Introduction
Chronic obstructive pulmonary disease (COPD) is
defined as a heterogeneous lung condition characterized by airway
limitation and respiratory symptoms resulting from airway
inflammation and emphysema (1).
Although >3 million individuals succumb to COPD annually
worldwide, global mortality rates might be underestimated, as
one-third of patients with COPD succumb to cardiovascular diseases
(2). The persistent prevalence of
COPD solidifies its status as a substantial public health concern
globally. In patients with COPD, pulmonary gas exchange anomalies
and airway limitations are important pathophysiological
characteristics. Damage to the pulmonary air-blood barrier is a
notable factor in abnormal gas exchange, directly contributing to
inefficiencies in gas exchange, exacerbating dyspnea and worsening
respiratory symptoms (3).
The pulmonary air-blood barrier is the key element
of the lungs, serving as the site for the exchange of oxygen and
carbon dioxide between the human body and the external environment
(4). It primarily consists of
alveolar epithelium cells and capillary endothelial cells, either
directly connected or separated by interstitial tissue (5). This barrier acts as a defense against
pathogen invasion and is important for maintaining normal lung
physiological function. Various pathogenic factors, including
cigarette smoke, pollution gases, bacteria and viruses, can induce
damage to the air-blood barrier. This damage increases
permeability, aggravates lung tissue edema and triggers an
inflammatory response (6).
Cigarette smoke is recognized as the most common
risk factor for the occurrence and progression of COPD (7,8).
Previous studies have shown that cigarette smoke inhalation can
lead to autophagy impairment in pulmonary vascular endothelial
cells, initiating aggresome formation, resulting in inflammatory
responses and senescence that contribute to the decline of
pulmonary function in patients with severe COPD (9,10).
Additionally, the injury and death of alveolar epithelium cells
induced by cigarette smoke worsens emphysema and airway remodeling
in COPD, promoting the development of the disease (11).
Bacterial infections also play a notable role in
damaging the pulmonary air-blood barrier in patients with COPD. The
total number of pathogenic bacteria associates with decreased lung
function in patients with COPD (12). For instance, Klebsiella
pneumoniae induces apoptosis of alveolar epithelial cells
through activation of the cytokine and IFN-γ-mediated signaling
pathways (13). Similarly,
Mycobacterium tuberculosis infection suppresses alveolar
epithelial cell autophagy, promotes inflammatory responses via
tissue inhibitor of metalloproteinases (TIMP)-2 suppression and
activates NF-κB (14).
Furthermore, bacterial infections induce rapid cell damage and
death of pulmonary microvascular endothelial cells by activating
pro-inflammatory caspase-1 (15).
It is therefore important to elucidate the relationship between
cigarette smoke, bacterial infection and the pulmonary air-blood
barrier.
The MAPK signaling pathway plays a central role in
the occurrence and progression of COPD. Its abnormal activation
drives disease progression by regulating pathological processes
such as the inflammatory response, oxidative stress and mucus
hypersecretion (16).
Specifically, p38 MAPK upregulates the expression of TNF-α and IL-6
through phosphorylating the transcription factors cyclic
AMP-dependent transcription factor ATF-2 and ETS domain-containing
protein Elk-1, thereby recruiting neutrophils and monocytes for
infiltration into lung tissues (17). The JNK pathway promotes the
differentiation of T helper 17 cells, which in turn leads to the
release of IL-17 to activate p38 MAPK, forming a positive
inflammatory feedback loop (18).
In addition, p38 MAPK upregulates the expression of matrix
metalloproteinases (MMPs) to degrade elastic fibers and collagen,
while simultaneously inhibiting the activity of TIMP, thus
facilitating the development of emphysema (19). In terms of mucus secretion,
cigarette smoke induces the transcription of the mucoprotein
(MUC)5AC gene by activating EGFR, which subsequently phosphorylates
ERK1/2 (20).
In the present study, a stable COPD rat model was
established through cigarette smoke inhalation and repetitive
bacterial infection to elucidate the effects of cigarette smoke and
bacterial infection on the pulmonary air-blood barrier and their
mechanisms of action. The present study evaluated pulmonary
function, pulmonary pathology, the ultrastructure of the air-blood
barrier and barrier integrity to assess injury in COPD rats.
Subsequently, inflammation, oxidative stress and the balance of
protease and antiprotease were compared between normal rats and
COPD model rats to clarify the potential mechanism of pulmonary
air-blood barrier damage caused by cigarette smoke inhalation and
repetitive bacterial infection.
Materials and methods
Chemicals and reagents
A total of 16 male Sprague-Dawley (SD) rats aged 6–7
weeks (220±20 g; cat no. 110011211105823815) were purchased from
Beijing Vital River Laboratory Animal Technology Co., Ltd. All mice
were raised at a controlled temperature (25°C) and humidity (50%)
under a 12 h light/dark cycle and provided free access to food and
water. The experimental protocols were approved by the Experimental
Animal Care and Ethics Committees of the First Affiliated Hospital
at Henan University of Traditional Chinese Medicine, Zhengzhou,
China (approval no. YFYDW2019031). Klebsiella pneumoniae
(cat. no. 46117-5a1) was obtained from the National Center for
Medical Culture Collections (https://www.cmccb.org.cn/htmls/index.html). The
cigarettes (Hongqi Canal® Filter tip) were purchased
from Henan Tobacco Industry (https://www.hatic.com/zhongyanAdmin/html/sy/).
Establishment of a COPD rat model
The COPD rat model was established through cigarette
smoke inhalation and repetitive bacterial infection (21). A total of 16 SD rats were randomly
divided into the control group and the model group. From weeks 1–8,
rats in the model group were exposed to cigarette smoke (3,000±500
ppm) for 40 min twice daily, and were given intranasal instillation
of Klebsiella pneumoniae suspension (6×108
Cfu/ml; 0.1 ml) once every five days (22,23).
In the experiment, cigarettes were placed in a sealed cigarette
box. The concentration of smoke was controlled by adjusting the
number of lit cigarettes and a smoke sensor was used to monitor the
concentration, ensuring that the smoke concentration was maintained
at 3,000±500 ppm. Rats in the control group were exposed to normal
saline (0.1 ml) once every 5 days. At the end of week 8, rats were
anesthetized after intraperitoneal injection of 2% pentobarbital
sodium at 40 mg/kg. Subsequently, under deep anesthesia, rats were
euthanized by abdominal aortic blood collection to induce rapid
blood loss and immediate death, aiming to minimize animal
suffering. Irreversible death was confirmed through the combined
evaluation of three important parameters: i) The cessation of
spontaneous thoracic wall movements, indicating respiratory arrest;
ii) the absence of cardiac pulsation upon palpation of the
precordial region, confirming cardiac arrest; and iii) the presence
of mydriasis.
Pulmonary function measurement
Pulmonary function was measured for both groups of
rats every four weeks from weeks 0–8, assessing tidal volume (TV),
peak expiratory flow (PEF) and 50% TV expiratory flow (EF50) using
unrestrained pulmonary function testing plethysmographs (B&E
Teksystems Ltd; http://www.bandetek.com/wzsy).
Pulmonary tissue histopathology
The lung tissues were immersed in a 4%
paraformaldehyde solution at room temperature for 12 h.
Subsequently, the tissues were cut, embedded in paraffin and
sectioned. The 4 µm lung tissue sections were stained with
hematoxylin at room temperature for 2 min and eosin at room
temperature for 15 sec, and observed using a light microscope
(Olympus Corporation). Mean alveolar numbers (MAN) and mean linear
intercept (MLI) were determined to assess the degree of alveolar
damage. Under a light microscope (magnification, ×200), six visual
fields were captured in each slice, and the MAN and MLI in a fixed
area of the visual field were measured. A cross was marked under
the visual field and the number of alveolar septa on the cross was
counted. MLI (µm)=L/Ns, where Ns is the number of alveolar septa
and L is the total length of the cross. MAN (/mm2)=Na/A,
where Na is the number of pulmonary alveoli in each visual field
and A is the area of the visual field.
Ultrastructure of lung tissue
The lung tissue was sectioned into small pieces
measuring 1×1×1 mm, fixed with 2.5% glutaraldehyde at 4°C for 24 h
and further fixed with 1% osmic acid at 4°C for 2 h. The tissue was
then cut, embedded in Epon218 (SPI-Pon 812R Epoxy Resin Monomer;
cat. no. 25068-38-6; SPI Supplies) and sectioned at a thickness of
70 nm. The ultrastructure of the air-blood barrier, capillary
endothelium, type I alveolar epithelial cells (AT I) and type II
alveolar epithelial cells (AT II) was observed using a JEM-1400
transmission electron microscope (JEOL, Ltd.). A total of three
fields of view were observed for each sample.
Immunohistochemical analysis
The lung tissues were immersed in a 4%
paraformaldehyde solution at room temperature for 12 h.
Subsequently, the tissues were cut, embedded in paraffin and
sectioned. Lung tissues were sectioned at a thickness of 4 µm, and
were dewaxed twice in xylene (20 min each), rehydrated through a
descending ethanol series (100, 95, 85 and 75%, 10 min each), and
rinsed in PBS three times for 5 min each. For antigen retrieval,
slides were immersed in citrate buffer (pH 6.0) and heated in a
microwave oven at high power for 5–8 min, followed by 5 min on low
power. The slides were then allowed to cool at room temperature for
40 min and rinsed again with PBS (three times, 5 min each).
Immunohistochemistry was conducted using a commercial
immunohistochemical kit (SA1022; Wuhan Boster Biological
Technology, Ltd.). Endogenous peroxidase activity was blocked by
incubating the sections with a peroxidase blocking solution for 15
min, followed by three washes with PBS (3 min each). Non-specific
binding was blocked by incubating the slides in 5% BSA (cat. no.
A8020; Beijing Solarbio Science & Technology Co., Ltd.) at 37°C
for 1 h. After blocking, the sections were incubated overnight at
4°C with the following primary antibodies: MUC5AC (1:500; cat. no.
85868-1-RR; Proteintech Group, Inc.), MUC5B (1:500; cat. no.
28118-1-AP; Proteintech Group, Inc.), zonula occludens (ZO)-1
(1:200; cat. no. AF5145; Affinity Biosciences) and claudin-5
(1:200; cat. no. AF5216; Affinity Biosciences). The following day,
the sections were washed three times in PBS (3 min each), then
incubated at 37°C for 1 h with HRP-conjugated goat anti-rabbit IgG
secondary antibody provided in commercial immunohistochemical kit
(cat. no. SA1022; Wuhan Boster Biological Technology, Ltd.). Signal
development was performed using the metal-enhanced DAB substrate
kit (cat. no. AR1027; Wuhan Boster Biological Technology, Ltd.) for
10 min at room temperature, followed by rinsing in distilled water.
Sections were counterstained with hematoxylin for 2 min with room
temperature and washed under running tap water for 10 min.
Differentiation was carried out in 1% hydrochloric acid ethanol for
3 sec, followed by an additional rinse under running water for 3
min. The stained slides were then dehydrated in ascending
concentrations of ethanol (75, 85, 95 and 100%, 5 min each),
cleared in xylene twice (5 min each) and mounted with neutral
resin. All stained sections were examined using a light microscope
(Olympus Corporation) for morphological analysis was performed
using Image-Pro Plus 6.0 software (Media Cybernetics, Inc.).
Immunofluorescence of lung tissue
The lung tissues were immersed in a 4%
paraformaldehyde solution at room temperature for 12 h.
Subsequently, the tissues were cut, embedded in paraffin and
sectioned. Lung tissues were sectioned at a thickness of 4 µm, and
were dewaxed twice in xylene (20 min each), rehydrated through a
descending ethanol series (100, 95, 85 and 75%, 10 min each), and
rinsed in PBS three times for 5 min each. For antigen retrieval,
slides were immersed in citrate buffer (pH 6.0) and heated in a
microwave oven at high power for 5–8 min, followed by 5 min on low
power. The slides were then allowed to cool at room temperature for
40 min and rinsed again with PBS (three times, 5 min each). The
slices were permeabilized with 0.2% Triton X-100, followed by
blocking in 3% BSA for 1 h. The slices were incubated with
target-specific primary antibodies overnight at 4°C: aquaporin
(AQP)-5 (1:1,000; cat. no. 20334-1-AP; Proteintech Group, Inc.),
surfactant protein-D (SP-D; 1:1,000; cat. no. DF13601; Affinity
Biosciences), CD31 (1:1,000; cat. no. AF6191; Affinity
Biosciences). After thorough PBS washing, the specimens were
exposed to fluorescein-conjugated secondary antibody (1:1,000;
Cy3-conjugated Goat Anti-Rabbit IgG(H+L); cat. no. SA00009-2;
Proteintech Group, Inc.; 1:1,000; Fluorescein (FITC)-conjugated
Goat Anti-Mouse IgG(H+L); cat. no. SA00003-1; Proteintech Group,
Inc.) in a dark place for 1 h at room temperature. Following three
additional PBS washes (3 times, 5 min each), the coverslips were
mounted onto glass slides with Fluoroshield medium containing DAPI.
Finally, a laser confocal microscope (LSM700; Carl Zeiss AG) was
used for detection.
Alcian blue/periodic acid-Schiff
(AB-PAS) staining
The lung tissues were immersed in a 4%
paraformaldehyde solution at room temperature for 12 h.
Subsequently, the tissues were cut, embedded in paraffin and
sectioned. The 4 µm lung tissue slices were stained with Alcian
Blue at room temperature for 15 min and with Schiff at room
temperature for 15 min. The lung tissue slices were observed using
a light microscope (Olympus Corporation). Positive staining was
considered indicative of goblet cells.
Enzyme-linked immunosorbent assay
(ELISA)
Lung tissue was homogenized in a phosphate-buffered
saline solution and centrifuged at 12,880 × g and 4°C for 10 min to
collect the supernatant. The secretion of IL-6 (Rat IL-6 ELISA Kit;
cat. no. E-EL-R0015; Elabscience Bionovation Inc.), IL-10 (Rat
IL-10 ELISA Kit; cat. no. E-EL-R0016; Elabscience Bionovation
Inc.), malondialdehyde (MDA) (MDA assay kit; cat. no. A003-1-2;
Nanjing Jiancheng Bioengineering Institute), total superoxide
dismutase (T-SOD) (SOD assay kit; cat. no. A001-3-2; Nanjing
Jiancheng Bioengineering Institute), MMP-9 (Rat MMP-9 ELISA Kit;
cat. no. E-EL-R3021; Elabscience Bionovation Inc.) and tissue
Inhibitors of metalloproteinase 1 (TIMP-1 ELISA Kit; cat. no.
E-EL-R0540; Elabscience Bionovation Inc.) in the lung tissue
homogenate was measured using ELISA kits, following the
manufacturer's instructions.
Western blot analysis
The protein levels of GAPDH (1:5,000, GAPDH
Polyclonal antibody, 10494-1-AP; Proteintech Group, Inc.), ZO-1
(1:1,000; cat. no. AF5145; Affinity Biosciences), SP-D (1:1,000;
cat. no. DF13601; Affinity Biosciences), claudin-5 (1:1,000; cat.
no. AF5216; Affinity Biosciences), angiopoietin (Ang)-2 (1:1,000;
cat. no. DF6137; Affinity Biosciences), p-p38 (1:1,000; cat. no.
AF4001; Affinity Biosciences), p38 (1:1,000, p38 MAPK Antibody,
AF6456; Affinity Biosciences), p-ERK (1:1,000; cat. no. AF1015;
Affinity Biosciences), ERK (1:1,000; cat. no. AF0155; Affinity
Biosciences), p-JNK (1:1,000; cat. no. AF3318; Affinity
Biosciences), JNK (1:1,000; cat. no. AF6318; Affinity Biosciences),
p-p65 (1:1,000; cat. no. AF2006; Affinity Biosciences), p65
(1:1,000; cat. no. AF5006; Affinity Biosciences), p-IκBα (1:1,000;
cat. no. AF2002; Affinity Biosciences) and IκBα (1:1,000; cat. no.
AF5002; Affinity Biosciences) in lung tissue were determined by
western blotting. Rat lung tissue (10 mg) was homogenized in 150 µl
of 1X RIPA Buffer (cat. no. R0020; Beijing Solarbio Science &
Technology Co., Ltd.), followed by centrifugation at 12,880 × g and
4°C for 10 min to collect the supernatant. The protein
concentration of the lung tissue homogenate was determined via the
BCA assay (BCA Protein Assay Kit; cat. no. PC0020; Beijing Solarbio
Science & Technology Co., Ltd.). The 30–40 µg lung tissue
protein samples were divided by 10% SDS-PAGE electrophoresis and
metastasized to PVDF membranes. Then 5% skimmed milk was used to
block the PVDF at room temperature for 1 h. Next, membranes were
incubated with their primary antibodies overnight at 4°C, and
secondary antibodies (1:5,000, HRP-conjugated Rabbit Anti-Goat
IgG(H+L); cat. no. SA00001-4; Proteintech Group, Inc.) at room
temperature for 1 h. The membranes were washed with 1X TBST with
0.1% Tween. The membranes were visualized using the Bio-Rad Imaging
System (ChemiDoc MP; Bio-Rad Laboratories, Inc.).
Statistical analysis
The experimental data were analyzed using SPSS v21.0
(IBM Corp.). Comparisons among groups were performed by unpaired
t-test or Mann-Whitney U test. If the data followed a normal
distribution, a Student's t-test was performed. If the data did not
follow a normal distribution, Mann-Whitney U test was used. All
experiments were independently repeated ≥3 times to ensure
reproducibility. The mean ± standard error of the mean was used for
data presentation. A P-value <0.05 was considered statistically
significant.
Results
Decline of pulmonary function and
damage of pulmonary pathology are induced by cigarette smoke
combined with Klebsiella pneumoniae infection
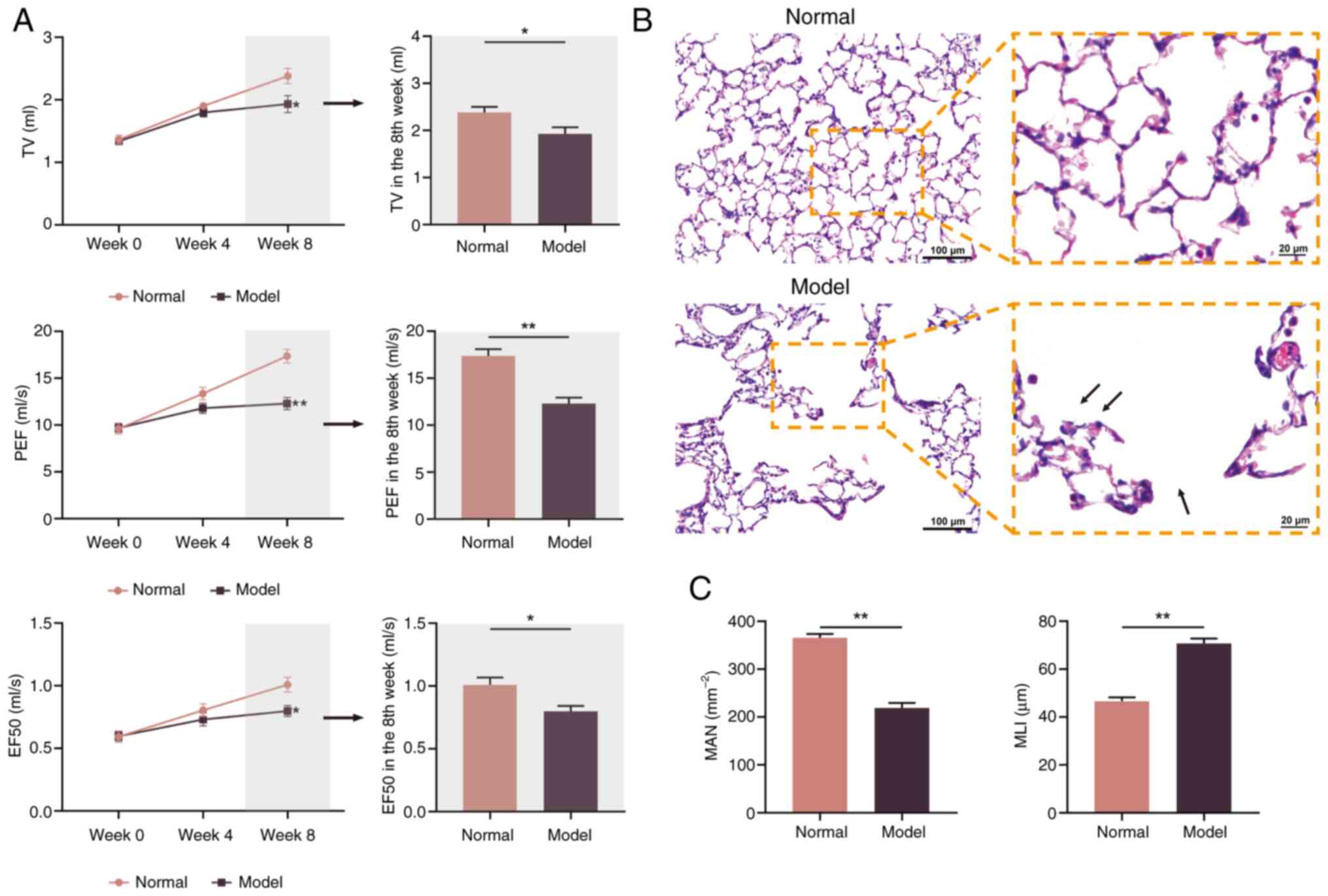
TV, PEF and EF50 are important indicators reflecting
the degree of dyspnea (24). TV,
PEF and EF50 significantly decreased with prolonged exposure to
cigarette smoke and bacterial infection (Fig. 1A). By week 8, these parameters
significantly decreased in COPD rats compared with normal rats
(P<0.05 and P<0.01).
MAN and MLI were used to assess the degree of
alveolar damage. By week 8, there was a notable increase in
alveolar rupture, fusion and thickening of the alveolar wall in
COPD rats compared with normal rats (Fig. 1B). The significant decrease in MAN
and increase in MLI observed in COPD rats compared with normal rats
indicated that pulmonary damage had occurred (both P<0.01;
Fig. 1C).
Cigarette smoke combined with
Klebsiella pneumoniae infection induces structural and functional
impairment of the air-blood barrier
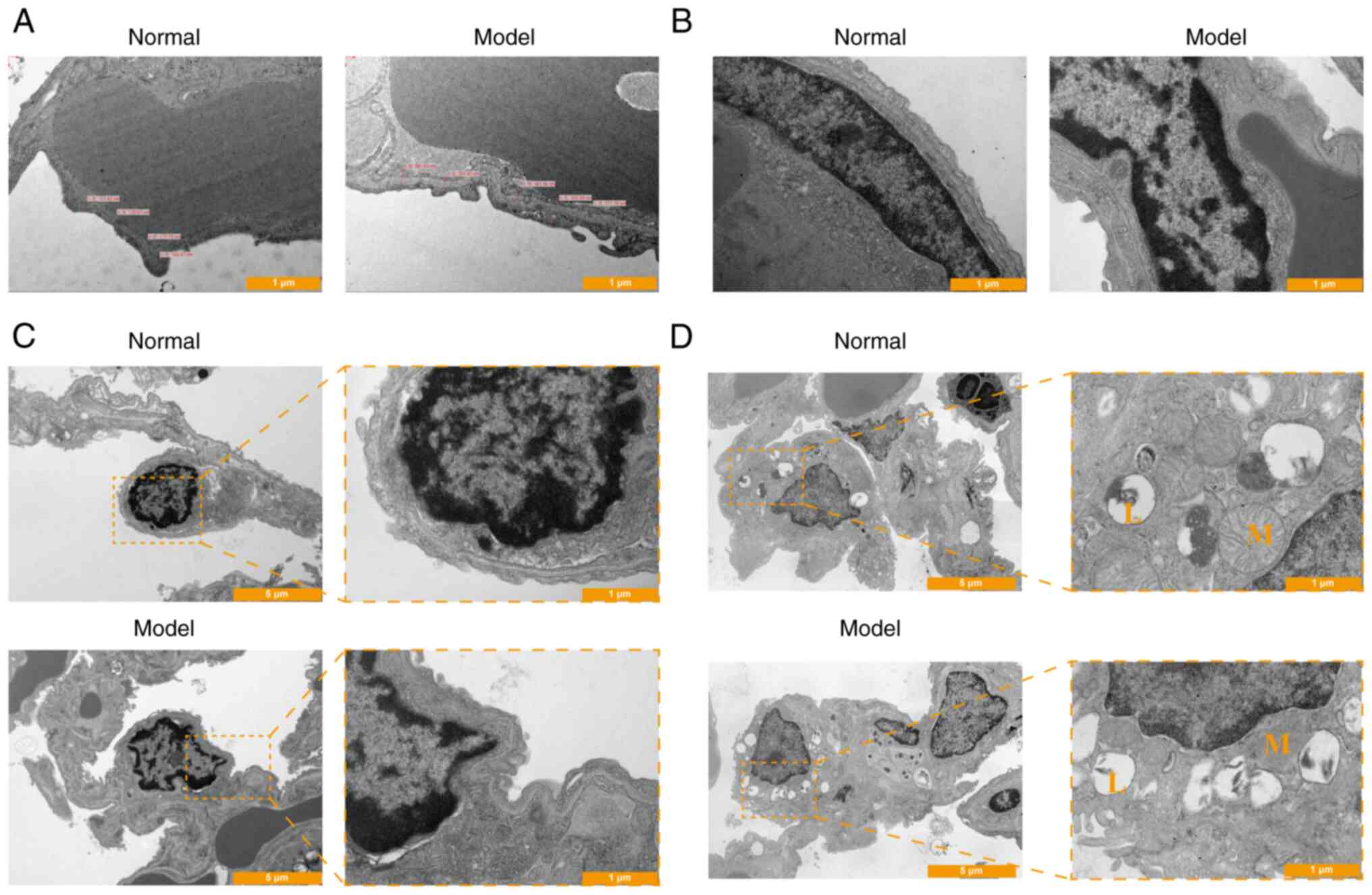
In week 8, the structural integrity of the
respiratory membrane in normal rats exhibited uniform thickness
with a clear structure (Fig. 2A).
The capillary endothelial cells appeared flat, with regular nuclear
shapes and uniform cell membrane thickness (Fig. 2B). Conversely, in the model group,
the present study observed unevenness in the structure of the
respiratory membrane, with blurred edges and noticeable thickening
(Fig. 2A). The capillary
endothelial space widened in the COPD model, displaying disordered
cell arrangement, irregular cell shapes, fuzzy membrane structures,
unclear edges and occasional cell debris (Fig. 2B).
The structure of AT I in normal rats was intact,
with a smooth surface, a thicker nucleus-containing part protruding
into the alveolar cavity and a thin non-nuclear cytoplasm
containing clear ribosomes and numerous pinocytic vesicles. In
model rats, the structure of AT I was blurred, with unclear edges,
a rough surface and a reduced number of vesicles for pinocytosis
(Fig. 2C). Immunohistochemical
analysis revealed a significant decrease in the expression of ZO-1
and claudin-5 in COPD rats compared with normal rats (P<0.01;
Fig. 3A). This low expression was
further supported by western blot analysis (P<0.05; Fig. 3B), indicating that cigarette smoke
and bacterial infection can damage the tight connections within the
air-blood barrier. These results suggested a notable increase in
structural damage to the air-blood barrier in COPD rats compared
with normal rats.
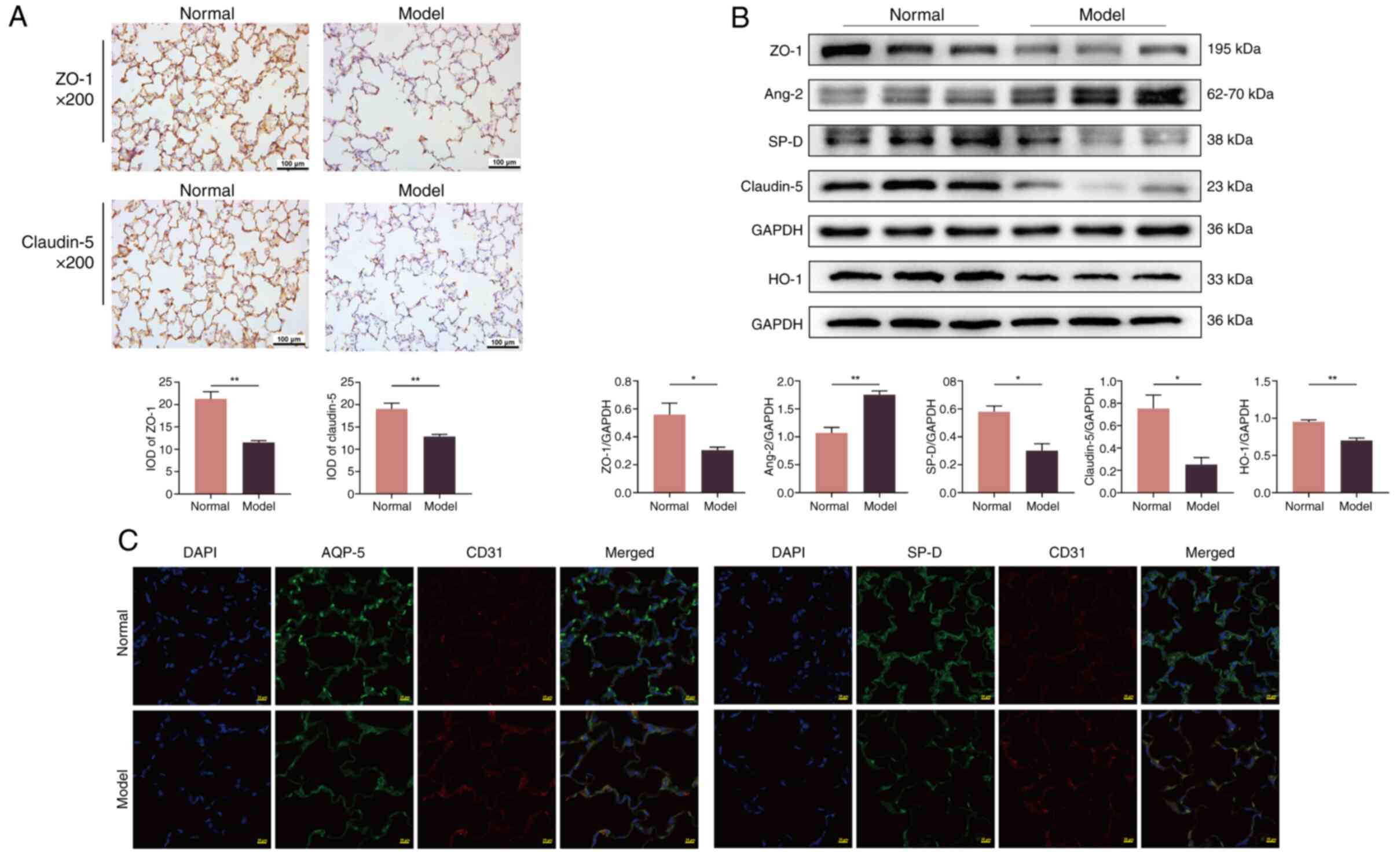 | Figure 3.Injury of the air-blood barrier
induced in all groups. (A) The expression level of ZO-1 and
claudin-5 in the lung tissue of rats was tested using
immunohistochemistry (×200 magnification). The values are expressed
as mean ± SE; (n=8). **P<0.01 vs. normal group. (B) Protein
expression of ZO-1, claudin-5, SP-D, HO-1 and Ang-2 in the lung
tissue tested via western blotting. The values are expressed as
mean ± SE; (n=3). *P<0.05 and **P<0.01 vs. normal group. (C)
The expression level of AQP-5, SP-D and CD31 in the lung tissue
tested using immunofluorescence (×200 magnification). ZO-1, zonula
occludens-1; SP-D, surfactant protein-D; HO-1, heme oxygenase-1;
Ang-2, angiopoietin 2; AQP-5, aquaporin-5; IOD, integrated optical
density. |
AT II had a clear structure in normal rats, with
neatly arranged villi, abundant lamellar bodies, numerous
mitochondria and well-defined mitochondrial ridges. Conversely, in
model rats, the structure of AT II was unclear, with lamellar
bodies detaching in the form of vacuoles and decreasing in number.
Additionally, mitochondria showed signs of pyknosis or swelling,
and the mitochondrial ridges appeared blurred (Fig. 2D).
The fluorescence intensity of AQP-5 and SP-D
markedly decreased in COPD rats compared with the normal group,
while that of CD31 increased notably (Fig. 3C). Furthermore, the protein
expression of SP-D (P<0.05) and heme oxygenase-1 (P<0.01)
significantly decreased, and the expression of Ang-2 increased
significantly (P<0.01) in COPD rats compared with normal rats
(Fig. 3B). These findings
indicated a significant decrease in the function of the air-blood
barrier in COPD rats compared with normal rats.
Cigarette smoke combined with
Klebsiella pneumoniae infection induces mucus hypersecretion,
inflammation and oxidative stress
In COPD rats, positive AB-PAS staining markedly
increased in the airways compared with normal rats, indicating
goblet cell proliferation (Fig.
4A). The expression of MUC5AC and MUC5B was significantly
elevated in COPD rats compared with normal rats (Fig. 4B). COPD rats also exhibited an
inflammatory response induced by cigarette smoke and bacterial
infection. When compared with normal rats, the secretion of IL-6
and TNF-α significantly increased in the lung tissue and serum of
COPD rats (P<0.05 and P<0.01), while that of IL-10 decreased
significantly in the lung tissue of COPD rats (P<0.05).
Similarly, the expression of MMP-9 increased and TIMP-1 decreased
significantly in the lung tissue of COPD rats (P<0.05 and
P<0.01), indicating abnormal extracellular matrix deposition and
the disruption of organizational structure.
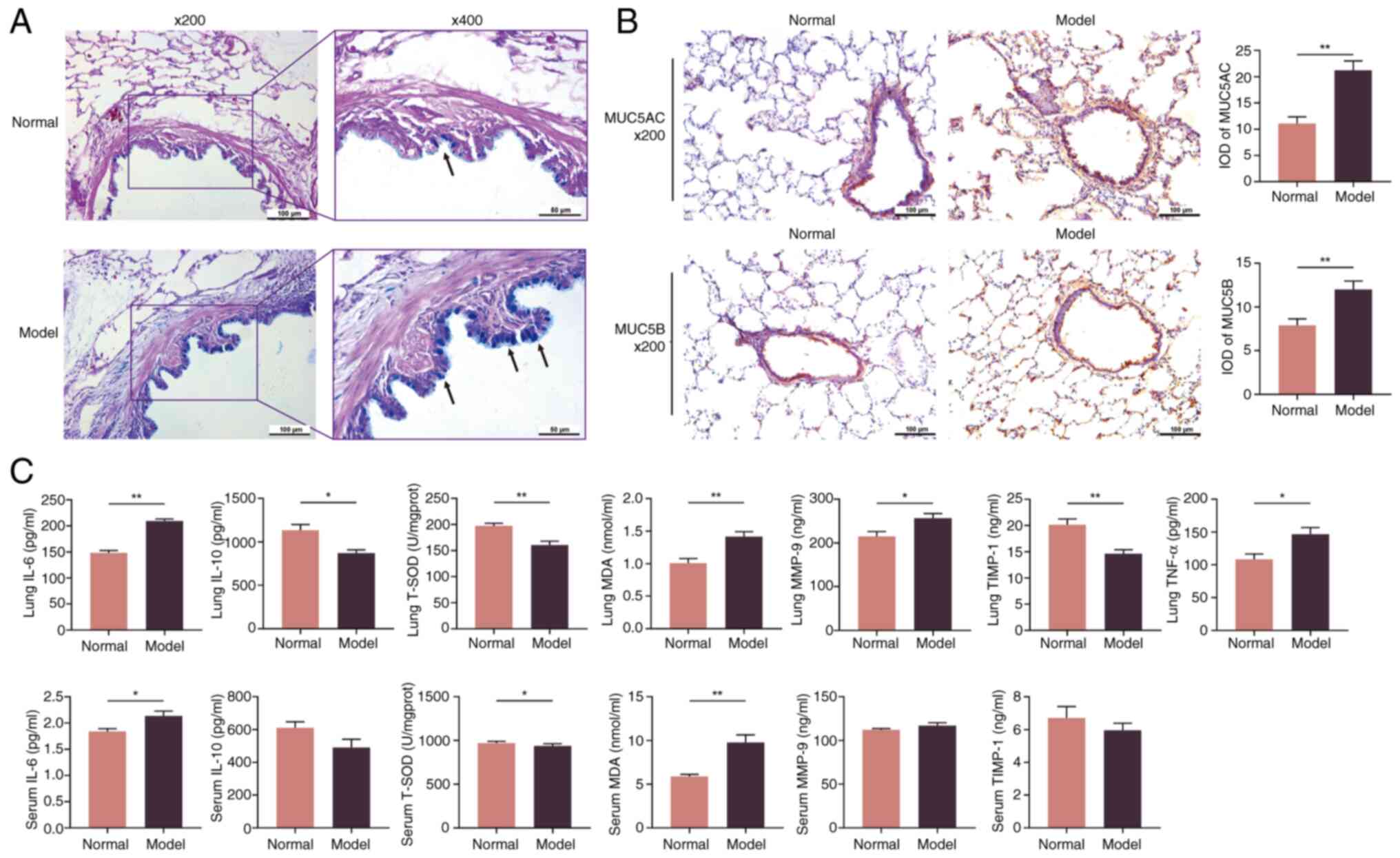 | Figure 4.Injury of mucus hypersecretion and
oxidative damage in all groups. (A) AB-PAS staining images of the
airway in all groups (AB-PAS; ×200 magnification and ×400
magnification). Arrows indicate goblet cells. (B) The expression
level of MUC5AC and MUC5B in the airway tested using
immunohistochemistry (×200 magnification). (C) The expression of
IL-6, IL-10, TNF-α, T-SOD, MDA, MMP-9 and TIMP-1 in lung tissue and
serum of rats in all groups. The values are expressed as mean ± SE;
(n=8). *P<0.05 and **P<0.01 vs. normal group. AB-PAS, Alcian
blue-periodic acid-schiff; MUC5AC, mucoprotein-5AC; MUC5B,
mucoprotein-5B; T-SOD, total superoxide dismutase; MDA,
malondialdehyde; MMP-9, matrix metalloproteinase 9; TIMP-1, tissue
inhibitor of metalloproteinases 2; IOD, integrated optical
density. |
Furthermore, oxidative stress was enhanced by
cigarette smoke and bacterial infection in COPD rats. The enzymatic
activity of T-SOD significantly decreased in both the lungs and
serum, while the expression of MDA significantly increased in COPD
rats compared with normal rats (P<0.05 and P<0.01) (Fig. 4C).
Cigarette smoke combined with
Klebsiella pneumoniae infection induces impairment of the air-blood
barrier via the MAPK/NF-κB/IκBα pathway
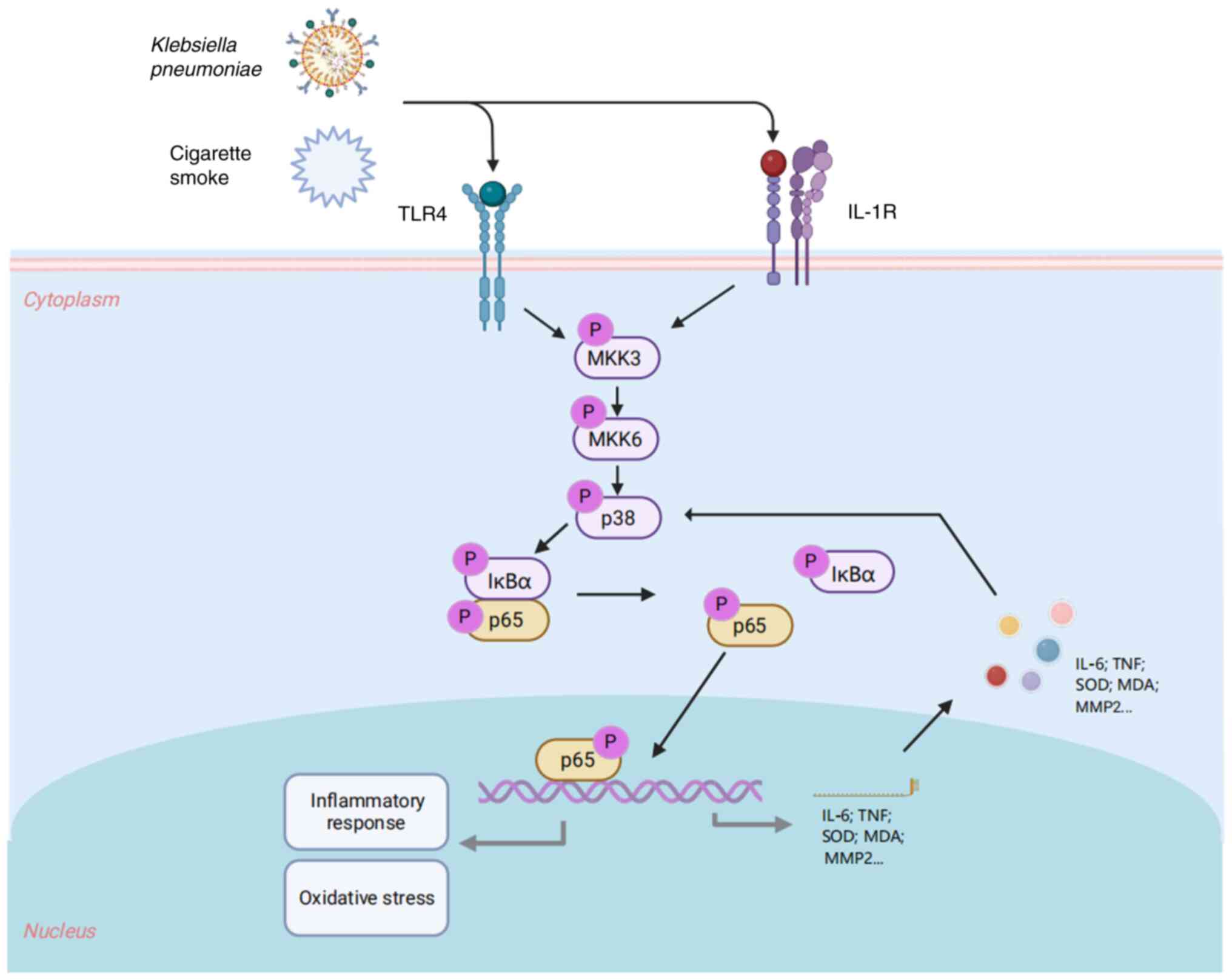
When compared with the normal group of rats, the
combination of cigarette smoke and Klebsiella pneumoniae
infection resulted in an elevation in the levels of p-p38, p-ERK,
p-JNK, p-p65 and p-IκBα proteins (all P<0.01), while having no
notable effect on the levels of total p38, ERK, JNK, p65 and IκBα
proteins (Fig. 5). This outcome
suggested that the combination of cigarette smoke and Klebsiella
pneumoniae infection may have led to damage to the air-blood
barrier by activating the MAPK/NF-κB/IκBα signaling pathway,
thereby promoting inflammatory responses and oxidative stress.
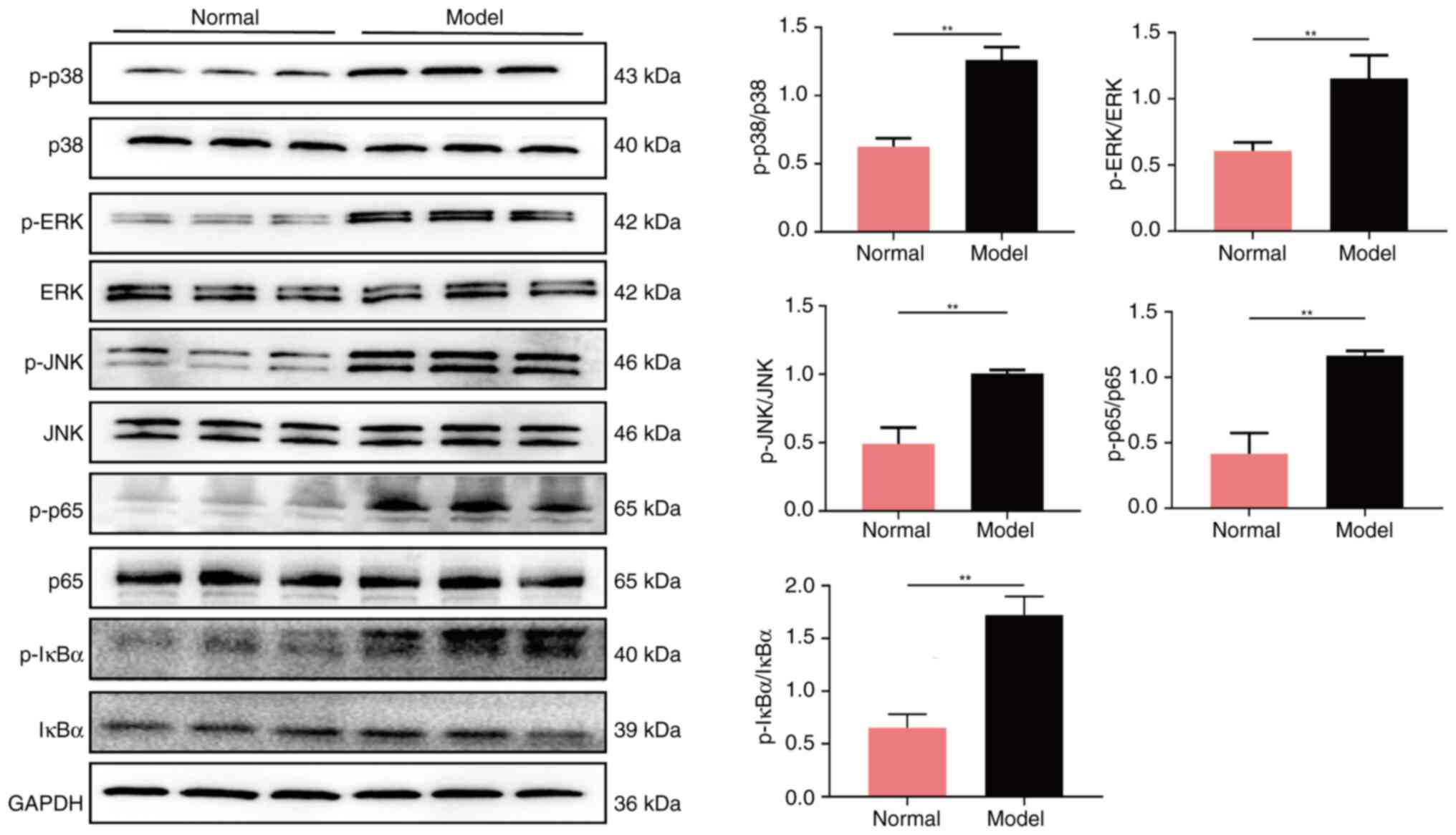 | Figure 5.Protein expression of p-p38, p38,
p-ERK, ERK, p-JNK, JNK, p-p65, p65, p-IκBα and IκBα in the lung
tissue tested using western blot analysis. **P<0.01 vs. normal
group. JNK, Janus kinase; p-, phosphorylated. |
Discussion
The air-blood barrier, also known as the
alveolar-capillary barrier, is an important component of the lung,
facilitating the exchange of oxygen and carbon dioxide between the
human body and the environment (25,26).
In the present study, a COPD rat model was established through
cigarette smoke inhalation and repetitive bacterial infection.
The pulmonary air-blood barrier plays a notable role
in gas and substance exchange, and its destruction leads to
irreversible and notable damage to respiratory function (27). The findings of the present study
revealed that cigarette smoke and bacterial infection resulted in
declined pulmonary function in model rats via deterioration in the
diffusion function of the alveolar-capillary barrier. The
heightened permeability of the pulmonary air-blood barrier may
contribute to alveolar edema, initiating a detrimental cycle of
tissue injury and disrupted balance of substance transport and gas
exchange (27).
Furthermore, the present study observed pathological
damage in the lungs of COPD rats, including alveolar rupture and
the formation of air cavities, which were indicative of compromised
structural integrity of the alveolar-capillary barrier. The
alveolar epithelial barrier, primarily composed of AT I and AT II
arranged in a continuous monolayer and interconnected by tight
junctions (28), plays an
important role in gas exchange. Conversely, the pulmonary
endothelial barrier, comprising endothelial cells, regulates the
entry of fluid and inflammatory components into the interstitium
(29). These barriers constitute
the pulmonary air-blood barrier. The hyperinflammatory environment
of the alveoli, including inflammatory cells, pro-inflammatory
cytokines, chemokines and other inflammatory mediators, can damage
the pulmonary epithelium and endothelium, leading to cell apoptosis
and disruption of intercellular junctions (30).
Additionally, the present study observed incomplete
structures of AT I and AT II in COPD rats. Furthermore, the
respiratory membrane in COPD rats showed marked thickening compared
with normal rats. Therefore, these structural damages to AT I, AT
II and the respiratory membrane contributed to the inefficiency of
gas exchange resulting from cigarette smoke inhalation and
repetitive bacterial infection. Additionally, the present study
observed mitochondrial swelling in the ultrastructure of alveolar
epithelial cells from COPD rats, which suggested that mitochondrial
dysfunction may have played an important role in the impairment of
the pulmonary air-blood barrier. However, the present study did not
further detect or evaluate mitochondrial function, nor analyze its
relationship with the impairment of the pulmonary air-blood
barrier. This is one of the limitations of the present study. In
our subsequent study, we will further detect reactive oxygen
species, mitochondrial respiratory chain synthases and apoptosis in
alveolar epithelial cells, aiming to explore how mitochondrial
damage and apoptosis in alveolar epithelial cells affect the
impairment of the pulmonary air-blood barrier and gas exchange in
COPD rats.
The functionality of the air-blood barrier relies on
the normal structure and properties of alveolar epithelial cells
and pulmonary capillary endothelial cells (PCECs) (31,32).
A previous study confirmed that injury to AT II cells induces
thickening of the air-blood barrier and damages intercellular
junctions, even in the presence of normal total intracellular
surfactant pools (33). Similarly,
endothelial function plays a notable role in maintaining normal
lung function by regulating osmotic pressure and barrier function
(34,35). In the present study, damage to
alveolar epithelial cells was also observed.
AQPs, a type of transmembrane channel protein
facilitating water transfer (36),
includes the protein AQP-5, a marker for AT I. AQP-5 contributes to
ion transport and the maintenance of lung fluid balance in AT I,
thereby preventing barrier disruption and edema (37). SP-D, synthesized and secreted by AT
II, is involved in the innate immune defense of the lung (38). SP-D aids in clearing infectious
pathogens and modulating the immune response by regulating multiple
inflammatory signaling pathways, such as the toll-like receptor
(TLR) 4 signaling pathway (39).
The present study aligns with these findings, as a decline in the
expression of AQP-5 and SP-D was observed, signifying the
hypofunctionality of AT I and AT II.
CD31, a member of the immunoglobulin superfamily of
cell adhesion molecules, serves as a molecular marker for PCECs. It
primarily inhibits normal cell apoptosis and participates in
platelet adhesion and aggregation in PCECs (40). Ang-2 is a ligand protein in the Ang
signaling pathway, belonging to a glycoprotein family that signals
through the transmembrane tyrosine-kinase-2 receptor (41). Ang-2 manifests notable antagonistic
effects to Ang-1 in pathological states, leading to vascular
homeostasis imbalance, seepage and the accelerated, abnormal
proliferation of PCECs (42). The
findings of the present study revealed reduced expression of CD31
and an increased expression of Ang-2 induced by cigarette smoke
inhalation and repetitive bacterial infection, indicating a
decrease in the functionality of PCECs.
Cellular connections are important indicators for
assessing the function and structure of the air-blood barrier.
ZO-1, a tight junction protein predominantly distributed between
epithelia and endothelia, regulates the paracellular permeability
of these cells (43).
ZO-1-deficient cells are capable of assembling functional barriers
under low-tension conditions, yet their tight junctions remain
impaired, characterized by a marked reduction and discontinuous
distribution in the recruitment of junctional components (44). Claudin-5, a member of the claudin
family, is the main structural determinant of the paracellular
endothelial barrier (45).
Claudin-5 promotes the sealing of tight junctions, reducing vessel
permeability and enhancing endothelial barrier function (46). In the present study, the
expressions of ZO-1 and claudin-5 were significantly decreased in
COPD rats compared with normal rats. Therefore, the present study
considered that not only the function but also the structure of the
lung air-blood barrier was compromised in COPD rats due to
cigarette smoke inhalation and repetitive bacterial infection.
Inflammation plays an important role in promoting
the pathological process of air-blood barrier injury, involving the
recruitment of numerous inflammatory cells and secretion of
inflammatory mediators, ultimately resulting in lung air-blood
barrier damage and notable inflammation in the lung (47,48).
Previous studies have demonstrated that regulating TLR4/NF-κB
signaling pathway activation can suppress the production of
pro-inflammatory cytokines, thus inhibiting the inflammatory
response and protecting the air-blood barrier (49,50).
Consistent with these findings, the results of the present study
revealed a strong inflammatory response, characterized by elevated
IL-6 and diminished IL-10 secretion, exacerbated by cigarette smoke
and bacterial infection, contributing to air-blood barrier injury.
Furthermore, the present study discovered mucus hypersecretion in
COPD rats, exemplified by MUC5AC and MUC5B, attributable to
cigarette smoke and bacterial infection. This may have exacerbated
the inflammatory response, further damaging the air-blood
barrier.
Another study verified that the inhibition of MMP
activity due to neutrophil depletion was associated with a
decreased insult to junction proteins and the alveolar-capillary
barrier (51). Furthermore, the
present study evaluated the expression of MMP-9 and TIMP-1,
demonstrating the high expression of MMP-9 and low expression of
TIMP-1 in lung tissue, although these changes in the serum were not
statistically significant. The abnormal expression of MMP-9 and
TIMP-1 in model rats suggested an imbalance of protease and
antiprotease, potentially induced by the inflammatory response in
the lung. Additionally, an imbalance of MMP-9 and TIMP-1 leads to
the rupture of alveoli and the occurrence of emphysema, resulting
in a decline in the diffusion function of the air-blood barrier
(52).
Oxidative stress is another important cause of lung
air-blood barrier damage. A study by Jia et al (25) reported particulate matter <2.5
µm (PM2.5)-induced air-blood barrier dysfunction. The study
observed that reducing oxidative stress via treatment with
N-acetyl-L-cysteine in a mouse model exposed to PM2.5 improved
air-blood barrier function and alleviated the effects of PM2.5. The
present study measured the enzymatic activity of T-SOD and the
protein level of MDA. The enzymatic activity of T-SOD significantly
decreased, and the protein level of MDA significantly increased in
rats after model establishment with cigarette smoke and bacterial
infection. These results indicate that oxidative stress, resulting
from cigarette smoke inhalation and repetitive bacterial infection,
was a notable cause of lung air-blood barrier damage. Therefore,
lung air-blood barrier damage caused by cigarette smoke and
bacterial infection predominantly resulted from an inflammatory
response, protease/antiprotease imbalance and oxidative stress.
In COPD, stimuli such as cigarette smoke and
lipopolysaccharides can activate pathways such as the EGFR/MAPK and
NF-κB pathways in airway epithelial cells, which induce goblet cell
hyperplasia and ciliary epithelial cell dysfunction. These changes
lead to excessive synthesis and impaired clearance of mucins,
directly causing airway obstruction and impairing the gas exchange
function of the pulmonary air-blood barrier (53). Additionally, the viscous and
excessive mucus facilitates bacterial colonization and accumulation
of inflammatory factors, which exacerbate chronic inflammation and
directly impair the structural integrity of the pulmonary air-blood
barrier (54). In the present
study, hypersecretion of airway mucus was also observed, which led
to the impairment of the pulmonary air-blood barrier.
In the present study, a combined approach of 8 weeks
of continuous cigarette smoke inhalation and repeated Klebsiella
pneumoniae infection was adopted. This approach simulated the
pathological environment of long-term smoke exposure and repeated
bacterial infection in patients with COPD, successfully
establishing a stable COPD rat model and providing a more
clinically relevant experimental vehicle for subsequent research.
Furthermore, the present study focused on the pulmonary air-blood
barrier. The present study conducted comprehensive analysis
covering the ultrastructural features and structural and functional
proteins of the barrier, fully elucidating the entire process from
structural damage to functional decline of the pulmonary air-blood
barrier. Furthermore, the present study provided evidence that the
combined effect of cigarette smoke and Klebsiella pneumoniae
infection activated the MAPK/NF-κB/IκBα signaling pathway. This
activation further regulated inflammatory responses, oxidative
stress and protease-antiprotease imbalance, ultimately inducing
damage to the pulmonary air-blood barrier.
Although the present study provided notable insights
into understanding the roles of cigarette smoke and bacterial
infection in the progression of COPD, the conclusions of the
present study still require further evidence to support these
conclusions. Primarily, the present study did not employ cigarette
smoke of different concentrations, different types of bacteria or
varying exposure durations to verify their effects. Furthermore,
the damage to the pulmonary air-blood barrier may have involved
multiple mechanisms; the application of multi-omics techniques can
aid in comprehensively dissecting these underlying mechanisms,
which will also serve as one of the starting points for our future
research.
Summarily, in the present study, a COPD rat model
was established through cigarette smoke inhalation and repetitive
bacterial infection. The pulmonary function and histopathology of
pulmonary tissues deteriorated in COPD model rats. The
ultrastructure of the air-blood barrier was compromised and the
structure and function of ATs and PCECs were impaired. Furthermore,
the secretion of inflammatory factors and mucus increased in model
rats due to cigarette smoke and bacterial infection. Similarly, a
protease/antiprotease imbalance and oxidative stress were observed
in COPD rats, resulting in damage to the lung air-blood barrier. In
summary, the function and structure of the air-blood barrier were
impaired in COPD model rats due to cigarette smoke inhalation and
repetitive bacterial infection, with underlying mechanisms likely
related to inflammation and oxidative stress regulated by the
MAPK/NF-κB/IκBα signaling pathway (Fig. 6).
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Key Research and
Development Program of China (grant no. 2023YFC3502600) and the
National Natural Sciences Foundation of China (grant nos. 82074406
and 81973822).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
YT, KX, and RL wrote the manuscript and conducted
the experiments. KL, XS and YL analyzed the results. YZ and ZQ
performed immunofluorescence and visualized all results. HD and XL
designed experiment and revised the manuscript. YT and XL confirmed
the authenticity of all the raw data. All authors read and approved
the final version of the manuscript.
Ethics approval and consent to
participate
All experimental protocols were approved by the
Experimental Animal Care and Ethics Committees of the First
Affiliated Hospital at Henan University of Traditional Chinese
Medicine, Zhengzhou, China (approval no. YFYDW2019031).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Global Initiative for Chronic Obstructive
Lung Disease, . Global strategy for the diagnosis, management,
prevention of Chronic Obstructive Pulmonary Disease. 2023.Available
from. http://www.goldcopd.org./
|
|
2
|
Rabe KF and Watz H: Chronic obstructive
pulmonary disease. Lancet. 389:1931–1940. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Neder JA, Kirby M, Santyr G, Pourafkari M,
Smyth R, Phillips DB, Crinion S, de-Torres JP and O'Donnell DE:
V̇/Q̇ Mismatch: A novel target for COPD treatment. Chest.
162:1030–1047. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Leiby KL, Raredon MSB and Niklason LE:
Bioengineering the Blood-gas Barrier. Compr Physiol. 10:415–452.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Weibel ER and Knight BW: A morphometric
study on the thickness of the pulmonary air-blood barrier. J Cell
Biol. 21:367–396. 1964. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Falcon-Rodriguez CI, Osornio-Vargas AR,
Sada-Ovalle I and Segura-Medina P: Aeroparticles, composition, and
lung diseases. Front Immunol. 7:32016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gao L, Zeng N, Yuan Z, Wang T, Chen L,
Yang D, Xu D, Wan C, Wen F and Shen Y: Knockout of Formyl peptide
Receptor-1 attenuates cigarette smoke-induced airway inflammation
in mice. Front Pharmacol. 12:6322252021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lin H, Wang C, Yu H, Liu Y, Tan L, He S,
Li Z, Wang C, Wang F, Li P and Liu J: Protective effect of total
Saponins from American ginseng against cigarette smoke-induced COPD
in mice based on integrated metabolomics and network pharmacology.
Biomed Pharmacother. 149:1128232022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vij N, Chandramani-Shivalingappa P, Van
Westphal C, Hole R and Bodas M: Cigarette smoke-induced autophagy
impairment accelerates lung aging, COPD-emphysema exacerbations and
pathogenesis. Am J Physiol Cell Physiol. 314:C73–C87. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zeng H, Kong X, Zhang H, Chen Y, Cai S,
Luo H and Chen P: Inhibiting DNA methylation alleviates cigarette
smoke extract-induced dysregulation of Bcl-2 and endothelial
apoptosis. Tob Induc Dis. 18:512020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Van Eeckhoutte HP, Donovan C, Kim RY,
Conlon TM, Ansari M, Khan H, Jayaraman R, Hansbro NG, Dondelinger
Y, Delanghe T, et al: RIPK1 kinase-dependent inflammation and cell
death contribute to the pathogenesis of COPD. Eur Respir J.
61:22015062023. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
D'Anna SE, Maniscalco M, Cappello F,
Carone M, Motta A, Balbi B, Ricciardolo FLM, Caramori G and Stefano
AD: Bacterial and viral infections and related inflammatory
responses in chronic obstructive pulmonary disease. Ann Med.
53:135–150. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Suresh A, Rao TC, Solanki S, Suresh MV,
Menon B and Raghavendran K: The holy basil administration
diminishes the NF-kB expression and protects alveolar epithelial
cells from pneumonia infection through interferon gamma. Phytother
Res. 36:1822–1835. 2022. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang B, Li H, Zhang J, Hang Y and Xu Y:
Activating transcription factor 3 protects alveolar epithelial type
II cells from Mycobacterium tuberculosis infection-induced
inflammation. Tuberculosis (Edinb). 135:1022272022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hardy KS, Tuckey AN, Renema P, Patel M,
Al-Mehdi AB, Spadafora D, Schlumpf CA, Barrington RA, Alexeyev MF,
Stevens T, et al: ExoU induces lung endothelial cell damage and
activates pro-inflammatory Caspase-1 during pseudomonas aeruginosa
infection. Toxins (Basel). 14:1522022. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lv R, Zhao Y, Wang X, He Y, Dong N, Min X,
Liu X, Yu Q, Yuan K, Yue H, et al: GLP-1 analogue liraglutide
attenuates CIH-induced cognitive deficits by inhibiting oxidative
stress, neuroinflammation, and apoptosis via the Nrf2/HO-1 and
MAPK/NF-κB signaling pathways. Int Immunopharmacol. 142:1132222024.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cai SY, Liu A, Xie WX, Zhang XQ, Su B, Mao
Y, Weng DG and Chen ZY: Esketamine mitigates mechanical
ventilation-induced lung injury in chronic obstructive pulmonary
disease rats via inhibition of the MAPK/NF-κB signaling pathway and
reduction of oxidative stress. Int Immunopharmacol. 139:1127252024.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang XF, Xiang SY, Lu J, Li Y, Zhao SJ,
Jiang CW, Liu XG, Liu ZB and Zhang J: Electroacupuncture inhibits
IL-17/IL-17R and post-receptor MAPK signaling pathways in a rat
model of chronic obstructive pulmonary disease. Acupunct Med.
39:663–672. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Marumo S, Hoshino Y, Kiyokawa H, Tanabe N,
Sato A, Ogawa E, Muro S, Hirai T and Mishima M: p38
mitogen-activated protein kinase determines the susceptibility to
cigarette smoke-induced emphysema in mice. BMC Pulm Med. 14:792014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jiang JJ, Chen SM, Li HY, Xie QM and Yang
YM: TLR3 inhibitor and tyrosine kinase inhibitor attenuate
cigarette smoke/poly I:C-induced airway inflammation and remodeling
by the EGFR/TLR3/MAPK signaling pathway. Eur J Pharmacol.
890:1736542021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li Y, Li SY, Li JS, Deng L, Tian YG, Jiang
SL, Wang Y and Wang YY: A rat model for stable chronic obstructive
pulmonary disease induced by cigarette smoke inhalation and
repetitive bacterial infection. Biol Pharm Bull. 35:1752–1760.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu L, Qin Y, Cai Z, Tian Y, Liu X, Li J
and Zhao P: Effective-components combination improves airway
remodeling in COPD rats by suppressing M2 macrophage polarization
via the inhibition of mTORC2 activity. Phytomedicine.
92:1537592021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jin F, Zhang L, Chen K, Miao Y, Liu Y,
Tian Y and Li J: Effective-component compatibility of Bufei Yishen
formula III combined with Electroacupuncture suppresses
inflammatory response in rats with chronic obstructive pulmonary
disease via regulating SIRT1/NF-κB signaling. Biomed Res Int.
2022:33607712022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li MY, Qin YQ, Tian YG, Li KC, Oliver BG,
Liu XF, Zhao P and Li JS: Effective-component compatibility of
Bufei Yishen formula III ameliorated COPD by improving airway
epithelial cell senescence by promoting mitophagy via the
NRF2/PINK1 pathway. BMC Pulm Med. 22:4342022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jia R, Wei M, Zhang X, Du R, Sun W, Wang L
and Song L: Pyroptosis participates in PM2.5-induced
air-blood barrier dysfunction. Environ Sci Pollut Res Int.
29:60987–60997. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yao XH, Luo T, Shi Y, He ZC, Tang R, Zhang
PP, Cai J, Zhou XD, Jiang DP, Fei XC, et al: A cohort autopsy study
defines COVID-19 systemic pathogenesis. Cell Res. 31:836–846. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Qiao Q, Liu X, Yang T, Cui K, Kong L, Yang
C and Zhang Z: Nanomedicine for acute respiratory distress
syndrome: The latest application, targeting strategy, and rational
design. Acta Pharm Sin B. 11:3060–3091. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Matthay MA, Zemans RL, Zimmerman GA, Arabi
YM, Beitler JR, Mercat A, Herridge M, Randolph AG and Calfee CS:
Acute respiratory distress syndrome. Nat Rev Dis Primers. 5:182019.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bhattacharya J and Matthay MA: Regulation
and repair of the alveolar-capillary barrier in acute lung injury.
Annu Rev Physiol. 75:593–615. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Thompson BT, Chambers RC and Liu KD: Acute
respiratory distress syndrome. N Engl J Med. 377:562–572. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang T, Liu C, Pan LH, Liu Z, Li CL, Lin
JY, He Y, Xiao JY, Wu S, Qin Y, et al: Inhibition of p38 MAPK
mitigates lung ischemia reperfusion injury by reducing Blood-air
barrier Hyperpermeability. Front Pharmacol. 11:5692512020.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Viola H, Washington K, Selva C, Grunwell
J, Tirouvanziam R and Takayama S: A High-throughput distal lung
air-blood barrier model enabled by density-driven underside
epithelium seeding. Adv Healthc Mater. 10:e21008792021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mühlfeld C, Neves J, Brandenberger C,
Hegermann J, Wrede C, Altamura S and Muckenthaler MU: Air-blood
barrier thickening and alterations of alveolar epithelial type 2
cells in mouse lungs with disrupted hepcidin/ferroportin regulatory
system. Histochem Cell Biol. 151:217–228. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li W, Long L, Yang X, Tong Z, Southwood M,
King R, Caruso P, Upton PD, Yang P, Bocobo GA, et al: Circulating
BMP9 protects the pulmonary endothelium during inflammation-induced
lung injury in mice. Am J Respir Crit Care Med. 203:1419–1430.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang Y, Xue L, Wu Y, Zhang J, Dai Y, Li F,
Kou J and Zhang Y: Ruscogenin attenuates sepsis-induced acute lung
injury and pulmonary endothelial barrier dysfunction via
TLR4/Src/p120-catenin/VE-cadherin signalling pathway. J Pharm
Pharmacol. 73:893–900. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Guibourdenche J, Bonnet-Serrano F, Younes
Chaouch L, Sapin V, Tsatsaris V, Combarel D, Laguillier C and
Grange G: Amniotic Aaquaporins (AQP) in normal and pathological
pregnancies: Interest in Polyhydramnios. Reprod Sci. 28:2929–2938.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Weber J, Rajan S, Schremmer C, Chao YK,
Krasteva-Christ G, Kannler M, Yildirim AÖ, Brosien M, Schredelseker
J, Weissmann N, et al: TRPV4 channels are essential for alveolar
epithelial barrier function as protection from lung edema. JCI
Insight. 5:e1344642020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Arroyo R and Kingma PS: Surfactant protein
D and bronchopulmonary dysplasia: A new way to approach an old
problem. Respir Res. 22:1412021. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sorensen GL: Surfactant protein D in
respiratory and non-respiratory diseases. Front Med (Lausanne).
5:182018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cheung KCP, Fanti S, Mauro C, Wang G, Nair
AS, Fu H, Angeletti S, Spoto S, Fogolari M, Romano F, et al:
Preservation of microvascular barrier function requires CD31
receptor-induced metabolic reprogramming. Nat Commun. 11:35952020.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Akwii RG, Sajib MS, Zahra FT and Mikelis
CM: Role of angiopoietin-2 in vascular physiology and
pathophysiology. Cells. 8:4712019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Nicolini G, Forini F, Kusmic C, Iervasi G
and Balzan S: Angiopoietin 2 signal complexity in cardiovascular
disease and cancer. Life Sci. 239:1170802019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Dai W, Nadadur RD, Brennan JA, Smith HL,
Shen KM, Gadek M, Laforest B, Wang M, Gemel J, Li Y, et al: ZO-1
regulates intercalated disc composition and atrioventricular node
conduction. Circ Res. 127:e28–e43. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Haas AJ, Zihni C, Krug SM, Maraspini R,
Otani T, Furuse M, Honigmann A, Balda MS and Matter K: ZO-1 guides
tight junction assembly and epithelial morphogenesis via
cytoskeletal tension-dependent and -independent functions. Cells.
11:37752022. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kakogiannos N, Ferrari L, Giampietro C,
Scalise AA, Maderna C, Ravà M, Taddei A, Lampugnani MG, Pisati F,
Malinverno M, et al: JAM-A Acts via C/EBP-α to promote claudin-5
expression and enhance endothelial barrier function. Circ Res.
127:1056–1073. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hashimoto Y, Campbell M, Tachibana K,
Okada Y and Kondoh M: Claudin-5: A pharmacological target to modify
the permeability of the blood-brain barrier. Biol Pharm Bull.
44:1380–1390. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Komiya K, Akaba T, Kozaki Y, Kadota JI and
Rubin BK: A systematic review of diagnostic methods to
differentiate acute lung injury/acute respiratory distress syndrome
from cardiogenic pulmonary edema. Crit Care. 21:2282017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Du L, Zhang J, Zhang X, Li C, Wang Q, Meng
G, Kan X, Zhang J and Jia Y: Oxypeucedanin relieves LPS-induced
acute lung injury by inhibiting the inflammation and maintaining
the integrity of the lung air-blood barrier. Aging (Albany NY).
14:6626–6641. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Peng LY, Yuan M, Shi HT, Li JH, Song K,
Huang JN, Yi PF, Fu BD and Shen HQ: Protective effect of
piceatannol against acute lung injury through protecting the
integrity of air-blood barrier and modulating the TLR4/NF-κB
signaling pathway activation. Front Pharmacol. 10:16132020.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Peng LY, Shi HT, Yuan M, Li JH, Song K,
Huang JN, Yi PF, Shen HQ and Fu BD: Madecassoside protects against
LPS-induced acute lung injury via inhibiting TLR4/NF-κB activation
and blood-air barrier permeability. Front Pharmacol. 11:8072020.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Sapoznikov A, Gal Y, Falach R, Sagi I,
Ehrlich S, Lerer E, Makovitzki A, Aloshin A, Kronman C and Sabo T:
Early disruption of the alveolar-capillary barrier in a
ricin-induced ARDS mouse model: Neutrophil-dependent and
-independent impairment of junction proteins. Am J Physiol Lung
Cell Mol Physiol. 316:L255–L268. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Uysal P and Uzun H: Relationship between
circulating Serpina3g, Matrix Metalloproteinase-9, and tissue
inhibitor of Metalloproteinase-1 and −2 with chronic obstructive
pulmonary disease severity. Biomolecules. 9:622019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Xu K, Ma J, Lu R, Shao X, Zhao Y, Cui L,
Qiu Z, Tian Y and Li J: Effective-compound combination of Bufei
Yishen formula III combined with ER suppress airway mucus
hypersecretion in COPD rats: Via EGFR/MAPK signaling. Biosci Rep.
43:BSR202226692023. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Rojas DA, Ponce CA, Bustos A, Cortés V,
Olivares D and Vargas SL: Pneumocystis exacerbates inflammation and
mucus hypersecretion in a murine, Elastase-induced-COPD model. J
Fungi (Basel). 9:4522023. View Article : Google Scholar : PubMed/NCBI
|