Chronic liver disease (CLD), characterized by
persistent hepatic inflammation and progressive fibrosis, poses a
formidable global health burden as a leading cause of mortality
(1). Major etiological drivers
include alcohol abuse, obesity-associated metabolic dysregulation
and chronic viral hepatitis (HBV/HCV). Notably, Non-alcoholic fatty
liver disease, now reclassified as metabolic dysfunction-associated
steatotic liver disease (MASLD) to reflect its metabolic etiology,
is a major chronic liver condition that accounts for >50% of CLD
cases (2). MASLD pathogenesis
intertwines with insulin resistance, leading to hepatocellular
lipid deposition, oxidative stress and cytokine-driven inflammation
(e.g., TNF-α, TGF-β) that culminate in steatohepatitis and
cirrhosis (3). Concurrently,
chronic HBV/HCV infections promote hepatocarcinogenesis through
viral persistence, immune evasion and metabolic reprogramming of
infected hepatocytes (4–6).
A unifying feature across CLD subtypes is metabolic
dysregulation, particularly in cholesterol, lipid and glucose
homeostasis, which both initiates and perpetuates disease
progression. For instance, HBV/HCV exploit host cholesterol
pathways for viral entry and inhibition of cholesterol synthesis
can reduce the infection of a variety of human viruses, including
HBV and HCV (7). MASLD-driven
hepatocellular carcinoma (HCC) thrives on oncogenic lipogenesis. In
the case of reduced nutritional availability, glucose metabolism
and lipid metabolism change due to the increased metabolic needs of
cancer cells and play an important role in the survival of cancer
cells (8–10). In summary, disorders of cholesterol
metabolism, lipid metabolism and glucose metabolism are not only
the causes of CLD, but also play an important role in its
pathological development. The interaction among these metabolic
pathways may lead to liver cell damage, inflammation and fibrosis,
affecting the prognosis and quality of life of patients. Therefore,
it is important to explore the interventions for these metabolic
abnormalities in the treatment of CLD.
Liver X receptors (LXRs) are one of the key
regulators of metabolic diseases and are also involved in the
development of CLD. The role of LXRs in CLD has a dual nature and
can play a protective role or show pathogenicity, which is more
severe in specific cases. Activation of LXRs helps regulate
cholesterol metabolism, reduces liver inflammation and protects
liver cells from damage. It has been reported that LXRs reduce
cholesterol levels by upregulating target genes related to reverse
cholesterol transport, cholesterol conversion to bile acids and
intestinal cholesterol absorption [such as ATP-binding cassette
transporter (ABCA1), ATP-binding cassette transporter G1 (ABCG1),
ATP-binding cassette transporter G5 (ABCG5), and ATP-binding
cassette transporter G8 (ABCG8), phospholipid transporters, ApoE
and cholesterol 7α-hydroxylase (CYP7A1)] (11). LXRs activation can effectively
reduce the inflammatory response by inhibiting the expression of
inflammatory mediators and inflammatory genes (12). However, when the disease is
accompanied by metabolic abnormalities, excessive activation of
LXRs can lead to lipid accumulation and further liver damage,
thereby aggravating the condition. LXRs also directly regulates the
expression of a number of genes involved in fatty acid synthesis,
including genes encoding sterol regulatory element-binding protein
1c (SREBP-1c), a major transcriptional regulator of fatty acid
synthesis. In addition, the net effect of LXRs activation is
increased levels of long-chain polyunsaturated fatty acids,
increased triglyceride synthesis and increased hepatic triglyceride
secretion in the form of very low-density lipoprotein granules
(13,14). In fact, LXRs ligands T0901317 and
GW3965 were originally designed to treat atherosclerosis, but they
can induce severe hypertriglyceridemia and fatty liver (15–17).
These two symptoms are common in MASLD (18). Due to the side effects of LXRs, the
development of LXRs agonists has been limited. Currently, there are
no FDA-approved drugs related to LXRs available for use, so it is
particularly important to study the regulatory mechanism of LXRs in
CLD. These observations underscore the therapeutic potential of
targeting metabolic regulators such as LXRs, which sit at the nexus
of lipid metabolism, inflammation and cellular proliferation. The
present review summarized the latest research progress on the role
of LXRs in different stages of CLD and its positive and negative
regulatory mechanisms.
Identified from hepatic cDNA libraries, LXRs derive
their name from predominant expression in the liver (19). As ligand-activated transcription
factors, LXRs heterodimerize with retinoid X receptors to bind LXRs
response elements in target genes. LXRs is a transcription factor
of the nuclear receptor superfamily, mainly including two subtypes:
LXRα (encoded by Nr1h3) and LXRβ (encoded by Nr1h2) (20). LXRα exhibits tissue-specific
enrichment in the liver, intestine and adipose tissue, whereas LXRβ
displays ubiquitous expression (21). Endogenous ligands include
oxysterols [such as 22(R)-hydroxycholesterol], cholesterol
derivatives that activate LXRs upon intracellular accumulation. For
example, oxysterols can directly bind to LXRα and LXRβ receptors,
activate these receptors and promote their binding to specific DNA
sequences, thereby regulating the expression of downstream
genes.
Cholesterol is an essential molecule required for
maintaining cell structure and is crucial for various normal
biological functions (22).
Cholesterol metabolism is tightly regulated in normal cells. Of
total cholesterol in the human body ~80% is typically synthesized
endogenously, while 20% is obtained through diet. It exists in free
form or as a cholesteryl ester and fatty acids (23). Excess cholesterol accumulation in
the liver or blood (hypercholesterolemia) can lead to pathological
consequences such as liver steatosis and atherosclerosis (24). In addition to atherosclerosis, to
which is most directly related, the accumulation of cholesterol in
cells can directly lead to abnormal mitochondrial function in liver
cells, which in turn triggers apoptosis or induces inflammatory
reactions in adjacent cells, eventually leading to hepatitis and
liver fibrosis (25). LXRs govern
cholesterol metabolism through three synergistic mechanisms
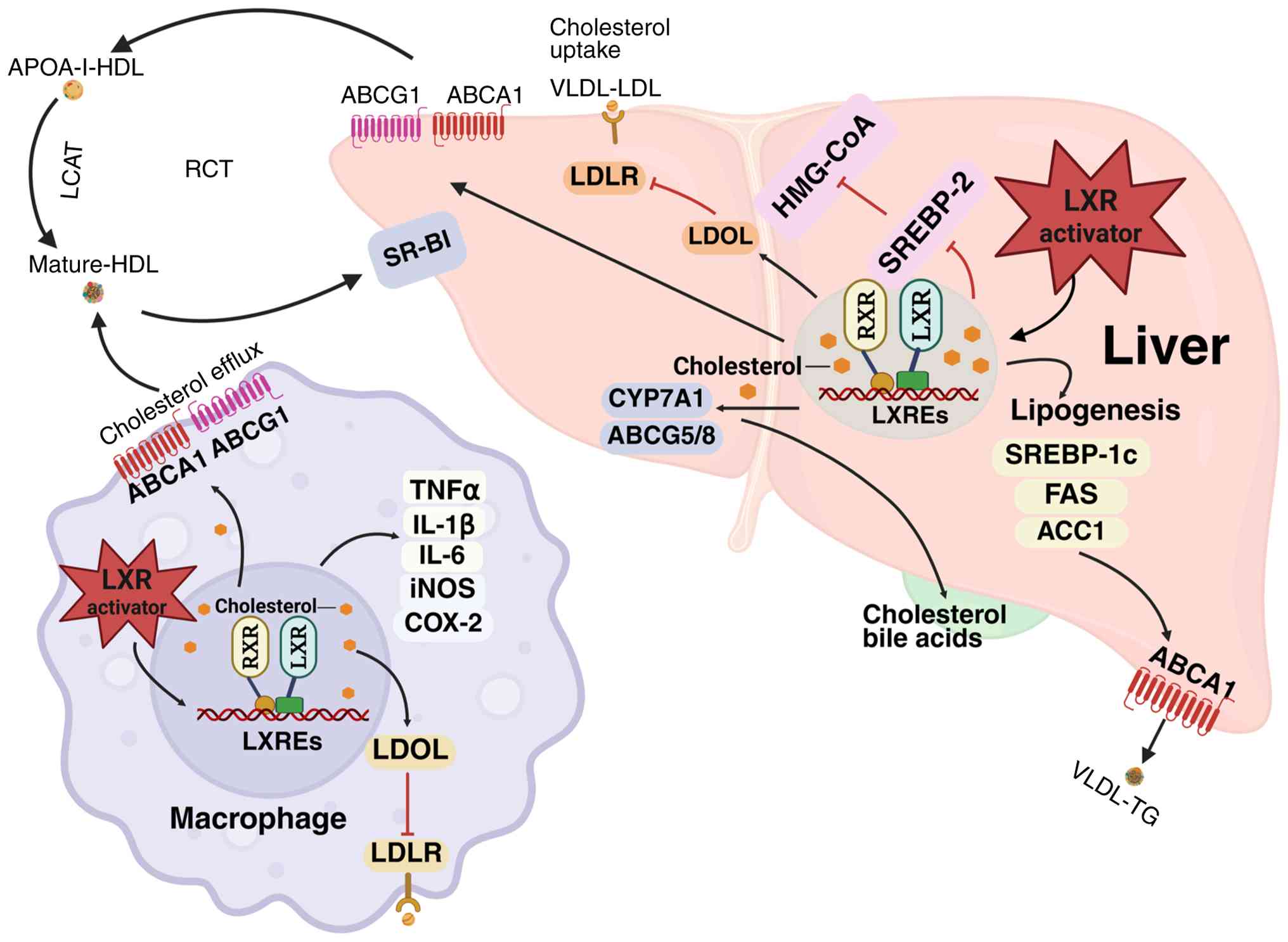
(Fig. 1): The first is inhibition
of de novo synthesis: LXRs activation inhibits SREBP-2, thus
reducing 3-Hydroxy-3-methylglutaryl-coenzyme A reductase
expression, curtailing cholesterol biosynthesis (26). The second is promotion of reverse
cholesterol transport (RCT): LXRs activation also reduces
cholesterol uptake and promotes its transport to high-density
lipoprotein (HDL) by regulating low-density lipoprotein receptor
(LDLR) and inducing ABCA1/ABCG1 transporter expression. HDL
transports cholesterol to the liver, which is further converted
into bile acids and excreted through bile (21,27–30).
The third is bile acid conversion: LXRs activation promotes the
conversion of cholesterol to bile acids by increasing the
expression of CYP7A1 and enhances cholesterol hydroxylation,
driving bile acid synthesis and fecal excretion (31–33).
CYP7A1 is a classical target gene of LXRs in rodents. However, in
humans, the response of CYP7A1 to LXRs activation is very weak or
even absent. In short, these pathways collectively prevent
cytotoxic cholesterol overload, which triggers mitochondrial
dysfunction, apoptosis and NLRP3 inflammasome activation in
hepatocytes and dysregulated LXRs signaling, thus links
hypercholesterolemia to MASLD progression and atherogenesis
(34).
Lipids are an important form of energy storage in
the body and it is also an important part of cell membranes and
signaling molecules. SREBP-1c is a core transcriptional activator
of fatty acid biosynthesis, which is mainly responsible for
maintaining lipid homeostasis and regulating fatty acid synthesis.
Its importance in lipid metabolism is self-evident (35). Studies in LXRs-deficient mice have
shown that the expression level of SREBP-1c mRNA in the liver is
markedly decreased and the mRNA levels of lipogenic enzymes
affected by it are also decreased (36). This indicates that LXRs are key
transcriptional regulators involved in lipid metabolism in the
liver and their adipogenic activity is mainly achieved by inducing
the transcription of the gene encoding SREBP-1c. Direct
transcriptional activation of SREBP-1c induces lipogenic enzymes,
such as acetyl-CoA carboxylase 1 and fatty acid synthase (FAS)
(36–38). The liver is an important organ of
insulin sensitivity and is essential for maintaining blood glucose
homeostasis. Insulin resistance can lead to changes in metabolic
gene expression and glucose metabolism disorders. LXRs not only
participates in cholesterol and lipid metabolism, but also plays an
important role in glucose metabolism (39). LXRs activation inhibits the
function of sodium-glucose cotransporter 2 (SGLT2) by reducing the
expression of SGLT2 protein. Suppression of SGLT2 attenuates renal
glucose reabsorption, implicating LXRs in systemic glucose
regulation (40). In addition,
inflammation is considered to be a major driver of the development
of a number of chronic diseases. LXRs also plays an important role
in the regulation of inflammation. LXRs inhibits inflammation
through two main mechanisms. The first is LXRs agonists repress
NF-κB via increasing the level of MyD88 mRNA alternative splicing
short form, thereby reducing the production of inflammatory
cytokines (such as TNF-α, IL-1β and IL-6) (41). The second is to enhance macrophages
cholesterol efflux, dampening pro-inflammatory cytokine production
(42). In general, this functional
pleiotropy positions LXRs as master regulators of
metabolic-inflammatory crosstalk, yet also underlies their
context-dependent duality in CLD pathogenesis.
The expression of HBV is not only related to liver
metabolism, but HBV also actively interferes with liver metabolic
pathways. This unique interaction between HBV and liver metabolism
is called the ‘metabolovirus’ model of HBV expression (43). Thus, regulation of the metabolism
of the host cell represents a viable approach to antiviral
intervention. Ezetimibe is an FDA-approved drug that inhibits liver
cholesterol renewal. It has been shown to inhibit HCV and HBV
infection by regulating liver cholesterol uptake (44). LXRs is the main transcriptional
regulator of a series of genes involved in cholesterol uptake,
transport, efflux and excretion (45). The pleiotropic effects of LXRs
agonist therapy may also contribute to antiviral activity. For
example, 22(S) -hydroxycholesterol, a LXRs ligand, is reported to
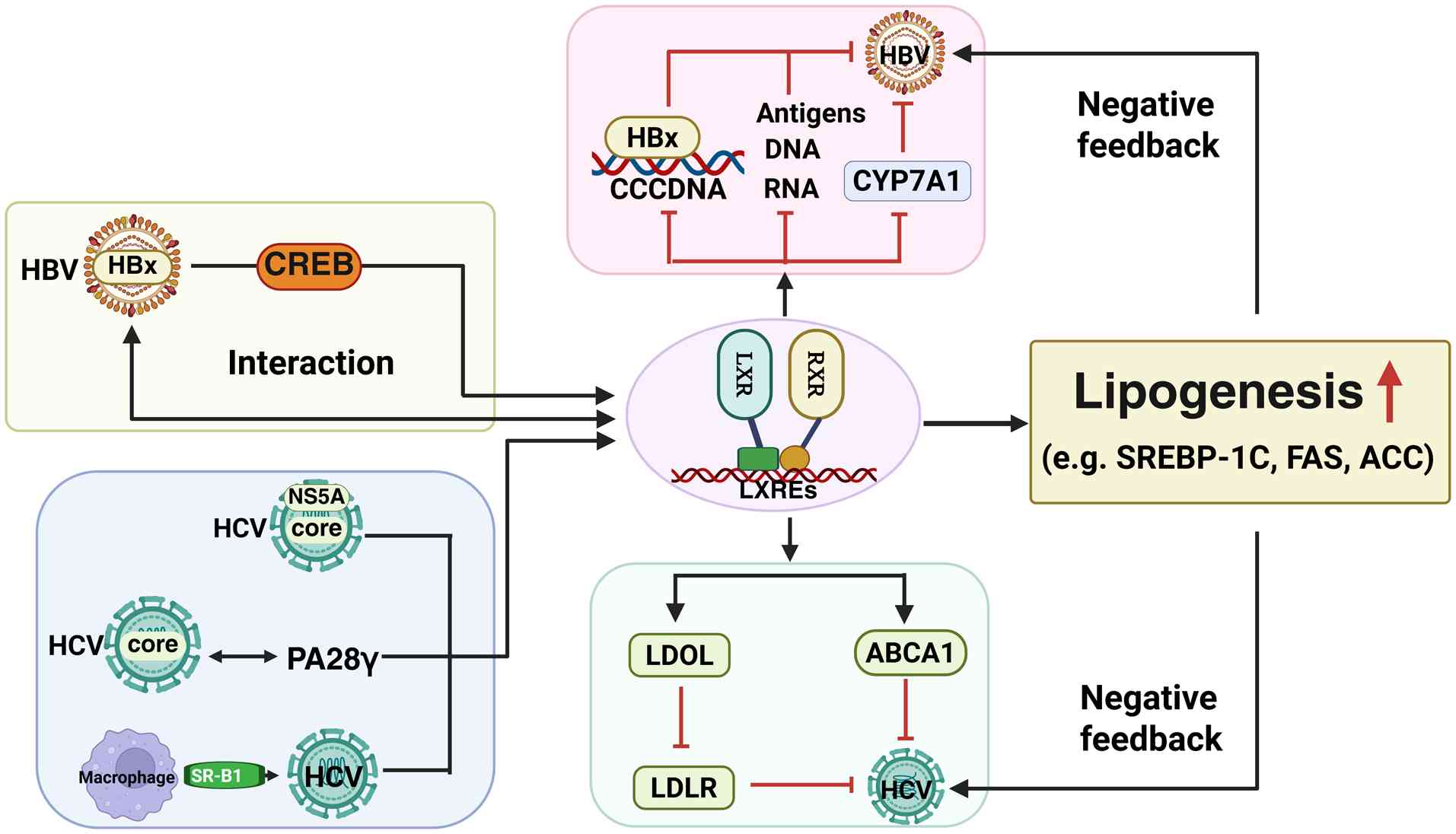
block HBV infection in dHepaRG cells (46). The activation of the LXRs pathway
in HBV-infected primary human hepatocytes can effectively inhibit
HBV, mainly through three mechanisms (Fig. 2): Reducing the production of viral
RNA, DNA and antigens; blocking the interaction between Hepatitis B
virus X protein (HBx) and viral replication template covalently
closed circular DNA (cccDNA) and reducing the transcription level
of cccDNA; and the destruction of the expression of downstream
LXRs' target gene CYP7A1 (47). In
addition to effectively inhibiting HBV replication, the role of
LXRs in HCV infection is also notable. LXRs can block and weaken
HCV infection through a variety of mechanisms, thereby enhancing
the host's antiviral ability. Although the exact mechanism that
regulates the entry of HCV into hepatocytes is unclear, LDLR has
been shown to be necessary for the entry of infectious HCV
particles. LXRs controls the ubiquitination and degradation of LDLR
by regulating the expression of inducible degrader of LDLR (LDOL),
thereby blocking HCV infection (48). It has been shown that the entry of
HCV cells requires cholesterol homeostasis and intact
cholesterol-rich membrane microdomains; however, a major regulator
of cellular cholesterol and phospholipid homeostasis is the ABCA1
transporter (49). LXRs agonist
GW3965 can inhibit HCV cell entry by activating LXRs and
upregulating the expression of ABCA1 (50).
In addition to inhibiting HCV replication, the
function of LXRs is also impaired by HCV. HCV NS5A and core
proteins promote intracellular lipid accumulation by enhancing the
expression and transcriptional activity of LXRα. This accumulation
of lipids is achieved by activating the LXRs response element,
leading to upregulation of genes associated with adipogenesis (such
as SREBP-1c, PPAR-γ and fatty acid synthase) (51). In the liver of HCV patients, LXRα
and its related adipogenic genes (such as PPARγ and SREBP-1c) and
inflammatory genes are abnormally increased, suggesting that LXRs
plays an important role in liver lipid accumulation (52). Specifically, HCV core protein
enhances the activity of LXRα/RXRα-dependent SREBP-1c promoter by
interacting with PA28γ, thereby promoting liver steatosis (53). Therefore, although LXRs activation
can inhibit HCV replication, it may also aggravate HCV-related
liver steatosis by promoting lipid accumulation. This dual role
reveals the complex role of LXRs in HCV infection: on the one hand,
it can be used as a potential target for antiviral therapy; on the
other hand, it may lead to abnormal lipid metabolism in
pathophysiology. In addition, HCV infection also leads to metabolic
and immune changes in liver macrophages, which further aggravates
lipid accumulation and inflammatory response and may develop into
steatohepatitis and fibrosis. Studies have shown that HCV RNA can
accumulate and persist in macrophages, although it does not express
viral proteins. After macrophages were exposed to HCV, the levels
of cholesterol efflux-related protein ABCA1 and SREBP-1c increased.
Macrophages recognize HCV through scavenger receptor B1 (SR-B1),
activate LXRα, lead to increased lipid and cholesterol and trigger
metabolic changes in HCV-related CLDs (54). This series of mechanisms indicates
the key role of LXRs in HCV infection and related liver diseases.
Similarly, HBV-related pathological mechanisms also involve the
role of LXRs. HBx is not only a key regulator of viral replication
and infection, but also interacts with a variety of signaling
pathways in host cells, which has a fresh impact on the
pathogenesis of liver (55). It
has been found that increased expression of HBx leads to lipid
accumulation in hepatocytes, which is mediated by SREBP1 and PPARγ
(56). It was further elucidated
the molecular mechanism of HBx-induced lipid accumulation in
hepatocytes. It has been shown that HBx interacts with LXRα,
enhances the binding of LXRα to LXRs response element (LXRE) and
then upregulates SREBP1 and FAS, resulting in an increase in lipid
in hepatocytes (57). In addition,
in HBV-related HCC, HBx also induces the transactivation of LXRα by
recruiting CREB-binding protein to the promoter of the target gene,
thereby activating the target gene of lipid production and further
aggravating lipid accumulation in hepatocytes (58). In summary, the role of LXRs in HBV
and HCV infection is very complex. Although the activation of LXRs
can inhibit viral replication, it can also lead to changes in lipid
metabolism, thus providing a more favorable living environment for
the virus. It is hoped that future research will help to further
understand the dual role of LXRs in viral infection in order to
develop new therapeutic strategies.
The pathogenesis of MASLD is closely linked to
insulin resistance, which leads to abnormal accumulation of free
fatty acids in hepatocytes (59).
This accumulation heightens the cells' sensitivity to oxidative
stress, mitochondrial dysfunction and endoplasmic reticulum stress,
prompting the secretion of inflammatory cytokines such as TNF-α,
TGF-β and monocyte chemoattractant protein-1 (MCP-1) (60). As previously reported, LXRs
activation can limit intracellular cholesterol levels by inducing
reverse cholesterol transport, increasing bile acid production and
inhibiting intestinal cholesterol absorption. In addition to its
key role in cholesterol homeostasis, LXRs is also an important
regulator of inflammatory gene expression. Ligand-activated LXRs
attenuates the induction of classical inflammatory genes [such as
inducible nitric oxide synthase (iNOS), cyclooxygenase-2, matrix
metalloproteinase-9 (MMP-9) and various chemokines] stimulated by
LPS, TNF-α and IL-1β (12). LXRs
plays a key regulatory role in MASLD (Fig. 3). Activation of LXRs can alleviate
the liver injury of MASLD by inhibiting JNK and PI3K signaling
pathways and reducing the expression of pro-inflammatory markers
such as TNF-α and iNOS (61). In
addition, the destruction of Ser196 phosphorylation (S196A) in LXRα
prevents cholesterol accumulation and reduces liver inflammation
and fibrosis. This mechanism helps to delay the progression of
MASLD. However, after disrupting S196A in LXRα, acetylation of the
key histone H3K27 can upregulate Ces1f and SREBP-1c and induce
hepatic steatosis (62). These
results indicate that LXRs plays an important role in regulating
inflammatory response and can inhibit liver inflammation and liver
fibrosis. However, activation of LXRs also promotes
adipogenesis.
Although LXRs may have a protective effect in some
ways, in the context of MASLD, the activation of LXRs often leads
to the disorder of lipid metabolism and promotes the aggravation of
the disease. The expression of LXRα, SREBP-1c, ACC and FAS in the
liver of MASLD patients is markedly upregulated. It is suggested
that LXRs and its downstream target genes are involved in lipid
metabolism of MASLD (63). LXRs
promotes adipogenesis by regulating key factors such as SREBP-1c
and carbohydrate response element-binding protein, which may lead
to hypertriglyceridemia and hepatic steatosis (64,65).
In addition to activating LXRs, the removal of inhibitory histone
markers histone H3 di-methylation at lysine 9 (H3K9me2) and H3K9me3
on LXRE can also upregulate the expression of LXRα-dependent
adipogenic genes, leading to triglyceride (TG) accumulation and
MASLD (66). Therefore, inhibition
of LXRs activity is considered to be a potential therapeutic
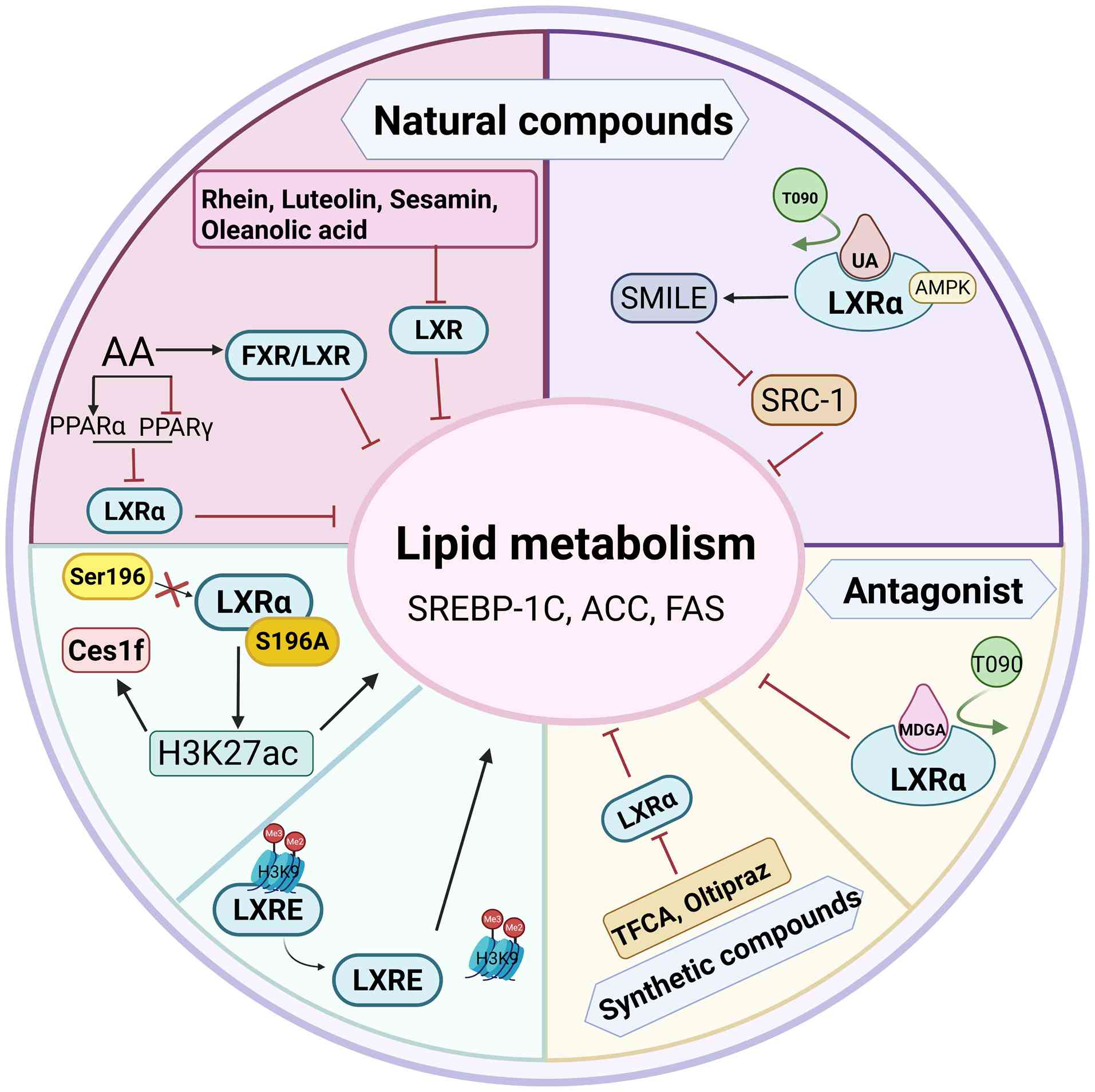
strategy in some cases (Table I).
As an antagonist of LXRs, meso-dihydroguaiaretic acid (MDGA)
inhibits activation of the LXRα ligand-binding domain by
competitively binding to the pocket for agonist T0901317, reduces
the transcriptional expression of lipogenesis-related LXRα
coactivator protein and downstream target genes involved in fatty
acid synthesis, and thereby reduced lipid accumulation in mice
administered with T0901317 or fed with a high-fat diet. These
results suggest that MDGA has the potential to attenuate
nonalcoholic steatosis mediated by selective inhibition of LXRα in
the liver of mice (67).
N-(4-trifluoromethylphenyl) 3,4-dimethoxycinnamamide is a compound
that inhibits the ligand binding domain of LXRα. It plays a role by
forming hydrogen bonds with Arg305 in the H5 region, affecting the
dissociation of thyroid hormone receptor-associated protein/vitamin
D receptor-interacting protein coactivator and the recruitment of
nuclear receptor corepressor to regulate the transcriptional
control exerted by LXRα, thereby attenuating LXRα-induced
adipogenesis and fatty liver (68).
A number of natural compounds have multiple
biological activities and can act on multiple metabolic pathways at
the same time to improve a number of aspects related to MASLD
(Table I), such as lipid
metabolism, inflammatory response and oxidative stress, which makes
them more effective in comprehensive treatment. Acanthoic acid (AA)
is a diterpene isolated from Acanthopanax koreanum Nakai
(Araliaceae), which has anti-inflammatory and hepatoprotective
effects. In a MASLD mouse model, AA markedly reduced hepatic
steatosis and fibrosis in MASLD mice and steatotic AML 12 cells by
activating FXR/LXRs to reduce the expression of SREBP-1 and target
genes, upregulate the expression of PPARα and downregulate the
expression of PPARγ (69). Natural
compounds may enhance the body's own metabolic capacity by
regulating endogenous metabolic pathways and signaling pathways,
thus playing a synergistic role in improving MASLD. Research shows
that 4,5-dihydroxyanthraquinone-2-carboxylic acid (Rhein) may
reduce hepatic TG accumulation via two pathways. First, Rhein
decreases de novo lipogenesis by directly suppressing
LXRs-mediated SREBP-1c expression. Second, Rhein improves MASLD and
related diseases through LXRs-mediated energy balance, metabolic
regulation pathways and immunomodulatory activities involved in
hepatic steatosis (70). Ursolic
acid (UA) is a plant triterpene compound that binds competitively
with T090 at the LXRα ligand binding domain that increases the
co-repressor protein SMILE and reduces the co-activator SRC 1
through AMPK, so as to reduce the expression of SREBP-1c, FAS and
ACC genes, thereby reducing liver lipid accumulation and improving
blood lipid levels. The RCT stimulation of LXRα is retained and
enhanced, providing a potential treatment for MASLD (71). Natural compounds such as luteolin,
sesamin and oleanolic acid have shown significant potential in
improving liver steatosis. Luteolin can not only inhibit the
activation of LXRs, but also further reduce lipid synthesis by
downregulating the expression of SREBP-1c, while maintaining
glycogen storage and helping to improve liver metabolism (72). Sesamin (73) and oleanolic acid (74) attenuate ligand-induced adipogenesis
through a similar mechanism, thereby reducing fat accumulation in
the liver. The study of these natural compounds provides an
important theoretical basis for the development of new therapeutic
strategies, especially in the treatment of nonalcoholic fatty liver
disease and other related diseases.
LXRs has been identified as a potential therapeutic
target for a variety of types of cancer, including breast cancer
(75), prostate cancer (76) and HCC (77–80).
Its mechanism of action involves promoting cholesterol metabolism
(75,76), interacting with TGF-β (78,79).
The activation of LXRβ can inhibit tumor growth and innate immune
response (81). Specifically
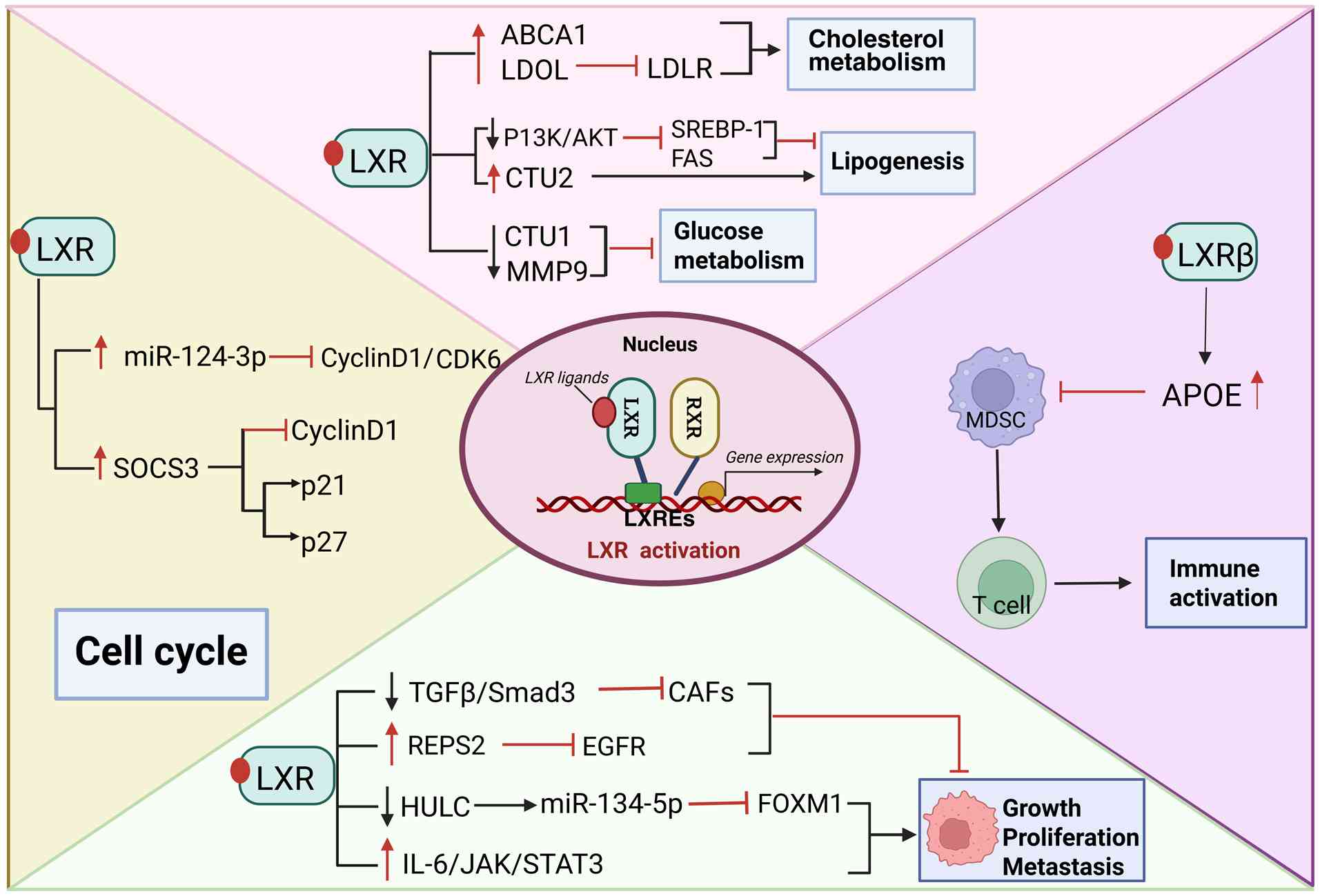
(Fig. 4), LXRs activation inhibits
tumor cell proliferation by promoting cholesterol catabolism
(80) and reducing intracellular
cholesterol levels (82).
Cholesterol plays a key role in cell proliferation in physiological
and tumor states and cancer cells need sufficient cholesterol to
support their rapid growth. A study has found that LDLR and ACAT
are overexpressed in tumor tissues of most patients with cancer,
which provides support for the rapid proliferation of cancer cells
(22). It has also been reported
that targeting LDLR with the LXRs agonist GW3965 causes
LDOL-mediated LDLR degradation and increased expression of the
ABCA1 cholesterol efflux transporter, resulting in decreased
cholesterol levels, which effectively promotes tumor cell death
(83). LXRα agonists limit the
expression of myofibroblast markers by antagonizing the TGFβ
signaling pathway at the transcriptional level, inhibiting the
differentiation of cancer-associated fibroblasts, thereby
inhibiting the growth of liver cancer cells (78). In addition, it has been found that
LXRs activation leads to LXRs target gene ApoE inhibiting
myeloid-derived suppressor cells survival through its effect on
LRP8 receptor, thereby weakening the immunosuppressive effect,
enhancing the activation of T cells and triggering a strong
anti-tumor response (81). Lipids
are mainly processed in the liver and play an important role in the
physiology of the organ and the pathological progression of a
number of diseases such as metabolic syndrome and HCC (84). Enhanced adipogenesis is important
for cancer cells because it provides lipids for membrane structural
units, post-translational modifications of proteins and energy
storage (85). In addition, LXRs
are activated by oxysterols, which can cause lipotoxicity in liver
cancer (86) and lipid production
promotes the occurrence and development of liver cancer (87). SREBP-1c was reported to be highly
expressed in liver cancer, prostate cancer and ovarian cancer
(88). In addition, FAS and ACC,
which are closely related to LXRs, have also been found in HCC and
various other human epithelial cancers and precancerous lesions
(89). Studies have found that
cytosolic thioureaase 2 (CTU2) is involved in lipogenesis by
directly promoting the synthesis of lipogenic proteins and have
determined that CTU2 is a LXRs target gene (90). By reducing the expression of CTU2
and LXRs ligands, this approach can induce tumor cell apoptosis and
inhibit cell proliferation to enhance the anti-tumor effect of HCC.
Bergapten acts as a LXRα/β agonist and inhibits HCC progression by
regulating PI3K/AKT and LDOL/LDLR signaling pathways. Moreover, it
downregulates SREBP 1 and FAS levels through AKT inhibition to
maintain lipid homeostasis, thereby exerting an anti-tumor effect
(91).
In HCC, a number of genes related to cell
proliferation [forkhead box protein M1 (FOXM1), suppressor of
cytokine signaling 3 (SOCS3)], invasion and migration are directly
regulated by LXRs. It has been found that activated LXRα can
downregulate the expression of HULC, that promotes the growth and
metastasis of HCC cells and which further upregulates the
expression level of miR-134-5p (inhibiting cancer cell
proliferation and invasion) and inhibits the expression of FOXM1
(affecting cell proliferation and migration), thereby inhibiting
the growth of liver cancer cells (92). The high expression of miR-124-3p in
LXRs-activated condition affects the formation of cyclin D1/CDK6
active complex by cyclin D1 and its binding partner CDK 6, thereby
regulating the cell cycle and inhibiting the progression of HCC
(93). Although LXRβ has
anticancer function, LXRα-mediated SOCS3 induction is the reason
for the anti-HCC effect of LXRs agonists (94). The methylation of SOCS3 gene
promoter region leads to its downregulation in HCC and the
re-expression of SOCS3 leads to apoptosis and cell cycle arrest
(95–97). The activation of LXRs upregulates
the expression of SOCS3 by enhancing the mRNA stability of SOCS3.
LXRs inhibits the occurrence and development of HCC by inducing
SOCS3 to downregulate cyclin D1 and downregulates p21 and p27
(98). Epidermal growth factor
receptor (EGFR) is a transmembrane tyrosine kinase receptor that
can be activated by a variety of ligands, thereby activating
multiple signaling pathways, promoting tumor cell proliferation and
metastasis and inhibiting apoptosis. It has been shown that the
activation of LXRs can promote the binding of LXRs protein to LXRE
in the REPS 2 promoter region, upregulate the expression of REPS 2
and inhibit EGF-mediated EGFR endocytosis and downstream activation
of AKT/NF-κB, p38MAPK and ERK1/2 signaling pathways, thereby
inhibiting the proliferation and migration of HCC cells and
exerting anti-tumor effects (99).
Malignant tumor cells increase glucose uptake by increasing the
expression of glucose transporter 1 (Glut1) to meet their high
energy needs (100). The
upregulation of Glut1 can also promote the activity of EGFR and
integrin signaling pathways, thereby enhancing the proliferation,
migration and invasion of tumor cells (101). However, activation of LXRs can
inhibit this process. Specifically, LXRs inhibits the progression
of HCC by downregulating the expression of Glut1 and tumor invasion
marker MMP9 and reducing the glucose content in HCC cells (102). Chronic activation of LXRα
promotes HCC at least in part by promoting the upregulation of
innate immunosuppressive factors caused by oxysterol accumulation,
as well as the IL-6/Janus kinase/STAT3 signaling and complement
pathways (103). If the
tumor-promoting effect caused by chronic activation of LXRs is
solved, its anti-tumor effect may be further improved. Tumor LXRs
expression is a prognostic marker for patients with HCC. The
average 5-year overall survival rate and average overall survival
time of patients with low LXRs expression are markedly lower than
those of patients with high LXRs levels (104). In general, LXRs play an important
role in the occurrence and development of liver cancer, but
research in this field is continues. Future research may reveal
more details about the relationship between LXRs and liver cancer
and may provide clues for the development of new strategies for the
treatment of HCC.
LXRs, as a core transcription factor connecting
metabolism and immunity, exhibit a complex role which is related to
the etiology, development stage and specific cell types of the
chronic diseases. For instance, LXRs is implicated throughout the
pathogenic progression from viral hepatitis to hepatic fibrosis and
eventually to HCC.
In viral hepatitis, LXRs primarily exerts a
protective regulatory role. In the early stage of infection, it can
directly interfere with the viral entry into hepatocytes. After
entering the stage of chronic inflammation and fibrosis, LXRs helps
mitigate excessive inflammatory responses and inhibits the
activation of hepatic stellate cells by promoting macrophage
cholesterol efflux (such as via its target gene ABCA1/ABCG1),
thereby delaying disease progression (105). However, persistent viruses can
impair this protective function of LXRs by interfering with the
host transcriptional program. Crucially, in chronic hepatitis B
progression, LXRα expression is specifically downregulated in
Kupffer cells, leading to disordered lipid metabolism and
subsequent activation of the Stat3 signaling pathway, which
promotes cancer stem cell formation. Upregulation of LXRα can
inhibit this process, revealing a key node of LXRs in suppressing
inflammation to cancer transition (106).
By contrast, the role of LXRs in the MASLD/MASH is
inherently more complex and context-dependent. In early disease
stages, activation of LXRα in hepatocytes mainly through
SREBP-1c-promotes de novo lipogenesis and directly
contributes to simple steatosis. As the disease progresses to
steatohepatitis and fibrosis, LXRs exerts anti-inflammatory and
anti-fibrotic effects by inducing antioxidant genes and suppressing
macrophage-mediated inflammation (107). However, sustained overactivation
of its lipogenic pathway can exacerbate hepatocyte lipotoxicity
(108). Notably, chronic
activation of intestine-specific LXRα (rather than hepatic LXRs)
has been shown to synergize with a high-cholesterol diet and
markedly accelerate the development of MASH-related HCC,
highlighting the spatial heterogeneity of LXRs function and an
alternative pathway driving carcinogenesis (109). When the disease enters the HCC
stage, the function of LXRs changes again and tends to exert tumor
inhibition through mechanisms such as reducing intratumoral
cholesterol levels, regulating immune microenvironment and
inhibiting proliferation-related genes.
Clinically, CLD often involves overlapping
etiologies (e.g., chronic viral hepatitis with concomitant hepatic
steatosis), which further complicates the regulation of the LXRs
signaling network. In cases of chronic viral hepatitis with hepatic
steatosis, viral interference may weaken LXRs-mediated
anti-inflammatory and cholesterol-efflux functions, whereas its
SREBP-1c-driven lipogenic activity can be amplified, collectively
exacerbating intrahepatic lipid accumulation and inflammatory
injury (58,110). It is hypothesized that under the
long-term combined attack of multiple etiologies, the LXRs pathway
may undergo ‘functional exhaustion’ or ‘selective inactivation’,
that is, its beneficial anti-inflammatory and anti-fibrosis
functions are gradually lost and the residual activity of harmful
lipid synthesis is exposed, thereby accelerating the evolution to
cirrhosis and liver cancer.
In summary, future studies should prioritize the
development of complex liver disease models that simulates
multi-factor interactions to precisely delineate the LXRs-regulated
networks in different etiological combinations and disease stages.
Building on such insights, designing cell-or pathway-selective LXRs
targeting strategies (e.g., tissue-targeted drugs that selectively
activate macrophage LXRs for anti-inflammatory purposes, but
simultaneously antagonize the lipid synthesis-promoting effects of
hepatocyte LXRs) has important translational medical significance
for restoring its homeostasis regulation function in the context of
complex liver diseases and achieving effective and precise
intervention.
The multiple roles of LXRs make it an important
regulator of CLD. Therapeutic targeting of LXRs holds promise for
mitigating hepatic steatosis through cholesterol efflux
potentiation, suppressing viral replication via lipid raft
disruption and restraining HCC progression by modulating oncogenic
lipogenesis. However, another feature of LXRs activation is indued
lipid production driven by SREBP-1c, which requires precise dose
control during LXRs targeted therapy. In order to improve the
precision in pharmacological intervention, future strategies should
take these aspects into account. Combinatorial regiments: For
example, coupling LXRs agonists with SGLT2 inhibitors to
counterbalance metabolic side effects. Oltipraz, a drug with
anti-fatty degeneration, may be combined with LXRs agonists to
counteract the side effects of LXRs activation. In addition, with
the discovery of some LXRs modulators, with the support of
artificial intelligence and machine learning technology, we should
accelerate the discovery and optimization of LXRs modulators. For
example, UA, as a natural compound, has demonstrated its potential
to inhibit harmful lipid production pathways while retaining
beneficial RCT activation. Third, developing tissue-specific LXRβ
agonists may avoid LXRα agonists-mediated systemic
hypertriglyceridemia. Based on the aforementioned strategies,
future research may unlock the full therapeutic potential of LXRs
axis in CLD.
Not applicable.
The present review was supported by the National Natural Science
Foundation of China (grant nos. 82000525 and 81873883) and Science
and Technology Support Plan for Youth Innovation of Colleges and
Universities of Shandong Province of China (grant no. 2021KJ106)
and Shandong Provincial Natural Science Foundation, China (grant
nos. ZR2023MH359 and ZR2023QH248). In addition, it was funded by
Youth Innovation Team Project for Talent Introduction and
Cultivation in Universities of Shandong Province.
Not applicable.
HY and XT provided the conceptual idea and design of
the present review, wrote the manuscript and contributed equally to
this work. XF, JZ, LW, XD and HZ performed the literature search
and analysis, were responsible for constructing the figures and
tables, and reviewed the manuscript. SL and ML provided valuable
guidance and revised the paper. Data authentication is not
applicable. All authors read and approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
De Siervi S, Cannito S and Turato C:
Chronic liver disease: Latest research in pathogenesis, detection
and treatment. Int J Mol Sci. 24:106332023. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rong L, Zou J, Ran W, Qi X, Chen Y, Cui H
and Guo J: Advancements in the treatment of non-alcoholic fatty
liver disease (NAFLD). Front Endocrinol (Lausanne). 13:10872602023.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yin X, Guo X, Liu Z and Wang J: Advances
in the diagnosis and treatment of non-alcoholic fatty liver
disease. Int J Mol Sci. 24:28442023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gregorio GV, Mieli-Vergani G and Mowat AP:
Viral hepatitis. Arch Dis Child. 70:343–348. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shafritz DA, Shouval D, Sherman HI,
Hadziyannis SJ and Kew MC: Integration of hepatitis B virus DNA
into the genome of liver cells in chronic liver disease and
hepatocellular carcinoma. Studies in percutaneous liver biopsies
and post-mortem tissue specimens. N Engl J Med. 305:1067–1073.
1981. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kiyosawa K, Sodeyama T, Tanaka E, Gibo Y,
Yoshizawa K, Nakano Y, Furuta S, Akahane Y, Nishioka K, Purcell RH,
et al: Interrelationship of blood transfusion, non-A, non-B
hepatitis and hepatocellular carcinoma: Analysis by detection of
antibody to hepatitis C virus. Hepatology. 12:671–675. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liou JW, Mani H and Yen JH: Viral
hepatitis, cholesterol metabolism, and cholesterol-lowering natural
compounds. Int J Mol Sci. 23:38972022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Anwar SD, Foster C and Ashraf A: Lipid
disorders and metabolic-associated fatty liver disease. Endocrinol
Metab Clin North Am. 52:445–457. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang S, Sheng F, Zou L, Xiao J and Li P:
Hyperoside attenuates non-alcoholic fatty liver disease in rats via
cholesterol metabolism and bile acid metabolism. J Adv Res.
34:109–122. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pope ED III, Kimbrough EO, Vemireddy LP,
Surapaneni PK, Copland JA III and Mody K: Aberrant lipid metabolism
as a therapeutic target in liver cancer. Expert Opin Ther Targets.
23:473–483. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Guo S, Li L and Yin H: Cholesterol
homeostasis and liver X Receptor (LXR) in Atherosclerosis.
Cardiovasc Hematol Disord Drug Targets. 18:27–33. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Joseph SB, Castrillo A, Laffitte BA,
Mangelsdorf DJ and Tontonoz P: Reciprocal regulation of
inflammation and lipid metabolism by liver X receptors. Nat Med.
9:213–219. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bischoff ED, Daige CL, Petrowski M, Dedman
H, Pattison J, Juliano J, Li AC and Schulman IG: Non-redundant
roles for LXRα and LXRβ in atherosclerosis susceptibility in low
density lipoprotein receptor knockout mice. J Lipid Res.
51:900–906. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Okazaki H, Goldstein JL, Brown MS and
Liang G: LXR-SREBP-1c-Phospholipid transfer protein axis controls
very low density lipoprotein (VLDL) Particle Size. J Biol Chem.
285:6801–6810. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hong C and Tontonoz P: Liver X receptors
in lipid metabolism: Opportunities for drug discovery. Nat Rev Drug
Discov. 13:433–444. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Joseph SB, McKilligin E, Pei L, Watson MA,
Collins AR, Laffitte BA, Chen M, Noh G, Goodman J, Hagger GN, et
al: Synthetic LXR ligand inhibits the development of
atherosclerosis in mice. Proc Natl Acad Sci USA. 99:7604–7609.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Schultz JR, Tu H, Luk A, Repa JJ, Medina
JC, Li L, Schwendner S, Wang S, Thoolen M, Mangelsdorf DJ, et al:
Role of LXRs in control of lipogenesis. Genes Dev. 14:2831–2838.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Deprince A, Haas JT and Staels B:
Dysregulated lipid metabolism links NAFLD to cardiovascular
disease. Mol Metab. 42:1010922020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lu TT, Repa JJ and Mangelsdorf DJ: Orphan
nuclear receptors as eLiXiRs and FiXeRs of sterol metabolism. J
Biol Chem. 276:37735–37738. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Martens N, Zhan N, Yam SC, Leijten FPJ,
Palumbo M, Caspers M, Tiane A, Friedrichs S, Li Y, van Vark-van der
Zee L, et al: Supplementation of seaweed extracts to the diet
reduces symptoms of Alzheimer's disease in the APPswePS1ΔE9 mouse
model. Nutrients. 16:16142024. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Repa JJ, Turley SD, Lobaccaro JA, Medina
J, Li L, Lustig K, Shan B, Heyman RA, Dietschy JM and Mangelsdorf
DJ: Regulation of Absorption and ABC1-mediated efflux of
cholesterol by RXR Heterodimers. Science. 289:1524–1529. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mayengbam SS, Singh A, Pillai AD and Bhat
MK: Influence of cholesterol on cancer progression and therapy.
Transl Oncol. 14:1010432021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cardoso D and Perucha E: Cholesterol
metabolism: A new molecular switch to control inflammation. Clin
Sci (Lond). 135:1389–1408. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Malhotra P, Gill RK, Saksena S and Alrefai
WA: Disturbances in cholesterol homeostasis and non-alcoholic fatty
liver diseases. Front Med (Lausanne). 7:4672020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Song Y, Liu J, Zhao K, Gao L and Zhao J:
Cholesterol-induced toxicity: An integrated view of the role of
cholesterol in multiple diseases. Cell Metab. 33:1911–1925. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Miserez AR, Muller PY, Barella L, Barella
S, Staehelin HB, Leitersdorf E, Kark JD and Friedlander Y:
Sterol-regulatory element-binding protein (SREBP)-2 contributes to
polygenic hypercholesterolaemia. Atherosclerosis. 164:15–26. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang L, Reue K, Fong LG, Young SG and
Tontonoz P: Feedback regulation of cholesterol uptake by the
LXR-IDOL-LDLR Axis. Arterioscler Thromb Vasc Biol. 32:2541–2546.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zelcer N, Hong C, Boyadjian R and Tontonoz
P: LXR regulates cholesterol uptake through Idol-dependent
ubiquitination of the LDL receptor. Science. 325:100–104. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kennedy MA, Barrera GC, Nakamura K, Baldán
Á, Tarr P, Fishbein MC, Frank J, Francone OL and Edwards PA: ABCG1
has a critical role in mediating cholesterol efflux to HDL and
preventing cellular lipid accumulation. Cell Metab. 1:121–131.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Leiva A, Verdejo H, Benítez ML, Martínez
A, Busso D and Rigotti A: Mechanisms regulating hepatic SR-BI
expression and their impact on HDL metabolism. Atherosclerosis.
217:299–307. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Peet DJ, Turley SD, Ma W, Janowski BA,
Lobaccaro JM, Hammer RE and Mangelsdorf DJ: Cholesterol and bile
acid metabolism are impaired in mice lacking the nuclear oxysterol
receptor LXR alpha. Cell. 93:693–704. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
El Roz A, Bard JM, Huvelin JM and Nazih H:
LXR agonists and ABCG1-dependent cholesterol efflux in MCF-7 breast
cancer cells: Relation to proliferation and apoptosis. Anticancer
Res. 32:3007–3013. 2012.PubMed/NCBI
|
|
33
|
Bensinger SJ, Bradley MN, Joseph SB,
Zelcer N, Janssen EM, Hausner MA, Shih R, Parks JS, Edwards PA,
Jamieson BD and Tontonoz P: LXR signaling couples sterol metabolism
to proliferation in the acquired immune response. Cell. 134:97–111.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Goicoechea L, Conde de la Rosa L, Torres
S, García-Ruiz C and Fernández-Checa JC: Mitochondrial cholesterol:
Metabolism and impact on redox biology and disease. Redox Biol.
61:1026432023. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xu X, So JS, Park JG and Lee AH:
Transcriptional control of hepatic lipid metabolism by SREBP and
ChREBP. Semin Liver Dis. 33:301–311. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Repa JJ, Liang G, Ou J, Bashmakov Y,
Lobaccaro JM, Shimomura I, Shan B, Brown MS, Goldstein JL and
Mangelsdorf DJ: Regulation of mouse sterol regulatory
element-binding protein-1c gene (SREBP-1c) by oxysterol receptors,
LXRalpha and LXRbeta. Genes Dev. 14:2819–2830. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yoshikawa T, Shimano H, Amemiya-Kudo M,
Yahagi N, Hasty AH, Matsuzaka T, Okazaki H, Tamura Y, Iizuka Y,
Ohashi K, et al: Identification of Liver X Receptor-Retinoid X
receptor as an activator of the sterol regulatory element-binding
protein 1c gene promoter. Mol Cell Biol. 21:2991–3000. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Horton JD, Shimomura I, Brown MS, Hammer
RE, Goldstein JL and Shimano H: Activation of cholesterol synthesis
in preference to fatty acid synthesis in liver and adipose tissue
of transgenic mice overproducing sterol regulatory element-binding
protein-2. J Clin Invest. 101:2331–2339. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Baranowski M: Biological role of liver X
receptors. J Physiol Pharmacol. 59 (Suppl 7):S31–S55. 2008.
|
|
40
|
Chonlaket P, Wongwan T and Soodvilai S:
Liver X receptor activation inhibits SGLT2-mediated glucose
transport in human renal proximal tubular cells. Exp Physiol.
103:250–260. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li N, Li Y, Han X, Zhang J, Han J, Jiang
X, Wang W, Xu Y, Xu Y, Fu Y and Si S: LXR agonist inhibits
inflammation through regulating MyD88 mRNA alternative splicing.
Front Pharmacol. 13:9736122022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rigamonti E, Chinetti-Gbaguidi G and
Staels B: Regulation of Macrophage Functions by PPAR-alpha,
PPAR-gamma, and LXRs in mice and men. Arterioscler Thromb Vasc
Biol. 28:1050–1059. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shlomai A and Shaul Y: The ‘metabolovirus’
model of hepatitis B virus suggests nutritional therapy as an
effective anti-viral weapon. Med Hypotheses. 71:53–57. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lucifora J, Esser K and Protzer U:
Ezetimibe blocks hepatitis B virus infection after virus uptake
into hepatocytes. Antiviral Res. 97:195–197. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang B and Tontonoz P: Liver X receptors
in lipid signalling and membrane homeostasis. Nat Rev Endocrinol.
14:452–463. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Iwamoto M, Watashi K, Tsukuda S, Aly HH,
Fukasawa M, Fujimoto A, Suzuki R, Aizaki H, Ito T, Koiwai O, et al:
Evaluation and identification of hepatitis B virus entry inhibitors
using HepG2 cells overexpressing a membrane transporter NTCP.
Biochem Biophys Res Commun. 443:808–813. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zeng J, Wu D, Hu H, Young JAT, Yan Z and
Gao L: Activation of the Liver X receptor pathway inhibits HBV
replication in primary human hepatocytes. Hepatology. 72:1935–1948.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zeng J, Wu Y, Liao Q, Li L and Chen X and
Chen X: Liver X receptors agonists impede hepatitis C virus
infection in an Idol-dependent manner. Antiviral Res. 95:245–256.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kapadia SB, Barth H, Baumert T, McKeating
JA and Chisari FV: Initiation of Hepatitis C virus infection is
dependent on cholesterol and cooperativity between CD81 and
scavenger receptor B type I. J Virol. 81:374–383. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Bocchetta S, Maillard P, Yamamoto M,
Gondeau C, Douam F, Lebreton S, Lagaye S, Pol S, Helle F,
Plengpanich W, et al: Up-Regulation of the ATP-binding cassette
transporter A1 inhibits hepatitis C virus infection. PLoS One.
9:e921402014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
García-Mediavilla MV, Pisonero-Vaquero S,
Lima-Cabello E, Benedicto I, Majano PL, Jorquera F,
González-Gallego J and Sánchez-Campos S: Liver X receptor
α-mediated regulation of lipogenesis by core and NS5A proteins
contributes to HCV-induced liver steatosis and HCV replication. Lab
Invest. 92:1191–1202. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lima-Cabello E, García-Mediavilla María V,
Miquilena-Colina María E, Vargas-Castrillón J, Lozano-Rodríguez T,
Fernández-Bermejo M, Olcoz José L, González-Gallego J,
García-Monzón C and Sánchez-Campos S: Enhanced expression of
pro-inflammatory mediators and liver X-receptor-regulated lipogenic
genes in non-alcoholic fatty liver disease and hepatitis C. Clin
Sci (Lond). 120:239–250. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Moriishi K, Mochizuki R, Moriya K,
Miyamoto H, Mori Y, Abe T, Murata S, Tanaka K, Miyamura T, Suzuki
T, et al: Critical role of PA28gamma in hepatitis C
virus-associated steatogenesis and hepatocarcinogenesis. Proc Natl
Acad Sci USA. 104:1661–1666. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Jennelle LT, Magoro T, Angelucci AR,
Dandekar A and Hahn YS: Hepatitis C virus alters macrophage
cholesterol metabolism through interaction with scavenger
receptors. Viral Immunol. 35:223–235. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Schollmeier A, Glitscher M and Hildt E:
Relevance of HBx for Hepatitis B Virus-associated pathogenesis. Int
J Mol Sci. 24:49642023. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kim KH, Shin HJ, Kim K, Choi HM, Rhee SH,
Moon HB, Kim HH, Yang US, Yu DY and Cheong J: Hepatitis B Virus X
protein induces hepatic steatosis via transcriptional activation of
SREBP1 and PPARgamma. Gastroenterology. 132:1955–1967. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Kim K, Kim Kook H, Kim Hyeong H and Cheong
J: Hepatitis B virus X protein induces lipogenic transcription
factor SREBP1 and fatty acid synthase through the activation of
nuclear receptor LXRalpha. Biochem J. 416:219–230. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Na TY, Shin YK, Roh KJ, Kang SA, Hong I,
Oh SJ, Seong JK, Park CK, Choi YL and Lee MO: Liver X receptor
mediates hepatitis B virus X protein-induced lipogenesis in
hepatitis B virus-associated hepatocellular carcinoma. Hepatology.
49:1122–1131. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Shiragannavar VD, Sannappa Gowda NG,
Puttahanumantharayappa LD, Karunakara SH, Bhat S, Prasad SK, Kumar
DP and Santhekadur PK: The ameliorating effect of withaferin A on
high-fat diet-induced non-alcoholic fatty liver disease by acting
as an LXR/FXR dual receptor activator. Front Pharmacol.
14:11359522023. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Nascè A, Gariani K, Jornayvaz FR and
Szanto I: NADPH oxidases connecting fatty liver disease, insulin
resistance and type 2 diabetes: Current knowledge and therapeutic
outlook. Antioxidants (Basel). 11:11312022. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Liu Y, Han X, Bian Z, Peng Y, You Z, Wang
Q, Chen X, Qiu D and Ma X: Activation of Liver X receptors
attenuates endotoxin-induced liver injury in mice with nonalcoholic
fatty liver disease. Dig Dis Sci. 57:390–398. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Becares N, Gage MC, Voisin M, Shrestha E,
Martin-Gutierrez L, Liang N, Louie R, Pourcet B, Pello OM, Luong
TV, et al: Impaired LXRα phosphorylation attenuates progression of
fatty liver disease. Cell Rep. 26:984–995.e6. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Higuchi N, Kato M, Shundo Y, Tajiri H,
Tanaka M, Yamashita N, Kohjima M, Kotoh K, Nakamuta M, Takayanagi R
and Enjoji M: Liver X receptor in cooperation with SREBP-1c is a
major lipid synthesis regulator in nonalcoholic fatty liver
disease. Hepatol Res. 38:1122–1129. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Cha JY and Repa JJ: The liver X Receptor
(LXR) and hepatic lipogenesis. J Biol Chem. 282:743–751. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Grefhorst A, Elzinga BM, Voshol PJ,
Plo¨sch T, Kok T, Bloks VW, van der Sluijs FH, Havekes LM, Romijn
JA, Verkade HJ and Kuipers F: Stimulation of lipogenesis by
pharmacological activation of the liver X receptor leads to
production of large, triglyceride-rich very low density lipoprotein
particles. J Biol Chem. 277:34182–34190. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Kim JH, Jung DY, Kim HR and Jung MH:
Histone H3K9 Demethylase JMJD2B plays a role in LXRα-Dependent
Lipogenesis. Int J Mol Sci. 21:83132020. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Sim WC, Park S, Lee KY, Je YT, Yin HQ,
Choi YJ, Sung SH, Park SJ, Park HJ, Shin KJ and Lee BH: LXR-α
antagonist meso-dihydroguaiaretic acid attenuates high-fat
diet-induced nonalcoholic fatty liver. Biochem Pharmacol.
90:414–424. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Sim WC, Kim DG, Lee KJ, Choi YJ, Choi YJ,
Shin KJ, Jun DW, Park SJ, Park HJ, Kim J, et al: Cinnamamides,
novel liver X receptor antagonists that inhibit ligand-induced
lipogenesis and fatty liver. J Pharmacol Exp Ther. 355:362–369.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Han X, Cui ZY, Song J, Piao HQ, Lian LH,
Hou LS, Wang G, Zheng S, Dong XX, Nan JX and Wu YL: Acanthoic acid
modulates lipogenesis in nonalcoholic fatty liver disease via
FXR/LXRs-dependent manner. Chem Biol Interact. 311:1087942019.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Sheng X, Wang M, Lu M, Xi B, Sheng H and
Zang YQ: Rhein ameliorates fatty liver disease through negative
energy balance, hepatic lipogenic regulation, and immunomodulation
in diet-induced obese mice. Am J Physiol Endocrinol Metab.
300:E886–E893. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Lin YN, Wang CCN, Chang HY, Chu FY, Hsu
YA, Cheng WK, Ma WC, Chen CJ, Wan L and Lim YP: Ursolic acid, a
novel liver X Receptor α (LXRα) antagonist inhibiting
ligand-induced nonalcoholic fatty liver and drug-induced
lipogenesis. J Agric Food Chem. 66:11647–11662. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Yin Y, Gao L, Lin H, Wu Y, Han X, Zhu Y
and Li J: Luteolin improves non-alcoholic fatty liver disease in
db/db mice by inhibition of liver X receptor activation to
down-regulate expression of sterol regulatory element binding
protein 1c. Biochem Biophys Res Commun. 482:720–726. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Tai TS, Tien N, Shen HY, Chu FY, Wang CCN,
Lu CH, Yu HI, Kung FP, Chuang HH, Lee YR, et al: Sesamin, a
Naturally occurring lignan, inhibits ligand-induced lipogenesis
through interaction with liver X Receptor Alpha (LXRα) and Pregnane
X Receptor (PXR). Evid Based Complement Alternat Med.
2019:94016482019. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Lin YN, Chang HY, Wang CCN, Chu FY, Shen
HY, Chen CJ and Lim YP: Oleanolic acid inhibits liver X receptor
alpha and pregnane X receptor to attenuate ligand-induced
lipogenesis. J Agric Food Chem. 66:10964–10976. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Hutchinson SA, Lianto P, Moore JB, Hughes
TA and Thorne JL: Phytosterols inhibit side-chain oxysterol
mediated activation of LXR in breast cancer cells. Int J Mol Sci.
20:32412019. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Krycer JR, Phan L and Brown AJ: A key
regulator of cholesterol homoeostasis, SREBP-2, can be targeted in
prostate cancer cells with natural products. Biochem J.
446:191–201. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Wan D, Yang Y, Liu Y, Cun X, Li M, Xu S,
Zhao W, Xiang Y, Qiu Y, Yu Q, et al: Sequential depletion of
myeloid-derived suppressor cells and tumor cells with a
dual-pH-sensitive conjugated micelle system for cancer
chemoimmunotherapy. J Control Release. 317:43–56. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Morén A, Bellomo C, Tsubakihara Y,
Kardassis D, Mikulits W, Heldin CH and Moustakas A: LXRα limits
TGFβ-dependent hepatocellular carcinoma associated fibroblast
differentiation. Oncogenesis. 8:362019. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Bellomo C, Caja L, Fabregat I, Mikulits W,
Kardassis D, Heldin CH and Moustakas A: Snail mediates crosstalk
between TGFβ and LXRα in hepatocellular carcinoma. Cell Death
Differ. 25:885–903. 2018.PubMed/NCBI
|
|
80
|
Wang Z, Yang X, Chen L, Zhi X, Lu H, Ning
Y, Yeong J, Chen S, Yin L, Wang X and Li X: Upregulation of
hydroxysteroid sulfotransferase 2B1b promotes hepatic oval cell
proliferation by modulating oxysterol-induced LXR activation in a
mouse model of liver injury. Arch Toxicol. 91:271–287. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Tavazoie MF, Pollack I, Tanqueco R,
Ostendorf BN, Reis BS, Gonsalves FC, Kurth I, Andreu-Agullo C,
Derbyshire ML, Posada J, et al: LXR/ApoE activation restricts
innate immune suppression in cancer. Cell. 172:825–840.e18. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Bovenga F, Sabbà C and Moschetta A:
Uncoupling nuclear receptor LXR and cholesterol metabolism in
cancer. Cell Metab. 21:517–526. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Guo D, Reinitz F, Youssef M, Hong C,
Nathanson D, Akhavan D, Kuga D, Amzajerdi AN, Soto H, Zhu S, et al:
An LXR agonist promotes glioblastoma cell death through inhibition
of an EGFR/AKT/SREBP-1/LDLR-Dependent Pathway. Cancer Discov.
1:442–456. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Alannan M, Fayyad-Kazan H, Trézéguet V and
Merched A: Targeting lipid metabolism in liver cancer.
Biochemistry. 59:3951–3964. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Menendez JA and Lupu R: Fatty acid
synthase and the lipogenic phenotype in cancer pathogenesis. Nat
Rev Cancer. 7:763–777. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Rudalska R, Harbig J, Snaebjornsson MT,
Klotz S, Zwirner S, Taranets L, Heinzmann F, Kronenberger T,
Forster M, Cui W, et al: LXRα activation and Raf inhibition trigger
lethal lipotoxicity in liver cancer. Nat Cancer. 2:201–217. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Calvisi DF, Wang C, Ho C, Ladu S, Lee SA,
Mattu S, Destefanis G, Delogu S, Zimmermann A, Ericsson J, et al:
Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling,
promotes development of human hepatocellular carcinoma.
Gastroenterology. 140:1071–1083.e5. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Guo D, Bell EH, Mischel P and Chakravarti
A: Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr
Pharm Des. 20:2619–2626. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Weidle UH, Scheuer W, Eggle D, Klostermann
S and Stockinger H: Cancer-related issues of CD147. Cancer Genomics
Proteomics. 7:157–169. 2010.PubMed/NCBI
|
|
90
|
Xue C, Wei Z, Zhang Y, Liu Y, Zhang S, Li
Q, Feng K, Yang X, Liu G, Chen Y, et al: Activation of CTU2
expression by LXR promotes the development of hepatocellular
carcinoma. Cell Biol Toxicol. 40:232024. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Pattanayak SP, Bose P, Sunita P, Siddique
MUM and Lapenna A: Bergapten inhibits liver carcinogenesis by
modulating LXR/PI3K/Akt and IDOL/LDLR pathways. Biomed
Pharmacother. 108:297–308. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
He J, Yang T, He W, Jiang S, Zhong D, Xu
Z, Wei Q, Zhang Y and Shi C: Liver X receptor inhibits the growth
of hepatocellular carcinoma cells via regulating
HULC/miR-134-5p/FOXM1 axis. Cell Signal. 74:1097202020. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Zhong D, Lyu X, Fu X, Xie P, Liu M, He F
and Huang G: Upregulation of miR-124-3p by Liver X receptor
inhibits the growth of hepatocellular carcinoma cells via
suppressing cyclin D1 and CDK6. Technol Cancer Res Treat.
19:15330338209674732020. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Bilotta MT, Abruzzese MP, Molfetta R,
Scarno G, Fionda C, Zingoni A, Soriani A, Garofalo T, Petrucci MT,
Ricciardi MR, et al: Activation of liver X receptor up-regulates
the expression of the NKG2D ligands MICA and MICB in multiple
myeloma through different molecular mechanisms. FASEB J.
33:9489–9504. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Niwa Y, Kanda H, Shikauchi Y, Saiura A,
Matsubara K, Kitagawa T, Yamamoto J, Kubo T and Yoshikawa H:
Methylation silencing of SOCS-3 promotes cell growth and migration
by enhancing JAK/STAT and FAK signalings in human hepatocellular
carcinoma. Oncogene. 24:6406–6417. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Jiang BG, Wang N, Huang J, Yang Y, Sun LL,
Pan ZY and Zhou WP: Tumor SOCS3 methylation status predicts the
treatment response to TACE and prognosis in HCC patients.
Oncotarget. 8:28621–28627. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Yuan K, Lei Y, Chen HN, Chen Y, Zhang T,
Li K, Xie N, Wang K, Feng X, Pu Q, et al: HBV-induced ROS
accumulation promotes hepatocarcinogenesis through Snail-mediated
epigenetic silencing of SOCS3. Cell Death Differ. 23:616–627. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Xiong H, Zhang Y, Chen S, Ni Z, He J, Li
X, Li B, Zhao K, Yang F, Zeng Y, et al: Induction of SOCS3 by liver
X receptor suppresses the proliferation of hepatocellular carcinoma
cells. Oncotarget. 8:64083–64094. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
He XY, Zhu MM, Zheng J, Wang CY, Zhao XK,
Zhang BT, Zhou DC, Zhang S, Yang XX, Duan YJ, et al: Liver X
receptor agonists exert antitumor effects against hepatocellular
carcinoma via inducing REPS2 expression. Acta Pharmacol Sin.
44:635–646. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Islam RA, Hossain S and Chowdhury EH:
Potential therapeutic targets in energy metabolism pathways of
breast cancer. Curr Cancer Drug Targets. 17:707–721. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Oh S, Kim H, Nam K and Shin I: Glut1
promotes cell proliferation, migration and invasion by regulating
epidermal growth factor receptor and integrin signaling in
triple-negative breast cancer cells. BMB Rep. 50:132–137. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Xiong T, Li Z, Huang X, Lu K, Xie W, Zhou
Z and Tu J: TO901317 inhibits the development of hepatocellular
carcinoma by LXRα/Glut1 decreasing glycometabolism. Am J Physiol
Gastrointest Liver Physiol. 316:G598–G607. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Xie Y, Sun R, Gao L, Guan J, Wang J, Bell
A, Zhu J, Zhang M, Xu M, Lu P, et al: Chronic activation of LXRα
sensitizes mice to hepatocellular carcinoma. Hepatol Commun.
6:1123–1139. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Long H, Guo X, Qiao S and Huang Q: Tumor
LXR expression is a prognostic marker for patients with
hepatocellular carcinoma. Pathol Oncol Res. 24:339–344. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Endo-Umeda K and Makishima M: Liver X
receptors regulate cholesterol metabolism and immunity in hepatic
nonparenchymal cells. Int J Mol Sci. 20:50452019. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Shi J, Li Q, Li J, Xi J, He Q, Zhou J,
Wang X, Song X, Li X, Yue X, et al: CHB-induced immune zonation
chaos elicited LXRα-mediated Lipid metabolism disorders in kupffer
cells to induce cancer stem cell formation. Adv Sci (Weinh).
13:e102752026. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Wang Z, Chen X, Li J, Chen M, Zhu H, Li X,
Yu W, Xia N, Zhang Y, Sun L, et al: Macrophages Atp6v0d2 regulates
XBP1-mediated cholesterol metabolism to suppress metabolic
dysfunction-associated steatohepatitis progression. Int
Immunopharmacol. 161:1150882025. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Lockhart SM, Muso M, Zvetkova I, Lam BYH,
Ferrari A, Schoenmakers E, Duckett K, Leslie J, Collins A,
Romartínez-Alonso B, et al: Damaging mutations in liver X
receptor-α are hepatotoxic and implicate cholesterol sensing in
liver health. Nat Metab. 6:1922–1938. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Piccinin E, Arconzo M, Pasculli E, Tricase
AF, Cultrera S, Bertrand-Michel J, Loiseau N, Villani G, Guillou H
and Moschetta A: Pivotal role of intestinal cholesterol and nuclear
receptor LXR in metabolic liver steatohepatitis and
hepatocarcinoma. Cell Biosci. 14:692024. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Kew MC: Hepatitis B virus × protein in the
pathogenesis of hepatitis B virus-induced hepatocellular carcinoma.
J Gastroenterol Hepatol. 26 (Suppl 1):S144–S152. 2011. View Article : Google Scholar
|