Introduction
Post-translational modifications (PTMs) operate at
the intersection of epigenetic regulation and cellular metabolism,
expanding proteomic diversity and fine-tuning biological function
by modulating protein activity, stability and intermolecular
interactions. Although histone acetylation and methylation have
been extensively studied, increasing attention has turned to lysine
acylations derived from metabolic intermediates (1,2).
Among these, lysine succinylation involves the covalent transfer of
a succinyl group from succinyl-CoA to lysine residues. In contrast
to relatively neutral modifications such as acetylation,
succinylation introduces a marked negative charge that offsets the
intrinsic positive charge of lysine (3,4).
This charge reversal induces substantial conformational changes,
affecting enzymatic activity, protein stability, intermolecular
interactions and subcellular localization.
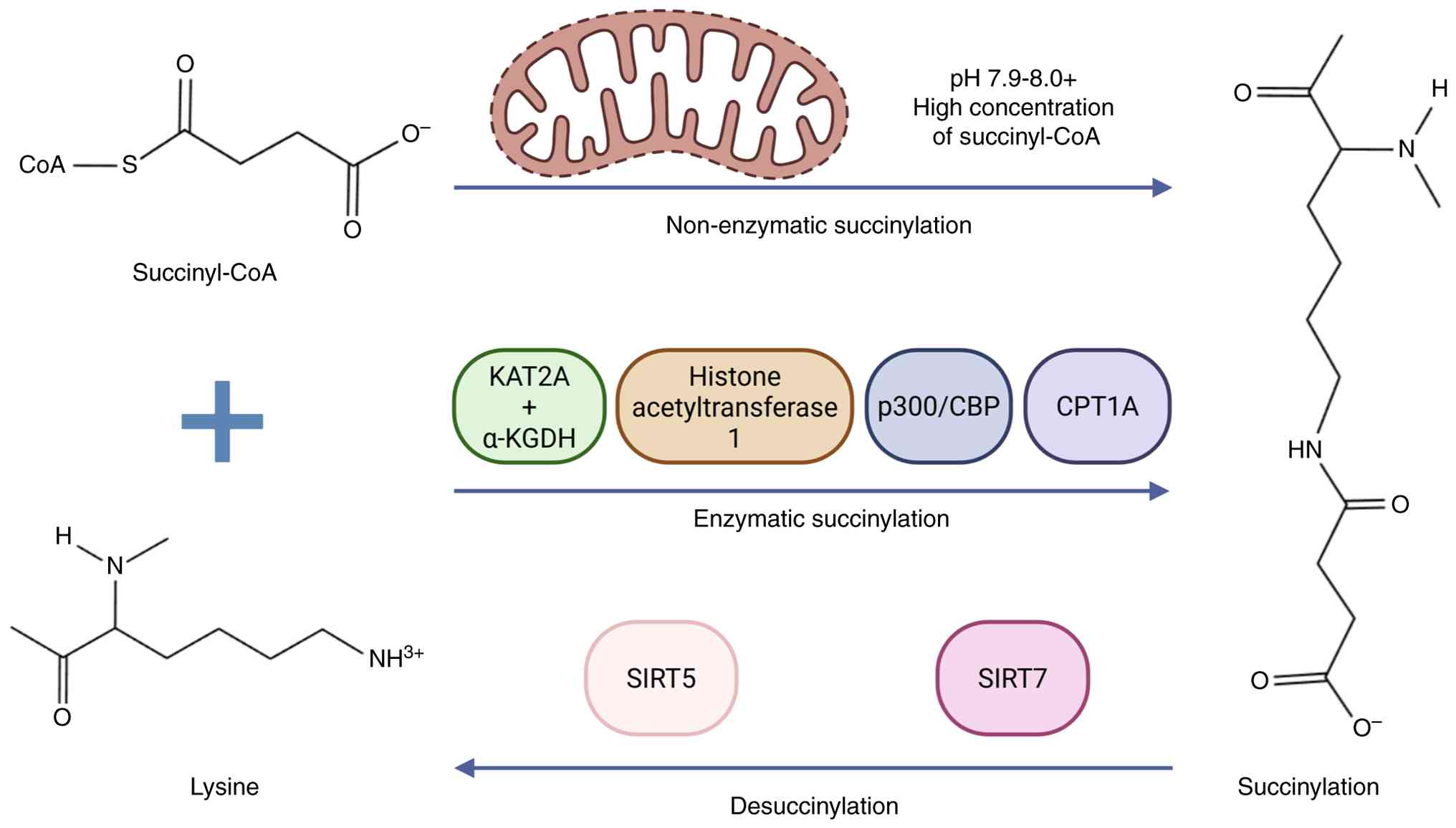
Succinylation is regulated through both enzymatic
and non-enzymatic mechanisms (Fig.
1). In the nucleus, lysine acetyltransferase 2A (KAT2A),
functioning together with the α-ketoglutarate dehydrogenase
(α-KGDH) complex, as well as canonical acetyltransferases such as
p300/CREB-binding protein, catalyse histone succinylation at sites
including H3K79 and H3K122, influencing transcriptional programs by
promoting open chromatin configurations that activate
pro-inflammatory and ferroptosis-related genes (5–8). In
the mitochondrial matrix, succinylation predominantly occurs
through non-enzymatic reactions. The mitochondrial microenvironment
promotes favourable biochemical conditions-specifically, an
alkaline pH and elevated local concentrations of succinyl-CoA-for
spontaneous succinyl group transfer, which occurs at rates
exceeding non-enzymatic acetylation (9,10).
As a result, mitochondrial proteins are particularly vulnerable to
this modification.
Given that non-enzymatic succinylation readily
occurs under physiological conditions, maintenance of cellular
homeostasis depends on efficient removal of succinyl groups by
dedicated ‘eraser’ enzymes. Sirtuin (SIRT)-5, an
NAD+-dependent class III lysine deacylase localized
primarily to mitochondria, serves as the principal desuccinylase
and displays strong catalytic selectivity for negatively charged
acyl modifications (11,12). Although SIRT7 has been reported to
desuccinylate specific histone residues (13), SIRT5 serves the dominant role in
counteracting the continual, spontaneous addition of succinyl
groups. Through this reversible regulation, SIRT5 modulates enzyme
function across key metabolic pathways, including the urea cycle
(argininosuccinate synthase 1), ketogenesis (HMGCS2), non-shivering
thermogenesis [uncoupling protein 1 (UCP1)], carbohydrate oxidation
through the pyruvate dehydrogenase complex (PDC) and fatty acid
β-oxidation (14–18). These actions preserve proteomic
integrity and sustain metabolic flexibility, limiting metabolic
dysfunction in preclinical models, such as ob/ob and diet-induced
obesity murine models (16,18).
Under physiological conditions, basal succinylation
levels are tightly regulated. However, metabolic stress, such as
that observed in diabetes and its associated complications, can
destabilize this equilibrium (19). Chronic hyperglycaemia and lipid
overload affect tricarboxylic acid (TCA) cycle flux by overwhelming
oxidative capacity and stalling intermediate turnover, leading to
intracellular accumulation of succinyl-CoA. In addition, oxidative
stress and NAD+ depletion in the diabetic environment
compromise SIRT5 activity by inducing transcriptional repression
and limiting its essential co-substrate, respectively. The
resulting imbalance between succinyl-CoA availability and
desuccinylase capacity promotes pathological protein
hypersuccinylation. This state establishes a feed-forward mechanism
in which hypersuccinylated enzymes exhibit diminished activity,
further aggravating metabolic inflexibility and perpetuating
‘metabolic memory’ (20,21).
Emerging evidence has implicated dysregulated
succinylation in a spectrum of metabolic disorders, including
impaired thermogenesis in obesity, diabetic cardiomyopathy (DbCM),
diabetic kidney disease (DKD) and diabetic retinopathy (DR)
(22,23). The present review consolidates
current mechanistic insights, elucidating how aberrant
succinylation associates metabolic imbalance with organ-specific
dysfunction (Fig. 2). Through
examining the context-dependent functions of SIRT5, the therapeutic
importance of targeting the succinyl-CoA/SIRT5 axis in diabetes and
its secondary complications are underscored.
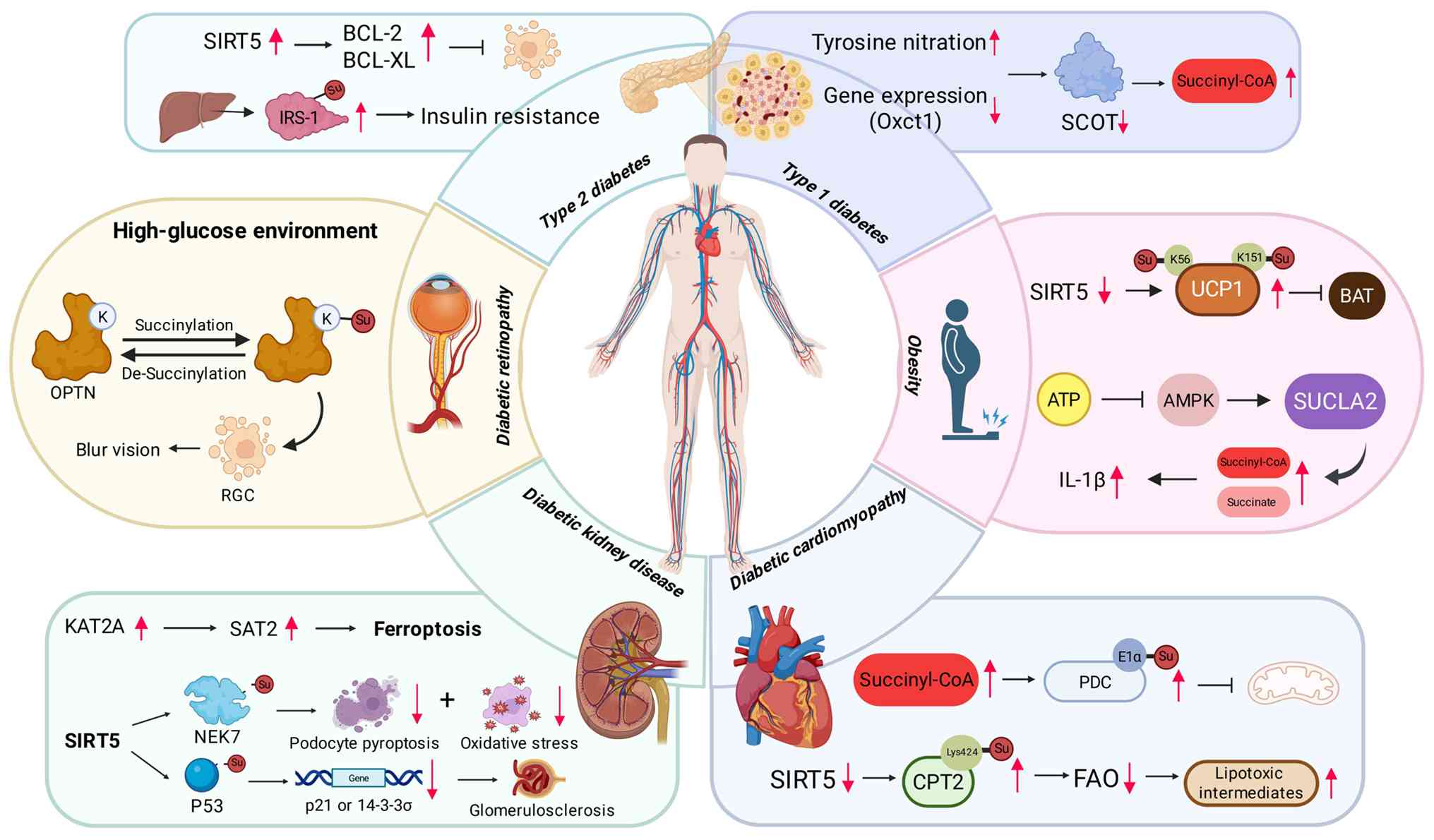 | Figure 2.Mechanism of succinylation in
diabetes and diabetic complications. Created with BioRender.com.
SIRT5, sirtuin 5; IRS-1, insulin receptor substrate 1; Oxct1,
3-oxoacid CoA-transferase 1; SCOT, succinyl-CoA:3-ketoacid CoA
transferase; OPTN, optineurin; RGC, retinal ganglion cells; UCP1,
uncoupling protein 1; BAT, brown adipose tissue; AMPK,
AMP-activated protein kinase; SUCLA2, succinyl-CoA synthetase
ADP-forming subunit β; KAT2A, lysine acetyltransferase 2A; SAT2,
spermidine/spermine N1-acetyltransferase family member 2; NEK7,
NIMA related kinase 7; PDC, pyruvate dehydrogenase complex; FAO,
fatty acid oxidation; CPT2, carnitine palmitoyltransferase 2. |
Dysregulated succinylation in diabetes
mellitus (DM)
DM is a chronic metabolic disease defined by
inadequate insulin secretion, impaired insulin sensitivity or a
combination of both, leading to sustained hyperglycaemia. Hallmark
clinical manifestations include polyphagia, polydipsia and
polyuria. DM comprises type 1 diabetes (T1D), T2D, gestational
diabetes and a number of less prevalent subtypes, with disease
susceptibility influenced by both genetic background and
environmental factors (24,25).
The global prevalence continues to escalate, with projections
indicating that ~783 million individuals will be affected by 2045
(26). DbCM, nephropathy and
retinopathy remain the principal causes of morbidity and mortality.
Persistent hyperglycaemia promotes chronic low-grade inflammation
and oxidative stress (27), with
data indicating that protein PTMs serve pivotal roles in regulating
the immune and metabolic circuits underlying diabetic complications
(28). As gestational diabetes and
certain uncommon variants often progress to T1D or T2D, they are
not further addressed in the present review.
T1D: Metabolic mismatch and
succinyl-CoA:3-ketoacid CoA transferase (SCOT) inhibition
T1D is an autoimmune disorder characterized by
progressive immune-mediated destruction of pancreatic β cells,
culminating in absolute insulin deficiency (29,30).
In the absence of endogenous insulin, glucose homeostasis is
markedly disrupted, resulting in chronic hyperglycaemia and
lifelong reliance on exogenous insulin therapy. The metabolic
consequences of T1D extend beyond impaired glucose regulation,
affecting lipid and amino acid metabolism, and increasing
vulnerability to systemic complications, including both
macrovascular and microvascular damage (31–33).
Although β-cell-directed autoimmunity defines T1D, the broader
metabolic alterations across peripheral tissues remain
insufficiently characterized.
Direct evidence associating lysine succinylation
with T1D is currently limited; however, findings from experimental
models have implicated the dysfunction of SCOT as a potential
mechanistic association (34).
SCOT, a central enzyme in ketone body utilization and a key
consumer of succinyl-CoA, exhibits reduced activity in T1D models
due to inhibitory tyrosine nitration and decreased 3-oxoacid
CoA-transferase 1 expression. This impairment is particularly
detrimental within the metabolic context of T1D. In contrast to
T2D, absolute insulin deficiency in T1D drives unrestrained
lipolysis and excessive hepatic ketogenesis. The resulting surge in
ketone body production necessitates efficient SCOT-mediated
turnover; however, suppression of SCOT activity compromises this
process. Such metabolic disequilibrium may limit succinyl-CoA
utilization, leading to its accumulation within the mitochondrial
matrix, potentially promoting pathological hypersuccinylation in
susceptible tissues, including the myocardium (35,36).
Emerging data suggested that mitochondrial
succinyl-CoA accumulation is not simply a metabolic by-product, but
may foster broader cellular dysfunction (37,38).
Beyond SCOT-dependent mechanisms, the characteristics of the T1D
inflammatory environment may directly affect succinylation
dynamics, specifically the balance between spontaneous modification
and SIRT5-mediated desuccinylation. β-cell destruction is
orchestrated by pro-inflammatory cytokines, including IL-1β and
IFN-γ, which promote pronounced oxidative stress and mitochondrial
injury in pancreatic islets (39,40).
Although direct validation in T1D remains limited, evidence from
related metabolic disorders, such as non-alcoholic fatty liver
disease (NAFLD) and metabolic cardiomyopathy, has indicated that
oxidative stress can attenuate SIRT5 activity, diminishing
de-succinylation capacity (41,42).
It may therefore be hypothesised that cytokine-driven redox
imbalance in T1D suppresses SIRT5 function, resulting in the
accumulation of hypersuccinylated mitochondrial proteins that
further compromise β-cell bioenergetics and viability.
In addition, protein succinylation may contribute to
the immunopathogenesis of T1D. PTMs can generate neoepitopes that
initiate or amplify autoimmune responses (43,44).
As aforementioned, succinylation induces notable structural and
physicochemical alterations in target proteins. These modifications
may theoretically generate neoantigens or modify antigen processing
and presentation by major histocompatibility complex molecules in β
cells. In comparison, analogous mechanisms have been established
for other PTMs, such as citrullination in autoimmune diseases
(45); whether aberrant
succinylation functions as a source of neoantigens in T1D remains
an important and unresolved question requiring further
investigation.
T2D: Insulin resistance and β-cell
dysfunction
T2D arises from the interplay between peripheral
insulin resistance and an insufficient compensatory increase in
insulin secretion (46,47). In the early stages, pancreatic β
cells increase insulin secretion to compensate for reduced insulin
sensitivity. This adaptive response depends on precise regulation
of glycolytic flux, TCA cycle activity and lipid metabolism in
response to glucose, incretin signalling and pharmacological
stimuli, such as metformin and sodium-glucose cotransporter-2
inhibitors (48). However,
persistent exposure to hyperglycaemia and elevated lipid levels
imposes chronic metabolic stress that progressively exhausts β-cell
reserve. The ensuing dysfunction is characterized by a depletion of
TCA cycle intermediates, including citrate, malate and
oxaloacetate, increased oxidative stress and impaired insulin
secretory capacity (49). Obesity
notably increases the risk of T2D by exacerbating insulin
resistance and sustaining low-grade inflammation, which further
affects metabolic homeostasis (50). Although classical disruptions in
TCA cycle dynamics and fatty acid oxidation (FAO), including
metabolic inflexibility and incomplete lipid oxidation, are well
documented (51,52), the contribution of emerging PTMs,
particularly protein succinylation, to β-cell stress adaptation
remains incompletely understood and merits further
investigation.
Experimental data have indicated that aberrant
succinylation, specifically pathological hyper-succinylation,
directly impairs β-cell function in T2D, a central event in disease
progression. In Goto-Kakizaki rats, a widely used T2D model, both
expression and activity of SCOT are diminished (53). Reduced SCOT function leads to
intracellular accumulation of succinyl-CoA, which adversely affects
β-cell function by impairing glucose-stimulated insulin secretion
(54). Targeted silencing of SCOT
in insulinoma cells has been shown to markedly attenuate
glucose-stimulated insulin secretion, underscoring its key
metabolic role. In addition, chronic glucolipotoxicity, a defining
feature of T2D, induces β-cell apoptosis (55,56).
Furthermore, experimental overexpression of SIRT5 counteracts this
process by upregulating mitochondrial anti-apoptotic proteins Bcl-2
and Bcl-XL, preventing cytochrome c release and inhibiting
downstream caspase-3/7 activation, preserving β-cell viability and
function (57). These findings
support the premise that excessive succinylation is detrimental to
β cells under diabetic stress conditions.
Beyond β-cell failure, dysregulated succinylation
also impairs insulin signalling in peripheral tissues. In hepatic
tissue from diet-induced obesity and diabetes models, abnormal
succinylation has been identified on numerous proteins integral to
insulin signalling pathways, including insulin receptor substrate
1. Proteomics analyses have mapped nine atypical succinylation
sites across 268 metabolically relevant hepatic proteins (58). Such modifications may disrupt
downstream signalling from the insulin receptor, reducing cellular
insulin responsiveness and aggravating systemic insulin resistance.
Pharmacological inhibition of SCOT using diphenylbutylpiperidines
(DPBPs) has been shown to improve glycaemic control in experimental
animal obesity models, specifically diet-induced obese mice.
However, their clinical utility is limited by marked off-target
effects. Namely, as potent dopamine D2 receptor antagonists, DPBPs
have been associated with extrapyramidal symptoms, sedation and
cardiac adverse events, including QT interval prolongation
(59,60). Despite these limitations, these
observations underscore the therapeutic potential of targeting the
succinyl-CoA metabolic axis in T2D (60).
At the molecular level, protein succinylation is
primarily determined by the balance between intracellular
succinyl-CoA availability and desuccinylase activity. In
T2D-associated metabolic stress, increased succinyl-CoA production
can overwhelm the regulatory capacity of SIRT5, leading to
widespread hypersuccinylation and subsequent metabolic impairment
(61). Enhancement of SIRT5
activity has been shown to ameliorate select metabolic
abnormalities, such as β-cell apoptosis, hepatic insulin resistance
and cardiac lipotoxicity.
Metabolic memory: A bridge to
epigenetic mechanisms
Subsequent to the acute enzymatic disturbances in
metabolic pathways, the present review will further discuss more
persistent epigenetic alterations that sustain cellular
dysfunction. The concept of ‘metabolic memory’ refers to the
continued progression of vascular complications even after
glycaemic control has been restored. Although this effect has
classically been attributed to oxidative stress and the
accumulation of advanced glycation end-products, emerging evidence
has identified aberrant lysine succinylation as a stable molecular
imprint capable of perpetuating dysfunction beyond the initial
hyperglycaemic exposure (62–64).
In contrast to transient phosphorylation,
succinylation produces a marked charge reversal and steric
constraint, changes that can stabilize metabolic enzymes in
maladaptive conformations (65).
During hyperglycaemia, excessive succinyl-CoA production exceeds
the desuccinylation capacity of SIRT5. Restoration of SIRT5
activity is often hindered not only by substrate overload but also
by NAD+ depletion and oxidative stress-induced
transcriptional repression (66).
This imbalance promotes sustained hypersuccinylation of
mitochondrial proteins, such as the PDC and carnitine
palmitoyltransferase 2 (CPT2), and contributes to cytosolic
succinate accumulation (16).
Elevated succinate functions as an oncometabolite that inhibits
prolyl hydroxylases, leading to stabilization of hypoxia-inducible
factor-1α and maintenance of a ‘pseudo-hypoxic’ state that persists
beyond the original metabolic insult (67,68).
Beyond its effects on metabolic enzymes,
succinylation associates altered metabolic flux with long-term
transcriptional reprogramming. Rather than requiring direct
transport of succinyl-CoA into the nucleus, recruitment of the
α-KGDH complex enables localized nuclear generation of succinyl-CoA
(8). This compartmentalized
metabolite pool supplies substrate for histone succinylation. By
altering chromatin structure, these epigenetic modifications
promote an open configuration at pro-inflammatory gene loci
(69). Therefore, prior metabolic
stress becomes embedded within the epigenome, sustaining pathogenic
transcriptional programs despite subsequent therapeutic
normalization of glycemia.
Pathological mechanisms in diabetic
complications
Obesity: Impaired thermogenesis and
macrophage polarization
Obesity is a complex metabolic disorder defined by
excessive adipose tissue accumulation resulting from chronic
positive energy balance (70). It
constitutes a major global health challenge and is a primary risk
factor for T2D, cardiovascular disease and NAFLD (71). Obesity induces marked metabolic
reprogramming in tissues, including adipose depots in the liver,
promoting hepatic steatosis and triggering endoplasmic reticulum
stress in adipocytes (72). These
disturbances contribute to systemic insulin resistance and broader
metabolic dysfunction (73–75).
Within this altered metabolic environment, widespread shifts in
PTMs occur, with protein succinylation emerging as a key regulatory
mechanism.
Brown adipose tissue (BAT), the principal site of
non-shivering thermogenesis, is particularly dependent on tightly
regulated succinylation dynamics (76,77).
Thermogenic capacity in BAT is mediated by UCP1, the stability and
activity of which are influenced by succinylation status.
SIRT5-mediated desuccinylation of UCP1 (notably, at residues K56
and K151) supports protein stability and thermogenic efficiency.
Under obesogenic conditions, reduced SIRT5 activity increases UCP1
succinylation, impairing BAT thermogenesis. The consequent decline
in energy expenditure promotes weight gain, highlighting the
therapeutic relevance of targeting the SIRT5/UCP1 regulatory axis
to restore metabolic flexibility in BAT.
In addition, a parallel succinyl-CoA-dependent
pathway operates in adipose tissue macrophages (ATMs), whereby
metabolic signalling directly promotes inflammatory activation. In
this setting, the glutaminolysis/AMP-activated protein kinase
(AMPK)/succinyl-CoA synthetase ADP-forming subunit β (SUCLA2) axis
drives pro-inflammatory responses. ATP generated through
glutaminolysis suppresses AMPK activity, limiting
phosphorylation-dependent inhibition of SUCLA2. Enhanced SUCLA2
activity further increases intracellular levels of succinyl-CoA and
succinate, stimulating excessive IL-1β production. This cascade
promotes inflammatory polarization of ATMs, suppresses thermogenic
function, reduces energy expenditure and exacerbates obesity
(78).
The metabolic consequences of increased succinyl-CoA
production are context dependent. When supplied as alternative
metabolic fuels, substrates that elevate succinyl-CoA may exert
beneficial effects. Dodecanedioic acid (DC12), a medium-chain
dicarboxylic acid, undergoes β-oxidation in tissues expressing the
acyl-CoA oxidase 1 isoform, generating succinyl-CoA that enters the
TCA cycle. Administration of DC12 has been shown to increase
whole-body energy expenditure, decrease adiposity, reduce hepatic
lipid accumulation and improve glucose tolerance, mitigating
obesity by enhancing mitochondrial substrate cycling (79).
The liver further illustrates the context-specific
nature of succinyl-CoA metabolism. Hepatic steatosis, a common
obesity-associated complication (80), has been shown to be notably
attenuated in ob/ob mice when hepatic SIRT5 activity is augmented
via genetic overexpression (18).
These findings underscore how the metabolic impact of SIRT5 is
highly dependent on tissue type and pathological context, rather
than uniformly protective or deleterious (Fig. 3).
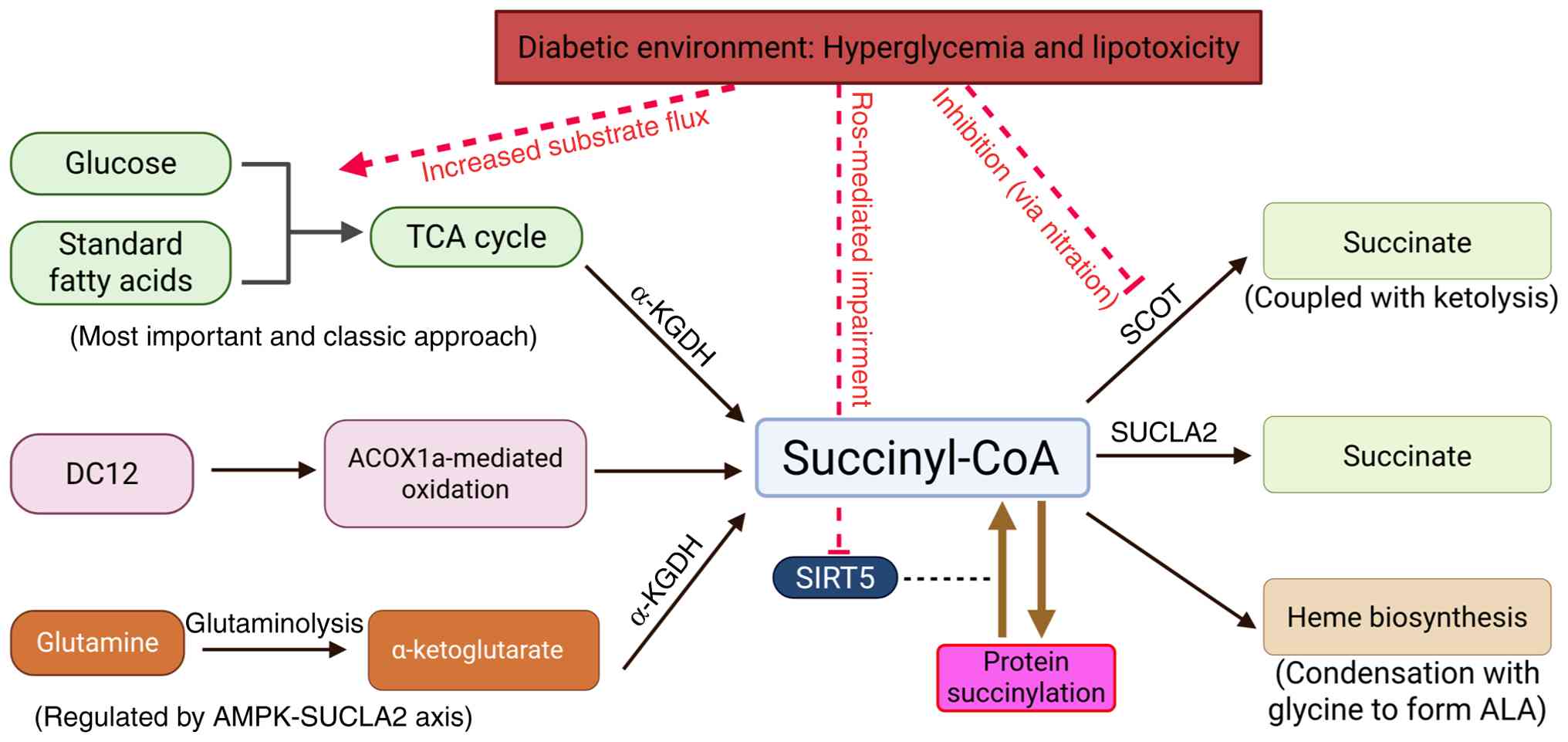 | Figure 3.Complexity of succinyl-CoA metabolism
and its dysregulation in the diabetic environment. Created with
BioRender.com. TCA, tricarboxylic acid; α-KGDH, α-ketoglutarate
dehydrogenase; DC12, dodecanedioic acid; ACOX1a, acyl-CoA oxidase
1; AMPK, AMP-activated protein kinase; SUCLA2, succinyl-CoA
synthetase ADP-forming subunit β; ROS, reactive oxygen species;
SCOT, succinyl-CoA:3-ketoacid CoA transferase; SIRT5, sirtuin 5;
ALA, δ-aminolevulinic acid. |
DbCM: Mitochondrial inflexibility and
lipotoxicity
Context-dependent metabolic consequences of
succinylation extend beyond adipose tissue to the cardiovascular
system. DbCM is a distinct myocardial disorder characterized by
structural and functional abnormalities in individuals with
diabetes, occurring independently of hypertension, coronary artery
disease or notable valvular pathology (81). Its development is largely driven by
metabolic disturbances, including impaired metabolic flexibility,
mitochondrial dysfunction and lipotoxicity, that culminate in
myocardial fibrosis, hypertrophy, and progressive impairment of
both diastolic and systolic function. Key pathogenic mechanisms
include altered substrate utilization, mitochondrial dysfunction,
oxidative stress and diminished ATP generation (82,83).
Ketone bodies represent an efficient energy
substrate for the heart, particularly under conditions of metabolic
stress. In the diabetic myocardium, tyrosine nitration-mediated
inhibition of SCOT reduces its enzymatic activity by 24–39%
(35). Impaired SCOT function
limits the conversion of ketone bodies into acetyl-CoA, aggravating
the energetic deficit characteristic of DbCM. As aforementioned,
although pharmacological inhibition of SCOT may transiently enhance
glucose tolerance, its potential cardiotoxic effects highlight the
need to maintain balanced ketone metabolism in the diabetic heart
(84).
The PDC, a key association between glycolysis and
the TCA cycle, represents an additional metabolic control point
disrupted in DbCM (85). Sustained
exposure of cardiomyocytes to hyperglycaemia and elevated fatty
acids increases intracellular succinyl-CoA concentrations,
promoting succinylation of the pyruvate dehydrogenase E1 subunit
α-1. This modification suppresses PDC activity, restricting
pyruvate flux into the TCA cycle and further compromising metabolic
flexibility, the capacity of the heart to alternate between glucose
and lipid substrates. Over time, this metabolic inflexibility
contributes to myocardial hypertrophy and diastolic dysfunction,
establishing a self-perpetuating cycle of metabolic stress,
excessive succinylation, PDC inhibition and progressive energetic
decline (17).
Regulatory control by SIRT5 is similarly disrupted
in DbCM. Mitochondrial impairment and oxidative stress can
attenuate SIRT5 activity, reducing desuccinylation efficiency and
exacerbating mitochondrial metabolic failure (12). SIRT5 also directly regulates CPT2,
a key enzyme in mitochondrial fatty acid β-oxidation (86). Deficiency of SIRT5 markedly
increases succinylation of CPT2 at lysine 424, suppressing its
enzymatic function and impairing FAO. Accumulation of long-chain
acylcarnitines subsequently promotes lipotoxic damage and worsens
cardiac dysfunction. By comparison, experimental SIRT5
overexpression has been shown to preserve CPT2 activity through
desuccinylation, enhancing FAO efficiency and mitigating lipotoxic
stress in the diabetic myocardium (61).
DKD: Epigenetic dysregulation and
renal cell injury
Similar to the myocardium, the kidneys are highly
susceptible to metabolic injury driven by aberrant succinylation,
particularly through epigenetic and microvascular pathways. DKD,
also termed diabetic nephropathy, is among the most prevalent
microvascular complications of diabetes and remains the leading
cause of chronic kidney disease and end-stage renal failure
worldwide, affecting ~50% of patients with T2D globally (87). It is characterized by progressive
damage to both glomerular filtration structures and renal tubular
compartments (88). Persistent
hyperglycaemia, frequently compounded by hypertension, initiates
structural and functional deterioration that culminates in
albuminuria, a clinical hallmark of DKD (89). As injury advances, the glomerular
filtration rate progressively declines, impairing the capacity of
the kidneys to eliminate metabolic waste, often leading to
irreversible renal failure (90).
Epigenetic dysregulation has emerged as a central
mechanism underlying the aberrant transcriptional programs observed
in DKD (91,92). Expression of the histone
succinyltransferase KAT2A is notably increased in affected kidneys.
KAT2A mediates succinylation at the spermidine/spermine
N1-acetyltransferase family member 2 (SAT2) promoter, enhancing its
transcriptional activation. Upregulated SAT2 expression exacerbates
renal injury by promoting iron-dependent ferroptosis and amplifying
inflammatory signalling, including increased production of IL-1β,
IL-6 and TNF-α (93,94). These processes accelerate
glomerular and tubular injury, positioning the KAT2A/H3K79
succinylation/SAT2 axis as a pivotal epigenetic driver of DKD
progression (95).
Succinylation dynamics similarly modulate
mitochondrial integrity in renal cells. Astragaloside IV, a
bioactive natural compound, confers renoprotective effects in part
through activation of CPT1A. Enhanced CPT1A activity restores
appropriate succinylation of 17β-hydroxysteroid dehydrogenase 10
(HSD17B10) at lysine 99 (96).
Properly succinylated HSD17B10 maintains the structural stability
and enzymatic function of mitochondrial ribonuclease P, a key
regulator of mitochondrial RNA processing (97), attenuating oxidative stress and
reducing tubular epithelial injury.
Podocyte dysfunction represents another hallmark of
DKD and desuccinylation appears protective in this setting. The
flavonoid quercetin-4′-O-β-D-glucopyranoside upregulates SIRT5
expression in podocytes (98).
SIRT5-mediated desuccinylation of NIMA related kinase 7 (NEK7), a
kinase key in activating the NLR family pyrin domain containing 3
(NLRP3) inflammasome, disrupts the NEK7/NLRP3 interaction (99). This interference suppresses
podocyte pyroptosis, reduces oxidative stress and helps preserve
podocyte structure and function (100).
However, SIRT5 activation does not uniformly confer
benefit. In mesangial cells under hyperglycaemic conditions, SIRT5
expression is elevated, promoting the desuccinylation of the tumour
suppressor p53. Although total p53 protein levels remain unchanged,
desuccinylation attenuates its transcriptional activity (101,102). Diminished p53 function reduces
expression of downstream targets such as p21 and 14-3-3σ,
facilitating mesangial cell proliferation and excessive
extracellular matrix deposition, including collagen and laminin
(103). These alterations
directly contribute to glomerulosclerosis and the progression of
DKD.
DR: Neurodegeneration and autophagic
flux
Although SIRT5 activity may aggravate pathological
remodelling in renal mesangial cells, its function in the retina
appears consistently protective. DR develops as chronic
hyperglycaemia progressively injures the retinal microvasculature
within the light-sensitive neural tissue at the posterior segment
of the eye. Structural weakening of retinal capillaries increases
vascular permeability, permitting leakage of plasma constituents or
blood into the retinal parenchyma. The resulting oedema and
microvascular damage contribute to progressive visual impairment
(104,105).
Aberrant protein succinylation has emerged as an
early contributor to neurodegenerative processes in DR. Under
hyperglycaemic conditions, increased succinylation of optineurin
(OPTN) at lysine 108 has been observed in experimental retinal
models. This modification is associated with disrupted autophagic
flux in retinal ganglion cells, compromising cellular homeostasis.
SIRT5-mediated desuccinylation of OPTN reverses this effect,
restores autophagic function and confers neuroprotection. These
findings delineate the SIRT5/OPTN axis as a central regulatory
mechanism and a promising therapeutic target in early-stage DR
(106). A comprehensive overview
of organ-specific regulatory mechanisms and their pathological
consequences is provided in Table
I.
 | Table I.Summary of dysregulated succinylation
targets and pathological consequences in diabetes and diabetic
complications. |
Table I.
Summary of dysregulated succinylation
targets and pathological consequences in diabetes and diabetic
complications.
| Target | Key substrate | Key regulator | Effect of aberrant
succinylation/desuccinylation | Pathological
consequences | Potential
therapeutic implications | (Refs.) |
|---|
| BAT | UCP1 (K56 and
K151) | SIRT5 decrease | Increased
succinylation reduces UCP1 stability and activity | Impaired
non-shivering thermogenesis; accelerated weight gain | Increase SIRT5
activity | (16) |
| Heart
(cardiomyocytes) | PDC (E1α
subunit) |
| Hypersuccinylation
suppresses the activity of PDC | Metabolic
inflexibility; myocardial hypertrophy | Restore
succinyl-CoA balance | (17) |
| Liver | IRS-1 | SIRT5 | Hypersuccinylation
disrupts downstream insulin signalling | Systemic insulin
resistance; hepatic steatosis | Increase SIRT5
activity | (18,58) |
| Pancreatic β
cells | Unspecified
mitochondrial proteins | SCOT decrease;
SIRT5 | Hypersuccinylation
increases cytochrome c release and caspase activation | β-cell apoptosis
and impaired insulin secretion | Increase SIRT5
activity; prevent SCOT inhibition | (53–58) |
| Heart
(cardiomyocytes) | CPT2 (K424) | SIRT5 decrease | Hypersuccinylation
inhibits the activity of CPT2 | Impaired FAO;
lipotoxic injury | Increase SIRT5
activity | (61,86) |
| ATMs | SUCLA2 (upstream
pathway) | AMPK-SUCLA2
axis | Elevated
succinyl-CoA results in excessive IL-1β production | Pro-inflammatory
ATM polarization; aggravated obesity | Modulate
glutaminolysis/AMPK/SUCLA2 axis | (78) |
| Kidney (epigenetic
level) | H3K79 | KAT2A increase | H3K79
hypersuccinylation enforces open chromatin at the SAT2
promoter | SAT2-driven
ferroptosis and inflammation (IL-1β, IL-6 and TNF-α) | Inhibit
KAT2A/epigenetic modulation | (93–95) |
| Kidney (tubular
cells) | HSD17B10 (K99) | CPT1A (upstream
activator) | Loss of proper
succinylation destabilizes mitochondrial RNase P | Oxidative stress;
tubular epithelial injury | Activate CPT1A to
reinstate proper succinylation | (96,97) |
| Kidney
(podocytes) | NEK7 | SIRT5 decrease | Hypersuccinylation
increases NEK7 interaction with NLRP3 | Inflammasome
activation; podocyte pyroptosis | Improve SIRT5
activity | (98–100) |
| Kidney (mesangial
cells) | p53 (K120) | SIRT5 increase
(paradoxical) | Excessive
desuccinylation dampens p53 transcriptional activity (p21,
14-3-3σ) | Mesangial cell
proliferation; extracellular matrix deposition | Organ-targeted
SIRT5 inhibition | (101–103) |
| RGCs | OPTN (K108) | SIRT5 decrease | Hypersuccinylation
impairs autophagic flux | RGC
neurodegeneration | Improve SIRT5
activity | (106) |
Future perspectives
Despite marked advances in characterizing the
succinylation landscape, a number of major obstacles remain before
these insights can be translated into clinical practice. Current
detection approaches rely predominantly on pan-succinyllysine
antibodies, which may exhibit cross-reactivity with other acyl
modifications (such as malonylation and glutarylation) (65,107). Furthermore, conventional mass
spectrometry workflows often lack the resolution to determine
absolute site-specific stoichiometry (108). The development of highly
selective chemical probes and improved quantitative proteomic
methodologies will therefore be important in discriminating
biologically meaningful, high-occupancy succinylation events from
low-abundance background modifications.
In addition, a static catalogue of succinylated
proteins does not adequately capture the dynamic nature of
metabolic disease. Future studies should aim to integrate
acyl-proteomics with metabolomics and metabolic flux analysis
within a multi-omics framework. Given that intracellular
succinyl-CoA concentrations fluctuate rapidly in response to
nutrient status, combining these datasets would enable direct
associations to be made between metabolic flux and the kinetics of
protein succinylation. Such an approach is key in defining causal
associations between metabolic overload and downstream signalling
perturbations in diabetes.
Therapeutic targeting of this axis similarly demands
precision. As aforementioned, SIRT5 exerts tissue-specific effects
that frequently oppose one another. Systemic activation of SIRT5 is
associated with adverse renal outcomes, including the promotion of
glomerular fibrosis. Accordingly, organ-selective delivery
strategies are imperative. Nanoparticle-based encapsulation of
SIRT5 modulators or antibody-drug conjugates directed toward
cell-specific surface markers may provide the spatial selectivity
required for safe intervention. Direct manipulation of the
intracellular succinyl-CoA pool is equally challenging, given its
indispensable roles in the TCA cycle and haem biosynthesis
(109). Future pharmacological
strategies must therefore recalibrate the succinyl-CoA/SIRT5
equilibrium without compromising mitochondrial bioenergetics or
inducing systemic metabolic toxicity.
At present, the pharmacological repertoire targeting
protein succinylation (Table II)
remains limited. Numerous SIRT5 inhibitors have been described
(such as the non-selective compound suramin and specific
mechanism-based thiosuccinyl peptides), yet the majority are
peptide-based or non-selective compounds with suboptimal
bioavailability or poor cellular permeability (110,111). There is a notable scarcity of
specific small-molecule SIRT5 activators. Efforts to enhance SIRT5
activity largely depend on specific NAD+ precursors
(such as nicotinamide mononucleotide and nicotinamide riboside)
that broadly stimulate SIRT function, lacking the specificity
needed to minimize off-target effects (112). To address this limitation, a
number of emerging specific small molecules, such as the selective
SIRT5 activator MC3138, have been developed to provide targeted
modulation (113,114). Despite these recent advancements,
caution is warranted. The majority of available modulators have not
progressed to clinical validation and interpreting reductions in
succinylation as an unequivocal therapeutic benefit without a
comprehensive safety assessment may be misleading. Emerging
strategies such as Proteolysis Targeting Chimeras offer an
alternative approach by selectively degrading hypersuccinylated
substrates, potentially circumventing the challenges of direct
enzymatic modulation (115,116).
| Table II.Overview of agents and compounds
affecting protein succinylation and their physiological
outcomes. |
Table II.
Overview of agents and compounds
affecting protein succinylation and their physiological
outcomes.
| Agent/compound | Mechanism of
action | Target
protein/pathway | Effect on
succinylation | Functional
outcome | (Refs.) |
|---|
| Pimozide | Inhibits SCOT
activity | SCOT | Increases (through
accumulation of succinyl-CoA) | Improves glycaemic
control in obesity models (discontinued due to its cardiac
toxicity) | (59–61) |
| DC12 | Increases
succinyl-CoA flux through | TCA cycle flux
β-oxidation | Increases (by
elevating succinyl-CoA production) | Increases energy
expenditure; reduces adiposity and hepatic lipids | (80) |
| Astragaloside
IV | Activates
CPT1A | HSD17B10 (through
CPT1A activation) | Restores
physiological levels (at lysine 99) | Mitigates oxidative
stress; renoprotective in DKD | (97) |
| QODG | Upregulates
expression of SIRT5 | NEK7 | Decreases (promotes
desuccinylation) | Inhibits NLRP3
inflammasome; reduces podocyte pyroptosis | (99) |
|
Suramin/thiosuccinyl peptides | Directly inhibits
SIRT5 enzymatic activity | SIRT5 | Increases (inhibits
desuccinylation) | Used primarily as
research tools; clinical utility limited by off-target effects or
poor bioavailability | (111, 112) |
| NAD+
precursors | Increases the
enzymatic activity of SIRT5 | Global SIRT
targets | Decreases (broad
desuccinylation) | Improves
mitochondrial function (non-specific, theoretical utility) | (113) |
| MC3138 | Directly activates
SIRT5 enzymatic activity | SIRT5 | Decreases (promotes
desuccinylation) | Emerging
SIRT5-selective small-molecule activator (preclinical
validation) | (114,115) |
| Lovastatin | Off-target
metabolic modulation |
Cytoskeleton-related proteins | Increases | Alters the
mechanical properties of breast cancer stem cells (potential
adverse effects) | (118) |
Unintended drug effects on the succinylome further
illustrate the complexity of therapeutic intervention. For example,
lovastatin, a widely prescribed lipid-lowering agent, has been
reported to induce succinylation of cytoskeleton-associated
proteins in breast cancer stem cells, altering their mechanical
properties (117). This
observation underscores the potential for pleiotropic and
off-target modulation of succinylation in non-intended tissues.
Accordingly, rigorous screening for unintended succinylation
changes should become an integral component of future drug
development pipelines.
Conclusion
Protein succinylation has developed from its initial
characterization as a metabolic intermediate to emerge as a key
regulator of diabetic pathology, orchestrating the important
interface between mitochondrial bioenergetics and cellular
signalling. Evidence summarized in the present review determined
that the breakdown of the succinyl-CoA/SIRT5 regulatory axis is a
primary driver of metabolic inflexibility and tissue injury.
However, the dichotomy of SIRT5 activity, indispensable in cardiac
homeostasis yet paradoxically pathogenic in renal mesangial cells,
challenges the viability of systemic enzymatic modulation.
Ultimately, understanding this context-dependent complexity is not
just a theoretical imperative but the prerequisite for developing
safe, precision-based therapeutics for diabetes and its
complications.
Acknowledgements
Not applicable.
Funding
The present review was supported by the President Foundation of
Nanfang Hospital, Southern Medical University (grant no. 2024A036),
the Medical Scientific Research Foundation of Guangdong (grant no.
B2025509) and the National College Students' Innovation and
Entrepreneurship Training Program, General Innovation Training
Project (grant no. 202512121008).
Availability of data and materials
Not applicable.
Authors' contributions
YX, FL, BL and QY contributed to the conception and
design of the present review. YX drafted the manuscript, and
participated in literature collation and manuscript editing. FL and
BL conceptualized the present review and contributed to the
writing. QY reviewed and revised the manuscript. All authors
commented on previous versions of the manuscript. Data
authentication is not applicable. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Authors' information
ORCID: Yeteng Xiong, 0009-0004-1759-9862.
References
|
1
|
Ling C and Rönn T: Epigenetics in human
obesity and type 2 diabetes. Cell Metab. 29:1028–1044. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li Y: Modern epigenetics methods in
biological research. Methods. 187:104–113. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li D, Zhang L, He Y, Zhou T, Cheng X,
Huang W and Xu Y: Novel histone post-translational modifications in
diabetes and complications of diabetes: The underlying mechanisms
and implications. Biomed Pharmacother. 156:1139842022. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shvedunova M and Akhtar A: Modulation of
cellular processes by histone and non-histone protein acetylation.
Nat Rev Mol Cell Biol. 23:329–349. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zorro Shahidian L, Haas M, Le Gras S,
Nitsch S, Mourão A, Geerlof A, Margueron R, Michaelis J, Daujat S
and Schneider R: Succinylation of H3K122 destabilizes nucleosomes
and enhances transcription. EMBO Rep. 22:e510092021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yazıcı E and McIntyre J: The complex
network of p300/CBP regulation: Interactions, posttranslational
modifications, and therapeutic implications. J Biol Chem.
301:1107152025. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hou X, Chen Y, Li X, Gu X, Dong W, Shi J
and Ji S: Protein succinylation: Regulating metabolism and beyond.
Front Nutr. 11:13360572024. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang Y, Guo YR, Liu K, Yin Z, Liu R, Xia
Y, Tan L, Yang P, Lee JH, Li XJ, et al: KAT2A coupled with the
α-KGDH complex acts as a histone H3 succinyltransferase. Nature.
552:273–277. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Simic Z, Weiwad M, Schierhorn A, Steegborn
C and Schutkowski M: The ɛ-amino group of protein lysine residues
is highly susceptible to nonenzymatic acylation by several
physiological Acyl-CoA thioesters. Chembiochem. 16:2337–2347. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wagner GR and Payne RM: Widespread and
enzyme-independent Nε-acetylation and Nε-succinylation of proteins
in the chemical conditions of the mitochondrial matrix. J Biol
Chem. 288:29036–29045. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang Y, Chen H and Zha X: Overview of
SIRT5 as a potential therapeutic target: Structure, function and
inhibitors. Eur J Med Chem. 236:1143632022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Guo L, Du Y, Li H, He T, Yao L, Yang G and
Yang X: Metabolites-mediated posttranslational modifications in
cardiac metabolic remodeling: Implications for disease pathology
and therapeutic potential. Metabolism. 165:1561442025. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shen R, Ruan H, Lin S, Liu B, Song H, Li L
and Ma T: Lysine succinylation, the metabolic bridge between cancer
and immunity. Genes Dis. 10:2470–2478. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang R, Fang J, Xie X, Carrico C, Meyer
JG, Wei L, Bons J, Rose J, Riley R, Kwok R, et al: Regulation of
urea cycle by reversible high-stoichiometry lysine succinylation.
Nat Metab. 6:550–566. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu Y, Ye X, Zhou Y, Cao X, Peng S, Peng Y,
Zhang X, Sun Y, Jiang H, Huang W, et al: Sodium butyrate activates
HMGCS2 to promote ketone body production through SIRT5-mediated
desuccinylation. Front Med. 17:339–351. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang G, Meyer JG, Cai W, Softic S, Li ME,
Verdin E, Newgard C, Schilling B and Kahn CR: Regulation of UCP1
and mitochondrial metabolism in brown adipose tissue by reversible
succinylation. Mol Cell. 74:844–857.e7. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Park S, Jeon JH, Min BK, Ha CM, Thoudam T,
Park BY and Lee IK: Role of the pyruvate dehydrogenase complex in
metabolic remodeling: Differential pyruvate dehydrogenase complex
functions in metabolism. Diabetes Metab J. 42:270–281. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Du Y, Hu H, Qu S, Wang J, Hua C, Zhang J,
Wei P, He X, Hao J, Liu P, et al: SIRT5 deacylates
metabolism-related proteins and attenuates hepatic steatosis in
ob/ob mice. EBioMedicine. 36:347–357. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bentley NL, Fiveash CE, Osborne B, Quek
LE, Ogura M, Inagaki N, Cooney GJ, Polly P, Montgomery MK and
Turner N: Protein hypoacylation induced by Sirt5 overexpression has
minimal metabolic effect in mice. Biochem Biophys Res Commun.
503:1349–1355. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang T, Qi F, Guo F, Shao M, Song Y, Ren
G, Linlin Z, Qin G and Zhao Y: An update on chronic complications
of diabetes mellitus: From molecular mechanisms to therapeutic
strategies with a focus on metabolic memory. Mol Med. 30:712024.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Berezin A: Metabolic memory phenomenon in
diabetes mellitus: Achieving and perspectives. Diabetes Metab
Syndr. 10 (Suppl 2):S176–S183. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mu F, Zhang H, Gong R, Lin R, Zhao M, Tao
X, Shang L, Xi M, Zhao J and Wang J: Lysine succinylation as a
metabolic switch in cardiovascular diseases: Mechanistic insights
and therapeutic perspectives. Redox Biol. 88:1039322025. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
He M, Wang Z, Miao Z, Zhao Y, Wei L, Zhang
L, Yin R, Wang Y and Yang L: Post-translational modifications in
diabetic kidney disease (Review). Int J Mol Med. 57:882026.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
American Diabetes Association Professional
Practice Committee, . 2. Diagnosis and classification of diabetes:
Standards of care in diabetes-2025. Diabetes Care. 48 (Suppl
1):S27–S49. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Diabetes Canada Clinical Practice
Guidelines Expert Committee, . Punthakee Z, Goldenberg R and Katz
P: Definition, classification and diagnosis of diabetes,
prediabetes and metabolic syndrome. Can J Diabetes. 42 (Suppl
1):S10–S15. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sun H, Saeedi P, Karuranga S, Pinkepank M,
Ogurtsova K, Duncan BB, Stein C, Basit A, Chan JCN, Mbanya JC, et
al: IDF diabetes atlas: Global, regional and country-level diabetes
prevalence estimates for 2021 and projections for 2045. Diabetes
Res Clin Pract. 183:1091192022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dionisio LM, Zheng Y and Cancelas JA:
Redox control in platelet activity and therapy. Antioxidants
(Basel). 14:12862025. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sharma C, Hamza A, Boyle E, Donu D and Cen
Y: Post-translational modifications and diabetes. Biomolecules.
14:3102024. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kawasaki E: Anti-islet autoantibodies in
type 1 diabetes. Int J Mol Sci. 24:100122023. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mameli C, Triolo TM, Chiarelli F, Rewers
M, Zuccotti G and Simmons KM: Lessons and gaps in the prediction
and prevention of type 1 diabetes. Pharmacol Res. 193:1067922023.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Arneth B, Arneth R and Shams M:
Metabolomics of type 1 and type 2 diabetes. Int J Mol Sci.
20:24672019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Forbes JM and Cooper ME: Mechanisms of
diabetic complications. Physiol Rev. 93:137–188. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Barrett EJ, Liu Z, Khamaisi M, King GL,
Klein R, Klein BEK, Hughes TM, Craft S, Freedman BI, Bowden DW, et
al: Diabetic microvascular disease: An endocrine society scientific
statement. J Clin Endocrinol Metab. 102:4343–4410. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Brahma MK, Ha CM, Pepin ME, Mia S, Sun Z,
Chatham JC, Habegger KM, Abel ED, Paterson AJ, Young ME and Wende
AR: Increased glucose availability attenuates myocardial ketone
body utilization. J Am Heart Assoc. 9:e0130392020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Turko IV, Marcondes S and Murad F:
Diabetes-associated nitration of tyrosine and inactivation of
succinyl-CoA:3-oxoacid CoA-transferase. Am J Physiol Heart Circ
Physiol. 281:H2289–H2294. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cong W, Ma W, Zhao T, Zhu Z, Wang Y, Tan
Y, Li X, Jin L and Cai L: Metallothionein prevents diabetes-induced
cardiac pathological changes, likely via the inhibition of
succinyl-CoA:3-ketoacid coenzyme A transferase-1 nitration at
Trp(374). Am J Physiol Endocrinol Metab. 304:E826–E835. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lancaster MS, Kim B, Doud EH, Tate MD,
Sharify AD, Gao H, Chen D, Simpson E, Gillespie P, Chu X, et al:
Loss of succinyl-CoA synthetase in mouse forebrain results in
hypersuccinylation with perturbed neuronal transcription and
metabolism. Cell Rep. 42:1132412023. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sadhukhan S, Liu X, Ryu D, Nelson OD,
Stupinski JA, Li Z, Chen W, Zhang S, Weiss RS, Locasale JW, et al:
Metabolomics-assisted proteomics identifies succinylation and SIRT5
as important regulators of cardiac function. Proc Natl Acad Sci
USA. 113:4320–4325. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li Y, Sun F, Yue TT, Wang FX, Yang CL, Luo
JH, Rong SJ, Xiong F, Zhang S and Wang CY: Revisiting the
antigen-presenting function of β cells in T1D pathogenesis. Front
Immunol. 12:6907832021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Apostolopoulou M, Lambadiari V, Roden M
and Dimitriadis GD: Insulin resistance in type 1 diabetes:
Pathophysiological, clinical, and therapeutic relevance. Endocr
Rev. 46:317–348. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhang S, Wu Z, Shi L, Yan S, Huang Z, Lu
B, Wang Z and Ji L:
2,3,5,4′-tetrahydroxy-stilbene-2-O-β-D-glucoside ameliorates NAFLD
via attenuating hepatic steatosis through inhibiting mitochondrial
dysfunction dependent on SIRT5. Phytomedicine. 99:1539942022.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu B, Che W, Zheng C, Liu W, Wen J, Fu H,
Tang K, Zhang J and Xu Y: SIRT5: A safeguard against oxidative
stress-induced apoptosis in cardiomyocytes. Cell Physiol Biochem.
32:1050–1059. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Alhamar G, Vinci C, Franzese V, Tramontana
F, Le Goux N, Ludvigsson J, Nissim A and Strollo R: The role of
oxidative post-translational modifications in type 1 diabetes
pathogenesis. Front Immunol. 16:15374052025. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhai Y, Wu K, Lin Q, Cao Z, Jia Y and Zhu
P: Post-translational modified neoantigens in autoimmune diseases:
Challenges of immune tolerance. Adv Sci (Weinh). 12:e017662025.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wu CY, Yang HY and Lai JH:
Anti-citrullinated protein antibodies in patients with rheumatoid
arthritis: Biological effects and mechanisms of immunopathogenesis.
Int J Mol Sci. 21:40152020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Blair M: Diabetes mellitus review. Urol
Nurs. 36:27–36. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Henning RJ: Type-2 diabetes mellitus and
cardiovascular disease. Future Cardiol. 14:491–509. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Preda A, Montecucco F, Carbone F, Camici
GG, Lüscher TF, Kraler S and Liberale L: SGLT2 inhibitors: From
glucose-lowering to cardiovascular benefits. Cardiovasc Res.
120:443–460. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Newsholme P, Rowlands J, Rose'Meyer R and
Cruzat V: Metabolic adaptions/reprogramming in islet beta-cells in
response to physiological stimulators-what are the consequences.
Antioxidants (Basel). 11:1082022. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Rohm TV, Meier DT, Olefsky JM and Donath
MY: Inflammation in obesity, diabetes, and related disorders.
Immunity. 55:31–55. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Koves TR, Ussher JR, Noland RC, Slentz D,
Mosedale M, Ilkayeva O, Bain J, Stevens R, Dyck JR, Newgard CB, et
al: Mitochondrial overload and incomplete fatty acid oxidation
contribute to skeletal muscle insulin resistance. Cell Metab.
7:45–56. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Jia G, Hill MA and Sowers JR: Diabetic
cardiomyopathy: An update of mechanisms contributing to this
clinical entity. Circ Res. 122:624–638. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hasan NM, Longacre MJ, Seed Ahmed M,
Kendrick MA, Gu H, Ostenson CG, Fukao T and MacDonald MJ: Lower
succinyl-CoA:3-ketoacid-CoA transferase (SCOT) and ATP citrate
lyase in pancreatic islets of a rat model of type 2 diabetes:
Knockdown of SCOT inhibits insulin release in rat insulinoma cells.
Arch Biochem Biophys. 499:62–68. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
MacDonald MJ, Longacre MJ, Langberg EC,
Tibell A, Kendrick MA, Fukao T and Ostenson CG: Decreased levels of
metabolic enzymes in pancreatic islets of patients with type 2
diabetes. Diabetologia. 52:1087–1091. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wu T, Shao Y, Li X, Wu T, Yu L, Liang J,
Zhang Y, Wang J, Sun T, Zhu Y, et al: NR3C1/Glucocorticoid receptor
activation promotes pancreatic β-cell autophagy overload in
response to glucolipotoxicity. Autophagy. 19:2538–2557. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Lytrivi M, Castell AL, Poitout V and Cnop
M: Recent insights into mechanisms of β-cell lipo- and
glucolipotoxicity in type 2 diabetes. J Mol Biol. 432:1514–1534.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wang Y, Liu Q, Huan Y, Li R, Li C, Sun S,
Guo N, Yang M, Liu S and Shen Z: Sirtuin 5 overexpression
attenuates glucolipotoxicity-induced pancreatic β cells apoptosis
and dysfunction. Exp Cell Res. 371:205–213. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Xu Y, Shi Z and Bao L: An expanding
repertoire of protein acylations. Mol Cell Proteomics.
21:1001932022. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Giunta R, Cheli G, Spaiardi P, Russo G and
Masetto S: Pimozide increases a delayed rectifier K+
conductance in chicken embryo vestibular hair cells. Biomedicines.
11:4882023. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Tabatabaei Dakhili SA, Greenwell AA, Yang
K, Abou Farraj R, Saed CT, Gopal K, Chan JSF, Chahade JJ, Eaton F,
Lee C, et al: The antipsychotic dopamine 2 receptor antagonist
diphenylbutylpiperidines improve glycemia in experimental obesity
by inhibiting succinyl-CoA:3-ketoacid CoA transferase. Diabetes.
72:126–134. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Wu M, Tan J, Cao Z, Cai Y, Huang Z, Chen
Z, He W, Liu X, Jiang Y, Gao Q, et al: Sirt5 improves
cardiomyocytes fatty acid metabolism and ameliorates cardiac
lipotoxicity in diabetic cardiomyopathy via CPT2 de-succinylation.
Redox Biol. 73:1031842024. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Cannito S, Giardino I, D'Apolito M,
Ranaldi A, Scaltrito F, Pettoello-Mantovani M and Piscazzi A: From
metabolic to epigenetic memory: The impact of hyperglycemia-induced
epigenetic signature on kidney disease progression and
complications. Genes (Basel). 16:14422025. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Trefely S, Lovell CD, Snyder NW and Wellen
KE: Compartmentalised acyl-CoA metabolism and roles in chromatin
regulation. Mol Metab. 38:1009412020. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Smestad J, Erber L, Chen Y and Maher LJ
III: Chromatin succinylation correlates with active gene expression
and is perturbed by defective TCA cycle metabolism. iScience.
2:63–75. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Hirschey MD and Zhao Y: Metabolic
regulation by lysine malonylation, succinylation, and
glutarylation. Mol Cell Proteomics. 14:2308–2315. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Katsyuba E, Romani M, Hofer D and Auwerx
J: NAD+ homeostasis in health and disease. Nat Metab.
2:9–31. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Tannahill GM, Curtis AM, Adamik J,
Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ,
Kelly B, Foley NH, et al: Succinate is an inflammatory signal that
induces IL-1β through HIF-1α. Nature. 496:238–242. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Selak MA, Durán RV and Gottlieb E: Redox
stress is not essential for the pseudo-hypoxic phenotype of
succinate dehydrogenase deficient cells. Biochim Biophys Acta.
1757:567–572. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Xie Z, Dai J, Dai L, Tan M, Cheng Z, Wu Y,
Boeke JD and Zhao Y: Lysine succinylation and lysine malonylation
in histones. Mol Cell Proteomics. 11:100–107. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Piché ME, Tchernof A and Després JP:
Obesity phenotypes, diabetes, and cardiovascular diseases. Circ
Res. 126:1477–1500. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Ferguson D and Finck BN: Emerging
therapeutic approaches for the treatment of NAFLD and type 2
diabetes mellitus. Nat Rev Endocrinol. 17:484–495. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Boden G, Cheung P, Salehi S, Homko C,
Loveland-Jones C, Jayarajan S, Stein TP, Williams KJ, Liu ML,
Barrero CA and Merali S: Insulin regulates the unfolded protein
response in human adipose tissue. Diabetes. 63:912–922. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Lee JH and Lee J: Endoplasmic reticulum
(ER) stress and its role in pancreatic β-cell dysfunction and
senescence in type 2 diabetes. Int J Mol Sci. 23:48432022.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Ajoolabady A, Lebeaupin C, Wu NN, Kaufman
RJ and Ren J: ER stress and inflammation crosstalk in obesity. Med
Res Rev. 43:5–30. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Ajoolabady A, Liu S, Klionsky DJ, Lip GYH,
Tuomilehto J, Kavalakatt S, Pereira DM, Samali A and Ren J: ER
stress in obesity pathogenesis and management. Trends Pharmacol
Sci. 43:97–109. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Argentato PP, de Cássia César H, Estadella
D and Pisani LP: Programming mediated by fatty acids affects
uncoupling protein 1 (UCP-1) in brown adipose tissue. Br J Nutr.
120:619–627. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Yang J, Zhang H, Parhat K, Xu H, Li M,
Wang X and Ran C: Molecular imaging of brown adipose tissue mass.
Int J Mol Sci. 22:94362021. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Peng C, Jiang H, Jing L, Yang W, Guan X,
Wang H, Yu S, Cao Y, Wang M, Ma H, et al: Macrophage SUCLA2 coupled
glutaminolysis manipulates obesity through AMPK. Nat Commun.
16:17382025. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Goetzman ES, Zhang BB, Zhang Y, Bharathi
SS, Bons J, Rose J, Shah S, Solo KJ, Schmidt AV, Richert AC, et al:
Dietary dicarboxylic acids provide a nonstorable alternative fat
source that protects mice against obesity. J Clin Invest.
134:e1741862024. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Osborne B, Bentley NL, Montgomery MK and
Turner N: The role of mitochondrial sirtuins in health and disease.
Free Radic Biol Med. 100:164–174. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Dillmann WH: Diabetic cardiomyopathy. Circ
Res. 124:1160–1162. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Lorenzo-Almorós A, Cepeda-Rodrigo JM and
Lorenzo Ó: Diabetic cardiomyopathy. Rev Clin Esp (Barc).
222:100–111. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Nakamura K, Miyoshi T, Yoshida M, Akagi S,
Saito Y, Ejiri K, Matsuo N, Ichikawa K, Iwasaki K, Naito T, et al:
Pathophysiology and treatment of diabetic cardiomyopathy and heart
failure in patients with diabetes mellitus. Int J Mol Sci.
23:35872022. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Greenwell AA, Tabatabaei Dakhili SA, Wagg
CS, Saed CT, Chan JSF, Yang K, Mangra-Bala IA, Stenlund MJ, Eaton
F, Gopal K, et al: Pharmacological inhibition of succinyl coenzyme
A: 3-ketoacid coenzyme A transferase alleviates the progression of
diabetic cardiomyopathy. J Am Heart Assoc. 13:e0326972024.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Guo B, Zhang F, Yin Y, Ning X, Zhang Z,
Meng Q, Yang Z, Jiang W, Liu M, Wang Y, et al: Post-translational
modifications of pyruvate dehydrogenase complex in cardiovascular
disease. iScience. 27:1106332024. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Schlaepfer IR and Joshi M: CPT1A-mediated
fat oxidation, mechanisms, and therapeutic potential.
Endocrinology. 161:bqz0462020. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Fenta ET, Eshetu HB, Kebede N, Bogale EK,
Zewdie A, Kassie TD, Anagaw TF, Mazengia EM and Gelaw SS:
Prevalence and predictors of chronic kidney disease among type 2
diabetic patients worldwide, systematic review and meta-analysis.
Diabetol Metab Syndr. 15:2452023. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Gupta S, Dominguez M and Golestaneh L:
Diabetic kidney disease: An update. Med Clin North Am. 107:689–705.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Jung CY and Yoo TH: Pathophysiologic
mechanisms and potential biomarkers in diabetic kidney disease.
Diabetes Metab J. 46:181–197. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Gaddy A, Elrggal M, Madariaga H, Kelly A,
Lerma E and Colbert GB: Diabetic kidney disease. Dis Mon.
71:1018482025. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Kato M and Natarajan R: Epigenetics and
epigenomics in diabetic kidney disease and metabolic memory. Nat
Rev Nephrol. 15:327–345. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Kuo FC, Chao CT and Lin SH: The dynamics
and plasticity of epigenetics in diabetic kidney disease:
Therapeutic applications Vis-à-Vis. Int J Mol Sci. 23:8432022.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Wang H, Liu D, Zheng B, Yang Y, Qiao Y, Li
S, Pan S, Liu Y, Feng Q and Liu Z: Emerging role of ferroptosis in
diabetic kidney disease: Molecular mechanisms and therapeutic
opportunities. Int J Biol Sci. 19:2678–2694. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Wu Y and Chen Y: Research progress on
ferroptosis in diabetic kidney disease. Front Endocrinol
(Lausanne). 13:9459762022. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Peng Q, Zhang H and Li Z: KAT2A-mediated
H3K79 succinylation promotes ferroptosis in diabetic nephropathy by
regulating SAT2. Life Sci. 376:1237462025. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Wang M, Li Q, Wang S, Zuo L, Hai Y, Yuan
S, Li X, Huang X, Yang C, Yao L, et al: Astragaloside IV protects
renal tubular epithelial cells against oxidative stress-induced
injury by upregulating CPT1A-mediated HSD17B10 lysine succinylation
in diabetic kidney disease. Phytother Res. 38:4519–4540. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Zhu L, Liu F, Shi C, Su X, Tan M, Xie S,
Yu M, Zou S, Tan Y, Xie S, et al: Hepatic micropeptide modulates
mitochondrial RNA processing machinery in hepatocellular carcinoma.
Mol Cell. 85:2303–2319.e7. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Wu M and Ye X:
Quercetin-4′-O-β-D-glucopyranoside inhibits podocyte injury by
SIRT5-mediated desuccinylation of NEK7. Clin Exp Pharmacol Physiol.
51:e139092024. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Sharif H, Wang L, Wang WL, Magupalli VG,
Andreeva L, Qiao Q, Hauenstein AV, Wu Z, Núñez G, Mao Y and Wu H:
Structural mechanism for NEK7-licensed activation of NLRP3
inflammasome. Nature. 570:338–343. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Zhu R, Bai X, Xu C, Qi W, Luo P, Wu M and
Luo M: Research progress on podocyte pyroptosis in diabetic
nephropathy. Curr Med Chem. 32:5772–5789. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Zhang J, Huang X, Zhang T, Gu C, Zuo W, Fu
L, Dong Y and Liu H: Antitumorigenic potential of
Lactobacillus-derived extracellular vesicles: p53 succinylation and
glycolytic reprogramming in intestinal epithelial cells via SIRT5
modulation. Cell Biol Toxicol. 40:662024. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Liu X, Rong F, Tang J, Zhu C, Chen X, Jia
S, Wang Z, Sun X, Deng H, Zha H, et al: Repression of p53 function
by SIRT5-mediated desuccinylation at Lysine 120 in response to DNA
damage. Cell Death Differ. 29:722–736. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Liu S, Xu Z, Li X, Zhang Y, Cai Z, Yin X
and Wang J: SIRT5 induces glomerular sclerosis in diabetic
nephropathy through p53. Int Immunopharmacol. 164:1153522025.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Pushparani DS, Varalakshmi J, Roobini K,
Hamshapriya P and Livitha A: Diabetic retinopathy-a review. Curr
Diabetes Rev. 21:43–55. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Kour V, Swain J, Singh J, Singh H and Kour
H: A review on diabetic retinopathy. Curr Diabetes Rev.
20:e2010232224182024. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Zhang Y, Li T, Cai X, Long D, Wang X, Liu
C and Wu Q: Sirt5-mediated desuccinylation of OPTN protects retinal
ganglion cells from autophagic flux blockade in diabetic
retinopathy. Cell Death Discov. 8:632022. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Zhang Z, Chen Y, Fang L, Zhao J and Deng
S: The involvement of high succinylation modification in the
development of prostate cancer. Front Oncol. 12:10346052022.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Meyer JG, D'Souza AK, Sorensen DJ, Rardin
MJ, Wolfe AJ, Gibson BW and Schilling B: Quantification of lysine
acetylation and succinylation stoichiometry in proteins using mass
spectrometric data-independent acquisitions (SWATH). J Am Soc Mass
Spectrom. 27:1758–1771. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Belot A, Puy H, Hamza I and Bonkovsky HL:
Update on heme biosynthesis, tissue-specific regulation, heme
transport, relation to iron metabolism and cellular energy. Liver
Int. 44:2235–2250. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Ekremoglu O and Koc A: The role of SIRT5
and p53 proteins in the sensitivity of colon cancer cells to
chemotherapeutic agent 5-fluorouracil. Mol Biol Rep. 48:5485–5495.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
He B, Du J and Lin H: Thiosuccinyl
peptides as Sirt5-specific inhibitors. J Am Chem Soc.
134:1922–1925. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Fiorentino F, Castiello C, Mai A and
Rotili D: Therapeutic potential and activity modulation of the
protein lysine deacylase sirtuin 5. J Med Chem. 65:9580–9606. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Barreca F, Aventaggiato M, Vitiello L,
Sansone L, Russo MA, Mai A, Valente S and Tafani M: SIRT5
activation and inorganic phosphate binding reduce cancer cell
vitality by modulating autophagy/mitophagy and ROS. Antioxidants
(Basel). 12:16352023. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Barreca F, Aventaggiato M, Cristina M,
Sansone L, Belli M, Lista MB, Francisci G, Valente S, Rotili D, Mai
A, et al: A combined SIRT5 activation and SIRT3 inhibition prevents
breast cancer spheroids growth by reducing HIF-1α and mitophagy.
Pharmaceuticals (Basel). 19:232025. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Xie Y, Cai N, Liu X, He L, Ma Y, Yan C,
Liang J, Ouyang SH, Luo A, He Y, et al: SIRT5: A potential target
for discovering bioactive natural products. J Nat Med. 79:441–464.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Ge J, Hsieh CY, Fang M, Sun H and Hou T:
Development of PROTACs using computational approaches. Trends
Pharmacol Sci. 45:1162–1174. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Zheng C, Yan S, Lu L, Yao H, He G, Chen S,
Li Y, Peng X, Cheng Z, Wu M, et al: Lovastatin inhibits EMT and
metastasis of triple-negative breast cancer stem cells through
dysregulation of cytoskeleton-associated proteins. Front Oncol.
11:6566872021. View Article : Google Scholar : PubMed/NCBI
|