Introduction
Cardiac fibrosis is characterized by excessive
deposition of extracellular matrix (ECM) proteins, representing a
common pathological endpoint in nearly all forms of heart disease,
including myocardial infarction, aortic stenosis, hypertrophic
cardiomyopathy and diabetic cardiomyopathy. This maladaptive
remodeling process directly contributes to heart failure
development and exacerbates adverse outcomes in established
patients with heart failure (1).
Cardiac fibroblasts (CFs) are the second most abundant cell
population in the heart after cardiomyocytes and are pivotal for
fibrogenesis through their exceptional plasticity and secretory
capacity. These cells dynamically transition between
differentiation states in response to cardiac injury, aging,
genetic predisposition and environmental factors, actively
secreting ECM structural proteins, proteolytic enzymes, growth
factors and cytokines that collectively drive pathological collagen
deposition (1). Despite
significant advances in identifying fibrogenic signaling pathways,
the absence of specific anti-fibrotic therapies highlights the
urgent need for novel mechanistic insights and therapeutic
targets.
The Developmental Origins of Health and Disease
(DOHaD), initially proposed by Barker, has gained significant
scientific recognition (2).
Maternal inflammatory conditions, including periodontitis,
urethritis and influenza, are common challenges during gestation.
This exposure creates an intrauterine environment characterized by
elevated proinflammatory cytokines and associated mediators,
potentially predisposing offspring to cardiovascular pathologies,
including hypertension and cardiac hypertrophy. Our previous
studies using lipopolysaccharides (LPS), a gram-negative bacterial
cell wall component, in pregnant rodent models have provided
supporting evidence (3–5). In these distinctive models, offspring
rats born to LPS-exposed dams revealed moderate elevation in
collagen synthesis without apparent cardiac dysfunction at 6 weeks
of age, yet developed significant left ventricular (LV) hypertrophy
by 8 months (4). Notably, prenatal
LPS-exposed mice demonstrated exacerbated cardiac fibrosis
responses to isoproterenol challenge as early as 4 weeks postnatal,
despite baseline fibrosis levels remaining comparable between the
exposed and control groups at this developmental stage (5). These observations highlight the need
to study the molecular mechanisms underlying the developmental
programming of cardiac vulnerability.
Dysregulation of intercellular communication is
associated with the pathological changes of fibrosis (6). Gap junctions (GJs), composed of
connexin proteins (Cxs), particularly Cx43 in cardiac tissue,
facilitate key cardiomyocyte-fibroblast crosstalk through ion
exchange and metabolite transfer (7,8).
Cx43-mediated communication modulates fibroblast activation,
phenotypic switching and pathological coupling with cardiomyocytes,
thereby covering all the key processes in fibrogenesis (7,8).
Notably, the autophagy pathway is an essential quality control
mechanism for cellular components, including connexins and has
emerged as a potential regulator of fibrotic progression (9,10).
The short half-life of Cx43 makes its turnover particularly
dependent on autophagic degradation, creating a potential
mechanistic association between these pathways in cardiac pathology
(10).
Epigenetic dysregulation, particularly DNA
methylation alterations, may be a critical mechanism underlying the
developmental programming of fibrosis. Our previous study
established that prenatal LPS exposure induces hypertension in
offspring associated with renal and vascular methylation changes
(11,12). This, coupled with emerging evidence
connecting DNA methylation to both autophagy regulation and Cx43
dysfunction, prompted us to hypothesize that methylation-involved
interactions between Cx43 and autophagy drive the developmental
programming of cardiac fibrosis. Therefore, the present study
systematically investigated these mechanistic relationships in a
well-characterized rat model of prenatal LPS exposure, aiming to
identify novel therapeutic targets for preventing maternal
inflammation-primed cardiac fibrosis.
Materials and methods
Animal treatment
A total of 16 female nulliparous, pregnant
Sprague-Dawley rats (age, 10 weeks; weight, 180–220 g) were
obtained from the Experimental Animal Center of Army Medical
University. Animals were individually housed in a controlled
environment at a constant temperature (24°C), relative humidity of
50±10%, under a 12-h light/dark cycle with ad libitum access
to standard laboratory chow and water. Following a 5-day
acclimation period, pregnant dams were randomly allocated to the
control (n=8) or LPS (n=8) treatment groups. Subjects received
intraperitoneal injections of either 0.5 ml saline (control) or
0.79 mg/kg LPS (MilliporeSigma) on gestational days 8, 10 and 12,
which is a critical window for embryonic cardiogenesis. Postpartum
litter parameters, including size and individual pup weights, were
recorded. To standardize nutritional availability, litters were
culled to eight neonates/dam until weaning at postnatal week 4.
Post-weaning offspring were group-housed by sex (4–5 animals/cage)
and given continuous access to food and water, under the same
controlled environmental conditions as those for the pregnant rats.
Body weights were monitored weekly from birth through the study
period. At 8 and 16 weeks of age, male offspring were randomly
selected for terminal procedures. After being deeply anesthetized
with sodium pentobarbital (50 mg/kg, i.p.), animals were sacrificed
by exsanguination via cardiac puncture while under deep anesthesia.
Mortality was confirmed by the cessation of respiration, loss of
corneal reflex and direct visual confirmation of cardiac arrest via
thoracotomy prior to tissue harvest (13). Blood and cardiac tissue samples
were then collected immediately for subsequent analyses.
Heart weight index and LV mass
index
The heart was perfused with ice-cold physiological
saline to remove any residual blood. Excess pericardial tissue and
the surrounding vasculature were carefully dissected away, followed
by moisture removal through gentle blotting with filter paper. The
total cardiac mass was recorded using an analytical balance. The LV
myocardium was surgically isolated from the cardiac base along the
interventricular groove. Morphometric indices were subsequently
calculated as the ratio of total heart weight (HW) or LV weight
(LVW) to body weight (BW), expressed as HW/BW and LVW/BW,
respectively.
Measurement of B-type Natriuretic
Peptide (BNP) and angiotensin II (Ang II) levels
Following a 30-min clotting period at room
temperature, whole blood samples were centrifuged at 3,000 × g for
15 min to isolate the serum. LV tissue specimens were immediately
homogenized in ice-cold phosphate-buffered saline (PBS; 50 mg
tissue/0.5 ml) containing 1% nonidet-P40 (Shanghai Biyuntian
Biotechnology Co., Ltd.) and a complete protease inhibitor cocktail
using a mechanical tissue homogenizer. The resulting homogenates
were centrifugated at 12,000 × g for 10 min at 4°C in a
refrigerated centrifuge to obtain clarified supernatants. Serum
concentrations of N-terminal pro-BNP (NT-proBNP; cat. no.
E-EL-R3023) and Ang II, along with myocardial tissue levels of Ang
II (cat. no. E-EL-R1430c), were quantified using commercial ELISA
kits (Elabscience Biotechnology Co., Ltd.) following manufacturer
protocols. All assays included appropriate quality controls and
standard curve validation.
Histological analysis
All procedures were performed at room temperature.
LV tissues were immersion-fixed in 4% paraformaldehyde for 24–48 h,
before paraffin embedding. Serial sections (4 µm) were prepared
using a rotary microtome. For basic histoarchitecture evaluation,
the sections were stained with hematoxylin and eosin (H&E;
hematoxylin staining for 5 min, eosin staining for 2 min). Collagen
fiber visualization was achieved using Masson's trichrome staining
performed following manufacturer specifications (Beijing Solarbio
Science & Technology Co., Ltd.). After overnight incubation in
potassium dichromate solution, the sections underwent ferric
hematoxylin staining for 6 min, followed by acid fuchsin staining
for 8 min, phosphomolybdic acid treatment for 2 min, and aniline
blue staining for 2 min. Prior to sectioning, the samples subjected
to sequential dehydration through a graded ethanol series
(70–100%), xylene clearing and paraffin infiltration. The stained
sections were imaged using bright-field microscopy (Nikon Eclipse
E100; Nikon Corporation). Quantitative assessment of myocardial
fibrosis was performed by calculating the collagen volume fraction
(CVF) through systematic random sampling of five non-overlapping
high-power fields (magnification, ×400) per section. CVF was
determined as the percentage ratio of the aniline blue-stained
collagen area to the total myocardial area using the image analysis
software (version 6.0; Media Cybernetics, Inc.).
Immunofluorescence staining
LV tissues embedded in OCT compound (OriGene
Technologies, Inc.) were cryosectioned into four sequential
10-µm-thick sections using a cryostat (Leica CM1950) and mounted on
poly-L-lysine-coated slides. For immunofluorescence detection, the
slides were first blocked with 10% normal goat serum (Life
Technologies; cat. no. 50062Z) in PBS containing 0.2% Tween-20 for
1 h at room temperature. Sections were incubated overnight at 4°C
with rabbit polyclonal anti-α-smooth muscle actin (α-SMA) antibody
(1:400 dilution; Abcam. cat. no. ab124964) in a humidified chamber.
After three PBS washes (5 min each), the slides were incubated with
Cy3-conjugated goat anti-rabbit IgG secondary antibody (1:400
dilution; Invitrogen; Thermo Fisher Scientific, Inc. A10520) for 1
h at room temperature, protected from light. Negative control
slides were processed identically, except for primary antibody
omission. Fluorescent images were captured using a laser-scanning
confocal microscope (Olympus FV3000; Olympus Corporation) with
excitation/emission wavelengths of 550/570 nm for Cy3
detection.
Neonatal rat cardiac fibroblasts
Isolation and treatment
CFs were isolated from neonatal Sprague-Dawley rats
(1–3 days postnatal) using established enzymatic digestion
protocols. Prior to sacrifice, neonatal pups were anesthetized by
hypothermia, which involved placement on a sterile ice-cold surface
for 3–5 min until unresponsive to a toe pinch, ensuring a surgical
plane of anesthesia (14).
Following confirmation of deep anesthesia, the pups were sacrificed
by rapid decapitation and immediately surface-sterilized through
two sequential 10-sec immersions in 75% ethanol. Ventricular tissue
was dissected free from atria, minced into 1 mm3
fragments and washed twice with Dulbecco's modified Eagle's Medium
(DMEM). Tissue fragments underwent five sequential digestions
(5-min intervals) in an enzymatic solution containing 0.5%
collagenase type I (Worthington Biochemical Corporation) and 0.05%
trypsin (Gibco; Thermo Fisher Scientific, Inc.) and maintained at
37°C in an oscillating water bath. After centrifugation at 300 × g
for 5 min, cell pellets were resuspended in complete culture medium
[DMEM supplemented with 10% fetal bovine serum (FBS;
HyClone™; Cytiva] and incubated under standard culture
conditions (37°C and 5% CO2). CFs were purified through
differential adhesion by discarding cardiomyocyte-containing
supernatant after 2 h of initial plating. Cells from third-passage
cultures were used for experiments following serum starvation (24 h
in DMEM without FBS). For the subsequent treatments, the cells were
divided into four groups: A control group receiving culture media
alone; an LPS group treated with 10 µg/ml LPS for 24 h; an LPS +
carbenoxolone (CBX) group co-treated with 10 µg/ml LPS and 400 µM
CBX (MilliporeSigma), a specific Cx43 gap junction inhibitor; an
LPS + all-trans retinoic acid (ATRA) group co-treated with 10 µg/ml
LPS and 10 µM ATRA (MilliporeSigma), a Cx43 agonist, for 24 h.
RNA preparation and reverse
transcription-quantitative (RT-q) PCR
Total RNA was isolated from LV tissue and cells
using the Total RNA Extraction kit (Tiangen Biotech, Co., Ltd.). RT
was performed using GoScript™ RT System (Promega
Corporation) according to the manufacturer's instructions. RT-qPCR
was carried out using Bestar® SYBR Green qPCR Mastermix
(DBI Biosciences. DBI-2043) according to the manufacturer's
instructions. The primers were designed by Primer software (Premier
5.0; Premier Biosoft International) based on published nucleotide
sequences for rat Cx43 (forward: 5′-GACTTCAGCCTCCAAGGAGTT-3′;
reverse: 5′-ACCCCAAGCTGACTCAACAG3-3′), rat LC3 (forward:
5′-CCCTGCTAACCCCCAATGTT-3′; reverse: 5′-GGGACATGACGACGTACACA′), rat
DNMT1 (forward: 5′-GCTGTTCCTTGTAGGCGAGT-3′; reverse:
5′-GGGGACTCAAACCTTGCGTA-3′), rat DNMT3A (forward:
5′-TGATGACGAGCCCGAGTATG-3′; reverse: 5′-GCCATCTCCGAACCACATGA-3′),
rat DNMT3B (forward: 5′-AATTACACGCAGGACGTGGT-3′; reverse:
5′-ACTGTTGCTGTTTCGGGTTC-3′) and rat β-actin (forward:
5′-CCATTGAACACGGCATTG-3′; reverse: 5′-TACGACCAGAGGCATACA-3′). The
PCR cycling conditions were as follows: initial denaturation at
95°C for 2 min, followed by 40 cycles of denaturation at 95°C for
10 sec, annealing at 60°C for 34 sec, and extension at 72°C for 30
sec. Final melting curve analysis (65–95°C, 0.5°C increments, 5 sec
per increment) was performed to confirm primer specificity. The
relative expression levels of target genes were calculated using
the 2−ΔΔCq method (15), with β-actin serving as the internal
reference gene. All experiments were independently replicated three
times, with each replicate containing six technical replicates.
Western blotting
The LV samples and cells were processed and western
blotting was conducted following the standard procedures. Briefly,
tissue and cells were lysed in RIPA lysis buffer (Beyotime
Institute of Biotechnology. P0013C) for 30 min on ice followed by
centrifugation at 12,000 × g for 15 min at 4°C to collect the
supernatant. The enhanced BCA protein assay kit (Beyotime
Biotechnology. cat. no. P0010) was used to determine the protein
concentration. Equal amounts of protein (30 µg per lane) were
separated by 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) and transferred onto 0.45 µm
polyvinylidene fluoride (PVDF) membranes (Millipore. IPVH00010).
The membranes were blocked with 5% non-fat dry milk (Santa Cruz
Biotechnology. sc-2325) diluted in Tris-buffered saline with
Tween-20 (TBST) at room temperature for 1 h, then incubated
overnight at 4°C with the following primary antibodies: anti-Cx43
(1:1,000; Abcam. ab11370), anti-microtubule-associated protein-1
light-chain (LC3; 1:1,000; Santa Cruz Biotechnology, Inc.
sc-271625), anti-DNMT1 (1:500; Novus Biologicals, LLC; cat. no.
NB100-56519), anti-DNMT3A (1:1,000; Novus Biologicals, LLC.
NB120-13888), anti-DNMT3B (1:1,000; Novus Biologicals, LLC.
NB300-516) and β-actin (1:1,000; Santa Cruz Biotechnology, Inc.,
USA. sc-47778) antibody were used. After three washes with TBST,
the membranes were incubated with horseradish peroxidase
(HRP)-conjugated goat anti-rabbit IgG (1:5,000; Zhongshan Golden
Bridge Bio Co., Ltd. ZB-2301) or HRP-conjugated goat anti-mouse IgG
(1:5,000; Invitrogen. A16084) at room temperature for 1 h. Protein
bands were visualized using an Enhanced Chemiluminescence (ECL)
Detection Kit (Thermo Fisher Scientific, Inc.) and imaged with a
ChemiDoc XRS+ System (Bio-Rad Laboratories, Inc.). Densitometric
analysis of the bands was performed using Image J software (Version
1.50i, NIH) with β-actin as the internal loading control.
Statistical analysis
Data are presented as mean ± standard deviation
(SD). Statistical analyses were performed with GraphPad Prism 9.0.0
software (Dotmatics). The Shapiro-Wilk test was adopted to evaluate
the normality of data distribution, while Levene's test was used to
examine the homogeneity of variance. An unpaired two-tailed
Student's t-test was employed for comparisons between two
independent groups. For multiple group comparisons, one-way ANOVA
was conducted, followed by Dunnett's post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Cardiac impairment in prenatally
LPS-exposed offspring rats
Newborns in the prenatal LPS exposure group
exhibited markedly reduced BW compared with controls at both birth
(P<0.01) and 1 week of age (P<0.01; Fig. 1). However, these LPS-exposed
offspring demonstrated persistent weight gain acceleration from 2
weeks onward (P<0.05 or P<0.01), despite comparable daily
feed intake between groups. Although postnatal catch-up growth can
compensate for intrauterine growth restriction, excessive
compensatory growth is strongly associated with adverse adult
health outcomes (16). The
findings revealed significant cardiac hypertrophy in LPS-exposed
offspring, as evidenced by elevated LVW/BW and HW/BW ratios at both
8 (P<0.05) and 16 weeks (P<0.01). Concurrently, markedly
increased circulating NT-proBNP levels, a ventricular stress
biomarker reflecting volume overload and cardiac dysfunction were
observed in 8-week (P<0.01) and 16-week-old (P<0.01)
LPS-exposed offspring (17). While
serum Ang II levels remained unaffected by prenatal LPS exposure,
substantial increases in LV Ang II concentration were detected at 8
weeks (P<0.05) and 16 weeks (P<0.01). As the primary RAS
agonist, Ang II mediates pressure overload-induced cardiac fibrosis
and hypertrophy.
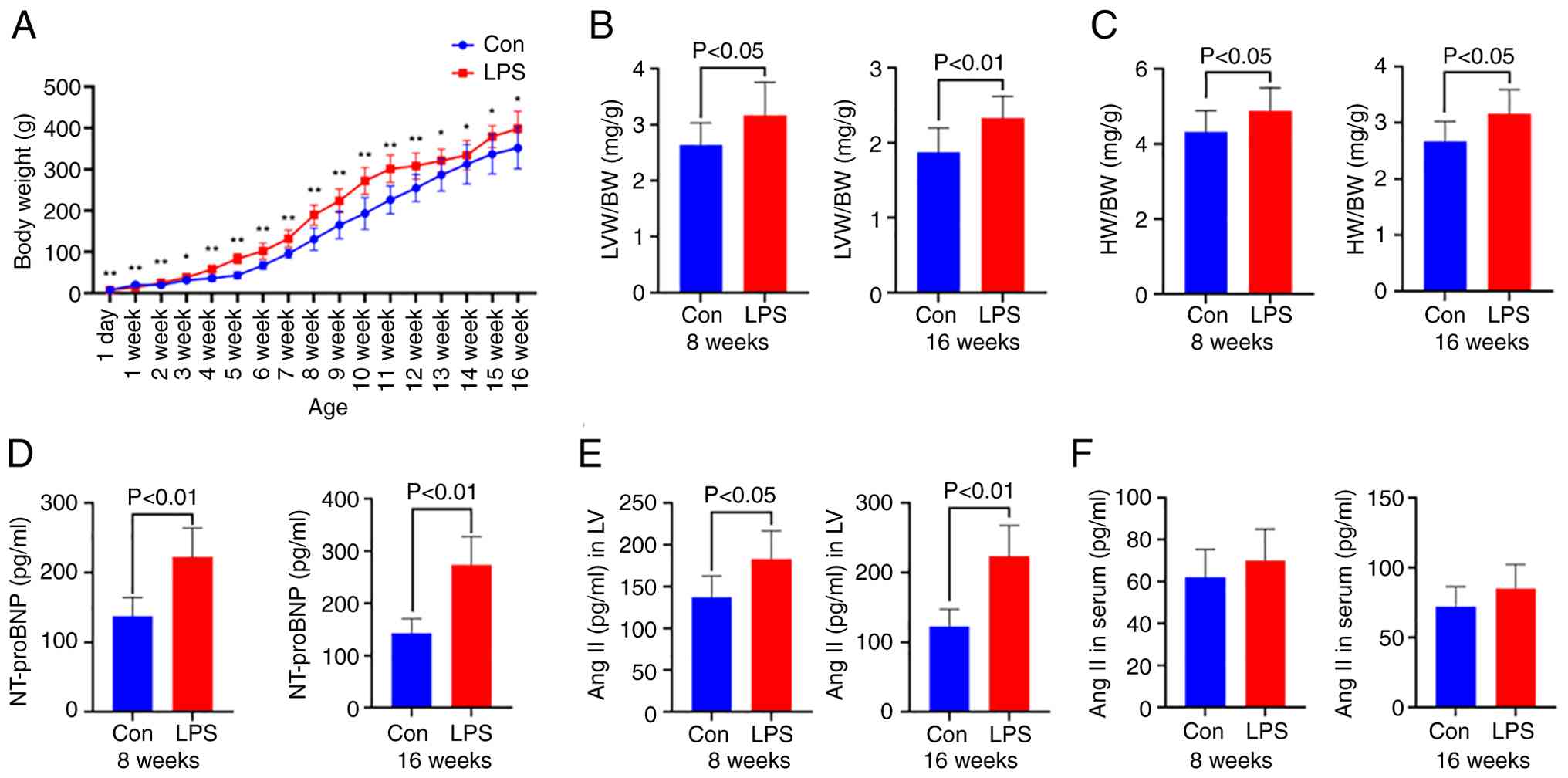 | Figure 1.BW of offspring rats from 1-day to
16-week-old (A). Heart damages in offspring at the age of 8 and 16
weeks, including the ratios (B) LVW/BW, (C) HW/BW and (D) NT-proBNP
level in serum. Concentration of Ang II in (E) left ventricle and
(F) serum. Data are presented as mean ± SD. n=10 in each group
(A-C) and n=7 in each group (D-F). *P<0.05, **P<0.01 vs Con.
BW, body weight; HW, heart weight; LVW, left ventricular weight;
NT-proBNP, N-terminal pro-brain natriuretic peptide; Ang II,
angiotensin II; Con, control; LPS, lipopolysaccharide. |
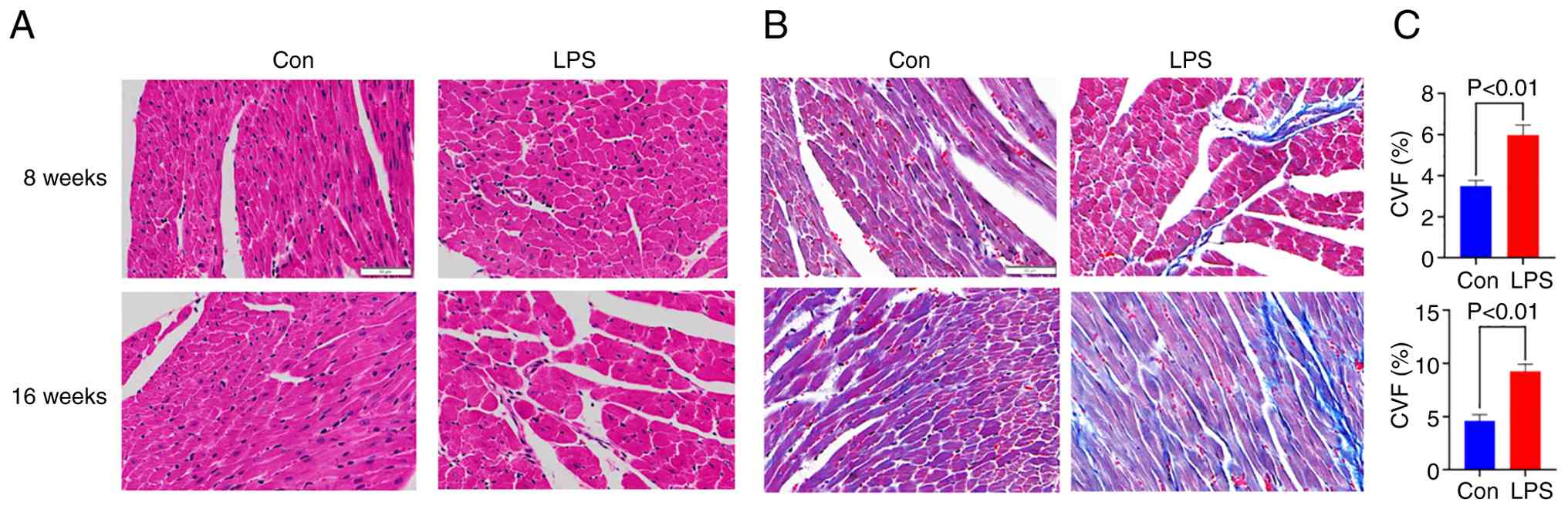
Heart histology alterations in
prenatally LPS-exposed offspring rats
To assess cardiac morphological alterations induced
by prenatal LPS exposure, myocardial architecture was examined
through histological analysis using H&E and Masson staining.
H&E staining revealed distinct pathological progression in
LPS-exposed offspring. Control specimens exhibited preserved
myocardial structure with tightly arranged cardiomyocytes.
Conversely, 8-week-old LPS-exposed offspring demonstrated
characteristic pathological features, including cellular edema,
intercellular space widening, myocardial fiber disruption, focal
necrosis and inflammatory cell infiltration. These pathological
manifestations exhibited age-dependent progression, with
16-week-old LPS-exposed offspring exhibiting exacerbated myocardial
degeneration characterized by extensive tissue damage and
pronounced inflammatory infiltration (Fig. 2A). Masson staining revealed
significant extracellular matrix remodeling in LPS-exposed groups.
Collagen deposition (blue-stained fibers) within the LV
interstitium was markedly increased compared with age-matched
controls, with 8-week-old LPS-exposed offspring showing initial
fibrosis that progressed substantially by 16 weeks of age. CVF
quantitative analysis confirmed these observations, demonstrating
statistically significant increases in the LPS-exposed groups at 8
weeks (P<0.01) and 16 weeks (P<0.01) compared with the
controls (Fig. 2B).
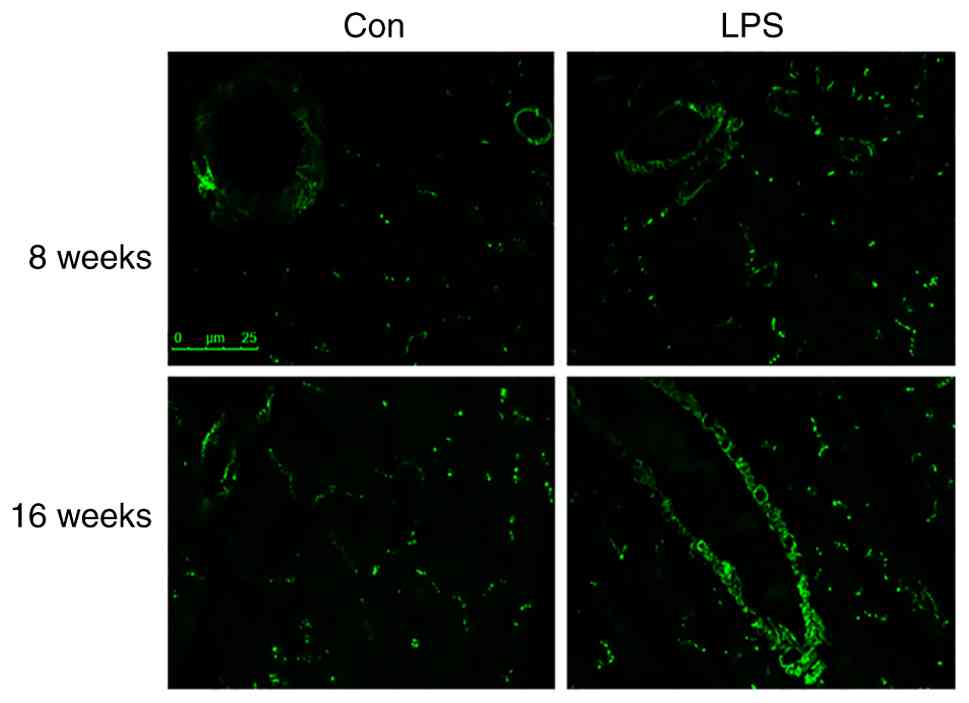
α-SMA immunolabeling in LV
samples
Complementing the histological findings,
immunofluorescence staining was used to study CF differentiation
into α-SMA-expressing myofibroblasts, a hallmark feature of
fibrotic progression. Prenatal LPS exposure markedly activated this
pathogenic transformation in the offspring myocardium. Compared
with controls, 8-week-old LPS-exposed offspring exhibited a
pronounced increase in α-SMA-positive cells in the LV, as evidenced
by the elevated fluorescent signal intensity and cluster density.
By 16 weeks of age, this activation progressed further, with
LPS-exposed offspring demonstrating expansive α-SMA-positive bands
and intensified fluorescence, indicative of advanced myofibroblast
aggregation and sustained fibrosis (Fig. 3).
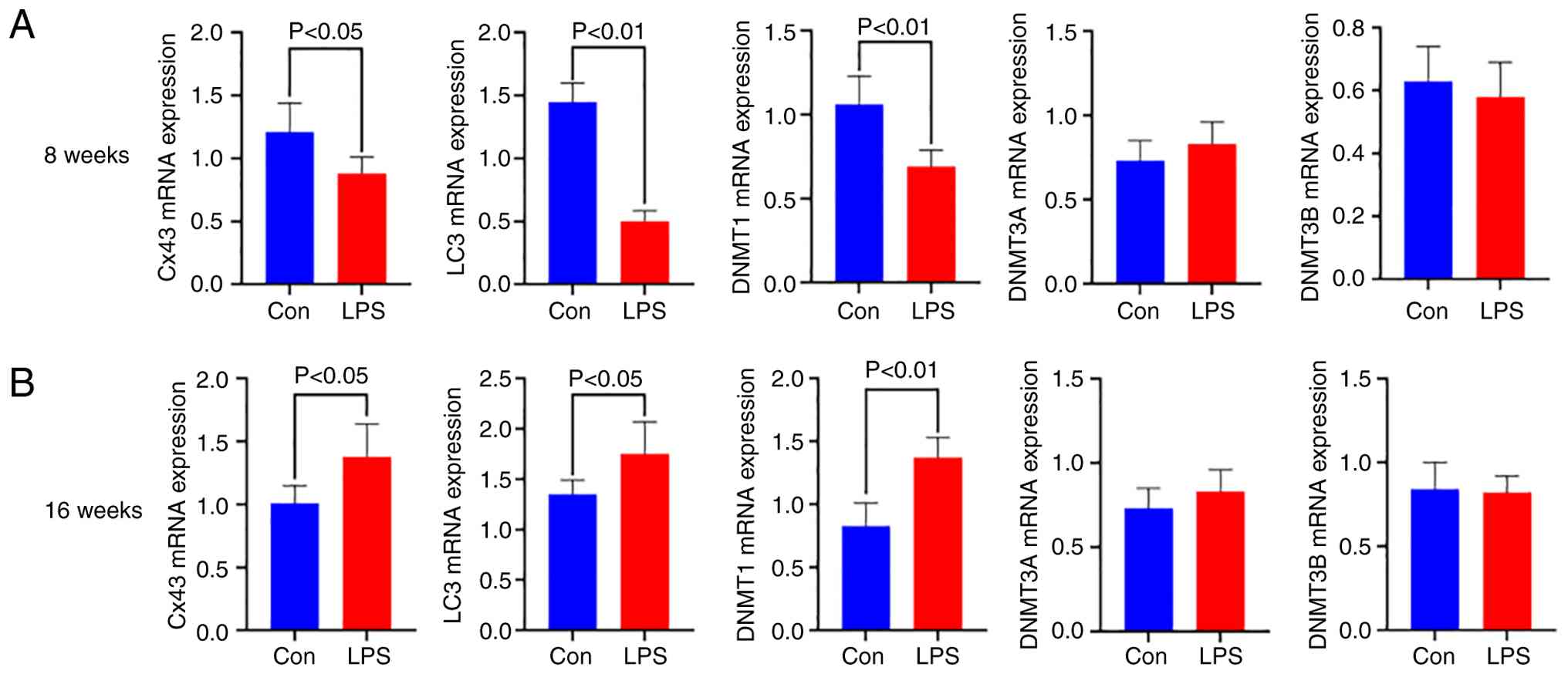
Cx43, LC3, DNMT1, DNMT3A and DNMT3B
mRNA expressions in LV samples
RT-qPCR analysis revealed distinct age-dependent
transcriptional alterations in the LPS-exposed offspring. Compared
with controls, 8-week-old offspring with prenatal LPS exposure
exhibited significant downregulation of Cx43 (P<0.05),
LC3 (P<0.01) and DNMT1 (P<0.01). However, these
trends were reversed at 16 weeks of age, with elevated mRNA levels
of Cx43 (P<0.05), LC3 (P<0.05) and DNMT1
(P<0.01). Conversely, non-significant intergroup differences
were observed in DNMT3A (P>0.05) or DNMT3B
(P>0.05) expression at either developmental stage, suggesting
isoform-specific regulatory mechanisms in DNA methylation (Fig. 4).
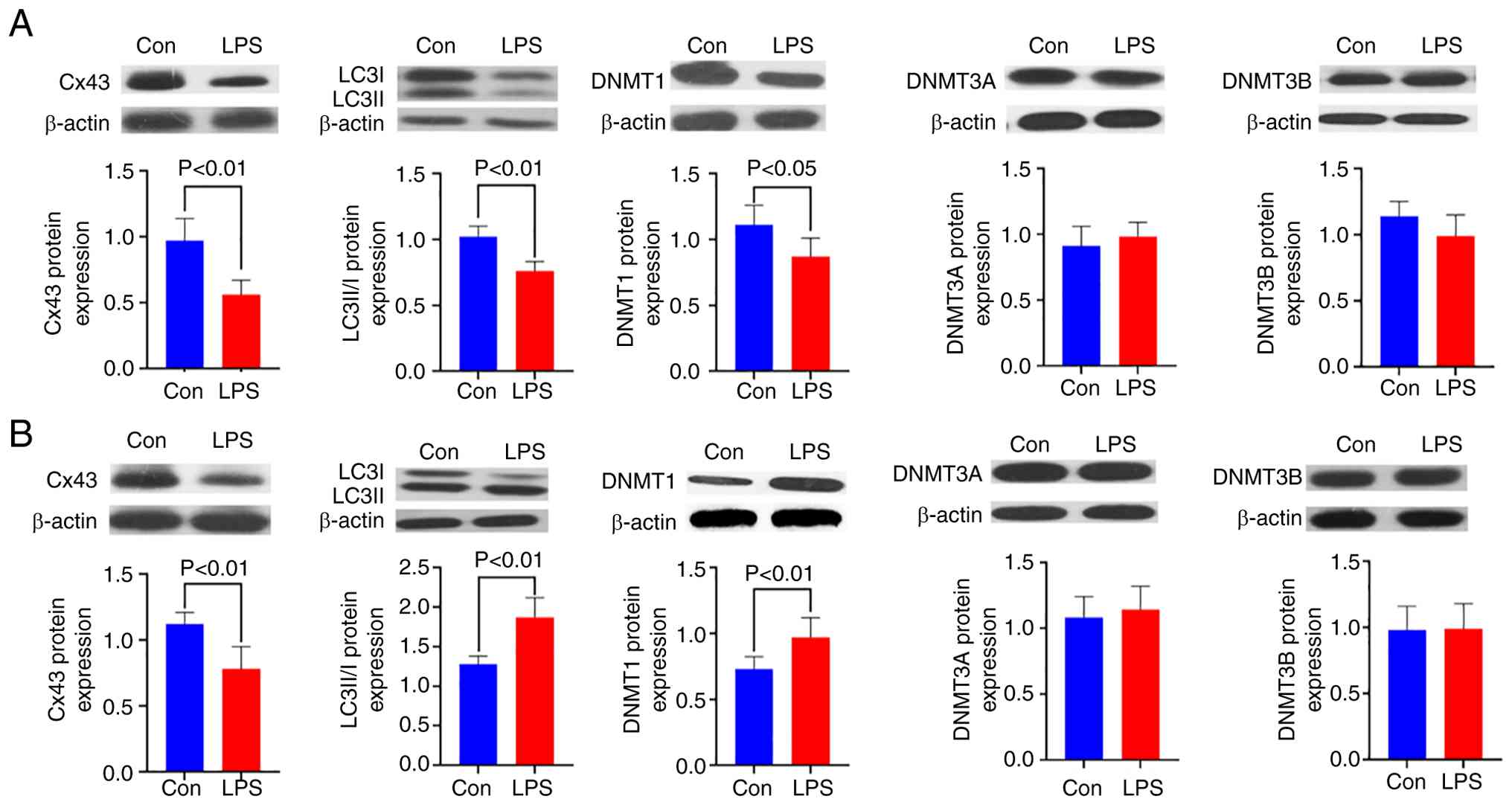
Cx43, LC3, DNMT1, DNMT3A and DNMT3B
Protein expressions in LV samples
Gap junctions, specialized plasma membrane domains
composed of intercellular channel arrays, are vital for electrical
and metabolic coupling between adjacent cardiomyocytes. Cx43 is the
predominant gap junction protein in the ventricular myocardium. The
present study revealed significant Cx43 downregulation in offspring
exposed to maternal LPS compared with controls, with pronounced
reductions observed at 8 weeks (P<0.01) and 16 weeks of age
(P<0.01). This persistent Cx43 depletion suggests sustained gap
junction impairment in the LV of prenatal LPS-exposed offspring.
The present study further demonstrated dynamic alterations in
cardiac autophagy. LC3 is a pivotal autophagy marker that exists in
two isoforms: cytoplasmic LC3-I and autophagosome-associated
LC3-II. At 8 weeks, prenatal LPS exposure resulted in significant
suppression of autophagy activity as evidenced by a markedly
decreased LC3II/I ratio (P<0.01) compared with controls.
Conversely, this ratio was substantially elevated in 16-week-old
LPS-exposed offspring (P<0.01), indicating the paradoxical
activation of autophagy pathways at later developmental stages.
Western blotting of LV tissues revealed a significant reduction in
DNMT1 expression at 8 weeks (P<0.05), followed by pronounced
upregulation at 16 weeks (P<0.01) compared with age-matched
controls. Conversely, neither DNMT3A (P>0.05) nor DNMT3B
(P>0.05) exhibited statistically significant differences between
the experimental groups at either time point. Notably, while Cx43
exhibited discordance between protein and mRNA expression patterns,
LC3 and DNMT1 demonstrated that the protein expression trends
closely mirrored their respective mRNA levels (Fig. 5).
mRNA and protein expression of Cx43,
LC3 and DNMT1 in rat primary cardiac fibroblasts
LPS stimulation was applied to rat primary CFs to
establish a pathological model mimicking the in vivo
conditions observed in prenatal LPS-exposed offspring. Following 24
h LPS stimulation, both mRNA and protein levels of Cx43 and LC3
markedly downregulated (P<0.01), whereas DNMT1 exhibited a
pronounced up-regulation at the transcriptional and translational
levels (P<0.05) compared with untreated controls. This molecular
profile resembled the pathological changes observed in prenatal
LPS-exposed offspring aged 8–16 weeks, establishing an in
vitro model that recapitulates the key molecular features of
developmental cardiac remodeling. To investigate Cx43-dependent
mechanisms, Cx43 was pharmacologically modulated using CBX as an
inhibitor and ATRA as an agonist. CBX treatment effectively
suppressed Cx43 expression (P<0.05) and further reduced LC3
levels at the mRNA (P<0.01) and protein (P<0.05) levels,
while also markedly attenuating DNMT1 expression (P<0.05 or
0.01) compared with LPS treatment alone. Cotreatment with LPS and
ATRA increased both mRNA (P<0.05) and protein (P<0.05)
expression of Cx43 compared with the LPS-stimulated group alone,
subsequently restoring the protein and mRNA expression levels of
DNMT1 and LC3 (P<0.05 or 0.01). These results demonstrated that
Cx43 regulates autophagy-related LC3 dynamics and DNMT1-mediated
epigenetic modifications during fibrotic progression, reinforcing
its position as an upstream node in this regulatory network
(Fig. 6).
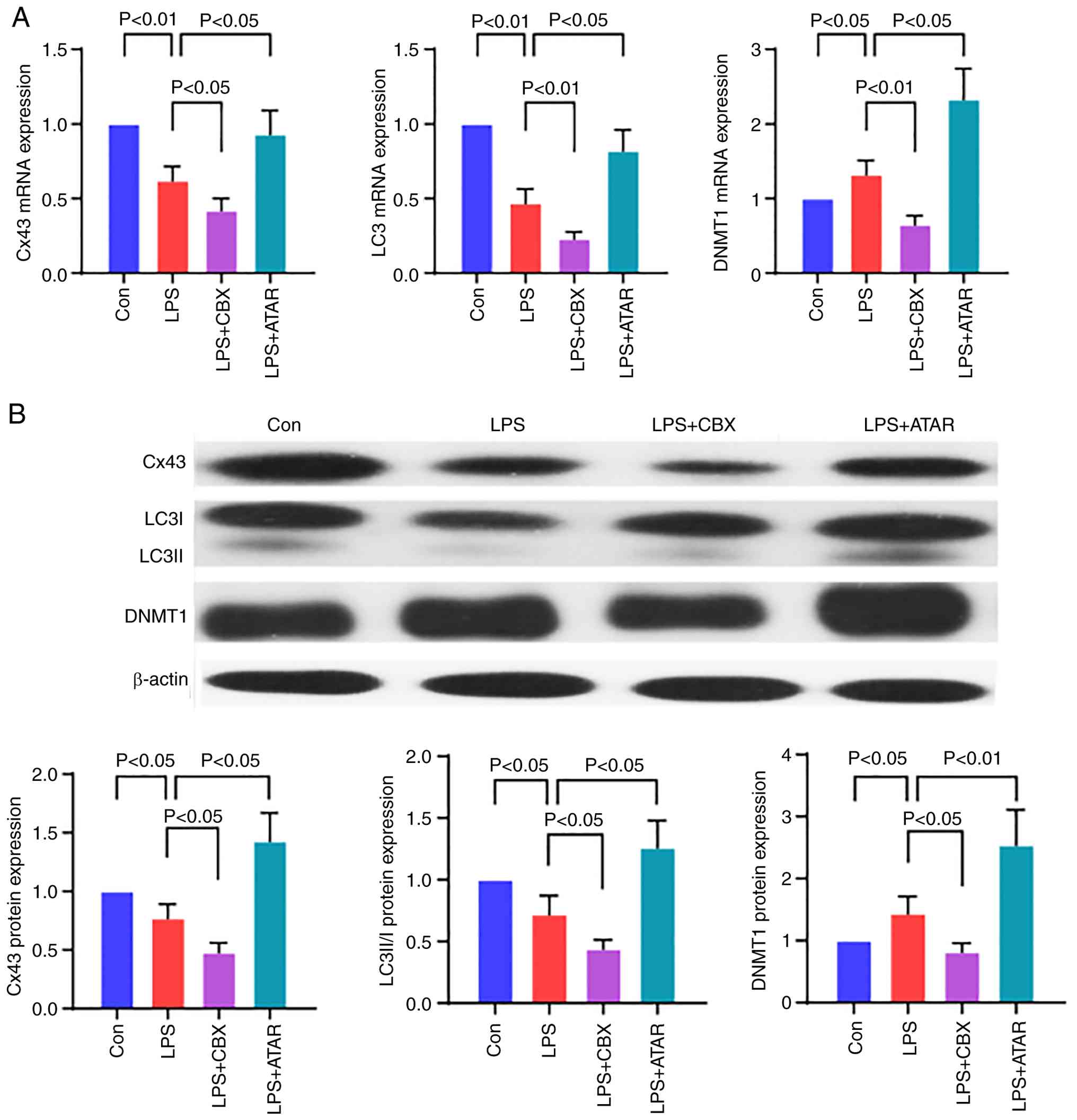 | Figure 6.Effect of Cx43 on the expression of
LC3 and DNMT1 in vitro. (A) mRNA and (B) protein expression
of Cx43, LC3 and DNMT1 in rat primary cardiac fibroblasts. Data are
presented as mean ± SD. n=3/group. Control group, rat primary
cardiac fibroblasts treated with culture media; LPS group, rat
primary cardiac fibroblasts treated with culture media containing
10 µg/ml LPS; LPS + CBX group, rat primary cardiac fibroblasts
treated with culture media containing 10 µg/ml LPS and 400 µM CBX;
LPS + ATRA group, rat primary cardiac fibroblasts treated with
culture media containing 10 µg/ml LPS and 10 µM all-trans retinoic
acid. Cx43, connexin 43; DNMT, DNA methyltransferase; LPS,
lipopolysaccharide; CBX, carbenoxolone; ATRA, all-trans retinoic
acid; Con, control; LPS, lipopolysaccharide. |
Discussion
The present study highlighted that maternal LPS
exposure during pregnancy may induce a cardiac fibrotic shift.
Notably, its key novel finding revealed age-dependent progressive
fibrotic alterations in the hearts of prenatally LPS-exposed
offspring rats, concomitant with reduced Cx43 expression. This
extends our previous observations (3–5,18).
Cx43, the predominant membrane protein forming ventricular GJs, is
essential for maintaining cardiac electrophysiological homeostasis.
Substantial evidence suggests that reduced Cx43 expression can
trigger excessive collagen deposition in aged and pathologically
remodeled hearts (7). Notably,
pharmacological interventions targeting fibrotic pathways,
including renin-angiotensin-aldosterone system inhibitors or TGF-β
receptor blockers, ameliorate cardiac fibrosis while restoring Cx43
expression. Similarly, GJ modifiers that upregulate Cx43 exhibit
anti-fibrotic effects, even in the contexts of TGF-β pathway
activation or pre-existing cardiovascular pathologies (19,20).
Although the mechanistic association between Cx43 dysregulation and
fibrosis remains incompletely understood, emerging studies have
proposed that Cx43 deficiency may disrupt cardiomyocyte-fibroblast
and fibroblast-fibroblast coupling, thereby promoting fibroblast
activation, proinflammatory responses and collagen overproduction
(7). Mechanistically, Cx43
downregulation via hemichannel blockade or autophagy-dependent
degradation activates the MAPK/ERK pathway, which synergizes with
Smad signaling to enhance collagen transcription and stabilize
Col1a1 mRNA (21). Our prior study
demonstrated persistent MAPK activation in the heart of LPS-exposed
offspring rats, particularly when challenged with secondary
stressors in adulthood (3).
Furthermore, Cx43 facilitates microtubule-dependent trafficking of
voltage-gated sodium channels (Nav1.5) to intercalated discs,
suggesting its critical role in regulating Nav1.5 membrane
localization and function. Therefore, Cx43 downregulation may
impair Nav1.5 distribution, contributing to fibrosis via
channel-independent mechanisms (22). Consistent with these findings, the
present study revealed that Cx43 mediated GJ disruption and
elevated α-SMA expression in the cardiac tissues of prenatally
LPS-exposed offspring. Collectively, the present study proposed a
novel paradigm in which intrauterine Cx43 suppression orchestrates
ventricular fibrosis via channel-dependent and independent
pathways, providing new insights into fibrotic pathogenesis. A key
finding of the present study is that maternal LPS exposure induced
localized, not systemic, RAS activation in the offspring heart,
which is in line with previous observations (18,23).
This localized activation is key, as Ang-II, the primary effector
peptide of the RAS-has been demonstrated in a rat isolated perfused
beating atrial model to directly downregulate Cx43 expression and
to induce atrial fibrosis (24,25).
Our previous studies further demonstrated that maternal LPS
exposure during gestation induces offspring cardiac abnormalities,
including localized inflammation, RAS activation, TGF-β
upregulation and hemodynamic overload, all of which are potential
contributors to Cx43 downregulation and GJ impairment (4,26).
Consequently, Cx43 reduction is a consequence of adverse fetal
programming and an active regulator of fibrotic progression.
Notably, while prenatal LPS exposure induced
upregulated Cx43 mRNA expression in 16-week-old offspring rats, its
protein levels were paradoxically decreased, suggesting
post-translational Cx43 degradation. This observation aligns with
the established role of autophagy in mediating protein turnover,
which is particularly prominent under pathological stress (10). Consistent with this, there was a
markedly elevated LC3-II/I ratio, a biomarker of autophagic flux,
in 16-week-old LPS-exposed offspring, indicating that Cx43
downregulation may result from hyperactivated autophagy. In
contrast to the 16-week cohort, 8-week-old LPS-exposed offspring
exhibited suppressed LC3-II/I ratios. This temporal divergence
suggests that autophagy may exert pleiotropic effects beyond Cx43
degradation in the present model. Functionally, autophagy is a
lysosome-dependent quality control system that eliminates damaged
cytoplasmic components, thereby maintaining homeostasis.
Dysregulation of this process, whether insufficient or excessive,
disrupts the myocardial protein/organelle balance, with autophagy
deficiency leading to toxic aggregate accumulation and
hyperactivation resulting in destructive self-digestion.
The relationship between autophagy and cardiac
fibrosis is still unclear. For instance, transverse aortic
constriction in mice for 8 weeks increased LV autophagy
concomitantly with fibrosis progression (27). Similarly, renovascular hypertension
induced myocardial autophagy specifically in domestic pigs with
moderate but not mild hypertension and autophagic activity
associated positively with fibrosis severity (28). Curcumin inhibited autophagy and
attenuated isoproterenol-induced fibrosis in rodent models
(9). However, endothelial-specific
autophagy suppression, either by a specific inhibitor or siRNA for
ATG5, exacerbates fibrotic responses in vitro and in
vivo (29). These apparent
contradictions likely stem from context-dependent variables,
including disease etiology, stage and intervention timing. In the
present experimental paradigm, prenatal inflammation primes
offspring rats for autophagy-induced cardiac fibrosis. Notably,
besides direct profibrotic effects, autophagy may indirectly
perpetuate fibrosis via Cx43 dysregulation, thereby establishing a
self-reinforcing loop.
The present study revealed dynamic changes in DNMT1
expression patterns in offspring rats exposed to prenatal LPS. The
mRNA and protein levels of DNMT1 in cardiac tissue exhibited an
initial decline at 8 weeks of age, followed by a rebound at 16
weeks. Conversely, DNMT3A and DNMT3B expression remained stable
across these developmental stages. These temporal variations in
DNMT1 activity may be key for cardiac fibrosis progression, given
that CpG hypomethylation is implicated in fibrogenic genes
transcriptional activation. This aligns with previous findings
demonstrating that DNMT1 suppression in TGF-β-stimulated cardiac
fibroblasts promotes α-SMA overexpression (30). Decreased DNMT1 levels enhance TGF-β
receptor I expression, exacerbating fibroblast differentiation and
fibrogenesis (30,31). Conversely, anti-fibrotic genes
hypermethylation mediated by DNMT3B upregulation, as observed in
SUN2 gene silencing during hepatic fibrosis (32), highlights the complex epigenetic
regulation of fibrosis. Notably, the concurrent dysregulation of
α-SMA, TGF-β and DNMT1 in the present model suggested a potential
mechanistic link to Cx43 modulation.
The present study observed discordant temporal
patterns between autophagy activity and DNA methylation dynamics
during age-dependent fibrosis progression. This paradoxical
relationship may reflect compensatory mechanisms that preserve
cardiac homeostasis in younger offspring, consistent with prior
reports that cardiac dysfunction in this model only manifests at 8
months of age (4). The present
findings further implied both impaired autophagy and aberrant
methylation patterns in Cx43 downregulation, highlighting their
synergistic roles in modulating cellular responses during fibrotic
remodeling. Emerging evidence positions Cx43 as a molecular nexus
integrating diverse signaling pathways, suggesting that its
interaction with epigenetic and autophagic regulators may operates
through interconnected networks rather than isolated pathways
(33). To clarify these
interactions, the present study used an in vitro LPS-treated
cardiac fibroblast model with pharmacological interventions using a
Cx43 inhibitor (CBX) and a Cx43 agonist (ATRA) (34,35),
providing direct evidence of the central regulatory role of Cx43 in
coordinating DNMT1 expression and autophagic activity. Cx43
inhibition blunted the LPS-triggered rise in DNMT1 but deepened the
suppression of LC3, whereas its activation rescued the expression
of both molecules. This dual effect aligns with the two established
mechanisms of Cx43 function. First, by serving as an ATP-release
channel that activates P2X7 purinergic receptors, Cx43 promotes
autophagy induction through multiple signaling cascades, a process
mechanistically associated with pressure overload-induced cardiac
hypertrophy (36–38). Second, the Cx43 downregulation
observed following prenatal inflammation engages MAPK signaling
pathways, considering that pharmacological inhibition of MAPK
restores DNMT1 expression in hypoxic cardiac progenitor cells
(3,39). Collectively, these bidirectional
manipulations confirm that Cx43 functions as an upstream modulator,
whose activity state dynamically influences DNMT1-mediated
epigenetic and autophagic responses in cardiac fibroblasts under
inflammatory stress.
Furthermore, the observed cardiac DNMT1 biphasic,
age-dependent regulation in vivo, characterized by
suppression at 8 weeks followed by upregulation at 16 weeks,
contrasts with its acute upregulation in LPS-stimulated cardiac
fibroblasts in vitro. This apparent discrepancy likely
originates from the fundamental differences in the experimental
context, timeframe and biological complexity between the two
models, which is a recognized challenge in translating in
vitro findings to developmental programming phenotypes
(40). In vivo findings
reflect a chronic developmental programming process initiated by
transient prenatal LPS exposure (41). Early DNMT1 downregulation may
represent an initial, maladaptive, or compensatory response in the
complex cardiac milieu, potentially facilitating the early
expression of fibrogenic genes. Its subsequent rebound at 16 weeks
could help stabilize the fibrotic phenotype or silencing protective
pathways during later-stage remodeling. Conversely, the in
vitro model captures the acute cellular response of isolated
fibroblasts to direct and sustained LPS stimulation, which mimics
immediate profibrotic activation and is marked by rapid DNMT1
induction (42). While this model
recapitulates the key molecular features of fibroblast activation,
it does not include the longer-term temporal evolution or
multicellular interactions inherent to the in vivo
programmed phenotype. Therefore, these two models are
complementary. The in vitro system elucidates the acute
cell-autonomous response, whereas the in vivo model reveals
the integrated stage-specific epigenetic dynamics within the
programmed heart.
The present study focused on elucidating the
consequences of maternal LPS exposure during gestation on offspring
cardiac fibrosis, revealing a significant association with aberrant
Cx43 expression patterns. While its findings established a
functional regulatory role for Cx43 activity, the precise
mechanistic relationships underlying its interaction with autophagy
and DNMT1-mediated epigenetic modifications warrant further
investigation. The complex regulatory landscape of Cx43, including
its hemichannel activity, phosphorylation status and subcellular
localization, must be systematically dissected to understand how
these specific facets differentially influence the fibrotic process
under inflammatory stress. Future studies using genetic tools and
pathway-specific inhibitors are crucial to map the exact downstream
signaling cascades (for example, MAPK/ERK) that transduce Cx43
activity into alterations in autophagic flux and DNA methylation
patterns.
Taken together, the data of the present study
integrated with existing literature propose that Cx43 deficiency
may serve as a molecular nexus connecting prenatal inflammatory
insults to the developmental programming of cardiac fibrosis. This
regulatory axis orchestrates a dynamic interplay between autophagic
flux and DNA methylation machinery, potentially establishing
persistent epigenetic imprints during cardiogenesis. These insights
advance a novel paradigm for understanding fibrosis pathogenesis,
in which early-life inflammatory challenges may prime the cardiac
epigenome via Cx43-mediated pathways. Consequently, targeted
interventions aimed at preserving Cx43 homeostasis during gestation
could emerge as preventive strategies against adult-onset
cardiovascular diseases by disrupting this developmental
programming cascade.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the National
Natural Science Foundation of China (grant no. 81573440) and the
Natural Science Foundation of Chongqing, China (grant no.
cstc2019jcyj-msxmX0609).
Availability of data and materials
The data and materials in the current study are
available from the corresponding author on reasonable request.
Authors' contributions
YL, YW and HGZ conceived and designed the research.
YW performed experiments. YW and YY analyzed data. YL, YW and YY
interpreted results of experiments. YL acquired funding. YW, YL and
HGZ confirm the authenticity of all the raw data. YW and YY
prepared figures. YW, YL and HGZ drafted the manuscript and YL
edited and revised the manuscript. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
The present study complied with the Guide for the
Care and Use of Laboratory Animals published by the US National
Institutes of Health (NIH Publication N.85-23, revised 1996;
http://www.nap.edu/readingroom/books/labrats/index.html)
and received ethical approval from the Army Medical University
Animal Care Committee (approval no. AMUWEC2020980).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Frangogiannis NG: Cardiac fibrosis.
Cardiovasc Res. 117:1450–1488. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Barker DJP: The origins of the
developmental origins theory. J Intern Med. 261:412–417. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang Q, Deng Y, Lai W, Guan X, Sun X, Han
Q, Wang F, Pan X, Ji Y, Luo H, et al: Maternal inflammation
activated ROS-p38 MAPK predisposes offspring to heart damages
caused by isoproterenol via augmenting ROS generation. Sci Rep.
6:301462016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wei Y, Du W, Xiong X, He X, Yi P, Deng Y,
Chen D and Li X: Prenatal exposure to lipopolysaccharide results in
myocardial remodelling in adult murine offspring. J Inflamm (Lond).
10:352013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cao D, Liu Y, Chen X, Liu J, Liu J, Lai W,
Li S, Wang W, Zhang W, Xiao D, et al: Activation of iNKT cells at
the maternal-fetal interface predisposes offspring to cardiac
injury. Circulation. 145:1032–1035. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Trovato-Salinaro A, Trovato-Salinaro E,
Failla M, Mastruzzo C, Tomaselli V, Gili E, Crimi N, Condorelli DF
and Vancheri C: Altered intercellular communication in lung
fibroblast cultures from patients with idiopathic pulmonary
fibrosis. Respir Res. 7:1222006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jansen JA, van Veen TAB, de Jong S, van
der Nagel R, van Stuijvenberg L, Driessen H, Labzowski R, Oefner
CM, Bosch AA, Nguyen TQ, et al: Reduced Cx43 expression triggers
increased fibrosis due to enhanced fibroblast activity. Circ
Arrhythm Electrophysiol. 5:380–390. 2012. View Article : Google Scholar
|
|
8
|
Cao L, Chen Y, Lu L, Liu Y, Wang Y, Fan J
and Yin Y: Angiotensin II upregulates fibroblast-myofibroblast
transition through Cx43-dependent CaMKII and TGF-β1 signaling in
neonatal rat cardiac fibroblasts. Acta Biochim Biophys Sin
(Shanghai). 50:843–852. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu R, Zhang HB, Yang J, Wang JR, Liu JX
and Lim CL: Curcumin alleviates isoproterenol-induced cardiac
hypertrophy and fibrosis through inhibition of autophagy and
activation of mTOR. Eur Rev Med Pharmacol Sci. 22:7500–7508.
2018.
|
|
10
|
Martins-Marques T, Catarino S, Zuzarte M,
Marques C, Matafome P, Pereira P and Girão H: Ischaemia-induced
autophagy leads to degradation of gap junction protein connexin43
in cardiomyocytes. Biochem J. 467:231–245. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang J, Cui J, Chen R, Deng Y, Liao X, Wei
Y, Li X, Su M, Yu J and Yi P: Prenatal exposure to
lipopolysaccharide alters renal DNA methyltransferase expression in
rat offspring. PLoS One. 12:e01692062017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Guan X, Dan GR, Yang Y, Ji Y, Lai WJ, Wang
FJ, Meng M, Mo BH, Huang P, You TT, et al: Prenatal inflammation
exposure-programmed hypertension exhibits multi-generational
inheritance via disrupting DNA methylome. Acta Pharmacol Sin.
43:1419–1429. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mohamed AS, Hosney M, Bassiony H,
Hassanein SS, Soliman AM, Fahmy SR and Gaafar K: Sodium
pentobarbital dosages for exsanguination affect biochemical,
molecular and histological measurements in rats. Sci Rep.
10:3782020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fish RE, Brown MJ, Danneman PJ and Karas
AZ: Anesthesia and analgesia in laboratory animals. 2nd edition.
London: Elsevier; 2008
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kelishadi R, Haghdoost AA, Jamshidi F,
Aliramezany M and Moosazadeh M: Low birthweight or rapid catch-up
growth: which is more associated with cardiovascular disease and
its risk factors in later life? A systematic review and
cryptanalysis. Paediatr Int Child Health. 35:110–123. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cao Z, Jia Y and Zhu B: BNP and NT-proBNP
as diagnostic biomarkers for cardiac dysfunction in both clinical
and forensic medicine. Int J Mol Sci. 20:18202019. View Article : Google Scholar
|
|
18
|
Hao XQ, Zhang HG, Yuan ZB, Yang DL, Hao LY
and Li XH: Prenatal exposure to lipopolysaccharide alters the
intrarenal renin-angiotensin system and renal damage in offspring
rats. Hypertens Res. 33:76–82. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Patin J, Castro C, Steenman M, Hivonnait
A, Carcouët A, Tessier A, Lebreton J, Bihouée A, Donnart A, Le
Marec H, et al: Gap-134, a Connexin43 activator, prevents
age-related development of ventricular fibrosis in
Scn5a+/− mice. Pharmacol Res. 159:1049222020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Stein M, Boulaksil M, Jansen JA, Herold E,
Noorman M, Joles JA, van Veen TAB, Houtman MJC, Engelen MA, Hauer
RNW, et al: Reduction of fibrosis-related arrhythmias by chronic
renin-angiotensin-aldosterone system inhibitors in an aged mouse
model. Am J Physiol Heart Circ Physiol. 299:H310–H321. 2010.
View Article : Google Scholar
|
|
21
|
Wu L, Wang Z, He X, Jiang Y, Pan R, Chen
S, Chen Y, Han Y, Yu H and Zhang T: GJA1 reverses arsenic-induced
EMT via modulating MAPK/ERK signaling pathway. Toxicol Appl
Pharmacol. 450:1161382022. View Article : Google Scholar
|
|
22
|
Jansen JA, Noorman M, Musa H, Stein M, de
Jong S, van der Nagel R, Hund TJ, Mohler PJ, Vos MA, van Veen TA,
et al: Reduced heterogeneous expression of Cx43 results in
decreased Nav1.5 expression and reduced sodium current that
accounts for arrhythmia vulnerability in conditional Cx43 knockout
mice. Heart Rhythm. 9:600–607. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gao M, Zhang X, Chen X, Mi C, Tang Y, Zhou
J and Li X: Prenatal exposure to lipopolysaccharide results in
local RAS activation in the adipose tissue of rat offspring. PLoS
One. 9:e1113762014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ding DZ, Jia YN, Zhang B, Guan CM, Zhou S,
Li X and Cui X: C-type natriuretic peptide prevents angiotensin
II-induced atrial connexin 40 and 43 dysregulation by activating
AMP-activated kinase signaling. Mol Med Rep. 20:5091–5099.
2019.PubMed/NCBI
|
|
25
|
Li X, Cui X, Zhou S, Xing DL, Piao HR,
Zhang QG, Zhao YQ and Liu LP: The novel ginsenoside AD2 prevents
angiotensin II-induced connexin 40 and connexin 43 dysregulation by
activating AMP kinase signaling in perfused beating rat atria. Chem
Biol Interact. 339:1094302021. View Article : Google Scholar
|
|
26
|
Chen X, Tang Y, Gao M, Qin S, Zhou J and
Li X: Prenatal exposure to lipopolysaccharide results in myocardial
fibrosis in rat offspring. Int J Mol Sci. 16:10986–10996. 2015.
View Article : Google Scholar
|
|
27
|
Zhang Y, Wang Z, Lan D, Zhao J, Wang L,
Shao X, Wang D, Wu K, Sun M, Huang X, et al: MicroRNA-24-3p
alleviates cardiac fibrosis by suppressing cardiac fibroblasts
mitophagy via downregulating PHB2. Pharmacol Res. 177:1061242022.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang X, Gibson ME, Li ZL, Zhu XY, Jordan
KL, Lerman A and Lerman LO: Autophagy portends the level of cardiac
hypertrophy in experimental hypertensive swine model. Am J
Hypertens. 29:81–89. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pan JA, Zhang H, Lin H, Gao L, Zhang HL,
Zhang JF, Wang CQ and Gu J: Irisin ameliorates doxorubicin-induced
cardiac perivascular fibrosis through inhibiting
endothelial-to-mesenchymal transition by regulating ROS
accumulation and autophagy disorder in endothelial cells. Redox
Biol. 46:1021202021. View Article : Google Scholar
|
|
30
|
He Y, Ling S, Sun Y, Sheng Z, Chen Z, Pan
X and Ma G: DNA methylation regulates α-smooth muscle actin
expression during cardiac fibroblast differentiation. J Cell
Physiol. 234:7174–7185. 2019. View Article : Google Scholar
|
|
31
|
Fu S, Sun L, Zhang X, Shi H, Xu K, Xiao Y
and Ye W: 5-Aza-2′-deoxycytidine induces human Tenon's capsule
fibroblasts differentiation and fibrosis by up-regulating TGF-β
type I receptor. Exp Eye Res. 165:47–58. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen X, Li WX, Chen Y, Li XF, Li HD, Huang
HM, Bu FT, Pan XY, Yang Y, Huang C, et al: Suppression of SUN2 by
DNA methylation is associated with HSCs activation and hepatic
fibrosis. Cell Death Dis. 9:10212018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fournier S, Clarhaut J, Cronier L and
Monvoisin A: GJA1-20k, a short isoform of Connexin43, from its
discovery to its potential implication in cancer progression.
Cells. 14:1802025. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Miura M, Nagano T, Murai N, Taguchi Y,
Handoh T, Satoh M, Miyata S, Miller L, Shindoh C and Stuyvers BD:
Effect of carbenoxolone on arrhythmogenesis in rat ventricular
muscle. Circ J. 80:76–84. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu Y, Wen Q, Chen XL, Yang SJ, Gao L, Gao
L, Zhang C, Li JL, Xiang XX, Wan K, et al: All-trans retinoic acid
arrests cell cycle in leukemic bone marrow stromal cells by
increasing intercellular communication through connexin 43-mediated
gap junction. J Hematol Oncol. 8:1102015. View Article : Google Scholar
|
|
36
|
Sun L, Gao J, Zhao M, Cui J, Li Y, Yang X,
Jing X and Wu Z: A novel cognitive impairment mechanism that
astrocytic p-connexin 43 promotes neuronic autophagy via activation
of P2X7R and down-regulation of GLT-1 expression in the hippocampus
following traumatic brain injury in rats. Behav Brain Res.
291:315–324. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Orioli E, De Marchi E, Giuliani AL and
Adinolfi E: P2X7 receptor orchestrates multiple signalling pathways
triggering inflammation, autophagy and metabolic/trophic responses.
Curr Med Chem. 24:2261–2275. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Higashikuni Y, Liu W, Numata G, Tanaka K,
Fukuda D, Tanaka Y, Hirata Y, Imamura T, Takimoto E, Komuro I and
Sata M: NLRP3 inflammasome activation through heart-brain
interaction initiates cardiac inflammation and hypertrophy during
pressure overload. Circulation. 147:338–355. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Su J, Fang M, Tian B, Luo J, Jin C, Wang
X, Ning Z and Li X: Hypoxia induces hypomethylation of the HMGB1
promoter via the MAPK/DNMT1/HMGB1 pathway in cardiac progenitor
cells. Acta Biochim Biophys Sin (Shanghai). 50:1121–1130. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lynch F, Lewis S, Macciocca I and Craig
JM: Epigenetics and DOHaD: How translation to predictive testing
will require a better public understanding. J Dev Orig Health Dis.
13:424–430. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Deng Y, Song L, Nie X, Shou W and Li X:
Prenatal inflammation exposure-programmed cardiovascular diseases
and potential prevention. Pharmacol Ther. 190:159–172. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu Z, Gao L, Kan C, Chen X, Shi K and
Wang W: DNMT1 methylation of LncRNA-ANRIL causes myocardial
fibrosis pyroptosis by interfering with the NLRP3/Caspase-1
pathway. Cell Mol Biol (Noisy-le-grand). 70:197–203. 2024.
View Article : Google Scholar : PubMed/NCBI
|