Introduction
Liver cancer ranks as the sixth most prevalent
malignancy, with hepatocellular carcinoma (HCC) comprising ~90% of
cases (1,2). Projections suggest that by 2030, HCC
will be responsible for >1 million deaths globally, positioning
it among the deadliest cancers (3). Notably, numerous patients are
diagnosed at an advanced stage, highlighting the need for reliable
methods for early detection and monitoring of HCC metastasis and
recurrence. Advancements in early diagnosis and management of HCC
have been substantial, with molecular detection techniques for
early detection gaining marked attention and moving toward clinical
application (4). Liquid biopsy, an
emerging non-invasive diagnostic approach, involves collecting
liquid samples to detect tumor-associated molecular markers,
providing insights into the tumor phenotype, genetics and
transcriptome (5). Key components
include circulating tumor cells (CTCs), extracellular vesicles
(EVs), circulating tumor DNA and RNA (6). Among these, CTCs and EVs have
garnered marked interest due to their stability and sensitivity,
making them the most promising liquid biopsy markers. CTCs are
malignant cells shed into the bloodstream from primary or
metastatic tumors, capable of invading distant organs via
the circulatory system, earning them the term disseminators of
malignancy (7,8). Some researchers propose that CTCs
have robust prognostic value, serving as tools for monitoring HCC
progression and guiding treatment post-resection of the primary
lesion (9,10). Based on their biological mechanisms
and size, EVs are typically categorized into exosomes,
microvesicles and apoptotic bodies, all playing pivotal roles in
tumorigenesis, metastasis and invasion. Among them, exosomes are
nanoscale vesicles with a diameter of 30–150 nm; they originate
from the endosomal system and are released into the extracellular
space upon fusion of multivesicular bodies with the plasma
membrane. Microvesicles have a size range of 100–1,000 nm and are
directly formed by budding off from the plasma membrane. Apoptotic
bodies are the largest EV subtype, with a diameter typically
ranging from 50 to 5,000 nm; they are formed at the end stage of
programmed cell death (apoptosis) and are generated by budding and
rupture of the apoptotic cell membrane (11,12);
in this context, they are collectively referred to as EVs.
Tumor-derived EVs, detectable in the blood of patients with
early-stage HCC, regulate recipient cell functions by reprogramming
signaling pathways. Cancer cell-derived EVs notably alter the tumor
microenvironment (TME) and facilitate the formation of
pre-metastatic niches (PMNs) (13). Furthermore, EVs have been shown to
protect CTCs during the metastatic process. Molecules carried by
EVs, such as nucleic acids, proteins and microRNAs (miRNAs),
contribute to the recurrence and metastasis of HCC via CTCs,
where CTCs are likened to seeds, and the cargo within EVs acts as
fertilizer, together influencing cancer cell behavior and promoting
metastasis. The present manuscript reviews the mechanisms
underlying EV and CTC-mediated HCC metastasis, as well as the role
of EVs in enhancing CTC-driven metastasis, to provide a
comprehensive understanding of HCC metastasis, recurrence and
prognosis (14) (Fig. 1).
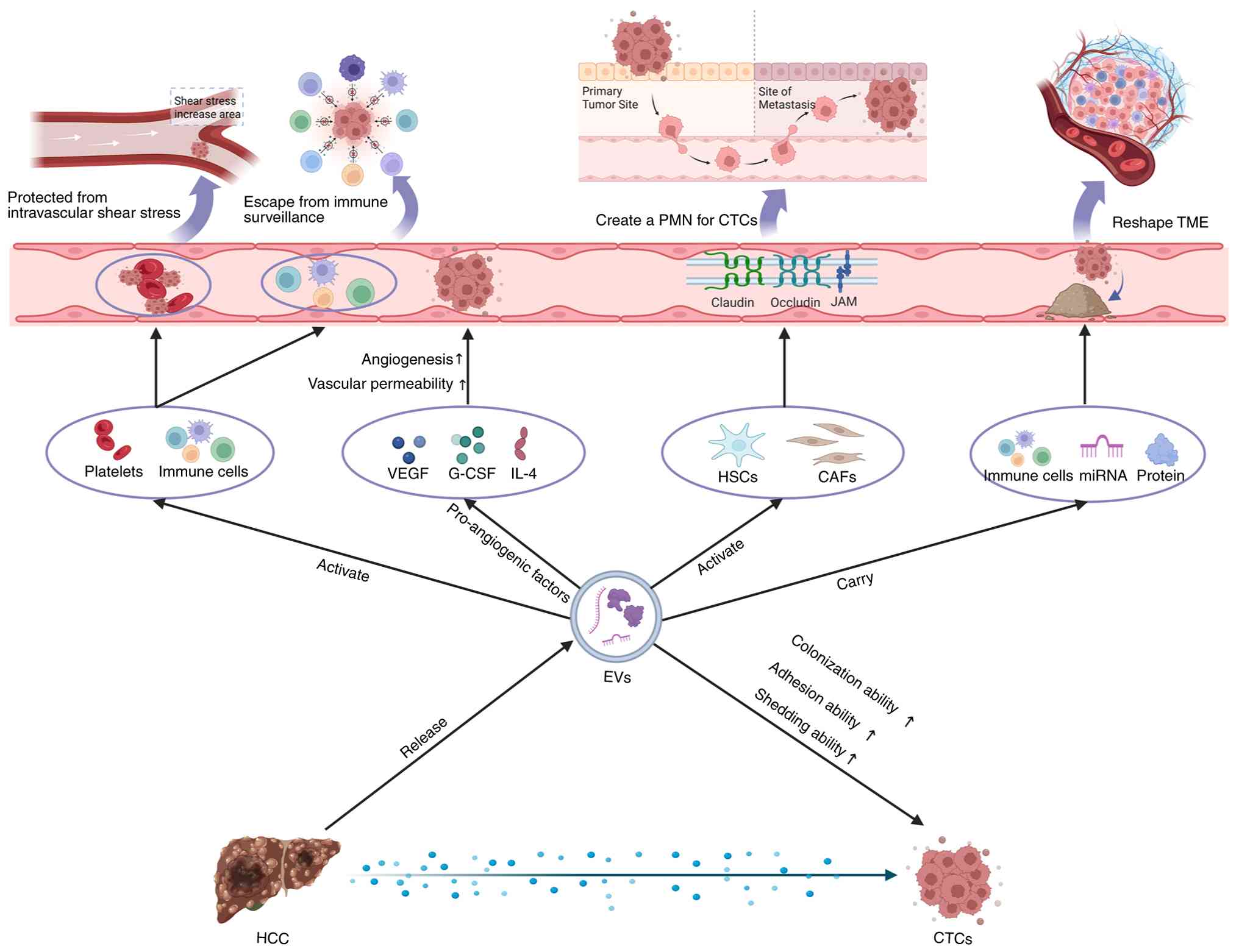 | Figure 1.Mechanism of EVs-mediated CTCs
promoting HCC metastasis. EVs protect CTCs by reducing
intravascular shear stress and evading immune surveillance, while
releasing cytokines to increase vascular permeability and enhance
the shedding ability of CTCs. Additionally, EVs can create
pre-metastatic niches for CTCs and remodel the TME, promoting the
colonization ability of CTCs. CTC, circulating tumor cells; TME,
tumor microenvironment; HSC, hepatic stellate cell; EVs,
extracellular vesicles; PMN, pre-metastatic niches; miRNA,
microRNA; CAFs, cancer associated fibroblasts; HCC, hepatocellular
carcinoma. |
Mechanism of EVs in HCC metastasis
EVs play a complex yet pivotal role in HCC
metastasis. Due to their high heterogeneity, distinct
subpopulations of EVs can precisely and collaboratively drive the
metastasis process. Their mechanisms encompass various aspects,
including PMN formation, modulation of the tumor immune
microenvironment (TIME), immune regulation and angiogenesis. The
present section summarizes the involvement of EVs in HCC recurrence
and metastasis (15–24) (Table
I).
 | Table I.Role of EVs in hepatocellular
carcinoma recurrence and metastasis. |
Table I.
Role of EVs in hepatocellular
carcinoma recurrence and metastasis.
| Cargo | Pathway | Mechanisms | (Refs.) |
|---|
| EV-CLTA | Up regulation of
BSG | Remodeling the
microvascular niche | (15) |
| miRNA-638 | Down regulation of
vascular endothelial-cadherin and ZO-1 | Remodeling the
microvascular niche | (16) |
| miRNA-1247-3p | Activation of
β1-integrin-NF-κB | Promote CAF
transformation | (17) |
| miRNA-92b | Inhibition of
CD69 | Inhibition of NK
cell activity | (18) |
| lncRNA PART1 | Up regulation of
TLR4 | Promote macrophage
M2 polarization | (19) |
| miRNA-15b | Inhibition of
Hippo | Promote macrophage
M2 polarization | (20) |
| EV-S100A10 | Up regulation of
EGFR | Promote EMT | (21) |
| miRNA-21 | Activation of
PDK1/AKT | Activation of CAFs
and promote angiogenesis | (22) |
| miRNA-21-5p | Activation of
YAP/β-catenin | CD8+T cell
depletion | (23) |
| miRNA-92a-2-5p | Activation of
PHLPP/p-AKT/β-catenin | Regulation of tumor
immune microenvironment | (24) |
PMN
The establishment of a tumor-induced
microenvironment in distant organs is needed for the colonization,
adaptation and survival of metastatic cells. This metastatic niche,
located in distant organs, is termed the PMN. The initial event in
HCC metastasis is the formation of the PMN, which may even occur
during the early stages of HCC development (25). EVs, secreted by various cell types,
serve as intercellular communication carriers; the unique receptors
and specific intercellular adhesion molecules on their surfaces
enable targeted recognition of recipient cells and facilitate
signal transduction. Subpopulations of EVs derived from different
cellular sources play distinct roles and collaborate in this
process. Specifically, integrins on the surface of tumor-derived
EVs are essential for their interactions with distant organs during
PMN formation (26); therefore,
EVs act as key mediators in the formation of PMNs.
A hallmark of aggressive malignancies is excessive
angiogenesis. Neovascularization is essential for providing oxygen,
nutrients and energy, all of which support cancer cell growth,
metastasis and the development of multidrug resistance (27). Remodeling of the microvasculature
is a prerequisite for PMN formation, including multi-step
collaborative processes such as increased vascular permeability,
neovascularization and functional reprogramming of endothelial
cells (28,29). The heterogeneity of EVs is
reflected in their precision in regulating endothelial cells. Small
EV subpopulations derived from HCC, rich in miRNA cargo, play a
central role in regulating endothelial cell function; for instance,
an EV subpopulation enriched with miR-183-5p activates the PI3K/AKT
pathway by downregulating SIK1, thereby enhancing endothelial cell
proliferation, migration, angiogenesis and vascular permeability,
ultimately promoting metastasis in vivo (30). By contrast, large EVs or apoptotic
body subpopulations may be more inclined to carry and deliver
intact protein growth factors, such as vascular endothelial growth
factor (VEGF) and fibroblast growth factor, directly providing
strong pro-proliferative signals to endothelial cells. For
instance, Golgi protein 73 secreted by EVs derived from HCC can
enhance VEGF production in HCC cells and bolster mitogenic
signaling in vascular endothelial cells, thereby promoting
angiogenesis in the TME (31).
Studies by Xu et al (15)
and Yokota et al (16)
established the key paradigm of HCC-derived EVs disrupting vascular
barriers, remodeling angiogenesis and initiating PMN by delivering
effector molecules such as basigin and miR-638, while
downregulating critical vascular endothelial junction proteins
(such as VE-cadherin, ZO-1). These findings are notable but
focusing solely on these functions risks oversimplifying a dynamic
and heterogeneous biological process. At the mechanistic level,
while individual effector molecules have been identified, little is
known about their upstream regulation; which tumor cell
subpopulations selectively load these cargos and under what
conditions? In addition, increased vascular permeability is only
the initial step in PMN formation. Furthermore, could the damaged
vascular endothelium release specific EVs or factors that feedback
and enhance the invasiveness of the primary tumor? In the context
of hepatocirrhosis, where HCC is already associated with
inflammation and vascular abnormalities, do tumor-derived EVs
exacerbate this ‘fragile foundation’ or initiate a novel
destructive program? Differentiating between these two scenarios is
essential for understanding the specificity of HCC metastasis.
In the metastatic niche, cancer-associated
fibroblasts (CAFs) play an active role in tumor metastasis
progression (32). Fang et
al (17) showed that
high-metastatic HCC cells have a greater ability to convert normal
fibroblasts into CAFs compared with low-metastatic HCC cells during
PMN formation. Fibroblasts induced by EVs from high-metastatic HCC
cells exhibit elevated expression of pro-inflammatory genes such as
interleukin-1β, transforming growth factor β (TGF-β) and collagen
types I, III and IV, which are critical for modulating the TME and
promoting carcinogenesis. Exosome microarray detection confirmed
the notable role of HCC-derived EVs in converting fibroblasts to
CAFs by activating the β1-integrin-NF-κB signaling within the PMN.
This research revealed that specific subpopulations of EVs released
by highly metastatic HCC cells express unique integrin combinations
on their surface, which dictate their targeting to specific organs
such as the liver and lungs. Upon uptake by resident cells in these
target organs, EVs release their cargo, such as miR-638, which
activates relevant signaling pathways, promoting angiogenesis,
inflammatory factor release and initiating PMN formation.
TIME
The TIME encompasses various immune cells,
extracellular immune factors, endothelial cells and CAFs, all
contributing to tumor survival (33). The recurrence and metastasis of HCC
is a multi-step process in which tumor cells not only evade
apoptosis and immune responses but also rely on immune cell
communication within the TIME (34). Immune cells, including
tumor-associated macrophages (TAMs) and regulatory T cells (Tregs),
accumulate within the TIME and facilitate the establishment of an
immunosuppressive environment. Conversely, immune cells such as
natural killer (NK) cells, CD8+ T cells, and
CD4+ T cells work together to counteract tumor-promoting
effects (35). EVs are abundant in
the immune microenvironment of HCC, acting as mediators of
intercellular communication. The transfer of RNA or proteins
via EVs between cells can influence the composition and
homeostasis of the TIME, contributing to immune escape and
promoting HCC metastasis and recurrence (36,37).
Numerous studies have confirmed that EV-mediated
crosstalk regulates immune cell activity within the HCC TIME. The
functional impairment of NK cells is recognized as a key mechanism
for tumor cells to evade immune surveillance. Liu et al
(38) demonstrated in an HCC mouse
model that EVs secreted by hepatocytes lacking
fructose-1,6-bisphosphatase 1 specifically target and infiltrate NK
cells, suppressing NK-mediated tumor surveillance and promoting
immune remodeling within the HCC TME. Exosomes secreted by HCC
cells, which carry major histocompatibility complex class I-related
chain A, may inhibit NK cell function by competitively suppressing
agonistic NKG2D receptor signaling, thereby reducing NK cell
cytotoxicity against HCC in vivo (39,40).
Additionally, microRNA carried by EVs derived from HCC can further
contribute to NK cell dysfunction and immune evasion. For instance,
miR-92b, mediated by HCC-EVs, downregulates the activation marker
CD69 on NK cells, impairing their activity (18).
Macrophages, notable players in the innate immune
response of the liver, are also involved in HCC proliferation,
metastasis, invasion and angiogenesis (41). TAMs are increasingly recognized as
essential components of the HCC TIME (42,43).
Tumor-derived factors can polarize macrophages into either the M1
phenotype, which exhibits anti-tumor activity, or the M2 phenotype,
which promotes tumor growth (44).
EVs derived from HCC cells promote macrophage activation and M2
polarization through various mechanisms, enabling tumors to evade
immune surveillance (45,46). For example, EVs secreted by HCC
deliver long non-coding (lnc)RNA PART1 to TAMs, upregulating TLR4
expression and thus inducing M2 polarization. Polarized M2
macrophages in turn promote HCC cell proliferation, metastasis and
tumor growth in vivo (19).
In another study, M2 polarization induced by HCC-EVs released
exosomes that transferred miR-15b to HCC cells, inhibiting the
Hippo pathway and promoting HCC proliferation, invasion and
metastasis by targeting large tumor suppressor kinase 1 (20). The T cell-mediated immune response
is central to cancer immunity, critical for immune surveillance and
tumor cell clearance (47). In the
TIME, T cell regeneration often leads to the deterioration of T
cell responses and HCC progression. EVs may modulate the function
and activity of CD4+ T cells, CD8+ T cells
and Tregs, influencing the homeostasis of TIME and contributing to
immune suppression, HCC progression and metastasis (19,48).
Gong et al (49)
demonstrated that norcholic acid increases programmed death ligand
1 (PD-L1) levels on the surface of HCC and its exosomes, notably
inhibiting CD4+ T cell function and inducing an
immunosuppressive microenvironment that promotes HCC proliferation,
invasion and metastasis. Furthermore, EVs from other immune cells
in HCC contribute to the depletion of CD8+ T cells,
another mechanism of immune evasion. For example, HCC-EVs carrying
miR-146a-5p induce macrophage polarization towards the
CD206+PD-L1+CD80+ M2 phenotype,
and these EVs-induced macrophages impair CD3+ T cell
function by upregulating expression of inhibitory receptors (such
as programmed cell death-1 and cytotoxic T-lymphocyte associated
protein 4) on co-cultured T cells (50). Another mechanism for creating an
immunosuppressive microenvironment in HCC is the recruitment of
Tregs to TIME. The acidic microenvironment in HCC promotes the
upregulation of miR-21 expression in EVs; miR-21 enhances Treg
proliferation and recruitment to TIME while reducing the proportion
of Th17 cells. The resulting imbalance between Th17 and Treg cells
leads to immune suppression, fostering HCC invasion and metastasis
(51,52).
CAFs are key components of the TIME, with increasing
evidence supporting their role in promoting HCC recurrence and
metastasis through immune suppression (53,54).
EVs carrying miR-20a-5p, released by CAFs, activate the
Wnt/β-catenin signaling pathway by downregulating LIM domain and
actin binding 1, thereby facilitating immune evasion and enhancing
HCC metastasis and invasion (55,56).
Under hypoxic conditions, CAFs inhibit the activity and
cytotoxicity of CD8+ T cells via an EV-dependent
mechanism, secreting EVs rich in circHIF1A and binding to HuR,
upregulating PD-L1 expression, which in turn promotes HCC cell
proliferation, migration and invasion. These findings suggest that
the interaction between CAFs and the TIME, mediated by EVs, is a
notable factor driving HCC progression (57). Further investigation into the
activation of CAFs by HCC-derived EVs may offer new insights into
the mechanisms underlying recurrence and metastasis, as well as
potential therapeutic strategies.
In conclusion, EVs do not promote HCC metastasis in
a general or indiscriminate manner. Their high heterogeneity,
reflected in the diversity of cellular origins, sizes and molecular
cargos, results in functionally specialized subpopulations. These
subpopulations collaborate with high precision in both space and
time; some prepare the environment by directing remote organs to
form the PMN, others remodel microvessels to facilitate transport
and others destroy the immune surveillance system along the way,
enabling immune evasion. Consequently, future research into liver
cancer metastasis and the development of EV-based liquid biopsy and
treatments must focus on identifying, isolating and analyzing key
pathogenic EV subpopulations, rather than considering EVs as a
homogenous entity.
Mechanism of CTCs in HCC metastasis
Epithelial-mesenchymal transition
(EMT)
EMT is a process in which epithelial cells lose
their adhesive properties and acquire mesenchymal traits, including
enhanced migration and invasion capabilities (58). This mechanism plays a marked role
in the metastasis of advanced HCC. EMT influences tumor cell
proliferation and metastasis through various mechanisms, including
the active release of CTCs from primary tumors, facilitating
distant metastasis and promoting HCC recurrence (59,60).
The activation of EMT in CTCs primarily occurs within the
bloodstream, involving dynamic processes related to immune evasion
and autophagy (61,62).
During EMT-mediated metastasis in HCC, CTCs
contribute to the degradation of the basement membrane and
extracellular matrix (ECM). Under hypoxic conditions and through
paracrine signaling, CTCs activate transcription factors, miRNAs
and other regulatory elements, leading to the disruption of tight
and adhesive cell connections and reorganization of the
cytoskeleton. This upregulates mesenchymal markers and
downregulates epithelial markers, enabling tumor cells to migrate
through the matrix and enter the bloodstream (63,64).
CTCs can be classified into three phenotypes based on EMT markers:
Epithelial, mesenchymal and mixed CTCs (65). A study of 33 patients with HCC
found that the number of epithelial CTCs was positively correlated
with tumor size, the number of mixed CTCs was positively correlated
with tumor number and the number of mesenchymal CTCs (M-CTCs) was
positively correlated with metastasis. Mixed CTCs are key players
in the EMT transition of HCC and contribute notably to intrahepatic
metastasis, whereas M-CTCs may serve as predictors of extrahepatic
metastasis (66). Both
experimental and clinical studies highlight the marked role of
EMT-associated CTCs in HCC metastasis and recurrence (67,68).
A study of 112 patients with HCC showed that elevated CTC counts
and a higher percentage of M-CTCs preceded the clinical detection
of HCC recurrence. Notably, BCAT1, a gene markedly upregulated in
these patients, may trigger the EMT process (69). Therefore, classifying CTCs based on
EMT characteristics could serve as an early predictor of HCC
recurrence, metastasis and survival (70). Furthermore, Lu et al
(71) demonstrated that platelets,
by binding to CTCs, induce autophagy via the AMPK/mTOR
signaling pathway, which promotes EMT and enhances the motility of
HCC cells.
The activation of EMT in CTCs may also be linked to
immune responses. Huang et al (72) confirmed through in vitro
experiments that there is a significant positive correlation
between the number of Treg cells, CTC count and vascular
infiltration, suggesting that Treg cells may serve as a key
regulator of CTC release. Treg cells can enhance the invasive
potential of HCC cells by secreting TGF-β1, thus triggering EMT.
Therefore, targeting Treg cells within the TME, for example,
through the depletion of Tregs via CCR8 targeting (73), or by inhibiting Treg-derived TGF-β1
signaling to suppress EMT (72),
may represent a promising therapeutic strategy to prevent CTC
release and, consequently, HCC metastasis and recurrence. Another
study found that lncRNA FOXD1-AS1 promotes HCC immune evasion
via PD-L1 and affects CTC production and metastasis in HCC
mouse models by upregulating the PI3K/AKT signaling pathway,
thereby regulating EMT and ultimately driving HCC proliferation,
invasion, and metastasis (74).
Formation of microvascular invasion
(MVI)
MVI, also known as microvascular tumor thrombus, is
characterized by the infiltration of malignant cells into the
microvessels surrounding the liver tumor. It is one of the notable
pathological features in HCC, strongly associated with poor
prognosis following surgical resection and transplantation, early
recurrence and intrahepatic metastasis (75). Studies suggest that CTCs released
by HCC can form vascular thrombi from individual cells, serving as
key components that further promote HCC recurrence and metastasis.
Preoperative CTC counting may serve as a predictive tool for MVI
and provide dynamic monitoring of HCC prognosis (9). Several mechanisms are thought to be
involved in this process.
Firstly, CTCs derived from HCC trigger a coagulation
response in peripheral blood, forming circulating tumor microemboli
(CTM), which facilitates CTC migration across the endothelial
barrier and metastasis both within and outside the liver. Studies
have shown that tissue factor is overexpressed in the peripheral
blood of patients with HCC and is closely associated with
extrahepatic metastasis, lymphatic spread and portal vein
thrombosis (76). Tissue factor,
notably expressed in cancer cells and CTCs, promotes the binding of
CTCs with coagulation factors, leading to the formation of clumps
composed of both tumor and non-tumor cell components, such as
platelets and fibrin. This coagulation response ultimately
facilitates the aggregation and adhesion of CTCs. When tissue
factor enters the bloodstream, it activates coagulation factor VII
and initiates the extrinsic coagulation pathway, leading to the
formation of CTMs in circulation (77). Moreover, once CTMs are formed,
tissue factor can regulate the proliferation and growth of HCC
cells by activating protease-activated receptors, making it a key
factor in reshaping the TME and promoting the formation and
progression of MVI.
Secondly, CTCs facilitate the dissemination and
colonization of HCC tumor cells by activating platelets, thereby
creating a favorable environment for tumor growth. Growing evidence
suggests that platelets play a pivotal role in promoting the
invasion and metastasis of HCC cells through multiple mechanisms.
The ability of platelets to protect tumor cells in circulation is
key to their support of hematogenous dissemination (78). CTCs induce platelet activation and
aggregation, with activated platelets binding to CTCs, shielding
them from the shear forces exerted by blood flow and vessel walls.
This protection is essential for the survival of CTCs and their
subsequent adhesion to the vascular endothelium at metastatic sites
(79). Moreover, CTC-induced
platelet activation also aids HCC cells in evading immune
surveillance. Sun et al (80) demonstrated that CTC-platelet
adhesion helps cancer cells escape NK cell-mediated innate immune
responses by upregulating the immune checkpoint CD155-TIGIT,
thereby promoting distant metastasis. Activated platelets also
secrete numerous growth factors, including TGF-β, VEGF and
platelet-derived growth factor. VEGF, secreted by CTCs, enhances
microvascular permeability by enlarging the spaces between adjacent
endothelial cells. The interaction between CTCs and platelets
markedly increases the likelihood of MVI (81). In summary, platelets activated by
CTCs not only protect the CTCs but also contribute to the formation
of new blood vessels in HCC by transporting cytokines that provide
nutrients essential for tumor cell growth (82,83).
Additionally, platelets regulate the integrity of tumor
vasculature, promoting CTC adhesion and transendothelial migration,
which leads to HCC metastasis and colonization. While the role of
platelets in HCC metastasis is well-documented, further research is
needed to elucidate the specific mechanisms underlying the
activation and maintenance of the CTC-platelet interaction.
Tumor cells consisting of two or more CTCs are
referred to as CTC clusters or CTMs. CTC clusters have been
proposed as indicators of tumor metastasis. Although the number of
CTC clusters in circulation is smaller than that of single CTCs,
their metastatic potential is notably higher, increasing by 23–50
times (84). Understanding the
formation mechanisms of CTC clusters is therefore critical for
elucidating the metastasis of HCC. Currently, two widely accepted
mechanisms explain the formation of CTC clusters; the first
involves tumor cells directly forming cluster-like structures that
invade microvessels after detaching from the primary tumor
(85). Aceto et al
(86) were the first to
demonstrate that CTC clusters originate from oligoclonal tumor cell
populations rather than from cell aggregation within blood vessels.
The authors also identified plakoglobin-dependent intercellular
adhesion as critical for CTC cluster formation. The second
mechanism suggests that tumor cells detach from the primary tumor
due to shear forces from blood flow, diffuse into blood vessels and
aggregate into clusters (87). Sun
et al (62) prospectively
measured CTCs in five key vascular sites in patients with HCC and
detected CTCs in tumor-feeding vessels in approximately half of the
patients. Notably, CTCs were found in the hepatic veins of 15
patients but not in the surrounding arteries, and CTC recurrence
was observed in peripheral veins in five of these patients. This
suggests that individual CTCs may aggregate again in the
bloodstream. While this contradicts conclusions made by Aceto et
al (86), it warrants further
investigation. The formation of CTC clusters is a multistage and
dynamic process, with mechanisms exhibiting spatiotemporal
heterogeneity. The two main pathways currently recognized are
collective shedding from the primary tumor and single-cell
re-aggregation in circulation, which likely to coexist and
complement each other. CTC clusters undergo a complex life cycle,
beginning with clonal collective shedding from the primary tumor,
followed by dynamic structural evolution in the challenging
environment of circulation, partially disintegrating and partially
reassembling. Ultimately, the metastasis efficiency may depend on
the ability of certain cell populations to maintain an aggregated
state or effectively reorganize within distant blood vessels. The
unique hemodynamics of the portal venous system in HCC, the
abundance of immune cells in the liver and the altered vascular
structure in cirrhosis may make the dynamic evolution of CTC
clusters particularly active in the intrahepatic circulation. This
may play a key role in the intrahepatic dissemination of HCC.
Mechanisms of EV-CTC interactions promoting
HCC metastasis
EVs enhance the shedding ability of
CTCs
At the onset of metastasis, EVs enhance CTC shedding
by promoting EMT and ECM remodeling, stimulating angiogenesis,
increasing vascular permeability and strengthening the adhesion
ability of CTCs. In advanced HCC, target organ metastasis is
common, with EMT activation playing a marked role in initiating
metastasis (88). Studies have
demonstrated that EVs are pivotal in the metastatic process of HCC,
inducing EMT activation and weakening the adhesion of tumor cells,
thus facilitating the shedding of tumor cells to form CTCs
(89). EVs regulate key genes (for
example, PRDM16 and LHX6) in the EMT pathway, activating the
Wnt/β-catenin, Notch and MAPK/ERK signaling pathways, among others,
to promote the detachment of CTCs from the primary tumor site and
enhance their shedding. Chen et al (90) observed that HCC cells treated with
EVs showed elevated expression of α-SMA and vimentin, along with
reduced expression of E-cadherin, promoting mesenchymal marker
expression. Western blot analysis confirmed that these processes
were mediated through the MAPK/ERK pathway, facilitating tumor cell
migration, chemotaxis and invasion. Further research has revealed
that EVs derived from hepatic stellate cells (HSCs) activate the
Notch signaling pathway in HCC cells via PRDM16, leading to
increased proliferation, migration, invasion and EMT progression
(91). Hypoxia-induced EVs in HCC
activate EMT through the Wnt/β-catenin signaling pathway, promoting
HCC cell proliferation, migration and invasion (92). Moreover, tumor-derived EVs can
directly or indirectly promote angiogenesis and increase vascular
permeability by releasing pro-angiogenic factors (for example, VEGF
and IL-8), which play a pivotal role in the formation of shed CTCs
at the primary tumor site (93).
Hypoxic HCC cell-derived EVs co-cultured with M2 macrophages
secrete higher levels of VEGF, granulocyte colony-stimulating
factor and IL-4, which further stimulate angiogenesis and increase
vascular permeability, enabling tumor cells to shed into
circulation as CTCs, thereby enhancing metastasis (94). Disruption of vascular barriers is a
pivotal step in CTC-mediated HCC metastasis, requiring the
breakdown of tight junctions and the integrity of vascular
endothelial cells (95).
Tumor-derived EVs enhance vascular permeability and promote CTC
production and shedding by transferring their contents to
endothelial cells. The protein nidogen 1, secreted by HCC-derived
EVs, increases the permeability of lung endothelial cells by
disrupting the integrity of the endothelial barrier and promoting
angiogenesis, raises tumor necrosis factor receptor 1 expression
and facilitates the formation of the PMN in the lungs (96).
In summary, EVs are key initiators driving HCC
metastasis, initiating EMT to internally transform tumor cells and
endow them with migratory potential. Simultaneously, EVs directly
target and downregulate key junction proteins (such as VE-cadherin
and p120-catenin) in endothelial cells by delivering specific
cargoes, such as miR-103. This disruption of tight junctions
compromises the vascular endothelial barrier, thereby reducing the
physical obstacles to CTC intravasation into the circulatory
system. This dual approach facilitates CTC generation and shedding,
driving HCC metastasis.
EVs protect CTCs and enhance their
adhesion ability
CTCs are notable drivers of distant metastasis in
HCC. The survival of CTCs in circulation faces notable challenges,
including the need to withstand intravascular shear stress and
evade immune surveillance. EVs can interact with various
hematopoietic and immune cells. For example, EVs induce platelet
activation by delivering CD97. Activated platelets can protect CTCs
from hemodynamic shear forces, thereby enhancing CTC survival in
the bloodstream. Additionally, EVs can induce neutrophils to form
neutrophil extracellular traps, which bind to CTCs and promote
their adhesion to blood vessels, helping CTCs survive and initiate
metastasis (97,98). EVs carry molecules that interact
with circulating immune cells, inducing immunosuppression and
further protecting CTCs from immune system attacks. Research has
demonstrated that PD-L1 is markedly expressed on CTCs, and EVs
promote immune escape by upregulating PD-L1 expression on CTCs,
thereby hindering CD8+ T cell-mediated immune responses
in HCC (99–101). The delivery of immune checkpoint
molecules such as PD-L1 by EVs is a recognized immune evasion
mechanism in various solid tumors. In HCC, however, this process
occurs within a unique liver immune microenvironment. In the
specific context of chronic inflammation caused by HBV/HCV
infection or non-alcoholic fatty liver disease and alcoholic liver
disease, Kupffer cells undergo marked functional remodeling and
spatial repositioning, while being in a ‘pre-activated’ immune
regulatory state, characterized by upregulation of immune
checkpoint molecules such as PD-L1 (102,103). This pre-existing immune tolerant
microenvironment in the liver, shaped by chronic inflammation,
makes Kupffer cells more sensitive to EVs from tumor sources.
Therefore, the EVs in HCC not only directly inhibit immune effector
cells, but also may synergistically amplify the immunosuppressive
effect by acting on these already sensitized Kupffer cells, thereby
achieving dual protection against CTCs; however, to the best of our
knowledge, there is currently no direct evidence that reveals
whether the specific mechanism of EV-mediated CTC survival
specifically occurs in HCC with different etiological backgrounds
such as HBV, HCV, non-alcoholic fatty liver disease or alcoholic
liver disease. Therefore, this hypothesis still requires targeted
verification through the design of rigorous experiments. EVs can
enhance CTC adhesion, facilitating their colonization in specific
organ microenvironments, highlighting the importance of the
PMN.
EVs reshape TME to facilitate the
colonization ability of CTCs
The TME of HCC is dynamic and complex, comprising
cancer cells, stromal cells, blood vessels, nerve fibers and
related decellularized components (104). EVs have emerged as key mediators
of intercellular communication within the TME, driving tumor
progression (105). Accumulating
evidence suggests that EVs transfer functional biological factors
to recipient cells, reshaping the TME by reprogramming both cancer
cell and stromal cell metabolism (106). Fang et al (17) demonstrated that EVs released by
metastatic HCC cells carry miR-1247-3p, which directly targets
B4GALT3 in fibroblasts. This activates the β1-integrin-NF-κB
signaling pathway, inducing the transformation of fibroblasts into
CAFs, and promotes lung metastasis in patients with HCC.
Furthermore, EVs contribute to the formation of a
PMN for CTCs, creating a favorable foundation for their
colonization. Tumor-derived EVs serve as key intercellular
messengers that recruit inhibitory immune cells (such as TAMs and
Tregs) to distant organs by carrying cytokines (such as TGF-β1 and
IL-10). These recruited immune cells suppress antitumor immunity
through functional reprogramming, including M2 polarization of
macrophages and enhancement of Treg-mediated suppression.
Concurrently, EVs promote the establishment of a PMN in distant
organs by increasing vascular permeability, activating fibroblasts
and remodeling the extracellular matrix. This coordinated
remodeling of the immunosuppressive and stromal microenvironment
facilitates the colonization and outgrowth of CTCs at distant sites
(107). EVs released by HCC cells
form a liver PMN rich in fibrotic stroma and immunosuppressive
cells by activating HSCs and recruiting myeloid-derived suppressor
cells; this creates an environment that promotes CTC colonization.
For example, TGF-β1-enriched EVs induce HSCs to secrete
fibronectin, thereby promoting CTC adhesion and invasion (108). Conversely, EVs released by CTCs
can further recruit immunosuppressive cells (such as Tregs) and
remodel the liver TME, forming a positive feedback loop. For
instance, CCL5 secreted by CTCs is transmitted to Kupffer cells
through EVs, stimulating the release of tumor necrosis factor-α,
exacerbating liver inflammation and fibrosis and accelerating HCC
metastasis (61).
In summary, EVs support CTCs by constructing a
dynamic, multi-layered protective network. In circulation,
platelet-derived EVs encapsulate CTCs, forming clusters that shield
them from shear stress, aiding their survival in the bloodstream.
Before colonization, EVs act as messengers, modifying the target
organ microenvironment and creating niches for CTCs. However, most
current research is based on in vitro models, and whether
the complex in vivo microenvironment fully aligns with this
protective model remains uncertain. Future research should focus on
longitudinal clinical cohort studies to dynamically monitor changes
in EV cargo and CTC characteristics in patients with HCC at various
stages, before and after treatment, to establish causal
relationships. Additionally, novel liquid biopsy strategies
targeting specific molecular markers of ‘CTC-protective EVs’ should
be developed to enable early and functional prediction of
metastatic risk.
Current status, challenges and clinical
value of EVs and CTCs in liver cancer detection
Tumor-derived EVs are increasingly recognized as a
promising liquid biopsy tool, with studies highlighting their
potential in detecting high-risk patients with HCC, yielding
promising results (109,110). However, the inherent
heterogeneity of EVs presents both a source of valuable information
and a major challenge for clinical application. The complex origins
of EVs in the blood of patients, coupled with signals from liver
cancer being potentially obscured by EVs from normal cells,
complicate their clinical translation. Consequently, researchers
have focused on developing more efficient methods for isolating
EVs, aiming to enhance their clinical utility. Despite these
advancements, a standardized protocol for EV isolation is still
lacking, and isolating specific EV subpopulations with high purity
remains a challenge, resulting in mixtures, signal dilution and
interference, thus limiting their clinical applicability (111).
Microfluidic separation technology, known for its
low sample consumption, high throughput and rapid reaction speed,
has emerged as a promising method for EV isolation in recent years
(112). This technology enables
the simultaneous sorting of different EV subpopulations based on
immune affinity and/or physical properties, showing promise for the
specific and efficient enrichment of HCC-derived EVs from the blood
of patients with cirrhosis, which is critical for distinguishing
tumor signals from benign inflammatory backgrounds. Such
advancements could markedly improve early diagnosis and monitoring
of minimal residual lesions (113). Sun et al (114) developed an EV click chip that
achieves high purification efficiency (90.2%) and recovery rate
(82.7%) for HCC-derived EVs. This method integrates: i) A
multi-labeled antibody cocktail targeting HCC-derived EVs; ii) a
nanostructured substrate to increase surface area for EV
interaction; and iii) a reversible capture/release system mediated
by click chemistry. Combined with reverse transcription droplet
digital PCR, this platform shows promising potential for early
detection of HCC. Wu et al (115) quantified Fascin-1-positive EVs in
patient plasma using nanoflow cytometry, finding that the levels of
Fascin-1-positive EVs in the plasma of patients with HCC were
notably higher than in healthy controls, with a positive
association with HCC staging. This demonstrates the potential of
Fascin-1-positive EVs as a novel liquid biopsy marker for
distinguishing early-stage HCC from healthy individuals.
Mechanistically, Fascin-1 may be selectively packaged into EVs by
HCC cells, where it can upregulate F-actin levels, promoting cancer
cell migration. Another research group developed a detection method
based on surface proteins of HCC-derived EVs to quantify three
subpopulations: EpCAM+ CD63+,
CD147+ CD63+ and GPC3+
CD63+ HCC-EVs. The HCC-derived EV score, calculated from
these subpopulations, is used to detect early HCC, showing a
sensitivity of 91% and specificity of 90% in distinguishing early
HCC from cirrhosis (116). Thus,
while EVs hold potential in screening high-risk HCC populations,
they should not replace existing detection methods but rather
complement them by providing dynamic molecular insights. Moving
forward, there is a need for the validation of marker combinations
in large prospective cohorts and the development of standardized,
high-throughput detection techniques suitable for mass
screening.
The heterogeneity of CTCs poses a marked challenge
for their reliable detection in bodily fluids, limiting the
clinical application of CTC-based assays. Technologies that capture
CTCs based on epithelial markers may overlook invasive
subpopulations that have undergone EMT, missing key targets and
complicating clinical detection. In patients with HCC, CTCs are
often present in low quantities, with only 0–86 CTCs detectable per
5 ml of peripheral blood (68).
Consequently, there is a need for more specific and sensitive CTC
capture technologies for further analysis.
Currently, no FDA-approved platform exists for
detecting HCC-related CTCs. Most CTC detection methods rely on
biological characteristics (antibody capture, such as EpCAM),
physical properties (such as size and density) or non-enriched
techniques, followed by immunocytochemical staining for CTC
characterization. Platforms focused on positive or negative
enrichment based on immune affinity can be further categorized into
magnetic and microfluidic devices. For instance, the CellSearch
platform uses magnetic beads to isolate EpCAM-labeled cells
(117); however, this technology
has notable limitations for HCC applications. The primary issue is
the mismatch between the epithelial markers it relies on and the
high heterogeneity and metastatic nature of HCC. Only 20–35% of HCC
tumor tissues express EpCAM (118), leading to the missed detection of
a large fraction of EpCAM-negative or low-expressing cells. A study
by Morris et al (119)
demonstrated that the Isolation by Size of Epithelial Tumor Cells
platform achieved a 100% CTC detection rate in patients with HCC,
whereas CellSearch detected only 28% of CTCs. Additionally,
CellSearch may fail to detect more aggressive M-CTCs, resulting in
false negatives, especially in early-stage HCC or in post-treatment
patients with low CTC counts. Single-cell RNA sequencing
(scRNA-seq) offers a powerful approach to uncover spatiotemporal
transcriptional dynamics related to tumor cell cycles and immune
evasion signals. Sun et al (61) used scRNA-seq to profile the
transcriptome of 113 single CTCs from four different vascular sites
in 10 patients with HCC. The authors identified chemokine CCL5 as a
key mediator of CTC immune evasion. The overexpression of CCL5 in
CTCs is regulated by the p38-MAX signaling pathway, which recruits
Tregs to promote immune evasion and facilitate CTC metastasis and
seeding. This highlights the potential of using scRNA-seq in the
future to develop a comprehensive molecular profile of HCC-related
CTCs. By identifying genes or protein markers, it may be possible
to create a standardized, clinically applicable multi-marker
detection panel.
Studies have highlighted the potential of CTCs in
predicting treatment efficacy and monitoring disease progression in
HCC, especially after liver resection. CanPatrol™, a technology
based on biophysical characteristics, employs a microfiltration
system to isolate and classify various CTC phenotypes. In a cohort
study of 115 patients with HCC, CTCs were isolated using the
CanPatrol enrichment method and classified via in situ
hybridization. The findings showed that M-CTCs were associated with
high Ki67 expression, both correlating with poor prognosis in
patients with HCC. The combination of M-CTCs and Ki67 expression
may serve as a critical prognostic marker for predicting overall
survival following liver resection (120). Since EMT is frequently linked
with tumor aggressiveness, understanding the biological connection
between EMT and metastasis warrants further mechanistic
investigation. Additionally, Chen et al (67) used the CanPatrol method to detect
total CTCs, M-CTCs and CTC-white blood cell (WBC) clusters in 136
patients with HCC. The simulated receiver operating characteristic
curve revealed an area under the curve of 0.821 for CTC-WBC
clusters (sensitivity 90.0%, specificity 93.7%), outperforming both
total CTCs (0.718) and M-CTCs (0.716). Furthermore, patients with
positive CTC-WBC clusters had a higher recurrence rate, with levels
rising 10 months before clinical detection of recurrence. While
these studies establish statistical associations between CTC
subtypes and HCC prognosis, the molecular mechanisms by which these
subtypes promote tumor proliferation, immune evasion and metastasis
remain insufficiently understood. Future research must include
expanded clinical validation, as well as the integration of models
such as organoids and in vivo imaging to explore the
biological roles of these CTC subpopulations. This will enable the
development of targeted clinical intervention strategies, advancing
from mere prognostic markers to actionable therapeutic guidance
tools.
Conclusions
The present manuscript reviews advances in
understanding the roles of EVs and CTCs in HCC metastasis and
recurrence. EVs enhance CTC shedding during metastasis and protect
CTCs from immune system attack. The bioactive molecules carried by
EVs promote CTC-mediated HCC recurrence and metastasis, with CTCs
acting as the initiators of metastatic spread and EVs facilitating
their colonization by remodeling the microenvironment.
In conclusion, EVs and CTCs work synergistically to
promote HCC metastasis and invasion through a multifaceted
mechanism. EVs act as messengers, paving the way for CTC
colonization by forming PMNs, remodeling the TME, transmitting
molecular signals and regulating immune responses. CTCs, as the
executors of metastasis, enable distant spread through processes
such as EMT, MVI and adaptive heterogeneity. Despite the marked
progress, several questions remain to be addressed. For instance,
it is still unclear whether CTCs release novel subpopulations of
EVs after colonizing distant sites, potentially remodeling
established metastatic foci. Further research is needed to explore
this aspect. Additionally, future therapeutic strategies could
focus on combining immune checkpoint inhibitors with
EV/CTC-targeted therapies, offering precise interventions to manage
HCC, prevent recurrence, and curb metastasis. These advancements
will deepen the understanding of liver cancer metastasis,
ultimately improving clinical outcomes for patients with HCC.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
All authors contributed to the study conception and
design. The study was conceived by ZW, while SY conducted the
literature search and data analysis. The manuscript was drafted
and/or critically revised by SY, WL and ZW. The first draft of the
manuscript was written by SY. Data authentication is not
applicable. All authors have read and approved the final version of
the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Vogel A, Meyer T, Sapisochin G, Salem R
and Saborowski A: Hepatocellular carcinoma. Lancet. 400:1345–1362.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249.
2021.PubMed/NCBI
|
|
3
|
Kim E and Viatour P: Hepatocellular
carcinoma: Old friends and new tricks. Exp Mol Med. 52:1898–1907.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lee YT, Fujiwara N, Yang JD and Hoshida Y:
Risk stratification and early detection biomarkers for precision
HCC screening. Hepatology. 78:319–362. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ma L, Guo H, Zhao Y, Liu Z, Wang C, Bu J,
Sun T and Wei J: Liquid biopsy in cancer current: Status,
challenges and future prospects. Signal Transduct Target Ther.
9:3362024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang JC, Hu JJ, Li YX, Luo W, Liu JZ and
Ye DW: Clinical applications of liquid biopsy in hepatocellular
carcinoma. Front Oncol. 12:7818202022. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pantel K and Speicher MR: The biology of
circulating tumor cells. Oncogene. 35:1216–1224. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ye Q, Ling S, Zheng S and Xu X: Liquid
biopsy in hepatocellular carcinoma: Circulating tumor cells and
circulating tumor DNA. Mol Cancer. 18:1142019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhou J, Zhang Z, Zhou H, Leng C, Hou B,
Zhou C, Hu X, Wang J and Chen X: Preoperative circulating tumor
cells to predict microvascular invasion and dynamical detection
indicate the prognosis of hepatocellular carcinoma. BMC Cancer.
20:10472020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
von Felden J, Schulze K, Krech T, Ewald F,
Nashan B, Pantel K, Lohse AW, Riethdorf S and Wege H: Circulating
tumor cells as liquid biomarker for high HCC recurrence risk after
curative liver resection. Oncotarget. 8:89978–89987. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Marar C, Starich B and Wirtz D:
Extracellular vesicles in immunomodulation and tumor progression.
Nat Immunol. 22:560–570. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kalluri R and LeBleu VS: The biology,
function, and biomedical applications of exosomes. Science.
367:eaau69772020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Buzas EI: The roles of extracellular
vesicles in the immune system. Nat Rev Immunol. 23:236–250. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fu Q, Zhang Q, Lou Y, Yang J, Nie G, Chen
Q, Chen Y, Zhang J, Wang J, Wei T, et al: Correction: Primary
tumor-derived exosomes facilitate metastasis by regulating adhesion
of circulating tumor cells via SMAD3 in liver cancer. Oncogene.
38:5740–5741. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu Y, Yao Y, Yu L, Zhang X, Mao X, Tey SK,
Wong SWK, Yeung CLS, Ng TH, Wong MY, et al: Clathrin light chain
A-enriched small extracellular vesicles remodel microvascular niche
to induce hepatocellular carcinoma metastasis. J Extracell
Vesicles. 12:e123592023. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yokota Y, Noda T, Okumura Y, Kobayashi S,
Iwagami Y, Yamada D, Tomimaru Y, Akita H, Gotoh K, Takeda Y, et al:
Serum exosomal miR-638 is a prognostic marker of HCC via
downregulation of VE-cadherin and ZO-1 of endothelial cells. Cancer
Sci. 112:1275–1288. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fang T, Lv H, Lv G, Li T, Wang C, Han Q,
Yu L, Su B, Guo L, Huang S, et al: Tumor-derived exosomal
miR-1247-3p induces cancer-associated fibroblast activation to
foster lung metastasis of liver cancer. Nat Commun. 9:1912018.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nakano T, Chen IH, Wang CC, Chen PJ, Tseng
HP, Huang KT, Hu TH, Li LC, Goto S, Cheng YF, et al: Circulating
exosomal miR-92b: Its role for cancer immunoediting and clinical
value for prediction of posttransplant hepatocellular carcinoma
recurrence. Am J Transplant. 19:3250–3262. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhou J, Che J, Xu L, Yang W, Zhou W and
Zhou C: Tumor-derived extracellular vesicles containing long
noncoding RNA PART1 exert oncogenic effect in hepatocellular
carcinoma by polarizing macrophages into M2. Dig Liver Dis.
54:543–553. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li J, Xue J, Ling M, Sun J, Xiao T, Dai X,
Sun Q, Cheng C, Xia H, Wei Y, et al: MicroRNA-15b in extracellular
vesicles from arsenite-treated macrophages promotes the progression
of hepatocellular carcinomas by blocking the LATS1-mediated Hippo
pathway. Cancer Lett. 497:137–153. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang X, Huang H, Sze KM, Wang J, Tian L,
Lu J, Tsui YM, Ma HT, Lee E, Chen A, et al: S100A10 promotes HCC
development and progression via transfer in extracellular vesicles
and regulating their protein cargos. Gut. 72:1370–1384. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhou Y, Ren H, Dai B, Li J, Shang L, Huang
J and Shi X: Hepatocellular carcinoma-derived exosomal miRNA-21
contributes to tumor progression by converting hepatocyte stellate
cells to cancer-associated fibroblasts. J Exp Clin Cancer Res.
37:3242018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pu J, Xu Z, Nian J, Fang Q, Yang M, Huang
Y, Li W, Ge B, Wang J and Wei H: M2 macrophage-derived
extracellular vesicles facilitate CD8+T cell exhaustion in
hepatocellular carcinoma via the miR-21-5p/YOD1/YAP/β-catenin
pathway. Cell Death Discov. 7:1822021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu G, Ouyang X, Sun Y, Xiao Y, You B, Gao
Y, Yeh S, Li Y and Chang C: The miR-92a-2-5p in exosomes from
macrophages increases liver cancer cells invasion via altering the
AR/PHLPP/p-AKT/β-catenin signaling. Cell Death Differ.
27:3258–3272. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fares J, Fares MY, Khachfe HH, Salhab HA
and Fares Y: Molecular principles of metastasis: A hallmark of
cancer revisited. Signal Transduct Target Ther. 5:282020.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hoshino A, Costa-Silva B, Shen TL,
Rodrigues G, Hashimoto A, Tesic Mark M, Molina H, Kohsaka S, Di
Giannatale A, Ceder S, et al: Tumour exosome integrins determine
organotropic metastasis. Nature. 527:329–335. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ye J, Gao X, Huang X, Huang S, Zeng D, Luo
W, Zeng C, Lu C, Lu L, Huang H, et al: Integrating Single-Cell and
spatial transcriptomics to uncover and elucidate GP73-mediated
Pro-angiogenic regulatory networks in hepatocellular carcinoma.
Research (Wash D C). 7:03872024.PubMed/NCBI
|
|
28
|
Fang Y, Cui W, Yang Y, Zhang X, Tian M,
Xie Z, Guo Y, Yuan W, Li Z and Yang S: Breaking the premetastatic
niche barrier: The role of endothelial cells and therapeutic
strategies. Theranostics. 15:6454–6475. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Okura GC, Bharadwaj AG and Waisman DM: The
role of extracellular proteases and extracellular matrix remodeling
in the Pre-Metastatic niche. Biomolecules. 15:16962025. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Han Y, Gong WS, Xing XS, Zhou H, Wang XL,
Xu Y, Zhou XL and Xue WL: miR-183-5p-enriched extracellular
vesicles promote the crosstalk between hepatocellular carcinoma
cell and endothelial cell via SIK1/PI3K/AKT and CCL20/CCR6
signaling pathways. Front Oncol. 15:15322392025. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu Y, Hu X, Zhou S, Sun T, Shen F and
Zeng L: Golgi Protein 73 Promotes Angiogenesis in Hepatocellular
Carcinoma. Research (Wash D C). 7:04252024.PubMed/NCBI
|
|
32
|
Kalluri R and Zeisberg M: Fibroblasts in
cancer. Nat Rev Cancer. 6:392–401. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li X, Ramadori P, Pfister D, Seehawer M,
Zender L and Heikenwalder M: The immunological and metabolic
landscape in primary and metastatic liver cancer. Nat Rev Cancer.
21:541–557. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ying Q, Liang L, Guo W, Zha R, Tian Q,
Huang S, Yao J, Ding J, Bao M, Ge C, et al: Hypoxia-inducible
microRNA-210 augments the metastatic potential of tumor cells by
targeting vacuole membrane protein 1 in hepatocellular carcinoma.
Hepatology. 54:2064–2075. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kurebayashi Y, Ojima H, Tsujikawa H,
Kubota N, Maehara J, Abe Y, Kitago M, Shinoda M, Kitagawa Y and
Sakamoto M: Landscape of immune microenvironment in hepatocellular
carcinoma and its additional impact on histological and molecular
classification. Hepatology. 68:1025–1041. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tu T, Budzinska MA, Maczurek AE, Cheng R,
Di Bartolomeo A, Warner FJ, McCaughan GW, McLennan SV and Shackel
NA: Novel aspects of the liver microenvironment in hepatocellular
carcinoma pathogenesis and development. Int J Mol Sci.
15:9422–9458. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liu WH, Ren LN, Wang X, Wang T, Zhang N,
Gao Y, Luo H, Navarro-Alvarez N and Tang LJ: Combination of
exosomes and circulating microRNAs may serve as a promising tumor
marker complementary to alpha-fetoprotein for early-stage
hepatocellular carcinoma diagnosis in rats. J Cancer Res Clin
Oncol. 141:1767–1778. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liu Z, You Y, Chen Q, Li G, Pan W, Yang Q,
Dong J, Wu Y, Bei JX, Pan C, et al: Extracellular vesicle-mediated
communication between hepatocytes and natural killer cells promotes
hepatocellular tumorigenesis. Mol Ther. 30:606–620. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Schmiedel D and Mandelboim O: NKG2D
Ligands-critical targets for cancer immune escape and therapy.
Front Immunol. 9:20402018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xiao W, Dong W, Zhang C, Saren G, Geng P,
Zhao H, Li Q, Zhu J, Li G, Zhang S and Ye M: Effects of the
epigenetic drug MS-275 on the release and function of
exosome-related immune molecules in hepatocellular carcinoma cells.
Eur J Med Res. 18:612013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Pollard JW: Tumour-educated macrophages
promote tumour progression and metastasis. Nat Rev Cancer. 4:71–78.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bao D, Zhao J, Zhou X, Yang Q, Chen Y, Zhu
J, Yuan P, Yang J, Qin T, Wan S and Xing J: Mitochondrial
fission-induced mtDNA stress promotes tumor-associated macrophage
infiltration and HCC progression. Oncogene. 38:5007–5020. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wu R, Xiong J, Zhou T, Zhang Z, Huang Z,
Tian S and Wang Y: Quercetin/Anti-PD-1 antibody combination therapy
regulates the gut microbiota, impacts macrophage immunity and
reshapes the hepatocellular carcinoma tumor microenvironment. Front
Biosci (Landmark Ed). 28:3272023. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gao J, Liang Y and Wang L: Shaping
polarization of Tumor-associated macrophages in cancer
immunotherapy. Front Immunol. 13:8887132022. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li X, Lei Y, Wu M and Li N: Regulation of
macrophage activation and polarization by HCC-derived exosomal
lncRNA TUC339. Int J Mol Sci. 19:29582018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cheng L, Liu J, Liu Q, Liu Y, Fan L, Wang
F, Yu H, Li Y, Bu L, Li X, et al: Exosomes from melatonin treated
hepatocellularcarcinoma cells alter the immunosupression status
through STAT3 pathway in macrophages. Int J Biol Sci. 13:723–734.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ma K, Xu Y, Cheng H, Tang K, Ma J and
Huang B: T cell-based cancer immunotherapy: Opportunities and
challenges. Sci Bull (Beijing). 70:1872–1890. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sun JF, Zhang D, Gao CJ, Zhang YW and Dai
QS: Exosome-Mediated MiR-155 transfer contributes to hepatocellular
carcinoma cell proliferation by targeting PTEN. Med Sci Monit Basic
Res. 25:218–228. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Gong Y, Li K, Qin Y, Zeng K, Liu J, Huang
S, Chen Y, Yu H, Liu W, Ye L and Yang Y: Norcholic acid promotes
tumor progression and immune escape by regulating farnesoid X
receptor in hepatocellular carcinoma. Front Oncol. 11:7114482021.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yin C, Han Q, Xu D, Zheng B, Zhao X and
Zhang J: SALL4-mediated upregulation of exosomal miR-146a-5p drives
T-cell exhaustion by M2 tumor-associated macrophages in HCC.
Oncoimmunology. 8:16014792019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Tian XP, Wang CY, Jin XH, Li M, Wang FW,
Huang WJ, Yun JP, Xu RH, Cai QQ and Xie D: Erratum: Acidic
microenvironment Up-Regulates exosomal miR-21 and miR-10b in
Early-stage hepatocellular carcinoma to promote cancer cell
proliferation and metastasis: Erratum. Theranostics. 11:6522–6523.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Yao SX, Zhang GS, Cao HX, Song G, Li ZT
and Zhang WT: Correlation between microRNA-21 and expression of
Th17 and Treg cells in microenvironment of rats with hepatocellular
carcinoma. Asian Pac J Trop Med. 8:762–765. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Baglieri J, Brenner DA and Kisseleva T:
The role of fibrosis and Liver-Associated fibroblasts in the
pathogenesis of hepatocellular carcinoma. Int J Mol Sci.
20:17232019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhu GQ, Tang Z, Huang R, Qu WF, Fang Y,
Yang R, Tao CY, Gao J, Wu XL, Sun HX, et al: CD36+
cancer-associated fibroblasts provide immunosuppressive
microenvironment for hepatocellular carcinoma via secretion of
macrophage migration inhibitory factor. Cell Discov. 9:252023.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Qi Y, Wang H, Zhang Q, Liu Z, Wang T, Wu Z
and Wu W: CAF-Released exosomal miR-20a-5p facilitates HCC
progression via the LIMA1-Mediated β-Catenin pathway. Cells.
11:38572022. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ruiz de Galarreta M, Bresnahan E,
Molina-Sánchez P, Lindblad KE, Maier B, Sia D, Puigvehi M, Miguela
V, Casanova-Acebes M, Dhainaut M, et al: β-Catenin activation
promotes immune escape and resistance to Anti-PD-1 therapy in
hepatocellular carcinoma. Cancer Discov. 9:1124–1141. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Shang H, Lu L, Fan M, Lu Y, Shi X and Lu
H: Exosomal circHIF1A derived from hypoxic-induced
carcinoma-associated fibroblasts promotes hepatocellular carcinoma
cell malignant phenotypes and immune escape. Int Immunopharmacol.
138:1122822024. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zhang CX, Huang RY, Sheng G and Thiery JP:
Epithelial-mesenchymal transition. Cell. 188:5436–5486. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Giannelli G, Koudelkova P, Dituri F and
Mikulits W: Role of epithelial to mesenchymal transition in
hepatocellular carcinoma. J Hepatol. 65:798–808. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Bonnomet A, Brysse A, Tachsidis A, Waltham
M, Thompson EW, Polette M and Gilles C: Epithelial-to-mesenchymal
transitions and circulating tumor cells. J Mammary Gland Biol
Neoplasia. 15:261–273. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Sun YF, Wu L, Liu SP, Jiang MM, Hu B, Zhou
KQ, Guo W, Xu Y, Zhong Y, Zhou XR, et al: Dissecting spatial
heterogeneity and the immune-evasion mechanism of CTCs by
single-cell RNA-seq in hepatocellular carcinoma. Nat Commun.
12:40912021. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Sun YF, Guo W, Xu Y, Shi YH, Gong ZJ, Ji
Y, Du M, Zhang X, Hu B, Huang A, et al: Circulating tumor cells
from different vascular sites exhibit spatial heterogeneity in
epithelial and mesenchymal composition and distinct clinical
significance in hepatocellular carcinoma. Clin Cancer Res.
24:547–559. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Pencheva N and Tavazoie SF: Control of
metastatic progression by microRNA regulatory networks. Nat Cell
Biol. 15:546–554. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Cayrefourcq L, Mazard T, Joosse S,
Solassol J, Ramos J, Assenat E, Schumacher U, Costes V, Maudelonde
T, Pantel K and Alix-Panabières C: Establishment and
characterization of a cell line from human circulating colon cancer
cells. Cancer Res. 75:892–901. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Wu S, Liu S, Liu Z, Huang J, Pu X, Li J,
Yang D, Deng H, Yang N and Xu J: Classification of circulating
tumor cells by epithelial-mesenchymal transition markers. PLoS One.
10:e01239762015. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Liu YK, Hu BS, Li ZL, He X, Li Y and Lu
LG: An improved strategy to detect the epithelial-mesenchymal
transition process in circulating tumor cells in hepatocellular
carcinoma patients. Hepatol Int. 10:640–646. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Chen J, Luo Y, Xi X, Li H, Li S, Zheng L,
Yang D and Cai Z: Circulating tumor cell associated white blood
cell cluster as a biomarker for metastasis and recurrence in
hepatocellular carcinoma. Front Oncol. 12:9311402022. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Chen J, Cao SW, Cai Z, Zheng L and Wang Q:
Epithelial-mesenchymal transition phenotypes of circulating tumor
cells correlate with the clinical stages and cancer metastasis in
hepatocellular carcinoma patients. Cancer Biomark. 20:487–498.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Qi LN, Xiang BD, Wu FX, Ye JZ, Zhong JH,
Wang YY, Chen YY, Chen ZS, Ma L, Chen J, et al: Circulating tumor
cells undergoing EMT provide a metric for diagnosis and prognosis
of patients with hepatocellular carcinoma. Cancer Res.
78:4731–4744. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Ou H, Huang Y, Xiang L, Chen Z, Fang Y,
Lin Y, Cui Z, Yu S, Li X and Yang D: Circulating tumor cell
phenotype indicates poor survival and recurrence after surgery for
hepatocellular carcinoma. Dig Dis Sci. 63:2373–2380. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Lu M, Gong X, Zhang YM, Guo YW, Zhu Y,
Zeng XB, Gao JH, Liu LM, Shu D, Ma R, et al: Platelets promote
primary hepatocellular carcinoma metastasis through TGF-β1-mediated
cancer cell autophagy. Cancer Lett. 600:2171612024. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Huang AH, Wang HB, Wu ZF, Wang YH, Hu B,
Jiang ZN, Jin M, Wang LB and Gao YB: Infiltrating regulatory T
cells promote invasiveness of liver cancer cells via inducing
epithelial-mesenchymal transition. Transl Cancer Res. 8:2405–2415.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Navarrete M, Costoya C, Galvez-Cancino F,
Peggs KS, Marabelle A and Quezada SA: Regulatory T cell depletion
in cancer: Challenges, opportunities, and future directions for
antibody development. Annu Rev Med. 77:239–251. 2026. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Guo BL, Zheng QX, Jiang YS, Zhan Y, Huang
WJ and Chen ZY: Long non-coding RNAFOXD1-AS1 modulated CTCs
epithelial-mesenchymal transition and immune escape in
hepatocellular carcinoma in vitro by sponging miR-615-3p. Cancer
Rep (Hoboken). 7:e20502024.PubMed/NCBI
|
|
75
|
Li K, Zhang R, Wen F, Meng F, Li Q, Hao A,
Yang B, Lu Z, Cui Y and Zhou M: Single-cell dissection of the
multicellular ecosystem and molecular features underlying
microvascular invasion in HCC. Hepatology. 79:1293–1309. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Zhou Q, Huang T, Wang YF, Zhou XB, Liang
LJ and Peng BG: Role of tissue factor in hepatocellular carcinoma
genesis, invasion and metastasis. Chin Med J (Engl). 124:3746–3751.
2011.PubMed/NCBI
|
|
77
|
Li H, Yu Y, Gao L, Zheng P, Liu X and Chen
H: Tissue factor: A neglected role in cancer biology. J Thromb
Thrombolysis. 54:97–108. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Gao JH, He AD, Liu LM, Zhou YJ, Guo YW, Lu
M, Zeng XB, Gong X, Lu YJ, Liang HF, et al: Direct interaction of
platelet with tumor cell aggravates hepatocellular carcinoma
metastasis by activating TLR4/ADAM10/CX3CL1 axis. Cancer Lett.
585:2166742024. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Liu Y, Zhang Y, Ding Y and Zhuang R:
Platelet-mediated tumor metastasis mechanism and the role of cell
adhesion molecules. Crit Rev Oncol Hematol. 167:1035022021.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Sun Y, Li T, Ding L, Wang J, Chen C, Liu
T, Liu Y, Li Q, Wang C, Huo R, et al: Platelet-mediated circulating
tumor cell evasion from natural killer cell killing through immune
checkpoint CD155-TIGIT. Hepatology. 81:791–807. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Lou XL, Sun J, Gong SQ, Yu XF, Gong R and
Deng H: Interaction between circulating cancer cells and platelets:
Clinical implication. Chin J Cancer Res. 27:450–460.
2015.PubMed/NCBI
|
|
82
|
Filippelli A, Del Gaudio C, Simonis V,
Ciccone V, Spini A and Donnini S: Scoping review on platelets and
tumor angiogenesis: Do we need more evidence or better analysis?
Int J Mol Sci. 23:134012022. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Zhang L, Yuan Y, Deng Y, Wang L and Chen
F: Platelet-circulating tumor cell crosstalk: A pivotal target in
cancer diagnosis and therapy (review). Oncol Rep.
55:22026.PubMed/NCBI
|
|
84
|
Zhang Q, Rong Y, Yi K, Huang L, Chen M and
Wang F: Circulating tumor cells in hepatocellular carcinoma:
Single-cell based analysis, preclinical models, and clinical
applications. Theranostics. 10:12060–12071. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Cheung KJ, Padmanaban V, Silvestri V,
Schipper K, Cohen JD, Fairchild AN, Gorin MA, Verdone JE, Pienta
KJ, Bader JS and Ewald AJ: Polyclonal breast cancer metastases
arise from collective dissemination of keratin 14-expressing tumor
cell clusters. Proc Natl Acad Sci USA. 113:E854–E863. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Aceto N, Bardia A, Miyamoto DT, Donaldson
MC, Wittner BS, Spencer JA, Yu M, Pely A, Engstrom A, Zhu H, et al:
Circulating tumor cell clusters are oligoclonal precursors of
breast cancer metastasis. Cell. 158:1110–1122. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Castro-Giner F and Aceto N: Tracking
cancer progression: From circulating tumor cells to metastasis.
Genome Med. 12:312020. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Yüregir Y, Kaçaroğlu D and Yaylacı S:
Regulation of hepatocellular carcinoma Epithelial-Mesenchymal
transition mechanism and targeted therapeutic approaches. Adv Exp
Med Biol. 1450:93–102. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Joseph JP, Harishankar MK, Pillai AA and
Devi A: Hypoxia induced EMT: A review on the mechanism of tumor
progression and metastasis in OSCC. Oral Oncol. 80:23–32. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Chen L, Guo P, He Y, Chen Z, Chen L, Luo
Y, Qi L, Liu Y, Wu Q, Cui Y, et al: HCC-derived exosomes elicit HCC
progression and recurrence by epithelial-mesenchymal transition
through MAPK/ERK signalling pathway. Cell Death Dis. 9:5132018.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Sun C, Xu W, Xia Y and Wang S: PRDM16 from
hepatic stellate cells-derived extracellular vesicles promotes
hepatocellular carcinoma progression. Am J Cancer Res.
13:5254–5270. 2023.PubMed/NCBI
|
|
92
|
Yu Y, Min Z, Zhou Zhihang, Linhong M, Tao
R, Yan L and Song H: Hypoxia-induced exosomes promote
hepatocellular carcinoma proliferation and metastasis via miR-1273f
transfer. Exp Cell Res. 385:1116492019. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Bai S, Wang Z, Wang M, Li J, Wei Y, Xu R
and Du J: Tumor-derived exosomes modulate primary site tumor
metastasis. Front Cell Dev Biol. 10:7528182022. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Yang Y, Wu T, Wang Y, Luo D, Zhao Z, Sun
H, Zhang M, Zhang B and Han B: Hypoxic tumour-derived exosomal
miR-1290 exacerbates the suppression of CD8+ T cells by promoting
M2 macrophage polarization. Immunology. 173:672–688. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Reymond N, d'Água BB and Ridley AJ:
Crossing the endothelial barrier during metastasis. Nat Rev Cancer.
13:858–870. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Mao X, Tey SK, Yeung CLS, Kwong EML, Fung
YME, Chung CYS, Mak LY, Wong DKH, Yuen MF, Ho JCM, et al: Nidogen
1-Enriched extracellular vesicles facilitate extrahepatic
metastasis of liver cancer by activating pulmonary fibroblasts to
secrete tumor necrosis factor receptor 1. Adv Sci (Weinh).
7:20021572020. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Pereira-Veiga T, Schneegans S, Pantel K
and Wikman H: Circulating tumor cell-blood cell crosstalk: Biology
and clinical relevance. Cell Rep. 40:1112982022. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Dudiki T, Veleeparambil M, Zhevlakova I,
Biswas S, Klein EA, Ford P, Podrez EA and Byzova TV: Mechanism of
Tumor-platelet communications in cancer. Circ Res. 132:1447–1461.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Wang X, Sun Q, Liu Q, Wang C, Yao R and
Wang Y: CTC immune escape mediated by PD-L1. Med Hypotheses.
93:138–139. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Zhao B, Zheng X, Wang Y, Cheng N, Zhong Y,
Zhou Y, Huang J, Wang F, Qi X, Zhuang Q, et al: Lnc-CCNH-8 promotes
immune escape by up-regulating PD-L1 in hepatocellular carcinoma.
Mol Ther Nucleic Acids. 35:1021252024. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Lin X, Shao H, Tang Y, Wang Q, Yang Z, Wu
H and Xing T: High expression of circulating exosomal PD-L1
contributes to immune escape of hepatocellular carcinoma and immune
clearance of chronic hepatitis B. Aging (Albany NY).
16:11373–11384. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Yuan F, Zhang W, Mu D and Gong J: Kupffer
cells in immune activation and tolerance toward HBV/HCV infection.
Adv Clin Exp Med. 26:739–745. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Wu D, Zhu H and Wang H: Extracellular
vesicles in Non-alcoholic fatty liver disease and alcoholic liver
disease. Front Physiol. 12:7074292021. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Chen C, Wang Z, Ding Y and Qin Y: Tumor
microenvironment-mediated immune evasion in hepatocellular
carcinoma. Frontiers in Immunology. 14:11333082023. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Gondaliya P, Sayyed AA, Driscoll J, Patel
K and Patel T: Extracellular vesicle RNA signaling in the liver
tumor microenvironment. Cancer Lett. 558:2160892023. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Gangoda L, Boukouris S, Liem M, Kalra H
and Mathivanan S: Extracellular vesicles including exosomes are
mediators of signal transduction: Are they protective or
pathogenic? Proteomics. 15:260–271. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Zhao L, Ma X and Yu J: Exosomes and
organ-specific metastasis. Mol Ther Methods Clin Dev. 22:133–147.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Yang X, Zhang Y, Zhang Y, Li H, Li L, Wu
Y, Chen X, Qiu L, Han J and Wang Z: Colorectal cancer-derived
extracellular vesicles induce liver premetastatic immunosuppressive
niche formation to promote tumor early liver metastasis. Signal
Transduct Target Ther. 8:1022023. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Lee YT, Tran BV, Wang JJ, Liang IY, You S,
Zhu Y, Agopian VG, Tseng HR and Yang JD: The role of extracellular
vesicles in disease progression and detection of hepatocellular
carcinoma. Cancers (Basel). 13:30762021. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
van Niel G, D'Angelo G and Raposo G:
Shedding light on the cell biology of extracellular vesicles. Nat
Rev Mol Cell Biol. 19:213–228. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Mukerjee N, Bhattacharya A, Maitra S, Kaur
M, Ganesan S, Mishra S, Ashraf A, Rizwan M, Kesari KK, Tabish TA
and Thorat ND: Exosome isolation and characterization for advanced
diagnostic and therapeutic applications. Mater Today Bio.
31:1016132025. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Fatima H, Chen X, Li Q, Nie X, Xie J and
Nie S: Advances in extracellular vesicle research: Tools and
techniques. Nanoscale Horiz. Feb 12–2026.doi: 10.1039/d5nh00734h
(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Zhang G, Huang X, Liu S, Xu Y, Wang N,
Yang C and Zhu Z: Demystifying EV heterogeneity: Emerging
microfluidic technologies for isolation and multiplexed profiling
of extracellular vesicles. Lab Chip. 25:1228–1255. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Sun N, Lee YT, Zhang RY, Kao R, Teng PC,
Yang Y, Yang P, Wang JJ, Smalley M, Chen PJ, et al: Purification of
HCC-specific extracellular vesicles on nanosubstrates for early HCC
detection by digital scoring. Nat Commun. 11:44892020. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Wu B, Wang Z, Wang G, Zhong Q, Wu Q, Li J,
Ma B, Tan X, Chen J, Wang Y and Zhang X: Quantification of
Fascin-1-Positive extracellular vesicles by nanoflow cytometry for
early detection of hepatocellular carcinoma in liquid biopsy. Int J
Med Sci. 22:1574–1584. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Sun N, Zhang C, Lee YT, Tran BV, Wang J,
Kim H, Lee J, Zhang RY, Wang JJ, Hu J, et al: HCC EV ECG score: An
extracellular vesicle-based protein assay for detection of
early-stage hepatocellular carcinoma. Hepatology. 77:774–788. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Rushton AJ, Nteliopoulos G, Shaw JA and
Coombes RC: A review of circulating tumour cell enrichment
technologies. Cancers (Basel). 13:9702021. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Schulze K, Gasch C, Staufer K, Nashan B,
Lohse AW, Pantel K, Riethdorf S and Wege H: Presence of
EpCAM-positive circulating tumor cells as biomarker for systemic
disease strongly correlates to survival in patients with
hepatocellular carcinoma. Int J Cancer. 133:2165–2171. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Morris KL, Tugwood JD, Khoja L, Lancashire
M, Sloane R, Burt D, Shenjere P, Zhou C, Hodgson C, et al:
Circulating biomarkers in hepatocellular carcinoma. Cancer
Chemother Pharmacol. 74:323–332. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Yang X, Ni H, Lu Z, Zhang J, Zhang Q, Ning
S, Qi L and Xiang B: Mesenchymal circulating tumor cells and Ki67:
Their mutual correlation and prognostic implications in
hepatocellular carcinoma. BMC Cancer. 23:102023. View Article : Google Scholar : PubMed/NCBI
|














