Cardiac arrest (CA) is defined by the sudden
cessation of cardiac mechanical activity due to various causes. As
a life-threatening emergency, which represents a leading global
driver of mortality and neurological disability. Each year,
>500,000 individuals undergo out-of-hospital resuscitation for
CA, with the annual incidence estimated at 30–97 per 100,000
population (1). While
cardiopulmonary resuscitation can lead to return of spontaneous
circulation (ROSC), patients often suffer from severe neurological
dysfunction. This impairment is driven by secondary cerebral
ischemia-reperfusion (IR) injury, which remains a marked
determinant of poor outcomes (2).
Cerebral IR injury is a multifaceted process involving oxidative
stress, calcium overload and mitochondrial dysfunction. These
pathways culminate in neuroinflammation, neuronal death and lasting
neurological deficits (3). Despite
progress, mechanistic insights remain incomplete, particularly
concerning inflammation-mediated cell death. Consequently,
investigating novel death pathways has become a research
priority.
Pyroptosis is a unique programmed cell death
pathway, which is molecularly mediated by inflammasomes and
gasdermin (GSDM) proteins. The core mechanism involves the
activation of inflammatory caspases, including caspase-1/4/5/11.
These enzymes cleave GSDM family members to form membrane pores,
facilitating the release of intracellular contents (4). In contrast to apoptosis, necroptosis
or ferroptosis, pyroptosis is characterized by cellular swelling
and membrane rupture; this process leads to the robust release of
pro-inflammatory cytokines (5).
Evidence suggests that pyroptosis is involved in diverse
conditions, including infection, autoimmune disease and
cardiovascular disorders. It also figures prominently in
neurodegenerative diseases, where it amplifies inflammation and
drives tissue injury. As a result, pyroptosis has become a primary
focus of translational research (6).
During cerebral IR injury, pyroptosis is excessively
activated, which triggers neuroinflammatory cascades, disrupts the
blood-brain barrier (BBB) and exacerbates neuronal loss, thereby
accelerating brain damage (7).
Experimental evidence indicates that inhibiting pyroptosis
mitigates tissue injury and confers neuroprotection (8). This offers a promising therapeutic
avenue for treating cerebral IR injury. Given its central role in
IR-induced brain damage, pyroptosis is now recognized as a
compelling therapeutic target (9).
Current interventions show promise, further underscoring its
translational value (10). In the
present review, the molecular mechanisms of pyroptosis and its
roles in cerebral IR injury were systematically summarized. The
potential therapeutic strategies were also explored to provide a
framework for both fundamental research and clinical translation.
While the present review primarily focused on brain injury
following CA/ROSC, evidence from focal ischemic stroke models (such
as middle cerebral artery occlusion) and in vitro systems
[oxygen-glucose deprivation/reoxygenation (OGD/R)] is integrated as
supportive data, particularly in mechanistic areas where
CA-specific evidence remains nascent.
To ensure a representative and comprehensive
overview of the field, a literature search was conducted across
PubMed (https://pubmed.ncbi.nlm.nih.gov/), Web of Science
(https://www.webofscience.com/) and
Scopus (https://www.scopus.com/) databases for
articles published through August 2025. The search strategy
involved various combinations of keywords, including ‘pyroptosis’,
‘cardiac arrest’, ‘cardiopulmonary resuscitation’, ‘cerebral
ischemia-reperfusion injury’ and ‘neuroinflammation’. Selection was
limited to peer-reviewed, English-language publication studies that
provided notable insights into the molecular cascades and
therapeutic potential of pyroptosis within the specific context of
post-arrest brain injury were prioritized.
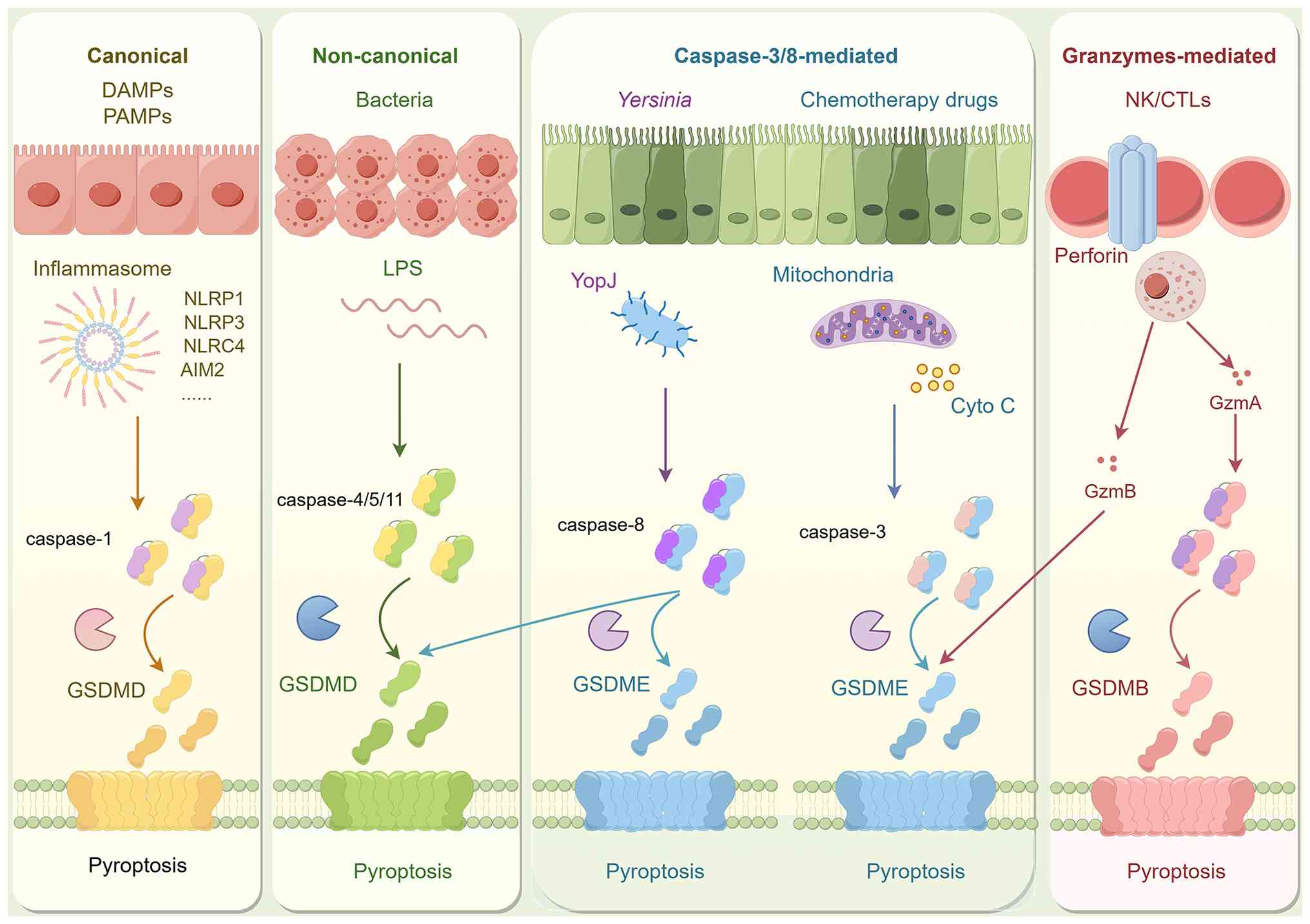
Further investigations revealed that pyroptosis is
not limited to the caspase-1 pathway. In mice, caspase-11 can
directly bind lipopolysaccharide (LPS) to induce pyroptosis. The
human homologs, caspase-4 and caspase-5, function similarly,
collectively constituting the non-canonical pathway (14). In 2015, Shi et al (15) and Kayagaki et al (16) independently identified GSDMD as the
key substrate of inflammatory caspases. Upon cleavage, the
N-terminal domain (NTD) of GSDMD inserts into the plasma membrane;
this process forms large non-selective pores that drive cellular
content release and robust inflammatory responses. This
breakthrough established the core execution mechanism of pyroptosis
and represented a major advancement in the field (15,16).
Subsequent research has elucidated the crosstalk
between pyroptosis and other cell death pathways. In 2017, Wang
et al (17) demonstrated
that caspase-3 can cleave GSDME, thereby converting apoptosis into
pyroptosis. As summarized in the 2018 recommendations of the
Nomenclature Committee on Cell Death, pyroptosis, which can also be
initiated by caspase-8 via GSDMD cleavage, was redefined as a
GSDM-dependent form of programmed cell death (18). This pathway is typically triggered
by the activation of inflammatory caspases. The GSDM family
comprises GSDMA, GSDMB, GSDMC, GSDMD, GSDME and GSDMF; among these
members, GSDMD is the most extensively characterized (19).
The canonical caspase-1-dependent pyroptotic pathway
is driven by the activation of inflammasomes. Inflammasomes are
cytosolic multiprotein complexes, which are composed of pattern
recognition receptors (PRRs), the adaptor protein
apoptosis-associated speck-like protein containing a caspase
recruitment domain (ASC) and pro-caspase-1. These complexes detect
pathogen-associated molecular patterns and host-derived
danger-associated molecular patterns (DAMPs) (20). Common PRRs include members of the
NOD-like receptor (NLR) family, such as NLRP1, NLRP3 and NLRC4, and
other sensors include AIM2-like receptors and pyrin. Among these,
the NLRP3 inflammasome is the most extensively characterized
(21). NLRP3 activation typically
follows a two-step model: First, a priming signal involving
microbial or endogenous factors activates NF-κB signaling to
upregulate NLRP3 and pro-IL-1β; second, an activation signal is
triggered by cellular events, such as potassium efflux,
mitochondrial dysfunction or lysosomal rupture (22). Upon activation, NLRP3 recruits ASC
and pro-caspase-1 to orchestrate inflammasome assembly.
Pro-caspase-1 then undergoes autocatalytic cleavage to generate
active caspase-1 (23), which
subsequently cleaves pro-IL-1β and pro-IL-18 into their mature
forms. These cytokines are then released through membrane pores to
propagate the inflammatory response. Simultaneously, caspase-1
cleaves GSDMD to release its NTD. This domain oligomerizes within
the plasma membrane to form pores, thereby disrupting osmotic
balance and ultimately precipitating cell rupture and pyroptosis
(24).
The non-canonical pyroptotic pathway operates
independently of canonical inflammasomes (25). In humans, this pathway is mediated
by caspase-4 and caspase-5, whereas caspase-11 serves as the
mediator in mice. In this pathway, LPS directly binds the caspase
recruitment domain (CARD) of caspase-4/5/11. This binding event
leads to their activation, and activated caspases subsequently
cleave GSDMD. This cleavage releases the pore-forming NTD, which
executes pyroptosis (26).
Notably, caspase-4/5/11 do not directly process pro-IL-1β or
pro-IL-18, but instead, GSDMD-mediated pore formation promotes
potassium efflux. This ion shift secondarily activates the NLRP3
inflammasome and caspase-1, thereby driving the maturation and
secretion of IL-1β and IL-18 (27).
Caspase-3 and caspase-8 are traditionally regarded
as key executioners of apoptosis; however, recent evidence
implicates them in pyroptosis via the cleavage of specific GSDM
family members (28). In cells
with high GSDME expression, caspase-3 cleaves the GSDME linker
region, which releases the pore-forming NTD and shifts the mode of
death from apoptosis to pyroptosis (29). Notably, GSDME expression levels
dictate the cell death outcome, where high expression favors
caspase-3-mediated pyroptosis and low expression favors apoptosis
(30). Additionally, during
Yersinia infection, the effector YopJ inhibits TAK1
signaling, which subsequently activates caspase-8. Activated
caspase-8 then cleaves both GSDMD and GSDME. This process liberates
their NTDs to form membrane pores and trigger pyroptosis (31). This pathway demonstrates that
caspase-8 can initiate pyroptosis independently of canonical
inflammasomes.
Gzms are the key effector molecules of cytotoxic T
lymphocytes and natural killer cells. They enter target cells via
perforin-mediated pores to induce cell death (32). Studies have elucidated their direct
role in pyroptosis regulation; GzmB can induce pyroptosis
indirectly through caspase-3 activation. Furthermore, it directly
cleaves GSDME at the same site to release the pore-forming NTD and
drive cell lysis (33). In
addition, GzmA was found to specifically cleave GSDMB at
Lys229/Lys244. This cleavage liberates the NTD to form pores,
establishing a caspase-independent pyroptotic pathway. Notably,
GSDMB is constitutively expressed in epithelial cells and certain
brain tissues, and its expression is markedly upregulated within
inflammatory microenvironments (34). These findings provide new
mechanistic insights and a theoretical basis for targeting
pyroptosis in cerebral IR injury. Fig.
1 illustrates the key molecules and pathways involved in
pyroptosis.
Moreover, the mitochondrial-GSDMD axis functions as
a pivotal amplifier of neuroinflammation during cerebral IR
(35). Specifically, the
N-terminal fragment of GSDMD (GSDMD-N) facilitates mitochondrial
membrane permeabilization, triggering the release of mitochondrial
DNA (mtDNA) into the cytosol (36). This translocation subsequently
engages the cyclin GMP-AMP synthase (cGAS)-stimulator of interferon
genes (STING) pathway, markedly exacerbating secondary neuroinjury
and inflammatory cascades following ROSC.
To ensure scientific rigor and distinguish between
pro-inflammatory signaling and terminal cell lysis, the following
three-stage experimental validation framework is proposed.
Focuses on transcriptional readiness with key
readouts of NF-κB nuclear translocation; mRNA and protein induction
of NLRP3 and pro-IL-1β.
Focuses on proteolytic processing with key readouts:
ASC speck formation; detection of cleaved caspase-1 (p20); and
quantification of mature IL-1β/IL-18 secretion.
Focuses on membrane perforation and cytolysis with
key readouts; Identification of GSDMD-N or GSDME-N fragments; LDH
release assays; and propidium iodide or ethidium homodimer
staining.
While Stage 2 markers indicate an active
inflammatory response, terminal pyroptosis must be confirmed by
Stage 3 markers (GSDMD cleavage and membrane rupture), which
represent the definitive hallmarks of lytic cell death.
Neurons are the most vulnerable cell type during
cerebral IR injury. Their heightened sensitivity to
ischemia-hypoxia largely determines the severity of neurological
deficits (37). Pyroptosis has
emerged as a distinct form of inflammatory programmed cell death,
which amplifies neuroinflammation through inflammasome activation
and cytokine release; this mechanism has been validated across
multiple neurological disorders, including Alzheimer's disease,
Parkinson's disease and traumatic brain injury (38). At the molecular level, NLRP3
inflammasome activation triggers caspase-1 activation and GSDMD
cleavage. These events lead to pore formation, neuronal swelling
and lysis. Such findings are well-demonstrated in both cerebral IR
injury and OGD/R models (39).
Moreover, genetic deletion or pharmacological inhibition of NLRP3,
caspase-1 or GSDMD markedly reduces neuronal death and attenuates
inflammation. These interventions also improve motor and behavioral
outcomes, supporting pyroptosis as a potential neuroprotective
target (40). Notably, neuronal
pyroptosis is not restricted to ischemic injury; it has also been
implicated in Parkinson's disease and Alzheimer's disease (AD),
suggesting it may represent a common neuronal response to diverse
insults (41). In summary,
elucidating the molecular mechanisms of neuronal pyroptosis in
cerebral IR injury deepens the understanding of pathology. It also
provides novel avenues for neuroprotective intervention (42).
Astrocytes are essential for maintaining ion
homeostasis, neurotransmitter metabolism and BBB integrity. They
provide notable neuronal support in the central nervous system.
Consequently, astrocyte dysfunction or death is linked to multiple
neurological disorders (43).
During cerebral IR injury, astrocytic pyroptosis occurs via two
primary pathways. These include inflammasome-mediated
caspase-1-dependent GSDMD cleavage and caspase-4/5/11 signaling.
Overexpression of CD73 inhibits astrocytic pyroptosis via the
adenosine A2B receptor/NF-κB signaling axis; this modulation
effectively mitigates brain injury (44). Following pyroptosis, astrocytes
release pro-inflammatory cytokines such as IL-1β and IL-18. This
release exacerbates local inflammation and neuronal damage. In AD
brains, astrocytic and neuronal pyroptosis have been linked to
amyloid-β deposition and neurodegeneration (45). Specifically, amyloid-β aggregates
act as DAMPs that trigger NLRP3 inflammasome activation; in turn,
the resulting pyroptosis and chronic neuroinflammation can further
promote amyloid-β production and aggregation, creating a
feed-forward pathological loop. Furthermore, astrocytic
inflammasome activation induces cell death and disrupts hippocampal
synaptic plasticity. It also impairs glutamate homeostasis through
IL-1 and IL-18 signaling, thereby aggravating excitotoxicity
(46). Astrocytic pyroptosis is
associated with early neurological deficits following brain injury.
Conversely, TNF-stimulated gene-6 alleviates this process by
suppressing NLRC4 inflammasome activation. This reduction in
pyroptosis alleviates brain edema and improves neurological
function (47). Astrocytic
pyroptosis is broadly involved in diverse neurological diseases;
therefore, targeting inflammasome, caspase or GSDM pathways offers
therapeutic promise. However, challenges remain in achieving
selectivity and clinical translation (48).
Microglia are the primary immune effector cells of
the central nervous system, where they activate rapidly in response
to cerebral IR injury. In this context, microglia mediate
inflammation and exacerbate neuronal damage. Furthermore, their
activation can impede long-term recovery (49). Beyond conventional immune
activation, microglia can undergo pyroptosis. This process
amplifies neuroinflammation through pore formation and cytokine
release. The interaction between interferon-inducible protein 204
and SUMO-specific protease 7 activates STING signaling. This
pathway induces both pyroptosis and mitochondrial dysfunction.
Notably, silencing these factors alleviates neurological deficits
(50). Mechanistically, microglial
pyroptosis is largely dependent on the NLRP3/caspase-1/GSDMD axis,
and ASC specks further amplify NLRP3 activity to promote
pyroptosis. This process accelerates α-synuclein aggregation and
neuronal degeneration, suggesting relevance to diverse
neurodegenerative diseases (51).
The pyroptotic release of cytosolic contents and inflammatory
mediators exacerbates neuroinflammation and secondary injury.
cGAS-STING activation drives NLRP3-mediated microglial pyroptosis,
which exacerbates pathology and motor deficits. Conversely,
pharmacological or genetic inhibition of this axis markedly
alleviates tissue damage (52). In
cerebral IR models, delayed administration [24 h post-transient
middle cerebral artery occlusion (tMCAO)] of the selective NLRP3
inhibitor MCC950 still reduces infarct size and improves
neurological outcomes. Although MCC950 is not cell-type specific,
its neuroprotective effects in cerebral IR models are largely
characterized by the suppression of microglial-mediated
neuroinflammation and cytokine release (53). In summary, microglial pyroptosis
represents a notable driver of cerebral IR injury and a promising
therapeutic target; however, its complex regulation and clinical
feasibility warrant further investigation (54).
Disruption of the BBB is a central pathological
event in cerebral IR injury; this breach further exacerbates
neuronal damage (55). As an
inflammasome-dependent death pathway, pyroptosis aggravates BBB
permeability via GSDM-mediated pore formation (56). The canonical NLRP3 inflammasome
pathway is a validated driver of IR pathology; caspase-1 activation
and IL-1β release promote inflammatory signaling. These events
directly injure endothelial cells and compromise BBB integrity
(57). Simultaneously, the
non-canonical pathway contributes to this dysfunction;
caspase-11-mediated GSDMD activation induces endothelial rupture
and the release of pro-inflammatory cytokines. This process spreads
inflammation and exacerbates BBB impairment (58), and oxidative stress further
amplifies BBB disruption during pyroptosis. It induces lipid
peroxidation and mitochondrial dysfunction, which enhance
inflammatory signaling and membrane injury (59). Astrocytes also actively participate
in this process; astrocytic secretion of CXCL10 promotes
endothelial pyroptosis via the CXCR3/cGAS/AIM2 pathway. This
mechanism leads to BBB breakdown, intensifying neuroinflammation
and aggravating brain injury (60). Together, these findings demonstrate
that pyroptosis contributes to BBB disruption through multiple
mechanisms. These insights highlight pyroptosis as a notable
therapeutic target in cerebral IR injury.
Pyroptosis in cerebral IR involves the synchronized
activation of both canonical (caspase-1) and non-canonical
(caspase-4/5/11) pathways. These cascades converge on
GSDMD-mediated membrane perforation, which serves as the definitive
execution step of lytic cell death.
Cellular heterogeneity within the neurovascular
unit. A key distinction exists within the neurovascular unit, while
neurons are the primary victims of terminal lytic death, microglia
and astrocytes function as ‘inflammatory amplifiers’. This glial
activation sustains the neuroimmune response and exacerbates BBB
instability, creating a feed-forward cycle of secondary
neuroinjury.
Future research should shift toward the development
of cell-specific modulators. The ultimate aim is to selectively
inhibit neuronal loss without compromising the essential trophic
and phagocytic support provided by glial cells during the recovery
phase.
Oxidative stress is a key driver of cell death and
tissue damage in cerebral IR injury. Together with mitochondrial
dysfunction and excitotoxicity, it forms the pathological basis of
injury in cerebral IR (61). In
cerebral IR models, reperfusion is characterized by a sharp rise in
reactive oxygen species (ROS) levels. These levels positively
associate with infarct volume and neurological deficits. Notably,
inhibition of NADPH oxidase 2 effectively reduces ROS production
and tissue injury (62).
Mechanistically, ROS promote NLRP3 inflammasome assembly by
inducing the dissociation of thioredoxin-interacting protein from
thioredoxin, which subsequently binds to the leucine-rich repeat
domain of NLRP3 to trigger its activation, and caspase-1
activation; this cascade leads to GSDMD cleavage and pyroptosis
(63). Conversely, ROS scavengers
or inflammasome inhibitors can partially reverse this injury.
Beyond direct effects, oxidative stress indirectly facilitates
inflammasome activation via mitochondrial dysfunction and calcium
dysregulation. These interactions form complex signaling networks
that represent potential therapeutic targets (64). In animal IR models,
nanomaterial-based antioxidant delivery effectively scavenges ROS.
This intervention suppresses the NLRP3/caspase-1/GSDMD pathway,
reducing neuronal pyroptosis and alleviating brain damage (65). These findings support antioxidant
interventions as viable neuroprotective strategies. Collectively,
oxidative stress drives neuronal pyroptosis via both direct and
indirect mechanisms (66).
Calcium homeostasis is essential for neuronal
survival and function. Its disruption triggers cell death via
mechanisms such as endoplasmic reticulum stress and mitochondrial
dysfunction (67). In cerebral IR
injury, excessive glutamate receptor activation and abnormal
calcium channel opening contribute to Ca2+ overload; the
reversal of Na+/Ca2+ exchange also plays a
role. These events collectively drive excitotoxicity and neuronal
injury (68). Calcium overload
activates the calcium-dependent cysteine protease calpain, which
subsequently liberates caspase-1 from the cytoskeleton to promote
its activation. This process leads to GSDMD cleavage and the
formation of pore-forming N-terminal fragments that drive canonical
pyroptosis (69). Aberrant
activation of the endoplasmic reticulum Ca2+ channel
inositol 1,4,5-triphosphate receptor 1 has been shown to enhance
Ca2+ efflux and induce pyroptosis via the
NLRP3/caspase-1 pathway (70). In
cerebral IR models, xanthine has been identified as a key
metabolite; it triggers endothelial Ca2+ overload and
induces GSDME-dependent pyroptosis, thereby exacerbating BBB
disruption and brain injury (71).
Thus, calcium dyshomeostasis acts as both a potent driver of
pyroptosis and a central pathological mechanism of IR injury.
Elucidating the cross-talk between Ca2+ imbalance and
pyroptosis will provide opportunities for identifying novel
therapeutic targets (72).
Mitochondrial dysfunction during cerebral IR leads
to marked energy failure, and also serves as an inflammatory
signaling hub that drives pyroptosis. In this context, the NLRP3
inflammasome acts as a notable bridge in stroke-associated
inflammation (73).
Mitochondria-derived danger signals include excessive mitochondrial
ROS, oxidized mtDNA leakage and abnormal mitochondrial permeability
transition pore opening. These signals induce or amplify NLRP3
inflammasome activation to promote pyroptotic cascades (74). Previous evidence demonstrates that
GSDMD forms pores in both the plasma and mitochondrial membranes;
this process facilitates mtDNA release and activates the cGAS-STING
pathway. This mechanism establishes a positive feedback loop
between inflammation and pyroptosis, thereby exacerbating tissue
damage (75). In cerebral IR
models, hyperactivation of dynamin-related protein 1 (Drp1) induces
mitochondrial dysfunction and GSDMD-mediated pore formation. This
aggravates neuronal pyroptosis and tissue injury. Conversely,
Apelin receptor early endogenous ligand mitigates this process by
suppressing Drp1 hyperactivation and NLRP3 signaling (76). Furthermore, enhancing PTEN-induced
putative kinase 1 (PINK1)/Parkin-dependent mitophagy markedly
inhibits pyroptosis. This suggests that the coupling of
mitochondrial quality control and pyroptosis represents a conserved
regulatory axis (77). Overall,
targeting mitochondrial function effectively suppresses
inflammation and pyroptosis, underscoring its central role in the
pathophysiology of cerebral IR injury (78).
The mitochondrial-pyroptotic triad: Oxidative
stress, calcium overload and mitochondrial dysfunction form a
self-perpetuating ‘triad’ that drives NLRP3 inflammasome
activation. This triad represents the most validated common pathway
across diverse cerebral IR models.
GSDMD-mediated feed-forward loops. Evidence
identifies mitochondrial GSDMD pores as a critical amplifier. These
pores allow mtDNA leakage, which directly amplifies intracellular
inflammatory signaling beyond the initial insult.
Identifying the ‘point of no return’. Mapping the
precise spatiotemporal sequence of these drivers is vital.
Determining the transition from reversible stress to irreversible
lysis will help identify the optimal therapeutic window for
interventions.
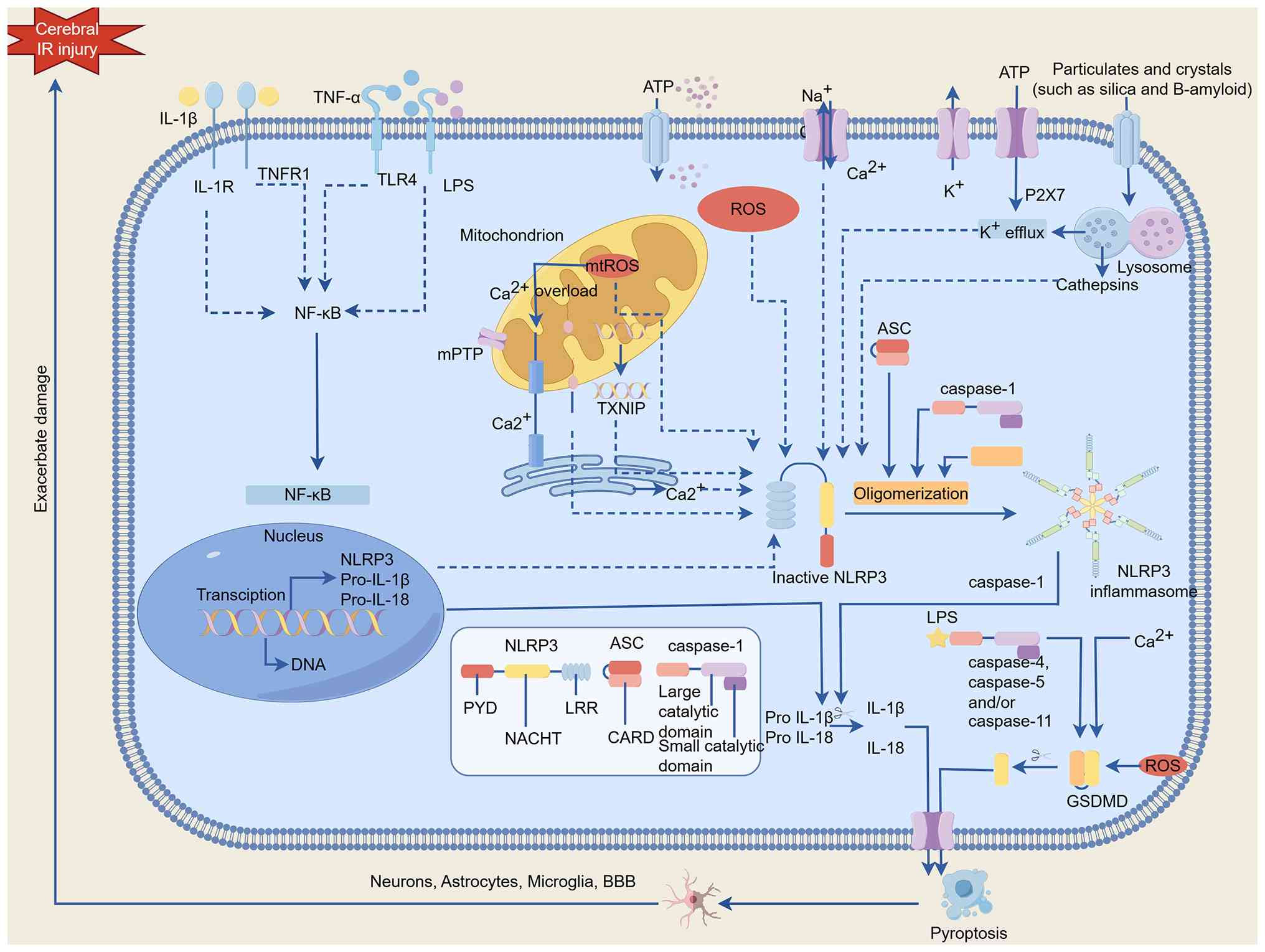
Pyroptosis serves as a pivotal driver and amplifier
of the neuroinflammatory response following cerebral IR injury.
Pyroptotic cells recognize endogenous danger signals and release
IL-1β, IL-18 and DAMPs. These events activate local immune
responses and trigger neuroinflammation (79). During cerebral IR, released DAMPs
activate the TLR4 signaling pathway, which amplifies inflammasome
activity and promotes pro-inflammatory cytokine production, further
propagating the inflammatory cascade (80). Moreover, GSDMD cleavage drives pore
formation in the plasma membrane to facilitate the release of
intracellular contents. The liberation of DAMPs and subsequent
membrane rupture are key steps in amplifying the inflammatory
response (81).
Evidence indicates that pyroptosis activates
microglial NLRP3 inflammasomes. This activation subsequently
induces A1-type astrocyte activation, generating a neurotoxic
response (82). The interaction
between NLRP3 and NF-κB is a marked amplification mechanism;
initial NLRP3 activation is mediated by IκB kinase β. This kinase
recruits inflammasome components to the trans-Golgi network to
sustain inflammatory progression (83). These inflammatory signals
exacerbate pyroptosis in endothelial cells and pericytes,
intensifies BBB disruption and facilitates leukocyte infiltration
(84). In mice IR models,
endothelial pyroptosis upregulates IL-1β, TNF-α and vascular cell
adhesion molecule-1. This promotes immune cell extravasation and
increases BBB permeability; conversely, inhibiting pyroptosis
reduces these factors and partially restores BBB integrity
(85).
Beyond localized cell death, pyroptosis orchestrates
the broader neuroimmune landscape by releasing DAMPs and
pro-inflammatory cytokines. These signals actively recruit
peripheral immune cells to the site of injury. For instance,
mediators such as high-mobility group box 1 (HMGB1) facilitate BBB
breakdown, allowing T cells to infiltrate the brain parenchyma
(86). This persistent neuroimmune
crosstalk drives the shift from acute phase inflammation to chronic
neuroinflammation. Ultimately, such chronic activation hinders
long-term neurorepair and functional recovery (87).
The contribution of pyroptosis to neuroinflammation
extends beyond amplification to include resolution. If pyroptotic
cells are not promptly cleared, persistent DAMP release drives
chronic inflammation and glial scar formation; these events
exacerbate neuronal injury (88).
Membrane repair mechanisms, including endosomal sorting complexes
required for transport-III (ESCRT-III) and nerve injury-induced
protein 1 (NINJ1), regulate the outcome of pyroptosis. These
systems determine whether inflammation resolves, thereby
influencing tissue repair and functional recovery (89). In summary, pyroptosis orchestrates
the neuroinflammatory cascade across the initiation, amplification
and resolution phases. This process aggravates cerebral IR injury
while providing a theoretical basis for novel therapeutic
interventions (90) (Fig. 2).
Apart from their fundamental roles in disease
pathogenesis, pyroptosis-associated molecules, particularly
N-terminal GSDMD-N and IL-18, are increasingly recognized as viable
biofluid biomarkers for quantifying the severity of ischemic
cerebral insults. Growing evidence indicates that increased
concentrations of these proteins in the serum or cerebrospinal
fluid reflect the degree of BBB permeability. Furthermore, these
biochemical signatures may function as reliable D indicators for
neurological outcomes, providing an objective measure of the extent
of brain tissue damage (91).
The acute-to-chronic transition: Pyroptosis dictates
the transition from acute injury to chronic neuroinflammation. This
pathological progression is balanced by membrane repair systems,
such as ESCRT-III and molecular executioners such as NINJ1.
Neuroimmune crosstalk and regeneration: Beyond
localized cell damage, pyroptotic DAMPs (such as HMGB1) facilitate
T-cell infiltration into the brain parenchyma. This sustained
neuroimmune crosstalk may create a hostile microenvironment that
markedly hinders long-term axonal regeneration.
Validation of biofluid biomarkers: A primary goal
for clinical translation is the systematic validation of biofluid
markers, specifically GSDMD-N and IL-18. Establishing the
prognostic value of these markers in serum or cerebrospinal fluid
is essential for the real-time monitoring of patients following
cardiac arrest.
In cerebral IR injury, pyroptosis and ferroptosis
are primary pathological forms of programmed cell death. These
pathways independently regulate neuronal damage but also interact
synergistically, and this crosstalk further exacerbates tissue
injury (92). Mechanistically,
iron overload and lipid peroxidation directly activate the NLRP3
inflammasome. This activation triggers caspase-1-mediated GSDMD
cleavage and pyroptosis (93).
Conversely, GSDMD pore formation facilitates Ca2+ influx
and ROS accumulation. These events amplify lipid peroxidation and
enhance ferroptotic susceptibility, thereby establishing a vicious
cycle (94). In cerebral IR
models, inhibiting ferroptosis suppresses lipid peroxidation via
the SLC7A11/GPX4/ACSL4 axis. This inhibition also downregulates the
caspase-1/GSDMD pathway, thereby attenuating pyroptosis and
improving neurological outcomes (95). Moreover, NLRP3-deficient mice
exhibit smaller infarct volumes and improved neurological scores.
These benefits are attributed to both pyroptosis suppression and
GPX4 upregulation. Reduced iron deposition and diminished lipid
peroxidation collectively mitigate ferroptosis in these models
(96). Autophagy, particularly
ferritinophagy, has emerged as a notable node linking ferroptosis
and pyroptosis by integrating iron homeostasis and inflammasome
activity (97). These insights
suggest that combined targeting of ferroptosis and pyroptosis may
provide a promising therapeutic strategy for cerebral IR
injury.
Autophagy is an essential degradative pathway which
maintains cellular homeostasis by clearing misfolded proteins and
damaged organelles to prevent the accumulation of cytotoxic factors
(98). In cerebral IR injury,
autophagy exerts a dual effect on pyroptosis. Moderate autophagy
activation alleviates mitochondrial dysfunction and oxidative
stress; this suppresses excessive inflammasome activation, thereby
reducing pyroptosis and conferring neuroprotection (99). For instance, an Astragalus-Angelica
herbal formulation activates autophagy via the AMPK/mTOR pathway.
This activation suppresses NLRP3-dependent pyroptosis and
attenuates cerebral IR injury (100). By contrast, excessive or
sustained autophagy can promote GSDMD cleavage and pro-inflammatory
cytokine release, which amplifies pyroptosis (101). In such settings, autophagy
inhibition mitigates inflammasome-mediated damage and improves
neurological function. Thus, autophagy exerts context-dependent
effects, acting as either protective or detrimental. This paradox
highlights its complexity as a therapeutic target. It underscores
the need to dissect the spatiotemporal dynamics of
autophagy-pyroptosis crosstalk across diverse cell types and
disease stages.
Apoptosis and pyroptosis frequently coexist and
interact during cerebral IR injury, and heir molecular signatures
are defined by distinct caspase family members (102). Specifically, caspase-3 and
caspase-9 drive classical apoptosis, whereas caspase-1, −4/5 and
−11 mediate pyroptosis through GSDMD or GSDME cleavage. Notably,
caspase-3-mediated cleavage of GSDME can convert apoptosis into
secondary pyroptosis, a lytic transition where cells initially
undergoing programmed apoptosis switch to a pro-inflammatory
phenotype due to GSDME-mediated membrane pore formation (103). Caspase-8 exhibits dual functions;
it triggers apoptosis through canonical pathways while also
directly cleaving GSDMD or promoting inflammasome activation. This
signaling bifurcation has been validated in TNF-α-induced cell
death and infection responses (104). Mitochondrial pathways further
integrate apoptosis and pyroptosis; BAX/BAK-mediated mitochondrial
outer membrane permeabilization induces Cytochrome c release to
drive apoptosis. Simultaneously, this process releases mtDNA to
activate the NLRP3 inflammasome and promote pyroptosis (105). Regulatory molecules such as
cellular FLICE-like inhibitory protein (c-FLIP), inhibitors of
apoptosis proteins and receptor-interacting
serine/threonine-protein kinase 1 (RIPK1) act as pivotal nodes in
this crosstalk. For example, c-FLIPL deficiency facilitates complex
II formation and promotes LPS-induced pyroptosis; this underscores
its role in dual-pathway regulation (106). Based on these insights,
pharmacological inhibition of caspases or GSDMs has been proposed
to attenuate both apoptosis- and pyroptosis-related injury.
However, efficacy remains context-dependent, warranting further
refinement using multi-omics approaches (107).
Both pyroptosis and necroptosis are pro-inflammatory
death pathways that often coexist in cerebral IR injury. These
processes are tightly interconnected through the PANoptosome
complex (108). Key molecular
convergence points include RIPK1, RIPK3, MLKL, caspase-8 and GSDMD,
which collectively determine cell fate. RIPK1 and RIPK3 activate
MLKL to initiate necroptosis; simultaneously, caspase-8 cleaves
GSDMD to induce pyroptosis (109). MLKL-mediated K+ efflux
has been shown to activate the NLRP3 inflammasome, which induces
GSDMD pore formation and couples necroptosis with pyroptosis
(110). Caspase-8 further
promotes IL-1β maturation, linking the two pathways in inflammatory
cell death. The emerging concept of PANoptosis (pyroptosis,
apoptosis, and necroptosis) emphasizes that these pathways are
co-regulated within the PANoptosome complex (111). Consequently, the inhibition of a
single pathway may be compensated by alternative mechanisms. In
cerebral IR models, pyroptosis and necroptosis jointly exacerbate
sterile inflammation and neuronal injury. Conversely, neural stem
cell transplantation mitigates these effects by downregulating
GSDMD and MLKL expression. This intervention ultimately improves
functional recovery (112,113).
These findings highlight the therapeutic potential of
simultaneously targeting pyroptotic and necroptotic pathways.
The PANoptosome and programmed cell death crosstalk:
Pyroptosis is no longer viewed in isolation but as part of the
‘PANoptosome’ complex, where it intersects with apoptosis,
necroptosis and ferroptosis. This integrated view reflects the
complex molecular interplay that dictates neuronal fate following
cerebral IR injury.
Autophagy as a cellular rheostat: Autophagy acts as
a complex rheostat in the context of ischemic injury. While
physiological autophagy confers neuroprotection by degrading
inflammasomes, excessive ‘autophagic stress’ can facilitate GSDMD
cleavage and accelerate lytic cell death.
Toward multi-node therapeutic inhibition: Future
therapies should transition toward ‘multi-node’ inhibition
strategies. Targeting the shared molecular hubs of the PANoptosome
may offer improved neuroprotection compared with inhibiting a
single death pathway alone, particularly given the compensatory
mechanisms inherent in programmed cell death.
The NLRP3 inflammasome is a central sensor in the
pyroptotic pathway. It recognizes intracellular danger signals to
mediate downstream caspase-1 activation and GSDMD cleavage. These
events ultimately lead to plasma membrane pore formation and
pro-inflammatory cytokine release (114). In a mouse cardiac
arrest/cardiopulmonary resuscitation (CA/CPR) model, selective
inhibition of NLRP3 using MCC950 administered post-ischemia notably
reduces NLRP3/caspase-1 activation to improve survival and
neurological deficit scores (115). Similarly, in a mouse model of
tMCAO, β-caryophyllene administered post-ischemia alleviates white
matter injury, and improves motor and cognitive functions by
suppressing NLRP3-mediated pyroptosis (116). In a rat CA/CPR model,
chrysophanol administered post-ischemia also mitigates cerebral IR
injury via the inhibition of the TRAF6/NLRP3 signaling pathway
(117). In a mouse tMCAO model,
pre-ischemic inhibition of the lipocalin-2/24p3R/NLRP3 axis
attenuates astrocyte pyroptosis and neuroinflammation by reducing
GSDMD-N and IL-1β levels (118).
Collectively, these findings establish NLRP3 as a pivotal mediator
of ischemic brain injury; however, its activation mechanisms remain
complex, with oxidative stress serving as a primary driver.
Oxidative stress is a primary driver of cerebral IR
injury, and ROS are recognized as major triggers for NLRP3
activation (119). In a mouse
distal middle cerebral artery occlusion model, administration of
hydrogen-rich saline, a potent ROS scavenger, both pre- and
post-ischemia effectively suppresses the ROS/NLRP3/caspase-1
pathway and reduces IL-1β levels, thereby improving neurological
deficit scores (120).
Additionally, in a mouse tMCAO model, OIP5 Antisense RNA 1
administered as a single dose post-ischemia promotes the
degradation of thioredoxin-interacting protein, which inhibits
oxidative stress-induced neuronal pyroptosis and alleviates
cerebral IR injury (121).
Likewise, methyl isoeugenol administered post-ischemia in a rat
tMCAO model activates the nuclear factor erythroid 2-related factor
2 (Nrf2)/NQO1/HO-1 antioxidant pathway, which suppresses ROS-driven
microglial pyroptosis and attenuates neuroinflammation (122). These findings underscore
oxidative stress modulation as an important therapeutic
strategy.
Calcium overload and mitochondrial dysfunction also
trigger aberrant NLRP3 activation (123). Dexmedetomidine reduces microglial
pyroptosis and neural injury in a rat permanent middle cerebral
artery occlusion (pMCAO) model when administered via post-ischemic
continuous infusion, a process mediated by the inhibition of the
P2X7R/NLRP3/caspase-1 pathway (124). By suppressing Drp1-mediated
mitochondrial fission, a single post-ischemic dose of FK866 in a
rat CA/CPR model ameliorates mitochondrial dysfunction, thereby
reducing both pyroptosis and neuroinflammation (125). Single post-ischemic
administration of Apelin-13-loaded macrophage membrane-encapsulated
nanoparticles in a mouse tMCAO model attenuates pyroptosis by
enhancing Sirtuin 3 activity and suppressing NLRP3 assembly
(126). These approaches
highlight novel strategies targeting calcium overload and
mitochondrial dysfunction.
Inflammatory propagation represents another notable
pathological process mediated by NLRP3-dependent pyroptosis
(127). Pre-ischemic
administration of indobufen and aspirin, alone or in combination
with clopidogrel or ticagrelor, attenuate pyroptosis in a rat tMCAO
model by inhibiting NF-κB/NLRP3 signaling (128). Knockdown of maternally expressed
gene 3 administered as a single pre-ischemic dose in a rat tMCAO
model inhibits the microRNA (miR)-145-5p/TLR4/NLRP3 axis, thereby
suppressing pyroptosis and conferring neuroprotection (129). In a rat tMCAO model, a single
pre-ischemic dose of calycosin reduces pyroptosis by suppressing
the HMGB1/NLRP3 signaling axis (130). Post-ischemic electroacupuncture
(EA) at the ST36 acupoint in a rat CA/CPR model alleviates neuronal
injury and inflammation by activating the α-7 nicotinic
acetylcholine receptor, which effectively suppresses microglial
pyroptosis (131). Furthermore,
in a mouse tMCAO model, a single post-ischemic dose of the STING
inhibitor C-176 reduces ischemic brain injury by disrupting the
STING-NLRP3 interaction and suppressing microglial pyroptosis
(132). Taken together, these
findings confirm the central role of NLRP3 in pyroptosis induction
and inflammatory amplification, underscoring its promise as a
translatable therapeutic target.
Caspase-1 is a notable component of the inflammasome
complex, which is responsible for the maturation of IL-1β and IL-18
and the initiation of pyroptosis (133). In a rat pMCAO model,
post-ischemic treatment with the caspase-1 inhibitor VX-765
ameliorates BBB disruption and neuroinflammation by inhibiting
caspase-1 and reducing pyroptosis, which ultimately leads to
improved neurological outcomes (134). AIM2 inflammasome-driven caspase-1
activation exacerbates brain injury and cognitive impairment.
Conversely, pre-ischemic treatment with the caspase-1 inhibitor
Ac-YVAD-CMK in a mouse tMCAO model improves cognitive function by
suppressing caspase-1-mediated pyroptosis (135). Additionally, miR-96-5p reduces
cerebral IR injury in a mouse tMCAO model when administered as a
single dose pre-ischemia primarily by downregulating caspase-1 and
suppressing pyroptosis (136).
Thus, targeting caspase-1 is a promising strategy to mitigate
IR-induced pyroptosis and neural injury.
GSDMD is the primary executioner of pyroptosis, and
cleavage of GSDMD liberates its NTD. This domain subsequently forms
pores in the plasma membrane, ultimately leading to cell rupture
(137). Administered
post-ischemia in a mouse tMCAO model, the GSDMD inhibitor
disulfiram reduces neutrophil infiltration and infarct size in
cerebral IR injury (91).
Similarly, in a mouse tMCAO model, genetic ablation (knockout) of
GSDMD decreases microglial pyroptosis in cerebral IR, a reduction
that markedly improves neurological function (138). Therefore, targeting the
caspase-1/GSDMD axis enables multi-level intervention. This
approach spans from inflammatory sensing to cellular rupture,
providing a systematic framework for modulating pyroptosis
(Table I) (91,115–118,120–122,124–126,128–132,134–136,138).
Beyond canonical pathways, modulating cell-specific
functional states and signaling networks represents a promising
therapeutic avenue. Microglia are the primary resident immune cells
of the brain where they play central roles in cerebral IR injury,
and their polarization state (M1 vs. M2) dictates the extent of
neuronal damage (139).
Intermittent θ-burst stimulation (iTBS), applied twice daily for 7
days post-ischemia in a mouse tMCAO model, reduces neuronal
pyroptosis by modulating microglial M1/M2 polarization (140). Via activation of the HACE1/Nrf2
axis, ETS variant transcription factor 5 administered as a single
pre-ischemic dose in a rat tMCAO model inhibits microglial M1
polarization and attenuates brain injury (141). Salvianolic acids for injection
administered daily post-ischemia in a rat tMCAO model exert
neuroprotective effects by promoting microglial M1-to-M2
polarization and inhibiting the NLRP3 inflammasome (142). Autophagy is a marked cellular
self-clearance mechanism, and its role in pyroptosis is complex and
often described as a double-edged sword. Both excessive and
insufficient autophagic activity can exacerbate injury (143). When administered as a single
post-ischemic dose in a rat tMCAO model, dexmedetomidine enhances
mitochondrial autophagy via the PINK1/Parkin pathway, thereby
reducing pyroptosis and protecting cerebral cells (144). Pre-ischemic combination therapy
involving EA and tigustilide in a rat tMCAO model activates the
PI3K/AKT/mTOR pathway, which inhibits autophagy and subsequently
reduces pyroptosis (145). These
studies suggest that regulating microglial activation and balancing
autophagy could provide new strategies for treating cerebral IR
injury.
As advanced drug delivery platforms, exosomes offer
promising strategies for the treatment of cerebral IR injury
(150). Post-ischemic treatment
with astrocyte-derived exosomal miR-378a-5p suppresses NLRP3
activation and alleviates neuronal pyroptosis in a rat tMCAO model
(151). Furthermore, a single
post-ischemic dose of bone marrow mesenchymal stem cell-derived
exosomes in a rat tMCAO model attenuate brain injury by modulating
microglial M1/M2 polarization and suppressing NLRP3 inflammasome
activation (152). Notably,
transferrin receptor-targeted nanomicelles delivering MCC950,
administered as a single post-ischemic dose in a Wistar rat model
of tMCAO, effectively inhibit NLRP3 activation and ameliorate
cerebral IR injury (153). A
cascade-type microglial pyroptosis inhibitor was developed to
integrate nanotechnology with antioxidative activity; a single
post-ischemic administration of this system in a rat tMCAO model
regulates the ROS/NLRP3/pyroptosis axis to mitigate
neuroinflammation (154).
Table II summarizes these
emerging biological regulators and delivery platforms, including
iTBS, transcription factors, ncRNAs and
exosome/nanotechnology-based systems, across various ischemia
models (140–142,144,145,147–149,151–154). These studies underscore the
therapeutic potential of exosome- and nanotechnology-based
interventions in cerebral IR injury.
The success of clinical translation hinges on BBB
permeability, optimal administration routes and precise dosing.
Small-molecule inhibitors, characterized by their inherent
lipophilicity, possess a distinct advantage in the acute CA/ROSC
setting due to rapid BBB penetration (155). While nanotechnology and
exosome-based platforms offer superior targeting specificity, they
must navigate challenges regarding off-target toxicity and
potential immunosuppressive risks (156). Biologics, conversely, are often
constrained by their large molecular weight, making BBB crossing a
notable hurdle (157). Given the
narrow therapeutic window in CA/ROSC, optimizing delivery protocols
during the hyper-acute phase is imperative to enhance overall
pharmacological feasibility.
CA remains a notable clinical challenge in
emergency medicine. In this context, cerebral IR injury is a
primary driver of high mortality and adverse neurological outcomes
(158). Pyroptosis is central to
the pathological progression of cerebral IR injury. Pyroptosis
drives neuronal damage by amplifying pro-inflammatory signals and
coordinating intercellular interactions. It also orchestrates
complex multi-pathway cross-regulation. This pathway integrates
various forms of programmed cell death; furthermore, it exhibits
cell type-specific roles across different populations in the brain.
Targeted interventions against pyroptosis show marked therapeutic
potential. These strategies provide novel insights for mitigating
cerebral IR injury and enhancing clinical recovery.
Despite these mechanistic advances, the inherent
constraints of current preclinical models require careful
consideration. The vast majority of the current understanding is
derived from rodent studies and in vitro cultures, which
often struggle to replicate the sophisticated physiological milieu
and the complex neurovascular unit of the human brain. Such
interspecies discrepancies represent a formidable barrier to
clinical translation, likely explaining why numerous
neuroprotective candidates with robust preclinical profiles fail to
yield therapeutic benefits in survivors of CA/ROSC.
Looking ahead, narrowing the current knowledge gaps
in cerebral IR injury requires a shift toward more human-centric
research paradigms. The integration of organoids derived from human
induced pluripotent stem cells with microfluidic BBB models
represents a notable step forward; these high-fidelity systems help
circumvent the inherent limitations of animal studies by more
accurately reflecting the human neural microenvironment. Parallel
to these structural models, the fusion of multiomics and artificial
intelligence provides a sophisticated toolkit for mapping the
intricate signaling nodes that govern pyroptotic networks, thereby
streamlining the identification of novel therapeutic targets
(159). Furthermore, evaluating
the clinical utility of circulating biomarkers, specifically
cleaved GSDMD and IL-18, is essential for establishing reliable
diagnostic and prognostic frameworks. Ultimately, the convergence
of these advanced biotechnologies is expected to expedite
high-throughput drug screening and accelerate the clinical
transition of neuroprotective strategies for survivors of cardiac
arrest (160).
Not applicable.
The present research was funded by the Henan Provincial Health
Commission Provincial-Ministerial Co-Construction Project (grant
no. SBGJ202102198).
Not applicable.
ZLi was responsible for the conceptualization of
the present study. ZLi and QM wrote the original draft of the
manuscript and created the figures. ZLi made the tables. ZLu
acquired the funding. ZLu, TY, ML, LF and JY wrote and edited the
review and, All authors have read and approved the final version of
the manuscript. Data authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Perkins GD, Callaway CW, Haywood K, Neumar
RW, Lilja G, Rowland MJ, Sawyer KN, Skrifvars MB and Nolan JP:
Brain injury after cardiac arrest. Lancet. 398:1269–1278. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sandroni C, Cronberg T and Sekhon M: Brain
injury after cardiac arrest: Pathophysiology, treatment, and
prognosis. Intensive Care Med. 47:1393–1414. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu DQ, Mei W, Zhou YQ and Xi H: Targeting
TRPM channels for cerebral ischemia-reperfusion injury. Trends
Pharmacol Sci. 45:862–867. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rao Z, Zhu Y, Yang P, Chen Z, Xia Y, Qiao
C, Liu W, Deng H, Li J, Ning P and Wang Z: Pyroptosis in
inflammatory diseases and cancer. Theranostics. 12:4310–4329. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ai Y, Meng Y, Yan B, Zhou Q and Wang X:
The biochemical pathways of apoptotic, necroptotic, pyroptotic, and
ferroptotic cell death. Mol Cell. 84:170–179. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sun Y, Li F, Liu Y, Qiao D, Yao X, Liu GS,
Li D, Xiao C, Wang T and Chi W: Targeting inflammasomes and
pyroptosis in retinal diseases-molecular mechanisms and future
perspectives. Prog Retin Eye Res. 101:1012632024. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sun R, Peng M, Xu P, Huang F, Xie Y, Li J,
Hong Y, Guo H, Liu Q and Zhu W: Low-density lipoprotein receptor
(LDLR) regulates NLRP3-mediated neuronal pyroptosis following
cerebral ischemia/reperfusion injury. J Neuroinflammation.
17:3302020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Huang X, Tan J, Ji Y, Luo J, Zhao Y and
Zhao J: BRCC3 mediates inflammation and pyroptosis in cerebral
ischemia/reperfusion injury by activating the NLRP6 inflammasome.
CNS Neurosci Ther. 30:e146972024. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Geng H, Tang J, Li Z, Zhang Y, Ye C, Zhang
Y, Li X, Li Y, Wang Y, Wang Y, et al: 14,15-EET maintains
mitochondrial homeostasis to inhibit neuronal pyroptosis after
ischemic stroke. Stroke. 56:1883–1896. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
She Y, Shao L, Jiao K, Sun R, Lang T, Long
H, Tang Y, Zhang W, Ding C and Deng C: Glycosides of buyang huanwu
decoction inhibits pyroptosis associated with cerebral
ischemia-reperfusion through Nrf2-mediated antioxidant signaling
pathway both in vivo and in vitro. Phytomed. 120:1550012023.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cookson BT and Brennan MA:
Pro-inflammatory programmed cell death. Trends Microbiol.
9:113–114. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ocansey DKW, Qian F, Cai P, Ocansey S,
Amoah S, Qian Y and Mao F: Current evidence and therapeutic
implication of PANoptosis in cancer. Theranostics. 14:640–661.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zychlinsky A, Prevost MC and Sansonetti
PJ: Shigella flexneri induces apoptosis in infected macrophages.
Nature. 358:167–169. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huang C, Li J, Wu R, Li Y and Zhang C:
Targeting pyroptosis for cancer immunotherapy: Mechanistic insights
and clinical perspectives. Mol Cancer. 24:1312025. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shi J, Zhao Y, Wang K, Shi X, Wang Y,
Huang H, Zhuang Y, Cai T, Wang F and Shao F: Cleavage of GSDMD by
inflammatory caspases determines pyroptotic cell death. Nature.
526:660–665. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kayagaki N, Stowe IB, Lee BL, O'Rourke K,
Li K, Warming S, Cuellar S, Haley B, Quiraoz M, Eriksson NO, et al:
Caspase-11 cleaves gasdermin D for non-canonical inflammasome
functions. Nature. 526:666–671. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang Y, Gao W, Shi X, Ding J, Liu W, He H,
Wang K and Shao F: Chemotherapy drugs induce pyroptosis through
caspase-3 cleavage of a gasdermin. Nature. 547:99–103. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Galluzzi L, Vitale I, Aaronson SA, Abrams
JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews
DW, et al: Molecular mechanisms of cell death: Recommendations of
the nomenclature committee on cell death 2018. Cell Death Differ.
25:486–541. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jiang X, Zhang X, Cai X, Li N, Zheng H,
Tang M, Zhu J, Su K, Zhang R, Ye N, et al: NU6300 covalently reacts
with cysteine-191 of gasdermin D to block its cleavage and
palmitoylation. Sci Adv. 10:eadi92842024. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang Y, Xu W and Zhou R: NLRP3
inflammasome activation and cell death. Cell Mol Immunol.
18:2114–2127. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ferrara F, Cordone V, Pecorelli A,
Benedusi M, Pambianchi E, Guiotto A, Vallese A, Cervellati F and
Valacchi G: Ubiquitination as a key regulatory mechanism for
O3-induced cutaneous redox inflammasome activation. Redox Biol.
56:1024402022. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Feng S, Wierzbowski MC, Hrovat-Schaale K,
Dumortier A, Zhang Y, Zyulina M, Baker PJ, Reygaerts T, Steiner A,
De Nardo D, et al: Mechanisms of NLRP3 activation and inhibition
elucidated by functional analysis of disease-associated variants.
Nat Immunol. 26:511–523. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dufies O, Doye A, Courjon J, Torre C,
Michel G, Loubatier C, Jacquel A, Chaintreuil P, Majoor A,
Guinamard RR, et al: Escherichia coli RhoGTPase-activating toxin
CNF1 mediates NLRP3 inflammasome activation via p21 activated
kinases-1/2 during bacteremia in mice. Nat Microbiol. 6:401–412.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
You Z, Huang X, Xiang Y, Dai J, Xu L,
Jiang J and Xu J: Ablation of NLRP3 inflammasome attenuates muscle
atrophy via inhibiting pyroptosis, proteolysis and apoptosis
following denervation. Theranostics. 13:374–390. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu Y, Pan R, Ouyang Y, Gu W, Xiao T, Yang
H, Tang L, Wang H, Xiang B and Chen P: Pyroptosis in health and
disease: Mechanisms, regulation and clinical perspective. Signal
Transduct Target Ther. 9:2452024. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Stoess C, Leszczynska A, Kui L and
Feldstein AE: Corrigendum: Pyroptosis and gasdermins-emerging
insights and therapeutic opportunities in metabolic
dysfunction-associated steatohepatitis. Front Cell Dev Biol.
12:14615812024. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xu Z, Kombe Kombe AJ, Deng S, Zhang H, Wu
S, Ruan J, Zhou Y and Jin T: NLRP inflammasomes in health and
disease. Mol Biomed. 5:142024. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang W, Zhu C, Liao Y, Zhou M, Xu W and
Zou Z: Caspase-8 in inflammatory diseases: A potential therapeutic
target. Cell Mol Biol Lett. 29:1302024. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hu Y, Liu Y, Zong L, Zhang W, Liu R, Xing
Q, Liu Z, Yan Q, Li W, Lei H and Liu X: The multifaceted roles of
GSDME-mediated pyroptosis in cancer: Therapeutic strategies and
persisting obstacles. Cell Death Dis. 14:8362024. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jiang M, Qi L, Li L and Li Y: The
caspase-3/GSDME signal pathway as a switch between apoptosis and
pyroptosis in cancer. Cell Death Discovery. 6:1122020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sarhan J, Liu BC, Muendlein HI, Li P,
Nilson R, Tang AY, Rongvaux A, Bunnell SC, Shao F, Green DR and
Poltorak A: Caspase-8 induces cleavage of gasdermin D to elicit
pyroptosis during yersinia infection. Proc Natl Acad Sci USA.
115:E10888–E10897. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hay ZLZ and Slansky JE: Granzymes: The
molecular executors of immune-mediated cytotoxicity. Int J Mol Sci.
23:18332022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tan X, Yang J, Li Y, Wan K, Feng S, Jing
X, Xie Z, Zhang L and Li W: Development of ZHPV16E7-granzyme B
immunoaffitoxin: Dual mechanisms targeting hpv16-positive cervical
cancer through epithelial-mesenchymal transition inhibition and
cell death. Front Immunol. 16:16167152025. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y,
Wang Y, Li D, Liu W, Zhang Y, et al: Granzyme a from cytotoxic
lymphocytes cleaves GSDMB to trigger pyroptosis in target cells.
Science. 368:eaaz75482020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li Q, Yang L, Wang K, Chen Z, Liu H, Yang
X, Xu Y, Chen Y, Gong Z and Jia Y: Oxidized mitochondrial DNA
activates the cGAS-STING pathway in the neuronal intrinsic immune
system after brain ischemia-reperfusion injury. Neurotherapeutics.
21:e003682024. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhao C, Liang F, Ye M, Wu S, Qin Y, Zhao
L, Zhang L, He J, Cen L and Lin F: GSDMD promotes neutrophil
extracellular traps via mtDNA-cGAS-STING pathway during lung
ischemia/reperfusion. Cell Death Discov. 9:3682023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Daniele SG, Trummer G, Hossmann KA,
Vrselja Z, Benk C, Gobeske KT, Damjanovic D, Andrijevic D, Pooth
JS, Dellal D, et al: Brain vulnerability and viability after
ischaemia. Nat Rev Neurosci. 22:553–572. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Han YH, Liu XD, Jin MH, Sun HN and Kwon T:
Role of NLRP3 inflammasome-mediated neuronal pyroptosis and
neuroinflammation in neurodegenerative diseases. Inflamm Res.
72:1839–1859. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhao B, Fei Y, Zhu J, Yin Q, Fang W and Li
Y: PAF receptor inhibition attenuates neuronal pyroptosis in
cerebral ischemia/reperfusion injury. Mol Neurobiol. 58:6520–6539.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yu Y, Liao X, Xing K, Xie Z, Xie N, He Y,
Huang Z, Tang X and Liu R: Genistein-3′-sodium sulfonate suppresses
NLRP3-mediated cell pyroptosis after cerebral ischemia. Metab Brain
Dis. 40:992025. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wu KJ, Wang WR, Cheng QH, Li H, Yan WZ,
Zhou FR and Zhang RJ: Pyroptosis in neurodegenerative diseases:
From bench to bedside. Cell Biol Toxicol. 39:2467–2499. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tuo QZ, Zhang ST and Lei P: Mechanisms of
neuronal cell death in ischemic stroke and their therapeutic
implications. Med Res Rev. 42:259–305. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Verkhratsky A, Butt A, Li B, Illes P,
Zorec R, Semyanov A, Tang Y and Sofroniew MV: Astrocytes in human
central nervous system diseases: A frontier for new therapies.
Signal Transduct Target Ther. 8:3962023. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhuang H, Lei W, Wu Q, Zhao S, Zhao Y,
Zhang S, Zhao N, Sun J and Liu Y: Overexpressed CD73 attenuates
GSDMD-mediated astrocyte pyroptosis induced by cerebral
ischemia-reperfusion injury through the A2B/NF-κB pathway. Exp
Neurol. 386:1151522025. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Moonen S, Koper MJ, Van Schoor E,
Schaeverbeke JM, Vandenberghe R, von Arnim CAF, Tousseyn T, De
Strooper B and Thal DR: Pyroptosis in Alzheimer's disease: Cell
type-specific activation in microglia, astrocytes and neurons. Acta
Neuropathol. 145:175–195. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zengeler KE, Hollis A, Deutsch TCJ,
Samuels JD, Ennerfelt H, Moore KA, Steacy EJ, Sabapathy V, Sharma
R, Patel MK, et al: Inflammasome signaling in astrocytes modulates
hippocampal plasticity. Immunity. 58:1519–1535.e11. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ding M, Jin L, Wei B, Cheng W, Liu W, Li X
and Duan C: Tumor necrosis factor-stimulated gene-6 ameliorates
early brain injury after subarachnoid hemorrhage by suppressing
NLRC4 inflammasome-mediated astrocyte pyroptosis. Neural Regener
Res. 19:1064–1071. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Oladapo A, Jackson T, Menolascino J and
Periyasamy P: Role of pyroptosis in the pathogenesis of various
neurological diseases. Brain Behav Immun. 117:428–446. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Shichita T, Ooboshi H and Yoshimura A:
Neuroimmune mechanisms and therapies mediating post-ischaemic brain
injury and repair. Nat Rev Neurosc. 24:299–312. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Guo T, Lai Y, Wu S, Lin C, Zhou X, Lin P,
Zheng M, Chen J and Lin F: IFI204 in microglia mediates traumatic
brain injury-induced mitochondrial dysfunction and pyroptosis via
SENP7 interaction. Cell Biol Toxicol. 41:892025. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zheng R, Yan Y, Dai S, Ruan Y, Chen Y, Hu
C, Lin Z, Xue N, Song Z, Liu Y, et al: ASC specks exacerbate
α-synuclein pathology via amplifying NLRP3 inflammasome activities.
J Neuroinflammation. 20:262023. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Xue YX, Chen YJ, Qin MZ, Shang FF, Lu YT,
Sun YH, Bian LG, Zhang A, Yu Y and Ding CY: Microglial STING
activation promotes neuroinflammation and pathological changes in
experimental mice with intracerebral haemorrhage. Acta Pharmacol
Sin. 46:2376–2392. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Bellut M, Bieber M, Kraft P, Weber ANR,
Stoll G and Schuhmann MK: Delayed NLRP3 inflammasome inhibition
ameliorates subacute stroke progression in mice. J
Neuroinflammation. 20:42023. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yang H, Xia Y, Ma Y, Gao M, Hou S, Xu S
and Wang Y: Inhibition of the cGAS-STING pathway: Contributing to
the treatment of cerebral ischemia-reperfusion injury. Neural
Regener Res. 20:1900–1918. 2024. View Article : Google Scholar
|
|
55
|
Qin C, Yang S, Chu YH, Zhang H, Pang XW,
Chen L, Zhou LQ, Chen M, Tian DS and Wang W: Signaling pathways
involved in ischemic stroke: Molecular mechanisms and therapeutic
interventions. Signal Transduction Targeted Ther. 7:2152022.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Bellut M, Papp L, Bieber M, Kraft P, Stoll
G and Schuhmann MK: NLPR3 inflammasome inhibition alleviates
hypoxic endothelial cell death in vitro and protects blood-brain
barrier integrity in murine stroke. Cell Death Dis. 13:202021.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Franke M, Bieber M, Kraft P, Weber ANR,
Stoll G and Schuhmann MK: The NLRP3 inflammasome drives
inflammation in ischemia/reperfusion injury after transient middle
cerebral artery occlusion in mice. Brain Behav Immun. 92:223–233.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wei C, Jiang W, Wang R, Zhong H, He H, Gao
X, Zhong S, Yu F, Guo Q, Zhang L, et al: Brain endothelial GSDMD
activation mediates inflammatory BBB breakdown. Nature.
629:893–900. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Xiao P, Huang H, Zhao H, Liu R, Sun Z, Liu
Y, Chen N and Zhang Z: Edaravone dexborneol protects against
cerebral ischemia/reperfusion-induced blood-brain barrier damage by
inhibiting ferroptosis via activation of nrf-2/HO-1/GPX4 signaling.
Free Radical Biol Med. 217:116–125. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Sheng W, Wu Z, Wei J, Wang J, Zhang S,
Ding Z, Zhong J, Deng D, Zhong Z, Yin Y, et al: Astrocyte-derived
CXCL10 exacerbates endothelial cells pyroptosis and blood-brain
barrier disruption via CXCR3/cGAS/AIM2 pathway after intracerebral
hemorrhage. Cell Death Discovery. 11:3732025. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Lan X, Wang Q, Liu Y, You Q, Wei W, Zhu C,
Hai D, Cai Z, Yu J, Zhang J and Liu N: Isoliquiritigenin alleviates
cerebral ischemia-reperfusion injury by reducing oxidative stress
and ameliorating mitochondrial dysfunction via activating the Nrf2
pathway. Redox Biol. 77:1034062024. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Yingze Y, Zhihong J, Tong J, Yina L, Zhi
Z, Xu Z, Xiaoxing X and Lijuan G: NOX2-mediated reactive oxygen
species are double-edged swords in focal cerebral ischemia in mice.
J Neuroinflammation. 19:1842022. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Shen S, He F, Cheng C, Xu B and Sheng J:
Uric acid aggravates myocardial ischemia-reperfusion injury via
ROS/NLRP3 pyroptosis pathway. Biomed Pharmacother. 133:1109902021.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Akbal A, Dernst A, Lovotti M, Mangan MSJ,
McManus RM and Latz E: How location and cellular signaling combine
to activate the NLRP3 inflammasome. Cell Mol Immunol. 19:1201–1214.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Fan L, Lin X, Hong L, Li L, Lin R, Ren T,
Tian J and Chen M: Simultaneous antioxidant and neuroprotective
effects of two-dimensional (2D) MXene-loaded isoquercetin for
ischemic stroke treatment. J Mater Chem B. 12:2795–2806. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Zhang M, Liu Q, Meng H, Duan H, Liu X, Wu
J, Gao F, Wang S, Tan R and Yuan J: Ischemia-reperfusion injury:
Molecular mechanisms and therapeutic targets. Signal Transduct
Target Ther. 9:122024. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Rahi V and Kaundal RK: Exploring the
intricacies of calcium dysregulation in ischemic stroke: Insights
into neuronal cell death and therapeutic strategies. Life Sci.
347:1226512024. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Neves D, Salazar IL, Almeida RD and Silva
RM: Molecular mechanisms of ischemia and glutamate excitotoxicity.
Life Sci. 328:1218142023. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Liu L, Wang Y, Wang X, Zhang G, Sha S,
Zhou R, Du Y, Wu C and Chen L: Transient receptor potential
vanilloid 4 blockage attenuates pyroptosis in hippocampus of mice
following pilocarpine-induced status epilepticus. Acta Neuropathol
Commun. 13:732025. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Mo G, Liu X, Zhong Y, Mo J, Li Z, Li D,
Zhang L and Liu Y: IP3R1 regulates Ca2+ transport and
pyroptosis through the NLRP3/Caspase-1 pathway in myocardial
ischemia/reperfusion injury. Cell Death Discovery. 7:312021.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Ye J, Bi X, Deng S, Wang X, Liu Z, Suo Q,
Wu J, Chen H, Wang Y, Qian K, et al: Hypoxanthine is a metabolic
biomarker for inducing GSDME-dependent pyroptosis of endothelial
cells during ischemic stroke. Theranostics. 14:6071–6087. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Zheng Y, Xu X, Chi F and Cong N:
Pyroptosis: A newly discovered therapeutic target for
ischemia-reperfusion injury. Biomolecules. 12:16252022. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Zhang X, Zeng W, Zhang Y, Yu Q, Zeng M,
Gan J, Zhang W, Jiang X and Li H: Focus on the role of mitochondria
in NLRP3 inflammasome activation: A prospective target for the
treatment of ischemic stroke (review). Int J Mol Med. 49:742022.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Newman LE and Shadel GS: Mitochondrial DNA
release in innate immune signaling. Annu Rev Biochem. 92:299–332.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Huang LS, Hong Z, Wu W, Xiong S, Zhong M,
Gao X, Rehman J and Malik AB: mtDNA Activates cGAS signaling and
suppresses the YAP-Mediated endothelial cell proliferation program
to promote inflammatory injury. Immunity. 52:475–486.e5. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Shen N, Kong L, Wang X, Zhang Y, Li R, Tao
C, Wang G, Xu P and Hu W: Elabela ameliorates neuronal pyroptosis
and mitochondrial fission via APJ/ZBP1 signaling in ischemic
stroke. Exp Neurol. 378:1148022024. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Yao M, Wang X, Lin H, Shu H, Xu Z, Tang L,
Guo W and Xu P: LncRNA Tug1 regulates post-stroke microglial
pyroptosis via PINK1/parkin-mediated mitophagy. Inflammation.
48:2677–2691. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Ghafouri-Fard S, Shoorei H, Poornajaf Y,
Hussen BM, Hajiesmaeili Y, Abak A, Taheri M and Eghbali A: NLRP3:
Role in ischemia/reperfusion injuries. Front Immunol.
13:9268952022. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Broz P: Pyroptosis: Molecular mechanisms
and roles in disease. Cell Res. 35:334–344. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Zhang M, Guo B, Zhang X, Han D, Lv L, Yan
X, Su C, Chai D, Zhao N, Yan X and Hu S: IFP35, a novel DAMP,
aggravates neuroinflammation following acute ischemic stroke via
TLR4/NF-κB/NLRP3 signaling. J Neuroinflammation. 22:1642025.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Schachter J, Guijarro A, Angosto-Bazarra
D, Pinilla M, Hurtado-Navarro L, Meunier E, Pérez-Oliva AB,
Schwarzbaum PJ and Pelegrin P: Gasdermin D mediates a fast
transient release of ATP after NLRP3 inflammasome activation before
ninjurin 1-induced lytic cell death. Cell Rep. 44:1152332025.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Li S, Fang Y, Zhang Y, Song M, Zhang X,
Ding X, Yao H, Chen M, Sun Y, Ding J, et al: Microglial NLRP3
inflammasome activates neurotoxic astrocytes in depression-like
mice. Cell Rep. 41:1115322022. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Schmacke NA, O'Duill F, Gaidt MM,
Szymanska I, Kamper JM, Schmid-Burgk JL, Mädler SC, Mackens-Kiani
T, Kozaki T, Chauhan D, et al: IKKβ primes inflammasome formation
by recruiting NLRP3 to the trans-golgi network. Immunity.
55:2271–2284.e7. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Hauptmann J, Johann L, Marini F, Kitic M,
Colombo E, Mufazalov IA, Krueger M, Karram K, Moos S, Wanke F, et
al: Interleukin-1 promotes autoimmune neuroinflammation by
suppressing endothelial heme oxygenase-1 at the blood-brain
barrier. Acta Neuropathol. 140:549–567. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Zheng ZJ, Zhu LZ, Qiu H, Zheng WYX, You
PT, Chen SH, Hu CL, Huang JR and Zhou YJ: Neferine inhibits BMECs
pyroptosis and maintains blood-brain barrier integrity in ischemic
stroke by triggering a cascade reaction of PGC-1α. Sci Rep.
14:144382024. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Li J, Wang Z, Li J, Zhao H and Ma Q:
HMGB1: A new target for ischemic stroke and hemorrhagic
transformation. Transl Stroke Res. 16:990–1015. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Cavalcanti RR, Almeida FM, Martinez AMB
and Freria CM: Neuroinflammation: Targeting microglia for
neuroprotection and repair after spinal cord injury. Front Immunol.
16:16706502025. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Clifford T, Finkel Z, Rodriguez B, Joseph
A and Cai L: Current advancements in spinal cord injury
research-glial scar formation and neural regeneration. Cells.
12:8532023. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Kayagaki N, Kornfeld OS, Lee BL, Stowe IB,
O'Rourke K, Li Q, Sandoval W, Yan D, Kang J, Xu M, et al: NINJ1
mediates plasma membrane rupture during lytic cell death. Nature.
591:131–136. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Ma H, Zhu Y, Zhao X, Jiang L, Yang J, Yang
T and Liu W: Pyroptosis: Inflammatory cell death mechanism and its
pathological roles in neurological diseases and injuries.
Apoptosis. 30:2057–2076. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Hu R, Liang J, Ding L, Zhang W, Wang Y,
Zhang Y, Zhang D, Pei L, Liu X, Xia Z, et al: Gasdermin D
inhibition ameliorates neutrophil mediated brain damage in acute
ischemic stroke. Cell Death Discovery. 9:502023. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Zhang Q, Jia M, Wang Y, Wang Q and Wu J:
Cell death mechanisms in cerebral Ischemia-reperfusion injury.
Neurochem Res. 47:3525–3542. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Kang R, Zeng L, Zhu S, Xie Y, Liu J, Wen
Q, Cao L, Xie M, Ran Q, Kroemer G, et al: Lipid peroxidation drives
gasdermin D-mediated pyroptosis in lethal polymicrobial sepsis.
Cell Host Microbe. 24:97–108.e4. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Wang C and Ruan J: Mechanistic insights
into gasdermin pore formation and regulation in pyroptosis. J Mol
Biol. 434:1672972022. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Huang Y, Yang J, Lu T, Shao C and Wan H:
Puerarin alleviates cerebral ischemia-reperfusion injury by
inhibiting ferroptosis through SLC7A11/GPX4/ACSL4 axis and
alleviate pyroptosis through caspase-1/GSDMD axis. Mol Neurobiol.
62:8931–8948. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Wang Z, Li Y, Ye Y, Zhu H, Zhang J, Wang
H, Lei J, Gu L and Zhan L: NLRP3 inflammasome deficiency attenuates
cerebral ischemia-reperfusion injury by inhibiting ferroptosis.
Brain Res Bull. 193:37–46. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Zhao P, Yin S, Qiu Y, Sun C and Yu H:
Ferroptosis and pyroptosis are connected through autophagy: A new
perspective of overcoming drug resistance. Mol Cancer. 24:232025.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Yamamoto H, Zhang S and Mizushima N:
Autophagy genes in biology and disease. Nat Rev Genet. 24:382–400.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Tang L, Zhang W, Liao Y, Wang W, Deng X,
Wang C and Shi W: Autophagy: A double-edged sword in
ischemia-reperfusion injury. Cell Mol Biol Lett. 30:422025.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Liu L, Wang R, Gao W, Hou X, Jin X, Zhao
Y, Zhou X and Zhang Y: Drug pairs of huangqi and dnggui alleviates
pyroptosis by promoting autophagy activity via AMPK/mTOR signaling
pathway in middle-cerebral artery occlusion/reperfusion in rats. J
Ethnopharmacol. 337:1189822025. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Liu H, Zhao Z, Wu T, Zhang Q, Lu F, Gu J,
Jiang T and Xue J: Inhibition of autophagy-dependent pyroptosis
attenuates cerebral ischaemia/reperfusion injury. J Cell Mol Med.
25:5060–5069. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Dho SH, Cho M, Woo W, Jeong S and Kim LK:
Caspases as master regulators of programmed cell death: Apoptosis,
pyroptosis and beyond. Exp Mol Med. 57:1121–1132. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Zhao M, Xian W, Liu W, Chen D, Wang S and
Cao J: Maresin1 alleviates neuroinflammation by inhibiting
caspase-3/GSDME-mediated pyroptosis in mice cerebral
ischemia-reperfusion model. J Stroke Cerebrovasc Dis.
33:1077892024. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Demarco B, Grayczyk JP, Bjanes E, Le Roy
D, Tonnus W, Assenmacher CA, Radaelli E, Fettrelet T, Mack V,
Linkermann A, et al: Caspase-8-dependent gasdermin D cleavage
promotes antimicrobial defense but confers susceptibility to
TNF-induced lethality. Sci Adv. 6:eabc34652020. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Yamazaki T and Galluzzi L: BAX and BAK
dynamics control mitochondrial DNA release during apoptosis. Cell
Death Differ. 29:1296–1298. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Muendlein HI, Jetton D, Connolly WM,
Eidell KP, Magri Z, Smirnova I and Poltorak A: cFLIPL protects
macrophages from LPS-induced pyroptosis via inhibition of complex
II formation. Science. 367:1379–1384. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Zhu C, Xu S, Jiang R, Yu Y, Bian J and Zou
Z: The gasdermin family: Emerging therapeutic targets in diseases.
Signal Transduction Targeted Ther. 9:872024. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Bertheloot D, Latz E and Franklin BS:
Necroptosis, pyroptosis and apoptosis: An intricate game of cell
death. Cell Mol Immunol. 18:1106–1121. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Lawlor KE, Murphy JM and Vince JE:
Gasdermin and MLKL necrotic cell death effectors: Signaling and
diseases. Immunity. 57:429–445. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Lei X, Chen Y, Lien E and Fitzgerald KA:
MLKL-driven inflammasome activation and caspase-8 mediate
inflammatory cell death in influenza a virus infection. Mbio.
14:e00110232023. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Gao J, Xiong A, Liu J, Li X, Wang J, Zhang
L, Liu Y, Xiong Y, Li G and He X: PANoptosis: Bridging apoptosis,
pyroptosis, and necroptosis in cancer progression and treatment.
Cancer Gene Ther. 31:970–983. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Yan WT, Yang YD, Hu XM, Ning WY, Liao LS,
Lu S, Zhao WJ, Zhang Q and Xiong K: Do pyroptosis, apoptosis, and
necroptosis (PANoptosis) exist in cerebral ischemia? Evidence from
cell and rodent studies. Neural Regen Res. 17:1761–1768. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Lisjak D, Alić I, Šimunić I and Mitrečić
D: Transplantation of neural stem cells improves recovery of
stroke-affected mice and induces cell-specific changes in GSDMD and
MLKL expression. Front Mol Neurosci. 17:14399942024. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Xiao Y, Zhao C, Tai Y, Li B, Lan T, Lai E,
Dai W, Guo Y, Gan C, Kostallari E, et al: STING mediates hepatocyte
pyroptosis in liver fibrosis by epigenetically activating the NLRP3
inflammasome. Redox Biol. 62:1026912023. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Jiang M, Li R, Lyu J, Li X, Wang W, Wang
Z, Sheng H, Zhang W, Karhausen J and Yang W: MCC950, a selective
NLPR3 inflammasome inhibitor, improves neurologic function and
survival after cardiac arrest and resuscitation. J
Neuroinflammation. 17:2562020. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Xin Q, Xu F, Ma Z and Wu J:
β-caryophyllene mitigates ischemic stroke-induced white matter
lesions by inhibiting pyroptosis. Exp Cell Res. 442:1142142024.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Xia P, Marjan M, Liu Z, Zhou W, Zhang Q,
Cheng C, Zhao M, Tao Y, Wang Z and Ye Z: Chrysophanol
postconditioning attenuated cerebral ischemia-reperfusion injury
induced NLRP3-related pyroptosis in a TRAF6-dependent manner. Exp
Neurol. 357:1141972022. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Li J, Xu P, Hong Y, Xie Y, Peng M, Sun R,
Guo H, Zhang X, Zhu W, Wang J, et al: Lipocalin-2-mediated
astrocyte pyroptosis promotes neuroinflammatory injury via NLRP3
inflammasome activation in cerebral ischemia/reperfusion injury. J
Neuroinflammation. 20:1482023. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Wang L, Ren W, Wu Q, Liu T, Wei Y, Ding J,
Zhou C, Xu H and Yang S: NLRP3 inflammasome activation: A
therapeutic target for cerebral ischemia-reperfusion injury. Front
Mol Neurosci. 15:8474402022. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Du Y, Chen L, Qiao H, Zhang L, Yang L,
Zhang P, Wang J, Zhang C, Jiang W, Xu R and Zhang X: Hydrogen-rich
saline-a novel neuroprotective agent in a mouse model of
experimental cerebral ischemia via the ROS-NLRP3 inflammasome
signaling pathway In vivo and In vitro. Brain Sci. 13:9392023.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Li Z, Pang Y, Hou L, Xing X, Yu F, Gao M,
Wang J, Li X, Zhang L and Xiao Y: Exosomal OIP5-AS1 attenuates
cerebral ischemia-reperfusion injury by negatively regulating TXNIP
protein stability and inhibiting neuronal pyroptosis. Int
Immunopharmacol. 127:1113102024. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Liu H, Chen W, He M, Nie L, Pan Y, Guan D,
Li Y, Wan T, Duan L, Yang C, et al: Methyl isoeugenol suppresses
NLRP3 inflammasome-mediated pyroptosis via activation of
Nrf2/NQO1/HO-1 signaling in cerebral ischemia-reperfusion injury.
Biochem Pharmacol. 237:1169472025. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Paik S, Kim JK, Silwal P, Sasakawa C and
Jo EK: An update on the regulatory mechanisms of NLRP3 inflammasome
activation. Cell Mol Immunol. 18:11412021. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Sun K, Zhang J, Yang Q, Zhu J, Zhang X, Wu
K, Li Z, Xie W and Luo X: Dexmedetomidine exerts a protective
effect on ischemic brain injury by inhibiting the
P2X7R/NLRP3/caspase-1 signaling pathway. Brain Res Bull. 174:11–21.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Zou X, Xie L, Wang W, Zhao G, Tian X and
Chen M: FK866 alleviates cerebral pyroptosis and inflammation
mediated by Drp1 in a rat cardiopulmonary resuscitation model. Int
Immunopharmacol. 89:1070322020. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Ma CS, Ma YP, Han B, Duan WL, Meng SC, Bai
M, Dong H, Zhang LY, Duan MY, Liu J, et al: Apelin-13-loaded
macrophage membrane-encapsulated nanoparticles for targeted
ischemic stroke therapy via inhibiting NLRP3 inflammasome-mediated
pyroptosis. Int J Nanomed. 19:9175–9193. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
An J, Ouyang L, Yu C, Carr SM, Ramprasath
T, Liu Z, Song P, Zou MH and Ding Y: Nicotine exacerbates
atherosclerosis and plaque instability via NLRP3 inflammasome
activation in vascular smooth muscle cells. Theranostics.
13:2825–2842. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Li F, Xu D, Hou K, Gou X, Lv N, Fang W and
Li Y: Pretreatment of indobufen and aspirin and their combinations
with clopidogrel or ticagrelor alleviates inflammasome mediated
pyroptosis via inhibiting NF-κB/NLRP3 pathway in ischemic stroke. J
Neuroimmune Pharmacol. 16:835–853. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Li L, Zha H, Miao W, Li C, Wang A, Qin S,
Gao S, Sheng L and Wang Y: LncRNA MEG3 promotes pyroptosis via
miR-145-5p/TLR4/NLRP3 axis and aggravates cerebral
ischemia-reperfusion injury. Metab Brain Dis. 40:2012025.
View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Zhang Y, Wu Q, Pan Y, Xin M, Wang W, Zhai
X, Zhou P, Zhang C and Wang Y: Intracerebroventricular calycosin
attenuates cerebral ischemia-reperfusion injury in rats via
HMGB1-dependent pyroptosis inhibition. Front Pharmacol.
16:15960872025. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Qin Y, Ma G, Xiao X, Liu Y, Zhao Z, Zhao
F, Guo F, Wang S, Sun X and Gao C: Electroacupuncture at ST36
treatment suppresses the microglial pyroptosis through activating
α7nAChR in a rat model of asphyxial cardiac arrest. J Inflammation
Res. 18:8705–8718. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Li W, Shen N, Kong L, Huang H, Wang X,
Zhang Y, Wang G, Xu P and Hu W: STING mediates microglial
pyroptosis via interaction with NLRP3 in cerebral ischaemic stroke.
Stroke Vasc Neurol. 9:153–164. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Zhang H, Ni W, Yu G, Geng Y, Lou J, Qi J,
Chen Y, Li F, Ye H, Ma H, et al: 3,4-dimethoxychalcone, a caloric
restriction mimetic, enhances TFEB-mediated autophagy and
alleviates pyroptosis and necroptosis after spinal cord injury.
Theranostics. 13:810–832. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Liang Y, Song P, Chen W, Xie X, Luo R, Su
J, Zhu Y, Xu J, Liu R, Zhu P, et al: Inhibition of caspase-1
ameliorates ischemia-associated blood-brain barrier dysfunction and
integrity by suppressing pyroptosis activation. Front Cell
Neurosci. 14:5406692020. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Kim H, Seo JS, Lee SY, Ha KT, Choi BT,
Shin YI, Ju Yun Y and Shin HK: AIM2 inflammasome contributes to
brain injury and chronic post-stroke cognitive impairment in mice.
Brain Behav Immun. 87:765–776. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Jin F, Jin L, Wei B, Li X, Li R, Liu W,
Guo S, Fan H and Duan C: miR-96-5p alleviates cerebral
ischemia-reperfusion injury in mice by inhibiting pyroptosis via
downregulating caspase 1. Exp Neurol. 374:1146762024. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Xia S, Zhang Z, Magupalli VG, Pablo JL,
Dong Y, Vora SM, Wang L, Fu TM, Jacobson MP, Greka A, et al:
Gasdermin D pore structure reveals preferential release of mature
interleukin-1. Nature. 593:607–611. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Wang K, Sun Z, Ru J, Wang S, Huang L, Ruan
L, Lin X, Jin K, Zhuge Q and Yang S: Ablation of GSDMD improves
outcome of ischemic stroke through blocking canonical and
Non-canonical inflammasomes dependent pyroptosis in microglia.
Front Neurol. 11:5779272020. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Zhao N, Hua W, Liu Q, Wang Y, Liu Z, Jin
S, Wang B, Pang Y, Qi J and Song Y: MALAT1 knockdown alleviates the
pyroptosis of microglias in diabetic cerebral ischemia via
regulating STAT1 mediated NLRP3 transcription. Mol Med. 29:442023.
View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Luo L, Liu M, Fan Y, Zhang J, Liu L, Li Y,
Zhang Q, Xie H, Jiang C, Wu J, et al: Intermittent theta-burst
stimulation improves motor function by inhibiting neuronal
pyroptosis and regulating microglial polarization via
TLR4/NFκB/NLRP3 signaling pathway in cerebral ischemic mice. J
Neuroinflammation. 19:1412022. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Meng D, Su Q, Jia K, Jin M, Liu W, Yang S
and Su Z: ETV5 improves ischemic stroke by targeting HACE1/Nrf2
axis to inhibit microglia M1 polarization and neuron pyroptosis.
Biochem Pharmacol. 240:1170572025. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Ma DC, Zhang NN, Zhang YN and Chen HS:
Salvianolic acids for injection alleviates cerebral
ischemia/reperfusion injury by switching M1/M2 phenotypes and
inhibiting NLRP3 inflammasome/pyroptosis axis in microglia in vivo
and in vitro. J Ethnopharmacol. 270:1137762021. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Bonam SR, Mastrippolito D, Georgel P and
Muller S: Pharmacological targets at the lysosomal autophagy-NLRP3
inflammasome crossroads. Trends Pharmacol Sci. 45:81–101. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Zhang J, Li R, Wang L and Ni S:
Dexmedetomidine activates mitophagy and protects against pyroptosis
in oxygen-glucose deprivation/reperfusion-induced brain damage via
PINK1/parkin pathway activation. J Bioenerg Biomembr. 57:183–195.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Qiu Z, Ma J, Zhang X, Jiao M and Zhi L:
Electroacupuncture combined with trigonelline inhibits pyroptosis
in cerebral ischemia-reperfusion by suppressing autophagy via the
PI3K/AKT/mTOR signaling pathway. Brain Res Bull. 221:1112002025.
View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Wei C, Shi M, Dong S, Li Z, Zhao B, Liu D,
Li G, Cen J, Yu L, Liang X and Shi L: SIRT5-related lysine
demalonylation of GSTP1 contributes to cardiomyocyte pyroptosis
suppression in diabetic cardiomyopathy. Int J Biol Sci. 20:585–605.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Liu Y, Jiang X, Zhang Y, Tong G, Tang K,
Gui Y, Wen L and Li C: miR-135a-5p alleviates cerebral
ischemia-reperfusion injury by inhibiting pyroptosis mediated
through the DDX3X/NLRP3 pathway. Exp Neurol. 385:1151272025.
View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Zhao Y, He X, Yang X, Hong Z, Xu Y, Xu J,
Zheng H, Zhang L, Zuo Z and Hu X: CircFndc3b mediates
exercise-induced neuroprotection by mitigating
microglial/macrophage pyroptosis via the ENO1/KLF2 axis in stroke
mice. Adv Sci (Weinh). 12:e24038182025. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Wang LQ, Zheng YY, Zhou HJ, Zhang XX, Wu P
and Zhu SM: LncRNA-fendrr protects against the ubiquitination and
degradation of NLRC4 protein through HERC2 to regulate the
pyroptosis of microglia. Mol Med. 27:392021. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Ding S, Kim YJ, Huang KY, Um D, Jung Y and
Kong H: Delivery-mediated exosomal therapeutics in
ischemia-reperfusion injury: Advances, mechanisms, and future
directions. Nano Converg. 11:182024. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Sun R, Liao W, Lang T, Qin K, Jiao K, Shao
L, Deng C and She Y: Astrocyte-derived exosomal miR-378a-5p
mitigates cerebral ischemic neuroinflammation by modulating
NLRP3-mediated pyroptosis. Front Immunol. 15:14541162024.
View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Liu X, Zhang M, Liu H, Zhu R, He H, Zhou
Y, Zhang Y, Li C, Liang D, Zeng Q and Huang G: Bone marrow
mesenchymal stem cell-derived exosomes attenuate cerebral
ischemia-reperfusion injury-induced neuroinflammation and
pyroptosis by modulating microglia M1/M2 phenotypes. Exp Neurol.
341:1137002021. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Prakash R, Vyawahare A, Sakla R, Kumari N,
Kumar A, Ansari MM, Kanika N, Jori C, Waseem A, Siddiqui AJ, et al:
NLRP3 inflammasome-targeting nanomicelles for preventing
ischemia-reperfusion-induced inflammatory injury. ACS Nano.
17:8680–8693. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Cheng Z, Shao W, Wei C, Zhang Y, Xiao R,
Zhang S, Zhang J, She Y, Pan C, Liu Q and Wang Q: Cascade-type
microglial pyroptosis inhibitors for enhanced treatment of cerebral
ischemia-reperfusion injury. ACS Nano. 19:10529–10548. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Wu D, Chen Q, Chen X, Han F, Chen Z and
Wang Y: The blood-brain barrier: Structure, regulation, and drug
delivery. Signal Transduct Target Ther. 8:2172023. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Meng W, Huang L, Guo J, Xin Q, Liu J and
Hu Y: Innovative nanomedicine delivery: Targeting tumor
microenvironment to defeat drug resistance. Pharmaceutics.
16:15492024. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Pardridge WM: Advanced Blood-brain barrier
drug delivery. Pharmaceutics. 15:932022. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Perkins GD, Neumar R, Hsu CH, Hirsch KG,
Aneman A, Becker LB, Couper K, Callaway CW, Hoedemaekers CWE, Lim
SL, et al: Improving outcomes after post-cardiac arrest brain
injury: A scientific statement from the international liaison
committee on resuscitation. Circulation. Jun 27–2024.(Epub ahead of
print). doi: 10.1161/CIR.0000000000001219. View Article : Google Scholar
|
|
159
|
Chen H, Shi Y, Yuan M, Li H and Liu X:
Integrating multi-omics and artificial intelligence fuels advanced
target identification and drug discovery. Biotechnol Adv.
87:1087852026. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Wan N, Shi J, Xu J, Huang J, Gan D, Tang
M, Li X, Huang Y and Li P: Gasdermin D: A potential new auxiliary
Pan-biomarker for the detection and diagnosis of diseases.
Biomolecules. 13:16642023. View Article : Google Scholar : PubMed/NCBI
|