Introduction
In 2023, the World Heart Federation released the
World Heart Report, which highlighted that cardiovascular diseases
(CVDs) will remain the primary cause of death globally for decades
(1). In 2023, Liu et al
(2) identified a novel form of
cell death termed disulfidptosis, which is triggered in cells with
high expression of the cystine transporter solute carrier family 7
member 11 (SLC7A11) under glucose deprivation. Elevated SLC7A11
levels increase cystine uptake; however, under glucose-deficient or
oxidative stress conditions, the depletion of nicotinamide adenine
dinucleotide phosphate (NADPH) impairs the reduction of cystine to
cysteine. This leads to disulfide stress (3), causing cytoskeletal dysfunction and,
ultimately, cell death.
Since disulfidptosis was first discovered in cancer
cells, existing studies have primarily focused on its role in
oncology (4,5). Nevertheless, emerging research has
suggested a potential link between disulfidptosis and CVDs
(6–9). Disulfide stress, a key step in
disulfidptosis, promotes antioxidant system collapse and reactive
oxygen species (ROS) accumulation, exacerbating oxidative stress
and potentially triggering cellular inflammation, autophagy and
other pathological processes through cascade signaling pathways,
worsening CVD progression (10).
Given these findings, the present review aimed to explore the
molecular mechanisms of disulfidptosis, emphasizing its
pathophysiological impact on CVDs, and discussing novel
perspectives for prevention and therapeutic intervention.
Search strategy
The present study is a narrative review, aiming to
provide a comprehensive and critical discussion on the current
research status, molecular mechanisms and potential implications of
the novel cell death modality disulfidptosis in the field of CVDs.
The PubMed database (https://pubmed.ncbi.nlm.nih.gov/) was searched using
the following search strategy: [‘disulfidptosis’ (Title/Abstract)
OR ‘disulfide stress’ (Title/Abstract) OR ‘SLC7A11’
(Title/Abstract) OR ‘xCT’ (Title/Abstract) OR ‘system Xc-’
(Title/Abstract)] AND [‘cardiovascular’ (Title/Abstract) OR ‘heart’
(Title/Abstract) OR ‘myocardial’ (Title/Abstract) OR
‘cardiomyocyte’ (Title/Abstract) OR ‘vascular smooth muscle cell’
(Title/Abstract) OR ‘endothelial cell’ (Title/Abstract) OR
‘fibroblast’ (Title/Abstract) OR ‘atherosclerosis’ (Title/Abstract)
OR ‘ischemia’ (Title/Abstract) OR ‘heart failure’
(Title/Abstract)]. The present review primarily focused on original
research articles, reviews, commentaries, and letters related to
the molecular mechanisms of disulfidptosis and its potential role
in CVDs, including myocardial infarction (MI), heart failure,
aortic dissection and hypertrophic cardiomyopathy. The search was
limited to articles published in English.
Disulfidptosis
SLC7A11: Core of disulfidptosis
SLC7A11regulates the uptake of extracellular cystine
concurrent with the secretion of intracellular glutamate (11), a subunit of the cystine/glutamate
antiporter system Xc-(xCT). Within the cell, cystine is reduced to
cysteine, which is the rate-limiting precursor for glutathione
(GSH) synthesis. GSH, a tripeptide composed of cysteine, glutamate
and glycine, is the primary intracellular antioxidant. GSH
effectively neutralizes ROS and maintains redox homeostasis
(12).
Under glucose-sufficient conditions, NADPH generated
via the pentose phosphate pathway (PPP) and
glycolysis-tricarboxylic acid cycle coupling facilitates the
reduction of cystine to cysteine, promoting GSH synthesis and
antioxidant defense (13).
However, glucose deprivation suppresses PPP activity, impairs NADPH
production and halts cystine reduction. This leads to intracellular
cystine accumulation (14).
SLC7A11 acts as the trigger switch for disulfidptosis as its
upregulation drives excessive cystine uptake. Disulfide stress is
directly induced by glucose deficiency (2), initiating a disulfidptosis cascade
(Fig. 1).
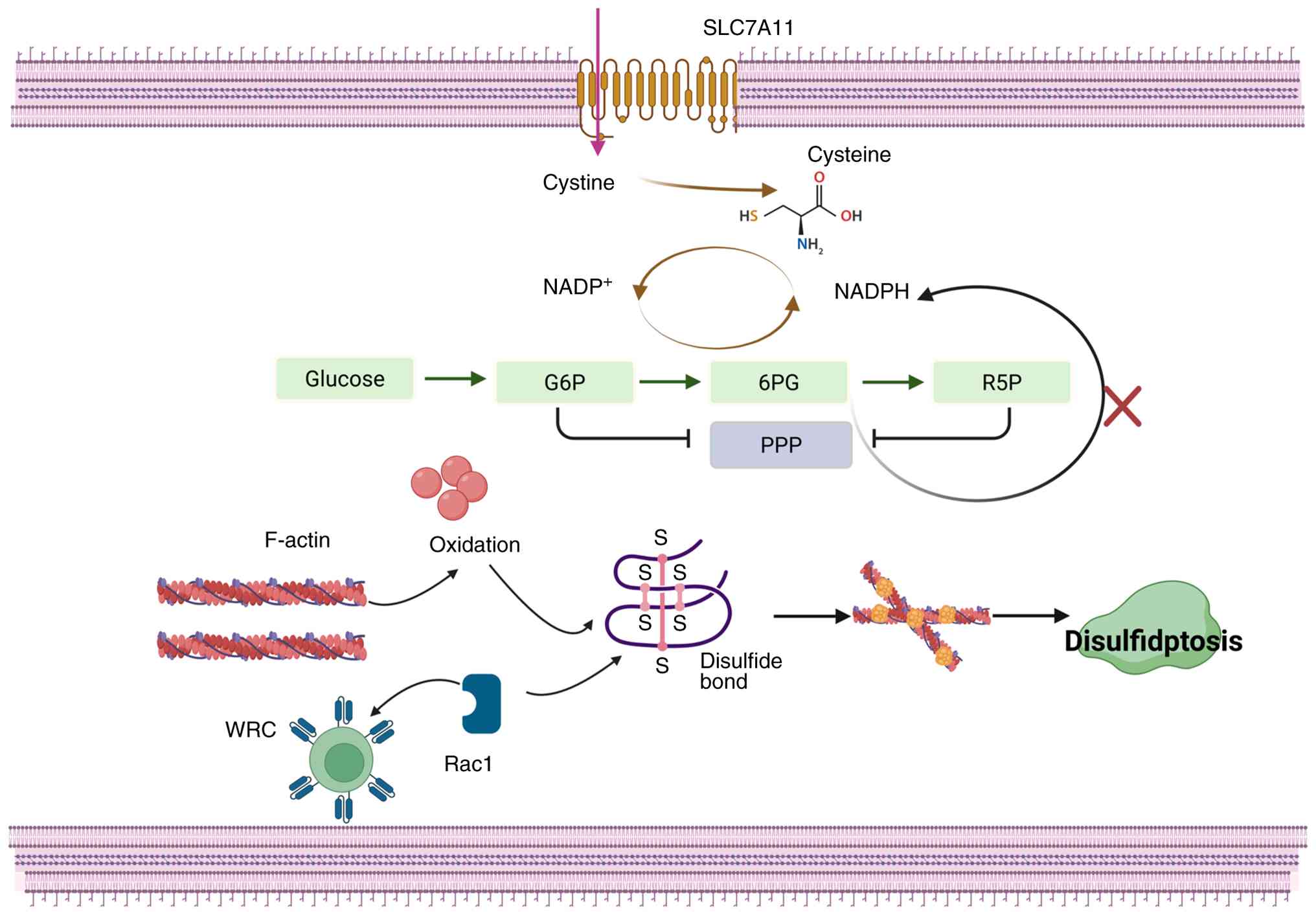 | Figure 1.Disulfidptosis mechanism. The cystine
transporter SLC7A11 imports extracellular cystine. Under
glucose-sufficient conditions, NADPH produced via the PPP and
glycolysis-tricarboxylic acid cycle coupling supports cystine
reduction to cysteine. Glucose deprivation suppresses PPP activity,
depleting NADPH, halting cystine reduction and leading to cystine
accumulation. The resulting disulfide stress causes spontaneous
oxidation of free sulfhydryl groups on F-actin, forming aberrant
intra- and intermolecular disulfide bonds. This crosslinking
disrupts the actin cytoskeleton, ultimately triggering cell death.
The WRC and Rac1 GTPase accelerate disulfidptosis by promoting
lamellipodia formation, which provides additional actin networks
for disulfide crosslinking in SLC7A11-upregulated cells. G6P,
glucose 6-phosphate; 6PG, 6-phosphogluconate; R5P, ribose
5-phosphate; WRC, WAVE regulatory complex; SLC7A11, solute carrier
family 7 member 11; F-actin, filamentous actin; NADPH, nicotinamide
adenine dinucleotide phosphate; PPP, pentose phosphate pathway. |
Actin cytoskeleton: The ultimate
target of disulfide crosslinking
The actin cytoskeleton is a biopolymeric network
composed primarily of microtubules, microfilaments and intermediate
filaments. It provides structural rigidity and is required for
diverse mechanical functions, including cell motility, shape
changes, division, mechanosensing and tension homeostasis (15). Actin maintains cytoskeletal
dynamics by forming filamentous actin (F-actin) (15). Key cysteine residues, such as
Cys374 in actin (16), and Cys988
and Cys1379 in non-muscle myosin heavy chain (MYH)9 (2), as well as other cytoskeletal
proteins, such as vinculin, are rich in free thiol (−SH) groups.
Under conditions of disulfide stress, such as SLC7A11 upregulation
and glucose deprivation, these-SH groups undergo spontaneous
oxidation, leading to aberrant intra- and intermolecular disulfide
bond formation in globular actin (17). This disrupts dynamic polymerization
and causes F-actin rigidification.
Further experiments have revealed that glucose
starvation induces F-actin retraction from the cell cortex and
stress fibers, physically detaching it from the plasma membrane.
This demonstrates that the actin network, a sensitive target, loses
its dynamic equilibrium due to disulfide crosslinking, ultimately
resulting in cytoskeletal collapse and cell death (2).
WAVE regulatory complex (WRC) complex
and Rac1: Amplifiers of disulfidptosis
In disulfidptosis, the WRC and Rac1 GTPase act as
synergistic amplifiers of cytoskeletal collapse, notably
exacerbating the lethal effects of disulfide stress. Rac1, a member
of the Rho GTPase family, serves as a fundamental regulator of the
actin cytoskeleton and serves important roles in cell motility,
polarity and migration (18). Rac1
has also been implicated in CVDs (19). The WRC is a pentameric complex
comprising Nck-associated protein 1 (NCKAP1), WAVE proteins such as
WAVE2, cytoplasmic FMR1-interacting protein (CYFIP)1/2, Abl
interactor 2 and protein BRICK1 (20). Rac1 activates WAVE proteins by
interacting with the WRC component CYFIP1/2, which subsequently
recruits and activates the actin-related protein 2/3 complex via
the WRC's VCA (verprolin-homology, cofilin-homology, acidic) domain
(21). This process drives F-actin
nucleation and lamellipodia formation (22).
Recent studies have revealed that NCKAP1-deficient
cells (UMRC6 cells) maintain normal SLC7A11 levels, cystine uptake
and NADP+:NADPH ratios, and exhibit attenuated glucose
starvation-induced disulfide bond formation, F-actin retraction and
plasma membrane detachment. Conversely, in in vitro
experiments using cancer cell lines with high SLC7A11 expression,
overexpression of a constitutively active Rac1 mutant promotes
lamellipodia formation and exacerbates disulfidptosis. These
findings demonstrate that Rac1/WRC-mediated lamellipodia formation
accelerates disulfidptosis, likely because the branched actin
networks within lamellipodia provide important substrates for
additional disulfide cross-linking between cytoskeletal proteins
(2).
Crosstalk between disulfidptosis and
ferroptosis
In the complex pathology of CVDs, disulfidptosis is
likely to form intricate dialogue networks with other cell death
modalities, collectively determining cellular fate and tissue
outcome. The present review focused primarily on the crosstalk
between disulfidptosis and ferroptosis, as disulfidptosis and
ferroptosis share the most direct and close relationship. Similar
to disulfidptosis, ferroptosis is notably linked to SLC7A11 and GSH
(23).
In 2003, Dolma et al (24) discovered a novel compound, erastin,
which could selectively eliminate tumor cells that expressed SV40
large T antigen (ST) and mutant Ras. This induced a new
non-apoptotic form of cell death. In 2008, Yang and Stockwell
(25) discovered two new
compounds: RSL5 and RSL3. Similar to erastin, these compounds
induced the iron-dependent, non-apoptotic cell death of tumor cells
containing mutant Ras, which could be inhibited by the iron
chelator deferoxamine and vitamin E. In 2012, the Stockwell
laboratory formally named this cell death process ferroptosis
(26).
Morphologically, ferroptosis is characterized by the
loss of plasma membrane integrity, cell membrane rupture,
mitochondrial shrinkage, rupture of the mitochondrial outer
membrane, reduction or loss of cristae, and increased membrane
thickening (27). At the molecular
level, the key cause of ferroptosis is the inactivation of GSH
peroxidase 4 (GPX4) (28). This
results in failure to clear lipid peroxides promptly, which are
subsequently catalyzed by divalent iron ions (Fe2+) via
the Fenton reaction (29),
ultimately damaging the integrity of the cell membrane system.
Initially, the inhibition of xCT on the cell
membrane, whose key component is SLC7A11, prevents cystine uptake.
This leads to intracellular cysteine depletion and an insufficient
amount of raw material for GSH synthesis (30). Consequently, GPX4 becomes
functionally stagnant due to the lack of its required cofactor
(30). Long-chain polyunsaturated
fatty acids (PUFAs), such as arachidonic acid, are recognized and
activated by acyl-CoA synthetase long chain family member 4
(31). Subsequently, under the
action of lysophosphatidylcholine acyltransferase 3 (32), they are esterified and incorporated
into membrane phospholipids. This results in cellular membranes,
especially plasma and mitochondrial membranes, being enriched with
PUFA-containing phospholipids (PUFA-PLs), which are notably
susceptible to oxidation due to their bis-allylic structures
(33). These membrane PUFA-PLs are
catalyzed by various oxidase systems, generating phospholipid
hydroperoxides (PL-PUFA-OOH) (34). Unstable intracellular
Fe2+, via the Fenton reaction, convert PL-PUFA-OOH into
highly reactive lipid radicals and lipid peroxyl radicals.
Ferroptosis occurs when the clearance capacity of GPX4 is
overwhelmed by the generation of lipid peroxides. In this process,
the inhibition of SLC7A11 is one of the primary causes of GPX4
inactivation (35).
Under glucose starvation conditions, high SLC7A11
expression steers cells toward disulfidptosis, whereas if SLC7A11
is inhibited, cells tend to undergo ferroptosis. Thus, SLC7A11 acts
as a common molecular switch for disulfidptosis and ferroptosis. In
this way, cell fate depends on SLC7A11 activity and glucose
availability (36).
Simultaneously, the depletion of GSH during disulfidptosis creates
a reduced cellular environment that is prone to ferroptosis.
Furthermore, the disruption of cell membrane integrity caused by
disulfidptosis might accelerate the diffusion of lipid peroxides
and, thus, the process of ferroptosis (37).
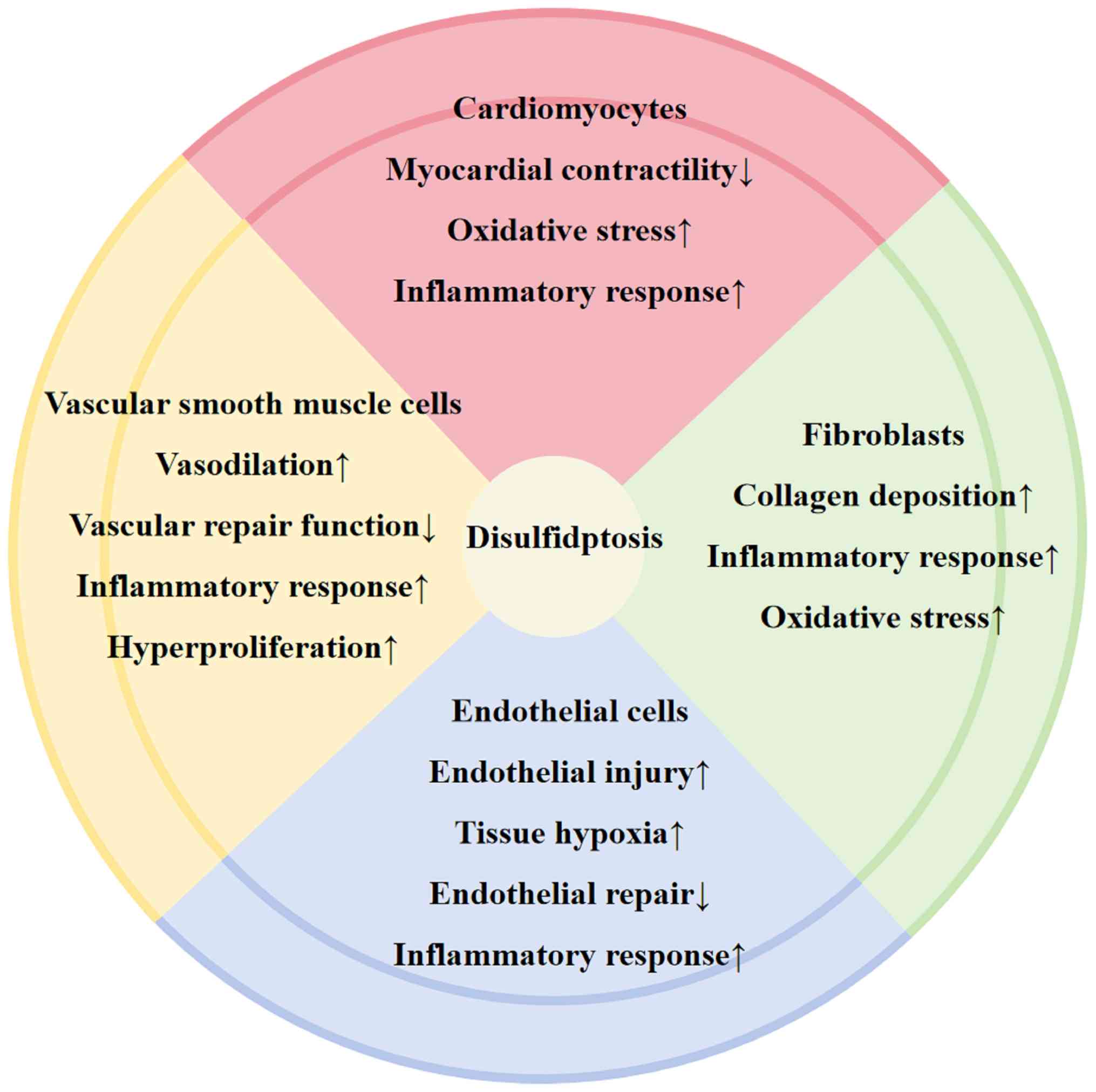
Cell types and disulfidptosis in the
cardiovascular system
The heart comprises intricately coordinated cell
types, including contractile cardiomyocytes, pacemaker cells,
supporting cells, immune cells and neural cells (38). Understanding the characteristics
and interaction mechanisms of disulfidptosis in these cells is
important for investigating CVD pathogenesis and developing
targeted therapies. The following section discusses the effects of
disulfidptosis on cardiomyocytes, vascular smooth muscle cells
(VSMCs), endothelial cells (ECs), fibroblasts and macrophages
(Fig. 2).
Cardiomyocytes
As terminally differentiated cells, cardiomyocytes
rely on stable oxidative metabolism and intact cytoskeletal
structure to maintain contractile function, making them notably
susceptible to disulfidptosis. Cardiac contraction depends on
cyclic contraction-relaxation generated by interactions between
actin-based thin filaments and myosin thick filaments (39). Actin is the cytoskeletal protein
most vulnerable to aberrant disulfide bonds in disulfidptosis,
leading to actin intermolecular cross-linking and disruption of
myofibrils in cardiomyocytes (40). Loss of disulfide homeostasis leads
to cytoskeletal disintegration, causing morphological alterations
in cardiomyocytes, such as shrinkage and fragmentation, that
directly impair contractility and cardiac function (41). Additionally, disulfidptosis
exacerbates oxidative stress and energy metabolism disorders
(42). Cardiomyocytes exhibit high
oxidative metabolism, generate ROS and require high GSH levels
(43). However, GSH depletion in
disulfidptosis induces oxidative stress in cardiomyocytes, damaging
mitochondria and ultimately rendering ATP production insufficient
to sustain contraction (44).
Concurrently, oxidative stress activates pro-inflammatory
signaling, such as the NF-κB pathway, in cardiomyocytes, promoting
the release of cytokines, such as TNF-α and IL-6, which exacerbate
local inflammation (45).
Disulfidptosis-induced cardiomyocyte death is irreversible. Dying
cells release damage-associated molecular patterns (DAMPs) that
recruit immune cells such as macrophages, triggering excessive
inflammation and secondary cardiomyocyte injury (46).
VSMCs
As the predominant cell type in the medial layer of
blood vessels, VSMCs maintain vascular tone and participate in
vascular remodeling, which includes processes such as contraction,
proliferation and migration; these processes depend on stable redox
homeostasis and dynamic cytoskeletal organization (47). The disulfide stress inherent to
disulfidptosis could directly conflict with the redox-dependent
activation mechanism of NADPH oxidase 1 (NOX1) in VSMCs.
Specifically, the aberrant intermolecular disulfide bonding that
characterizes disulfidptosis may competitively disrupt or
dysregulate the precise formation of intermolecular disulfide bonds
between protein disulfide isomerase and p47phox, a known
prerequisite for NOX1 activation. If NOX1 activity is thereby
compromised, the resulting alteration in cellular redox balance
could, in turn, affect the NADPH-mediated reduction of cystine to
cysteine. This potential crosstalk suggests a mechanism whereby
disulfidptosis might influence cellular redox signaling and
cyst(e)ine metabolism through interference with NOX1 function
(48). Disulfidptosis may markedly
disrupt vascular homeostasis by targeting important physiological
processes in VSMCs. VSMC contraction relies on phosphorylation of
myosin light chain (MLC), which is mediated by MLC kinase (MLCK)
(49). During disulfidptosis, GSH
depletion elevates the intracellular oxidative potential, leading
to the oxidation of free-SH groups in MLCK and the formation of
abnormal disulfide bonds (50).
This results in reduced or abolished enzymatic activity, impaired
MLC phosphorylation and consequent vascular dysfunction,
manifesting as excessive vasodilation or regional blood flow
disturbances due to uneven contractility (51).
VSMC migration, proliferation and injury repair
require the dynamic remodeling of cytoskeletal components,
including microtubules and microfilaments (52). During disulfidptosis, the oxidation
of -SH groups in tubulin and actin promotes aberrant protein
polymerization through intermolecular disulfide bonding. For
example, actin filaments may irreversibly cross-link into bundled
structures, causing cytoskeletal rigidity (2). This stiffness inhibits VSMC migratory
capacity and compromises vascular repair mechanisms (53). Dying VSMCs release DAMPs that
recruit inflammatory cells, such as macrophages and monocytes, and
initiate local inflammation (54).
The cytokines secreted by these inflammatory cells, such as
platelet-derived growth factor (PDGF) and TGF-β, act on surviving
vascular smooth muscle cells (VSMCs), promoting their transition to
a synthetic phenotype and enhancing proliferation, thereby
exacerbating vascular wall thickening. PDGF and TGF-β primarily
drive the phenotypic switch of VSMCs from a ‘contractile’ to a
‘synthetic’ state by regulating gene expression, intracellular
signaling and cytoskeletal remodeling (55).
ECs
ECs are important for cardiovascular homeostasis,
serving as a selective barrier between blood and tissues, and
regulating vascular tone, anticoagulation, inflammatory responses
and tissue perfusion (56). As
their functions are highly dependent on an intact cytoskeleton and
a precise redox balance, ECs may represent another notable target
for the pathological effects of disulfidptosis. The following
section extrapolates potential dysfunctions in ECs based on the
core biochemical mechanisms of disulfidptosis, aberrant protein
disulfide crosslinking and reductive power failure.
Members of the Rho family of guanine nucleotide
exchange factors act as key regulators of Rho GTPase activity and
differentially activate small GTPases, such as Rac1, a protein
implicated in disulfidptosis. These Rho GTPases are primarily
involved in actin cytoskeleton remodeling. In ECs, they modulate
junctional stability, and serve important roles in angiogenesis and
the maintenance of endothelial barrier integrity (57). Tight and adherens junctions form
the structural basis of endothelial barrier integrity. It can be
logically extrapolated that the pervasive oxidative-reductive
imbalance and protein disulfide stress occurring during
disulfidptosis could target the important sulfhydryl-rich domains
of the relevant junctional proteins. If abnormal intra- or
intermolecular disulfide crosslinking occurs, it directly disrupts
the structure and function of junctional complexes, leading to
abnormal increases in endothelial permeability (58). The consequence is plasma
extravasation, which causes tissue edema and provides a pathway for
the extravasation of inflammatory cells, such as neutrophils,
exacerbating local inflammation (59). ECs regulate vasodilation primarily
through nitric oxide (NO) production, which is catalyzed by
endothelial NO synthase (eNOS). The activity of eNOS is strictly
dependent on the reduced state of certain amino acid residues
(60). Therefore, it is reasonable
to hypothesize that the GSH depletion and ROS accumulation
accompanying disulfidptosis alter the redox modification status of
eNOS, impairing its function and leading to decreased NO
bioavailability (61,62). Insufficient NO generation triggers
abnormal microvascular constriction and reduced blood perfusion,
such as in the myocardial or cerebral microcirculation, aggravating
tissue hypoxia and promoting disease progression (63).
A healthy endothelium maintains an anticoagulant
phenotype through the surface expression of molecules such as
thrombomodulin (64).
Extrapolating from the general outcome of cell damage caused by
disulfidptosis: EC death or dysfunction exposes the pro-thrombotic
subendothelial matrix and activates platelets and the coagulation
cascade, predisposing patients to local thrombosis and exacerbating
ischemia (65). Simultaneously,
dying or stressed ECs secrete DAMPs, such as high mobility group
box 1. These molecules can activate innate immune cells, including
macrophages and neutrophils, amplifying the release of
pro-inflammatory cytokines, such as IL-1β and TNF-α, via pathways
such as the NF-κB pathway, establishing a cycle of endothelial
damage, inflammation and thrombosis (65). Although the endothelium possesses a
certain capacity for regeneration and repair, it has been
extrapolated that disulfidptosis may hinder this process through
two potential mechanisms: i) Abnormal crosslinking of cytoskeletal
proteins, such as actin and tubulin, could directly impair the
migratory capacity of surviving ECs toward the injury site; and ii)
the persistent oxidative stress and inflammatory microenvironment
triggered by disulfidptosis may suppress the mobilization, homing
and differentiation of endothelial progenitor cells (66). Prolonged impairment of repair
leaves the vascular wall continuously exposed to detrimental
stimuli, creating conditions conducive to abnormal VSMC
proliferation, lipid depositions and pathological vascular
remodeling (67).
Fibroblasts
Fibroblasts are the central cells responsible for
producing and remodeling the extracellular matrix (ECM), and their
dysfunction is a notable factor in the fibrosis of tissues such as
the myocardium and blood vessels. The ECM is a dynamic network
composed of collagen, including types I, III and IV, fibronectin,
laminin, elastin and proteoglycans. An imbalance between ECM
synthesis and degradation is key to the development of fibrosis
(68).
Based on the core characteristics of disulfidptosis,
involving the collapse of intracellular reductive power, which is
caused by factors such as GSH depletion and a decreased
NADPH:NADP+ ratio, and the consequent risk of widespread
protein oxidation and aberrant disulfide bond formation, a testable
scientific hypothesis can be proposed: This abnormal redox
environment may disrupt the normal metabolic balance of ECM in
fibroblasts. The synthesis and secretion of ECM components such as
collagen is a multistep, intricate process involving enzymes such
as prolyl hydroxylase, which catalyzes post-translational
modifications in the endoplasmic reticulum (69). It is hypothesized that the activity
of these enzymes could be impaired if their important sulfhydryl
groups are oxidized, or if the function of the cytoskeletal and
vesicular transport systems, such as tubulin-dependent dynein
transport, that are responsible for intracellular trafficking were
hindered due to abnormal protein crosslinking. Theoretically, this
could affect the proper processing and secretion of procollagen
(70).
ECM degradation is primarily performed by the matrix
metalloproteinase (MMP) family. Their catalytic centers contain
conserved cysteine residues within the zinc-binding domain, which
must remain in a reduced state for enzymatic activity (71). Therefore, it is reasonable to
speculate that the highly oxidative environment induced by
disulfidptosis could oxidize these key sulfhydryl groups,
potentially inhibiting MMP activity. If the synthesis and
degradation pathways are concurrently disrupted, it could
theoretically lead to increased net deposition of ECM, which is a
hallmark of fibrosis (72).
Another potential link between disulfidptosis and the fibrotic
process may be established through abnormal fibroblast activation
and the inflammatory microenvironment. Oxidative stress induces
fibroblast activation and transformation into highly secretory
myofibroblasts (73).
Consequently, it has been hypothesized that the GSH depletion and
ROS accumulation accompanying early-stage disulfidptosis may
activate signaling pathways such as the NF-κB pathway within
fibroblasts, promoting the expression of pro-fibrotic factors such
as TGF-β. This could drive fibroblasts toward a pathological
phenotype in a paracrine or autocrine manner (31). When disulfidptosis reaches the
threshold of cellular collapse, fibroblast lysis and the subsequent
release of DAMPs can recruit and activate immune cells such as
macrophages, initiating and perpetuating local chronic
inflammation. Notably, chronic inflammation is a key driver of
fibrosis progression (74).
Evidence for disulfidptosis in CVDs
In a recent study, a natural resin extracted from
Dracaena cochinchinensis demonstrated anti-inflammatory and
neuroprotective properties against ischemic brain injury. Molecular
docking analysis suggested that this resin may downregulate genes
associated with disulfidptosis, including Flna, Iqgap1, Tln1
and Myh9. This indicates that its neuroprotective effects
may be achieved through modulating these genes and thereby
potentially influencing the disulfidptosis pathway. However,
whether disulfidptosis is directly involved in mediating the
resin's effects requires further experimental confirmation
(75).
In the context of type A aortic dissection (TAAD), a
recent study utilized machine learning to analyze
disulfidptosis-related genes. Compared with healthy controls,
CAPZB, PDLIM1, and MYH10 were identified as three hub genes, all of
which exhibited lower expression levels in TAAD samples. This study
also revealed marked differences in immune cell infiltration in
TAAD tissues. These associative findings suggested that alterations
in the expression of disulfidptosis-related genes may have been
linked to changes in the immune microenvironment of TAAD; however,
the specific causal mechanisms underlying this association warrant
further investigation (8).
Regarding ischemic cardiomyopathy (IC), Tan et
al (7) utilized bioinformatics
and machine learning approaches to identify MYH9, NUBPL, MYL6,
MYH10 and NCKAP1 as potential diagnostic biomarkers.
These genes were associated with processes such as myocardial
structure and immune cell infiltration. A diagnostic model based on
these five genes demonstrated high predictive accuracy across
multiple datasets, which was supported by preliminary validation in
a mouse model of IC. Notably, this study primarily provided
associative evidence at the gene expression level and has not
directly confirmed the occurrence of disulfidptosis as a cell death
event in IC models (7).
Furthermore, studies on other CVDs, such as heart failure and
hypertrophic cardiomyopathy, have observed changes in the
expression of disulfidptosis-related genes, implying a potential
association between disulfidptosis and CVDs (6,9,76).
In summary, thus far, research has predominantly
focused on bioinformatics analyses and associative validation of
expression changes in disulfidptosis-related genes across various
CVDs. These findings offer important evidence and hypotheses that
disulfidptosis may be involved in the pathological processes of
CVDs. However, it must be emphasized that the association between
these pathologies at the gene expression level does not equate to
the functional activation of this cell death program. There is
still a lack of key experimental evidence that directly confirms
the occurrence of disulfidptosis, such as by detecting
characteristic protein disulfide cross-linking and cytoskeletal
collapse, in CVD models, both in vivo and in vitro.
Therefore, future research should move beyond associative analyses
and focus on verifying the specific mechanistic role of
disulfidptosis in CVDs through direct molecular and functional
experiments (Table I) (6–9,75).
 | Table I.Summary of bioinformatics and
experimental studies linking disulfidptosis to specific
cardiovascular diseases. |
Table I.
Summary of bioinformatics and
experimental studies linking disulfidptosis to specific
cardiovascular diseases.
| First author,
year | Study model or
source of evidence | Key findings | Evidence strength
and rationale | (Refs.) |
|---|
| Fan et al,
2024 | Bioinformatics:
Publicly available datasets from patients with HCM. | Discovered
differential expression of disulfidptosis-related genes in HCM and
constructed an associated risk model. | Medium:
Bioinformatics associations provided preliminary evidence for a
transcriptional link between disulfidptosis and HCM. | (6) |
| Tan et al,
2025 | 1. Bioinformatics:
Human | 1. Identified a
diagnostic gene signature, including | Medium: Strong
bioinformatics associations with | (7) |
|
| myocardial tissue
transcriptomic | MYH9, NUBPL, MYL6,
MYH10 and NCKAP1, | diagnostic
potential were observed and preliminary |
|
|
| data for IC from
the GEO database. | related to
disulfidptosis in IC. | in vivo
mRNA-level validation was obtained in a |
|
|
| 2. In vivo
validation: Mouse model | 2. Observed
concordant mRNA expression trends | disease model.
Direct evidence of protein-level |
|
|
| of IC. | for some key genes
in mouse myocardial tissue. | modification and a
functional demonstration of |
|
|
|
|
| disulfide
disulfidptosis in cardiomyocytes was |
|
|
|
|
| lacking. |
|
| Wang et al,
2025 | Bioinformatics:
Transcriptomic data | Identified
disulfidptosis-related genes, such as | Medium:
Bioinformatics associations suggested a | (8) |
|
| from patients with
TAAD vs. normal | CAPZB,
PDLIM1 and MYH10, which were | potential link
between disulfidptosis-related gene |
|
|
| aortic
tissues. | differentially
expressed in TAAD, and associated | expression and TAAD
pathology. Requires |
|
|
|
| with immune cell
infiltration and metabolic alterations. | functional
validation to establish causality. |
|
| Zhao et al,
2024 | Bioinformatics:
Publicly available | Revealed that
disulfidptosis-related genes were | Medium:
Bioinformatics associations indicated a | (9) |
|
| datasets of
patients with HF. | associated with
characteristics of the immune | potential role of
disulfidptosis-related pathways in |
|
|
|
| microenvironment in
HF. | the immune
landscape of HF. |
|
| Xu et al,
2025 | 1. In vivo:
Mouse model of ischemic | 1. Dragon's blood
resin treatment alleviated | Weak and indirect:
Indirect protective evidence in | (75) |
|
| brain injury. | brain injury. | a related ischemic
model and computational |
|
|
| 2. In
silico: Molecular docking study. | 2. Predicted that
the active components of the resin | speculation on the
mechanism was provided. |
|
|
|
| bound to and
modulated disulfidptosis-related proteins, such as FLNA, IQGAP1,
TLN1 and Myh9. | The study did not
directly measure or modulate disulfidptosis itself, leaving the
proposed mechanism hypothetical. |
|
Therapeutic targets and intervention
strategies
Directly inhibiting SLC7A11 activity may be an
effective approach for ameliorating disulfidptosis. SLC7A11 is the
specific subunit of the xCT transporter on the cell membrane that
is responsible for exchanging extracellular cystine for
intracellular glutamate. Sulfasalazine, which is similar to the
known xCT inhibitor erastin, effectively inhibits the expression
and transport function of xCT, leading to GSH depletion in
MDA-MB-231 and MDA-MB-468 cells (triple-negative breast cancer cell
lines) (77). This mechanism has
been shown to improve heart failure (78).
Furthermore, supplementing NAD+
represents another therapeutic strategy. NAD is a notable coenzyme
required for cellular energy metabolism and redox homeostasis
(79). Supplementation with NAD
precursors can markedly increase myocardial NAD levels, improving
cardiac function (80–82). Also, activators of malic enzyme can
promote NADPH generation (83),
which has been shown to alleviate pulmonary hypertension (84,85).
As disulfide bond formation is central to
disulfidptosis, reducing the formation of aberrant disulfide bonds
is important for disease mitigation. For example, the disulfide
reductant dithiothreitol can reduce abnormal disulfide bonds, but
its application is limited because of its strong odor and toxicity
(86). Thioredoxin-interacting
protein (TXNIP) is a novel molecular target that enhances
endogenous disulfide reductase capacity (87). A single study has shown that
cardiomyocyte-specific TXNIP C247S mutation knock-in mice exhibit
higher survival rates and smaller infarct sizes following MI than
control mice. Inhibition of thioredoxin by TXNIP promotes
mitochondrial antioxidant capacity in cardiomyocytes, protecting
the heart from MI-induced oxidative damage. This protective
mechanism involves potent regulation of the thioredoxin system via
a disulfide bond exchange mechanism in adult mouse cardiomyocytes
(87).
Additionally, inhibiting actin cross-linking to
protect the cytoskeleton represents another strategy to suppress
disulfidptosis. Reportedly, in HL-1 murine atrial cardiomyocytes
exposed to 2% ethanol, the cytoskeleton was markedly disrupted
during apoptosis and the anti-apoptotic transcriptional coactivator
Yes-associated protein (YAP) was inactivated. Conversely, the
retrovirus-induced expression of constitutively active YAP and
jasplakinolide-mediated stabilization of the actomyosin
cytoskeleton prevented cardiomyocyte cell death (88).
Discussion and conclusions
The present review systematically elaborated on the
core molecular mechanisms of disulfidptosis, a novel form of
regulated cell death, and explored its potential role in CVDs from
the perspective of different cell types within the cardiovascular
system. The core of disulfidptosis lies in the lethal imbalance
between SLC7A11-mediated cystine metabolism and glucose-dependent
NADPH regeneration. The resulting disulfide stress directly affects
the cytoskeletal system, which maintains cell morphology and
function, and ultimately leads to cellular collapse. For example,
disulfidptosis can: i) Impair cardiomyocyte contractility; ii)
exacerbate inflammatory responses; iii) promote the synthetic
phenotype switch, proliferation and migration of VSMCs; iv)
aggravate vascular wall thickening; v) disrupt endothelial barrier
function; vi) activate platelets and the coagulation cascade,
triggering thrombosis and worsening tissue ischemia; and vii)
induce myofibroblast generation, leading to excessive ECM
deposition and accelerated fibrosis. Given that glucose starvation
is a primary trigger for disulfidptosis and pathological scenarios
such as myocardial ischemia-reperfusion injury, the hypoglycemic
environment within atherosclerotic plaques and diabetic
cardiomyopathy represent typical glucose starvation conditions; as
such, the present review hypothesized a close relationship between
CVDs and disulfidptosis.
Particularly noteworthy is the mirror-image
crosstalk between disulfidptosis and ferroptosis. These cell death
modalities share SLC7A11 as a common molecular switch, yet their
outcomes diverge based on the cellular metabolic environment; high
SLC7A11 expression under glucose starvation steers cells toward
disulfidptosis, whereas its inhibition directs cells toward
ferroptosis. This nuanced relationship suggests that in
pathological contexts with marked metabolic fluctuations, such as
myocardial ischemia-reperfusion injury, these two death modalities
may coexist or occur sequentially, collectively amplifying tissue
damage. To definitively establish the role of disulfidptosis in
CVDs, future research should focus on the development and
application of direct experimental biomarkers. Key approaches
should include, but are not limited to: i) Employing non-reducing
gel electrophoresis to detect aberrant disulfide crosslinking in
cytoskeletal proteins, such as actin; ii) monitoring the NADPH:GSH
ratio to confirm reductive power collapse; iii) assessing SLC7A11
protein levels and cystine transport activity; and iv) rigorously
distinguishing disulfidptosis from ferroptosis through
morphological observations, for example F-actin staining, combined
with specific inhibitors. Establishing and standardizing these
detection criteria should be a notable research priority for the
next phase in this field.
Although bioinformatics analyses have revealed
notable alterations in key disulfidptosis-related genes in CVDs,
such as IC and TAAD, providing preliminary evidence for their
association with disulfidptosis, it is important to recognize the
apparent limitations of current studies. The primary challenge is
that the majority of evidence remains indirect, lacking definitive
proof of disulfidptosis occurrence in CVD models, such as in
vivo animal models or clinical samples, including the direct
detection of aberrant disulfide bonds in cytoskeletal proteins (for
example, actin), within myocardial or vascular tissues.
However, studies suggest that small molecule
inhibitors directly targeting SLC7A11 activity or NAD+
supplementation could be potential therapeutic strategies for
mitigating disulfidptosis in CVDs. Their combined use may alleviate
the adverse effects of NADPH depletion. Additionally, disulfide
reductants, such as TXNIP, and the inhibition of actin
cross-linking have shown potential in ameliorating the progression
of CVDs. Therefore, this strategy represents a promising research
topic.
Future investigations should explore the interaction
mechanisms between disulfidptosis and ferroptosis by utilizing
single-cell sequencing and multi-omics integration to identify
novel therapeutic targets, providing novel options for the
diagnosis and treatment of CVDs. Furthermore, it is important to
precisely evaluate the contribution of disulfidptosis across a
broader range of CVD models, such as hypertensive heart disease,
myocarditis and heart failure, to clarify whether it acts as a
primary driver or synergistic disruptor.
Acknowledgements
The figures in this article were created using
BioRender (biorender.com).
Funding
The present review was supported by the Advantageous Discipline
of Shanghai Putuo District Health Commission (grant no. 2023ysxk01)
and the Natural Science Foundation of Shanghai Municipality (grant
no. 23ZR1456500).
Availability of data and materials
Not applicable.
Authors' contributions
XJ, ZC, XD and ZL designed the article and wrote the
manuscript. Data authentication is not applicable. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
World Heart Federation (WHF), . 2023 World
Heart Report. 2023.
|
|
2
|
Liu X, Nie L, Zhang Y, Yan Y, Wang C,
Colic M, Olszewski K, Horbath A, Chen X, Lei G, et al: Actin
cytoskeleton vulnerability to disulfide stress mediates
disulfidptosis. Nat Cell Biol. 25:404–414. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
He Y, Chen Y, Song W, Zhu L, Dong Z and Ow
DW: A Pap1-Oxs1 signaling pathway for disulfide stress in
Schizosaccharomyces pombe. Nucleic Acids Res. 45:106–114. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zheng P, Zhou C, Ding Y and Duan S:
Disulfidptosis: A new target for metabolic cancer therapy. J Exp
Clin Cancer Res. 42:1032023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mao C, Wang M, Zhuang L and Gan B:
Metabolic cell death in cancer: Ferroptosis, cuproptosis,
disulfidptosis, and beyond. Protein Cell. 15:642–660. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fan H, Tan X, Xu S, Zeng Y, Zhang H, Shao
T, Zhao R, Zhou P, Bo X, Fan J, et al: Identification and
validation of differentially expressed disulfidptosis-related genes
in hypertrophic cardiomyopathy. Mol Med. 30:2492024. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tan X, Xu S, Zeng Y, Yu F, Qin Z, Zhang G,
Fan J, Bo X, Tang J, Fan H, et al: A novel disulfidptosis-related
diagnostic gene signature and differential expression validation in
ischaemic cardiomyopathy. J Cell Mol Med. 29:e704752025. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang D, Wang C, Liu H, Zhang Z, Li M, Ge
X, Bi A, Gao C, Tian X, Liu K, et al: Integrated bioinformatic
analysis of immune infiltration and disulfidptosis related gene
subgroups in type A aortic dissection. Sci Rep. 15:137192025.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhao L, Zhang J, Song Q, Dai C, Qin Y and
Li A: Comprehensive analysis of disulfidptosis-related genes and
the immune microenvironment in heart failure. Front Cell Dev Biol.
12:15168982024. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Qian S, Chen G, Li R, Ma Y, Pan L and Wang
X and Wang X: Disulfide stress and its role in cardiovascular
diseases. Redox Biol. 75:1032972024. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Koppula P, Zhuang L and Gan B: Cystine
transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient
dependency, and cancer therapy. Protein Cell. 12:599–620. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yu X, Liu Z, Yu Y, Qian C, Lin Y, Jin S,
Wu L and Li S: Hesperetin promotes diabetic wound healing by
inhibiting ferroptosis through the activation of SIRT3. Phytother
Res. 38:1478–1493. 2024. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tang S, Yong J, Yan J, Peng T, Long F and
Chen H: Composition of Polygonatum zanlanscianense Pamp.
Steam and leaf phenolic extract and its protective mechanism on
t-BHP-induced oxidative damage of HepG2 cells. Molecules.
28:74872023. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ying M, You D, Zhu X, Cai L, Zeng S and Hu
X: Lactate and glutamine support NADPH generation in cancer cells
under glucose deprived conditions. Redox Biol. 46:1020652021.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen X, Roeters SJ, Cavanna F, Alvarado J
and Baiz CR: Crowding alters F-actin secondary structure and
hydration. Commun Biol. 6:9002023. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sobierajska K, Skurzynski S, Stasiak M,
Kryczka J, Cierniewski CS and Swiatkowska M: Protein disulfide
isomerase directly interacts with β-actin Cys374 and regulates
cytoskeleton reorganization. J Biol Chem. 289:5758–5773. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Matyushenko AM, Artemova NV, Shchepkin DV,
Kopylova GV, Nabiev SR, Nikitina LV, Levitsky DI and Bershitsky SY:
The interchain disulfide cross-linking of tropomyosin alters its
regulatory properties and interaction with actin filament. Biochem
Biophys Res Commun. 482:305–309. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bailly C, Degand C, Laine W, Sauzeau V and
Kluza J: Implication of Rac1 GTPase in molecular and cellular
mitochondrial functions. Life Sci. 342:1225102024. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang Y, Wang S, Lei M, Boyett M, Tsui H,
Liu W and Wang X: The p21-activated kinase 1 (Pak1) signalling
pathway in cardiac disease: From mechanistic study to therapeutic
exploration. Br J Pharmacol. 175:1362–1374. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Alekhina O, Burstein E and Billadeau DD:
Cellular functions of WASP family proteins at a glance. J Cell Sci.
130:2235–2241. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lebensohn AM and Kirschner MW: Activation
of the WAVE complex by coincident signals controls actin assembly.
Mol Cell. 36:512–524. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Begemann A, Sticht H, Begtrup A, Vitobello
A, Faivre L, Banka S, Alhaddad B, Asadollahi R, Becker J, Bierhals
T, et al: New insights into the clinical and molecular spectrum of
the novel CYFIP2-related neurodevelopmental disorder and impairment
of the WRC-mediated actin dynamics. Genet Med. 23:543–554. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Guo Y, Chen X, Hu J, Su Y, Yin F and Liu
X: The dual role of SLC7A11 in tumor drug resistance: Mechanisms,
challenges, and therapeutic potential. Am J Cancer Res.
15:4516–4532. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dolma S, Lessnick SL, Hahn WC and
Stockwell BR: Identification of genotype-selective antitumor agents
using synthetic lethal chemical screening in engineered human tumor
cells. Cancer Cell. 3:285–296. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang WS and Stockwell BR: Synthetic lethal
screening identifies compounds activating iron-dependent,
nonapoptotic cell death in oncogenic-RAS-harboring cancer cells.
Chem Biol. 15:234–245. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang D, Gong G, Song J, Chen J, Wang S, Li
J and Wang G: Ferroptosis-mediated osteoclast-osteoblast crosstalk:
Signaling pathways governing bone remodeling in osteoporosis. J
Orthop Surg Res. 20:8882025. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bersuker K, Hendricks JM, Li Z, Magtanong
L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, et al:
The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit
ferroptosis. Nature. 575:688–692. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhao M, Jin Z, Xia C, Chen S, Zeng L, Qin
S and He Q: Inhibition of free heme-catalyzed Fenton-like reaction
prevents non-alcoholic fatty liver disease by hepatocyte-targeted
hydrogen delivery. Biomaterials. 301:1222302023. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dong Y, Wu F, Liu K, Yue Y, Shen X, Qu Z,
Yu S and Du W: Kindlin-2/Otub1/Slc7a11 axis improved cardiac
ischemia reperfusion injury by inhibiting cardiomyocyte
ferroptosis. Antioxid Redox Signal. 43:727–744. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xu W, Deng K and Pei L: P4HA3 depletion
induces ferroptosis and inhibits colorectal cancer growth by
stabilizing ACSL4 mRNA. Biochem Pharmacol. 233:1167462025.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bai X, Kang J, Wei S, Wang Y, Liu Y, Yuan
B, Lu Q, Li H, Yan J, Yang X and Chang J: A pH responsive
nanocomposite for combination sonodynamic-immunotherapy with
ferroptosis and calcium Ion overload via SLC7A11/ACSL4/LPCAT3
pathway. Exploration (Beijing). 5:202400022024. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shahtout JL, Eshima H, Ferrara PJ, Maschek
JA, Cox JE, Drummond MJ and Funai K: Inhibition of the skeletal
muscle Lands cycle ameliorates weakness induced by physical
inactivity. J Cachexia Sarcopenia Muscle. 15:319–330. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fei W, Chen D, Tang H, Li C, Zheng W, Chen
F, Song Q, Zhao Y, Zou Y and Zheng C: Targeted GSH-exhausting and
hydroxyl radical self-producing manganese-silica nanomissiles for
MRI guided ferroptotic cancer therapy. Nanoscale. 12:16738–16754.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang CH, Yan YJ and Luo Q: The molecular
mechanisms and potential drug targets of ferroptosis in myocardial
ischemia-reperfusion injury. Life Sci. 340:1224392024. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Qiu H, Liu J, Shao N, Zhao J, Chen C,
Jiang Y, Zhao X and Xu L: SLC7A11 as a bridge between ferroptosis
and disulfidptosis: A promising target for tumor treatment. Cell
Commun Signal. 23:4602025. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang M, Zheng H, Jin H, Zhu X, Liu S,
Chen Y, Zhang H and Zhang S: Regulating SLC7A11/GSH/GPX4 axis by
glucose dyshomeostasis to simultaneously promote disulfidptosis,
cuproptosis and ferroptosis. Bioact Mater. 54:744–758.
2025.PubMed/NCBI
|
|
38
|
Farah EN, Hu RK, Kern C, Zhang Q, Lu TY,
Ma Q, Tran S, Zhang B, Carlin D, Monell A, et al: Spatially
organized cellular communities form the developing human heart.
Nature. 627:854–864. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Little M, Risi CM, Larrinaga TM, Summers
MD, Nguyen T, Smith GE Jr, Atherton J, Gregorio CC, Kostyukova AS
and Galkin VE: Interaction of cardiac leiomodin with the native
cardiac thin filament. PLoS Biol. 23:e30030272025. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Leng W, Li Y, Liang X, Yuan L, Li X and
Gao R: Thermo-reversible gelation and enhanced umami perception of
myofibrillar proteins induced by protein-glutaminase-mediated
deamidation. Food Chem. 471:1428022025. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Valencia DA, Koeberlein AN, Nakano H,
Rudas A, Patel AA, Harui A, Spencer C, Nakano A and Quinlan ME:
Human formin FHOD3-mediated actin elongation is required for
sarcomere integrity in cardiomyocytes. Elife. 13:RP1040482025.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Qin H, Xu J, Yue Y, Chen M, Zhang Z, Xu P,
Zheng Y, Zeng H, Weng J, Yang J and Yu F: Disulfidptosis-related
gene signatures as prognostic biomarkers and predictors of
immunotherapy response in HNSCC. Front Immunol. 15:14566492024.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li Y and Li M: Dihydromyricetin protects
against Hypoxia/reoxygenation injury in cardiomyocytes by
activating miR-34a-mediated notch1 pathway. Cardiovasc Toxicol.
25:294–305. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Huang YJ, Tong HY, Huang XJ, Xiao XC, Dong
Y and Iqbal MS: Anshen-Buxin-Liuwei pill, a Mongolian medicinal
formula could alleviate cardiomyocyte hypoxia/reoxygenation injury
via mitochondrion pathway. Mol Biol Rep. 49:885–894. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wu W, Wang J, Wang G, Wang F, Yang Y, Liu
Z, Song Q, Chen S and Chen H: Monotropein inhibits MMP9-mediated
cardiac oxidative stress, inflammation, matrix degradation and
apoptosis in a mouse and cell line models of septic cardiac injury.
Mol Biol Rep. 52:3292025. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Dutta A, Das M, Ghosh A and Rana S:
Molecular and cellular pathophysiology of circulating
cardiomyocyte-specific cell free DNA (cfDNA): Biomarkers of heart
failure and potential therapeutic targets. Genes Dis. 10:948–959.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Barron JT, Gu L and Parrillo JE: NADH/NAD
redox state of cytoplasmic glycolytic compartments in vascular
smooth muscle. Am J Physiol Heart Circ Physiol. 279:H2872–H2878.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gimenez M, Veríssimo-Filho S, Wittig I,
Schickling BM, Hahner F, Schürmann C, Netto LES, Rosa JC, Brandes
RP, Sartoretto S, et al: Redox activation of Nox1 (NADPH Oxidase 1)
Involves an intermolecular disulfide bond between protein disulfide
isomerase and p47phox in vascular smooth muscle cells. Arterioscler
Thromb Vasc Biol. 39:224–236. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kang H, Liu J, Sun A, Liu X, Fan Y and
Deng X: Vascular smooth muscle cell glycocalyx mediates shear
stress-induced contractile responses via a Rho kinase (ROCK)-myosin
light chain phosphatase (MLCP) pathway. Sci Rep. 7:420922017.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Gao WD, Murray CI, Tian Y, Zhong X, DuMond
JF, Shen X, Stanley BA, Foster DB, Wink DA, King SB, et al:
Nitroxyl-mediated disulfide bond formation between cardiac
myofilament cysteines enhances contractile function. Circ Res.
111:1002–1011. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Martinsen A, Baeyens N, Yerna X and Morel
N: Rho kinase regulation of vasopressin-induced calcium entry in
vascular smooth muscle cell: Comparison between rat isolated aorta
and cultured aortic cells. Cell Calcium. 52:413–421. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Tang DD and Gerlach BD: The roles and
regulation of the actin cytoskeleton, intermediate filaments and
microtubules in smooth muscle cell migration. Respir Res.
18:542017. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Li T, He M, Li Z, Wang D, Xu Y, Wu W and
Yan Y: Metformin inhibits aortic atherosclerosis in mice by
regulating actin skeleton in vascular smooth muscle cells. Nan Fang
Yi Ke Da Xue Xue Bao. 39:1357–1363. 2019.(In Chinese). PubMed/NCBI
|
|
54
|
Yang H, Luo YY, Zhang LT, He KR and Lin X:
Extracellular histones induce inflammation and senescence of
vascular smooth muscle cells by activating the AMPK/FOXO4 signaling
pathway. Inflamm Res. 71:1055–1066. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Chigareva I, Karelova A, Zeinalova N,
Abdulkhadzhiev A, Isaev A, Kurbanov G, Israpilov I, Dagaeva I,
Dashaeva M, Petchina A, et al: Phenotypic switching of vascular
smooth muscle cells: Key mechanism in atherosclerosis progression.
Georgian Med News. 54–58. 2025.PubMed/NCBI
|
|
56
|
Kotlyarov S: Immune function of
endothelial cells: Evolutionary aspects, molecular biology and role
in atherogenesis. Int J Mol Sci. 23:97702022. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Kempers L, Driessen AJM, van Rijssel J,
Nolte MA and van Buul JD: The RhoGEF Trio: A protein with a wide
range of functions in the vascular endothelium. Int J Mol Sci.
22:101682021. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wu CY, Sharma A, Edwards JD, Liu PP,
Kapral MK, Herrmann N, Wu CF, Podolsky S, Swardfager W and Shah BR:
Cardiovascular effectiveness and safety of SGLT2 inhibitors vs DPP4
inhibitors by dementia status: A cohort study of older adults with
diabetes. Diabetologia. 69:386–398. 2026. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ma Y, Yang X, Chatterjee V, Meegan JE,
Beard RS Jr and Yuan SY: Role of neutrophil extracellular traps and
vesicles in regulating vascular endothelial permeability. Front
Immunol. 10:10372019. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Yao L, Li F, Ni J, Wei M, Li T, Shi J and
Tian J: Effect of bazi bushen capsule on D-Galactose-induced human
endothelial cell senescence through PI3K/Akt/eNOS signaling
pathway. Aging Med (Milton). 8:258–266. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Han D, Ding B, Zheng P, Yuan M, Bian Y,
Chen H, Wang M, Chang M, Kheraif AAA, Ma P, et al: NADPH
Oxidase-like nanozyme for High-efficiency tumor therapy through
increasing glutathione consumption and blocking glutathione
regeneration. Adv Healthc Mater. 13:e23033092024. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Janaszak-Jasiecka A, Płoska A, Wierońska
JM, Dobrucki LW and Kalinowski L: Endothelial dysfunction due to
eNOS uncoupling: Molecular mechanisms as potential therapeutic
targets. Cell Mol Biol Lett. 28:212023. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Carlström M, Weitzberg E and Lundberg JO:
Nitric oxide signaling and regulation in the cardiovascular system:
Recent advances. Pharmacol Rev. 76:1038–1062. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Iba T, Helms J, Okada H, Nagakari K, Sato
K, Ferrer R and Levy JH: Damage-associated molecular patterns,
immunothrombosis, and intravascular inflammation in sepsis: A
narrative integrative review. Semin Thromb Hemost. Dec 31–2025.doi:
10.1055/a-2776-5999 (Epub ahead of print).
|
|
65
|
Yan B, Yu X, Cai X, Huang X, Xie B, Lian
D, Chen J, Li W, Lin Y, Ye J and Li J: A review: The significance
of Toll-like receptors 2 and 4, and NF-κB signaling in endothelial
cells during atherosclerosis. Front Biosci (Landmark Ed).
29:1612024. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Larionov A, Hammer CM, Fiedler K and
Filgueira L: Dynamics of endothelial cell diversity and plasticity
in health and disease. Cells. 13:12762024. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Li F, Kumar S, Pokutta-Paskaleva A, Kang
DW, Kim C, Raykin J, Omojola V, Hoffmann C, Zhao F, Teichmann M, et
al: Endothelial cell (EC)-specific Ctgf/Ccn2 expression increases
EC reprogramming and atherosclerosis. Matrix Biol. 136:102–110.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Alam P, Stiens SM, Bowles HJ, Bui H and
Bowles DK: Yoda1 inhibits TGFβ-induced cardiac fibroblast
activation via a BRD4-dependent pathway. Cells. 14:10282025.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Gelse K, Pöschl E and Aigner T:
Collagens-structure, function, and biosynthesis. Adv Drug Deliv
Rev. 55:1531–1546. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Farhan H, Zahoor M, Tauer JT and Högler W:
The endoplasmic reticulum proteostasis network and bone disease.
Trends Mol Med. Jul 10–2025.doi: 10.1016/j.molmed.2025.06.005 (Epub
ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Taherkhani S, Sheibani M,
Mohammadkhanizadeh A, Virag JAI, de Castro Braz L and Azizi Y:
Metalloproteinases (MMPs) in hypertensive disorders: Role,
function, pharmacology, and potential strategies to mitigate
pathophysiological changes. Front Pharmacol. 16:15592882025.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
He X, Xu B, Fang A, Li X, Huang Z, Qin S,
Xiao W, Li G, Tian M, Fan N, et al: A cascade-responsive
nanoplatform with tumor cell-specific drug burst release for
chemotherapy. Acta Biomater. 162:120–134. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Zou X, Wu T, Lin J, Su T, Xiao H, Ni C, Hu
L, Lin W, Chen W, Ye RD and Xiang L: SAA3 deficiency exacerbates
intestinal fibrosis in DSS-induced IBD mouse model. Cell Death
Discov. 11:252025. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Bhattacharyya S, Midwood KS, Yin H and
Varga J: Toll-Like Receptor-4 signaling drives persistent
fibroblast activation and prevents fibrosis resolution in
scleroderma. Adv Wound Care (New Rochelle). 6:356–369. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Xu C, Hang H, Li W, Meng Y, Zhao H, Liu C
and Zhang R: Dragon's Blood modulates disulfidptosis-related genes
to alleviate ischemic brain injury in mice. Neurochem Res.
50:1852025. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Moroni F, Gertz Z and Azzalini L: Relief
of ischemia in ischemic cardiomyopathy. Curr Cardiol Rep.
23:802021. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Long Y, Xu Z, Yu J, Hu X, Xie Y, Duan X,
Li N, Yan Y, Wang Y and Qin J: Targeting xCT with sulfasalazine
suppresses triple-negative breast cancer growth via inducing
autophagy and coordinating cell cycle and proliferation. Anticancer
Drugs. 35:830–843. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Chen C, Zhang X, Zheng C, Gao Z, Jiang X,
Bai Y and Meng Y: Sulfasalazine exacerbates angiotensin II-induced
cardiac remodelling by activating Akt signal pathway. Clin Exp
Pharmacol Physiol. 49:776–783. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Smith HB, Lee K, Freeman MJ, Stevenson DM,
Amador-Noguez D and Sauer JD: Listeria monocytogenes requires
DHNA-dependent intracellular redox homeostasis facilitated by Ndh2
for survival and virulence. Infect Immun. 91:e00022232023.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Li S, Rodrigues PG, Chakraborty AD,
Correia C, Schouten EM, Strömstedt M, Löfgren L, Persson M,
Fredlund L, Rohman M, et al: Nicotinamide-N-methyltransferase
inhibition improves cardiac function and structure in a heart
failure with preserved ejection fraction mouse model. Pharmacol
Res. 217:1078202025. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Szarvas Z, Reyff ZA, Peterfi A, Pinto CB,
Owens CD, Kaposzta Z, Mukli P, Pinaffi-Langley A, Adams CA, Muranyi
M, et al: Effects of NAD+ supplementation with oral
nicotinamide riboside on vascular health and cognitive function in
older adults with peripheral artery disease: Results from a pilot
4-week open-label clinical trial. J Pharmacol Exp Ther.
392:1036072025. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Yu F, Zhao H, Luo L and Wu W: Nicotinamide
adenine dinucleotide supplementation to alleviate heart failure: A
mitochondrial dysfunction perspective. Nutrients. 17:18552025.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Yoshida T, Kawabe T, Cantley LC and
Lyssiotis CA: Discovery and characterization of a novel allosteric
Small-molecule Inhibitor of NADP+-Dependent malic enzyme
1. Biochemistry. 61:1548–1553. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Luo Y, Qi X, Zhang Z, Zhang J, Li B, Shu
T, Li X, Hu H, Li J, Tang Q, et al: Inactivation of malic enzyme 1
in endothelial cells alleviates pulmonary hypertension.
Circulation. 149:1354–1371. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Rao RJ and Chan SY: Mediating metabolism:
Inhibition of malic Enzyme 1 (ME1) restores endothelial
bioenergetics and adenosine signaling in pulmonary hypertension.
Circulation. 149:1372–1374. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Fang JT, Wang ST, Wang H and Fang WJ: A
novel peptide mapping method utilizing cysteine as a reducing
agent. Pharm Res. 42:173–184. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Nakayama Y, Mukai N, Wang BF, Yang K,
Patwari P, Kitsis RN and Yoshioka J: Txnip C247S mutation protects
the heart against acute myocardial infarction. J Mol Cell Cardiol.
155:36–49. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Noritake K, Aki T, Kimura M, Funakoshi T,
Unuma K and Uemura K: Restoration of YAP activation rescues HL-1
cardiomyocytes from apoptotic death by ethanol. J Toxicol Sci.
42:545–551. 2017. View Article : Google Scholar : PubMed/NCBI
|