Introduction
Cardiomyocyte apoptosis serves a pivotal role in the
pathogenesis of heart failure (HF) (1), a condition characterized by notable
systolic and diastolic dysfunction that compromises the ability of
the heart to pump and fill with blood effectively (2). A key contributor to this dysfunction
is the reduced oxygen utilization capacity of the failing heart,
leading to an inability to meet systemic metabolic demands
(3). This metabolic imbalance
initiates remodeling processes that exacerbate cardiac dysfunction
and accelerate HF progression. Hypoxia-induced cardiomyocyte
apoptosis serves a pivotal role in driving HF progression (4).
Excessive reactive oxygen species (ROS) production
is a central mechanism underlying hypoxia-induced myocardial
injury. Elevated ROS levels cause oxidative damage to cellular
components, including lipids, proteins and DNA, thereby impairing
membrane integrity and cellular function (5). Under normal physiological conditions,
mitochondria are equipped with antioxidant systems to maintain ROS
at controlled levels (6). However,
hypoxia disrupts mitochondrial function, resulting in a decreased
mitochondrial membrane potential (MMP) and the release of
cytochrome c, further exacerbating mitochondrial damage
(7). This mitochondrial
dysfunction impairs the respiratory chain, markedly reducing ATP
synthesis, depleting cellular energy and promoting apoptosis
(8). These cascading events
highlight the critical role of preserving mitochondrial integrity
and regulating ROS to maintain cardiomyocyte survival under hypoxic
stress.
Visinin-like protein 1 (VSNL1), a member of the
neuronal calcium-sensing protein family, is highly expressed in the
sinoatrial node, which regulates the pacemaker activity of the
heart (9,10). Emerging evidence suggests that
VSNL1 serves a crucial role in several biological processes,
including cardiac pacing, cell proliferation, tumor progression and
neural signaling pathways (10–13).
Notably, VSNL1 has been identified as a key anti-apoptotic factor
in several cell types, such as colorectal cancer cells, where its
downregulation has been reported to induce apoptosis and inhibit
cell proliferation (12).
Similarly, silencing of VSNL1 in H295R cells has been reported to
increase susceptibility to calcium-induced apoptosis (14). However, to the best of our
knowledge, the role of VSNL1 in the regulation of myocardial
apoptosis remains unexplored and its potential effects in cardiac
cells have yet to be determined.
Therefore, the present study established a
hypoxia-induced myocardial cell model to assess cardiomyocyte
apoptosis, and the changes in ROS levels, the MMP and markers of
cellular damage. The expression levels of VSNL1 were modulated to
evaluate its role in hypoxia-induced myocardial apoptosis, and the
underlying molecular mechanisms were explored. The findings provide
novel insights into potential therapeutic targets for myocardial
protection and suggest promising clinical strategies for the
treatment of cardiovascular diseases.
Materials and methods
Cell treatment
The human AC16 cardiomyocyte cell line (cat. no.
MZ-4038; Ningbo Mingzhou Biotechnology Co., Ltd.) was employed in
the present study. The cells were cultured in DMEM (Shanghai Basal
Media Technologies Co., Ltd.) supplemented with 10% fetal bovine
serum (Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin-streptomycin (Biosharp Life Sciences) at 37°C in a
humidified atmosphere containing 5% CO2. To induce
apoptosis, AC16 cardiomyocytes were subjected to hypoxic conditions
in a modular incubator (Thermo Fisher Scientific, Inc.) with a gas
mixture of 94% N2, 5% CO2 and 1%
O2 for 24 h at 37°C. The control group was maintained
under normoxic conditions with 95% air and 5% CO2. When
time-series data were required, parallel cultures were exposed for
0 h, 10 min, 1, 6 or 24 h.
Lactate dehydrogenase (LDH) release
assay
Cells were exposed to hypoxic conditions (94%
N2, 5% CO2, 1% O2) at 37°C for 24
h. Following treatment of cells from each group, the cell culture
supernatants were collected. The supernatants were then combined
with an LDH assay working solution (cat. no. C0016; Beyotime
Institute of Biotechnology) and the resulting mixture was incubated
at room temperature in the dark for 30 min. After incubation, the
absorbance was measured at 490 nm to determine the LDH
activity.
Measurement of creatine kinase-MB
(CK-MB) levels
The concentration of CK-MB in the cell supernatants
was determined using an ELISA kit (cat. no. H197-1-2; Nanjing
Jiancheng Bioengineering Institute) according to the manufacturer's
instructions.
ROS level assessment
Cells were seeded in 6-well plates at a density of
5×105 cells/well and cultured at 37°C in a humidified
atmosphere containing 5% CO2. After overnight
incubation, the cells were exposed to hypoxic conditions (94%
N2, 5% CO2, 1% O2) at 37°C for 24
h. After removing the culture medium,
2′-7′-dichlorodihydrofluorescein diacetate (Beyotime Institute of
Biotechnology) diluted in serum-free DMEM was added to the cells
and cells were incubated for 20 min at 37°C. Following three washes
with serum-free DMEM, ROS levels were observed under a fluorescence
microscope (Olympus Corporation) and analyzed using ImageJ software
(version 1.53e; National Institutes of Health, USA). Single-cell
suspensions were then collected and analyzed using a NovoCyte
Penteon flow cytometer (Agilent Technologies, Inc.) with
NovoExpress software (version 1.5.0; Agilent Technologies, Inc.)
(15).
Measurement of malonaldehyde (MDA)
levels
MDA levels were determined using a commercial assay
kit (cat. no. S0131; Beyotime Institute of Biotechnology). Briefly,
cells were lysed on ice with RIPA buffer (cat. no. P0013C; Beyotime
Institute of Biotechnology), and after centrifugation at 11,100 × g
for 15 min at 4°C, the supernatant was collected for subsequent
analysis. MDA concentrations in the supernatant were quantified at
532 nm using a microplate reader (PT3502PC; Beijing Potenov Xinqiao
Technology Co., Ltd.), according to the manufacturer's protocol.
MDA levels were normalized to the protein concentration. Protein
concentration was determined using the BCA protein assay kit (cat.
no. P0010; Beyotime Institute of Biotechnology) according to the
manufacturer's instructions.
VSNL1 lentivirus infection
For lentivirus-induced overexpression of VSNL1, AC16
cells were seeded at a density of 1×104 cells/well in
96-well plates (100 µl/well) and were incubated overnight at 37°C
in a 5% CO2 incubator. The recombinant lentiviral vector
pHBLV-CMV-MCS-3FLAG-EF1-ZsGreen-T2A-PURO (Hanbio Biotechnology Co.,
Ltd.) was used to generate the virus using a 3rd generation
lentiviral packaging system. Lentiviral particles were produced by
co-transfecting 293T cells (cat. no. CRL-3216; American Type
Culture Collection) with 4 µg VSNL1 plasmid, 3 µg packaging plasmid
(pCMV-dR8.91) and 1 µg envelope plasmid (pCMV–VSV-G) (Hanbio
Biotechnology Co., Ltd.) at a ratio of 4:3:1 using
Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.). Transfection was performed at 37°C for 48 h.
Viral supernatants were collected at 48 and 72 h post-transfection,
filtered through a 0.45-µm membrane, and stored at −80°C.
For infection, the virus was thawed on ice, diluted
in fresh medium and added to AC16 cells at 30–50% confluence at a
multiplicity of infection of 10. On day 1 post-infection (~24 h),
the virus-containing medium was removed and replaced with fresh
complete medium. The cells were incubated at 37°C for an additional
48 h, and then harvested at 72 h post-infection for further
experiments. Stable cell lines were selected with medium containing
2 µg/ml puromycin (Sigma-Aldrich; Merck KGaA) for 7 days, followed
by maintenance in medium with 1 µg/ml puromycin.
Cell transfection
Cell transfection using Lipofectamine 3000
(Invitrogen; Thermo Fisher Scientific, Inc.) resulted in the
downregulation of natriuretic peptide receptor B (NPRB) and VSNL1
in AC16 cardiomyocytes. The small interfering RNAs (siRNAs) used in
the present study included a sequence targeting human VSNL1
(si-VSNL1; 5′-CGACCCTTCCATTGTATTA-3′), a sequence targeting human
NPRB (si-NPRB; 5′-GACGACCCATCCTGTGATA-3′) and a non-targeting
negative control (cat. no. siN0000001-1-5); all purchased from
Guangzhou RiboBio Co., Ltd. The specific sequence of non-targeting
negative control was not provided by the supplier. Prior to
transfection, the original culture medium was replaced with fresh
high-glucose DMEM supplemented with 10% FBS and 1%
penicillin-streptomycin. The transfection mixture was prepared by
mixing 50 nM siRNA with 5 µl Lipofectamine 3000 in Opti-MEM reduced
serum medium (Gibco; Thermo Fisher Scientific, Inc.). To form a
lipoplex, the transfection mixture was incubated at room
temperature for 15 min. After 15 min, the transfection mixture was
added to the cells, and the cells were continuously cultured in a
37°C incubator with 5% CO2 without replacing the medium
containing the transfection mixture for 6 h. After 6 h of
incubation with the transfection mixture, the medium was aspirated
and replaced with fresh complete high-glucose DMEM (10% FBS, 1%
penicillin-streptomycin), and the cells were further cultured under
the same conditions until 48 h post-transfection (i.e., 42 h after
medium replacement). The subsequent experiments were performed at
this 48 h time point.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was isolated from AC16 cells using
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. The RNA concentration
was determined using a NanoDrop® spectrophotometer
(Thermo Fisher Scientific, Inc.). For complementary DNA synthesis,
RNA was incubated at 42°C for 15 min, followed by a 5-sec heat
treatment at 85°C, as described in the manufacturer's instructions
of the TransScript® Green Two-Step qRT-PCR SuperMix kit
(Thermo Fisher Scientific, Inc.). The primer sequences used for
human AC16 cells were as follows: VSNL1 forward,
5′-TATGACCTGGATGGTGATGGCAAG-3′ and reverse,
5′-AATCTTGTCTACTCGCTGCTCAGG-3′; 2′,3′-cyclic nucleotide 3′
phosphodiesterase (CNP) forward, 5′-ATGGTGTCGGCTGACGCTTAC-3′ and
reverse, 5′-CGTTCGTGGTTGGTGTCATCAAG-3′; NPRB forward,
5′-CTGCGCATGGAACAGTATGC-3′ and reverse, 5′-GGGTGCTCTCTGCTGACAAT-3′;
BAX forward, 5′-CCCCCGAGAGGTCTTTTTCC-3′ and reverse,
5′-TGTCCAGCCCATGATGGTTC-3′; BCL-2 forward,
5′-GGGATTCCTGCGGATTGACA-3′ and reverse, 5′-TGCATAAGGCAACGATCCCA-3′;
and β-actin forward, 5′-GGGAAATCGTGCGTGACATTAAG-3′ and reverse,
5′-TGTGTTGGCGTACAGGTCTTTG-3′. Each primer mix was loaded into a
96-well fluorescent RT-PCR plate, and RT-PCR was performed using
the CFX Connect system (Bio-Rad Laboratories, Inc.). The
thermocycling parameters were as follows: Initial denaturation step
at 94°C for 30 sec, followed by 40 cycles of 5 sec at 94°C and 30
sec at 60°C. mRNA expression levels were determined using the
2−ΔΔCq method (16).
All experiments were independently replicated three times.
Western blot analysis
Total proteins were extracted from both AC16
cardiomyocytes and mouse cardiac tissues using RIPA lysis buffer
containing protease and phosphatase inhibitors (cat. no. P0039;
Beyotime Institute of Biotechnology). For myocardial tissue
samples, freshly collected heart tissue was weighed and immediately
homogenized in ice-cold RIPA buffer using a mechanical homogenizer.
Following homogenization, the samples were incubated on ice for 30
min and then centrifuged at 11,100 × g for 15 min at 4°C. For cell
samples, cultured cells were washed twice with PBS, lysed directly
in RIPA buffer and centrifuged under the same conditions. The
supernatants containing total proteins were collected, and protein
concentrations were determined using a BCA protein assay kit
(Thermo Fisher Scientific, Inc.). Equal amounts of protein (30 µg)
were separated by SDS-PAGE on 10% gels and transferred onto PVDF
membranes. The membranes were blocked in 5% milk at room
temperature for 1 h, and incubated overnight at 4°C with primary
antibodies. The antibodies used were as follows: VSNL1 (1:400; cat.
no. A2797; ABclonal Biotech Co., Ltd.), NPRB (1:1,000; cat. no.
55113-1-AP; Proteintech Group, Inc.), CNP (1:1,000; cat. no.
13427-1-AP; Proteintech Group, Inc.), BCL-2 (1:1,000; cat. no.
26593-1-AP; Proteintech Group, Inc.), BAX (1:1,000; cat. no.
50599-2-IG; Proteintech Group, Inc.) and β-actin (1:10,000; cat.
no. 66009-1-IG; Proteintech Group, Inc.). Subsequently, the
membranes were incubated with HRP-conjugated secondary antibodies,
including anti-rabbit (1:5,000; cat. no. SA00001-2; Proteintech
Group, Inc.) and anti-mouse secondary antibodies (1:5,000; cat. no.
SA00001-1; Proteintech Group, Inc.) for 1 h at room temperature.
After antibody binding, chemiluminescent detection was performed
using BeyoECL Moon (cat. no. P0018FS; Beyotime Institute of
Biotechnology) and band visualization was performed on the Tanon
4600 Chemiluminescence Imaging System (Tanon Science and Technology
Co., Ltd.).
Analysis of the VSNL1 expression
profile
To assess the expression profile of VSNL1 across
various normal human tissues, data were retrieved from the Human
Protein Atlas (HPA) database (https://www.proteinatlas.org). This freely accessible
online resource offers extensive information on human protein
expression in numerous normal tissues and cell lines. Relevant data
on VSNL1 expression were obtained to analyze its tissue-specific
distribution and expression pattern.
MMP assessment
The MMP was evaluated using the Enhanced MMP Assay
Kit with JC-1 (Beyotime Institute of Biotechnology). After removing
the culture medium, cells in 6-well plates were incubated with a
mixture of 1 ml cell culture medium and 1 ml JC-1 staining solution
for 20 min at 37°C. Following incubation, cells were washed twice
with JC-1 staining buffer and then visualized under a fluorescence
microscope (Olympus Corporation). Image analysis was performed
using ImageJ software (version 1.8.0_172; National Institutes of
Health).
Intracellular calcium measurement
Intracellular Ca2+ levels were measured
using the Calcium Assay Kit (cat. no. S1063S; Beyotime Institute of
Biotechnology) according to the manufacturer's instructions.
Briefly, AC16 cells were lysed and centrifuged at 12,000 × g for 30
min at 4°C to collect supernatants. Standards and samples were
added to a 96-well plate, followed by the assay working solution.
After incubation at room temperature for 5–10 min in the dark,
absorbance was measured at 575 nm. Calcium concentrations were
calculated from a standard curve.
Co-immunoprecipitation (Co-IP)
experiment
To assess whether there is a protein-protein
interaction between VSNL1 and CNP, Co-IP technology was applied in
AC16 cells in the present study. Initially, AC16 cells grown to
80–90% confluence in 10-cm dishes were treated with 500 µl lysis
buffer per dish. The lysis buffer contained 1% NP-40, 150 mM NaCl,
50 mM Tris-HCl (pH 7.5) and protease inhibitors (1 mM PMSF and 1
µg/ml aprotinin; cat. no. P1049; Beyotime Institute of
Biotechnology). The cells were then incubated on ice for 30 min.
The lysate was then centrifuged at 12,000 × g for 15 min at 4°C,
and the supernatant was collected for further analysis. For each IP
reaction, 500 µg total protein (equivalent to 200 µl supernatant)
was incubated overnight at 4°C with either specific primary
antibodies against VSNL1 (2 µg; cat. no. A2797; ABclonal Biotech
Co., Ltd.) or CNP (2 µg; cat. no. 13427-1-AP; Proteintech Group,
Inc.), or with isotype control IgG antibodies (2 µg; cat. no.
AC005; ABclonal Biotech Co., Ltd.) as a negative control. To
capture antibody-protein immunocomplexes, 50 µl 50% (v/v) Protein
A/G agarose bead slurry (equivalent to 25 µl packed agarose beads;
cat. no. 20421; Thermo Fisher Scientific, Inc.) was added to each
IP reaction mixture, which was then gently rotated at 4°C for 4–6 h
to allow sufficient bead-antibody-antigen complex formation. The
immunoprecipitates were washed five times with ice-cold wash buffer
(same formulation as the lysis buffer without the protease
inhibitors) to remove nonspecific binding proteins. Immunocomplexes
were isolated by centrifugation at 3000 × g for 5 min at 4°C to
pellet the beads, followed by aspiration of the supernatant. The
pellets were resuspended in 30 µl 2X SDS sample buffer and boiled
at 95°C for 10 min to elute the proteins from the beads. The eluted
products were separated by SDS-PAGE on 10% gels, transferred onto a
PVDF membrane and analyzed by western blotting, as aforementioned,
to assess the co-precipitation of CNP and VSNL1. Co-precipitation
of CNP with VSNL1 would suggest a potential protein-protein
interaction between the two.
Establishment of acute myocardial
infarction (AMI) model
A total of 30 male C57BL/6 mice (age, 8–10 weeks;
weight, 22–25 g) were purchased from Beijing Vital River Laboratory
Animal Technology Co., Ltd.; Charles River Laboratories, and were
used in the present study. Mice were housed in a specific
pathogen-free facility under the following controlled conditions:
Temperature 22±2°C, relative humidity 50±10% and a 12-h light/dark
cycle (lights on at 7:00 a.m. and off at 7:00 p.m.). Animals had
free access to standard laboratory chow and sterile water
throughout the experimental period.
Mice were randomly assigned to two experimental
groups: An AMI group or a sham-operated control group. The AMI
group underwent left anterior descending (LAD) coronary artery
ligation at the proximal one-third segment to induce ischemia,
while the sham group received the same surgical procedure without
ligation. All procedures were performed under general anesthesia
using isoflurane (3% for induction and 1.5–2% for maintenance). AMI
was induced by permanent ligation of the LAD coronary artery with a
6–0 silk suture. In the sham-operated group, the LAD coronary
artery was passed with the suture needle but not ligated, serving
as a surgical control.
All animal protocols were approved by the Ethics
Committee of Shanghai University of Medicine and Health Sciences
(Shanghai, China; approval no. 2021-SZR-05-410482198512239314) and
the present study conformed to institutional and national
guidelines for the care and use of laboratory animals.
For sacrifice, mice were confirmed to be under deep
anesthesia with isoflurane, followed by the intraperitoneal
injection of 150 mg/kg pentobarbital sodium. Mortality was verified
by the absence of heartbeat, respiration and reflex responses.
Echocardiographic assessment
Transthoracic echocardiography was performed 24 h
after the establishment of the AMI model using the Vevo 2100
high-resolution imaging system (FUJIFILM VisualSonics, Inc.)
equipped with a 30-MHz transducer. Mice were anesthetized with 3%
isoflurane for induction and 1.5% isoflurane for maintenance and
then positioned supine on a temperature-controlled platform. After
hair removal in the precordial region, a pre-warmed ultrasound gel
was applied to ensure optimal acoustic coupling. M-mode images were
captured from the parasternal long-axis view at the level of the
papillary muscles to evaluate cardiac function.
Assessment of myocardial infarct size
using 2,3,5-triphenyltetrazolium chloride (TTC) staining
To evaluate infarct size, hearts were harvested
immediately after euthanasia. The isolated hearts were first rinsed
in cold saline and then incubated in 1% TTC solution prepared in
Tris buffer at 37°C for 15 min.
Statistical analysis
Statistical analysis was performed using GraphPad
Prism 8.0 software (Dotmatics). Data are presented as the mean ±
standard deviation from at least three independent experiments.
Two-tailed unpaired Student's t-test was used for two-group
comparisons; one-way ANOVA followed by Tukey's post hoc test was
used for multiple-group comparisons. P<0.05 was considered to
indicate a statistically significant difference.
Results
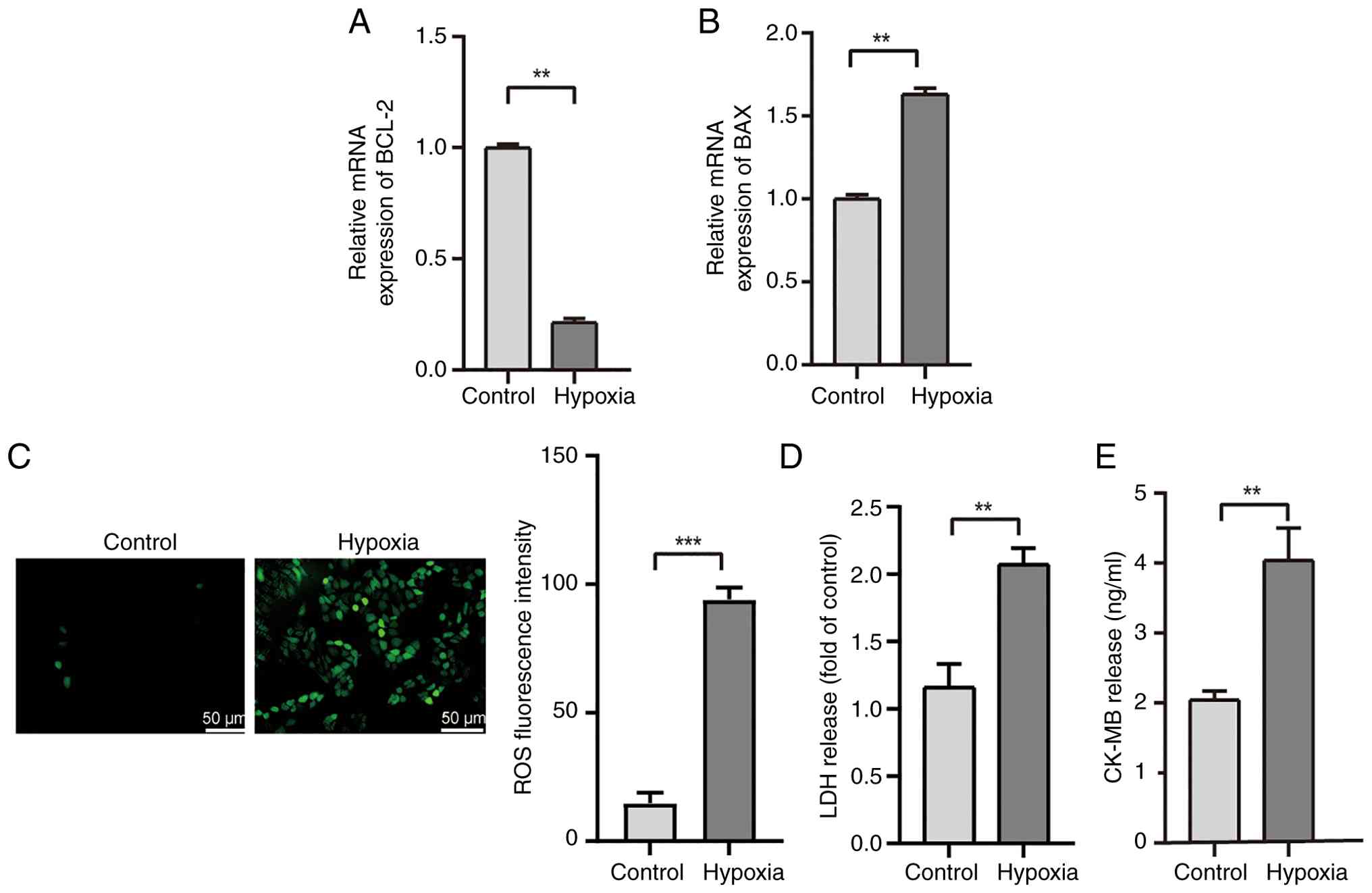
Hypoxia induces apoptosis in
cardiomyocytes
To assess the effects of hypoxia on apoptosis and
cellular damage, a hypoxia-induced cardiomyocyte model was
established. AC16 cardiomyocytes, exposed to hypoxia for 24 h,
exhibited significant downregulation of BCL-2 mRNA expression and
upregulation of BAX mRNA expression. The results indicated that
prolonged hypoxia activated apoptotic pathways (Fig. 1A and B).
As excessive production of ROS can trigger oxidative
stress, damage proteins, lipids and DNA in cells, and further
activate apoptosis signaling pathways (17), the present study analyzed
intracellular ROS levels and the release of myocardial injury
markers LDH and CK-MB (Fig. 1C-E).
Hypoxia exposure resulted in a substantial increase in ROS levels,
accompanied by pronounced elevations in LDH and CK-MB release,
signifying severe oxidative stress and cellular injury.
Collectively, these findings indicated that hypoxia
may induce oxidative stress and subsequent apoptosis. This
established a robust experimental model for further mechanistic
studies on the role of VSNL1 in cardiomyocyte apoptosis under
hypoxic conditions.
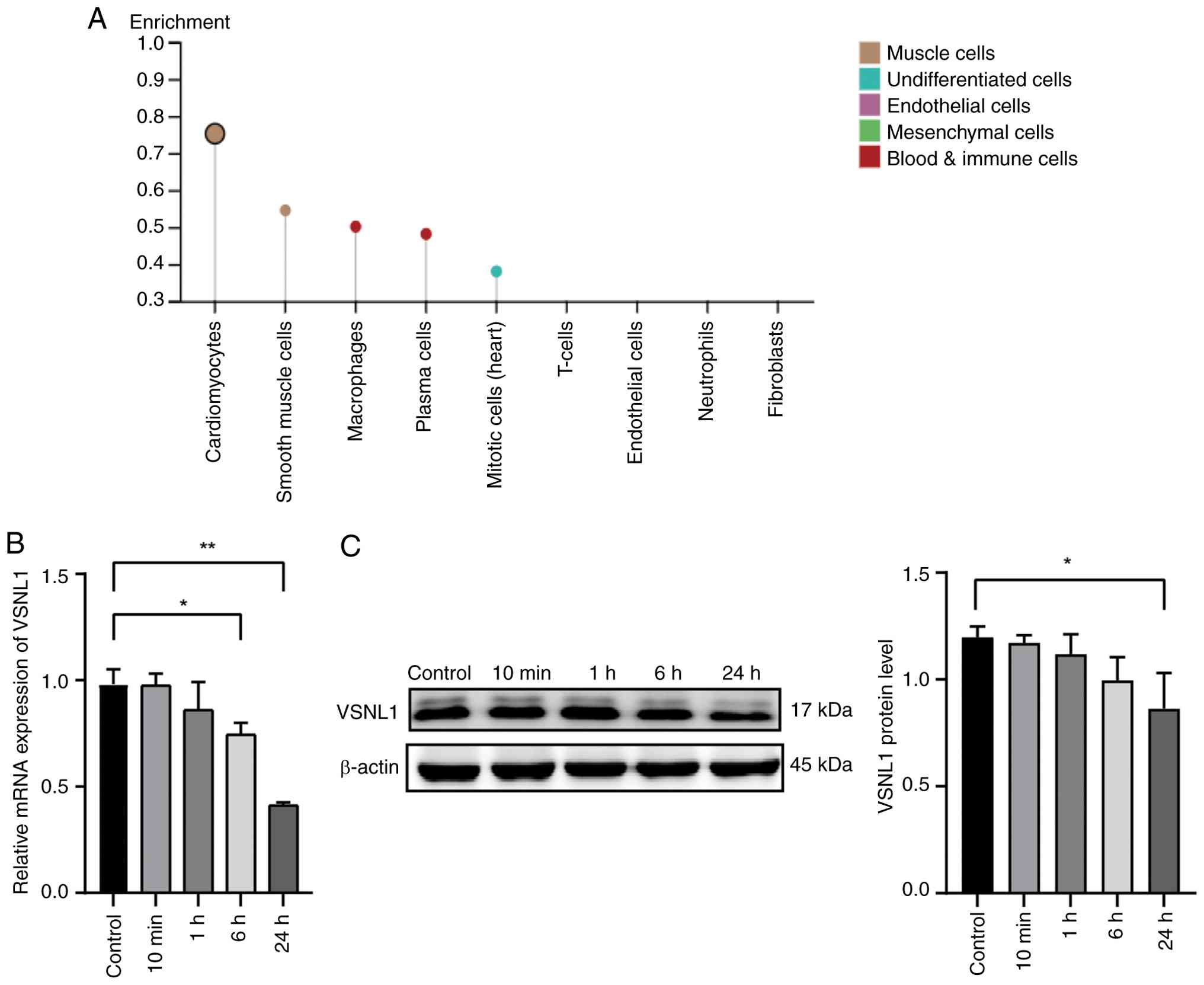
Hypoxia induces downregulation of
VSNL1 expression in cardiomyocytes
To evaluate the expression profile of VSNL1 in
several normal human tissues, data from the HPA database
(https://www.proteinatlas.org) were
analyzed (Fig. 2A). The results
revealed that VSNL1 expression was markedly higher in
cardiomyocytes compared with in other tissues. Subsequently, AC16
cells were exposed to hypoxic conditions for 0 min, 10 min, 1 h, 6
h and 24 h, and the mRNA and protein expression levels of VSNL1
were assessed using qPCR and western blotting, respectively. The
results revealed a significant reduction in VSNL1 expression after
24 h of hypoxia (Fig. 2B and
C).
These findings suggested that hypoxia markedly
downregulated VSNL1 expression in cardiomyocytes, potentially
indicating its involvement in the pathophysiology of
hypoxia-induced cardiac injury.
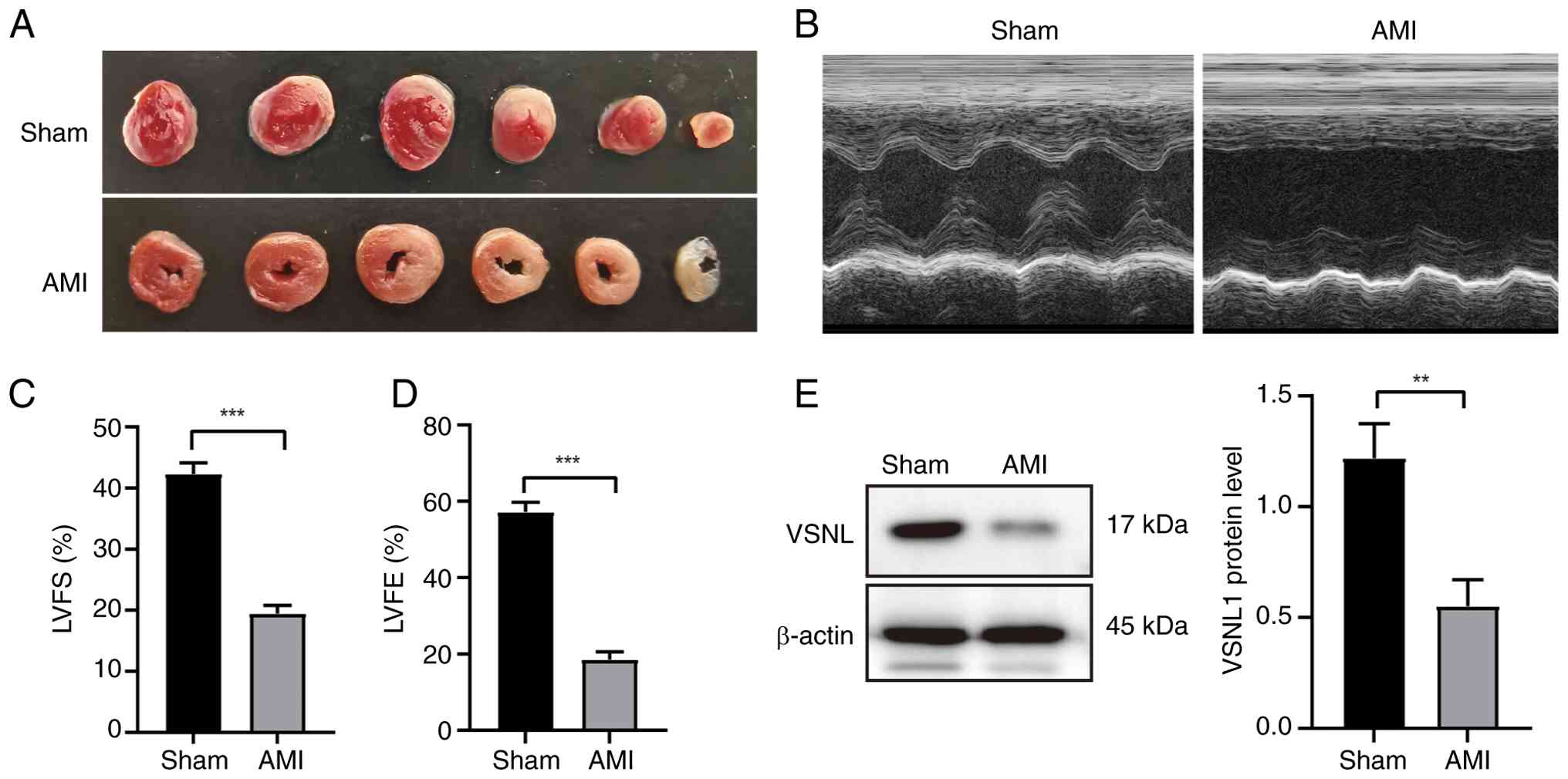
Establishment and validation of the
AMI mouse model
To further assess whether the aforementioned
regulation also occurs in vivo, an AMI model was established
in mice via permanent ligation of the LAD coronary artery. Mice
were randomly assigned to an AMI group or a sham-operated control
group. Successful induction of infarction was confirmed by TTC
staining, which showed pale infarcted regions in AMI mice and
red-stained viable myocardium in sham controls (Fig. 3A). Echocardiographic assessment
revealed significant reductions in the left ventricular ejection
fraction and fractional shortening in AMI mice compared with the
sham group (Fig. 3B-D), indicating
impaired cardiac function. Consistent with the in vitro
findings, western blot analysis demonstrated that VSNL1 expression
was markedly decreased in the myocardial tissue of AMI mice
(Fig. 3E), supporting its
potential role in the pathogenesis of ischemic heart injury.
VSNL1 inhibits cardiomyocyte apoptosis
under hypoxic conditions
Subsequently, the role of VSNL1 in regulating
hypoxia-induced cardiomyocyte apoptosis was assessed using VSNL1
overexpression and silencing. qPCR and western blot analyses
demonstrated that lentivirus-mediated VSNL1 overexpression markedly
increased VSNL1 RNA and protein levels in AC16 cells (Fig. 4A and B), whilst siRNA-mediated
silencing of VSNL1 markedly reduced VSNL1 protein expression
(Fig. 4G). Following hypoxia
treatment, VSNL1 overexpression markedly upregulated the
anti-apoptotic protein BCL-2 and downregulated the pro-apoptotic
protein BAX (Fig. 4C). By
contrast, VSNL1 silencing decreased BCL-2 levels and increased BAX
protein levels, suggesting that VSNL1 inhibits apoptosis activation
and exerts protective effects on cardiomyocytes (Fig. 4H).
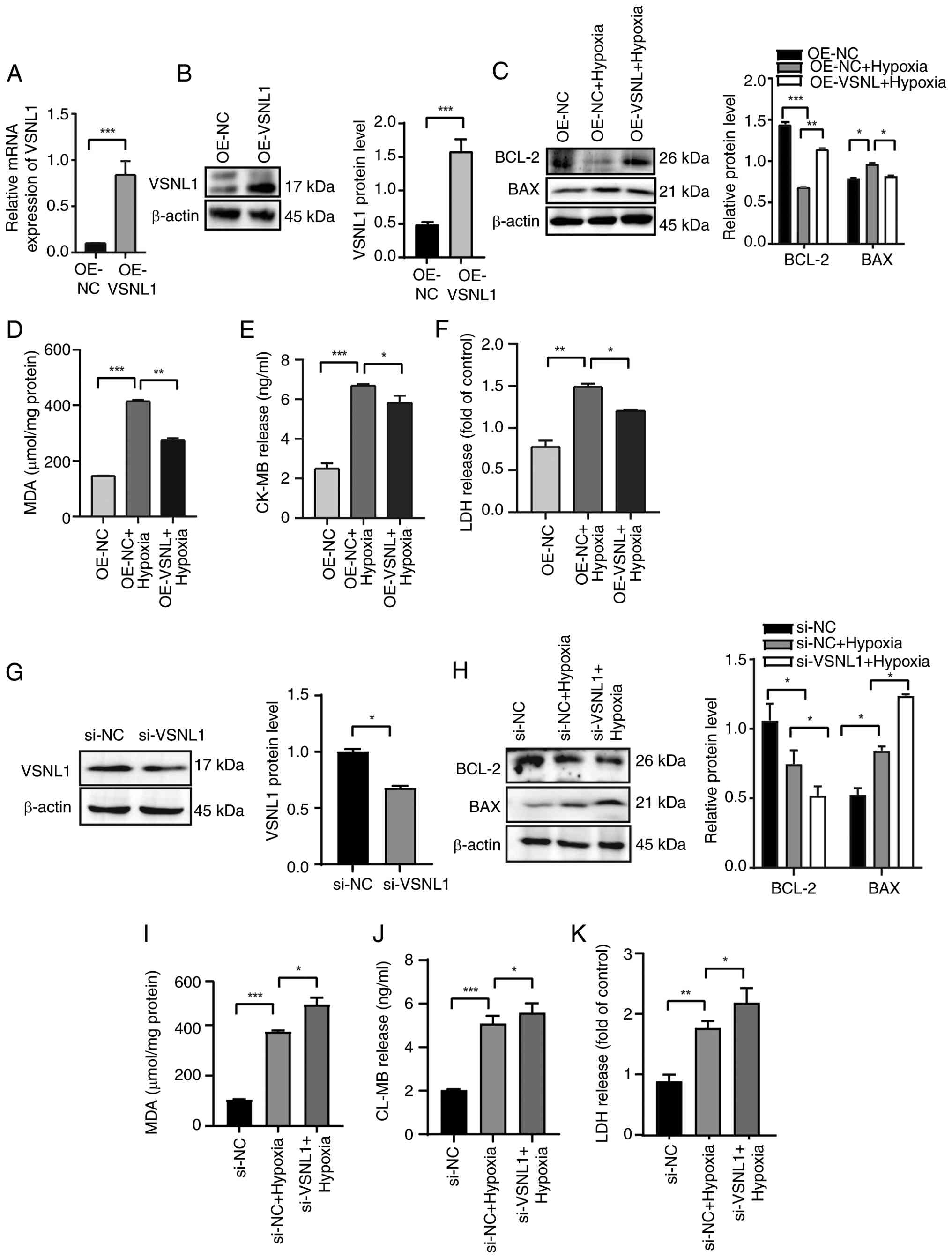 | Figure 4.Regulatory role of VSNL1 in
hypoxia-induced cardiomyocyte apoptosis. (A and B) AC16
cardiomyocytes were infected with a lentivirus overexpressing
VSNL1. VSNL1 expression was assessed by (A) quantitative PCR and
(B) western blotting. (C) Western blot analysis of BCL-2 and BAX
protein expression in AC16 cells following VSNL1 overexpression,
along with semi-quantification of BCL-2 and BAX protein levels. The
levels of (D) MDA, (E) CK-MB and (F) LDH were measured using
specific assay kits after VSNL1 overexpression. (G) VSNL1
expression in AC16 cells post-transfection with siRNA was assessed
by western blotting. (H) Western blot analysis of BCL-2 and BAX
protein expression following VSNL1 silencing, with
semi-quantification of BCL-2 and BAX protein levels. The levels of
(I) MDA, (J) CK-MB and (K) LDH were measured using specific assay
kits following VSNL1 silencing. *P<0.05, **P<0.01,
***P<0.001. VSNL1, visinin-like protein 1; OE-NC, overexpression
negative control; OE-VSNL1, VSNL1 overexpression; siRNA, small
interfering RNA; si-NC, siRNA negative control; si-VSNL1, VSNL1
knockdown via siRNA; MDA, malonaldehyde; CK-MB, creatine kinase-MB;
LDH, lactate dehydrogenase. |
To further evaluate whether VSNL1 modulates
cardiomyocyte injury related to apoptosis, intracellular levels of
MDA, CK-MB and LDH were measured. The results revealed that VSNL1
overexpression markedly reduced the release of these markers
(Fig. 4D-F), whereas VSNL1
silencing exacerbated hypoxia-induced cardiomyocyte damage,
demonstrated by markedly elevated levels of MDA, CK-MB and LDH
(Fig. 4I-K).
VSNL1 alleviates hypoxia-induced
oxidative stress during the early phase of myocardial
apoptosis
Hypoxia-induced cellular damage is closely
associated with elevated ROS levels, a hallmark of oxidative stress
(18). ROS serve as key mediators
of mitochondria-mediated apoptosis (19,20),
characterized by excessive ROS production and loss of MMP (21). To evaluate the role of VSNL1 in
alleviating intracellular oxidative stress, ROS accumulation was
quantified and visualized using fluorescence microscopy and flow
cytometry. The results revealed that the hypoxia-treated cells
exhibited a significant increase in green fluorescence intensity
(Fig. 5A and B), indicative of
excessive ROS accumulation. Notably, overexpression of VSNL1
markedly reduced ROS levels compared with those in the hypoxia
group. Conversely, silencing of VSNL1 using siRNA exacerbated
oxidative stress, as demonstrated by the increase in ROS levels
(Fig. 5C and D). These findings
highlighted the role of VSNL1 in alleviating hypoxia-induced
oxidative stress during the early phase of myocardial
apoptosis.
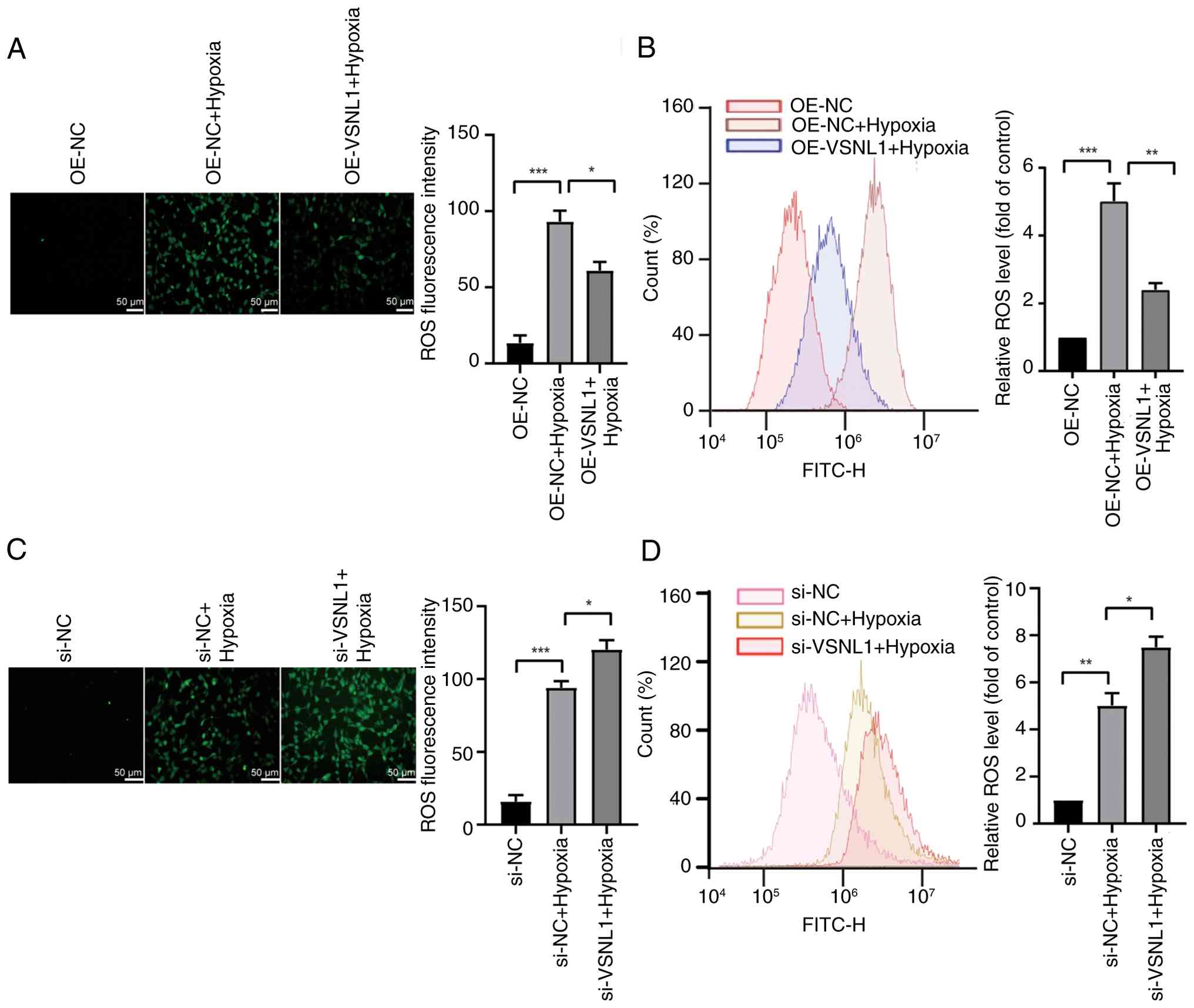 | Figure 5.VSNL1 alleviates hypoxia-induced
oxidative stress. Representative fluorescence images of ROS in
hypoxia-treated AC16 cells following VSNL1 (A) overexpression or
(C) knockdown. Scale bar, 50 µm. Quantification of ROS levels in
hypoxia-treated AC16 cells assessed by flow cytometry after VSNL1
(B) overexpression or (D) knockdown. *P<0.05, **P<0.01,
***P<0.001. VSNL1, visinin-like protein 1; ROS, reactive oxygen
species; OE-NC, overexpression negative control; OE-VSNL1, VSNL1
overexpression; siRNA, small interfering RNA; si-NC, siRNA negative
control; si-VSNL1, VSNL1 knockdown via siRNA. |
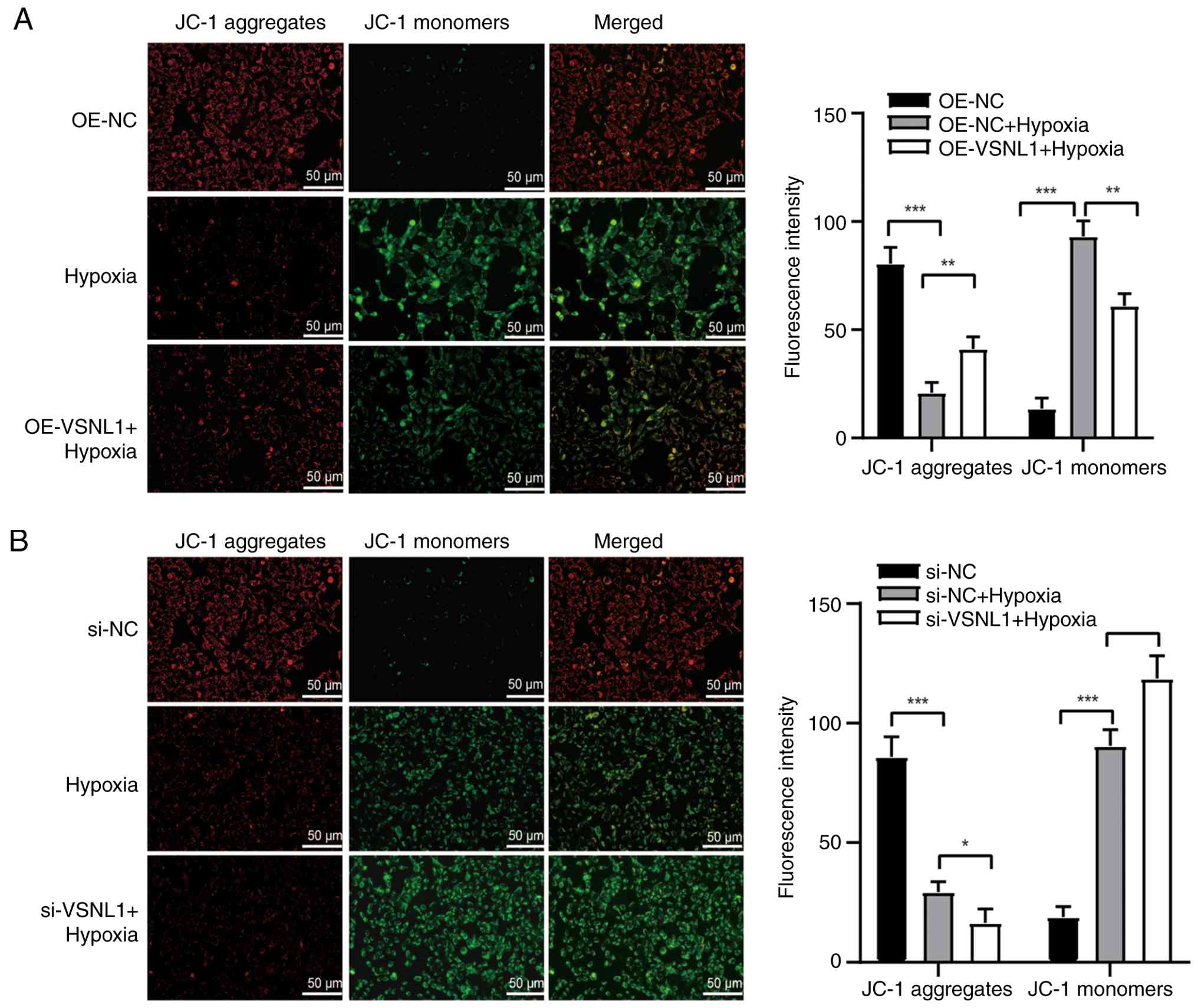
VSNL1 preserves the MMP in
hypoxia-induced myocardial apoptosis
The loss of MMP is a pivotal event that increases
mitochondrial outer membrane permeability, representing an early
step in the intrinsic apoptotic pathway (22). To evaluate whether VSNL1 influences
mitochondrial depolarization, the MMP was measured using JC-1 dye,
which detects changes in the MMP via fluorescence shifts. Under
hypoxic conditions, a marked increase in JC-1 monomers emitting
green fluorescence was observed, along with a near absence of JC-1
aggregates emitting red fluorescence, indicating a substantial loss
of MMP. Overexpression of VSNL1 markedly mitigated this effect,
reducing green fluorescence and partially restoring red
fluorescence, suggesting that VSNL1 overexpression alleviated the
loss of MMP (Fig. 6A). By
contrast, cells treated with si-VSNL1 under hypoxic conditions
exhibited decreased red fluorescence and increased green
fluorescence compared with the hypoxia-only group, confirming a
pronounced reduction in MMP (Fig.
6B). These findings underscored the essential role of VSNL1 in
maintaining the MMP and mitochondrial function, as well as its
regulatory effect in attenuating hypoxia-induced myocardial
apoptosis.
Mechanistic study of VSNL1-mediated
regulation of the CNP/NPRB signaling pathway
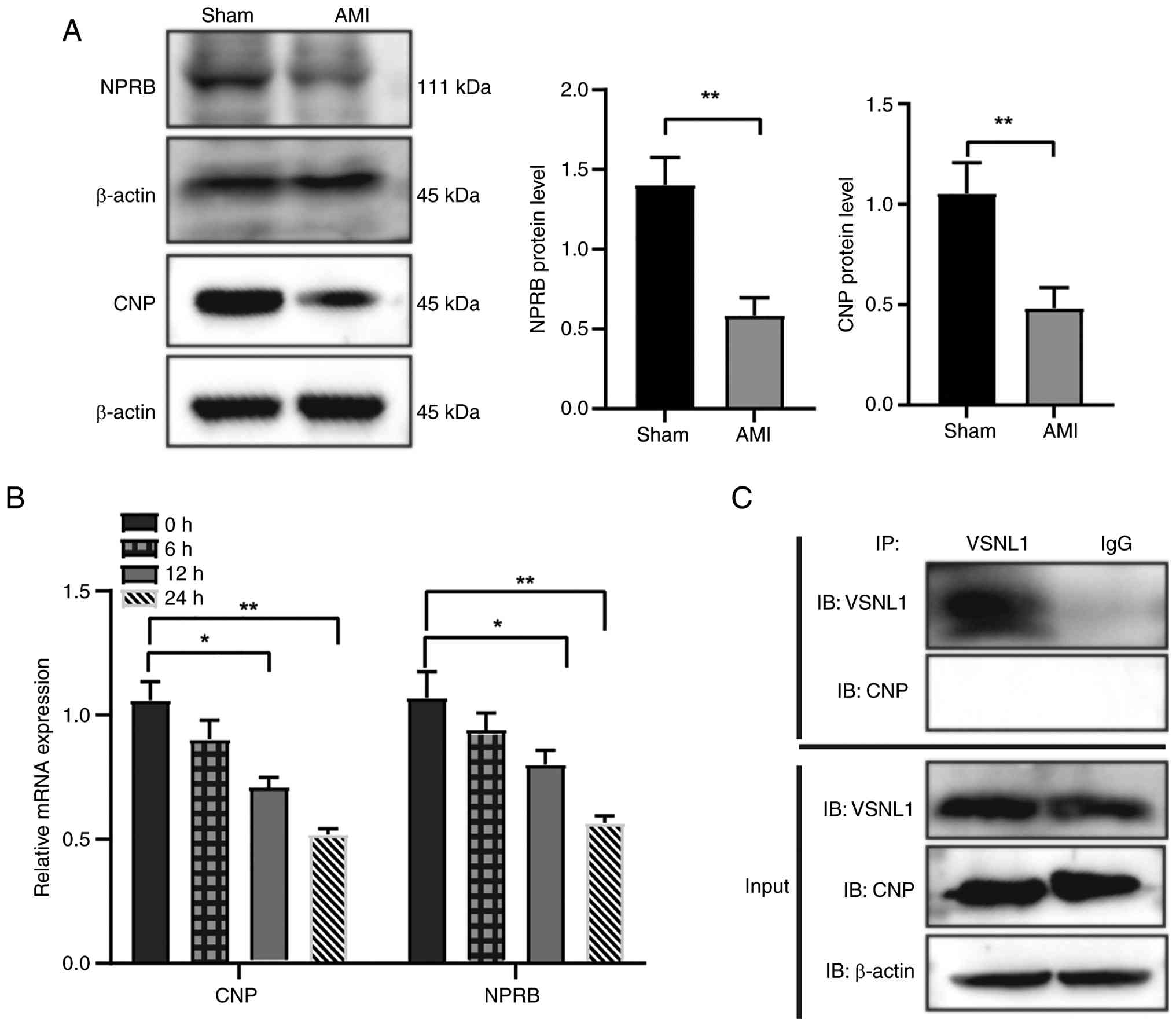
To assess the expression changes of the CNP/NPRB
signaling pathway during the acute phase of AMI, the present study
employed a mouse model of AMI and performed western blot analysis
of myocardial tissues from the Sham and AMI groups (24 h post-AMI).
The results revealed significant downregulation of CNP protein
expression in the AMI group compared with the Sham group. In
addition, the expression levels of NPRB were significantly reduced
in the AMI group (Fig. 7A). This
suggests that suppression of the CNP/NPRB pathway may serve a
critical role in the early pathological process of AMI.
To further evaluate the dynamic changes of this
signaling pathway under ischemic conditions, an in vitro AMI
model was established using AC16 cardiomyocytes. Cells were
harvested at multiple time points and qPCR analysis was performed.
The results demonstrated a continuous decline in mRNA expression
levels of both CNP and NPRB over time. Notably, their expression
levels at 24 h post-simulation were markedly reduced compared with
those at 0 h, supporting the sustained inhibition of the CNP/NPRB
pathway during myocardial ischemia (Fig. 7B).
To determine whether VSNL1 regulates this pathway
via direct protein-protein interaction, a Co-IP assay was performed
in AC16 cells. Experimental groups included the VSNL1-IP group, IgG
control group and total protein (Input) group. Immunoblotting was
performed using anti-VSNL1 and anti-CNP antibodies, and the results
revealed that both VSNL1 and CNP proteins were markedly expressed
in the Input group, indicating sufficient protein extraction. In
the VSNL1-IP group, VSNL1 was successfully precipitated using the
anti-VSNL1 antibodies, confirming the validity of the Co-IP system.
By contrast, no CNP band was detected in the VSNL1
immunoprecipitated complex, indicating that VSNL1 does not form a
stable, direct protein-protein complex with CNP under the
experimental conditions used. Additionally, no specific bands were
observed in the IgG control group, excluding the possibility of
nonspecific binding (Fig. 7C).
In summary, although VSNL1 may participate in
cardioprotective processes by modulating the CNP/NPRB signaling
pathway, its regulatory mechanism is more likely to be mediated
through signaling modulation rather than direct physical
interaction.
VSNL1 mediates hypoxia-induced
cardiomyocyte apoptosis via the CNP/NPRB pathway
The CNP/NPRB signaling pathway is known for its
protective anti-inflammatory and anti-apoptotic effects,
particularly in cardiac ischemia-reperfusion injury (23,24).
To assess whether VSNL1 regulates apoptosis through this pathway,
the present study evaluated its effect on CNP and NPRB expression.
The results demonstrated that hypoxia markedly downregulated CNP
and NPRB mRNA and protein levels, whereas VSNL1 overexpression
restored their expression (Fig. 8A and
B). Conversely, VSNL1 knockdown exacerbated their
downregulation (Fig. 8C).
Hypoxia-induced Ca2+ overload is closely associated with
cardiomyocyte damage and dysfunction (25). Given the role of VSNL1 in calcium
signaling, it was hypothesized that it maintains calcium
homeostasis via the CNP/NPRB pathway, which regulates cGMP levels
and calcium dynamics critical for myocardial function (26). Supporting this, the findings of the
present study demonstrated that VSNL1 overexpression markedly
reduced intracellular Ca2+ levels in hypoxia-exposed
AC16 cells (Fig. 8D).
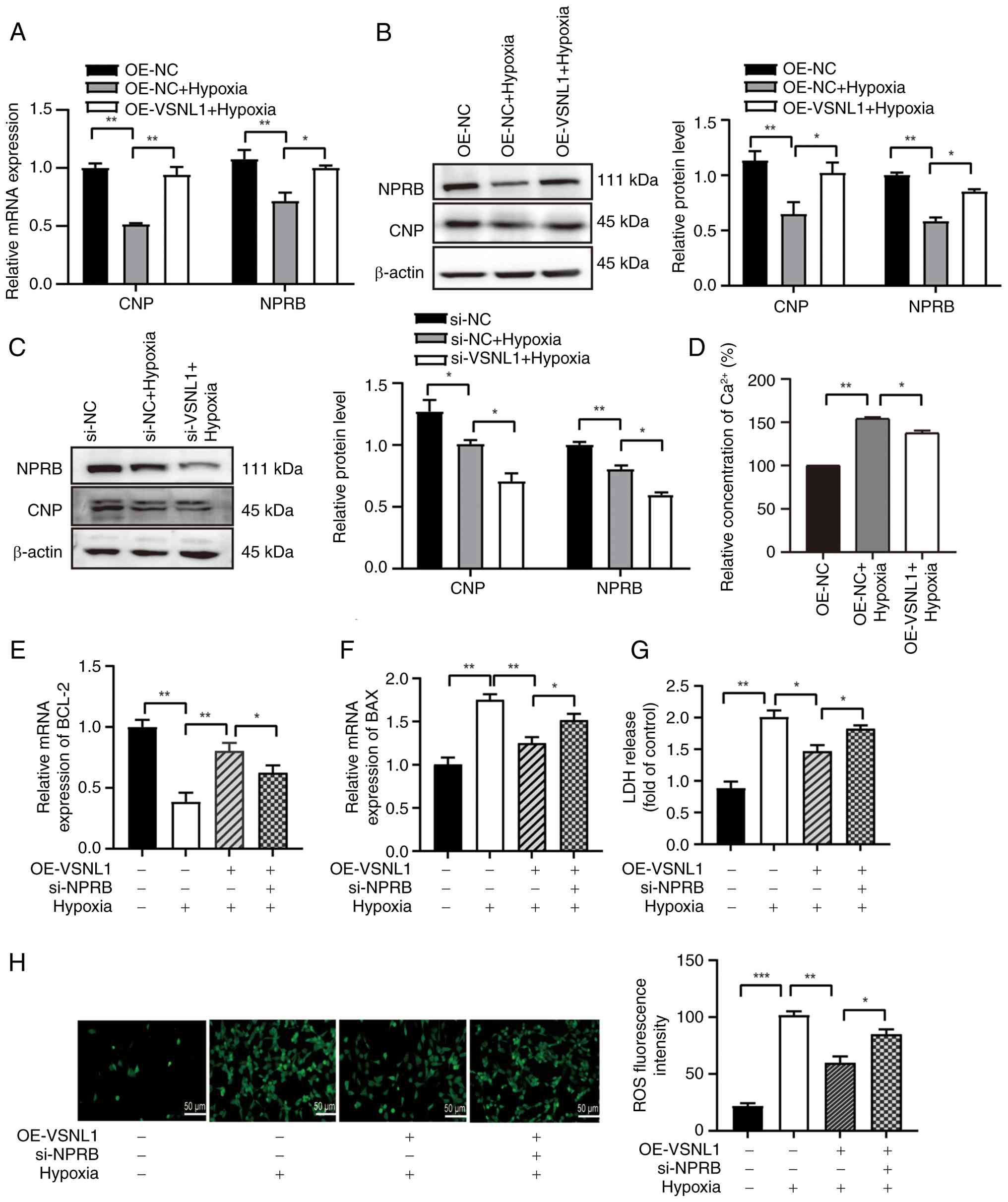 | Figure 8.VSNL1 regulates hypoxia-induced
apoptosis via the CNP/NPRB pathway in cardiomyocytes. (A) CNP and
NPRB expression in hypoxia-exposed AC16 cardiomyocytes following 24
h of VSNL1 overexpression, assessed by qPCR. (B) Protein expression
levels of CNP and NPRB in hypoxia-exposed cells and control cells
with VSNL1 overexpression, analyzed by western blotting. (C)
Protein expression levels of CNP and NPRB following VSNL1 knockdown
in hypoxic cells, measured by western blotting. (D) Intracellular
Ca2+ levels in hypoxia-injured and control cells, with
or without VSNL1 overexpression measured after 24 h. (E) BCL-2 and
(F) BAX mRNA expression following si-NPRB knockdown and OE-VSNL1
treatment in hypoxia-induced AC16 cardiomyocytes, assessed by qPCR.
(G) LDH release was measured in treated AC16 cardiomyocytes, where
the treatment consisted of VSNL1 overexpression, NPRB silencing and
hypoxic exposure. (H) ROS levels were measured. Scale bar, 50 µm.
*P<0.05; **P<0.01; ***P<0.001. VSNL1, visinin-like protein
1; CNP, 2′,3′-cyclic nucleotide 3′ phosphodiesterase; NPRB,
natriuretic peptide receptor B; qPCR, quantitative PCR; OE-NC,
overexpression negative control; OE-VSNL1, VSNL1 overexpression;
siRNA, small interfering RNA; si-NC, siRNA negative control;
si-VSNL1, VSNL1 knockdown via siRNA; si-NPRB, NPRB knockdown via
siRNA; LDH, lactate dehydrogenase; ROS, reactive oxygen
species. |
To further evaluate the role of the CNP/NPRB pathway
in the VSNL1-mediated anti-apoptotic mechanism, NPRB was silenced
in VSNL1-overexpressing AC16 cells under hypoxic conditions. The
results revealed that NPRB knockdown markedly interrupted the
anti-apoptotic effect of VSNL1 by decreasing BCL-2 expression while
increasing BAX expression (Fig. 8E and
F). Furthermore, NPRB silencing exacerbated cell injury
compared with VSNL1 overexpression alone, as indicated by increased
LDH release and elevated ROS levels (Fig. 8G and H). To confirm the efficiency
of si-NPRB transfection, western blot analysis was performed in
cells transfected with si-NPRB alone (without other treatments),
and successful knockdown of NPRB was confirmed (Fig. S1). Taken together, these findings
highlighted the critical role of CNP/NPRB as a downstream effector
of VSNL1 in mitigating hypoxia-induced cardiomyocyte apoptosis.
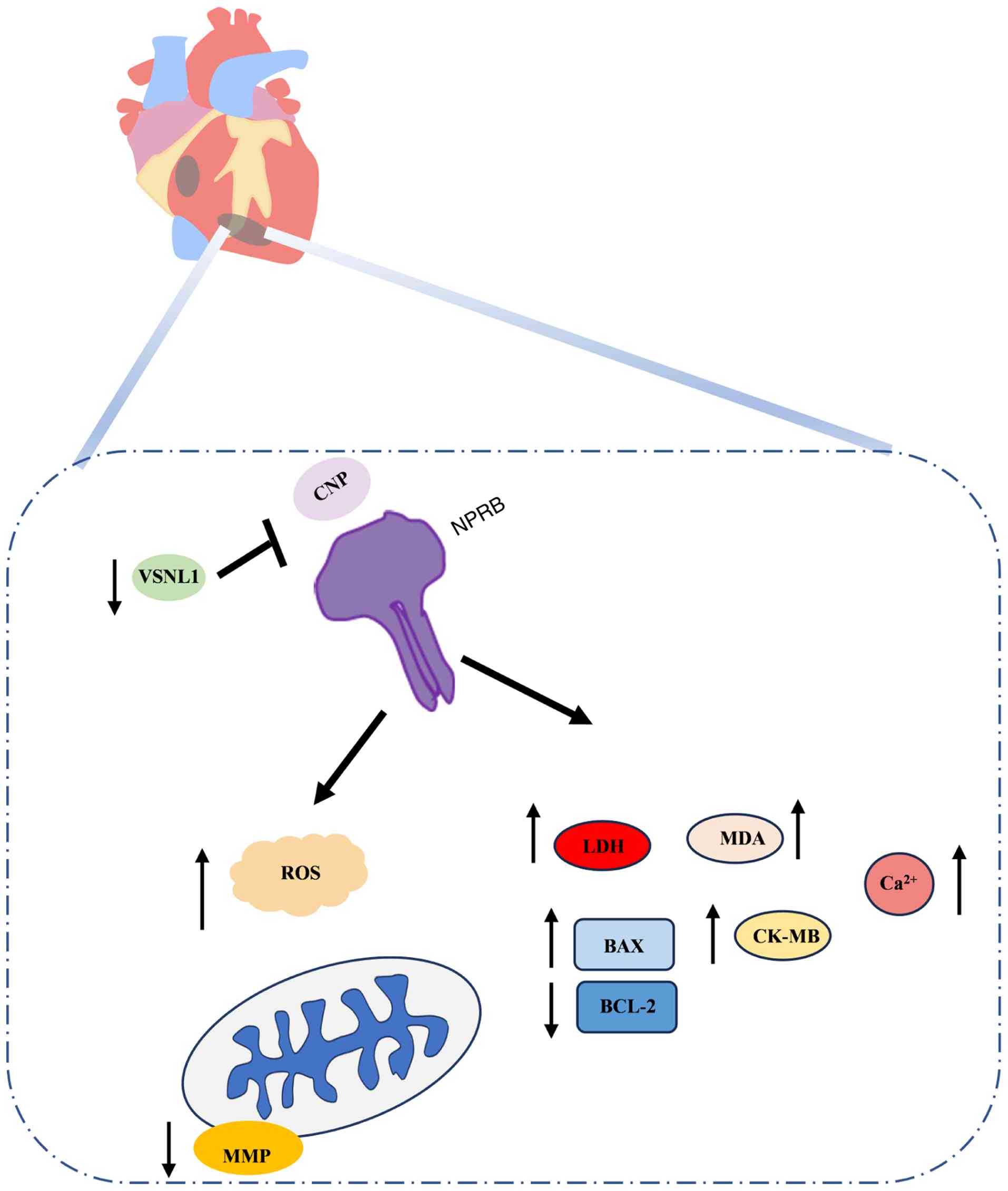
Discussion
The present study revealed downregulation of VSNL1
in both hypoxia-treated AC16 cardiomyocytes and myocardial tissue
from an AMI mouse model, suggesting its potential role in
ischemia-induced cardiac injury. Overexpression of VSNL1
effectively mitigated hypoxia-induced cardiomyocyte apoptosis and
alleviated associated cellular injury. Mechanically, VSNL1
decreased excessive ROS production and preserved the MMP in the
early stages of apoptosis, through the regulation of CNP/NPRB
signaling. The protective effect of VSNL1 on cardiomyocyte
apoptosis was further demonstrated by silencing of VSNL1, which
exacerbated apoptosis. The results further demonstrated that VSNL1
attenuated hypoxia-induced cardiomyocyte apoptosis and elucidated
the relevant signaling pathways. These findings provided compelling
evidence that VSNL1 protects cardiomyocytes from hypoxia-induced
apoptosis by modulating the CNP/NPRB signaling pathway (Fig. 9).
Consistent with previous findings that hypoxia
induces apoptosis in cardiomyocytes (27,28),
the data from the present study demonstrated that hypoxia triggered
apoptosis in AC16 cells. Emerging evidence highlights the role of
VSNL1 in cardiac development (29), an area of growing interest in
cardiology research. During early embryonic development, VSNL1 is
predominantly expressed in the crescent-shaped heart region and, as
development progresses, its expression shifts to become more
concentrated in the atrial precursors, the venous system and the
vascular plexus surrounding pulmonary structures (10). This dynamic expression pattern
suggests that VSNL1 serves an important regulatory role in heart
formation and structural establishment. In pathological contexts,
downregulation of VSNL1 has been observed in myocardial infarction,
potentially linked to oxidative stress or ischemic remodeling.
However, its specific molecular and cellular roles in the heart
remain incompletely understood (30). Aligning with these findings, the
present study demonstrated a time-dependent decrease in VSNL1
expression in hypoxia-treated AC16 cardiomyocytes. Furthermore,
VSNL1 knockdown markedly exacerbated apoptosis under hypoxic
conditions, whereas its overexpression alleviated these effects.
These results underscore the pivotal role of VSNL1 in protecting
cardiomyocytes from hypoxic injury by regulating apoptosis.
CNP, a key member of the natriuretic peptide family,
regulates cardiovascular functions primarily by stimulating cGMP
production through its receptor NPRB (31). Despite its low expression levels in
the heart, CNP exerts notable paracrine effects on vascular tone,
blood pressure, inflammation, angiogenesis and cardiomyocyte
function (32–34). Through the NPRB signaling pathway,
CNP provides cardioprotection by enhancing contractility, reducing
fibrosis and stabilizing cardiac electrophysiology (35–37).
Notably, CNP expression is markedly reduced in failing ventricles,
with its levels associated with disease severity and prognosis in
conditions such as ischemic and dilated cardiomyopathies (38). However, the precise mechanisms
through which the CNP/NPRB pathway regulates cardiomyocyte
apoptosis and maintains calcium homeostasis in HF remain poorly
understood. In the present study, it was demonstrated that hypoxic
conditions markedly reduced the expression levels of both CNP and
NPRB in AC16 cardiomyocytes. Notably, VSNL1 overexpression
activated the CNP/NPRB signaling pathway, whereas VSNL1 knockdown
inhibited this pathway. Additionally, siRNA-mediated knockdown of
NPRB demonstrated that the reduction of NPRB expression partially
compromised the anti-apoptotic effects of VSNL1 overexpression,
providing novel insights into the role of the CNP/NPRB signaling
pathway in regulating apoptosis in cardiomyocytes.
In HF, dysregulation of Ca2+ leads to
abnormal myocardial contraction and diastole, disrupting cardiac
function (39). CNP negatively
regulates protein kinase A and calcium/calmodulin-dependent protein
kinase II activity through activation of the cGMP signaling
pathway, while affecting Ca2+ transient amplitude,
demonstrating a complex Ca2+ processing regulation
(38). Furthermore, knockdown of
the calcium-sensing protein VSNL1 has been previously reported to
downregulate key calcium-handling proteins [such as
sarcoplasmic/endoplasmic reticulum calcium ATPase 2 (SERCA2) and
ryanodine receptor 2 (RyR2)] and voltage-dependent Ca2+
channels, indicating that VSNL1 may modulate myocardial
Ca2+ homeostasis, possibly in synergy with the CNP/NPRB
pathway (40). The present study
demonstrated that hypoxic injury resulted in increased calcium ion
levels, whereas overexpression of VSNL1 alleviated hypoxia-induced
calcium overload. Therefore, it was hypothesized that calcium ions
serve as downstream effectors of the CNP/NPRB pathway, potentially
mediated by cGMP-dependent regulation of calcium-handling proteins,
which warrants further investigation.
Although the present study did not directly assess
the downstream effectors of the CNP/NPRB signaling axis,
accumulating evidence strongly supports the existence of a
canonical CNP/NPRB/cGMP/protein kinase G (PKG) cascade mediating
cardioprotective effects through regulation of oxidative stress,
calcium homeostasis and mitochondrial integrity (41–44).
Upon activation by CNP, NPRB stimulates membrane-bound guanylyl
cyclase activity, elevating intracellular cGMP levels, which in
turn activate PKG, which is a key serine/threonine kinase involved
in cardiomyocyte survival (41).
PKG exerts anti-apoptotic effects by inhibiting mitochondrial
permeability transition pore opening through phosphorylation of
targets such as cyclophilin D and voltage dependent anion channel
1, thereby preserving mitochondrial function and suppressing
cytochrome c-mediated apoptosis (42).
PKG serves a critical role in calcium handling by
enhancing SERCA2a activity, inhibiting phospholamban and RyR2
function, and modulating Na+/Ca2+ exchanger
activity. These actions collectively promote efficient sarcoplasmic
reticulum Ca2+ reuptake, prevent cytosolic calcium
overload and maintain contractility under pathological conditions
such as ischemia-reperfusion injury (43,44).
While the current study primarily focused on the
upstream regulation and anti-apoptotic effects of VSNL1 via the
CNP/NPRB axis, elucidating the downstream signaling events is
essential to comprehensively understand the mechanism. As a future
research direction, it is intended to investigate the activation
status of PKG and the expression or modulation of calcium-handling
proteins in response to VSNL1 regulation, using both molecular and
functional assays. These future studies will aim to clarify whether
the CNP/NPRB-mediated cardioprotection involves cGMP signaling and
calcium homeostasis pathways.
In the present study, overexpression of VSNL1
resulted in reduced ROS accumulation, stabilization of the MMP and
attenuation of calcium dysregulation, which are phenotypes that
closely mirror the known downstream effects of PKG activation. As
VSNL1 is a neuronal calcium sensor protein with EF-hand motifs and
high calcium affinity (45), it is
plausible that VSNL1 synergizes with the CNP/NPRB/cGMP/PKG
signaling axis to fine-tune calcium flux and mitochondrial
function, particularly within specialized microdomains such as the
sarcoplasmic reticulum-mitochondria interface.
It can be hypothesized that VSNL1 enhances the
cardioprotective effects of CNP/NPRB signaling through the
following pathways: i) Facilitating intracellular calcium buffering
to mitigate calcium overload; ii) interacting with calcium-handling
proteins to stabilize their expression or phosphorylation; and iii)
serving as a scaffolding protein that spatially couples PKG with
its downstream effectors. While direct biochemical validation
remains necessary, the current literature provides a robust
theoretical framework supporting the involvement of the cGMP/PKG
axis in mediating the cardioprotective effects observed with VSNL1
overexpression (13,46). These insights establish a plausible
mechanistic link between VSNL1 and natriuretic peptide signaling
and highlight VSNL1 as a potential therapeutic node for ischemic
heart disease.
In conclusion, the present study highlighted the
role of VSNL1 in alleviating hypoxia-induced cardiomyocyte
apoptosis and the potential for developing novel therapeutic
strategies, such as VSNL1 agonists, offering promising clinical
implications. Nonetheless, an important limitation should be noted:
While the AC16 cell line is of human origin, its phenotype differs
from that of mature cardiomyocytes. To strengthen the reliability
and clinical relevance of the findings of the present study, future
research should validate these results using experimental animal
models that more closely replicate the physiological conditions of
the human heart, as well as primary cardiomyocytes. Such approaches
will provide stronger evidence for translating these insights into
therapeutic strategies.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was funded by the Shanghai Science and
Technology Innovation Action Plan (grant nos. 21S11901700 and
22010502600), the Shanghai Natural Science Foundation (grant no.
21ZR1428400) and the Clinical Research Project of Pudong New Area
Health Commission (Grant No. 2025-PWYC-14), Shanghai, China.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
YL and XC were responsible for the conceptualization
and design of the study. Material preparation, data collection and
analysis were performed by XY, WF, RC and LM. The manuscript was
drafted by XY, XC and YL, who also critically revised it for
important intellectual content. YL and XC confirm the authenticity
of all the raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
All animal experimental procedures in the present
study were reviewed and approved by the Ethics Committee of
Shanghai University of Medicine and Health Sciences (approval no.
2021-SZR-05-410482198512239314; Shanghai, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Yu X, Yang Y, Chen T, Wang Y, Guo T, Liu
Y, Li H and Yang L: Cell death regulation in myocardial toxicity
induced by antineoplastic drugs. Front Cell Dev Biol.
11:10759172023. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen XJ, Liu SY, Li SM, Feng JK, Hu Y,
Cheng XZ, Hou CZ, Xu Y, Hu M, Feng L and Xiao L: The recent advance
and prospect of natural source compounds for the treatment of heart
failure. Heliyon. 10:e271102024. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jiang M, Fan X, Wang Y and Sun X: Effects
of hypoxia in cardiac metabolic remodeling and heart failure. Exp
Cell Res. 432:1137632023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen Z, Zhang S, Guo C, Li J and Sang W:
Downregulation of miR-200c protects cardiomyocytes from
hypoxia-induced apoptosis by targeting GATA-4. Int J Mol Med.
39:1589–1596. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang X, Hu C, Kong CY, Song P, Wu HM, Xu
SC, Yuan YP, Deng W, Ma ZG and Tang QZ: FNDC5 alleviates oxidative
stress and cardiomyocyte apoptosis in doxorubicin-induced
cardiotoxicity via activating AKT. Cell Death Differ. 27:540–555.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ma L, Liu M, Liu C, Zhang H, Yang S, An J,
Qu G, Song S and Cao Q: Research progress on the mechanism of the
antitumor effects of cannabidiol. Molecules. 29:19432024.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li C, Liu X, Li J, Lai J, Su J, Zhu B, Gao
B, Li Y and Zhao M: Selenomethionine inhibited HADV-induced
apoptosis mediated by ROS through the JAK-STAT3 signaling pathway.
Nutrients. 16:19662024. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hao M, Liu Y, Chen P, Jiang H and Kuang
HY: Astragaloside IV protects RGC-5 cells against oxidative stress.
Neural Regen Res. 13:1081–1086. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tage H, Yamaguchi K, Nakagawa S, Kasuga S,
Takane K, Furukawa Y and Ikenoue T: Visinin-like 1, a novel target
gene of the Wnt/β-catenin signaling pathway, is involved in
apoptosis resistance in colorectal cancer. Cancer Med.
12:13426–13437. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ola R, Lefebvre S, Braunewell KH, Sainio K
and Sariola H: The expression of Visinin-like 1 during mouse
embryonic development. Gene Expr Patterns. 12:53–62. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
He C, Liu W, Xiong Y, Wang Y, Pan L, Luo
L, Tu Y, Song R and Chen W: VSNL1 promotes cell proliferation,
migration, and invasion in colorectal cancer by binding with
COL10A1. Ann Clin Lab Sci. 52:60–72. 2022.PubMed/NCBI
|
|
12
|
Aiba T, Hijiya N, Akagi T, Tsukamoto Y,
Hirashita Y, Kinoshita K, Uchida T, Nakada C, Kurogi S, Ueda Y, et
al: Overexpression of VSNL1 enhances cell proliferation in
colorectal carcinogenesis. Pathobiology. 91:121–131. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Braunewell KH, Brackmann M, Schaupp M,
Spilker C, Anand R and Gundelfinger ED: Intracellular neuronal
calcium sensor (NCS) protein VILIP-1 modulates cGMP signalling
pathways in transfected neural cells and cerebellar granule
neurones. J Neurochem. 78:1277–1286. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Williams TA, Monticone S, Crudo V, Warth
R, Veglio F and Mulatero P: Visinin-like 1 is upregulated in
aldosterone-producing adenomas with KCNJ5 mutations and protects
from calcium-induced apoptosis. Hypertension. 59:833–839. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tan BL, Norhaizan ME and Chan LC:
Manilkara Zapota (L.) P. Royen leaf water extract induces
apoptosis in human hepatocellular carcinoma (HepG2) Cells via
ERK1/2/Akt1/JNK1 signaling pathways. Evid Based Complement Alternat
Med. 2018:78265762018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chang X, Zhang T, Wang J, Liu Y, Yan P,
Meng Q, Yin Y and Wang S: SIRT5-related desuccinylation
modification contributes to quercetin-induced protection against
heart failure and high-glucose-prompted cardiomyocytes injured
through regulation of mitochondrial quality surveillance. Oxid Med
Cell Longev. 2021:58768412021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Peng J, Yang Z, Li H, Hao B, Cui D, Shang
R, Lv Y, Liu Y, Pu W, Zhang H, et al: Quercetin reprograms
immunometabolism of macrophages via the SIRT1/PGC-1α signaling
pathway to ameliorate lipopolysaccharide-induced oxidative damage.
Int J Mol Sci. 24:55422023. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang Y, Chen G, Zhuang X and Guo M:
Inhibition of growth of colon tumors and proliferation of HT-29
cells by Warburgia ugandensis extract through mediating
G0/G1 cell cycle arrest, cell apoptosis, and
intracellular ROS generation. Oxid Med Cell Longev.
2021:88076762021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Owari T, Tanaka N, Nakai Y, Miyake M, Anai
S, Kishi S, Mori S, Fujiwara-Tani R, Hojo Y, Mori T, et al:
5-Aminolevulinic acid overcomes hypoxia-induced radiation
resistance by enhancing mitochondrial reactive oxygen species
production in prostate cancer cells. Br J Cancer. 127:350–363.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Deng F, Wang S, Cai S, Hu Z, Xu R, Wang J,
Feng D and Zhang L: Inhibition of caveolae contributes to propofol
preconditioning-suppressed microvesicles release and cell injury by
hypoxia-reoxygenation. Oxid Med Cell Longev. 2017:35421492017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ma J, Yu W, Wang Y, Cao G, Cai S, Chen X,
Yan N, Yuan Y, Zeng H, Fleenor DL, et al: Neuroprotective effects
of C-type natriuretic peptide on rat retinal ganglion cells. Invest
Ophthalmol Vis Sci. 51:3544–3553. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jin X, Zhang Y, Li X, Zhang J and Xu D:
C-type natriuretic peptide ameliorates ischemia/reperfusion-induced
acute kidney injury by inhibiting apoptosis and oxidative stress in
rats. Life Sci. 117:40–45. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ruhr IM, Shiels HA, Crossley DA II and
Galli GLJ: Developmental programming of sarcoplasmic reticulum
function improves cardiac anoxia tolerance in turtles. J Exp Biol.
227:jeb2474342024. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Abuzaanona A and Lanfear D:
Pharmacogenomics of the natriuretic peptide system in heart
failure. Curr Heart Fail Rep. 14:536–542. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hu D, Gu Y, Wu D, Zhang J, Li Q, Luo J, Li
S, Yuan Z and Zhu B: Icariside II protects cardiomyocytes from
hypoxia-induced injury by upregulating the miR-7-5p/BTG2 axis and
activating the PI3K/Akt signaling pathway. Int J Mol Med.
46:1453–1465. 2020.PubMed/NCBI
|
|
27
|
Xie X, Ji Q, Han X, Zhang L and Li J:
Knockdown of long non-coding RNA TTTY15 protects cardiomyocytes
from hypoxia-induced injury by regulating let-7b/MAPK6 axis. Int J
Clin Exp Pathol. 13:1951–1961. 2020.PubMed/NCBI
|
|
28
|
Fan W, Yang C, Hou X, Wan J and Liao B:
Novel insights into the sinoatrial node in single-cell RNA
sequencing: From developmental biology to physiological function. J
Cardiovasc Dev Dis. 9:9862022.
|
|
29
|
Buttgereit J, Qadri F, Monti J,
Langenickel TH, Dietz R, Braunewell KH and Bader M: Visinin-like
protein 1 regulates natriuretic peptide receptor B in the heart.
Regul Pept. 161:51–57. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mei C, Kang Y, Zhang C, He C, Liao A and
Huang D: C-type natriuretic peptide plays an anti-inflammatory role
in rat epididymitis induced by UPEC. Front Cell Infect
Microbiol. 11:7118422021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nakagawa Y and Nishikimi T: CNP, the third
natriuretic peptide: Its biology and significance to the
cardiovascular system. Biology (Basel). 11:9862022.PubMed/NCBI
|
|
32
|
Bubb KJ, Aubdool AA, Moyes AJ, Lewis S,
Drayton JP, Tang O, Mehta V, Zachary IC, Abraham DJ, Tsui J and
Hobbs AJ: Endothelial C-type natriuretic peptide is a critical
regulator of angiogenesis and vascular remodeling. Circulation.
139:1612–1628. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Moyes AJ and Hobbs AJ: C-type Natriuretic
peptide: A multifaceted paracrine regulator in the heart and
vasculature. Int J Mol Sci. 20:22812019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pernomian L, Prado AF, Silva BR, Azevedo
A, Pinheiro LC, Tanus-Santos JE and Bendhack LM: C-type natriuretic
peptide induces anti-contractile effect dependent on nitric oxide,
oxidative stress, and NPR-B activation in sepsis. Front Physiol.
7:2262016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dorey TW, Liu Y, Jansen HJ, Bohne LJ,
Mackasey M, Atkinson L, Prasai S, Belke DD, Fatehi-Hassanabad A,
Fedak PWM and Rose RA: Natriuretic peptide receptor B protects
against atrial fibrillation by controlling atrial cAMP via
phosphodiesterase 2. Circ Arrhythm Electrophysiol. 16:e0121992023.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dorey TW, Mackasey M, Jansen HJ, McRae MD,
Bohne LJ, Liu Y, Belke DD, Atkinson L and Rose RA: Natriuretic
peptide receptor B maintains heart rate and sinoatrial node
function via cyclic GMP-mediated signalling. Cardiovasc Res.
118:1917–1931. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cachorro E, Günscht M, Schubert M, Sadek
MS, Siegert J, Dutt F, Bauermeister C, Quickert S, Berning H,
Nowakowski F, et al: CNP promotes antiarrhythmic effects via
phosphodiesterase 2. Circ Res. 132:400–414. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dabravolski SA, Sadykhov NK, Kartuesov AG,
Borisov EE, Sukhorukov VN and Orekhov AN: Interplay between
Zn2+ homeostasis and mitochondrial functions in
cardiovascular diseases and heart ageing. Int J Mol Sci.
23:68902022. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liang D, Xue J, Geng L, Zhou L, Lv B, Zeng
Q, Xiong K, Zhou H, Xie D, Zhang F, et al: Cellular and molecular
landscape of mammalian sinoatrial node revealed by single-cell RNA
sequencing. Nat Commun. 12:2872021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
O'Rourke B, Van Eyk JE and Foster DB:
Mitochondrial protein phosphorylation as a regulatory modality:
Implications for mitochondrial dysfunction in heart failure.
Congest Heart Fail. 17:269–282. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Potter LR, Abbey-Hosch S and Dickey DM:
Natriuretic peptides, their receptors, and cyclic guanosine
monophosphate-dependent signaling functions. Endocr Rev. 27:47–72.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Inserte J and Garcia-Dorado D: The
cGMP/PKG pathway as a common mediator of cardioprotection:
Translatability and mechanism. Br J Pharmacol. 172:1996–2009. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Takimoto E, Champion HC, Li M, Belardi D,
Ren S, Rodriguez ER, Bedja D, Gabrielson KL, Wang Y and Kass DA:
Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and
reverses cardiac hypertrophy. Nat Med. 11:214–222. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhazykbayeva S, Budde H, Kaçmaz M, Zemedie
Y, Osman H, Hassoun R, Jaquet K, Akin I, El-Battrawy I and Herwig
M: Exploring PKG signaling as a therapeutic avenue for pressure
overload, ischemia, and HFpEF. Expert Opin Ther Targets.
28:857–873. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Dai QQ, Wang YY, Jiang YP, Li L and Wang
HJ: VSNL1 promotes gastric cancer cell proliferation and migration
by regulating P2X3/P2Y2 receptors and is a clinical indicator of
poor prognosis in gastric cancer patients. Gastroenterol Res Pract.
2020:72419422020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Brackmann M, Schuchmann S, Anand R and
Braunewell KH: Neuronal Ca2+ sensor protein VILIP-1 affects cGMP
signalling of guanylyl cyclase B by regulating clathrin-dependent
receptor recycling in hippocampal neurons. J Cell Sci.
118:2495–2505. 2005. View Article : Google Scholar : PubMed/NCBI
|