Introduction
Cerebral aneurysms (CAs) are a pathological
condition characterized by a bulging or weakened region in the
arterial wall of the brain, which increases the risk of rupture
(1). The rupture of CAs typically
leads to the compression of surrounding brain tissue, potentially
impairing basic brain functions (2). Aneurysmal subarachnoid hemorrhage
(aSAH) is responsible for ~5% of all strokes and carries a high
mortality rate ranging between 22 and 50% (3). With advancements in scientific
technologies, the detection rate of CAs has gradually increased.
Common diagnostic techniques for CAs involve magnetic resonance
imaging and computed tomography scans, and angiography (4). For smaller, asymptomatic aneurysms, a
conservative approach focused on observation is often employed
(5). Surgical interventions, such
as aneurysm clipping and endovascular embolization, are commonly
used to seal off larger or symptomatic aneurysms and prevent
rupture (6). Notably, control of
blood pressure is widely recognized as a key factor in preventing
intracranial aneurysm (IA) rupture; therefore, blood pressure
management and adjunctive treatments to prevent thrombosis are
frequently used as therapeutic strategies (7).
Mitochondrial damage can lead to disturbances in
energy metabolism, impairing the normal function of vascular smooth
muscle cells and compromising the stability of the vessel wall
(8). The formation of CAs is often
associated with vascular wall remodeling; this process involves
smooth muscle cell migration and proliferation, as well as the
upregulation of matrix metalloproteinases (MMPs), all contributing
to vascular wall expansion (9).
Mitochondrial damage impairs the normal function of these cells,
particularly affecting the proliferation, migration and matrix
synthesis of vascular smooth muscle cells. Research has suggested
that mitochondrial repair represents a potential pharmacological
target for cerebral ischemia. It offers new therapeutic directions
for related diseases, including transient ischemic attacks,
aneurysms and strokes (10). Zhao
et al (11) demonstrated
that microRNA-29a, by targeting Mcl-1, can regulate the
mitochondrial apoptotic pathway and promote the progression of CAs,
making it a potential therapeutic target for aneurysm treatment.
Furthermore, Wang et al (12) utilized Mendelian randomization
analysis to identify mitochondrial-associated proteins, such as
CCDC90B, tRNA PusA and AIF1, as factors linked to an increased risk
of CAs. Notably, these factors were particularly associated with
aSAH. These outcomes provide novel perspectives into the
pathogenesis and potential treatment strategies for CAs.
Lactate dehydrogenase A (LDHA) encodes the A
subunit of LDH, an enzyme that facilitates anaerobic glycolysis; it
catalyzes the reversible conversion of pyruvate to lactate while
simultaneously reducing NADH to NAD (13). A previous study showed that LCN2
promotes vascular remodeling in pulmonary hypertension (PH) via the
Akt-hypoxia-inducible factor 1α (HIF-1α)-LDHA axis (14). Furthermore, Wu et al
(15) showed that LDHA-mediated
lactate generation promotes pulmonary vasculature remodeling in PH
by activating the Akt pathway. This finding highlights the critical
function of LDHA in vascular remodeling and metabolic reprogramming
in PH. Previous research has also explored the effects of lactate
metabolism-related genes, LDHA and vascular endothelial growth
factor A (VEGFA), on vascular endothelial cells (VECs) under
oxidative stress (16). Oxidative
stress promotes a metabolic shift towards glycolysis by regulating
LDHA expression, glycolytic activity and lactate production.
Upregulation of LDHA can enhance cell survival and reduce
apoptosis, whereas knockdown of VEGFA leads to the opposite
effects. Notably, oxidative stress contributes to the formation and
rupture of CAs by damaging endothelial cells and promoting the
transformation of smooth muscle cells into an inflammatory
phenotype. It also initiates inflammatory responses and activates
MMPs through the generation of free radicals (17). The present study initially exposed
VECs to hydrogen peroxide (H2O2)-induced
oxidative stress and then subjected them to oxygen-glucose
deprivation/reperfusion (OGD/R) treatment to mimic
ischemia-reperfusion damage. This approach allows for the modeling
of the pathological conditions of VECs in CAs. A previous study has
shown that LDHA can exacerbate myocardial ischemia-reperfusion
injury by inducing the lactylation of NOD-, LRR- and pyrin
domain-containing protein 3 (18).
The current study aimed to examine the function of
LDHA in the pathophysiology of CAs, particularly focusing on its
impacts on mitochondrial function, oxidative stress and metabolic
reprogramming in VECs. Given the critical role of oxidative stress
and mitochondrial damage in aneurysm progression, it was
hypothesized that LDHA overexpression may mitigate these adverse
effects. This protective effect could potentially be achieved by
enhancing antioxidant defense, preserving mitochondrial integrity
and promoting glycolytic activity. Additionally, the potential
participation of the HIF-1α pathway in mediating the protective
effects of LDHA under oxidative conditions was explored. The
findings of the present study may provide novel insights into the
molecular mechanisms underlying aneurysm development. They could
also identify potential therapeutic targets for CAs and related
disorders, such as subarachnoid hemorrhage.
Materials and methods
Cell line and culture
The human umbilical VEC-derived cell line EA.hy926
(cat. no. SCSP-5285) was provided by The Cell Bank of Type Culture
Collection of The Chinese Academy of Sciences. The VECs were
cultivated in Dulbecco's Modified Eagle Medium (DMEM; Gibco; Thermo
Fisher Scientific, Inc.) supplemented with 1%
penicillin-streptomycin (Gibco; Thermo Fisher Scientific, Inc.) and
10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.) in a
humidified atmosphere containing 5% CO2 at 37°C. The
cells were passaged when they reached 80–90% confluence and the
medium was changed every 2 days. All experiments were conducted
using cells between passages 3 and 5 after thawing. To investigate
the roles of HIF-1α signaling and glycolysis, the cells were
treated with 20 µM PX478 (MedChemExpress), a HIF-1α inhibitor, for
20 h at 37°C, or 2 mM 2-deoxy-D-glucose (2-DG; MedChemExpress), a
glycolysis inhibitor, for 24 h at 37°C, under oxidative stress
conditions induced by preconditioning with 0.5 mM
H2O2 for 12 h at 37°C, followed by OGD/R (6-h
OGD at 37°C and 24-h reoxygenation at 37°C) [hereafter referred to
as 0.5 mM H2O2 (OGD/R)].
Oxidative stress and
ischemia-reperfusion injury model generation in VECs
The pathogenesis of CAs is multifactorial, with
oxidative stress being a key factor. Oxidative stress can lead to
endothelial dysfunction, inflammation and vascular wall remodeling,
all of which contribute to the formation and rupture of aneurysms
(19). Oxidative stress is often
induced by hemodynamic changes, such as turbulent blood flow and
wall shear stress (WSS), which are common in aneurysmal regions
(20). Additionally,
ischemia-reperfusion injury, a condition where blood flow is
temporarily interrupted and then restored, is a notable contributor
to oxidative stress and mitochondrial dysfunction in CAs. The OGD/R
model is widely used to simulate ischemia-reperfusion injury in
vitro; this model mimics the conditions of transient cerebral
ischemia followed by reperfusion, which is relevant to the
pathophysiology of CAs (21,22).
The present study employed a combined treatment of 0.5 mM
H2O2 and OGD/R to induce oxidative stress in
VECs, thereby mimicking ischemia-reperfusion injury. VECs, as
critical cellular components of the cerebral arterial wall, serve a
pivotal role in maintaining vascular integrity and their
dysfunction is closely associated with aneurysm pathogenesis. VECs
were subjected to oxidative stress via treatment with 0.5 mM
H2O2 (OGD/R) to mimic ischemic conditions, as
previously described by Wu et al (16). OGD/R involved incubating cells in
glucose-free DMEM under hypoxic conditions (1% O2) for
24 h, followed by reoxygenation with complete DMEM under normoxic
conditions (21% O2) for 24 h.
Cell transfection
VECs were plated in 24-well plates at a density of
2×105 cells/well to achieve optimal confluence for
transfection. To regulate LDHA expression, VECs were transfected
with an LDHA overexpression plasmid (pcDNA-3.1-LDHA) or with an
empty pcDNA-3.1 vector (negative control) (both from Shanghai
GenePharma Co., Ltd.). The cells were transfected with 500 ng
plasmid in 24-well plates using Lipofectamine® 3000
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) at 37°C and 5%
CO2 for 6 h according to the manufacturer's
instructions. VECs were harvested 48 h post-transfection for
further analysis.
Experimental groups
For subsequent experiments, VECs were divided into
six groups: i) Control (untreated VECs); ii) 0.5 mM
H2O2 (OGD/R); iii) 0.5 mM
H2O2 (OGD/R) + vector (empty pcDNA-3.1
vector); iv) 0.5 mM H2O2 (OGD/R) + over-LDHA
(pcDNA-3.1-LDHA plasmid); v) 0.5 mM H2O2
(OGD/R) + over-LDHA + PX478 (20 µM PX478 treatment); vi) 0.5 mM
H2O2 (OGD/R) + over-LDHA + 2-DG (2 mM 2-DG
treatment).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) assay
Total RNA was extracted from VECs using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's guidelines. cDNA synthesis
was performed using the PrimeScript RT Reagent Kit (Takara Bio,
Inc.) at 37°C for 15 min, followed by 85°C for 5 sec, according to
the manufacturer's instructions. On a StepOnePlus Real-Time PCR
System (Applied Biosystems; Thermo Fisher Scientific, Inc.), qPCR
was performed to measure gene expression levels using the SYBR
Green PCR Master Mix (Takara Bio, Inc.). The thermocycling
conditions were as follows: Initial denaturation at 95°C for 30
sec, followed by 40 cycles at 95°C for 5 sec and 60°C for 30 sec.
Gene expression was normalized to the internal control gene
β-actin. The 2−ΔΔCq technique was implemented to
determine relative expression levels (23). The primer sequences used for
amplification are listed in Table
I.
 | Table I.Primer sequences for reverse
transcription-quantitative polymerase chain reaction. |
Table I.
Primer sequences for reverse
transcription-quantitative polymerase chain reaction.
| Target | Sequence,
5′-3′ |
|---|
| LDHA | Forward:
TGTCTCTGGCAAAGTGGATATCTT |
|
| Reverse:
GACCCACCCATGACAGCTTA |
| Caspase-3 | Forward:
TCCTAGCGGATGGGTGCTAT |
|
| Reverse:
TCCAGGGATATTCCAGAGTCCA |
| Caspase-8 | Forward:
TCACCTTGTGTCTGAGCTGG |
|
| Reverse:
TGACCAACTCAAGGGCTCAG |
| Caspase-9 | Forward:
AGGCCCCATATGATCGAGGA |
|
| Reverse:
TCGACAACTTTGCTGCTTGC |
| HK2 | Forward:
TTGTTGCATGAAACTCCGGC |
|
| Reverse:
GCTAACTTCGGCCACAGGAT |
| PGM5 | Forward:
CTAGCAGGCCCTGGAATCAG |
|
| Reverse:
TTCTCCCACCCACCTTCTCC |
| PKM | Forward:
GTGATGTGGCCAATGCAGTC |
|
| Reverse:
CTTGAGGCTCGCACAAGTTC |
| OGDH | Forward:
ATCGTGTCACCGACAGGAAC |
|
| Reverse:
CGGAACTCCATGCTGGAACA |
| DLD | Forward:
ACAGCGGAAAAATGCAGAGC |
|
| Reverse:
ATCCTCCAGGACCAGAACCT |
| SUCLG2 | Forward:
ATGGGATCACCAAAGCCTGC |
|
| Reverse:
GGGCGACGTTCTCTTGGATA |
| β-actin | Forward:
AGCTCACCATGGATGATGATATCGC |
|
| Reverse:
CACATAGGAATCCTTCTGACCCAT |
Western blotting (WB)
Protein lysates from VECs were prepared using RIPA
lysis buffer (Thermo Fisher Scientific, Inc.) with phosphatase and
protease inhibitors, and the BCA Protein Assay Kit (Thermo Fisher
Scientific, Inc.) was utilized to calculate the protein
concentration. Equal amounts of protein (20–40 µg) were denatured
at 95°C for 5 min in SDS sample buffer and were separated by
SDS-polyacrylamide gel electrophoresis on 10–12% polyacrylamide
gels, depending on the molecular weight of the target proteins. The
proteins were then transferred onto polyvinylidene fluoride
membranes (MilliporeSigma) using a wet transfer system at 4°C.
Following transfer, the membranes were blocked with 5% non-fat milk
in TBST buffer (20 mM Tris-HCl, 150 mM NaCl, 0.1% Tween-20, pH 7.6)
for 1 h at room temperature to reduce nonspecific binding.
Subsequently, the membranes were incubated overnight at 4°C with
primary antibodies against the following proteins: LDHA (1:1,000;
cat. no. 2012; Cell Signaling Technology, Inc.), caspase-3
(1:5,000; cat. no. ab32351; Abcam), caspase-9 (1:2,000; cat. no.
ab202068; Abcam), caspase-8 (1:1,000; cat. no. ab32397; Abcam),
cytochrome c (Cyt-c; 1:5,000; cat. no. ab133504; Abcam),
Mcl-1 (1:1,000; cat. no. ab32087; Abcam), HIF-1α (1:1,000; cat. no.
ab51608; Abcam), sodium-calcium exchanger 1 (NCX1; 1:1,000; cat.
no. ab177952; Abcam), sodium-proton exchanger 1 (NHE1; 1:1,000;
cat. no. ab67313; Abcam) and monocarboxylate transporter 4 (MCT4;
1:1,000; cat. no. ab308528; Abcam), with β-actin (1:1,000; cat. no.
ab8227; Abcam) used as an internal loading control for whole-cell
and cytoplasmic proteins, and Lamin B1 (1:20,000; cat. no.
12987–1-AP; Wuhan Sanying Biotechnology) used as a nuclear loading
control. After primary antibody incubation, the membranes were
washed three times with TBST (10 min each) and were then incubated
with horseradish peroxidase-conjugated goat anti-rabbit IgG (H+L)
secondary antibody (1:10,000; cat. no. ab6721; Abcam) for 1 h at
room temperature. After washing again three times in TBST, the
protein bands were visualized using enhanced chemiluminescence
detection reagent (Beyotime Institute of Biotechnology), and images
were captured using a chemiluminescence imaging system (Bio-Rad
Laboratories, Inc.). Band intensities were semi-quantified using
ImageJ software (version 1.5.2; National Institutes of Health).
The nuclear and cytoplasmic proteins were extracted
using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo
Fisher Scientific, Inc.) following the manufacturer's
instructions.
Flow cytometry
For flow cytometry, VECs were detached utilizing a
trypsin-EDTA solution (Gibco; Thermo Fisher Scientific, Inc.),
followed by a wash with phosphate-buffered saline (PBS). The cells
(1×106) were subsequently stained with an Annexin
V-fluorescein isothiocyanate/propidium iodide apoptosis detection
kit (Beyotime Institute of Biotechnology), in accordance with the
manufacturer's instructions. A FACSCalibur flow cytometer (BD
Biosciences) was applied for flow cytometric analysis, and FlowJo
software (version 10.8.0; BD Biosciences) was used to process and
interpret the data.
Observation of mitochondrial
morphology
After being cleaned with PBS, VECs were fixed for 48
h at 4°C with 2.5% glutaraldehyde in phosphate buffer (pH 7.0).
After three washes with PBS, the cells were post-fixed with 1%
osmium tetroxide in phosphate buffer for 1 h at room temperature.
The samples were then washed three times with PBS. After
dehydrating the samples with ethanol solution (30–100%), they were
transferred to anhydrous acetone for 20 min at room temperature.
Subsequently, absolute acetone and the Spurr resin mixture were
mixed 1:1(v/v) and allowed to permeate VECs for 1 h at room
temperature. The infiltration process continued with a 3-h
incubation in a 1:3 mixture of acetone and resin, followed by
embedding in pure Spurr resin overnight at room temperature. The
embedded samples were placed in capsules containing the embedding
medium and polymerized for ~9 h at 70°C. Ultrathin sections (60–90
nm) were cut from the resin-embedded blocks using a Leica Ultracut
microtome (EM UC7; Leica Biosystems). To improve contrast, the
sections were first stained for 15 min with 1% uranyl acetate and
then for another 15 min with alkaline lead citrate. The stained
sections were examined under a transmission electron microscope
(HT7700; Hitachi, Ltd.) at ×2,500 magnification. Mitochondrial
morphology was evaluated for structural damage, such as changes in
size, swelling and shape.
Immunofluorescence (IF) staining
VECs were seeded onto glass coverslips in 24-well
plates at 5×104 cells/well and allowed to adhere
overnight. Following treatment, the cells were fixed with 4%
paraformaldehyde for 15 min at room temperature and permeabilized
with 0.1% Triton X-100 in PBS for 10 min at room temperature.
Non-specific binding was blocked by incubating the cells in 5%
bovine serum albumin (BSA; Beyotime Institute of Biotechnology) in
PBS for 1 h at room temperature. Subsequently, the cells were
treated overnight at 4°C with anti-HIF-1α primary antibodies
(1:500; cat. no. ab51608; Abcam) diluted in 1% BSA blocking buffer.
After washing with PBS, the cells were incubated at room
temperature for 1 h with Alexa Fluor® 488-conjugated
anti-rabbit IgG (1:200; cat. no. ab150077; Abcam). DAPI (1 µg/ml)
was employed to counterstain the nuclei for 5 min. Coverslips were
mounted on slides with a sealing agent, and images were captured
utilizing a DMi8 fluorescence microscope (Leica Biosystems).
Fluorescence intensity and colocalization were analyzed using
ImageJ software (version 1.5.2; National Institutes of Health).
Representative images are displayed and the experiment was
conducted in triplicate.
Measurement of reactive oxygen species
(ROS), malondialdehyde (MDA) and glutathione (GSH) levels
The ROS, MDA and GSH assay kits (cat nos. E004-1-1,
A003-1-2 and A006-2-1, respectively) were purchased from Nanjing
Jiancheng Bioengineering Institute, and were conducted in
compliance with the manufacturer's guidelines. VECs were plated at
a density of 5×105 cells/well in 6-well plates and were
cultured overnight for adherence. For ROS detection, the cells were
incubated with 10 µM DCFH-DA fluorescent probe, provided in the ROS
assay kit, in the dark for 30 min at 37°C. Subsequently, the cells
were washed twice with PBS, and fluorescence intensity was detected
using a microplate reader. Furthermore, MDA levels were quantified
to assess lipid peroxidation. After treatment, cell lysates were
prepared by adding RIPA buffer supplemented with protease
inhibitors, and were centrifuged at 12,000 × g for 15 min at 4°C.
The supernatants were processed according to the kit's
instructions, and absorbance was measured at 532 nm using a
spectrophotometer. VECs were lysed and centrifuged under the same
conditions to evaluate GSH levels. Following the kit's
instructions, the supernatants were incubated with the supplied
reaction mixture, and absorbance was measured at 405 nm to
determine GSH concentrations. Using a BCA protein assay kit, the
total protein content in the lysates was employed to standardize
all of the outcomes.
Determination of pyruvate, lactate,
and succinate levels
To assess the levels of pyruvate, lactate and
succinate in VECs under oxidative stress, the cells were seeded in
6-well plates (2×105 cells/well; n=3 wells per
experiment) and were first exposed to 0.5 mM
H2O2 (OGD/R) treatment, then transfected to
induce LDHA overexpression. The culture supernatants were collected
48 h after transfection, clarified by centrifugation at 1,500 × g
for 10 min at 4°C, and stored at −80°C until analysis, and their
pyruvate, lactate and succinate concentrations were analyzed
utilizing corresponding commercial assay kits according to the
manufacturer's recommendations. Pyruvate and lactate assay kits
were purchased from Beijing Solarbio Science & Technology Co.,
Ltd. (cat nos. BC2205 and BC2235, respectively), while the
succinate assay kit (cat no. MAK184) was obtained from
MilliporeSigma. The levels of pyruvate were measured at 340 nm,
lactate at 340 nm and succinate at 570 nm, using a
spectrophotometer (BioTeke Corporation). All measurements were
conducted in triplicate and the findings were normalized to protein
concentration, which was detected by the BCA assay.
Assessment of mitochondrial membrane
potential (MMP)
MMP in cells subjected to OGD/R treatment was
evaluated utilizing the cationic dye JC-1 (cat. no. T3168;
Invitrogen; Thermo Fisher Scientific, Inc.), following the
manufacturer's guidelines. Briefly, after OGD/R treatment, the
cells were washed twice with PBS to remove any residual medium.
Next, VECs were cultured with JC-1 at a final concentration of 10
µg/ml in serum-free medium for 30 min at 37°C, ensuring proper
penetration of the dye into the cells. JC-1 accumulates in the
mitochondria in a potential-dependent manner, exhibiting red
fluorescence when aggregated in the mitochondria of cells with high
membrane potential, and green fluorescence when dissociated in
cells with low membrane potential. Following incubation, the JC-1
working solution was removed, and the cells were washed twice with
PBS to eliminate any unbound dye. To reduce any background
fluorescence and to ensure clear imaging, washing steps were
performed gently. Fluorescence images were then captured using a
fluorescence microscope (Leica Biosystems) equipped with
appropriate filters to detect both red and green fluorescence
emissions. Specifically, red fluorescence was observed at 540 nm
for excitation and 590 nm for emission, whereas green fluorescence
was measured at 485 nm for excitation and 535 nm for emission.
Images were captured at ×40 magnification, and five randomly
selected fields per sample were analyzed to ensure representative
results.
Measurement of extracellular
acidification rate (ECAR)
ECAR was analyzed utilizing a Seahorse XF Analyzer
(Agilent Technologies, Inc.) to evaluate metabolic shifts in VECs
following H2O2 and OGD/R treatment. VECs were
sequentially exposed to glucose (10 mM), oligomycin A (Oligo, 1 µM)
and 2-DG (50 mM) from the Seahorse XF Glycolysis Stress Test Kit
(cat. no. 103020-100; Agilent Technologies, Inc.) at 37°C; after
each injection, ECAR was recorded for three cycles (mix 3 min,
measure 3 min each) to assess glycolytic function. Real-time data
were collected and plotted as ECAR over time to visualize the
cellular metabolic response to the treatments.
Intracellular Ca2+
measurement in VECs
To measure intracellular Ca2+ levels in
VECs under oxidative stress with LDHA overexpression, the cells
were seeded in 6-well plates at 70% confluence. Cells were first
subjected to 0.5 mM H2O2 (OGD/R), then
immediately transfected with either an empty vector or an LDHA
overexpression plasmid using Lipofectamine 3000 and incubated for
48 h post-transfection. Cells were collected and resuspended in 500
µl calcium assay buffer. The cell suspension was then scraped on
ice and centrifuged at 13,000 × g for 10 min at 4°C to obtain the
supernatant. Intracellular Ca2+ concentration was
determined using the Calcium Detection Assay Kit (cat. no.
ab102505; Abcam) according to the manufacturer's instructions, and
absorbance was read at 575 nm on a microplate reader. The resulting
data were used to assess any alterations in Ca2+
homeostasis due to LDHA overexpression under oxidative stress
conditions.
Measurement of NAD+ and
NADH levels
NAD+ and NADH concentrations were
assessed using the NAD+/NADH Quantification Colorimetric
Assay Kit (cat no. ab65348; Abcam) according to the manufacturer's
protocol. After plating VECs at a density of 5×105
cells/well in 6-well plates, they were lysed utilizing RIPA buffer
(Beyotime Institute of Biotechnology), containing phosphatase and
protease inhibitors, and rinsed with PBS. The lysates were then
homogenized in 100 µl NAD+ or NADH extraction buffer.
The absorbance of the supernatant was detected at 450 nm utilizing
a microplate reader, and the NAD+ and NADH levels were
calculated from the standard curve.
Measurement of NADPH content
NADPH levels in VECs following OGD/R treatment were
measured using the NADPH Assay Kit (cat no. S0179; Beyotime
Institute of Biotechnology) according to the manufacturer's
protocol. VECs were seeded in 6-well plates at 5×105
cells/well and allowed to adhere overnight. Following treatment
with the extraction buffer, cells were centrifuged at 10,000 × g
for 10 min at 4°C. The resulting supernatant was incubated for 30
min at 60°C to degrade NADP+. The supernatant was then
cooled on ice, combined with a working solution and incubated for
20 min at 37°C. The absorbance was measured at 450 nm using a
microplate reader.
Determination of glucose-6-phosphate
dehydrogenase (G6PDH) activity
The activity of G6PDH in cells was measured using a
commercial G6PDH Assay Kit (cat no. S0189; Beyotime Institute of
Biotechnology) as per the manufacturer's instructions. For 10 min,
cells (5×105 cells/well; 6-well plates) were centrifuged
at 12,000 × g at 4°C after being treated with the extraction
solution. The supernatant (50 µl) was then transferred to a 96-well
plate, followed by the addition of 50 µl G6PDH working solution,
and was incubated in the dark for 10 min at room temperature. The
absorbance was determined at 450 nm using a microplate reader.
Statistical analysis
R language (version 4.3.1; R Foundation for
Statistical Computing; http://www.r-project.org/) was used to examine the
data. The data are presented as the mean ± SD and all experiments
were performed in triplicate. The Student's t-test was used to
assess differences between two groups, while one-way ANOVA with
Tukey's post hoc test determined differences among multiple groups.
P<0.05 was considered to indicate significant differences.
Results
Oxidative stress induces apoptosis and
mitochondrial pathway activation in VECs
Flow cytometric analysis revealed a significant
increase in apoptosis in VECs following treatment with 0.5 mM
H2O2 (OGD/R) compared with that in the
control group (Fig. 1A). To
explore the molecular mechanisms, the expression levels of
apoptosis-associated factors, including caspase-3, caspase-8 and
caspase-9, were assessed using RT-qPCR and WB. The results
demonstrated a marked upregulation of these factors following
treatment with 0.5 mM H2O2 (OGD/R) compared
with those in the control group (Fig.
1B-D). Subsequent WB of mitochondrial pathway-related proteins
showed that treatment with 0.5 mM H2O2
(OGD/R) conditions increased Cyt-c expression while suppressing
Mcl-1 expression (Fig. 1E and F).
These findings collectively indicated that oxidative stress may
promote apoptosis in VECs, accompanied by the activation of
mitochondrial apoptosis-related pathways.
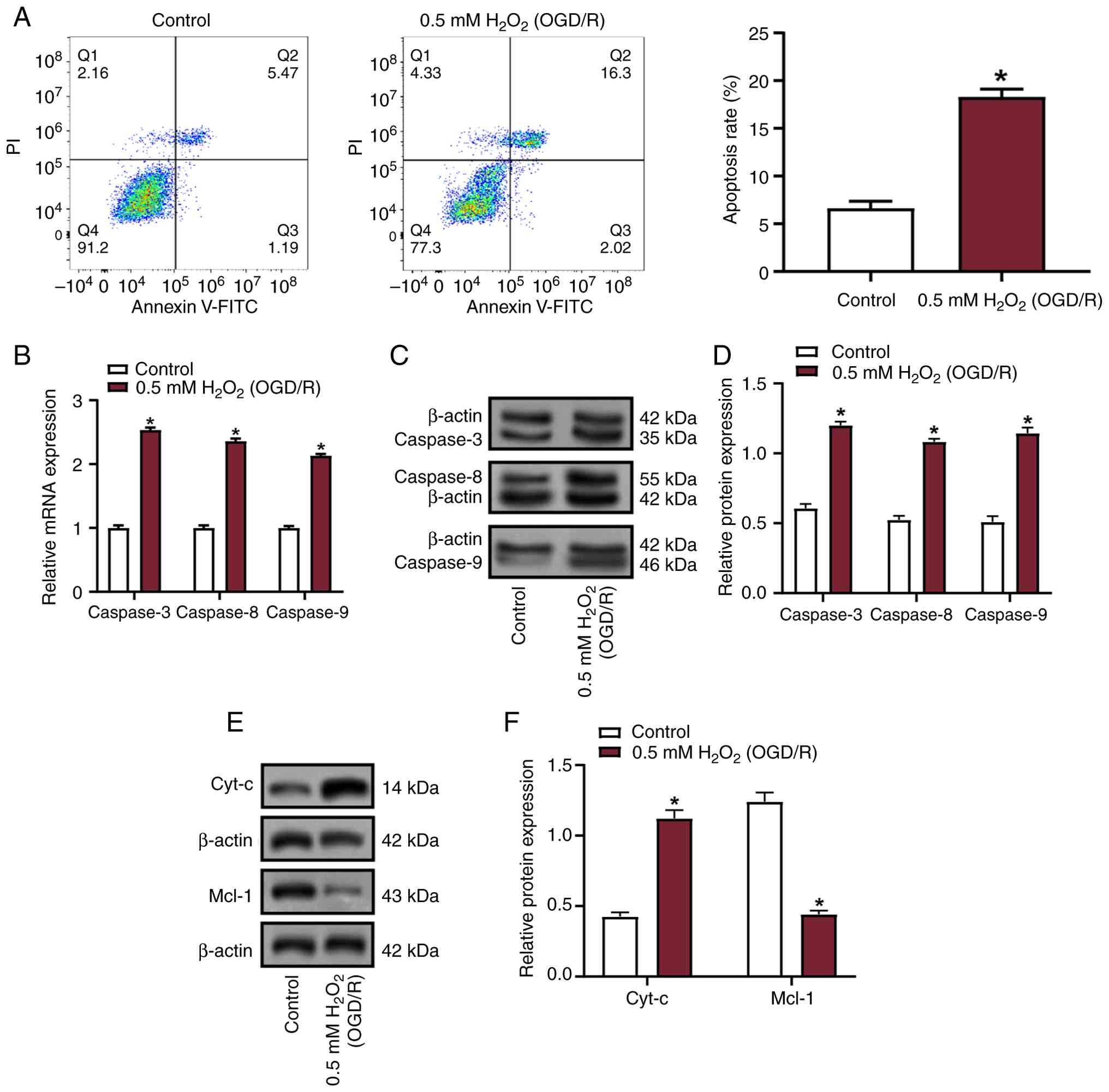 | Figure 1.Analysis of apoptosis and expression
of apoptosis-related proteins in VECs under oxidative stress
conditions. (A) Apoptosis rate of VECs treated with 0.5 mM
H2O2 under OGD/R conditions, assessed using
flow cytometry. (B) Relative mRNA expression levels of caspase-3,
caspase-8 and caspase-9 in VECs treated with 0.5 mM
H2O2 (OGD/R) compared with the control group,
measured by reverse transcription-quantitative polymerase chain
reaction. mRNA expression levels were normalized to β-actin. (C) WB
of caspase-3, caspase-8 and caspase-9 protein expression in VECs
treated with 0.5 mM H2O2 (OGD/R). The images
show representative protein bands with β-actin as the loading
control. (D) Semi-quantification of WB data. Protein expression
levels are normalized to β-actin. (E) WB of mitochondrial
pathway-related proteins Cyt-c and Mcl-1 in VECs treated with 0.5
mM H2O2 (OGD/R). (F) Protein expression was
normalized to β-actin, and the results are shown as relative
expression levels compared with the control group. Data are
presented as the mean ± SD of three independent experiments.
*P<0.05 vs. control. H2O2, hydrogen
peroxide; OGD/R, oxygen-glucose deprivation/reperfusion; VECs,
vascular endothelial cells; WB, western blotting; PI, propidium
iodide; FITC, fluorescein isothiocyanate; Cyt-c, cytochrome
c. |
LDHA overexpression mitigates
oxidative stress-induced apoptosis and mitochondrial damage in
VECs
To investigate the connection between LDHA
expression, apoptosis and mitochondrial damage, VECs subjected to
0.5 mM H2O2 (OGD/R) conditions were
transfected with an LDHA overexpression plasmid. The transfection
efficiency was verified by RT-qPCR and WB, which revealed a
significant increase in LDHA expression following transfection
(Fig. 2A-C). The effects of LDHA
overexpression on mitochondrial morphology were further examined
using transmission electron microscopy at ×2,500 magnification. In
the 0.5 mM H2O2 (OGD/R) group, mitochondria
displayed notable structural damage, including varying sizes,
marked swelling, a rounded shape, vacuolar degeneration, and
severely reduced, fragmented or absent cristae (Fig. 2D). By contrast, LDHA overexpression
improved mitochondrial morphology, with less swelling and
better-preserved cristae. WB revealed that 0.5 mM
H2O2 (OGD/R) treatment increased the
expression levels of the pro-apoptotic proteins caspase-3 and
Cyt-c, while reducing the expression levels of the anti-apoptotic
protein Mcl-1 (Fig. 2E and F).
Notably, LDHA overexpression reversed these effects,
reducing caspase-3 and Cyt-c levels, while reversing Mcl-1
expression. Flow cytometric analysis further demonstrated that 0.5
mM H2O2 (OGD/R) treatment significantly
promoted apoptosis in VECs; however, LDHA overexpression
effectively mitigated the pro-apoptotic effects of oxidative stress
(Fig. 2G). These findings
indicated that upregulation of LDHA under oxidative stress
conditions could alleviate OGD/R-induced apoptosis and
mitochondrial damage in VECs.
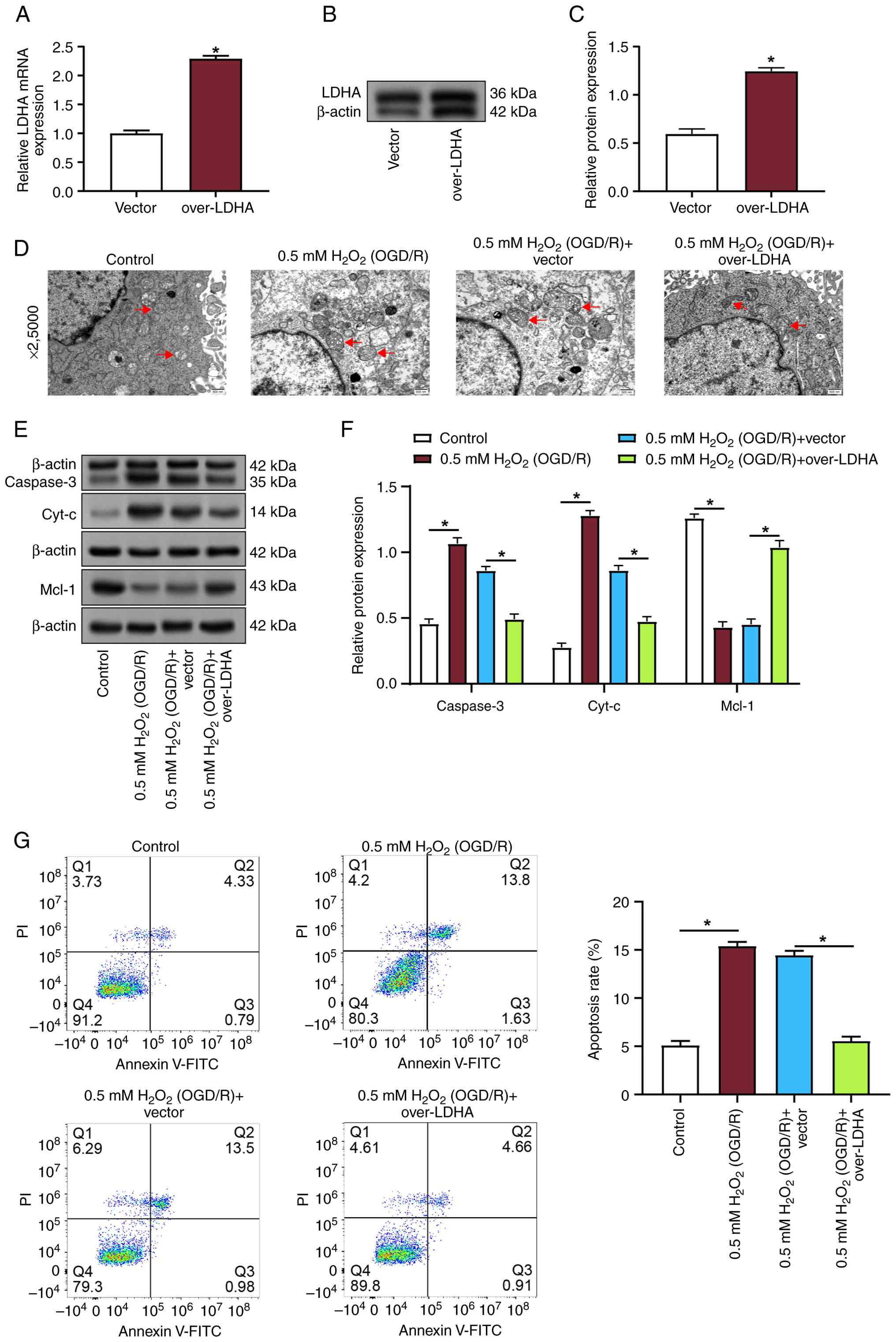 | Figure 2.LDHA overexpression mitigates
apoptosis and mitochondrial damage in OGD/R-treated VECs. (A)
Reverse transcription-quantitative polymerase chain reaction
analysis of LDHA mRNA in VECs transfected with vector or over-LDHA.
(B) Representative western blot analysis of LDHA with β-actin as
loading control. (C) Semi-quantification of LDHA protein expression
relative to β-actin. (D) Transmission electron microscopy images of
VECs at ×2,500 magnification showing mitochondrial morphology. Red
arrows indicate mitochondria (swollen and with disrupted cristae
under OGD/R conditions). Scale bar: 500 nm. (E) Western blot
analysis of caspase-3, Cyt-c and Mcl-1 in VECs from the indicated
groups; β-actin was used as the loading control. (F) Densitometric
semi-quantification of relative protein expression normalized to
β-actin for caspase-3, Cyt-c and Mcl-1. (G) Flow cytometric
analysis of the effects of LDHA overexpression on apoptosis levels
in VECs subjected to H2O2 (OGD/R) treatment.
Data are presented as the mean ± SD of three independent
experiments. *P<0.05 vs. vector or as indicated.
H2O2, hydrogen peroxide; OGD/R,
oxygen-glucose deprivation/reperfusion; VECs, vascular endothelial
cells; LDHA, lactate dehydrogenase A; WB, western blotting; PI,
propidium iodide; FITC, fluorescein isothiocyanate; Cyt-c,
cytochrome c. |
LDHA overexpression reduces oxidative
stress and preserves MMP in VECs under OGD/R conditions
To investigate the function of LDHA in oxidative
stress, the levels of ROS, GSH and MDA were detected in VECs
treated with 0.5 mM H2O2 (OGD/R) and
overexpressing LDHA. Compared with those in the control group, 0.5
mM H2O2 (OGD/R) treatment resulted in
significant increases in MDA and ROS, along with a reduction in
GSH; however, LDHA overexpression reversed these effects, and
restored MDA, ROS and GSH levels (Fig.
3A-C). MMP was next assessed using the JC-1 fluorescent dye.
Control cells predominantly exhibited red fluorescence, indicating
intact MMP (Fig. 3D). By contrast,
0.5 mM H2O2 (OGD/R)-treated cells showed a
marked increase in green fluorescence, indicating mitochondrial
depolarization. LDHA overexpression, however, resulted in a higher
red/green fluorescence ratio in contrast to the two control groups
and the 0.5 mM H2O2 (OGD/R) + vector group,
suggesting that LDHA overexpression mitigated the loss of MMP
induced by oxidative stress. Taken together, these results
demonstrated that LDHA overexpression may reduce ROS generation and
help maintain MMP during OGD/R-induced oxidative damage in
VECs.
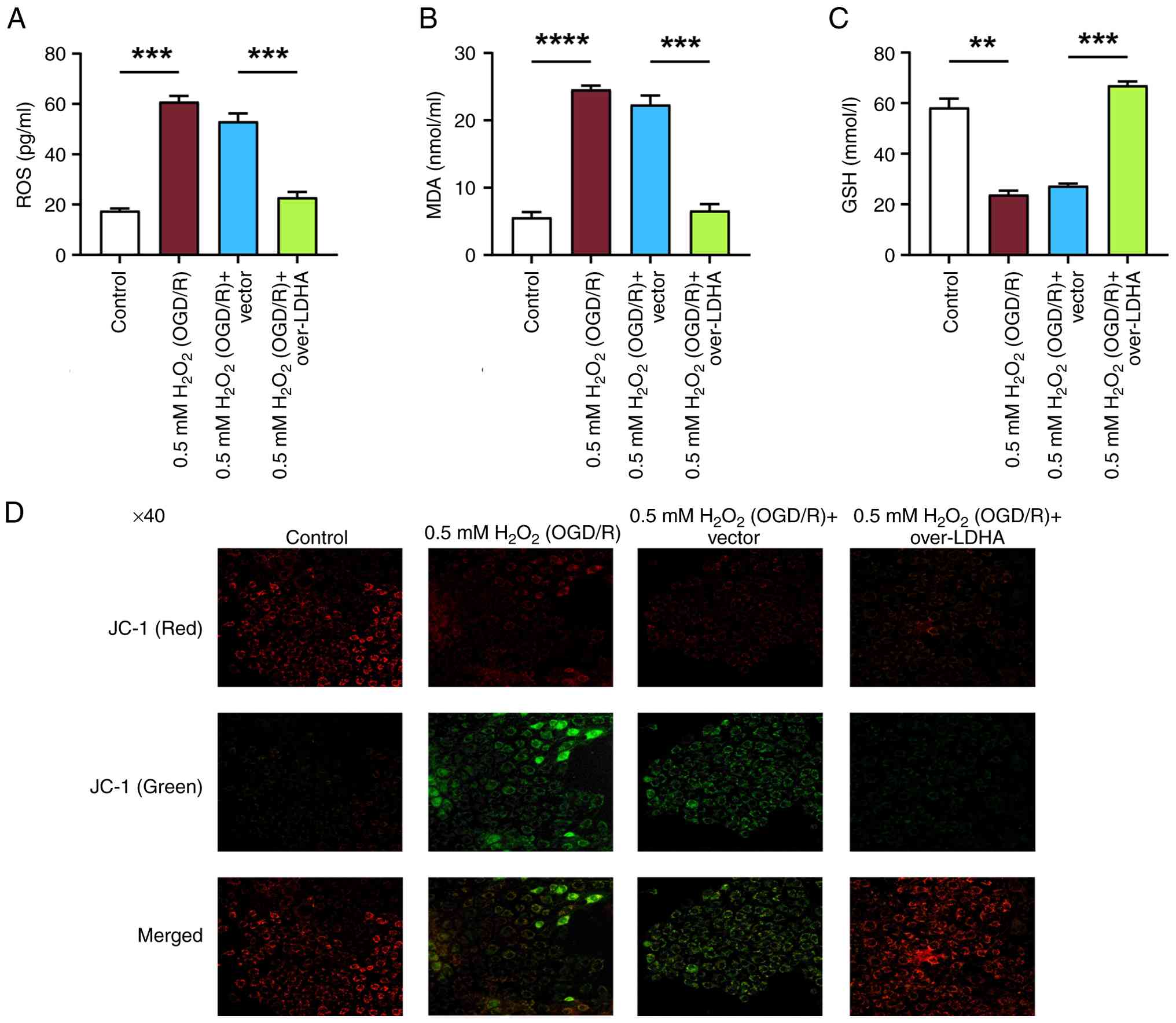 | Figure 3.Effect of LDHA overexpression on
oxidative stress markers and MMP in VECs under OGD/R conditions.
Levels of (A) ROS, (B) MDA and (C) GSH in VECs following 0.5 mM
H2O2 (OGD/R) treatment with or without LDHA
overexpression. (D) MMP analysis using JC-1 fluorescent dye in VECs
treated with 0.5 mM H2O2 (OGD/R) and
overexpressing LDHA. Cells were assessed for red and green
fluorescence, where red fluorescence indicates intact MMP and green
fluorescence indicates depolarized MMP. Magnification, ×40. Data
are presented as the mean ± SD of three independent experiments.
**P<0.01, ***P<0.001 and
****P<0.0001. H2O2, hydrogen
peroxide; OGD/R, oxygen-glucose deprivation/reperfusion; VECs,
vascular endothelial cells; LDHA, lactate dehydrogenase A; ROS,
reactive oxygen species; MDA, malondialdehyde; GSH, glutathione;
MMP, mitochondrial membrane potential. |
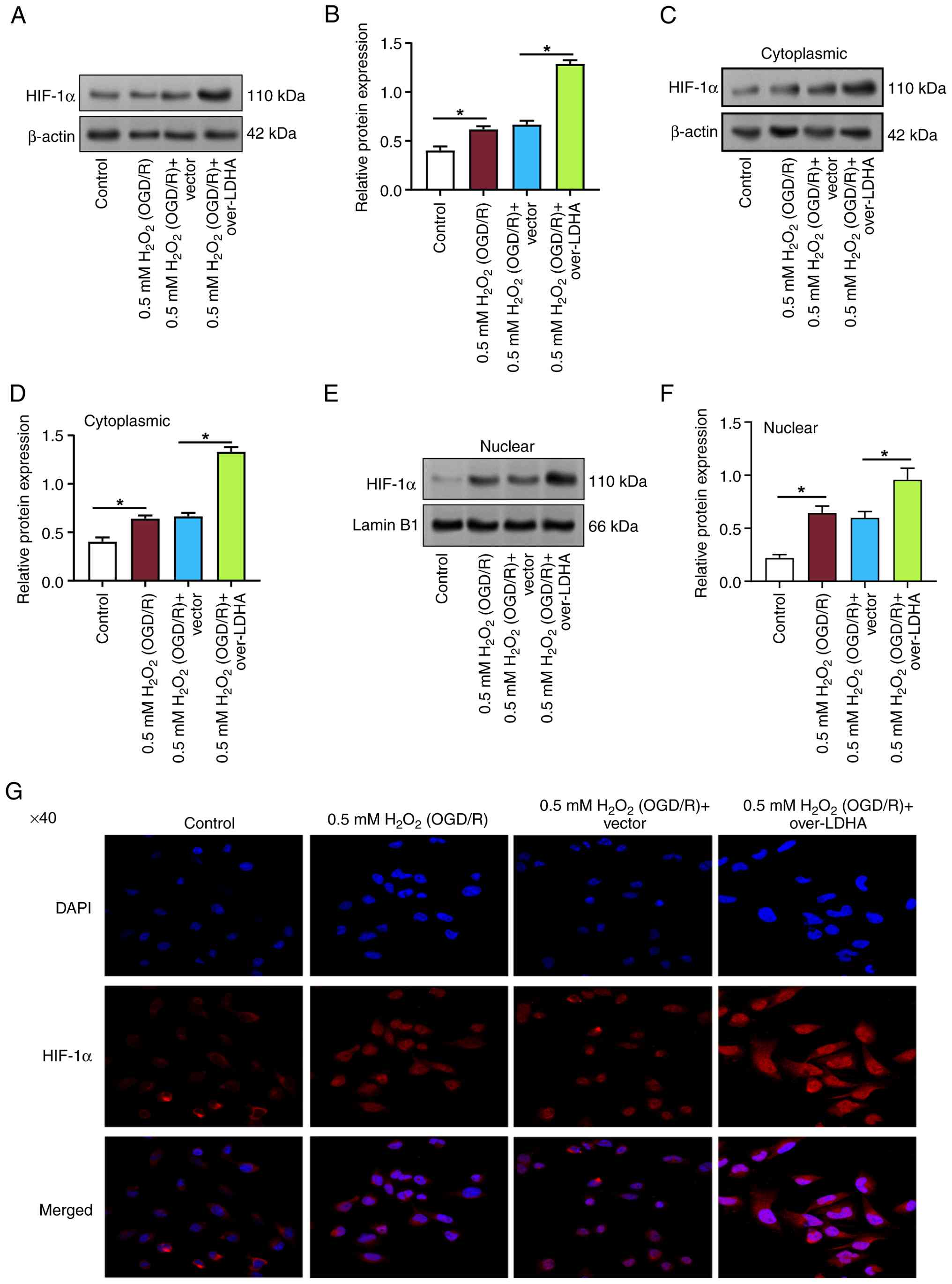
LDHA overexpression enhances HIF-1α
expression and nuclear translocation in VECs under OGD/R
conditions
In the OGD/R model, HIF-1α activity is essential for
the adaptation of cells to hypoxic conditions and for promoting
cell survival. To further investigate the effects of 0.5 mM
H2O2 (OGD/R) and LDHA overexpression on
HIF-1α expression and localization in VECs, WB was employed. The
outcomes indicated that compared with in the control group, the
expression levels of total HIF-1α were increased in VECs treated
with 0.5 mM H2O2 (OGD/R) (Fig. 4A and B). Moreover, LDHA
overexpression further enhanced the expression of HIF-1α compared
with that in the 0.5 mM H2O2 (OGD/R) + vector
group. Further analysis revealed that both cytoplasmic and nuclear
HIF-1α levels were elevated in the 0.5 mM
H2O2 (OGD/R) group compared with in the
control group (Fig. 4C-F). LDHA
overexpression also promoted the accumulation of HIF-1α in both the
cytoplasm and nucleus, compared with that in the 0.5 mM
H2O2 (OGD/R) + vector group. IF staining was
used to assess the cellular localization of HIF-1α. Under control
conditions, HIF-1α was primarily localized in the cytoplasm
(Fig. 4G). After 0.5 mM
H2O2 (OGD/R) treatment, HIF-1α expression was
markedly upregulated. Notably, HIF-1α expression was further
elevated in VECs transfected with an LDHA overexpression plasmid
following OGD/R. These findings suggested that LDHA upregulation
may promote HIF-1α nuclear translocation in VECs under OGD/R
conditions, potentially contributing to cellular adaptation and
survival under oxidative stress.
LDHA alleviates OGD/R-induced VEC
apoptosis and mitochondrial damage via HIF-1α nuclear
translocation
To further explore the role of LDHA in apoptosis and
mitochondrial damage, the effects of LDHA overexpression and the
HIF-1α inhibitor (PX478) on VECs exposed to 0.5 mM
H2O2 (OGD/R) were investigated by flow
cytometry. The outcomes demonstrated that LDHA overexpression
significantly suppressed apoptosis induced by 0.5 mM
H2O2 (OGD/R) in VECs; however, treatment with
PX478 reversed the protective effect of LDHA overexpression on
apoptosis (Fig. 5A). A ROS assay
kit was subsequently employed to quantify ROS levels. Compared with
those in the 0.5 mM H2O2 (OGD/R) + vector
group, LDHA overexpression resulted in a marked reduction in ROS
levels (Fig. 5B). By contrast,
treatment with PX478 diminished the effect of LDHA overexpression
on ROS levels (Fig. 5B). The MMP
was assessed using JC-1 fluorescent dye. In 0.5 mM
H2O2 (OGD/R)-treated cells, green
fluorescence was markedly enhanced, indicating mitochondrial
depolarization (Fig. 5C). By
contrast, LDHA overexpression improved the red/green fluorescence
ratio, suggesting a preserved MMP; however, this effect was
attenuated when PX478 was applied, indicating that the protective
role of LDHA overexpression in MMP preservation was partially
dependent on HIF-1α signaling. To directly assess the effect on
HIF-1α expression, WB was performed to detect HIF-1α protein
expression levels in the cytoplasm and nucleus under OGD/R
conditions. LDHA overexpression significantly elevated HIF-1α
expression in both compartments, whereas PX478 treatment led to a
notable decrease in HIF-1α levels (Fig. 5D-G). These findings collectively
suggested that LDHA overexpression could alleviate OGD/R-induced
mitochondrial damage in VECs, likely through the promotion of
HIF-1α nuclear translocation.
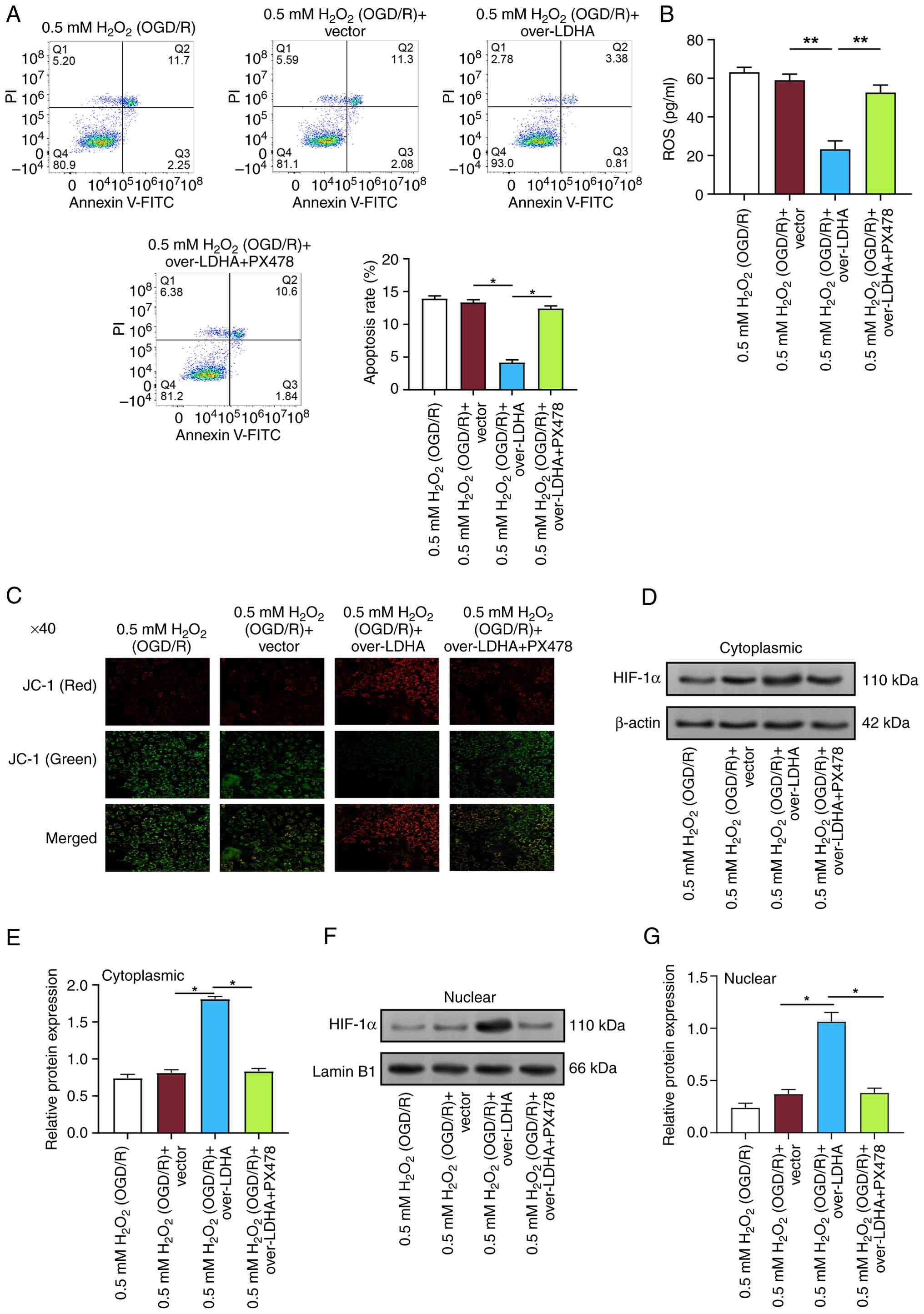 | Figure 5.LDHA overexpression alleviates
OGD/R-induced apoptosis and mitochondrial damage in VECs via HIF-1α
nuclear translocation. (A) Flow cytometric analysis of apoptosis in
VECs treated with 0.5 mM H2O2 (OGD/R) and
overexpressing LDHA, with or without the HIF-1α inhibitor PX478.
(B) ROS levels measured using a ROS assay kit in VECs treated with
0.5 mM H2O2 (OGD/R) and overexpressing LDHA,
with or without PX478. (C) MMP was assessed by JC-1 fluorescent dye
in VECs treated with 0.5 mM H2O2 (OGD/R) and
overexpressing LDHA, with or without PX478. The images show
representative red (intact MMP) and green (depolarized MMP)
fluorescence. Magnification, ×40. (D-G) WB of cytoplasmic and
nuclear HIF-1α expression in VECs treated with 0.5 mM
H2O2 (OGD/R) and overexpressing LDHA, with or
without PX478. (D) Representative cytoplasmic HIF-1α western blot
analysis; β-actin was used as a cytoplasmic control. (E)
Semi-quantification of cytoplasmic HIF-1α in (D), normalized to
β-actin. (F) Representative nuclear HIF-1α western blot analysis;
Lamin B1 was used as a nuclear control. (G) Semi-quantification of
nuclear HIF-1α in (F), normalized to Lamin B1. Data are presented
as the mean ± SD of three independent experiments. *P<0.05 and
**P<0.01. H2O2, hydrogen
peroxide; OGD/R, oxygen-glucose deprivation/reperfusion; VECs,
vascular endothelial cells; WB, western blotting; PI, propidium
iodide; FITC, fluorescein isothiocyanate; LDHA, lactate
dehydrogenase A; HIF-1α, hypoxia-inducible factor 1α; ROS, reactive
oxygen species; MMP, mitochondrial membrane potential. |
LDHA overexpression enhances
glycolytic activity and modulates metabolic pathways in VECs under
OGD/R conditions
Pyruvate, lactate and succinate serve important
roles in cellular energy metabolism, directly affecting
mitochondrial function and ATP synthesis (24). To further explore the impact of
LDHA overexpression on cellular metabolism, the levels of pyruvate,
lactate and succinate were measured in VECs treated with 0.5 mM
H2O2 (OGD/R) and overexpressing LDHA. The
results showed that LDHA overexpression increased pyruvate and
lactate levels, while reducing succinate levels in VECs subjected
to 0.5 mM H2O2 (OGD/R) (Fig. 6A-C). Subsequently, the expression
levels of key glycolytic genes [hexokinase 2 (HK2),
phosphoglucomutase 5 (PGM5), pyruvate kinase M (PKM) and LDHA] were
assessed in VECs by RT-qPCR. The data revealed that LDHA
overexpression significantly promoted the expression levels of HK2,
PGM5, PKM and LDHA compared with those in the 0.5 mM
H2O2 (OGD/R) + vector group, indicating
enhanced glycolytic activity in VECs (Fig. 6D). Further analysis of
tricarboxylic acid (TCA) cycle-related genes [oxoglutarate
dehydrogenase (OGDH), dihydrolipoamide dehydrogenase (DLD) and
succinyl-CoA synthetase β subunit (SUCLG2)] in VECs treated with
0.5 mM H2O2 (OGD/R) and overexpressing LDHA
showed that LDHA overexpression suppressed the expression levels of
OGDH and DLD, while increasing SUCLG2 expression compared with
those in the 0.5 mM H2O2 (OGD/R) + vector
group (Fig. 6E). ECAR was measured
to assess the dynamic metabolic changes in VECs. Upon glucose
addition, all groups showed a rapid increase in ECAR, confirming
the activation of glycolysis (Fig.
6F). The 0.5 mM H2O2 (OGD/R) + LDHA
overexpression group exhibited higher ECAR increase compared with
that in the 0.5 mM H2O2 (OGD/R) + vector and
0.5 mM H2O2 (OGD/R) groups. Following Oligo
treatment, all groups showed a marked ECAR peak, indicating
enhanced glycolytic activity. Notably, the ECAR in the 0.5 mM
H2O2 (OGD/R) + LDHA overexpression group was
higher than that in the other groups, suggesting that LDHA
overexpression strongly enhanced glycolysis. After the addition of
the glycolysis inhibitor 2-DG, ECAR was significantly decreased in
all groups, returning to baseline levels. These results
collectively demonstrated that LDHA overexpression may enhance
glucose metabolism in VECs, promoting glycolysis and modulating
metabolic pathways following OGD/R-induced damage.
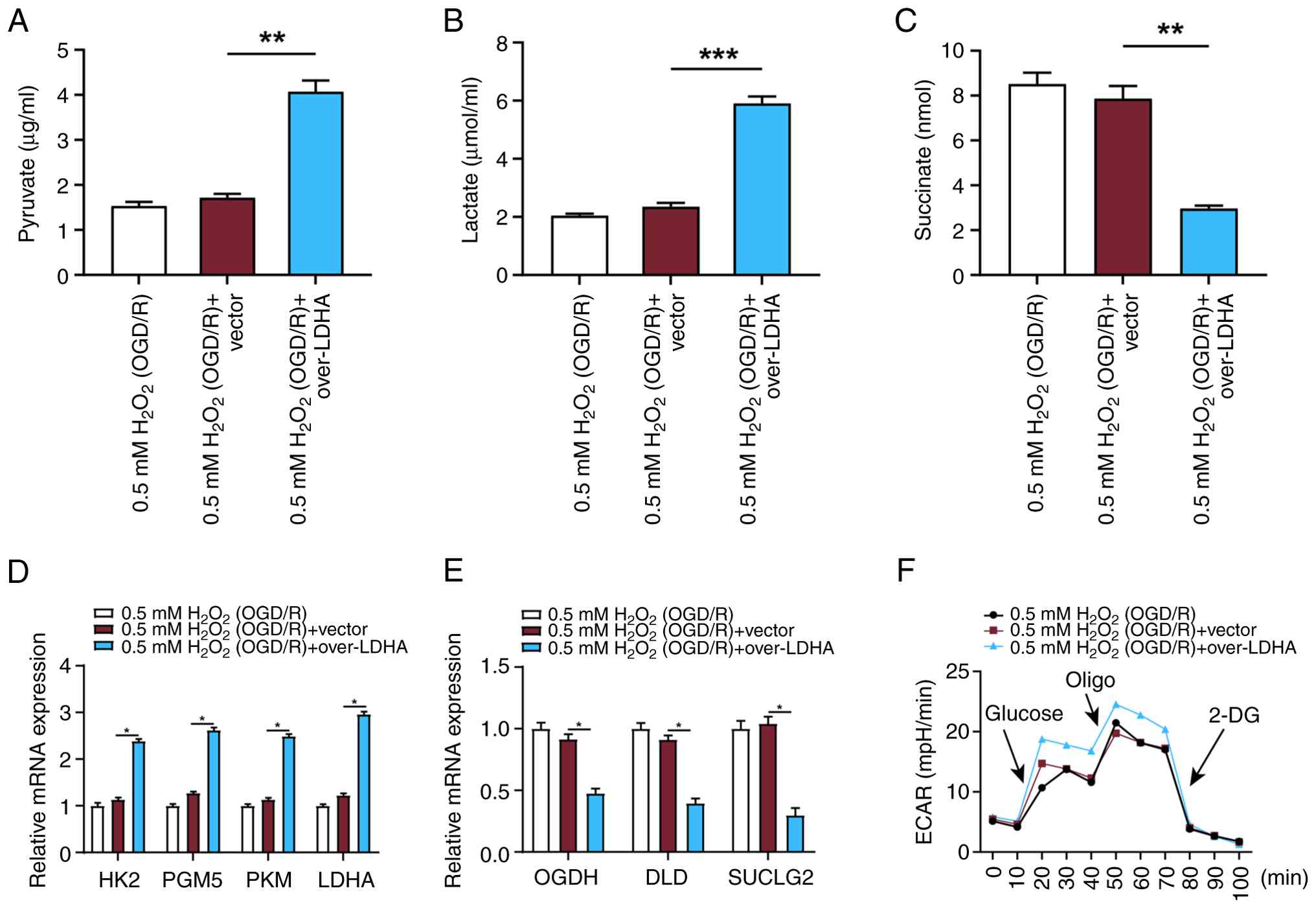 | Figure 6.LDHA overexpression enhances
glycolytic activity and modulates metabolic pathways in VECs under
OGD/R conditions. (A) Pyruvate, (B) lactate and (C) succinate
levels measured in VECs treated with 0.5 mM
H2O2 (OGD/R) and overexpressing LDHA. (D)
Expression levels of key glycolytic genes (HK2, PGM5, PKM and LDHA)
were assessed by RT-qPCR in VECs treated with 0.5 mM
H2O2 (OGD/R) and overexpressing LDHA. (E)
Expression levels of tricarboxylic acid cycle-related genes (OGDH,
DLD and SUCLG2) were assessed by RT-qPCR in VECs treated with 0.5
mM H2O2 (OGD/R) and overexpressing LDHA. (F)
ECAR was measured to assess metabolic changes in VECs. Data are
presented as the mean ± SD of three independent experiments.
*P<0.05, **P<0.01 and ***P<0.001.
H2O2, hydrogen peroxide; OGD/R,
oxygen-glucose deprivation/reperfusion; VECs, vascular endothelial
cells; LDHA, lactate dehydrogenase A; HK2, hexokinase 2; PGM5,
phosphoglucomutase 5; PKM, pyruvate kinase M; OGDH, oxoglutarate
dehydrogenase; DLD, dihydrolipoamide dehydrogenase; SUCLG2,
succinate-CoA ligase β subunit; ECAR, extracellular acidification
rate; Oligo, oligomycin; 2-DG, 2-deoxy-D-glucose; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction. |
LDHA alleviates oxidative
stress-induced mitochondrial damage via glycolytic reprogramming in
VECs
To further investigate whether LDHA protects against
oxidative stress-induced mitochondrial injury through glycolytic
reprogramming, LDHA-overexpressing VECs we treated with or without
the glycolysis inhibitor 2-DG under 0.5 mM
H2O2 (OGD/R) conditions. WB showed that
compared with LDHA overexpression alone, the addition of 2-DG
markedly increased the expression levels of the mitochondrial
apoptosis-related proteins caspase-3 and Cyt-c, while reducing the
expression of the anti-apoptotic protein Mcl-1 (Fig. 7A-D). The current study further
evaluated oxidative stress markers. Compared with LDHA
overexpression alone, treatment with 2-DG significantly increased
ROS and MDA levels, while GSH levels were decreased (Fig. 7E-G). These findings support that
LDHA overexpression may mitigate oxidative stress-induced
mitochondrial damage in VECs, likely through enhancing glycolysis
and maintaining redox homeostasis.
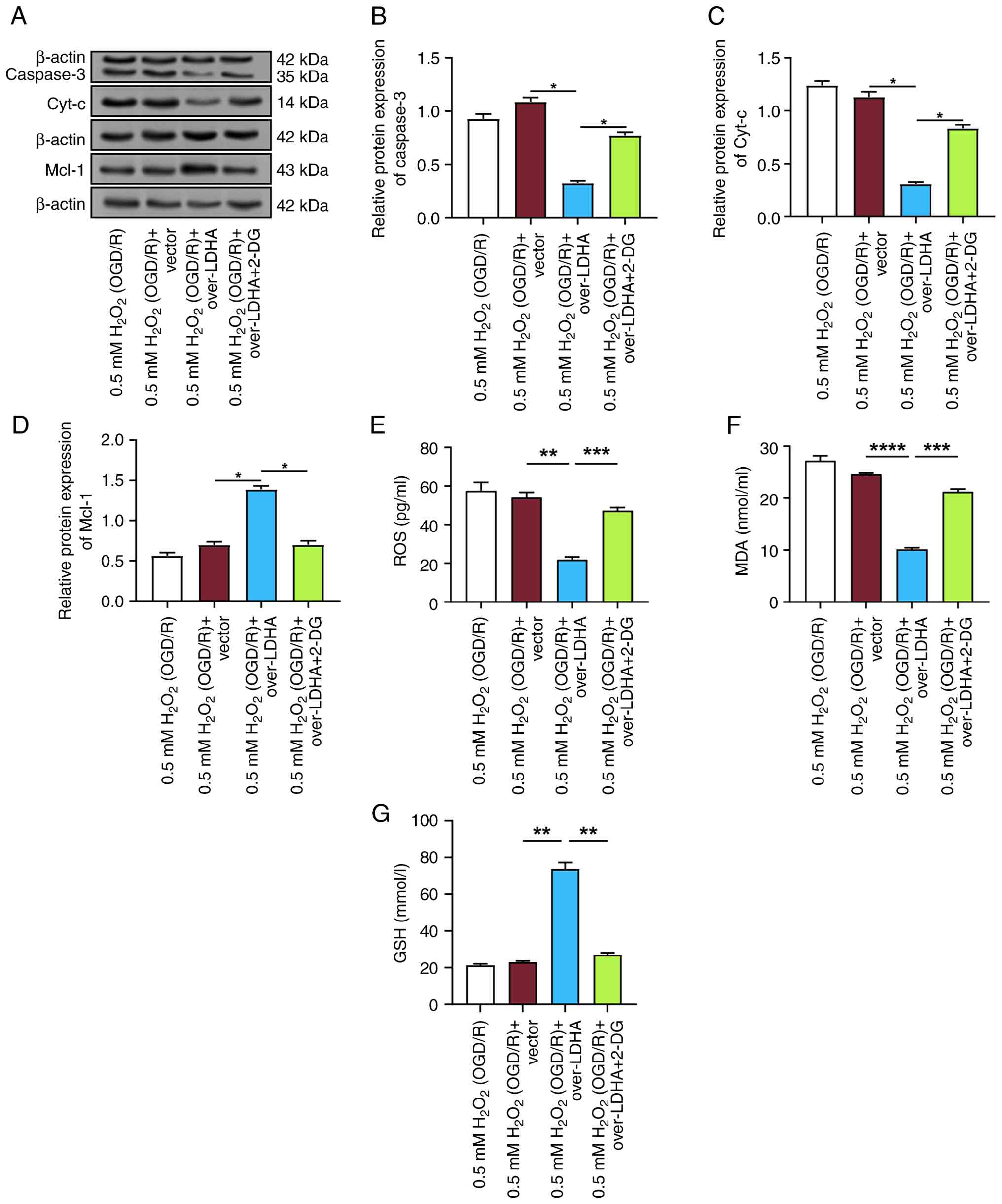 | Figure 7.LDHA protects against oxidative
stress-induced mitochondrial damage through glycolytic
reprogramming in VECs. (A) Representative WB images of caspase-3,
Cyt-c and Mcl-1 protein expression in VECs treated with 0.5 mM
H2O2 (OGD/R) and overexpressing LDHA, with or
without 2-DG. Semi-quantification of (B) caspase-3, (C) Cyt-c and
(D) Mcl-1 protein levels normalized to internal controls.
Measurement of (E) ROS, (F) MDA and (G) GSH levels in each group.
Data are presented as the mean ± SD from three independent
experiments. *P<0.05, **P<0.01, ***P<0.001
and ****P<0.0001. H2O2,
hydrogen peroxide; OGD/R, oxygen-glucose deprivation/reperfusion;
VECs, vascular endothelial cells; LDHA, lactate dehydrogenase A;
2-DG, 2-deoxy-D-glucose; WB, western blotting; ROS, reactive oxygen
species; MDA, malondialdehyde; GSH, glutathione; Cyt-c, cytochrome
c. |
LDHA overexpression induces lactic
acid production without causing acidosis in VECs
A previous study demonstrated that in response to
LDHA knockdown, lactate levels are increased (16). This has been attributed to the
compensatory upregulation of LDHB activity under disease
conditions, which may still drive lactate accumulation.
Subsequently, the potential effects of LDHA overexpression on
intracellular Na+ and Ca2+ homeostasis were
assessed. WB of transmembrane transporter proteins (NHE1 and NCX1)
revealed no significant changes in their expression levels
following LDHA overexpression under 0.5 mM
H2O2 (OGD/R) conditions (Fig. 8A and B). Similarly, calcium assay
measurements showed no significant alterations in intracellular
Ca2+ concentration following LDHA overexpression
(Fig. 8C). In addition, the
expression levels of the lactate transporter MCT4 were examined by
WB. The results indicated that LDHA overexpression upregulated MCT4
protein levels (Fig. 8D and E).
Collectively, these data demonstrated that LDHA overexpression may
increase lactate production in VECs without intracellular acidosis,
as evidenced by unchanged NHE1/NCX1 expression and cytosolic
Ca2+ together with upregulated MCT4 indicating enhanced
lactate efflux.
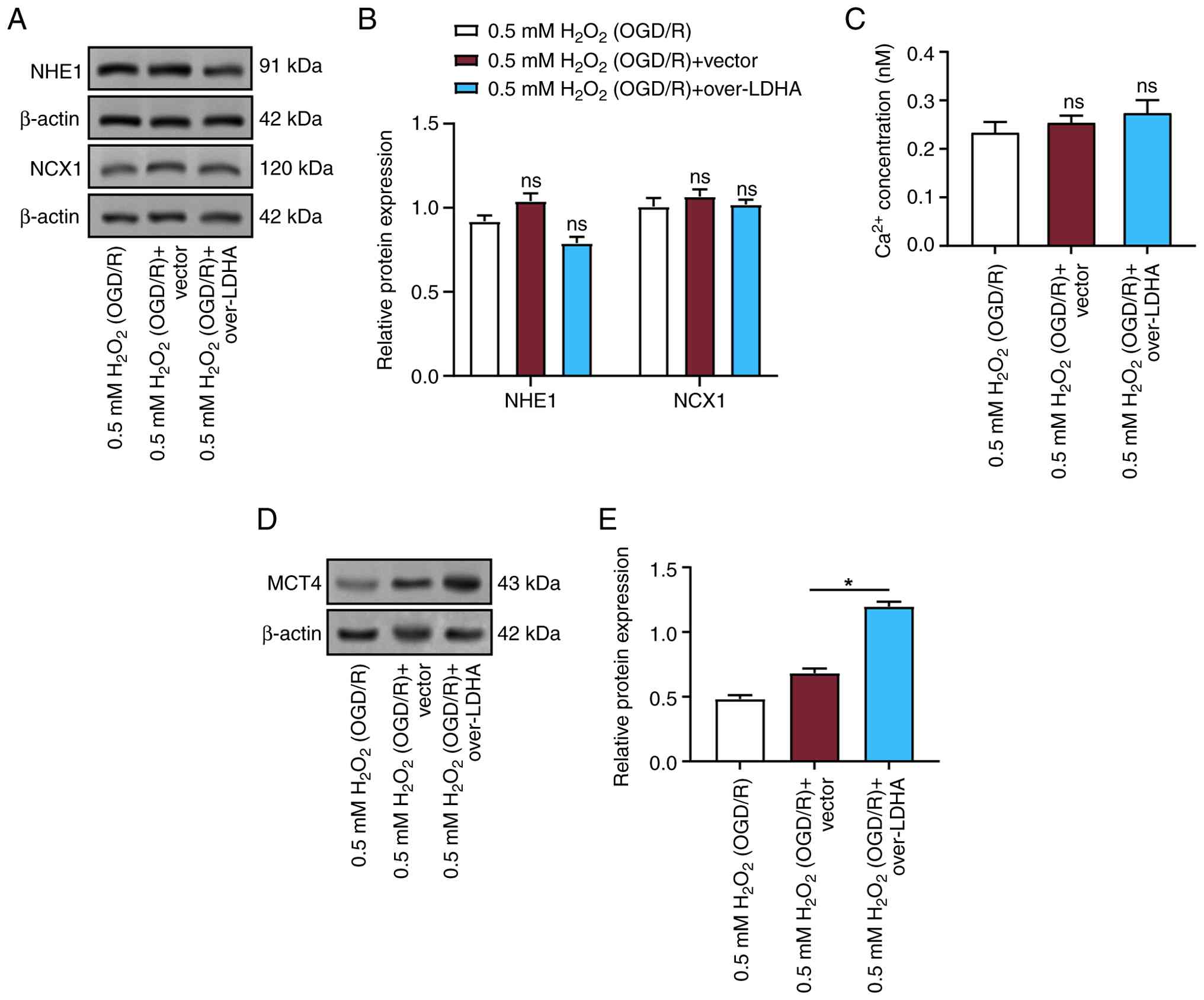 | Figure 8.LDHA overexpression induces lactic
acid production without causing acidosis in VECs. (A)
Representative WB showing protein levels of NHE1 and NCX1 in VECs
treated with 0.5 mM H2O2 (OGD/R), 0.5 mM
H2O2 (OGD/R) + vector, or 0.5 mM
H2O2 (OGD/R) + LDHA overexpression. (B)
Semi-quantification of NHE1 and NCX1 protein expression. (C)
Intracellular Ca2+ concentration measured in VECs after
LDHA overexpression under 0.5 mM H2O2 (OGD/R)
conditions. (D) WB of MCT4 expression in VECs after LDHA
overexpression under 0.5 mM H2O2 (OGD/R)
conditions. (E) Semi-quantification of MCT4 protein expression in
VECs treated with 0.5 mM H2O2 (OGD/R) and
overexpressing LDHA. Data are presented as the mean ± SD of three
independent experiments. *P<0.05; ns, not significant.
H2O2, hydrogen peroxide; OGD/R,
oxygen-glucose deprivation/reperfusion; VECs, vascular endothelial
cells; LDHA, lactate dehydrogenase A; NHE1, sodium-proton exchanger
1; NCX1, sodium-calcium exchanger 1; MCT4, monocarboxylate
transporter 4; WB, western blotting. |
LDHA overexpression enhances
antioxidant defense and maintains redox balance in VECs under OGD/R
conditions
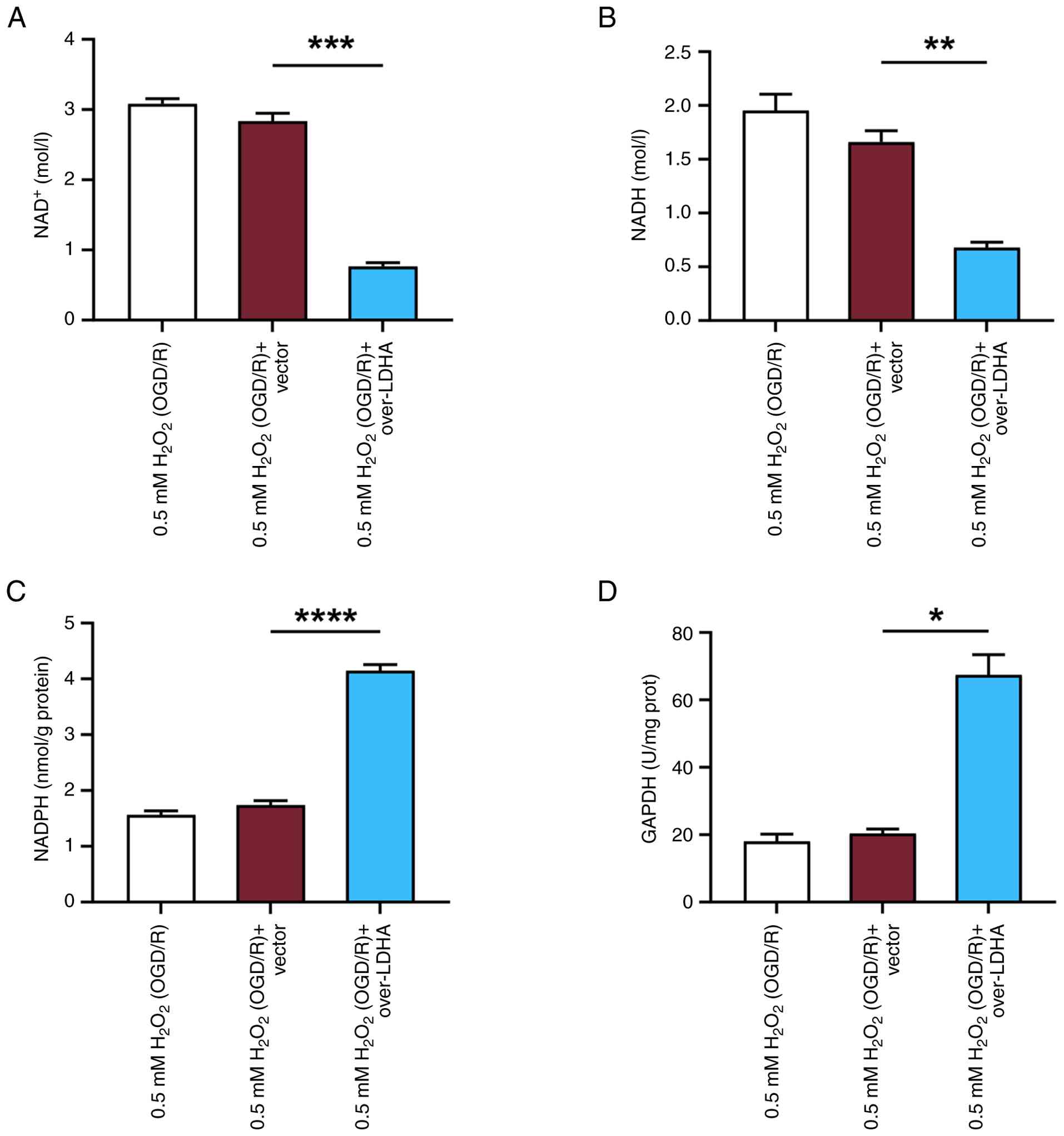
To further explore the mechanism by which LDHA
overexpression strengthens antioxidant defense and maintains redox
balance, the levels of NAD+ and NADH were measured in
VECs treated with 0.5 mM H2O2 (OGD/R) and
overexpressing LDHA. Using the NAD+/NADH assay kit, it
was revealed that LDHA overexpression reduced both NAD+
and NADH levels in VECs subjected to OGD/R-induced damage (Fig. 9A and B). It has previously been
shown that NADH interacts with LDHA to generate electrons, which
could promote ROS production (25). Therefore, the reduction of ROS
observed in the current study may be due to the decreased
interaction between NADH and LDHA following overexpression. To
investigate whether LDHA overexpression influences the activation
of the pentose phosphate pathway (PPP) during OGD/R damage, NADPH
levels and G6PDH activity were assessed in VECs. The results
revealed that LDHA overexpression significantly increased both
NADPH levels and G6PDH activity compared with those in the 0.5 mM
H2O2 (OGD/R) + vector group (Fig. 9C and D). These outcomes indicated
that LDHA overexpression may enhance activation of the PPP in VECs
during oxidative stress induced by OGD/R.
Discussion
Endothelial cells are essential for the progression
and formation of aneurysms through mechanisms such as maintaining
vascular integrity, regulating vasomotion and participating in
inflammatory responses (26).
Morel et al (27) reported
that both low and excessively high WSS in CAs can alter endothelial
cell morphology. Low WSS was shown to downregulate cytoskeletal
proteins and upregulate extracellular matrix proteins, whereas
extremely high WSS induced the opposite effects. These changes may
contribute to vascular remodeling in aneurysms. Additionally, Zhu
et al (28) indicated that
high mobility group box 1 (HMGB1) can induce oxidative stress and
inflammation in endothelial cells exposed to WSS, whereas HMGB1
knockdown can attenuate this process. During apoptosis, Cyt-c is
typically released from mitochondria into the cytoplasm when
mitochondrial membrane permeability increases. It then binds with
Apaf-1 to form apoptosomes. By blocking the permeability of the
outer membrane of the mitochondria and stopping the release of
Cyt-c, Mcl-1 can alter the susceptibility of the cell to apoptotic
signals (29). Huang et al
(30) noted that FOXO1 reduces
Mcl-1 expression by suppressing its transcription, thereby
promoting apoptosis, inflammation and inhibition of cell
proliferation in human brain vascular smooth muscle cells. The
current study observed significant apoptosis in VECs following
treatment with 0.5 mM H2O2 (OGD/R), alongside
increased expression levels of apoptosis-related proteins,
including caspase-3, caspase-9 and caspase-8. Additionally,
mitochondrial pathway-related proteins were affected, with elevated
Cyt-c expression and decreased levels of Mcl-1 detected following
treatment with 0.5 mM H2O2 (OGD/R),
indicating that oxidative stress can promote VEC apoptosis by
activating the mitochondrial apoptotic pathway.
Oxidative stress-induced ROS can oxidize lipids,
leading to mitochondrial membrane damage (31). This damage disrupts mitochondrial
permeability, causes a loss of membrane potential, and subsequently
impairs ATP synthesis. Mitochondrial damage will aggravate
oxidative stress, forming a vicious cycle. Tavris et al
(32) revealed that vascular
smooth muscle cells in abdominal aortic aneurysms exhibit
mitochondrial dysfunction and increased oxidative stress, leading
to abnormal mitochondrial morphology, enhanced ROS production,
weakened antioxidant responses and aggravated DNA damage. Nguyen
et al (33) investigated
the function of mitochondrial oxidative stress in various types of
brain injuries, both traumatic and non-traumatic. This previous
study highlighted the reliance of the brain on mitochondrial
function to meet its oxidative demands. However, the effects of ROS
varied across different injury types. Chen et al (34) emphasized the critical function of
mitochondrial dysfunction and necrotic apoptosis in the formation
of CAs. This previous study observed upregulation of these
processes in monocytes/macrophages and vascular smooth muscle
cells, suggesting potential new targets for the diagnosis and
treatment of IAs. The current study discovered that LDHA
overexpression alleviated apoptosis and mitochondrial damage
induced by oxidative stress in VECs. Specifically, LDHA
overexpression improved mitochondrial morphology, reduced swelling
and damage, and suppressed the upregulation of caspase-3 and Cyt-c,
while restoring Mcl-1 expression. Additionally, LDHA overexpression
significantly reduced ROS and MDA levels, restored GSH levels and
protected MMP from oxidative stress-induced damage. These outcomes
imply that LDHA overexpression has a protective function in
mitigating oxidative stress and mitochondrial dysfunction in
VECs.
In response to OGD/R, the levels of the essential
transcription factor HIF-1 are typically increased (35). HIF-1α and HIF-1β are the two
subunits that make up HIF-1 (36).
Under hypoxic conditions, HIF-1α degradation is inhibited,
resulting in increased stability and accumulation within the cell.
The accumulation of HIF-1α enables its dimerization with HIF-1β to
form an active HIF-1 complex. This complex translocates to the
nucleus and activates the expression of several target genes
(37). Kalashnyk et al
(38) reported that α7 nicotinic
acetylcholine receptors are upregulated in hepatocytes and U373
cells (human glioblastoma/astrocytoma cell line). These receptors
interact with HIF-1α and influence its translocation, particularly
under hypoxic conditions. Papandreou et al (39) reported that HIF-1 induces pyruvate
dehydrogenase kinase 1 (PDK1), which suppresses mitochondrial
function and reduces mitochondrial oxygen consumption. Although
HIF-1 promotes glycolysis, it paradoxically inhibits mitochondrial
oxygen utilization. This inhibition of oxygen utilization
paradoxically increases intracellular oxygen tension, thereby
reducing cell death in hypoxic environments. Han et al
(40) demonstrated that Axl
promotes M1 macrophage polarization through the STAT1/HIF-1α
signaling pathway; by contrast, inhibiting Axl can reduce M1
macrophage infiltration and prevent IA rupture. PX478, as an
inhibitor targeting HIF-1α, can effectively block HIF-1α-mediated
transcriptional activation by altering the stability of HIF-1α. In
the present study, overexpression of LDHA under OGD/R conditions
enhanced the expression and nuclear translocation of HIF-1α,
helping VECs adapt to the hypoxic environment and improving cell
survival. Furthermore, LDHA overexpression mitigated OGD/R-induced
cell apoptosis and mitochondrial damage, whereas treatment with the
HIF-1α inhibitor PX478 reversed the protective effects of LDHA
overexpression. These findings indicated that LDHA may exert its
protective role through the HIF-1α signaling pathway by alleviating
oxidative stress-induced mitochondrial damage and apoptosis.
In the brain, glucose serves as the main oxidative
substrate for energy synthesis. Energy dysregulation may be
indicated by mitochondrial injury, which would impact glucose
metabolism. Gusdon et al (41) reported that after aSAH, patients
exhibit a shift in metabolism toward glycolysis, characterized by
elevated levels of glycolytic metabolites and reduced levels of
oxidative metabolites. Dai et al (42) highlighted the function of LDHA in
T-cell energy metabolism, emphasizing its crucial involvement in
glycolysis. Specifically, LDHA was shown to promote lactate
production, exhibit non-classical enzymatic activity and contribute
to oxidative stress in immune-related diseases, such as Listeria
monocytogenes and murine cytomegalovirus infections. Tian et
al (43) discovered that VGLL4
can alleviate neurodegeneration in models of Alzheimer's disease
(AD), including AD model mice and AD model cells, by upregulating
LDHA expression and enhancing lactate production. NHE1 and NCX1 are
two distinct transmembrane transporters that serve key roles in
cellular ion homeostasis and signal transduction. Yin et al
(44) showed that limonene
protects against endothelial dysfunction in type 2 diabetes by
inhibiting the TRPM2/NHE1 signaling pathway, reducing oxidative
stress and mitigating mitochondrial damage. Glycolysis generates
energy by breaking down glucose, whereas the TCA cycle produces a
greater amount of ATP through a more complete oxidative process.
The present study demonstrated that LDHA overexpression enhanced
pyruvate and lactate levels, while reducing succinate levels. These
findings suggested a potential inhibition of mitochondrial
function. In addition, LDHA overexpression significantly promoted
the expression of glycolytic genes and enhanced glycolytic
activity, while suppressing the expression of TCA cycle-related
genes. After LDHA-overexpressing cells were treated with 2-DG, the
expression of mitochondrial apoptosis-related proteins caspase3 and
Cyt-c was increased, whereas the expression of Mcl-1 was decreased.
Furthermore, ROS and MDA levels were elevated, and GSH levels were
reduced. These findings further suggested that LDHA may protect
mitochondria from oxidative stress-induced endothelial cell injury
through glycolytic reprogramming. Although lactate levels increased
following LDHA overexpression, no acidosis was observed, and there
were no significant changes in Na+ and Ca2+
homeostasis or transporter expression. In addition, LDHA
overexpression upregulated the lactate transporter MCT4, which may
help alleviate lactate accumulation.
During cellular respiration, NAD+ and
NADH serve crucial roles. Metabolic processes such as glycolysis,
the TCA cycle and oxidative phosphorylation all depend on the
exchange between NAD+ and NADH (45). Zhao et al (46) demonstrated that NAD+
activates the sirtuin 1/peroxisome proliferator-activated receptor
γ coactivator 1α pathway. This activation was shown to improve
cognitive function in rats with chronic cerebral ischemia, suppress
neuroinflammation, protect mitochondria and reduce ROS production.
Lee et al (47) revealed
that supplements enhancing the NAD/NADH ratio and mitochondrial
function can provide protection to brain endothelial cells against
oxidative stress induced by OGD. These supplements also improved
tight junction protein expression and alleviated damage in a mouse
model of intracerebral hemorrhage. The PPP is an important cellular
metabolic pathway; while similar to glycolysis, the PPP serves a
different primary function. Zhang et al (48) conducted a bioinformatics analysis
to identify four oxidative stress-related diagnostic biomarkers
(FLVCR2, SDSL, TBC1D2, SLC31A1). This previous study demonstrated
that these biomarkers were associated with oxidative stress, ROS
accumulation, abnormal glucose metabolism and immune suppression
infiltration in IAs. These findings provide new insights for
clinical management. G6PDH is a key enzyme in the PPP, catalyzing
the transformation of G6P to 6-phosphoglucono-δ-lactone,
accompanied by the production of NADPH. Studies have shown that
G6PD deficiency reduces NADPH levels in microglia, triggering
oxidative stress and functional damage. Supplementation with
metabolites and small molecules has been shown to increase NADPH
production and improve microglial function (49,50).
Wiśniewski et al (51)
noted that, after aSAH, individuals with delayed cerebral ischemia
(DCI) exhibited higher F2-isoprostane (IsoP) levels and lower G6PD
levels. Both changes were associated with the occurrence and
prognosis of DCI. Reduced G6PD may impair antioxidant responses and
elevate F2-IsoP levels. In the present study, overexpression of
LDHA in VECs subjected to OGD/R treatment led to a decrease in
NAD+ and NADH levels. It also reduced ROS generation,
while increasing NADPH levels and G6PDH activity. This could
enhance the activity of the PPP, thereby improving the cellular
antioxidant defense capacity.
HIF-1α is a pivotal transcription factor that
orchestrates cellular responses to hypoxia and oxidative stress.
The current study demonstrated that LDHA overexpression enhanced
HIF-1α expression and nuclear translocation of HIF-1α. However, the
mechanisms by which LDHA affects the downstream targets of HIF-1α
to protect blood vessels remain to be investigated. Notably, HIF-1α
upregulates VEGF, a key angiogenic factor that promotes endothelial
cell survival, proliferation and migration, thereby maintaining
vascular integrity and reducing aneurysm progression risk (52,53).
In addition, HIF-1α induces the expression of PDK1, which redirects
pyruvate metabolism towards lactate production, enhances glycolytic
activity and reduces mitochondrial ROS generation (54). This metabolic reprogramming
supports energy homeostasis and mitigates oxidative injury under
hypoxic stress. Furthermore, HIF-1α modulates the balance of Bcl-2
family proteins by upregulating anti-apoptotic proteins, such as
Bcl-2 and Bcl-xL, while downregulating pro-apoptotic Bax, thus
protecting VECs from oxidative stress-induced apoptosis (55). HIF-1α also induces antioxidant
enzymes, such as superoxide dismutase and catalase, neutralizes ROS
and upregulates MCT4, facilitating lactate efflux and preventing
acidosis, maintaining cellular pH homeostasis (56). Collectively, these downstream
targets contribute to the protective effects observed in VECs under
oxidative stress. By promoting glycolysis, enhancing angiogenesis,
inhibiting apoptosis and improving antioxidant defenses, HIF-1α may
have a central role in maintaining vascular integrity and
preventing CA progression. Future research should further elucidate
the interactions between LDHA and HIF-1α, as well as their
downstream proteins, to develop targeted interventions for CAs and
related vascular disorders.
The present study demonstrated that LDHA
overexpression promoted the activation and nuclear translocation of
HIF-1α, which is known to regulate downstream pathways involved in
metabolic reprogramming and vascular protection. Consistent with
this, it was observed that LDHA overexpression enhanced the
expression of glycolytic enzymes, such as HK2, PGM5 and PKM,
increased the expression of the lactate transporter MCT4, elevated
NADPH production and enhanced G6PDH activity. Although the current
data suggested that HIF-1α activation may contribute to these
changes, the direct regulatory relationships between HIF-1α and
these downstream proteins require further experimental
validation.
In conclusion, the outcomes of the current study
present compelling evidence suggesting that LDHA overexpression
mitigates oxidative stress-induced apoptosis and mitochondrial
damage in VECs under OGD/R conditions. By enhancing glycolytic
activity and promoting the nuclear translocation of HIF-1α, LDHA
could contribute to cellular adaptation and survival under
oxidative stress conditions. Furthermore, LDHA overexpression may
activate the PPP, boosting antioxidant defense and maintaining
redox balance. These effects collectively support the notion that
LDHA serves a protective role in VECs under oxidative damage,
potentially offering a novel therapeutic target for conditions such
as ischemic stroke and aSAH, where mitochondrial dysfunction and
oxidative stress are central to pathogenesis.
Acknowledgements
Not applicable.
Funding
This work was supported by the Songjiang District Science and
Technology Key Project (grant no. 2025SJKJGG085).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
BD, LY, AY and YZ conceived and designed the
research. BD and LY performed the experiments and acquired the
data, analyzed and interpreted the data, and drafted the
manuscript. BD performed statistical analysis. AY and YZ revised
the manuscript for important intellectual content. BD and LY
confirm the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Abbate PM, Hasan AH, Venier A, Vauclin V,
Pizzuto S, Sgreccia A, Di Maria F, Coskun O, Mizutani K and Rodesch
G: Consoli: The cerebral arterial wall in the development and
growth of intracranial aneurysms. App Sci. 12:59642022. View Article : Google Scholar
|
|
2
|
Tawk RG, Hasan TF, D'Souza CE, Peel JB and
Freeman WD: Diagnosis and treatment of unruptured intracranial
aneurysms and aneurysmal subarachnoid hemorrhage. Mayo Clinic
Proceedings; Elsevier: 2021, View Article : Google Scholar
|
|
3
|
Weiland J, Beez A, Westermaier T, Kunze E,
Sirén AL and Lilla N: Neuroprotective strategies in aneurysmal
subarachnoid hemorrhage (aSAH). Int J Mol Sci. 22:54422021.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Allaw S, Khabaz K, Given TC, Montas DM,
Alcazar-Felix RJ, Srinath A, Kass-Hout T, Carroll TJ, Hurley MC and
Polster SP: A review of intracranial aneurysm imaging modalities,
from CT to state-of-the-art MR. Am J Neuroradiol. 46:1082–1092.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sahlein DH, Gibson D, Scott JA, DeNardo A,
Amuluru K, Payner T, Rosenbaum-Halevi D and Kulwin C: Artificial
intelligence aneurysm measurement tool finds growth in all
aneurysms that ruptured during conservative management. J
Neurointerv Surg. 15:766–770. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
de Liyis BG, Surya SC and Tini K:
Effectivity and safety of endovascular coiling versus microsurgical
clipping for aneurysmal subarachnoid hemorrhage: A systematic
review and meta-analysis. Clin Neurol Neurosurg. 236:1080582024.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huang X, Wang Z, Shen Z, Lei F, Liu YM,
Chen Z, Qin JJ, Liu H, Ji YX, Zhang P, et al: Projection of global
burden and risk factors for aortic aneurysm-timely warning for
greater emphasis on managing blood pressure. Ann Med. 54:553–564.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Salnikova D, Orekhova V, Grechko A,
Starodubova A, Bezsonov E, Popkova T and Orekhov A: Mitochondrial
dysfunction in vascular wall cells and its role in atherosclerosis.
Int J Mol Sc. 22:89902021. View Article : Google Scholar
|
|
9
|
Wang Z, Ma J, Yue H, Zhang Z, Fang F, Wang
G, Liu X and Shen Y: Vascular smooth muscle cells in intracranial
aneurysms. Microvasc Res. 149:1045542023. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kaur MM and Sharma S: Mitochondrial repair
as potential pharmacological target in cerebral ischemia.
Mitochondrion. 63:23–31. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhao W, Zhang H and Su JY: MicroRNA-29a
contributes to intracranial aneurysm by regulating the
mitochondrial apoptotic pathway. Mol Med Rep. 18:2945–2954.
2018.PubMed/NCBI
|
|
12
|
Wang S, Wang J, Niu Z, Zhang K, Yang T,
Hou S and Lin N: Causal relationship between
mitochondrial-associated proteins and cerebral aneurysms: A
Mendelian randomization study. Front Neurol. 15:14050862024.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lin Y, Wang Y and Li PF: Mutual regulation
of lactate dehydrogenase and redox robustness. Front Physiol.
13:10384212022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang G, Liu S, Kong X, Jiao H, Tong F, Guo
Z, Zhang M, Guan X, Ren N, Li W, et al: Lipocalin-2 induced LDHA
expression promotes vascular remodelling in pulmonary hypertension.
Cell Prolif. 57:e137172024. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wu D, Wang S, Wang F, Zhang Q, Zhang Z and
Li X: Lactate dehydrogenase A (LDHA)-mediated lactate generation
promotes pulmonary vascular remodeling in pulmonary hypertension. J
Transl Med. 22:7382024. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu J, Lu L, Dai B and Yu A: Unraveling the
role of LDHA and VEGFA in oxidative stress: A pathway to
therapeutic interventions in cerebral aneurysms. Biomol Biomed.
25:360–374. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Renu K, Gopalakrishnan AV and Madhyastha
H: Is periodontitis triggering an inflammatory response in the
liver, and does this reaction entail oxidative stress? Odontology.
113:889–902. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fang L, Yu Z, Qian X, Fang H and Wang Y:
LDHA exacerbates myocardial ischemia-reperfusion injury through
inducing NLRP3 lactylation. BMC Cardiovasc Disord. 24:6512024.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Luo L, Ma X, Kong D, Dai Y, Li T, Yu H,
Liu J, Li M, Xu Y, Xiang G, et al: Multiomics integrated analysis
and experimental validation identify TLR4 and ALOX5 as oxidative
stress-related biomarkers in intracranial aneurysms. J
Neuroinflammation. 21:2252024. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sheinberg DL, McCarthy DJ, Elwardany O,
Bryant JP, Luther E, Chen SH, Thompson JW and Starke RM:
Endothelial dysfunction in cerebral aneurysms. Neurosurg Focus.
47:E32019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Uchida K, Yasunaga H, Sumitani M,
Horiguchi H, Fushimi K and Yamada Y: Effects of remifentanil on
in-hospital mortality and length of stay following clipping of
intracranial aneurysm: A propensity score-matched analysis. J
Neurosurg Anesthesiol. 26:291–298. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ling C, Yang Y, Hu X, Cai M, Wang H and
Chen C: Phoenixin-14 alleviates inflammatory smooth muscle
cell-induced endothelial cell dysfunction in vitro. Cytokine.
157:1559732022. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Glancy B, Kane DA, Kavazis AN, Goodwin ML,
Willis WT and Gladden LB: Mitochondrial lactate metabolism: History
and implications for exercise and disease. J Physiol. 599:863–888.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wu J, Gu X, Zhang J, Mi Z, He Z, Dong Y,
Ge W, Ghimire K, Rong P, Wang W and Ma X: 4-OI Protects MIN6 cells
from oxidative stress injury by reducing LDHA-Mediated ROS
generation. Biomolecules. 12:12362022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bonafiglia QA, Bendeck M and Gotlieb AI:
Vascular pathobiology: Atherosclerosis and large vessel disease.
Cardiovascular Pathology. Elsevier; pp. 65–306. 2022
|
|
27
|
Morel S, Schilling S, Diagbouga MR,
Delucchi M, Bochaton-Piallat ML, Lemeille S, Hirsch S and Kwak BR:
Effects of low and high aneurysmal wall shear stress on endothelial
cell behavior: Differences and similarities. Front Physiol.
12:7273382021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhu H, Zeng Y, Tan J, Li M and Zhao Y:
HMGB1 induced oxidative stress and Inflammation in endothelial
cells exposed to Impinging Flow. Cerebrovasc Dis. 53:437–48. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mustafa M, Ahmad R, Tantry IQ, Ahmad W,
Siddiqui S, Alam M, Abbas K, Moinuddin Hassan MI, Habib S and Islam
S: Apoptosis: A comprehensive overview of signaling pathways,
morphological changes, and physiological significance and
therapeutic implications. Cells. 13:18382024. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Huang J, Hong L, Shen B, Zhou Y, Lan J and
Peng Y: FOXO1 represses MCL1 transcription to regulate the function
of vascular smooth muscle cells in intracranial aneurysm. Exp Brain
Res. 240:2861–2870. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kowalczyk P, Sulejczak D, Kleczkowska P,
Bukowska-Ośko I, Kucia M, Popiel M, Wietrak E, Kramkowski K,
Wrzosek K and Kaczyńska K: Mitochondrial oxidative stress-A
causative factor and therapeutic target in many diseases. Int J Mol
Sci. 22:133842021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tavris BS, Peters AS, Böckler D and
Dihlmann SL: Mitochondrial dysfunction and increased DNA damage in
vascular smooth muscle cells of abdominal aortic aneurysm
(AAA-SMC). Oxid Med Cell Longev. 2023:62379602023. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nguyen A, Patel AB, Kioutchoukova IP, Diaz
MJ and Lucke-Wold B: Mechanisms of mitochondrial oxidative stress
in Brain Injury: From pathophysiology to therapeutics. Oxygen.
3:163–178. 2023. View Article : Google Scholar
|
|
34
|
Chen B, Xie K, Zhang J, Yang L, Zhou H,
Zhang L and Peng R: Comprehensive analysis of mitochondrial
dysfunction and necroptosis in intracranial aneurysms from the
perspective of predictive, preventative, and personalized medicine.
Apoptosis. 28:1452–1468. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang H, Liu X, Yang F, Cheng D and Liu W:
Overexpression of HIF-1α protects PC12 cells against OGD/R-evoked
injury by reducing miR-134 expression. Cell Cycle. 19:9902020.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yfantis A, Mylonis I, Chachami G,
Nikolaidis M, Amoutzias GD, Paraskeva E and Simos G:
Transcriptional response to hypoxia: The role of HIF-1-associated
co-regulators. Cells. 12:7982023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lee SH, Golinska M and Griffiths JR:
HIF-1-independent mechanisms regulating metabolic adaptation in
hypoxic cancer cells. Cells. 10:23712021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kalashnyk O, Lykhmus O, Koval L, Uspenska
K, Obolenskaya M, Chernyshov V, Komisarenko S and Skok M: α7
Nicotinic acetylcholine receptors regulate translocation of HIF-1α
to the cell nucleus and mitochondria upon hypoxia. Biochem Biophys
Res Commun. 657:35–42. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Papandreou I, Cairns RA, Fontana L, Lim AL
and Denko NC: HIF-1 mediates adaptation to hypoxia by actively
downregulating mitochondrial oxygen consumption. Cell metabolism.
3:187–197. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Han Y, Li G, Zhang Z, Zhang X, Zhao B and
Yang H: Axl promotes intracranial aneurysm rupture by regulating
macrophage polarization toward M1 via STAT1/HIF-1α. Front Immunol.
14:11587582023. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gusdon AM, Fu C, Putluri V, Paz AS, Chen
H, Ren X, Hassan MK, Dash P, Coarfa C, Putluri N, et al: Early
systemic glycolytic shift after aneurysmal subarachnoid hemorrhage
is associated with functional outcomes. Neurocrit Care. 37:724–734.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dai M, Wang L, Yang J, Chen J, Dou X, Chen
R, Ge Y and Lin Y: LDHA as a regulator of T cell fate and its
mechanisms in disease. Biomed Pharmacother. 158:1141642023.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Tian Q, Li J, Wu B, Wang J, Xiao Q, Tian
N, Yi L, Luo M, Li Z, Pang Y, et al: Hypoxia-sensing VGLL4 promotes
LDHA-driven lactate production to ameliorate neuronal dysfunction
in a cellular model relevant to Alzheimer's disease. FASEB J.
37:e232902023. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yin YL, Wang HH, Gui ZC, Mi S, Guo S, Wang
Y, Wang QQ, Yue RZ, Lin LB, Fan JX, et al: Citronellal attenuates
oxidative Stress-induced mitochondrial damage through TRPM2/NHE1
pathway and effectively inhibits endothelial dysfunction in type 2
diabetes mellitus. Antioxidants. 11:22412022. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gupta R and Gupta N, Gupta R and Gupta N:
Glycolysis and gluconeogenesis. Fund Bacterial Physiol Metabol.
267–87. 2021. View Article : Google Scholar
|
|
46
|
Zhao Y, Zhang J, Zheng Y, Zhang Y, Zhang
XJ, Wang H, Du Y, Guan J, Wang X and Fu J: NAD+ improves cognitive
function and reduces neuroinflammation by ameliorating
mitochondrial damage and decreasing ROS production in chronic
cerebral hypoperfusion models through Sirt1/PGC-1α pathway. J
Neuroinflammation. 18:2072021. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lee MJ, Zhu J, An J, Choi HJ, Pyo CH and
Heo JY: The increase of Nad/Nadh ratio in mitochondria alleviates
intracerebral hemorrhage injury by enhancing neurovascular unit
integrity. IBRO Neurosci Rep. 15:S5982023. View Article : Google Scholar
|
|
48
|
Zhang J, Duan P, Nie B, Zhang Z, Shi R,
Liu Q, Wang S, Xu T and Tian J: Identification and verification of
the oxidative Stress-related signature markers for intracranial
Aneurysm-applied bioinformatics. Front Biosci (Landmark Ed).
29:2942024. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Mondal A, Mukherjee S, Upadhyay P, Saxena
I, Pati S and Singh S: Enhancing NADPH to restore redox homeostasis
and lysosomal function in G6PD-deficient microglia. bioRxiv.
2024:6079182024.
|
|
50
|
Mondal A, Munan S, Saxena I, Mukherjee S,
Upadhyay P, Gupta N, Dar W, Samanta A, Singh S and Pati S: G6PD
deficiency mediated impairment of iNOS and lysosomal acidification
affecting phagocytotic clearance in microglia in response to
SARS-CoV-2. Biochim Biophys Acta Mol Basis Dis. 1870:1674442024.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wiśniewski K, Popęda M, Price B,
Bieńkowski M, Fahlström A, Drummond K and Adamides AA:
Glucose-6-phosphate dehydrogenase and 8-iso-prostaglandin F2α as
potential predictors of delayed cerebral ischemia after aneurysmal
subarachnoid hemorrhage. J Neurosurg. 139:698–707. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhang YM, Miao ZM, Chen YP, Song ZB, Li
YY, Liu ZW, Zhou GC, Li J, Shi LL, Chen Y, et al: Ononin promotes
radiosensitivity in lung cancer by inhibiting HIF-1α/VEGF pathway.
Phytomedicine. 125:1552902024. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Li PP, He L, Zhang LM, Qin XM and Hu JP:
Naoluo xintong decoction ameliorates cerebral Ischemia-reperfusion
injury by promoting angiogenesis through activating the HIF-1α/VEGF
signaling pathway in rats. Evid Based Complement Alternat Med.
2022:93414662022.PubMed/NCBI
|
|
54
|
Chen J, Zhang M, Liu Y, Zhao S, Wang Y,
Wang M, Niu W, Jin F and Li Z: Histone lactylation driven by
mROS-mediated glycolytic shift promotes hypoxic pulmonary
hypertension. J Mol Cell Biol. 14:mjac0732023. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ai X, Yu P, Luo L, Sun J, Tao H, Wang X
and Meng X: Berberis dictyophylla F. inhibits angiogenesis and
apoptosis of diabetic retinopathy via suppressing
HIF-1α/VEGF/DLL-4/Notch-1 pathway. J Ethnopharmacol.
296:1154532022. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Lee DC, Sohn HA, Park ZY, Oh S, Kang YK,
Lee KM, Kang M, Jang YJ, Yang SJ, Hong Y, et al: A Lactate-induced
response to hypoxia. Cell. 161:595–609. 2015. View Article : Google Scholar : PubMed/NCBI
|