pasquale V, Moles A and Pavone LM:
Cathepsins in the pathophysiology of mucopolysaccharidoses: New
perspectives for therapy. Cells. 9:9792020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hu ML, Liao QZ, Liu BT, Sun K, Pan CS,
Wang XY, Yan L, Huo XM, Zheng XQ, Wang Y, et al: Xihuang pill
ameliorates colitis in mice by improving mucosal barrier injury and
inhibiting inflammatory cell filtration through network regulation.
J Ethnopharmacol. 319:1170982024. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dong L, Xie J, Wang Y, Jiang H, Chen K, Li
D, Wang J, Liu Y, He J, Zhou J, et al: Mannose ameliorates
experimental colitis by protecting intestinal barrier integrity.
Nat Commun. 13:48042022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu C, Yao Q, Hu T, Cai Z, Xie Q, Zhao J,
Yuan Y, Ni J and Wu QQ: Cathepsin B deteriorates diabetic
cardiomyopathy induced by streptozotocin via promoting
NLRP3-mediated pyroptosis. Mol Ther Nucleic Acids. 30:198–207.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhao S, Gong Z, Zhou J, Tian C, Gao Y, Xu
C, Chen Y, Cai W and Wu J: Deoxycholic acid triggers NLRP3
inflammasome activation and aggravates DSS-induced colitis in mice.
Front Immunol. 7:5362016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu M, Wen H, Zuo L, Song X, Geng Z, Ge S,
Ge Y, Wu R, Chen S, Yu C and Gao Y: Bryostatin-1 attenuates
intestinal ischemia/reperfusion-induced intestinal barrier
dysfunction, inflammation, and oxidative stress via activation of
Nrf2/HO-1 signaling. FASEB J. 37:e229482023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhao Y, Zhuang Y, Shi J, Fan H, Lv Q and
Guo X: Cathepsin B induces kidney diseases through different types
of programmed cell death. Front Immunol. 16:15353132025. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kip AM, Grootjans J, Manca M, Hadfoune MH,
Boonen B, Derikx JPM, Biessen EAL, Olde Damink SWM, Dejong CHC,
Buurman WA and Lenaerts K: Temporal transcript profiling identifies
a role for unfolded protein stress in human gut
ischemia-reperfusion injury. Cell Mol Gastroenterol Hepatol.
13:681–694. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang Z, Li Z, Feng D, Zu G, Li Y, Zhao Y,
Wang G, Ning S, Zhu J, Zhang F, et al: Autophagy induction
ameliorates inflammatory responses in intestinal
Ischemia-reperfusion through inhibiting NLRP3 inflammasome
activation. Shock. 52:387–395. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nadatani Y, Watanabe T, Shimada S, Otani
K, Tanigawa T and Fujiwara Y: Microbiome and intestinal
ischemia/reperfusion injury. J Clin Biochem Nutr. 63:26–32. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhenzhen L, Wenting L, Jianmin Z, Guangru
Z, Disheng L, Zhiyu Z, Feng C, Yajing S, Yingxiang H, Jipeng L, et
al: miR-146a-5p/TXNIP axis attenuates intestinal
ischemia-reperfusion injury by inhibiting autophagy via the
PRKAA/mTOR signaling pathway. Biochem Pharmacol. 197:1148392022.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cong R, Yang J, Zhou J, Shi J, Zhu Y, Zhu
J, Xiao J, Wang P, He Y and He B: The potential role of protein
tyrosine phosphatase, receptor type C (CD45) in the intestinal
ischemia-reperfusion injury. J Comput Biol. 27:1303–1312. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang XY, Liang HS, Hu JJ, Wan YT, Zhao J,
Liang GT, Luo YH, Liang HX, Guo XQ, Li C, et al: Ribonuclease
attenuates acute intestinal injury induced by intestinal ischemia
reperfusion in mice. Int Immunopharmacol. 83:1064302020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang J, Wu Y, Xu Y, Jia J, Xi W, Deng H
and Tu W: Dexmedetomidine resists intestinal ischemia-reperfusion
injury by inhibiting TLR4/MyD88/NF-κB signaling. J Surg Res.
260:350–358. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu C, Cai Z, Hu T, Yao Q and Zhang L:
Cathepsin B aggravated doxorubicin-induced myocardial injury via
NF-κB signalling. Mol Med Rep. 22:4848–4856. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wu J, Chen S, Wu P, Wang Y, Qi X, Zhang R,
Liu Z, Wang D and Cheng Y: Cathepsin B/HSP70 complex induced by
Ilexsaponin I suppresses NLRP3 inflammasome activation in
myocardial ischemia/reperfusion injury. Phytomedicine.
105:1543582022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sun C, Cao N, Wang Q, Liu N, Yang T, Li S,
Pan L, Yao J, Zhang L, Liu M, et al: Icaritin induces resolution of
inflammation by targeting cathepsin B to prevents mice from
ischemia-reperfusion injury. Int Immunopharmacol. 116:1098502023.
View Article : Google Scholar
|
|
27
|
Morinaga Y, Yanagihara K, Nakamura S,
Hasegawa H, Seki M, Izumikawa K, Kakeya H, Yamamoto Y, Yamada Y,
Kohno S and Kamihira S: Legionella pneumophila induces cathepsin
B-dependent necrotic cell death with releasing high mobility group
box1 in macrophages. Respir Res. 11:1582010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Menzel K, Hausmann M, Obermeier F,
Schreiter K, Dunger N, Bataille F, Falk W, Scholmerich J, Herfarth
H and Rogler G: Cathepsins B, L, and D in inflammatory bowel
disease macrophages and potential therapeutic effects of cathepsin
inhibition in vivo. Clin Exp Immunol. 146:169–180. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu Y, Ji T, Jiang H, Chen M, Liu W, Zhang
Z and He X: Emodin alleviates intestinal ischemia-reperfusion
injury through antioxidant stress, anti-inflammatory responses, and
anti-apoptosis effects via Akt-mediated HO-1 upregulation. J
Inflamm (Lond). 21:252024. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li L, Zhou C, Li T, Xiao W, Yu M and Yang
H: Interleukin-28A maintains the intestinal epithelial barrier
function through regulation of claudin-1. Ann Transl Med.
9:3652021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Brooks TA, Hawkins BT, Huber JD, Egleton
RD and Davis TP: Chronic inflammatory pain leads to increased
blood-brain barrier permeability and tight junction protein
alterations. Am J Physiol Heart Circ Physiol. 289:H738–H743. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Adwas AA, Elsayed A, Azab AE and Quwaydir
FA: Oxidative stress and antioxidant mechanisms in human body. J
Appl Biotechnol Bioeng. 6:43–47. 2019.
|
|
33
|
Wang G, Yao J, Li Z, Zu G, Feng D, Shan W,
Li Y, Hu Y, Zhao Y and Tian X: miR-34a-5p inhibition alleviates
intestinal ischemia/reperfusion-induced reactive oxygen species
accumulation and apoptosis via activation of SIRT1 signaling.
Antioxid Redox Signal. 24:961–973. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li F, Wang X, Deng Z, Zhang X, Gao P and
Liu H: Dexmedetomidine reduces oxidative stress and provides
neuroprotection in a model of traumatic brain injury via the PGC-1α
signaling pathway. Neuropeptides. 72:58–64. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hou M, Chen F, He Y, Tan Z, Han X, Shi Y,
Xu Y and Leng Y: Dexmedetomidine against intestinal
ischemia/reperfusion injury: A systematic review and meta-analysis
of preclinical studies. Eur J Pharmacol. 959:1760902023. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ustundag B, Kazez A, Demirbag M, Canatan
H, Halifeoglu I and Ozercan IH: Protective effect of melatonin on
antioxidative system in experimental ischemia-reperfusion of rat
small intestine. Cell Physiol Biochem. 10:229–236. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sambuy Y, De Angelis I, Ranaldi G, Scarino
M, Stammati A and Zucco F: The Caco-2 cell line as a model of the
intestinal barrier: Influence of cell and culture-related factors
on Caco-2 cell functional characteristics. Cell Biol Toxicol.
21:1–26. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Meunier V, Bourrie M, Berger Y and Fabre
G: The human intestinal epithelial cell line Caco-2;
pharmacological and pharmacokinetic applications. Cell Biol
Toxicol. 11:187–194. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Deng F, Lin ZB, Sun QS, Min Y, Zhang Y,
Chen Y, Chen WT, Hu JJ and Liu KX: The role of intestinal
microbiota and its metabolites in intestinal and extraintestinal
organ injury induced by intestinal ischemia reperfusion injury. Int
J Biol Sci. 18:39812022. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Moretti J and Blander JM: Increasing
complexity of NLRP3 inflammasome regulation. J Leukoc Biol.
109:561–571. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ito H, Kimura H, Karasawa T, Hisata S,
Sadatomo A, Inoue Y, Yamada N, Aizawa E, Hishida E, Kamata R, et
al: NLRP3 inflammasome activation in lung vascular endothelial
cells contributes to intestinal ischemia/reperfusion-induced acute
lung injury. J Immunol. 205:1393–1405. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lyu H, Ni H, Huang J, Yu G, Zhang Z and
Zhang Q: VX-765 prevents intestinal ischemia-reperfusion injury by
inhibiting NLRP3 inflammasome. Tissue Cell. 75:1017182022.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cai Z, Xu S and Liu C: Cathepsin B in
cardiovascular disease: Underlying mechanisms and therapeutic
strategies. J Cell Mol Med. 28:e700642024. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li Y, Xia X, Niu Z, Wang K, Liu J and Li
X: hCeO2@ Cu5.4O nanoparticle alleviates inflammatory responses by
regulating the CTSB-NLRP3 signaling pathway. Front Immunol.
15:13440982024. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Xu W, Huang Y and Zhou R: NLRP3
inflammasome in neuroinflammation and central nervous system
diseases. Cell Mol Immunol. 22:341–355. 2025. View Article : Google Scholar : PubMed/NCBI
|