Introduction
Pulmonary fibrosis, a common end-stage manifestation
of various interstitial lung diseases, arises from incompletely
elucidated etiologies involving diverse risk factors such as
environmental exposures, smoking, genetic predisposition and
autoimmune disorders. This etiological complexity hinders the
identification of viable therapeutic targets (1). Idiopathic pulmonary fibrosis (IPF)
refers to pulmonary fibrosis of unknown cause. Its pathological
core involves excessive deposition of extracellular matrix (ECM),
which disrupts alveolar architecture, leading to the formation of
extensive fibrotic scars and ultimately irreversible loss of lung
function. As the most prevalent interstitial lung disease, IPF
predominantly affects middle-aged and elderly individuals and
carries a poor prognosis. Data indicate that untreated IPF patients
have a median survival of only 2–6 years following diagnosis
(2,3). Epidemiological studies as of 2020
report a global IPF incidence ranging from 1–13 per 100,000
person-years and a prevalence of 3 to 45 per 100,000 population.
The highest incidence and prevalence rates of IPF are observed in
three regions: Korea, Canada and the US (4). A meta-analysis of global studies
estimated 3- and 5-year cumulative survival rates for IPF patients
at 61.8 and 45.6%, respectively (5). Therefore, developing precise and
effective treatments for pulmonary fibrosis is imperative.
In recent years, the therapeutic focus for IPF has
shifted from anti-inflammatory strategies to antifibrotic agents
and comorbidity management (6).
However, no reliable cure exists. Current pharmacologic options for
IPF are limited. Although nintedanib and pirfenidone are US
FDA-approved for IPF and can modestly slow disease progression
(7). Moreover, a significant
proportion of patients on long-term treatment with these drugs
experience severe gastrointestinal adverse effects. Lung
transplantation remains the only curative intervention, but it is
not widely accessible due to limitations in donor organ
availability, economic constraints and challenges in postoperative
care. Consequently, there is a pressing need to develop novel,
low-toxicity and high-efficiency antifibrotic therapies to
alleviate symptoms and improve survival.
S1P signaling has been extensively investigated in
the context of lung diseases. Previous reviews by Ebenezer et
al (8) and Mohammed and
Harikumar et al (9)
summarize the roles of S1P signaling across a broad spectrum of
pulmonary disorders, with a primary focus on immune regulation,
inflammatory responses and vascular-related functions. By contrast,
the present review focusedon pulmonary fibrosis and provides a
mechanism-oriented synthesis of recent advances regarding the roles
of S1P signaling ininflammation, fibroblast-to-myofibroblast
transition (FMT), epithelial-to-mesenchymal transition (EMT),
autophagy and oxidative stress, with the aim of clarifying its
functional relevance and potential therapeutic implications in
pulmonary fibrosis.
Overview of S1P
S1P is a bioactive lipid mediator, primarily
synthesized and secreted into the circulation by erythrocytes and
platelets. As a critical intercellular signaling molecule, S1P
regulates essential physiological processes, such as cell
proliferation, survival, migration, apoptosis, angiogenesis and
inflammation. Through the modulation of these core cellular
functions, S1P plays a significant role in complex
pathophysiological conditions, including immune response,
angiogenesis and fibrosis (10,11).
S1P is synthesized through a stepwise enzymatic pathway.
Sphingomyelin is first hydrolyzed to ceramide (Cer) by
sphingomyelinase. Cer is then converted to sphingosine (Sph) by
ceramidase. Finally, Sph is phosphorylated by Sph kinases (SphKs)
to generate S1P (12). This
intracellular synthesis primarily depends on two key rate-limiting
enzymes, SphK1 and SphK2. Although SphK1 and SphK2 share high
sequence homology, they exhibit distinct differences in subcellular
localization, tissue distribution and biological functions
(13,14). SphK1 is predominantly cytosolic but
can translocate to the plasma membrane or nucleus to exert its
catalytic activity, thereby promoting cell proliferation and
survival (15,16). By contrast, SphK2 is primarily
localized within organelles such as the nucleus and endoplasmic
reticulum and is more closely associated with inhibiting DNA
synthesis and inducing apoptosis (17).
Cellular S1P levels are maintained by a tightly
regulated dynamic balance between its synthesis and degradation.
S1P degradation proceeds via two principal pathways: Reversible
dephosphorylation back to Sph by S1P phosphatases (SPP) and
irreversible catabolization to hexadecenal and
phosphor-ethanolamine by S1P lyase (S1PL) (18–20).
In tissues, low S1P concentrations are maintained due to efficient
degradation by S1PL. By contrast, erythrocytes and platelets lack
S1PL, resulting in the high S1P concentration characteristic of
blood. S1P mediates signaling through both intracellular and
extracellular mechanisms. While its extracellular signaling is
well-established, its intracellular roles remain less defined.
Intracellularly, S1P can function as a second messenger, directly
targeting proteins such as histone deacetylase (HDAC) to influence
Ca2+ homeostasis, gene transcription and protein
modification (21). S1P can act
via autocrine mechanisms or be exported to the extracellular milieu
by transporters, including specific ATP-binding cassette (ABC)
transporters and spinster homolog 2 (Spns2) transporters. Once
extracellular, S1P binds to five S1PRs on the target cell surface,
initiating diverse downstream signaling cascades (22–24).
Extracellular S1P signaling via S1PRs has been
extensively characterized. S1PRs are expressed on nearly all cell
types, with particularly high levels found on immune cells such as
neutrophils, dendritic cells, natural killer cells and macrophages
(11). S1PR1, highly expressed on
endothelial and immune cells, promotes cell proliferation,
migration, angiogenesis and immune regulation through pathways
including mitogen-activated protein kinase (MAPK), phosphoinositide
3-kinase (PI3K)/protein kinase B (AKT) and extracellular
signal-regulated kinase 1/2 (ERK1/2) (25). S1PR2 and S1PR3 are predominantly
expressed on fibroblasts and epithelial cells, where they regulate
cell proliferation and extracellular matrix synthesis. S1PR4 is
found in hematopoietic and lymphoid tissues, as well as the lung
and is implicated in immune regulation and tumor suppression. S1PR5
is primarily expressed in the central nervous system and is
involved in neuromodulation. To date, research on S1PRs in fibrotic
diseases has largely focused on S1PR1-3, with the roles of S1PR4
and S1PR5 being less explored (Fig.
1).
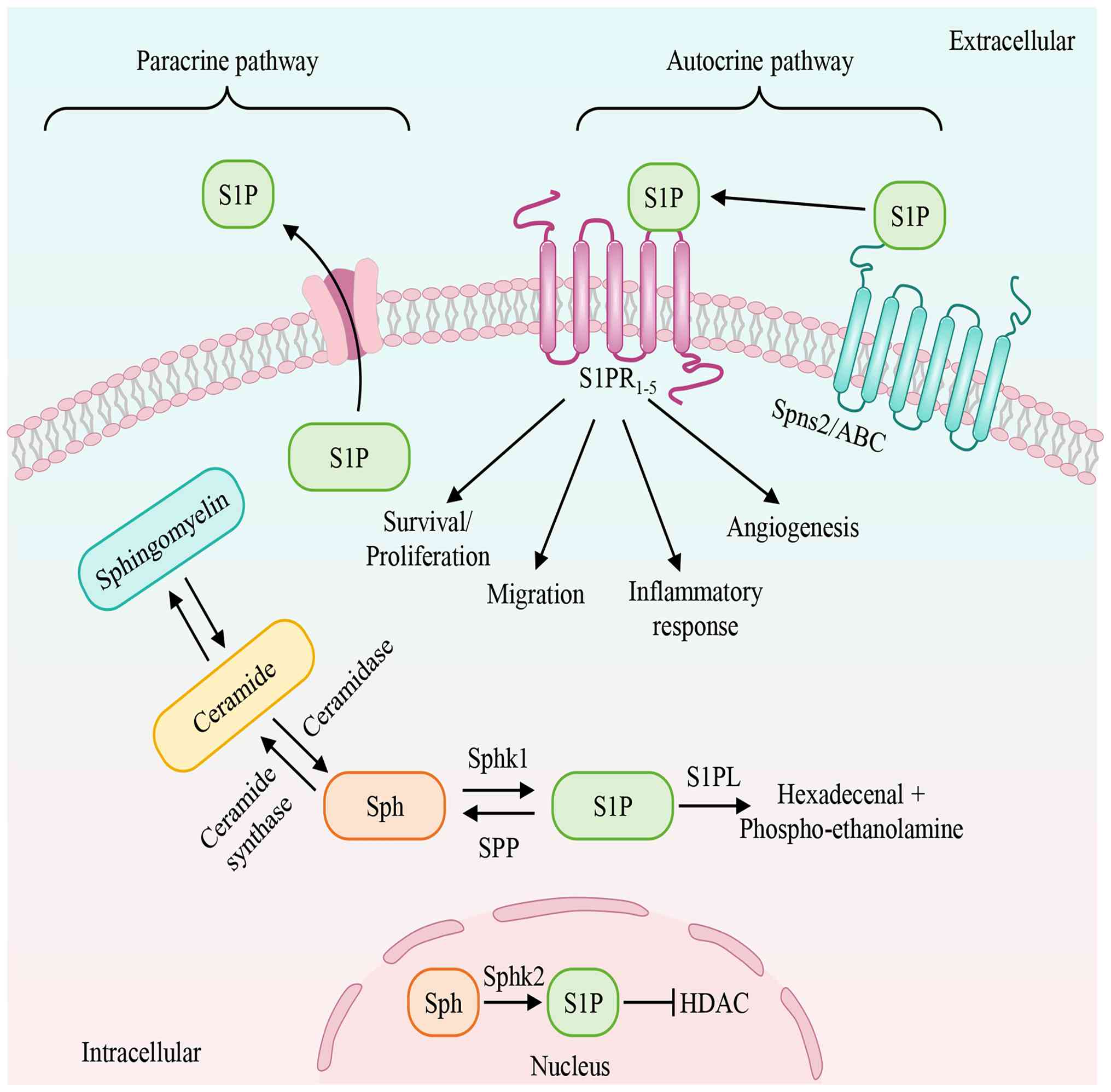 | Figure 1.Synthesis, degradation and transport
of S1P. This figure depicts the intracellular synthesis,
degradation and transport of S1P, leading to extracellular
signaling. Sphingomyelinases catalyzes the conversion of
sphingomyelin to produce Cer, which is then converted into Sph by
ceramidase. S1P can be generated by SphK1 and SphK2, while S1P
produced by SphK2 in the nucleus inhibits HDAC activity. Following
S1P generation, SPL degrades it into phosphor-ethanolamine and
hexadecenal, or it is exported out of the cell by Spns2 and ABC,
acting on extracellular S1PRs via autocrine or paracrine
mechanisms. By binding to different receptors, S1P regulates
numerous physiological and pathological processes. ‘→’ represents
‘activation’; ‘—|’ represents ‘inhibition’.S1P,
sphingosine-1-phosphate; Sph, sphingosine; SphK1, sphingosine
kinase 1; SphK2 sphingosine kinase 2; HDAC, histone deacetylase;
S1PRs, sphingosine-1-phosphate receptors; Spns2, spinster homolog
2; ABC, ATP-binding cassette. |
Molecular mechanisms of S1P in pulmonary
fibrosis
Massive fibroblast proliferation and ECM
accumulation constitute the fundamental pathological hallmarks of
pulmonary fibrosis (26). Repeated
injury to alveolar epithelial cells initiates an inflammatory
response that recruits inflammatory cells to the site of damage.
These infiltrating inflammatory cells subsequently generate and
secrete abundant pro-fibrotic factors, thereby stimulating
fibroblast proliferation, promoting their differentiation into
myofibroblasts and ultimately leading to excessive ECM deposition
(27). Disruption of the balance
between tissue injury and repair results in lung scarring and
progressive architectural distortion, which impairs gas exchange
and manifests as overt pulmonary fibrosis.
S1P is a crucial bioactive sphingolipid metabolite
that functions as both an extracellular and intracellular signaling
mediator. It has been shown to regulate physiological and
pathophysiological processes in a variety of diseases. For
instance, in malignancies, S1P accelerates disease progression by
remodeling the tumor microenvironment and promoting angiogenesis,
invasion and metastasis (28,29).
In brain injury pathology, S1P participates in modulating
neuroinflammatory responses and neuronal survival (30). Similarly, in cardiovascular and
pulmonary diseases, S1P is recognized as a key molecular driver of
disease progression (31,32). In recent years, attention on the
role of S1P in fibrotic diseases has steadily increased.
Accumulating evidence indicates that S1P plays a central
pro-fibrotic role in the fibrosis of multiple tissues and organs,
including the lung, liver and kidney (33–35).
The SphK1/S1P/S1PRs signaling axis has been identified as a
critical pathway mediating pathological injury in various diseases,
including tissue fibrosis (36),
thereby offering important insights for mechanistic studies and
therapeutic target discovery in fibrotic diseases.
The dysregulated role of S1P in IPF, a clinically
refractory form of pulmonary fibrosis, has been preliminarily
established. Clinical study reveals that S1P levels are such as
elevated in bronchoalveolar lavage fluid (BALF) and lung tissues
from IPF patients compared with healthy controls and its expression
correlates inversely with key pulmonary function parameters;
specifically, diffusion capacity for carbon monoxide, expiratory
volume and lung volumes (37).
Investigations using clinical specimens and animal models further
confirm that S1P concentrations are markedly increased in plasma,
lung tissue and BALF from pulmonary fibrosis patients and
bleomycin-induced mice, with this upregulation closely linked to
the severity of lung function impairment. Notably, SphK1 expression
is also upregulated in fibrotic lungs; selective silencing of SphK1
not only effectively reduces endogenous S1P levels but also such as
attenuates the severity of bleomycin-induced pulmonary fibrosis and
decreases mortality in mice (38).
Although current studies suggest a potential pivotal
role for S1P in the pathogenesis of pulmonary fibrosis, the precise
molecular mechanism through which S1P regulates fibrotic
progression remains incompletely understood. The present
reviewsynthesizedrecent advances in the understanding of S1P in IPF
development, to provide a theoretical foundation for further
elucidating the pathological mechanisms of pulmonary fibrosis and
facilitating the development of targeted therapeutic strategies
(Fig. 2; Table I).
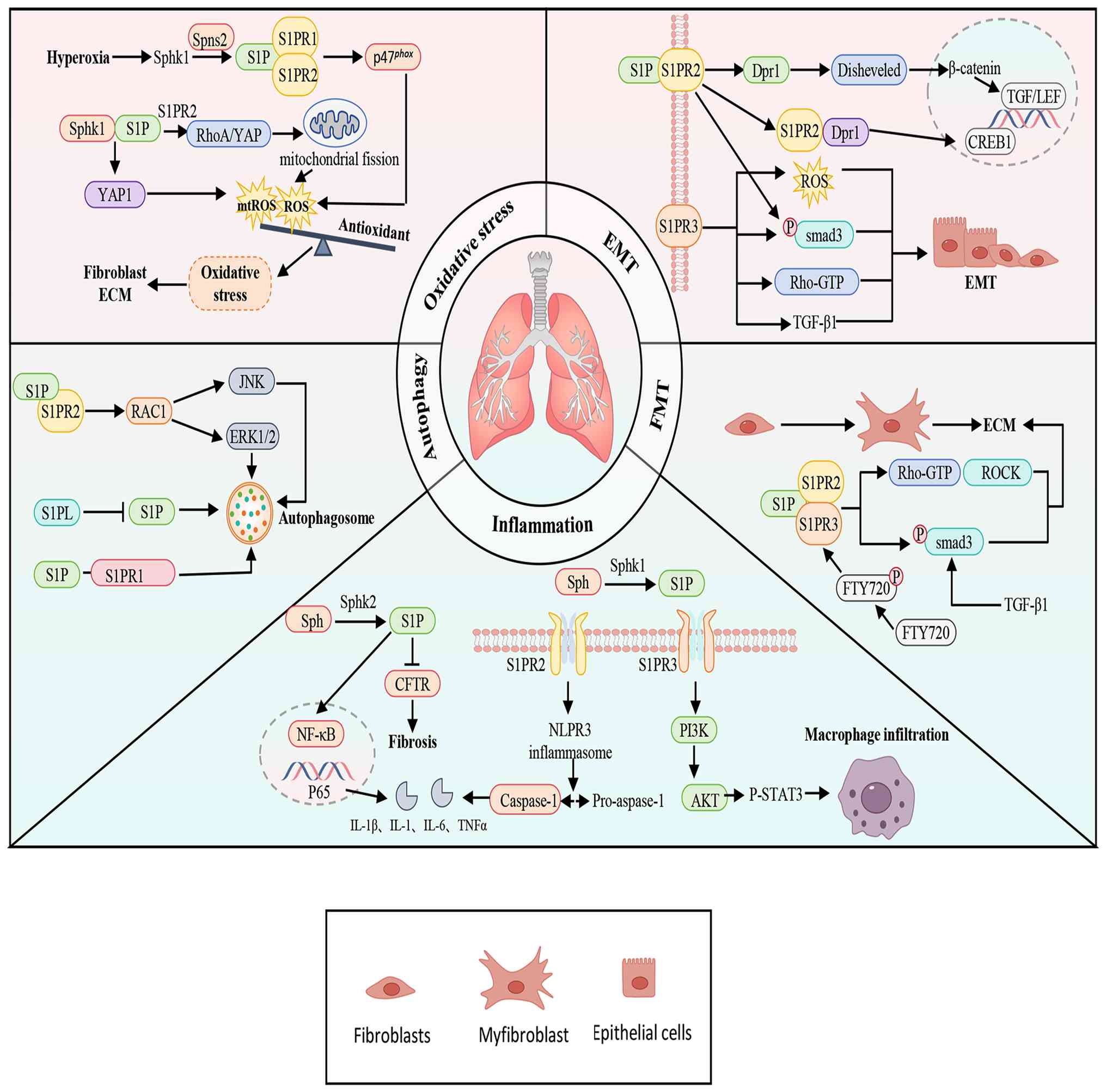 | Figure 2.Mechanisms of S1P in Pulmonary
Fibrosis. S1P drives the progression of pulmonary fibrosis by
regulating multiple key pathological processes, including
inflammatory responses, FMT, EMT, autophagy and oxidative stress.
‘→’ represents ‘activation’; ‘—|’ represents ‘inhibition’. S1P,
Sph1-phosphate; Sph, sphingosine; SphK1, Sph kinase 1; CFTR, cystic
fibrosis transmembrane conductance regulator; NF-κB, nuclear factor
κB; NLRP3, NOD-like receptor family pyrin domain-containing 3;
IL-1β, interleukin-1β; TNF-α, tumor necrosis factor α; PI3K/AKT,
phosphoinositide 3-kinase/protein kinase B; STAT3, signal
transducer and activator of transcription 3; FMT,
fibroblast-to-myofibroblast transition; ECM, extracellular matrix;
ROCK1, Rho-associated coiled-coil containing protein kinase;
TGF-β1, transforming growth factor-beta 1; EMT,
epithelial-to-mesenchymal transition; Dpr1, Dapper1; TCF/LEF,
T-cell factor/lymphoid enhancer-binding factor; CREB1, cyclic
AMP-responsive element-binding protein 1; ROS, reactive oxygen
species; mtROS, Mitochondrial reactive oxygen species: JNK, c-Jun
N-terminal kinase; RhoA, Ras homolog family member A; YAP,
Yes-associated protein. |
 | Table I.List of molecular mechanism of S1P
regulating pulmonary fibrosis. |
Table I.
List of molecular mechanism of S1P
regulating pulmonary fibrosis.
| First author/s,
year | Phenotype | Molecular | Target | Experimental
model | (Refs.) |
|---|
| Xiong et al,
2023 | Inflammation | S1PR1 | Smad2 | Mice | (48) |
|
|
|
| Smad3 | HPMECs |
|
|
|
|
| RhoA/ROCK1 |
|
|
| Qiu et al,
2024 |
| S1PR3 | PI3K/Akt- | Mice, Mouse | (51) |
| Chen et al,
2023 |
| SphK2 | STAT3 | BMDMs | (56) |
|
|
|
| CFTR, | Mice, |
|
|
|
|
| NF-κB-p65 | - |
|
| Urata et al,
2005 | FMT | S1PR2/3 | RhoA/ROCK | Mice, WI-38 | (64) |
| Sobel et al,
2013, |
| S1PR2/3 | Smad3 | Mice, | (52,67) |
| Keller et
al, 2007 |
|
|
| Human lung |
|
|
|
|
|
| fibroblasts |
|
| Milara et
al, 2012 | EMT | S1PR2/3 | Smad3, | -, | (37) |
|
|
|
| RhoA-GTP | ATII, A549 |
|
| Mu et al,
2025 |
| S1PR2 | Dpr1, CREB1 | Mice, | (74) |
|
|
|
|
| BEAS-2B, A549 |
|
| Huang and
Natarajan, | Autophagy | S1PL | - | Mice, Human | (82) |
| 2015 |
|
|
| lung
fibroblasts |
|
| Goel et al,
2021 |
| S1PR1 | - | Mice, HLMVECs | (83) |
| Liu et al,
2021 |
| S1PR2 | RAC1/JNK, | Mice, | (84) |
|
|
|
| RAC1/ERK1/2 | MLE-12 |
|
| Milara et
al, 2012 | Oxidative | S1PR2/3 | RhoA-GTP | -, | (37) |
|
| stress |
|
| ATII, A549 |
|
| Harijith et
al, 2016 |
| SphK1 | p47phox | Mice, HLMVECs | (95) |
| Zhou et al,
2023 |
| S1PR2 | RhoA/YAP | Mice, MLE-12 | (96) |
|
|
|
| Fis1, Drp1 |
|
|
| Huang et al,
2020 |
| SphK1 | Hippo/YAP | Mice, Mouse primary
fibroblasts | (98) |
Inflammation
Inflammation is a critical defense mechanism for
maintaining organismal homeostasis, primarily functioning to
eliminate harmful stimuli and initiate tissue repair. The classical
inflammatory response comprises four core components: The
inflammatory trigger, the corresponding receptor, the release of
inflammatory mediators and the resulting response in target tissues
(39). Under physiological
conditions, the inflammatory response is rapidly activated upon
tissue injury to initiate repair. However, if dysregulated and
sustained, it can progress into a chronic state, thereby promoting
the development of various chronic inflammatory diseases (40). At the molecular level, immune cells
secrete multiple pro-inflammatory cytokines, such as interleukin-1β
(IL-1β), IL-6 and tumor necrosis factor-alpha (TNF-α), upon
inflammatory initiation. These cytokines regulate the expression
and function of inflammation-related genes by binding to receptors
on target cells and activating multiple intracellular signaling
pathways, including nuclear factor κB (NF-κB) (41,42).
A distinct phase transition in inflammation occurs during pulmonary
fibrosis progression, characterized by a persistent inflammatory
response and abnormal fibrous tissue proliferation. This phase
features the substantial release of various inflammatory factors
and chemokines, which damage alveolar epithelial and endothelial
cells. The resulting increase in vascular permeability triggers
extensive exudation of inflammatory mediators (43). These mediators further amplify the
local inflammatory response and activate the inflammatory cascade,
ultimately stimulating the excessive proliferation of fibroblasts
and myofibroblasts. This leads to disproportionate ECM deposition,
thereby disrupting alveolar architecture and normal physiological
functions (44).
S1P is a key regulator of inflammatory responses and
exhibits context-dependent bidirectional effects, exerting either
pro-inflammatory or anti-inflammatory actions depending on specific
S1PR binding profiles and the tissue environment. This dual nature
has made S1P a central focus in research on various
inflammation-related diseases (45–47).
Increased vascular permeability is a critical early step in
inflammation. Restoring S1P-S1PR1 signaling enhances vascular
barrier integrity, limiting the extravasation of inflammatory cells
and cytokines and thereby attenuates tissue inflammation and injury
in pulmonary fibrosis. Therefore, modulating vascular
permeabilityrepresents a promising therapeutic strategy for
treating and halting pulmonary fibrosis progression. Among S1PRs,
S1PR1 is the predominant receptor expressed on pulmonary
endothelial cells. Studies show that S1PR1-knockout mice display
severe impairment of the pulmonary endothelial barriers, which
aggravates pulmonary fibrosis. Conversely, augmenting S1PR1
expression restores endothelial tight junctions, suppresses
inflammatory cell infiltration andmarkedly reduces collagen
deposition. Mechanistically, S1PR1 activation inhibits transforming
growth factor-β1 (TGF-β1)-mediated activation of Smad 2/3 and Ras
homolog family member A (RhoA)/Rho-associated coiled-coil
containing protein kinase 1 (ROCK1) signaling pathways, thereby
preserving pulmonary vascular endothelial barrier integrity and
limiting fibrotic progression (48). Another study further indicates that
the loss of S1PR1 expression and function after acute injury
impedes tissue repair, ultimately promoting chronic inflammation
and fibrosis (49), underscoring
the protective role of S1PR1 in maintaining lung tissue integrity.
S1P also modulates fibrotic processes by regulating inflammatory
effector cells such as macrophages. For example, S1P can promote
the synthesis of pro-inflammatory and pro-fibrotic mediators,
including IL-1β and TGF-β (50).
In bleomycin-induced pulmonary fibrosis models, macrophages show
elevated S1PR3 expression. Knockdown of S1PR3 inhibits M2-type
macrophage polarization via the PI3K/Akt-signal transducer and
activator of transcription 3 (STAT3) pathway. Since M2 macrophages
secrete various growth factors and cytokines such as TGF-β, which
stimulate fibroblast proliferation and collagen synthesis, reduced
M2 polarization alleviates pulmonary fibrosis (51). The dual pro- and anti-inflammatory
roles of S1P highlight its complex regulation in pulmonary
fibrosis. Sobel et al (52)
found that in pulmonary fibroblasts, S1P receptor agonists activate
Smad-independent PI3K/Akt and ERK1/2 pathways via S1PR2/S1PR3,
stimulating extracellular matrix synthesis and exerting a
pro-fibrotic effect. By contrast, specific knockout of S1PR1 in
pulmonary vascular endothelial cells disrupts tight junctions,
increases vascular permeability and exacerbates bleomycin-induced
pulmonary fibrosis (53),
indicating that S1P actions depend on cell type and receptor
subtype. Furthermore, S1P concentration and exposure duration
critically shape its effects. At low concentrations (<1 µM or 85
µg/kg), S1P protects the endothelium from inflammatory damage,
whereas high levels (>5-10 µM) can disrupt the endothelial
barrier in vitro, probably via S1PRs other than S1PR1
(54). Similarly, short-term
administration of S1P1 agonists prevents vascular leakage in acute
lung injury models, but prolonged exposure leads to functional
antagonism at S1PR1 and worsens post-injury vascular leakage
(55). These findings underscore
that the influence of S1P on pulmonary inflammation is both
concentration- and time-dependent.
The NF-κB signaling pathway constitutes a key target
through which S1P mediates cross-regulation between inflammation
and fibrosis. In a cigarette smoke-induced mouse model of chronic
obstructive pulmonary disease, deletion of SphK2 reduced S1P
production, enhanced cystic fibrosis transmembrane conductance
regulator activity and indirectly suppressed the expression and
nuclear translocation of NF-κBsubunit p65 (56). Together, these changes alleviated
small airway fibrosis and inflammatory responses. Moreover, S1P can
promote the priming and activation of the NOD-like receptor family
pyrin domain-containing 3 (NLRP3) inflammasome. In a liver fibrosis
model, S1P activated the NLRP3 inflammasome in a
concentration-dependent manner and stimulated the secretion of
IL-1β and IL-18. By contrast, specific knockdown of S1PR2 markedly
inhibited NLRP3 inflammasome activation, thereby delaying hepatic
inflammation and fibrosis progression (57), suggesting that the S1P-NLRP3 axis
may play a similar pro-inflammatory role in pulmonary fibrosis. In
summary, by engaging receptors such as S1PR1/2/3, S1P activates
critical signaling pathways including PI3K/Akt-STAT3, NF-κB and the
NLRP3 inflammasome. These pathways modulate the secretion of
pro-inflammatory mediators and the infiltration of inflammatory
cells, thereby driving fibrotic progression. Different S1PRs
mediate distinct downstream cascades, eliciting varied cellular
responses at the tissue level; together, these integrated effects
ultimately shape the pathological outcome of fibrosis. Importantly,
the seemingly contradictory profibrotic and antifibrotic effects of
S1P reported across different experimental models can be explained
by differences in target cell types, S1P receptor subtype
specificity, ligand concentration and disease stage or exposure
timing and therefore do not represent true inconsistencies.
FMT
FMTrepresents a central pathological event in
pulmonary fibrosis development. Under the synergistic influence of
inflammatory factors and growth factors, fibroblasts initiate a
phenotypic transformation program and differentiate into
myofibroblasts, which in turn drive aberrant synthesis and
deposition of ECM (58,59). It is now widely accepted that FMT
constitutes the primary route for myofibroblast accumulation in
lung tissue; The canonical myofibroblast marker α-smooth muscle
actin (α-SMA) is routinely used to distinguish these cells from
untransformed fibroblasts (60,61).
As the main effector cells responsible for excessive ECM
deposition, myofibroblasts can be activated across diverse
pathological microenvironments, making FMT a key therapeutic target
in anti-fibrotic interventions (62).
In recent years, the regulatory role of S1P in FMT
has attracted increasing research attention, with multiple studies
confirming its involvement through diverse molecular mechanisms
(63). Specifically, S1P can
selectively bind to S1PR2 and S1PR3 on the cell membrane,
triggering Rho-GTPase-mediated signaling (64). This initiates a Rho-kinase-mediated
cascade that ultimately alters lung fibroblast morphology and
function, conferring myofibroblast-like characteristics. Evidence
indicates that S1P induces fibroblasts to adopt a myofibroblast
phenotype via trans-activation of the TGF-β1 signaling pathway,
wherein SphK1 serves as a molecular bridge linking TGF-β1 and S1P
signaling (65). One study showed
that TGF-β1 stimulation markedly upregulates SphK1 expression in
lung fibroblasts; activated SphK1 then translocates from the
cytoplasm to the plasma membrane, where it catalyzes sphingosine
phosphorylation to generate S1P. The newly synthesized S1P binds to
S1PR2 and S1PR3, activates Rho/Rho kinase pathways and accelerates
fibroblast-to-myofibroblast differentiation (66). Furthermore, studies on fingolimod
(FTY720) support S1P's role in regulating FMT. FTY720 is
phosphorylated intracellularly to its active form FTY720-P, which
binds to S1PR2/S1PR3, activates the downstream Smad3 signaling
pathway andultimately induces FMT (52,67,68).
Notably, the Smad3 pathway operates as a downstream branch of S1P
receptors, parallel to the aforementioned Rho kinase pathway, with
both being regulated by upstream TGF-β1 signals. Collectively, the
S1P signaling axis regulates FMT, which subsequently contributes to
the pathological progression of pulmonary fibrosis.
Epithelial-to-mesenchymal transition
(EMT)
EMTis a pivotal process in tissue repair and a
critical mechanism in the pathogenesis of pulmonary fibrosis. As a
reversible biological process, EMT involves the loss of epithelial
polarity and the acquisition of a mesenchymal phenotype. This
transition is characterized by downregulation of epithelial markers
such as E-cadherin and upregulation of mesenchymal markers,
including N-cadherin and vimentin (69,70).
EMT has been consistently observed in lung tissues, both in
vitro using lung epithelial cell models and in vivo in
animal studies (71). Notably, S1P
levels show a significant positive correlation with EMT in
pulmonary fibrosis (72,73), suggesting that S1P may contribute
to fibrosis by modulating EMT. In vitro experiments
conducted by Milara et al (37) using A549 cells demonstrated that
S1P drives EMT primarily through its specific receptors S1PR2 and
S1PR3. Receptor activation triggers multiple downstream signaling
events, including SMAD3 phosphorylation, RhoA-GTP activation,
increased oxidative stress and elevated production and release of
TGF-β1. The same study further indicated that TGF-β1-induced EMT
was partly mediated via the S1P/SPHK1 axis, pointing to functional
crosstalk between the TGF-β1 signaling pathway and the S1P/SPHK1
axis. This crosstalk mechanism indicates that S1P not only directly
induces EMT, but also further amplifies the pro-fibrotic effects of
TGF-β1 itself by promoting its release. In another study,
stimulation of A549 cells with either TGF-β or S1P induced similar
fibroblast-like morphological changes, accompanied by increased
expression of the mesenchymal marker cluster of differentiation 90
and decreased expression of the epithelial marker epithelial cell
adhesion molecule. Immunofluorescence confirmed EMT induction,
implying that S1P and TGF-β exert comparable effects in this
process. Importantly, treatment with the TGF-β receptor inhibitor
LY2109761, namely the antagonists of TGF-β Type I and Type II,
markedly attenuated S1P-induced EMT, suggesting that S1P-dependent
EMT requires TGF-β signaling downstream (72). These findings collectively indicate
that targeting the S1P/TGF-β axis may offer a promising therapeutic
strategy for EMT-associated pulmonary fibrosis. Further elucidating
the mechanism, Mu et al (74) reported that S1PR2 agonists strongly
promote EMT in human bronchial epithelial cells. S1PR2 appears to
modulate EMT through two parallel pathways: i) By interacting with
Dapper1 (Dpr1) to inhibit Dishevelled degradation, leading to
β-catenin nuclear accumulation and activation of T-cell
factor/lymphoid enhancer-binding factor-dependent transcription of
EMT-related genes; and ii) by forming and S1PR2-Dpr1 complex that
translocates to the nucleus and binds to and activates cyclic
AMP-responsive element-binding protein 1, thereby enhancing the
expression of EMT-associated transcription factors (74). Although this mechanism has been
established in vitro, its relevance in vivo,
particularly its clinical implications, requires further
investigation. In summary, current evidence supports the
hypothesisthat S1P contributes to the pathological progression of
pulmonary fibrosis, at least in part, through the regulation of
EMT.
Autophagy
Autophagy is a highly conserved catabolic process in
eukaryotic cells, primarily involving the lysosome-mediated
degradation of abnormal proteins, damaged organelles and other
dysfunctional cellular components, thereby maintaining
intracellular homeostasis (75).
S1P and its receptors are known to interfere with multiple stages
of autophagy, including initiation, nucleation, elongation,
maturation, membrane fusion and substrate degradation and
ultimately modulate autophagic activity through various signaling
and metabolic pathways (76).
As research on the pathogenesis of pulmonary
fibrosis advances, dysregulated autophagy has been recognized as
one of its key pathological features (77). In the fibrotic lung
microenvironment, autophagic activity is typically suppressed. This
is evidenced by decreased expression of autophagic markers LC3 and
Beclin-1, alongside a significant reduction in autophagosome
numbers observed by transmission electron microscopy (78). Such inhibition of autophagy can
accelerate pulmonary fibrosis by aggravating alveolar epithelial
cell injury, promoting myofibroblast activation and enhancing
abnormal ECM deposition (79,80).
Emerging evidence indicates that S1P-mediated
regulation of autophagy represents a critical pathway in
fibrogenesis (81). S1PL is a
central enzyme controlling S1P synthesis and degradation. Notably,
S1PL expression is upregulated in lung tissues from patients with
IPF and in bleomycin-induced pulmonary fibrosis models.
Mechanistically, S1PL overexpression enhances autophagy by
increasing LC3 and Beclin-1 expression while reducing TGF-β-induced
intracellular S1P accumulation in lung fibroblasts. Conversely,
S1PL knockdown exacerbates bleomycin-induced pulmonary fibrosis in
mice. These findings collectively suggest that enhancing S1PL
activity to downregulate S1P signaling may offer a therapeutic
strategy for pulmonary fibrosis (82). Further supporting this hypothesis,
studies in human lung microvascular endothelial cells from smokers
showed reduced expression of both S1PR1 and autophagy-related
proteins. Knockdown of S1PL elevates intracellular S1P levels and
promotes autophagy, whereas inhibition of S1PR1 suppresses
autophagic activity (83).
Together, these results highlight S1PR1 as a key nodal molecule in
S1P-mediated regulation of autophagy.
However, the role of S1P in autophagy is not
unidirectional. Although most evidence suggests that S1P promotes
disease progression by suppressing autophagy, it can also exert
opposite effects in specific contexts. In a murine asthma model
(84), S1P binding to S1PR2 was
found to activate autophagy, whereas S1PR2 antagonism suppressed
autophagic activity in lung tissue. This inhibitory effect could be
reversed by a Ras-related C3 botulinum toxin substrate 1 (RAC1)
inhibitor. Furthermore, alterations in c-Jun N-terminal kinase
(JNK) and ERK1/2 pathway activity was associatedwith changes in
autophagy-related protein levels, indicating that the S1P-S1PR2
axis activates downstream JNK and ERK1/2 pathways via RAC1
stimulation, thereby promoting autophagy (84). Thus, the bidirectional regulation
of autophagy by S1P likely depends on receptor subtypes, target
cell types and the specific tissue microenvironment. For instance,
in lung microvascular endothelial cells from chronic smokers,
decreased S1PR1 expression positively correlated with reduced
levels of autophagic markers. suggesting that S1P1 downregulation
inhibits the autophagy pathway. Knockdown of S1PR1 suppressed
S1P-induced autophagy, suggesting that S1P-mediated positive
regulation of autophagy requires S1PR1 (83). In pulmonary epithelial cells,
S1P/S1PR3 signaling has been shown to help prevent sepsis by
activating the ERK1/2 and p38 pathways; a mechanism thought to
restrain excessive autophagy and thereby preserve pulmonary
epithelial barrier integrity (85). The precise molecular mechanisms
through which S1P regulates autophagy in pulmonary fibrosis remain
incompletely understood and warrant further targeted
investigation.
Overall, these observations suggest that the
apparently bidirectional effects of S1P on autophagy arise from
context-dependent S1PR signaling, particularly differences in
receptor subtype engagement, target cell identity and local
pulmonary microenvironment, rather than true inconsistencies among
studies.
Oxidative stress
Oxidative stress plays a critical role in the
pathogenesis of pulmonary fibrosis and correlates strongly with
disease severity. Reactive oxygen species (ROS) serve as the
central effector molecules driving fibrosis-related
cytopathological events (86).
Oxidative stress occurs when ROS production exceeds the capacity of
the endogenous antioxidant defense system. This
oxidation-antioxidation imbalance leads to cellular structural and
functional abnormalities. In mammalian cells, ROS are primarily
generated through cellular oxidative metabolism, with the NADPH
oxidase (NOX) family and mitochondria constituting the two major
sources (87). Elevated ROS levels
directly damage alveolar epithelial cells and compromise alveolar
barrier integrity. Moreover, ROS act as signaling molecules that
trigger FMT and ECM deposition (88,89).
Clinical evidence further supports the involvement of oxidative
stress in IPF. Markers of oxidative stress are markedly elevated in
exhaled breath condensate, bronchoalveolar lavage fluid and
diseased lung tissue from IPF patients, directly linking oxidative
stress to IPF pathogenesis (90,91).
S1P modulates oxidative stress through multiple
molecular mechanisms, thereby acting as a key signaling node
linking redox imbalance to pulmonary fibrosis. Studies have shown
that oxidative stress signals can activate SphK1, leading to
increased intracellular S1P levels and establishing a positive
feedback loop termed the ‘oxidative stress-SphK1-S1P’ axis
(92–94). Evidence from a hyperoxia-induced
lung injury model demonstrates that S1P regulates NOX-mediated ROS
generation. Hyperoxia markedly upregulates SphK1 expression in lung
tissue, thereby elevating S1P production. Mechanistically, S1P is
exported via the Spns2 transporter and subsequently activates
endothelial S1PR1/2. This receptor activation modulates the
activity of the NOX subunit p47phox, driving substantial ROS
production. Notably, SphK1 knockout such as reduced ROS levels and
attenuated lung injury (95). At
the receptor level, Zhou et al (96) reported that knockdown of S1pr2
inhibits S1P binding of S1P to S1PR2, thereby regulating the
RHOA/YAP pathway. This in turn influences the expression of
downstream mediators such as connective tissue growth factor and
cysteine-rich angiogenic inducer 61 and remodels mitochondrial
dynamics by promoting mitochondrial fusion and mitofusin expression
while suppressing fission. Concomitantly, it downregulates
phosphorylation of fission 1 and dynamin-related protein 1, reduces
mitochondrial ROS production and alleviates pulmonary fibrosis.
The production of mitochondrial ROS (mtROS) is a
critical contributor to pulmonary fibrosis (97). SphK1/S1P regulation of mtROS has
been shown to involve the Hippo signaling pathway. Specifically,
S1P activates the transcriptional co-activator YAP1, which
undergoes dephosphorylation and nuclear translocation, directly
initiating transcription of genes involved in mtROS synthesis and
leading to mtROS overproduction. Inhibition of SphK1 attenuated
YAP1 nuclear translocation and mtROS generation (98). Furthermore, ROS accumulation can
amplify fibrotic responses via crosstalk with the TGF-β1 pathway.
Milara et al (37)
demonstrated that S1P directly promotes ROS production through
S1PR2/3 activation. The elevated ROS then further stimulates TGF-β1
signaling, ultimately inducing EMT and driving fibrosis. In
summary, S1P contributes to pulmonary fibrosis by regulating
NOX-derived and mitochondrial ROS production and by participating
in ROS-TGF-β1 crosstalk. These mechanisms provide new insights into
the central role of S1P in fibrotic pathogenesis. Notably, most
mechanistic insights into S1P-mediated regulation of oxidative
stress in pulmonary fibrosis are derived from animal models and
in vitro experiments; therefore, their relevance to human
pulmonary fibrosis requires further clinical validation.
Therapeutic strategies for targeting S1P in
pulmonary fibrosis
Given the established role of S1P-related signaling
as a potential therapeutic target, the development of specific
inhibitors has become a major focus in pharmacological and
pharmaceutical research. A variety of natural and synthetic
compounds have been shown to modulate S1P. Among these, the
polyphenolic flavonoid quercetin is considered one of the most
potent SphK1 inhibitors. Zhang et al (99) reported that quercetin blocks
S1P-mediated pro-fibrotic signaling by inhibiting SphK1 expression.
Conversely, upregulation of SphK1 attenuates quercetin's
anti-fibrotic effects, further confirming that its action depends
on the SphK1/S1P axis. Myricetin targets serine
palmitoyltransferase to inhibit de novo sphingolipid biosynthesis
and suppresses SphK1 activation, thereby alleviating
radiation-induced pulmonary fibrosis (100). Moreover, fenugreek alkaloid
(trigonelline), a natural plant-derived alkaloid with diverse
pharmacological properties, reduces SphK1 activity and S1P
production, blocks phosphorylation and nuclear translocation of
YAP/transcriptional coactivator with PDZ-binding motif (TAZ) in the
Hippo signaling pathway and consequently suppresses cell
proliferation and matrix deposition, ultimately mitigating
pulmonary fibrosis (101).
In a similar vein, Huang et al (98) observed that PF543 inhibits SphK1
activation and thereby prevents nuclear translocation of YAP1. This
reduces co-localization between YAP1 and ferroptosis suppressor
protein 1in fibroblasts, attenuates fibroblast activation and
alleviates pulmonary fibrosis. Notably, the SphK1 inhibitor
(SKI-349) suppresses SphK1 activity, downregulates S1P levels and
attenuates fibroblast aggregation and neutrophil infiltration,
leading to amelioration of pulmonary fibrosis (102). Additionally, the S1PR1 agonist
IMMH002 helps maintain endothelial barrier integrity, thereby
reducing inflammatory responses and collagen accumulation, which
effectively mitigates fibrotic progression (49). Inhibitors targeting S1PR2 (such as
GLPG2938, S118, JTE-013) and S1PR3 (such as TY-52156, CAY10444) can
modulate inflammatory responses and EMT by antagonizing their
respective receptors, thus intervening in pulmonary fibrosis
development (53,74,96,103). Collectively, these studies
provide experimental support for developing combined therapeutic
regimens that pair S1P-pathway inhibitors with anti-fibrotic
natural products (Table II).
Although the pathogenic role of S1P in pulmonary fibrosis is well
documented, a key translational challenge remains: how to precisely
harness its regulatory potential and tune its activity to achieve
optimal therapeutic outcomes.
| Table II.List of the therapeutic effects of
S1P pathway-targeting drugs in pulmonary fibrosis. |
Table II.
List of the therapeutic effects of
S1P pathway-targeting drugs in pulmonary fibrosis.
| First author/s,
year | Compound | Target | Mechanism of
action | Therapeutic
effect | (Refs.) |
|---|
| Hao et al,
2023 | IMMH002 | S1PR1 | Activate S1PR1 to
protect the integrity of the endothelial barrier | Reducing
inflammatory responses and collagen deposition to improve pulmonary
fibrosis | (49) |
| Qiu et al,
2024 | TY-52156 | S1PR3 | Antagonizes
S1PR3, | Reducing TGF-β1
release, | (51) |
|
| CAY10444 |
| inhibiting
activation of the PI3K/AKT-STAT3 signaling pathway | decrease collagen
deposition |
|
| Mu et al,
2025 | S118 | S1PR2 | Inhibits binding to
Dpr1, reduces β-catenin accumulation, and blocks nuclear
translocation of the S1PR2 | Suppressing
inflammatory responses and EMT formation | (74) |
| Zhou et al,
2023 | JTE-013 | S1PR2 | Antagonizes S1PR2,
inhibits mitochondrial membrane potential elevation, reactive
oxygen species production, and apoptosis, and suppresses the
expression of collagen | Reducinglung
inflammation and fibrosis | (96) |
| Huang et al,
2020 | PF543 | SphK1 | Inhibiting SphK1
suppresses YAP1 nuclear translocation, reduces mtROS, and decreases
the expression of fibronectin and α-SMA. | Reducing the
colocalization of YAP1 and FSP1 in fibroblasts, thereby decreasing
fibroblast activation | (98) |
| Zhang et al,
2018 | Quercetin | SphK1 | Inhibition of
SphK1/S1P signaling | Reducing collagen
deposition | (99) |
| Gorshkova et
al, 2012 | Myristicin | SphK1 | Inhibition of SphK1
activation | Alleviating
radiation-induced pulmonary fibrosis | (100) |
| Zeyada et
al, 2024 | Fenugreek
alkaloid | SphK1 | Reducing SphK1
activity decreases S1P production, thereby inhibiting the
phosphorylation and nuclear translocation of YAP/TAZ | Inhibiting cell
proliferation and matrix deposition alleviates pulmonary
fibrosis | (101) |
| Lu et al,
2025 | SKI-349 | SphK1 | Inhibit SphK1 to
downregulate S1P expression | Reducingfibroblast
accumulation and neutrophil infiltration | (102) |
| Mammoliti et
al, 2021 | GLPG2938 | S1PR2 | Antagonizes S1PR2,
preventing S1P from interacting with its receptor | Reducing
inflammatory response and mitigate lung tissue damage | (103) |
A comprehensive understanding of the upstream
regulatory mechanisms governing S1P is critical for elucidating its
roles in physiological and pathological contexts, as well as for
developing targeted therapeutic strategies. S1P expression is
regulated at multiple levels, with microRNAs (miRNAs) emerging as
important modulators. Dysregulation of specific miRNAs can offer
valuable insights into diseases associated with aberrant S1P
signaling. For example, miR124, miR125b, miR133b and miR130a can
downregulate key components of the S1P pathway, including SphK1,
SGPL1, S1PR1 and S1PR2, by directly targeting their respective
genes (104). In cancer,
miR-495-3p inhibits tumor proliferation by acting on SphK1
(105); similarly, miR-92a
(106), miR302-367 (107) and miR363 (108) have been shown to target and
suppress S1PR1 expression; while miR-148a targets S1PR1 to suppress
hepatocyte metastasis (109).
These miRNA-mediated regulations not only affect the transcription
and translation of S1P-associated molecules but also modulate their
functions in metabolism, inflammation and fibrosis. Further
investigation of these mechanisms will provide a stronger
theoretical foundation for developing targeted S1P therapies and
enhance their translational potential.
Clinical application of S1P signaling
Given the critical involvement of the SphK-S1P
signaling axis in various diseases, SphK inhibitors and S1P
receptor antagonists represent promising therapeutic candidates for
pulmonary fibrosis. A review of clinical trial databases, including
PubMed, ClinicalTrials.gov andthe WHO international clinical trials
registry platform, reveals that most registered trials targeting
S1P have focused on neurological autoimmune and inflammatory
conditions; particularly multiple sclerosis (MS), ulcerative
colitis and Crohn's disease. Nevertheless, modulators of S1P
receptors have achieved notable clinical success, with several
agents already approved and in clinical use. FTY720 (fingolimod)
stands out as the first approved non-selective S1P receptor
modulator for multiple sclerosis treatment. It is a structural
analogue of endogenous S1P that, after oral administration, is
phosphorylated in vivo to FTY720 phosphate. This active
metabolite acts as an agonist at S1PR1, S1PR3, S1PR4 and S1PR5 but
exhibits low affinity for S1PR2. Its binding to S1PR1 induces
receptor internalization and degradation, resulting in functional
antagonism that inhibits lymphocyte egress and attenuates
inflammatory responses (110).
Phase III trial demonstrated that FTY720 markedly reduces the
annualized relapse rate and delays disability progression in
patients with MS (111,112). Due to its S1PR1-targeted
activity, FTY720 has also been explored as a potential therapeutic
for IPF. Preclinical studies indicate its ability to ameliorate
fibrosis in cardiac, renal and hepatic tissues (113–115). Though limited to animal model
studies, findings regarding its effect on pulmonary fibrosis are
conflicting. One study indicated that FTY720 reduces pulmonary
fibrosis by inhibiting the TGF-β1/TGF-β-activated kinase 1/P38MAPK
pathway and promoting autophagy (116). Conversely, long-term
administration of FTY720 was shown to exacerbate bleomycin-induced
pulmonary fibrosis and vascular leakage in mice andthe effect is
potentially mediated through S1PR3 activation (55). This discrepancy appears to be dose-
and regimen-dependent. Short-term, low-dose FTY720 predominantly
activates S1PR1, thereby enhancing endothelial barrier function and
attenuating pulmonary edema and fibrosis (117). By contrast, prolonged or
high-dose exposure leads to S1PR1 desensitization, shifting the
dominant signaling toward the S1PR3-mediated pathway (118). This latter promotes vascular
permeability, inflammatory cell infiltration and fibrotic
responses. Thus, the net effects of FTY720 in pulmonary fibrosis
depend critically on the dosing strategy and the resulting
receptor-activation profile. Taken together, the divergent effects
of FTY720 on pulmonary fibrosis are best explained by dose- and
duration-dependent shifts in S1PR signaling, particularly the
balance between S1PR1 and S1PR3, rather than indicating genuine
disagreement in the literature.
MS serves as a prototypical autoimmune disorder,
characterized by T cell-driven aberrant immune attacks (119). FTY720 achieves its therapeutic
effect in MS primarily by modulating lymphocyte trafficking,
thereby inhibiting immune cell infiltration into the central
nervous system (110). However,
it also induces a pronounced reduction in peripheral CD4+ T cells,
which may exacerbate immunosuppression (120), a potentially serious risk for
patients with pre-existing immune compromise or chronic infections.
By contrast, IPF is not solely an immune-mediated condition. Beyond
its immunomodulatory properties, the clinical use of FTY720 in IPF
is complicated by the patient population profile: IPF predominantly
affects middle-aged and elderly individuals, who often have
cardiovascular comorbidities (such as hypertension, coronary heart
disease) and exhibit pulmonary vascular remodeling and right
ventricular dysfunction (121,122). These factors may amplify the risk
of adverse reactions to S1P-targeting agents. FTY720 is known to
induce bradycardia (123), which
could worsen pre-existing cardiac output limitations in IPF
patients and thereby trigger or aggravate dyspnea. Consequently,
despite its potential anti-fibrotic benefits, the clinical
translation of FTY720 for IPF remains markedly constrained by
cardiovascular safety concerns.
Second-generation selective S1PR modulators, such as
siponimod, ozanimod and ponesimod, have been developed to improve
safety profiles while maintaining therapeutic efficacy through
optimized receptor selectivity (124,125). Ozanimod acts as a potent agonist
of S1PR1 and S1PR5; siponimod is an antagonist of both receptors;
and ponesimod functions as a selective S1PR1 antagonist (126). Ozanimod is clinically approved
for reducing immune-mediated inflammation in MS
andmoderate-to-severe ulcerative colitis (127) and is under investigation for
Crohn's disease (128), systemic
lupus erythematosus (129) and
coronavirus disease 2019 (COVID-19) (130). In an in vitro study using
TGF-β1-stimulated human lung fibroblasts to investigate S1PR
agonist-mediated fibrotic responses, ponesimod induced less
extracellular matrix synthesis than FTY720, likely due to its
weaker activation of S1PR3. This suggests that receptor-specific
modulators such as ozanimod, siponimod and ponesimod may offer a
therapeutic advantage over FTY720 in pulmonary fibrosis.
Nevertheless, these agents also carry risks. Although higher
receptor selectivity may reduce cardiovascular side effects,
ozanimod is associated with potential hepatotoxicity, mainly
reflected in elevated transaminases (131); a particular concern for IPF
patients who may have concurrent liver impairment (132). Moreover, in clinical trials for
colitis and MS, ozanimod administration was linked to measurable
declines in lung function (133).
Consequently, caution should be exercised when prescribing ozanimod
to patients with severe respiratory disorders or pulmonary
fibrosis. Similarly, siponimod and ponesimod have been reported to
reduce forced expiratory volume in early-stage MS trials (134), which could further burden
respiration in IPF patients. Therefore, future clinical
applications must involve careful patient stratification based on
cardiovascular and hepatic function, as well as concurrent
medications. Priority should be given to agents with higher
receptor selectivity to minimize off-target effects. Throughout
treatment, rigorous monitoring of heart rate, cardiac and hepatic
function and pulmonary parameters is essential to ensure patient
safety.
Several other S1PR1 modulators are currently under
clinical investigation, including ceralifimod (ONO-4641), cenerimod
(ACT-334441), etrasimod (APD334), amiselimod (MT-1303), VPC01091
and VPC23019a (135). These
agents primarily exert immunosuppressive effects through receptor
antagonism. Although clinically effective, they are often
associated with adverse reactions such as bradycardia and impaired
endothelial barrier function. Targeting S1P transport may offer an
alternative therapeutic approach. Spns2 transport inhibitors can
such as lower extracellular S1P levels by blocking its export. For
example, the Spns2 inhibitor (SLF1081851) attenuated inflammation
and improved fibrosis in a renal fibrosis model (136). Moreover, the Spns2 inhibitor
(SLF80821178) was reported not to induce bradycardia or impair
pulmonary endothelial barrier function, while exerting only a mild
effect on lymphocytes (95,137).
These findings highlight Spns2 as a promising alternative target
for pulmonary fibrosis therapy.
To date, only a limited number of inhibitors
targeting S1P-metabolic enzymes (such as SphKs and SPL) and S1P
receptor agonists/antagonists have been developed, with most
studies still confined to preclinical stages. In the future,
combination strategies that pair S1P-pathway inhibitors with
existing first-line anti-fibrotic agents (such as nintedanib,
pirfenidone), natural compounds, signaling-pathway inhibitors (such
as NF-κB, YAP/TAZ, ROCK or TGF-β receptor inhibitors),
antioxidants, or anti-inflammatory drugs hold considerable promise.
Such multi-targeted approaches could overcome the limitations of
monotherapy and provide more effective, precise treatment options
for pulmonary fibrosis. Importantly, the translation of S1P-based
combination therapies into human trials requires careful
consideration of drug safety and toxicity, disparities between
animal models and human pathophysiology and interindividual
variability in treatment responses. Clinical trial designs should
therefore account for these potential factors and incorporate
rigorous safety and toxicity assessments to minimize potential harm
to healthy tissues.
Conclusion and outlook
S1P is a bioactive sphingolipid that plays a broad
role in fundamental cellular processes, including cell
proliferation, differentiation, migration and inflammation. S1P
exerts a central pro-fibrotic function in IPF by modulating key
pathological pathways such as inflammatory responses, EMT, FMT,
autophagy and oxidative stress. Although substantial evidence has
elucidated the contribution of S1P to pulmonary fibrosis,
translating this knowledge into effective S1P-targeted therapies
remains challenging. Several important questions remain unresolved.
The cell type-specific functions of different S1P receptor
subtypes, as well as their potential interactions, are not fully
understood, with S1PR2 and S1PR3 being of special interest.
Clarifying these issues will require the generation of
cell-specific knockout mouse models to determine how the loss of
S1P signaling from distinct cellular compartments affects the
development and progression of pulmonary fibrosis. Moreover, the
role of S1P export mediated by the transporter Spns2 in lung
fibrosis has yet to be elucidated. It remains unclear which cell
types are responsible for Spns2-dependent S1P release in the lung
and how this transport process contributes to the regulation of the
local fibrotic microenvironment. Single-cell transcriptomic
approaches could be used to identify Spns2-expressing cell
populations, followed by the establishment of cell-type-specific
Spns2 knockout mice to further dissect the underlying molecular
mechanisms. The present review has summarized the involvement of
S1P signaling in pulmonary fibrosis, systematically analyzed its
underlying mechanisms and discussed current therapeutic strategies
aimed at the S1P pathway and its associated molecules. These
insights provide a valuable foundation for identifying novel
therapeutic targets and developing new treatment approaches for
IPF.
In the future, therapeutic strategies may evolve
from single-target modulation toward integrated and multi-pathway
interventions that concurrently address several core mechanisms of
pulmonary fibrosis. For instance, combining single-cell sequencing
with S1P signaling analysis could elucidate its cell-type-specific
distribution and activity, thereby revealing precise cellular
targets for intervention. Further investigation is warranted to
uncover new mechanistic insights and translate them into innovative
treatment strategies for S1P-driven pulmonary fibrosis.
Acknowledgements
Not applicable.
Funding
The present review was supported by the Guangxi Science and
Technology Base and Talent Project (grant no. Guike
AD22035221).
Availability of data and materials
Not applicable.
Authors' contributions
YQ and CT conceived the study and drafted the
manuscript. GL and ST provided critical revision and supervised the
work. Data authentication is not applicable. All authors reviewed
and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ABC
|
ATP-binding cassette
|
|
BALF
|
bronchoalveolar lavage fluid
|
|
Cer
|
ceramide
|
|
Dpr1
|
Dapper1
|
|
ECM
|
extracellular matrix
|
|
EMT
|
epithelial-to-mesenchymal
transition
|
|
ERK1/2
|
extracellular signal-regulated kinase
1/2
|
|
FMT
|
fibroblast-to-myofibroblast
transition
|
|
HDAC
|
histone deacetylase
|
|
IL-1β
|
interleukin-1beta
|
|
IL-6
|
interleukin-6
|
|
IPF
|
idiopathic pulmonary fibrosis
|
|
JNK
|
c-Jun N-terminal kinase
|
|
MAPK
|
mitogen-activated protein kinase
|
|
MS
|
multiple sclerosis
|
|
mtROS
|
mitochondrial reactive oxygen
species
|
|
NF-κB
|
nuclear factor kappa-B
|
|
NLRP3
|
NOD-like receptor family pyrin
domain-containing 3
|
|
PI3K/AKT
|
phosphoinositide 3-kinase/protein
kinase B
|
|
RAC1
|
Ras-related C3 botulinum toxin
substrate 1
|
|
RhoA/ROCK1
|
Ras homolog family member
A/Rho-associated coiled-coil containing protein kinase 1
|
|
ROS
|
reactive oxygen species
|
|
Sph
|
sphingosine
|
|
S1P
|
SPh1-phosphate
|
|
S1PL
|
S1P lyase
|
|
S1PRs
|
S1P receptors
|
|
SphK1
|
Sph kinase 1
|
|
Spns2
|
spinster homolog 2
|
|
SPP
|
S1P phosphatase
|
|
STAT3
|
signal transducer and activator of
transcription 3
|
|
TAZ
|
transcriptional coactivator with
PDZ-binding motif
|
|
TGF-β1
|
transforming growth factor β1
|
|
TNF-α
|
tumor necrosis factor α
|
|
YAP
|
Yes-associated protein
|
References
|
1
|
Wang L, Huang J, Tan G, Zhang Y, Xia M,
Kang Z, Xiao Y and Lei M: Trends and hotspots in pulmonary fibrosis
biomarker research: A bibliometric analysis. Front Med (Lausanne).
12:15413642025. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Podolanczuk AJ, Thomson CC, Remy-Jardin M,
Richeldi L, Martinez FJ, Kolb M and Raghu G: Idiopathic pulmonary
fibrosis: State of the art for 2023. Eur Respir J. 61:22009572023.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jeganathan N, Corte TJ and Spagnolo P:
Editorial: Epidemiology and risk factors for interstitial lung
diseases. Front Med (Lausanne). 11:13848252024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Maher TM, Bendstrup E, Dron L, Langley J,
Smith G, Khalid JM, Patel H and Kreuter M: Global incidence and
prevalence of idiopathic pulmonary fibrosis. Respir Res.
22:1972021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zheng Q, Cox IA, Campbell JA, Xia Q,
Otahal P, de Graaff B, Corte TJ, Teoh AKY, Walters EH and Palmer
AJ: Mortality and survival in idiopathic pulmonary fibrosis: A
systematic review and meta-analysis. ERJ Open Res. 8:00591–2021.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Richeldi L, Collard HR and Jones MG:
Idiopathic pulmonary fibrosis. Lancet. 389:1941–1952. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
King TE Jr, Bradford WZ, Castro-Bernardini
S, Fagan EA, Glaspole I, Glassberg MK, Gorina E, Hopkins PM,
Kardatzke D, Lancaster L, et al: A phase 3 trial of pirfenidone in
patients with idiopathic pulmonary fibrosis. N Engl J Med.
370:2083–2092. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ebenezer DL, Fu P and Natarajan V:
Targeting sphingosine-1-Phosphate signaling in lung diseases.
Pharmacol Ther. 168:143–157. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mohammed S and Harikumar KB: Sphingosine
1-Phosphate: A novel target for lung disorders. Front Immunol.
8:2962017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hait NC, Allegood J, Maceyka M, Strub GM,
Harikumar KB, Singh SK, Luo C, Marmorstein R, Kordula T, Milstien S
and Spiegel S: Regulation of histone acetylation in the nucleus by
sphingosine-1-phosphate. Science. 325:1254–1257. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schwalm S, Manaila R, Oftring A, Schaefer
L, von Gunten S and Pfeilschifter J: The contribution of the
sphingosine 1-phosphate signaling pathway to chronic kidney
diseases: Recent findings and new perspectives. Pflugers Arch.
476:1845–1861. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhao T, Ding T, Sun Z, Shao X, Li S, Lu H,
Yuan JH and Guo Z: SPHK1/S1P/S1PR pathway promotes the progression
of peritoneal fibrosis by mesothelial-mesenchymal transition. FASEB
J. 38:e234172024. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Natarajan V, Dudek SM, Jacobson JR,
Moreno-Vinasco L, Huang LS, Abassi T, Mathew B, Zhao Y, Wang L,
Bittman R, et al: Sphingosine-1-phosphate, FTY720, and
sphingosine-1-phosphate receptors in the pathobiology of acute lung
injury. Am J Respir Cell Mol Biol. 49:6–17. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Serra M and Saba JD: Sphingosine
1-phosphate lyase, a key regulator of sphingosine 1-phosphate
signaling and function. Adv Enzyme Regul. 50:349–362. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Khan SA, Goliwas KF and Deshane JS:
Sphingosipids in lung pathology in the coronavirus disease era: A
review of sphingolipid involvement in the pathogenesis of lung
damage. Front Physiol. 12:7606382021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jozefczuk E, Guzik TJ and Siedlinski M:
Significance of sphingosine-1-phosphate in cardiovascular
physiology and pathology. Pharmacol Res. 156:1047932020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pitson SM, Xia P, Leclercq TM, Moretti PA,
Zebol JR, Lynn HE, Wattenberg BW and Vadas MA:
Phosphorylation-dependent translocation of sphingosine kinase to
the plasma membrane drives its oncogenic signalling. J Exp Med.
201:49–54. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhao M, Bian R, Xu X, Zhang J, Zhang L and
Zheng Y: Sphingolipid metabolism and signalling pathways in heart
failure: From molecular mechanism to therapeutic potential. J
Inflamm Res. 18:5477–5498. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhao Y, Gorshkova IA, Berdyshev E, He D,
Fu P, Ma W, Su Y, Usatyuk PV, Pendyala S, Oskouian B, et al:
Protection of LPS-induced murine acute lung injury by
sphingosine-1-phosphate lyase suppression. Am J Respir Cell Mol
Biol. 45:426–435. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Saba JD: Fifty years of lyase and a moment
of truth: Sphingosine phosphate lyase from discovery to disease. J
Lipid Res. 60:456–463. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fan X, Liu L, Shi Y, Guo F, He X, Zhao X,
Zhong D and Li G: Recent advances of the function of sphingosine
1-phosphate (S1P) receptor S1P3. J Cell Physiol. 236:1564–1578.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Maceyka M, Harikumar KB, Milstien S and
Spiegel S: Sphingosine-1-phosphate signaling and its role in
disease. Trends Cell Biol. 22:50–60. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sun G, Wang B, Wu X, Cheng J, Ye J, Wang
C, Zhu H and Liu X: How do sphingosine-1-phosphate affect immune
cells to resolve inflammation? Front Immunol. 15:13624592024.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li J, Fan Y, Tu W, Wu L, Pan Y, Zheng M,
Qu Y and Cao L: Sphingosine-1-phosphate in the regulation of
diabetes mellitus: A scientometric study to an in-depth review.
Front Endocrinol (Lausanne). 15:13776012024. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kunkel GT, Maceyka M, Milstien S and
Spiegel S: Targeting the sphingosine-1-phosphate axis in cancer,
inflammation and beyond. Nat Rev Drug Discov. 12:688–702. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Glass DS, Grossfeld D, Renna HA, Agarwala
P, Spiegler P, Kasselman LJ, Glass AD, DeLeon J and Reiss AB:
Idiopathic pulmonary fibrosis: Molecular mechanisms and potential
treatment approaches. Respir Investig. 58:320–335. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yu Q, Zhu D, Zou Y, Wang K, Rao P and Shen
Y: Catalpol attenuates pulmonary fibrosis by inhibiting ang II/AT1
and TGF-β/Smad-mediated epithelial mesenchymal transition. Front
Med (Lausanne). 9:8786012022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li RZ, Wang XR, Wang J, Xie C, Wang XX,
Pan HD, Meng WY, Liang TL, Li JX, Yan PY, et al: The key role of
sphingolipid metabolism in cancer: New therapeutic targets,
diagnostic and prognostic values, and anti-tumor immunotherapy
resistance. Front Oncol. 12:9416432022. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Satyananda V, Oshi M, Tokumaru Y, Maiti A,
Hait N, Matsuyama R, Endo I and Takabe K: Sphingosine 1-phosphate
(S1P) produced by sphingosine kinase 1 (SphK1) and exported via
ABCC1 is related to hepatocellular carcinoma (HCC) progression. Am
J Cancer Res. 11:4394–4407. 2021.PubMed/NCBI
|
|
30
|
Zhang W, Li Y, Li F and Ling L:
Sphingosine-1-phosphate receptor modulators in stroke treatment. J
Neurochem. 162:390–403. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Theilmeier G, Schmidt C, Herrmann J, Keul
P, Schäfers M, Herrgott I, Mersmann J, Larmann J, Hermann S,
Stypmann J, et al: High-density lipoproteins and their constituent,
sphingosine-1-phosphate, directly protect the heart against
ischemia/reperfusion injury in vivo via the S1P3 lysophospholipid
receptor. Circulation. 114:1403–1409. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ziegler AC and Gräler MH: Barrier
maintenance by S1P during inflammation and sepsis. Tissue Barriers.
9:19400692021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang F and Lu Y: The Sphingosine
1-Phosphate axis: An emerging therapeutic opportunity for
endometriosis. Reprod Sci. 30:2040–2059. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pyne NJ, Dubois G and Pyne S: Role of
sphingosine 1-phosphate and lysophosphatidic acid in fibrosis.
Biochim Biophys Acta. 1831:228–238. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Donati C, Cencetti F, Bernacchioni C,
Vannuzzi V and Bruni P: Role of sphingosine 1-phosphate signalling
in tissue fibrosis. Cell Signal. 78:1098612021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yang L, Yue S, Yang L, Liu X, Han Z, Zhang
Y and Li L: Sphingosine kinase/sphingosine 1-phosphate (S1P)/S1P
receptor axis is involved in liver fibrosis-associated
angiogenesis. J Hepatol. 59:114–123. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Milara J, Navarro R, Juan G, Peiró T,
Serrano A, Ramón M, Morcillo E and Cortijo J:
Sphingosine-1-phosphate is increased in patients with idiopathic
pulmonary fibrosis and mediates epithelial to mesenchymal
transition. Thorax. 67:147–156. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Huang LS, Berdyshev E, Mathew B, Fu P,
Gorshkova IA, He D, Ma W, Noth I, Ma SF, Pendyala S, et al:
Targeting sphingosine kinase 1 attenuates bleomycin-induced
pulmonary fibrosis. FASEB J. 27:1749–1760. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Medzhitov R: Inflammation 2010: new
adventures of an old flame. Cell. 140:771–776. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng
J, Li Y, Wang X and Zhao L: Inflammatory responses and
inflammation-associated diseases in organs. Oncotarget.
9:7204–7218. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kaminska B: MAPK signalling pathways as
molecular targets for Anti-inflammatory therapy-from molecular
mechanisms to therapeutic benefits. Biochim Biophys Acta.
1754:253–262. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu T, Zhang L, Joo D and Sun SC: NF-κB
signaling in inflammation. Signal Transduct Target Ther.
2:170232017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jayant G, Kuperberg S, Somnay K and
Wadgaonkar R: The role of sphingolipids in regulating vascular
permeability in idiopathic pulmonary fibrosis. Biomedicines.
11:17282023. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Moss BJ, Ryter SW and Rosas IO: Pathogenic
mechanisms underlying idiopathic pulmonary fibrosis. Annu Rev
Pathol. 17:515–546. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jo H, Shim K and Jeoung D: The crosstalk
between FcεRI and sphingosine signaling in allergic inflammation.
Int J Mol Sci. 23:138922022. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yang J, Tang X, Li B and Shi J:
Sphingosine 1-phosphate receptor 2 mediated early stages of
pancreatic and systemic inflammatory responses via NF-kappa B
activation in acute pancreatitis. Cell Commun Signal. 20:1572022.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zheng Z, Zeng YZ, Ren K, Zhu X, Tan Y, Li
Y, Li Q and Yi GH: S1P promotes inflammation-induced tube formation
by HLECs via the S1PR1/NF-κB pathway. Int Immunopharmacol.
66:224–235. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Xiong W, Chen S, Xiang H, Zhao S, Xiao J,
Li J, Liu Y, Shu Z, Ouyang J, Zhang J, et al: S1PR1 attenuates
pulmonary fibrosis by inhibiting EndMT and improving endothelial
barrier function. Pulm Pharmacol Ther. 81:1022282023. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hao M, Fu R, Tai J, Tian Z, Yuan X, Chen
Y, Wang M, Jiang H, Ji M, Lai F, et al: S1PR1 serves as a viable
drug target against pulmonary fibrosis by increasing the integrity
of the endothelial barrier of the lung. Acta Pharm Sin B.
13:1110–1127. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Di Paolo A, Vignini A, Alia S, Membrino V,
Delli Carpini G, Giannella L and Ciavattini A: Pathogenic role of
the Sphingosine 1-Phosphate (S1P) pathway in common gynecologic
disorders (GDs): A possible novel therapeutic target. Int J Mol
Sci. 23:135382022. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Qiu H, Liu J, You J, Zhou O, Hao C, Shu Y,
Ma D, Zou W, Zhang L, Liu E, et al: Inhibition of sphingosine
1-phosphate receptor 3 ameliorates bleomycin-induced pulmonary
fibrosis by suppressing macrophage M2 polarization. Genes Dis.
12:1012442024. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sobel K, Menyhart K, Killer N, Renault B,
Bauer Y, Studer R, Steiner B, Bolli MH, Nayler O and Gatfield J:
Sphingosine 1-phosphate (S1P) receptor agonists mediate
pro-fibrotic responses in normal human lung fibroblasts via S1P2
and S1P3 receptors and Smad-independent signaling. J Biol Chem.
288:14839–14851. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Knipe RS, Spinney JJ, Abe EA, Probst CK,
Franklin A, Logue A, Giacona F, Drummond M, Griffith J, Brazee PL,
et al: Endothelial-specific loss of Sphingosine-1-Phosphate
receptor 1 increases vascular permeability and exacerbates
Bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol.
66:38–52. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Sammani S, Moreno-Vinasco L, Mirzapoiazova
T, Singleton PA, Chiang ET, Evenoski CL, Wang T, Mathew B, Husain
A, Moitra J, et al: Differential effects of sphingosine 1-phosphate
receptors on airway and vascular barrier function in the murine
lung. Am J Respir Cell Mol Biol. 43:394–402. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Shea BS, Brooks SF, Fontaine BA, Chun J,
Luster AD and Tager AM: Prolonged exposure to sphingosine
1-phosphate receptor-1 agonists exacerbates vascular leak,
fibrosis, and mortality after lung injury. Am J Respir Cell Mol
Biol. 43:662–673. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chen Y, Zhang Y, Rao C, Huang J and Qing
Q: Deletion of sphingosine kinase 2 attenuates cigarette
smoke-mediated chronic obstructive pulmonary disease-like symptoms
by reducing lung inflammation. Biomol Biomed. 23:259–270. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Hou L, Yang L, Chang N, Zhao X, Zhou X,
Dong C, Liu F, Yang L and Li L: Macrophage sphingosine 1-Phosphate
Receptor 2 blockade attenuates liver inflammation and fibrogenesis
triggered by NLRP3 inflammasome. Front Immunol. 11:11492020.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Xia C, Cheng L, Zhao W, Chang A, Wang Z,
Liu H, Pan X, Li W, Koji S, Li Z, et al: LncRNA SYISL promotes
fibroblast myofibroblast transition via miR-23a-mediated TRIOBP
regulation. Cell Mol Life Sci. 82:2142025. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Panagiotidis GD, Vasquez-Pacheco E, Chu X,
Seeger W, El Agha E, Bellusci S and Lingampally A: Revisiting
pulmonary fibrosis: Inflammatory dynamics of the
lipofibroblast-to-inflammatory lipofibroblast-to-activated
myofibroblast reversible switch. Front Immunol. 16:16095092025.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Mo C, Xie S, Liu B, Zhong W, Zeng T, Huang
S, Lai Y, Deng G, Zhou C, Yan W, et al: Indoleamine 2,3-dioxygenase
1 limits hepatic inflammatory cells recruitment and promotes bile
duct ligation-induced liver fibrosis. Cell Death Dis. 12:162021.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Ju X, Wang K, Wang C, Zeng C, Wang Y and
Yu J: Regulation of myofibroblast dedifferentiation in pulmonary
fibrosis. Respir Res. 25:2842024. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Zhao T and Su Y: Mechanisms and
therapeutic potential of myofibroblast transformation in pulmonary
fibrosis. J Respir Biol Transl Med. 2:100012025.PubMed/NCBI
|
|
63
|
Kawashima T, Yamazaki R, Matsuzawa Y,
Yamaura E, Takabatake M, Otake S, Ikawa Y, Nakamura H, Fujino H and
Murayama T: Contrary effects of sphingosine-1-phosphate on
expression of α-smooth muscle actin in transforming growth factor
β1-stimulated lung fibroblasts. Eur J Pharmacol. 696:120–129. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Urata Y, Nishimura Y, Hirase T and
Yokoyama M: Sphingosine 1-phosphate induces alpha-smooth muscle
actin expression in lung fibroblasts via Rho-kinase. Kobe J Med
Sci. 51:17–27. 2005.PubMed/NCBI
|
|
65
|
Takuwa Y, Ikeda H, Okamoto Y, Takuwa N and
Yoshioka K: Sphingosine-1-phosphate as a mediator involved in
development of fibrotic diseases. Biochim Biophys Acta.
1831:185–192. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Kono Y, Nishiuma T, Nishimura Y, Kotani Y,
Okada T, Nakamura S and Yokoyama M: Sphingosine kinase 1 regulates
differentiation of human and mouse lung fibroblasts mediated by
TGF-beta1. Am J Respir Cell Mol Biol. 37:395–404. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Keller CD, Rivera Gil P, Tölle M, van der
Giet M, Chun J, Radeke HH, Schäfer-Korting M and Kleuser B:
Immunomodulator FTY720 induces myofibroblast differentiation via
the lysophospholipid receptor S1P3 and Smad3 signaling. Am J
Pathol. 170:281–292. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Sobel K, Monnier L, Menyhart K, Bolinger
M, Studer R, Nayler O and Gatfield J: FTY720 phosphate activates
Sphingosine-1-Phosphate receptor 2 and selectively couples to
Gα12/13/Rho/ROCK to induce myofibroblast contraction. Mol
Pharmacol. 87:916–927. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Hill C, Li J, Liu D, Conforti F, Brereton
CJ, Yao L, Zhou Y, Alzetani A, Chee SJ, Marshall BG, et al:
Autophagy inhibition-mediated epithelial-mesenchymal transition
augments local myofibroblast differentiation in pulmonary fibrosis.
Cell Death Dis. 10:5912019. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Wei J, Zhao Q, Yang G, Huang R, Li C, Qi
Y, Hao C and Yao W: Mesenchymal stem cells ameliorate
silica-induced pulmonary fibrosis by inhibition of inflammation and
epithelial-mesenchymal transition. J Cell Mol Med. 25:6417–6428.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Kim KK, Kugler MC, Wolters PJ, Robillard
L, Galvez MG, Brumwell AN, Sheppard D and Chapman HA: Alveolar
epithelial cell mesenchymal transition develops in vivo during
pulmonary fibrosis and is regulated by the extracellular matrix.
Proc Natl Acad Sci USA. 103:13180–13185. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Riemma MA, Cerqua I, Romano B, Irollo E,
Bertolino A, Camerlingo R, Granato E, Rea G, Scala S, Terlizzi M,
et al: Sphingosine-1-phosphate/TGF-β axis drives epithelial
mesenchymal transition in asthma-like disease. Br J Pharmacol.
179:1753–1768. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Huang LS, Berdyshev EV, Tran JT, Xie L,
Chen J, Ebenezer DL, Mathew B, Gorshkova I, Zhang W, Reddy SP, et
al: Sphingosine-1-phosphate lyase is an endogenous suppressor of
pulmonary fibrosis: Role of S1P signalling and autophagy. Thorax.
70:1138–1148. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Mu SY, Xu R, Wu XF, Cheng YY, Sun ZM, Liu
HT, Shao HB, Zhang XN, Zhang XN, Yang M, et al: Inhibition of
sphingosine-1-phosphate receptor-2 attenuates idiopathic pulmonary
fibrosis by preventing its binding to dapper1 in bronchial
epithelial cells. Br J Pharmacol. 183:957–972. 2026. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Lu G, Wang Y, Shi Y, Zhang Z, Huang C, He
W, Wang C and Shen HM: Autophagy in health and disease: From
molecular mechanisms to therapeutic target. MedComm (2020).
3:e1502022. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Xiao S, Peng K, Li C, Long Y and Yu Q: The
role of sphingosine-1-phosphate in autophagy and related disorders.
Cell Death Discov. 9:3802023. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Sosulski ML, Gongora R, Danchuk S, Dong C,
Luo F and Sanchez CG: Deregulation of selective autophagy during
aging and pulmonary fibrosis: The role of TGFβ1. Aging Cell.
14:774–783. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Patel AS, Lin L, Geyer A, Haspel JA, An
CH, Cao J, Rosas IO and Morse D: Autophagy in idiopathic pulmonary
fibrosis. PLoS One. 7:e413942012. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Lv X, Li K and Hu Z: Autophagy and
pulmonary fibrosis. Adv Exp Med Biol. 1207:569–579. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Hill C and Wang Y: Autophagy in pulmonary
fibrosis: Friend or foe? Genes Dis. 9:1594–1607. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Tatler AL and Jenkins G:
Sphingosine-1-phosphate metabolism: Can its enigmatic lyase promote
the autophagy of fibrosis? Thorax. 70:1106–1107. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Huang LS and Natarajan V: Sphingolipids in
pulmonary fibrosis. Adv Biol Regul. 57:55–63. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Goel K, Beatman EL, Egersdorf N, Scruggs
A, Cao D, Berdyshev EV, Schweitzer KS and Petrache I: Sphingosine 1
phosphate (S1P) Receptor 1 is decreased in human lung microvascular
endothelial cells of smokers and mediates S1P effect on autophagy.
Cells. 10:12002021. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Liu H, Li L, Chen Z, Song Y, Liu W, Gao G,
Li L, Jiang J, Xu C, Yan G and Cui H: S1PR2 inhibition attenuates
allergic asthma possibly by regulating autophagy. Front Pharmacol.
11:5980072021. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Weigel C, Hüttner SS, Ludwig K, Krieg N,
Hofmann S, Schröder NH, Robbe L, Kluge S, Nierhaus A, Winkler MS,
et al: S1P lyase inhibition protects against sepsis by promoting
disease tolerance via the S1P/S1PR3 axis. EBioMedicine.
58:1028982020. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Jelic MD, Mandic AD, Maricic SM and
Srdjenovic BU: Oxidative stress and its role in cancer. J Cancer
Res Ther. 17:22–28. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Veith C, Boots AW, Idris M, van Schooten
FJ and van der Vliet A: Redox imbalance in idiopathic pulmonary
fibrosis: A role for oxidant Cross-talk between NADPH oxidase
enzymes and mitochondria. Antioxid Redox Signal. 31:1092–1115.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Toussaint O, Medrano EE and von Zglinicki
T: Cellular and molecular mechanisms of stress-induced premature
senescence (SIPS) of human diploid fibroblasts and melanocytes. Exp
Gerontol. 35:927–945. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Raish M, Ahmad A, Ahmad Ansari M, Ahad A,
Al-Jenoobi FI, Al-Mohizea AM, Khan A and Ali N: Sinapic acid
ameliorates bleomycin-induced lung fibrosis in rats. Biomed
Pharmacother. 108:224–231. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Psathakis K, Mermigkis D, Papatheodorou G,
Loukides S, Panagou P, Polychronopoulos V, Siafakas NM and Bouros
D: Exhaled markers of oxidative stress in idiopathic pulmonary
fibrosis. Eur J Clin Invest. 36:362–367. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Bargagli E, Penza F, Vagaggini C, Magi B,
Perari MG and Rottoli P: Analysis of carbonylated proteins in
bronchoalveolar lavage of patients with diffuse lung diseases.
Lung. 185:139–144. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Lan T, Liu W, Xie X, Xu S, Huang K, Peng
J, Shen X, Liu P, Wang L, Xia P and Huang H: Sphingosine kinase-1
pathway mediates high glucose-induced fibronectin expression in
glomerular mesangial cells. Mol Endocrinol. 25:2094–2105. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Maceyka M, Milstien S and Spiegel S:
Shooting the messenger: Oxidative stress regulates
sphingosine-1-phosphate. Circ Res. 100:7–9. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Martín-Montañez E, Pavia J, Valverde N,
Boraldi F, Lara E, Oliver B, Hurtado-Guerrero I, Fernandez O and
Garcia-Fernandez M: The S1P mimetic fingolimod phosphate regulates
mitochondrial oxidative stress in neuronal cells. Free Radic Biol
Med. 137:116–130. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Harijith A, Pendyala S, Ebenezer DL, Ha
AW, Fu P, Wang YT, Ma K, Toth PT, Berdyshev EV, Kanteti P and
Natarajan V: Hyperoxia-induced p47phox activation and ROS
generation is mediated through S1P transporter Spns2, and
S1P/S1P1&2 signaling axis in lung endothelium. Am J Physiol
Lung Cell Mol Physiol. 311:L337–L351. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Zhou J, Song Y, Wang X, Li X, Liu C, Tian
C, Wang C, Li L, Yan G and Cui H: JTE-013 alleviates pulmonary
fibrosis by affecting the RhoA/YAP pathway and mitochondrial
Fusion/fission. Pharmaceuticals (Basel). 16:14442023. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Harijith A, Pendyala S, Reddy NM, Bai T,
Usatyuk PV, Berdyshev E, Gorshkova I, Huang LS, Mohan V, Garzon S,
et al: Sphingosine kinase 1 deficiency confers protection against
hyperoxia-induced bronchopulmonary dysplasia in a murine model:
Role of S1P signaling and Nox proteins. Am J Pathol. 183:1169–1182.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Huang LS, Sudhadevi T, Fu P, Punathikannan
PK, Ebenezer DL, Ramchandran R, Putherickal V, Cheresh P, Zhou G,
Ha AW, et al: Sphingosine Kinase 1/S1P signaling contributes to
pulmonary fibrosis by activating Hippo/YAP pathway and
mitochondrial reactive oxygen species in lung fibroblasts. Int J
Mol Sci. 21:20642020. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Zhang X, Cai Y, Zhang W and Chen X:
Quercetin ameliorates pulmonary fibrosis by inhibiting SphK1/S1P
signaling. Biochem Cell Biol. 96:742–751. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Gorshkova I, Zhou T, Mathew B, Jacobson
JR, Takekoshi D, Bhattacharya P, Smith B, Aydogan B, Weichselbaum
RR, Natarajan V, et al: Inhibition of serine palmitoyltransferase
delays the onset of radiation-induced pulmonary fibrosis through
the negative regulation of sphingosine kinase-1 expression. J Lipid
Res. 53:1553–1568. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Zeyada MS, Eraky SM and El-Shishtawy MM:
Trigonelline mitigates bleomycin-induced pulmonary inflammation and
fibrosis: Insight into NLRP3 inflammasome and SPHK1/S1P/Hippo
signaling modulation. Life Sci. 336:1222722024. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Lu F, Wang G, Yang X, Luo J, Ma H, Pan L,
Yao Y and Xie K: SPHK1-S1p Signaling Drives Fibrocyte-mediated
pulmonary fibrosis: Mechanistic insights and therapeutic potential.
Pharmaceuticals (Basel). 18:8592025. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Mammoliti O, Palisse A, Joannesse C, El
Bkassiny S, Allart B, Jaunet A, Menet C, Coornaert B, Sonck K, Duys
I, et al: Discovery of the S1P2 Antagonist GLPG2938
(1-[2-Ethoxy-6-(trifluoromethyl)-4-pyridyl]-3-[[5-methyl-6-[1-methyl-3-(trifluoromethyl)pyrazol-4-yl]pyridazin-3-yl]methyl]urea),
a preclinical candidate for the treatment of idiopathic pulmonary
fibrosis. J Med Chem. 64:6037–6058. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Allameh A, Atashbasteh M, Mortaz E, Naeeni
B and Jafari-Khorchani M: Down-regulation of key regulatory factors
in sphingosine-1-phosphate (S1P) pathway in human lung fibroblasts
transfected with selected microRNAs. Mol Biol Res Commun.
13:201–209. 2024.PubMed/NCBI
|
|
105
|
Arora S, Singh P, Tabassum G, Dohare R and
Syed MA: miR-495-3p regulates sphingolipid metabolic reprogramming
to induce Sphk1/ceramide mediated mitophagy and apoptosis in NSCLC.
Free Radic Biol Med. 189:71–84. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Bonauer A, Carmona G, Iwasaki M, Mione M,
Koyanagi M, Fischer A, Burchfield J, Fox H, Doebele C, Ohtani K, et
al: MicroRNA-92a controls angiogenesis and functional recovery of
ischemic tissues in mice. Science. 324:1710–1713. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Pi J, Tao T, Zhuang T, Sun H, Chen X, Liu
J, Cheng Y, Yu Z, Zhu HH, Gao WQ, et al: A
MicroRNA302-367-Erk1/2-Klf2-S1pr1 pathway prevents tumor growth via
restricting angiogenesis and improving vascular stability. Circ
Res. 120:85–98. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Zhou P, Huang G, Zhao Y, Zhong D, Xu Z,
Zeng Y, Zhang Y, Li S and He F: MicroRNA-363-mediated
downregulation of S1PR1 suppresses the proliferation of
hepatocellular carcinoma cells. Cell Signal. 26:1347–1354. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Zhang SL and Liu L: MicroRNA-148a inhibits
hepatocellular carcinoma cell invasion by targeting
sphingosine-1-phosphate receptor 1. Exp Ther Med. 9:579–584. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Brinkmann V: FTY720 (fingolimod) in
Multiple Sclerosis: Therapeutic effects in the immune and the
central nervous system. Br J Pharmacol. 158:1173–1182. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Diouf I, Malpas CB, Sharmin S, Roos I,
Horakova D, Kubala Havrdova E, Patti F, Shaygannejad V, Ozakbas S,
Eichau S, et al: Effectiveness of multiple disease-modifying
therapies in relapsing-remitting multiple sclerosis: Causal
inference to emulate a multiarm randomised trial. J Neurol
Neurosurg Psychiatry. 94:1004–1011. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Irmak Ön B, Havla J and Mansmann U:
Multivariable prognostic prediction of efficacy and safety outcomes
and response to fingolimod in people with relapsing-remitting
multiple sclerosis. Mult Scler Relat Disord. 93:1062472025.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Ahmed N, Linardi D, Muhammad N, Chiamulera
C, Fumagalli G, Biagio LS, Gebrie MA, Aslam M, Luciani GB, Faggian
G, et al: Sphingosine 1-phosphate receptor modulator fingolimod
(FTY720) attenuates myocardial fibrosis in Post-heterotopic heart
transplantation. Front Pharmacol. 8:6452017. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Tian T, Zhang J, Zhu X, Wen S, Shi D and
Zhou H: FTY720 ameliorates renal fibrosis by simultaneously
affecting leucocyte recruitment and TGF-β signalling in
fibroblasts. Clin Exp Immunol. 190:68–78. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Baweja S, Kumari A, Negi P, Tomar A,
Tripathi DM, Mourya AK, Rastogi A, Subudhi PD, Thangariyal S, Kumar
G, et al: Hepatopulmonary syndrome is associated with low
sphingosine-1-phosphate levels and can be ameliorated by the
functional agonist fingolimod. J Hepatol. 79:167–180. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Jin Y, Liu W, Gao G, Song Y, Liu H, Li L,
Zhou J, Yan G and Cui H: Effect of FTY-720 on pulmonary fibrosis in
mice via the TGF-β1 signaling pathway and autophagy. Biomol Ther
(Seoul). 31:434–445. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Müller HC, Hocke AC, Hellwig K, Gutbier B,
Peters H, Schönrock SM, Tschernig T, Schmiedl A, Hippenstiel S,
N'Guessan PD, et al: The Sphingosine-1 Phosphate receptor agonist
FTY720 dose dependently affected endothelial integrity in vitro and
aggravated ventilator-induced lung injury in mice. Pulm Pharmacol
Ther. 24:377–385. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Oo ML, Chang SH, Thangada S, Wu MT, Rezaul
K, Blaho V, Hwang SI, Han DK and Hla T: Engagement of
S1P1-degradative mechanisms leads to vascular leak in mice. J Clin
Invest. 121:2290–2300. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Bittner S and Wiendl H:
Neuroimmunotherapies Targeting T cells: From pathophysiology to
therapeutic applications. Neurotherapeutics. 13:4–19. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Chun J and Hartung HP: Mechanism of action
of oral fingolimod (FTY720) in multiple sclerosis. Clin
Neuropharmacol. 33:91–101. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Üzer F, Akanlar S and Çilli A: Real-life
data in the treatment and follow-up of idiopathic pulmonary
fibrosis: A single-center study. Tuberk Toraks. 71:347–355. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Guenther A, Tello S, Schoppe MC,
Pons-Kuehnemann J, Seeger W, Stiben J, Tello K, Molina Molina M,
Vancheri C, Crestani B and Krauss E: Pulmonary Hypertension drives
prognosis in idiopathic pulmonary fibrosis: Insights from the
European IPF registry. J Clin Med. 14:73522025. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Bellanca CM, Augello E, Mariottini A,
Bonaventura G, La Cognata V, Di Benedetto G, Cantone AF, Attaguile
G, Di Mauro R, Cantarella G, et al: Disease modifying strategies in
multiple sclerosis: New rays of hope to combat disability? Curr
Neuropharmacol. 22:1286–1326. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Roy R, Alotaibi AA and Freedman MS:
Sphingosine 1-Phosphate receptor modulators for multiple sclerosis.
CNS Drugs. 35:385–402. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Li J, Huang Y, Zhang Y, Liu P, Liu M,
Zhang M and Wu R: S1P/S1PR signaling pathway advancements in
autoimmune diseases. Biomol Biomed. 23:922–935. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Bravo GÁ, Cedeño RR, Casadevall MP and
Ramió-Torrentà L: Sphingosine-1-Phosphate (S1P) and S1P signaling
pathway modulators, from current insights to future perspectives.
Cells. 11:20582022. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Sands BE, D'Haens G, Panaccione R,
Regueiro M, Ghosh S, Hudesman D, Ahmad HA, Mehra D, Wu H, Jain A,
et al: Ozanimod in patients with moderate to severe ulcerative
colitis naive to advanced therapies. Clin Gastroenterol Hepatol.
22:2084–2095.e4. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Harris S, Feagan BG, Hanauer S, Vermeire
S, Ghosh S, Yan J, Wu C, Hu Y, Maddux R, Wolf DC and D'Haens G:
Ozanimod differentially impacts circulating lymphocyte subsets in
patients with moderately to severely active Crohn's disease. Dig
Dis Sci. 69:2044–2054. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Pérez-Jeldres T, Alvarez-Lobos M and
Rivera-Nieves J: Targeting Sphingosine-1-Phosphate signaling in
Immune-mediated diseases: Beyond multiple sclerosis. Drugs.
81:985–1002. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Lellouche F, Blais-Lecours P, Maltais F,
Sarrazin JF, Rola P, Nguyen T, Châteauvert N and Marsolais D:
Ozanimod therapy in patients with COVID-19 requiring oxygen
support: A randomized Open-label pilot trial. Chest. 165:810–819.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Paik J: Ozanimod: A review in ulcerative
colitis. Drugs. 82:1303–1313. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Cocconcelli E, Tonelli R, Abbati G,
Marchioni A, Castaniere I, Pelizzaro F, Russo FP, Vegetti A,
Balestro E, Pietrangelo A, et al: Subclinical liver fibrosis in
patients with idiopathic pulmonary fibrosis. Intern Emerg Med.
16:349–357. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Sands BE, Schreiber S, Blumenstein I,
Chiorean MV, Ungaro RC and Rubin DT: Clinician's Guide to using
ozanimod for the treatment of ulcerative colitis. J Crohns Colitis.
17:2012–2025. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Coyle PK, Freedman MS, Cohen BA, Cree BAC
and Markowitz CE: Sphingosine 1-phosphate receptor modulators in
multiple sclerosis treatment: A practical review. Ann Clin Transl
Neurol. 11:842–855. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
McGinley MP and Cohen JA: Sphingosine
1-phosphate receptor modulators in multiple sclerosis and other
conditions. Lancet. 398:1184–1194. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Tanaka S, Zheng S, Kharel Y, Fritzemeier
RG, Huang T, Foster D, Poudel N, Goggins E, Yamaoka Y, Rudnicka KP,
et al: Sphingosine 1-phosphate signaling in perivascular cells
enhances inflammation and fibrosis in the kidney. Sci Transl Med.
14:eabj26812022. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Hisano Y, Kobayashi N, Yamaguchi A and
Nishi T: Mouse SPNS2 functions as a sphingosine-1-phosphate
transporter in vascular endothelial cells. PLoS One. 7:e389412012.
View Article : Google Scholar : PubMed/NCBI
|














