Introduction
Calcific aortic valve disease (CAVD) is the most
prevalent valvular heart disease. Aortic valve (AV) calcification
causes restricted valve opening, leading to an unfavorable
prognosis marked by complications, including congestive heart
failure and sudden cardiac death. Therapeutic options for CAVD
remain scarce despite its clinical importance due to the poor
elucidation of mechanisms underlying its pathogenesis. Currently,
surgical or transcatheter AV replacement represent the only
standard treatment strategies for CAVD. Notably, patients with
chronic kidney disease (CKD) demonstrate a higher prevalence of
CAVD (28–85%) and severe aortic stenosis (6–13%) than patients
without CKD (1,2). CKD is characterized by the
progressive loss of renal function, causing uremic toxin
accumulation that contributes to cardiovascular complications.
Among these toxins, p-cresyl sulfate (PCS), a gut
microbiota-generated p-cresol-derived metabolite, has been
implicated in kidney deterioration and cardiovascular diseases
(3–5). Circulating PCS levels increase as
renal function decreases (6);
however, the role of PCS in CKD-induced valvular calcification
remains ambiguous.
The klotho/sirtuin-1 (SIRT1) pathway has previously
been implicated in the pathogenesis of renal-related diseases
(7–11). SIRT1 upregulation has been
associated with endoplasmic reticulum stress inhibition and
vascular calcification attenuation in CKD (12). CKD progression is notably
associated with klotho downregulation. Previous studies have
demonstrated that uremic toxins promote klotho suppression and the
upregulation of hypoxia-inducible factor-1α (HIF-1α) (13,14),
a transcription factor that has been shown to be rapidly degraded
within minutes under normoxic conditions via the
ubiquitin-proteasome system (15).
HIF-1α further suppresses klotho expression by activating p53
(16,17). These findings suggest that PCS, as
a uremic toxin, can directly impact klotho/SIRT1 and HIF-1α
signaling in valvular interstitial cells (VICs).
Klotho deficiency has been demonstrated to
upregulate runt-related transcription factor 2 (RUNX2), a key
driver of osteogenic gene transcription, in AVs. Furthermore,
klotho deficiency has been shown to promote AV fibrosis,
potentially via the AMP-activated protein kinase α/RUNX2 pathway
(18). Recombinant klotho has been
shown to attenuate hyperphosphatemia-induced osteogenic responses
in human aortic VICs (19).
Additionally, our previous study demonstrated that an osteogenic
medium reduced klotho expression in aortic VICs (20), highlighting the important role of
klotho in RUNX2 regulation. However, the role of klotho in
CKD-associated CAVD remains to be fully elucidated.
SIRT1 activation has been demonstrated to mitigate
klotho deficiency-induced vascular disease (21). In human calcified aortic stenosis,
SIRT1 protein levels are reduced within AV leaflets (22). One study on aged apolipoprotein
E−/− mice has demonstrated that resveratrol, a SIRT1
activator, inhibits AV calcification and AV stenosis progression
(23). As an
NAD+-dependent class III deacetylase, SIRT1 regulates
inflammation, cell death and metabolism through deacetylation
(24,25). Furthermore, SRT1720, another SIRT1
activator, has been reported to reduce vascular smooth muscle cell
calcification by inhibiting the RUNX2 pathway (26).
The present study aimed to investigate whether PCS
directly enhanced VIC calcification, thus contributing towards
CKD-associated CAVD, and aimed to explore the therapeutic potential
of the klotho/SIRT1 pathway in VIC osteogenesis.
Materials and methods
Isolation and culture of porcine
VICs
As previously described (20), AV leaflets were obtained from
6-month-old pig hearts purchased from a commercial slaughterhouse
(Yahsen Frozen Foods, Co., Ltd.). AVs were digested with
collagenase I (250 U/ml; MilliporeSigma) for 30 min with gentle
shaking at 37°C to remove endothelial cells, and then were
subjected to a second digestion for 12 h with collagenase I (250
U/ml) under gentle rocking at 37°C to isolate VICs. As previously
described, isolated VICs were cultured in a DMEM/F12 medium
(MilliporeSigma) (20). The
Institutional Animal Care and Use Committee (IACUC) of Taipei
Medical University (Taipei, Taiwan) reviewed and approved the use
of AV tissues and primary VICs in the present study (approval no.
LAC 2020-0332).
Induction of osteogenesis in aortic
VICs and associated treatments
Aortic VICs (1×105/well in the 6-well
plate) were cultured in DMEM/F12 medium in a cell culture incubator
at 37°C for 7 days in the presence or absence of PCS (10 and 100
µM; APExBIO Technology LLC). The present study selected a PCS
concentration of 100 µM to reflect the comparatively high PCS
levels observed in advanced CKD (27). To clarify the underlying mechanism
of PCS activity on VIC calcification, VICs were also incubated with
PX-478 (0.5 µM; cat. no. HY-10231; MedChemExpress), recombinant
klotho (100 pM; cat. no. APA798Hu02; Cloud-Clone Corp. CCC) and the
SIRT1 activator SRT1720 (1 mM; cat. no. CAS 925434-55-5;
Calbiochem; Merck KGaA) at 37°C and refreshed every 24 h before
further analysis.
Calcification assessment
Alizarin Red S (ARS) staining was performed in
isolated VICs to visualize and quantify calcium deposition. VICs
were fixed with 4% paraformaldehyde for 15 min and incubated with
ARS staining solution for 1 h, both at room temperature.
Subsequently, the excess dye was removed and the sample was washed
with distilled water. ARS-stained cells were imaged using an
Olympus CKX41 fluorescence microscope (Olympus Corporation).
Calcification was subsequently quantified using ImageJ software
(version 1.53t, National Institutes of Health).
Western blot analysis
Proteins extracted from aortic VICs were extracted
using RIPA lysis buffer (MilliporeSigma) supplemented with protease
and phosphatase inhibitors. Protein concentrations were determined
using a BCA protein assay kit (Thermo Fisher Scientific, Inc.).
Equal amounts of protein (30 µg per lane) were separated on 5–12%
gradient gels using SDS-PAGE and transferred onto a PVDF membrane.
The membranes were blocked with 10% non-fat milk in PBS for 1 h at
room temperature and incubated overnight at 4°C with primary
antibodies against RUNX2 (1:2,000 dilution; cat. no. 12556; Cell
Signaling Technology), acetylated-NF-κB (1:2,000 dilution;
acetyl-NF-κB; cat. no. 3045; Cell Signaling Technology, Inc.),
NF-κB (1:3,000 dilution; cat. no. 8242; Cell Signaling Technology,
Inc.), HIF-1α (1:1,000 dilution; cat. no. NB100-479; Novus
Biologicals), klotho (1:1,000 dilution; cat. no. LS-C145689;
LifeSpan BioSciences, Inc.) and β-actin (1:6,600 dilution; cat. no.
ab6276; Abcam). After washing, membranes were incubated with
HRP-conjugated goat anti-rabbit IgG-HRP (cat. no. 7074; Cell
Signaling Technology, Inc.) or goat anti-mouse IgG-HRP (cat. no.
7076; Cell Signaling Technology, Inc.) secondary antibodies for 1 h
at room temperature. Protein bands were visualized using an
enhanced chemiluminescence kit (Thermo Fisher Scientific). Band
intensities were quantified using ImageJ software (version 1.53,
NIH) and normalized to β-actin.
PCS-induced CKD rat model development
and klotho recombinant protein administration
Male Wistar rats aged 8 weeks (~250 g; n=13) were
used in this study. The animals were obtained from BioLASCO Taiwan
Co., Ltd. Prior to experimentation, the rats were allowed to
acclimatize for at least 1 week under controlled housing
conditions. Animals were maintained in a specific pathogen-free
animal facility at a temperature of 19–21°C with 40–70% relative
humidity and a 12-h light/12-h dark cycle. Rats had ad
libitum access to food and water, and were fed a standard
laboratory diet (LabDiet® 5001; PMI Nutrition
International). All animals were housed in standard cages under
routine veterinary monitoring throughout the experimental period.
The rats were divided into the following three groups: Control rats
(n=4), PCS-induced CKD model rats (n=5) and PCS-induced CKD rats
(n=4) treated with rat klotho recombinant protein (0.01 mg/kg/day;
cat. no. RPH757Ra02; Arp Inc.). To induce the CKD model, rats were
subject to intraperitoneal injection of PCS (50 mg/kg/day; APExBIO
Technology LLC) for 4 weeks; rats were anesthetized with isoflurane
prior to injections, which was administered by inhalation at a flow
rate of 1 l/min (induction, 5%; maintenance, 2% with oxygen). The
CKD control group was injected with the same volume of isotonic
saline for the following 8 weeks. The CKD + klotho group also
received 0.01 mg/kg klotho protein dissolved in 0.01 mol/l PBS via
intraperitoneal injection once every 2 days for a total of 8 weeks
following the 4-week PCS treatment. After 12 weeks, all rats were
euthanized via administration of 5% isoflurane in oxygen until
breathing had ceased. Upon observation of a loss of righting reflex
and the absence of a response to a toe pinch, bilateral thoracotomy
was performed as a secondary physical method of euthanasia to
ensure that rats had been successfully sacrificed. Death was
confirmed by auscultation to detect the cessation of respiration
and the absence of a heartbeat for ≥5 min, as well as by
confirmation of the absence of a corneal reflex. A bilateral
thoracotomy was then performed and the heart tissue was extracted.
The animal experiments performed in the present study were approved
by the IACUC of Taipei Medical University (approval no. LAC
2023-0087).
Immunohistochemistry (IHC)
AVs isolated from rats were fixed in 4%
paraformaldehyde and embedded in paraffin at room temperature for 2
h, followed by routine paraffin embedding. Paraffin-embedded
tissues were sectioned at 3-µm thickness and mounted on FRC-13
slides (Matsunami Glass). Sections were deparaffinized in Surgipath
xylene (cat. no. 3803665; Leica Biosystems) twice for 10 min each,
and rehydrated through a descending ethanol series consisting of
100, 95, 80 and 70% Surgipath reagent alcohol (cat. no. 3803686;
Leica Biosystems) for 5 min each, followed by washing in tap water
for 5 min.
Heat-induced epitope retrieval was performed using
citrate antigen retrieval buffer (cat. no. CBB500; Scytek
Laboratories, Inc.) for 30 min, after which sections were washed
with TBST wash buffer (cat. no. TBT999; Scytek Laboratories, Inc.)
for 5 min. Endogenous peroxidase activity was blocked using
Peroxidase Block (cat. no. RE7157; Novolink; Leica Biosystems) for
10 min at room temperature, followed by TBST washes twice for 3 min
each. Non-specific binding was blocked using Protein Block solution
(cat. no. RE7158; Novolink; Leica Biosystems) for 10 min at room
temperature, followed by TBST washes twice for 3 min each.
Sections were incubated with a rabbit monoclonal
anti-RUNX2 antibody (cat. no. 12556; Cell Signaling Technology,
Inc.), diluted 1:30 in antibody dilution buffer (cat. no. ADB250;
Roche Tissue Diagnostics), overnight at 4°C in a humidified
chamber. After primary antibody incubation, slides were washed with
TBST twice for 3 min each. Signal detection was performed using the
Polink-2 Plus HRP Rabbit DAB Kit [cat. no. D39-6; GBI (Labs) Ltd.]
according to the manufacturer's instructions. Briefly, sections
were incubated with Rabbit Antibody Enhancer for 15 min at room
temperature, washed with TBST twice for 3 min, and subsequently
incubated with Polymer-HRP for Rabbit for 15 min at room
temperature.
Immunoreactivity was visualized using DAB chromogen
(cat. no. RE7270-K; Novolink; Lecia Biosystems) for 5 min at room
temperature. Slides were rinsed in tap water for 5 min,
counterstained with Surgipath Hematoxylin Gill II (cat. no.
3801522; Leica Biosystems) for 5 min and washed again in tap water
for 5 min. Sections were then dehydrated in 100% ethanol twice for
5 min each, cleared in xylene twice for 5 min each, and mounted
using Surgipath Micromount mounting medium (cat. no. 3801731; Leica
Biosystems). RUNX2 expression was quantified using ImageJ (version
1.53t, National Institutes of Health) under an Olympus CKX41
fluorescence microscope (Olympus Corporation).
Statistical analysis
All parameters are expressed as the mean ± SEM.
Statistical analyses were performed using unpaired Student's t-test
between two groups or one-way analysis of variance for multiple
groups. Post-hoc analysis was performed using Tukey's multiple
comparisons test. Statistical analyses were performed using
Graphpad Prism 9 (Dotmatics). P<0.05 was considered to indicate
a statistically significant difference.
Results
Effects of PCS on VIC
calcification
The present study incubated VICs with 10 and 100 µM
PCS to investigate the effect of PCS on VIC calcification, as well
as its impact on RUNX2 expression. The present study observed that,
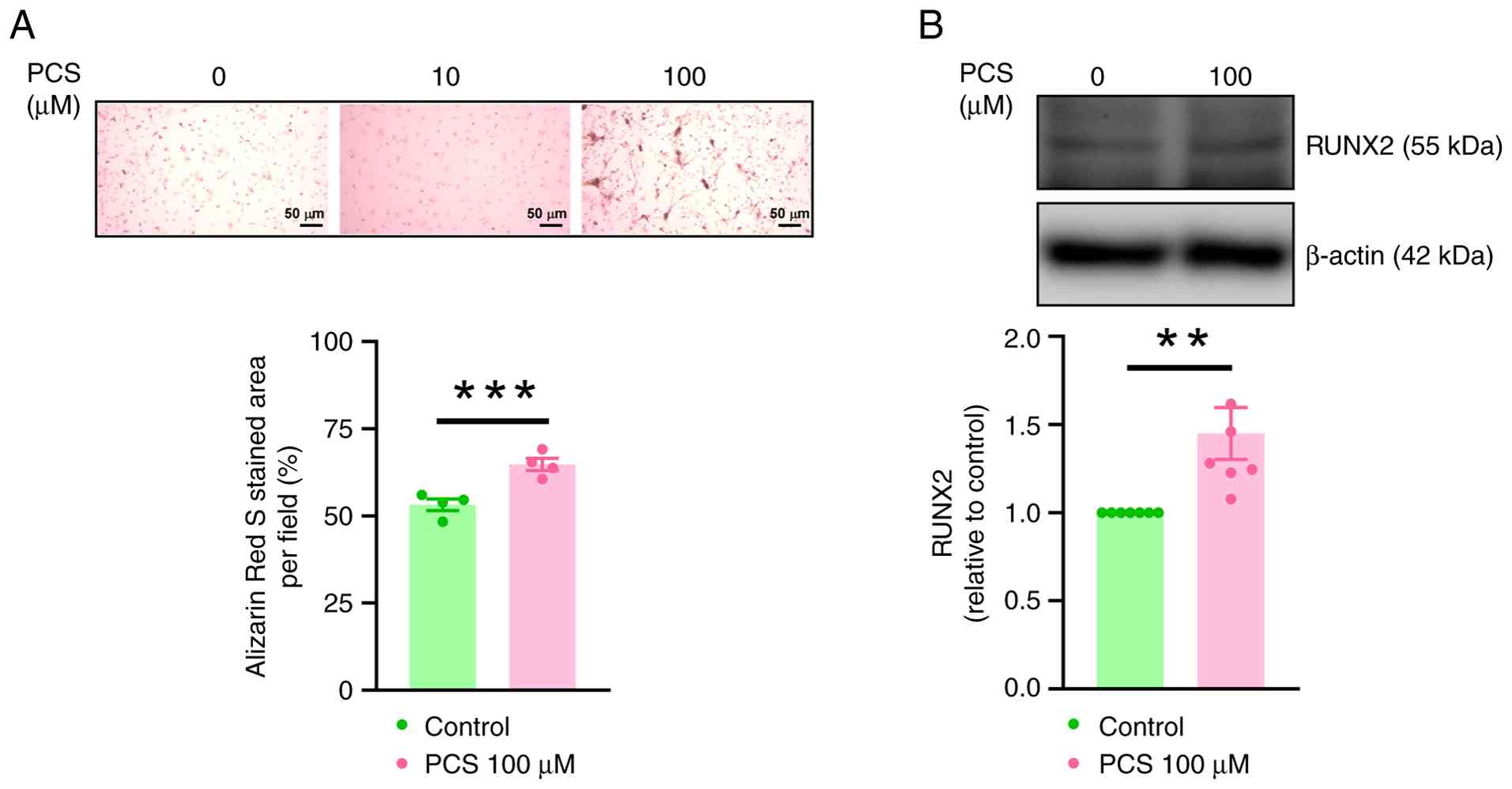
compared with untreated VICs, incubation with 100 µM PCS
significantly increased VIC calcification (Fig. 1A) and RUNX2 protein expression
(Fig. 1B). Furthermore, treatment
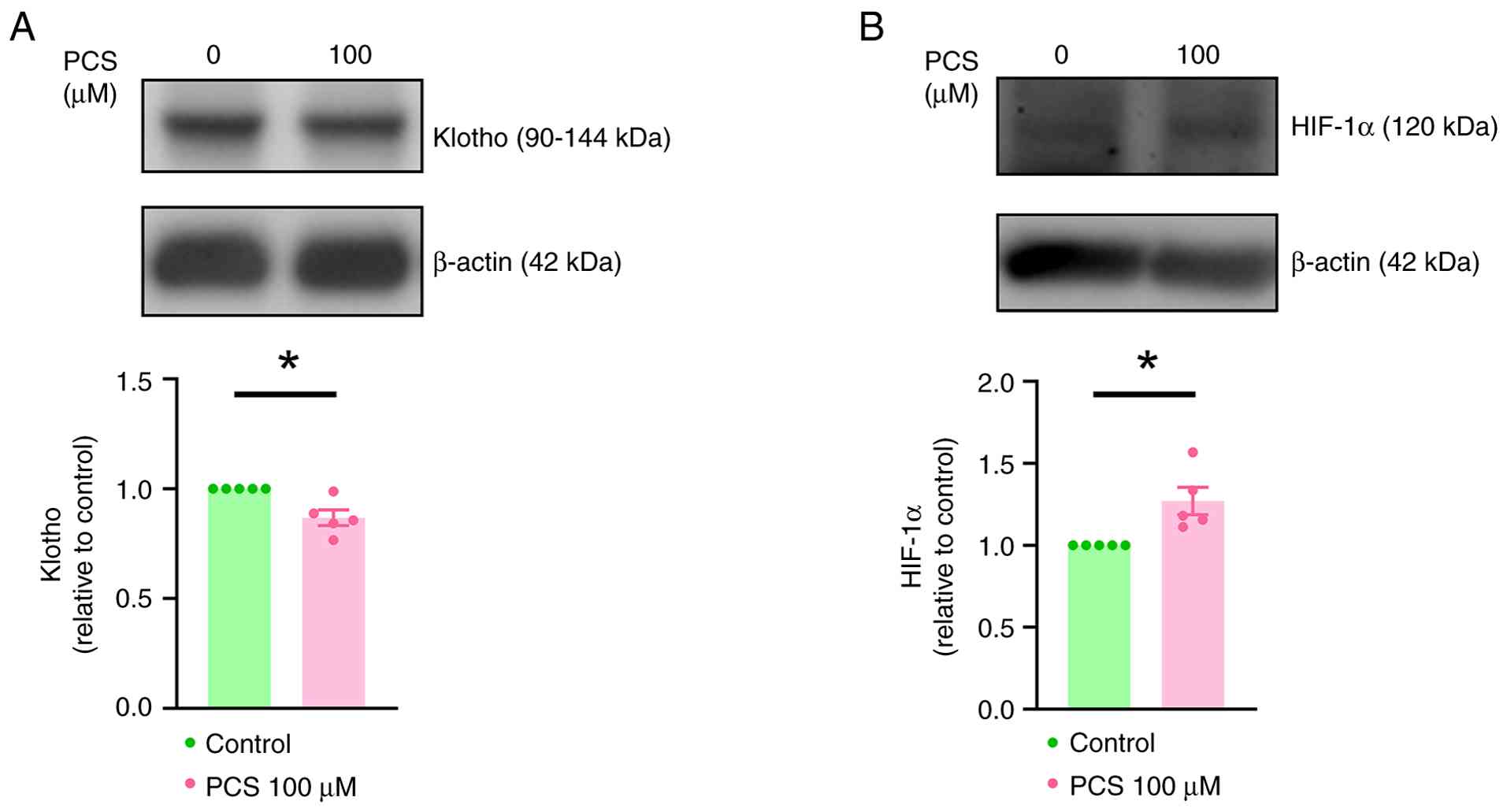
with PCS significantly decreased klotho expression and
significantly increased HIF-1α expression in VICs (Fig. 2).
Effects of a HIF-1α inhibitor,
recombinant klotho and SIRT1 activator on VIC calcification
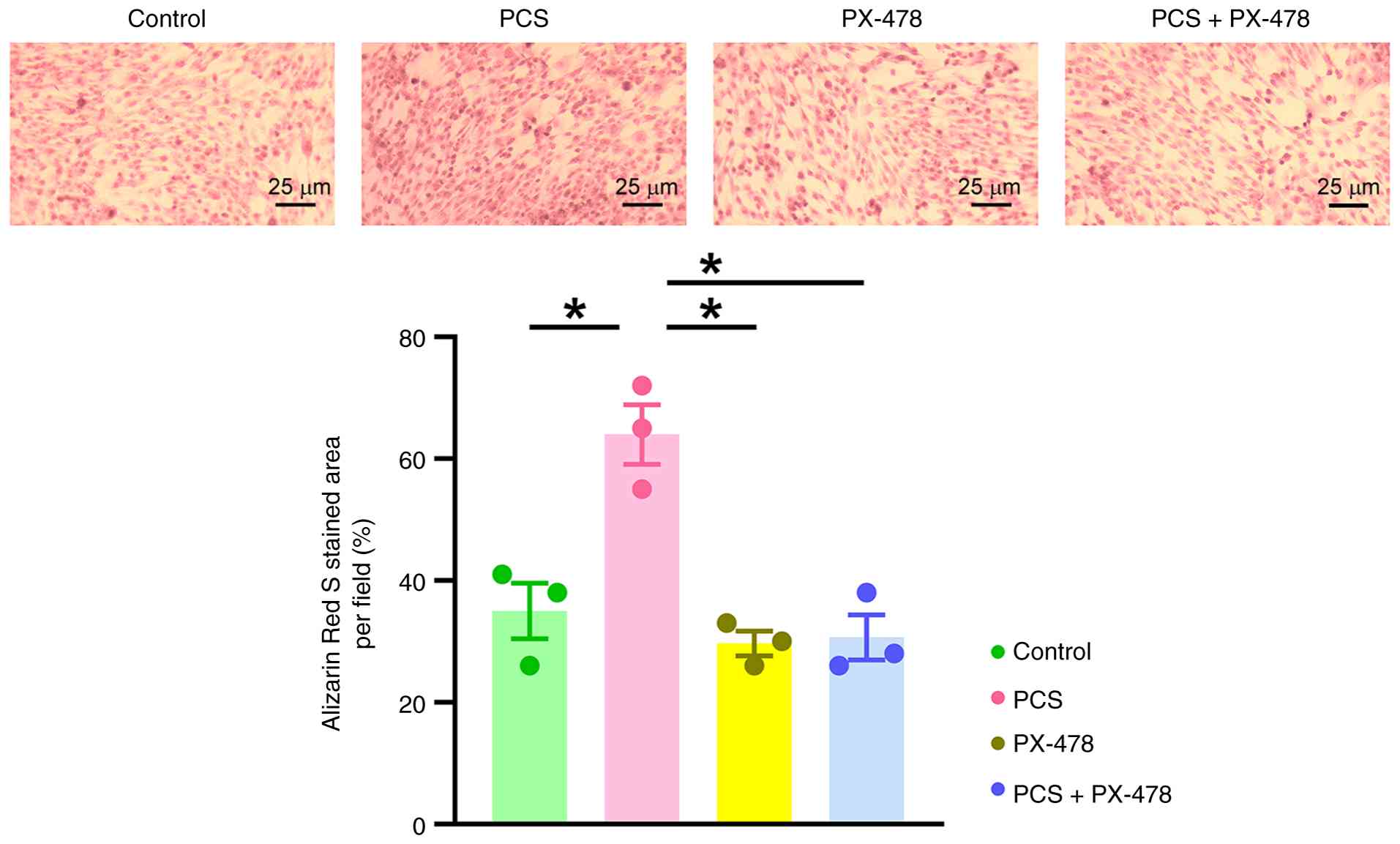
The present study investigated whether HIF-1α or
klotho modulated the effects of PCS in VICs. Treatment with the
HIF-1α inhibitor PX-478 (0.5 µM) was shown to significantly reduce
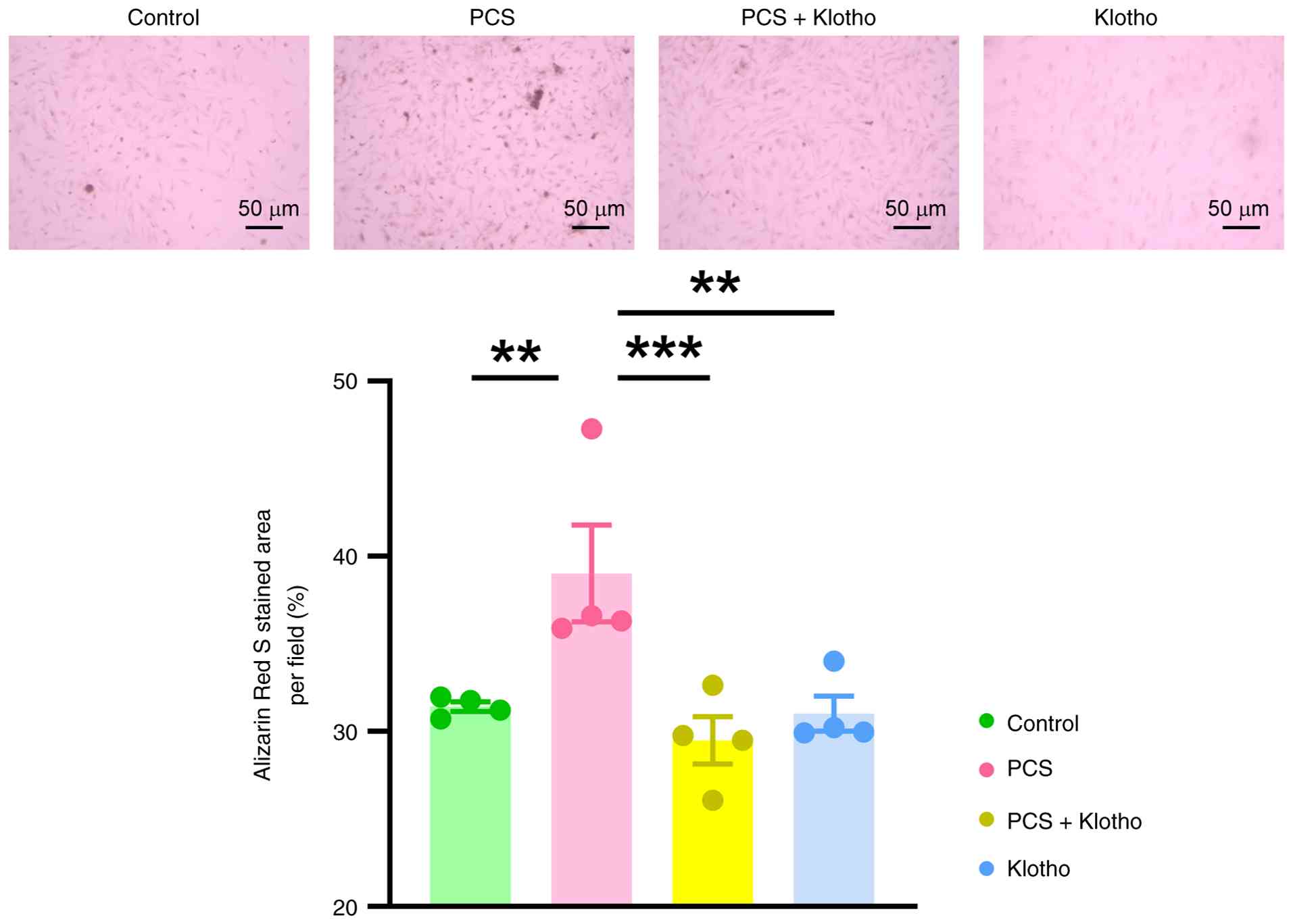
PCS-mediated calcification of VICs (Fig. 3). Additionally, klotho (100 pM)
administration significantly attenuated PCS-induced calcification
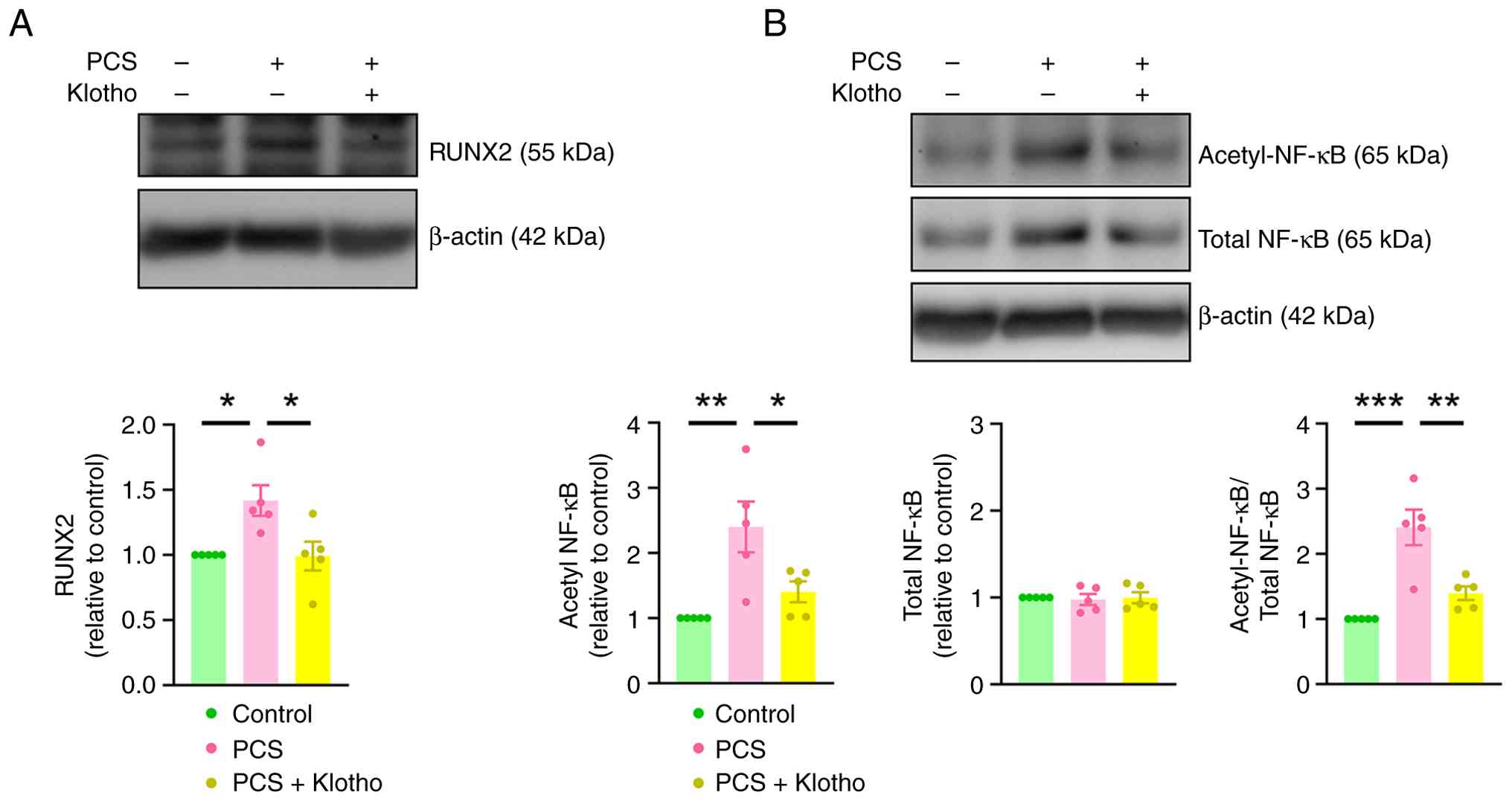
by 24.4% (Fig. 4). Furthermore,
klotho administration significantly attenuated PCS-induced
increases in the expression levels of RUNX2 and acetyl-NF-κB/total
NF-κB in VICs (Fig. 5).
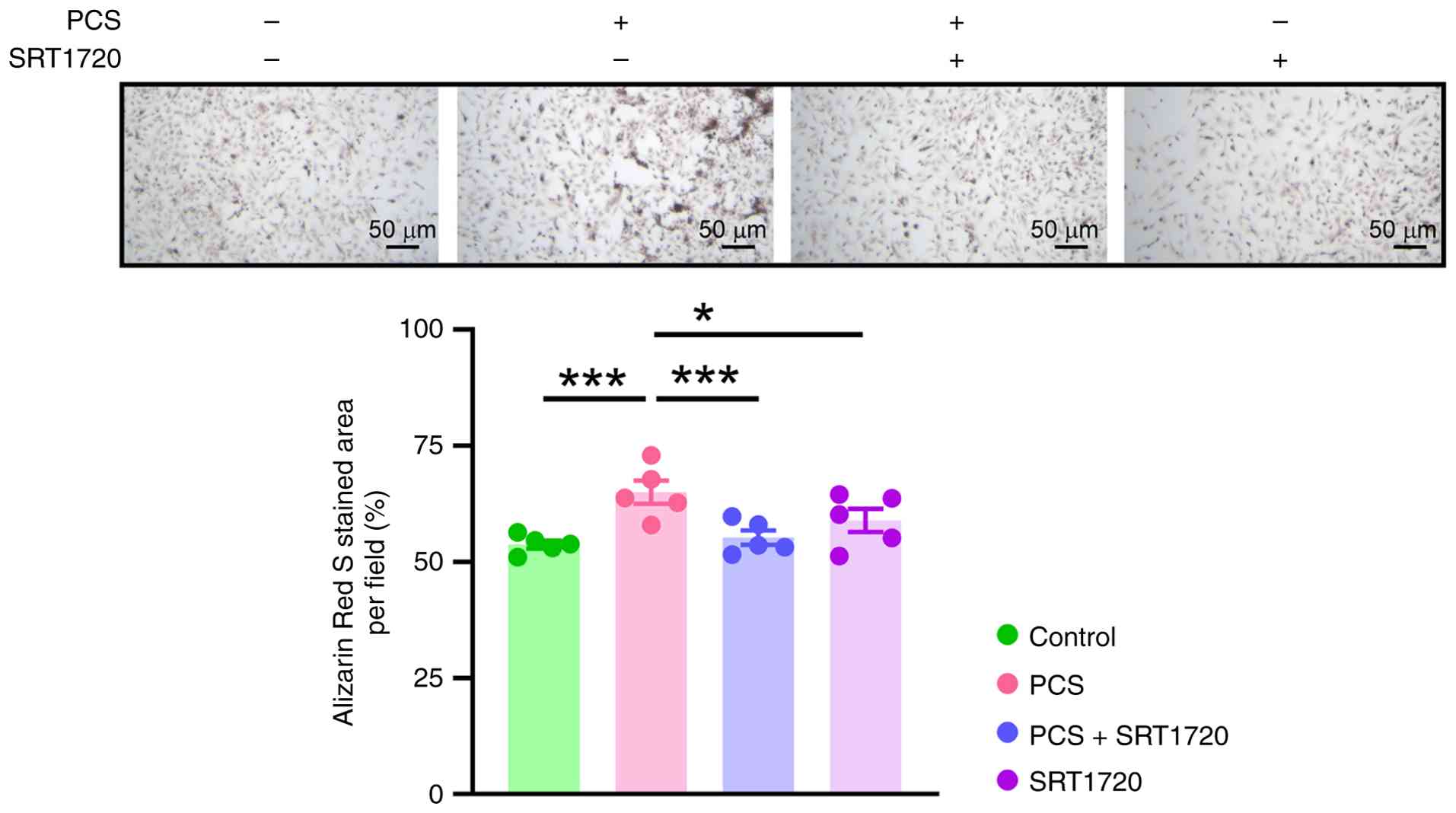
Similarly, the present study examined the effects of
SIRT1 on PCS-treated VICs. ARS staining revealed that SRT1720 (1
mM), a SIRT1 activator, significantly reduced PCS-induced VIC
calcification (Fig. 6).
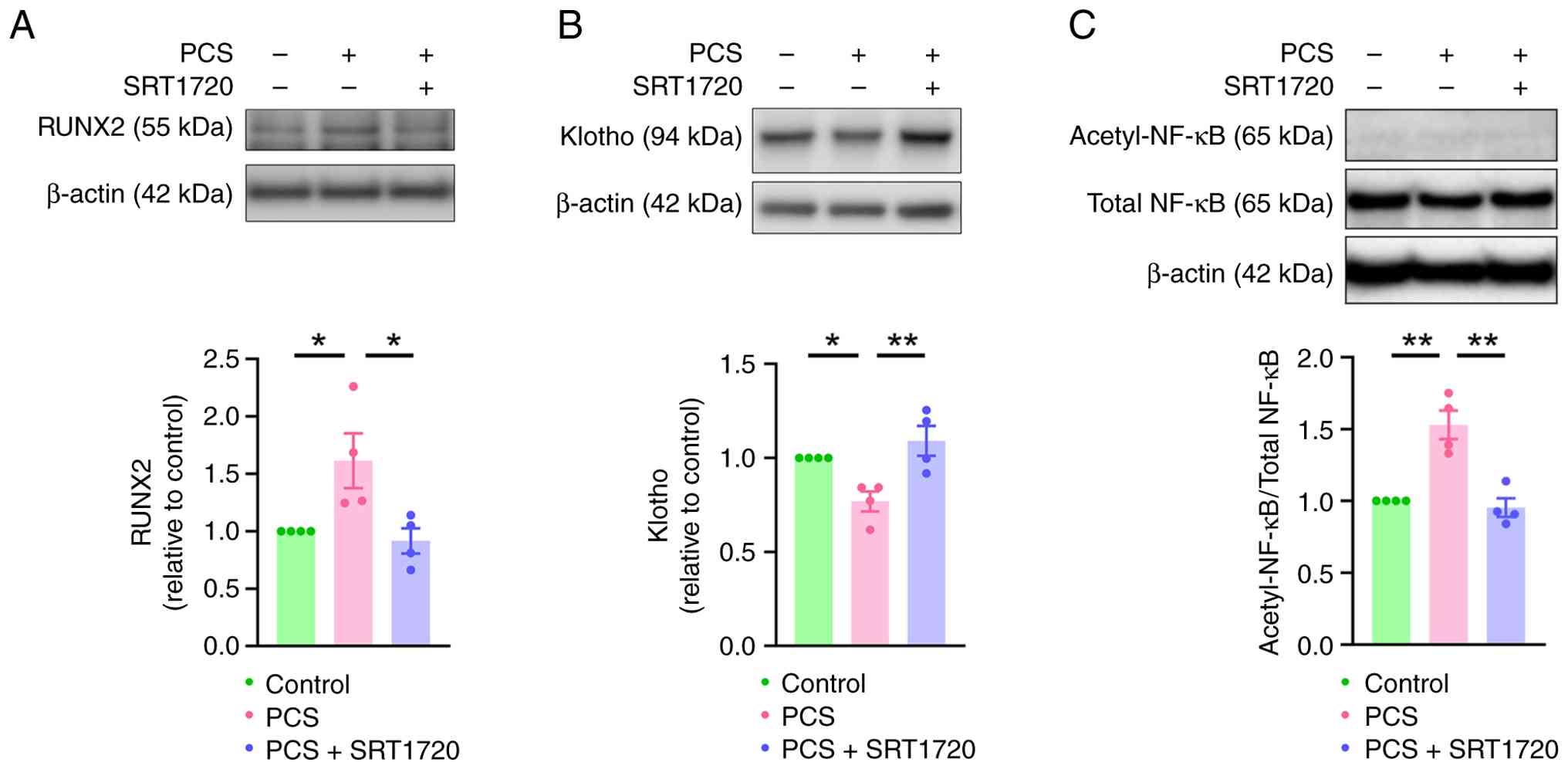
Furthermore, SRT1720 significantly attenuated PCS-mediated
downregulation of klotho and significantly reduced PCS-enhanced
RUNX2 expression in VICs (Fig. 7).
PCS administration also resulted in the elevation of NF-kB
acetylation compared with the control group, which was
significantly mitigated by co-treatment with klotho. These findings
suggested that activation of the klotho/SIRT1 axis was important
for preventing PCS-induced VIC calcification.
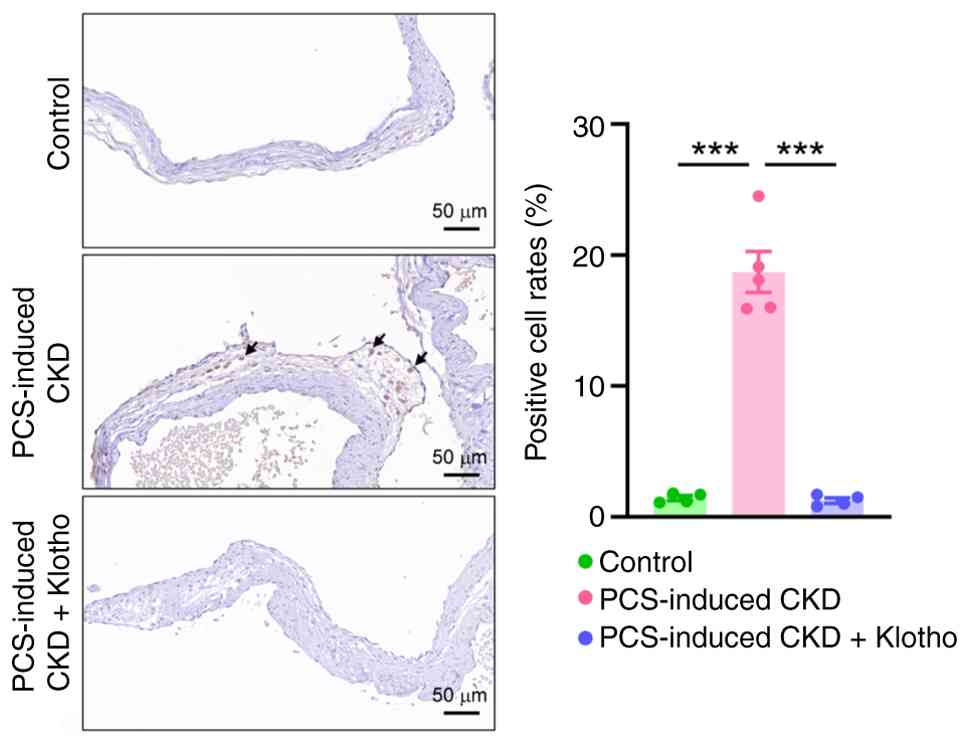
Klotho ameliorates CKD-mediated RUNX2
upregulation in PCS-treated AVs
The present study developed a PCS-induced rat model
of CKD to evaluate whether klotho played a role in the CKD-mediated
upregulation of RUNX2. Rats were treated with recombinant klotho
protein for a total of 8 weeks. IHC revealed that PCS-induced CKD
model rats exhibited a significant increase in RUNX2 expression in
the AV leaflet (Fig. 8). Treatment
with recombinant klotho mitigated the CKD-mediated upregulation of
RUNX2 in PCS-induced model rats.
Discussion
Elevated levels of circulating PCS have previously
been associated with an increased risk of cardiovascular disease in
patients with CKD (22).
Furthermore, CKD occurrence is associated with increased HIF-1α
expression (28–30). PCS, a uremic toxin, is associated
with the pathogenesis of arterial stiffness in CKD (31). A PCS concentration of 100 µM was
used in the in vitro protocols of the present study in order
to reflect the high physiological levels of PCS observed in CKD.
The high physiological levels of PCS in CKD have been suggested to
promote cellular stress, contributing to conditions such as AV
stenosis (32). To the best of our
knowledge, the present study demonstrated for the first time that
PCS upregulated HIF-1α expression levels in VICs. Previous studies
have demonstrated that HIF-1α upregulates RUNX2 expression in stem
cells by activating vascular endothelial growth factor (33), which may further contribute to
RUNX2 upregulation in VICs (34).
The results of the present study indicated that inhibition of
HIF-1α activity significantly ameliorated PCS-mediated
calcification of VICs.
In patients with end-stage kidney disease, plasma
PCS concentrations have been reported to range from 115 to >500
µM, whereas healthy individuals demonstrate concentration levels of
15–35 µM (35). In the VIC culture
system used in the present study, 100 µM PCS was selected for CKD
modelling as it was considered within the upper pathophysiological
range of serum PCS concentrations observed in advanced CKD, and
this aligned with PCS concentrations used in a previous in
vitro study of PCS-induced toxicity in vascular and renal
cells, which typically range from 20–500 µM (36).
Furthermore, the results of the present study
revealed that PCS administration reduced klotho expression and
increased HIF-1α expression, which suggested that this effect may
be mediated by upregulating HIF-1α. Mechanistically, a previous
study has established that HIF-1α-dependent p53 activation
suppresses klotho expression (17). Under hypoxic conditions (low oxygen
availability), HIF-1α is stabilized and translocates into the
nucleus. HIF-1α activation has been shown to suppress klotho
activity by inducing the expression of microRNAs (miRs), such as
miR-34a, and other mediators that reduce klotho levels, fostering a
feedback loop associating oxygen availability, klotho and HIF-1α
(33). Although no knockdown
experiments were performed in the present study to isolate this
specific pathway, existing literature and parallel observations in
our previous study strongly support this mechanism (34). Specifically, PCS treatment
simultaneously upregulated HIF-1α and downregulated klotho, whereas
klotho supplementation was shown to decrease the osteogenic
phenotype. These findings suggested that the PCS-induced
simultaneous upregulation of HIF-1α and suppressed klotho
expression resulted in the observed calcification of VICs.
The observed attenuation of PCS-induced VIC
calcification resulting from klotho supplementation suggested that
PCS-mediated klotho downregulation significantly promoted VIC
calcification. Consistent with the results of a previous study
(37), the present study also
demonstrated reduced klotho with increased HIF-1α in VICs. Klotho
deficiency may contribute, at least in part, to the HIF-1α
activation. The in vivo experiments of the present study
revealed that klotho administration reduced PCS-mediated
upregulation of RUNX2 in the AVs of CKD model rats. The present
study focused on the effects of PCS on the expression of RUNX2,
which is an important driver of osteogenic transdifferentiation in
AVs. The present study identified that, in CKD model rats that were
treated with PCS for 4 weeks, PCS upregulated RUNX2 expression in
rat AVs; however, no calcium deposition was detected via the
Alizarin red S staining. This result likely reflects the model's
relatively early stage, as CKD-associated metabolic disturbances
often precede overt mineral deposition. The results of the present
study also demonstrated that klotho supplementation mitigated the
PCS-induced upregulation of RUNX2 in the AVs of rats. Accordingly,
future studies should analyze a broader panel of histological
markers, such as HIF-1α or klotho, and extend the duration of CKD
model induction.
Furthermore, klotho deficiency has been demonstrated
to aggravate inflammation by activating the NF-κB/RUNX2 axis
(38,39). Given that NF-κB activation is a key
driver of CAVD pathogenesis (39),
klotho has been shown to play a protective role by preventing NF-κB
nuclear translocation and reducing its transcriptional activity
(26). Soluble klotho
administration in klotho-mutant mice and membrane-bound klotho
overexpression in cultured cells have been shown to suppress NF-κB
signaling and its downstream effects (40). Our previous study also demonstrated
that the incubation of aortic VICs in an osteogenic culture medium
reduced klotho expression (20).
Furthermore, PCS has been reported to reduce klotho expression in
renal tubules by inducing klotho gene hypermethylation (13). Previous findings have further
suggested that klotho can mitigate the deleterious effects of PCS
on the heart (35). Collectively,
these findings have provided substantial evidence that the
PCS/NF-κB/RUNX2 axis markedly contributes to klotho suppression and
VIC calcification. Notably, the present study demonstrated that
klotho supplementation attenuated PCS-induced VIC calcification,
highlighting the important role of klotho deficiency in both in
vivo (CKD-induced) and in vitro (PCS-induced) settings.
The results of the present study suggested that PCS significantly
contributed to AV calcification in CKD.
A previous study has demonstrated that klotho
deficiency inhibits SIRT1 activity and that pharmacological
activation of SIRT1 with SRT1720 can ameliorate klotho
deficiency-induced arterial stiffness (21). Furthermore, SRT1720 has been
demonstrated to suppress HIF-1α expression both in vivo and
in vitro (41). A study by
Szymanska et al (42)
reported that treatment with resveratrol or SRT2104, a
SIRT1-specific activator, notably decreased HIF-1α levels.
Additionally, SIRT1-mediated deacetylation of HIF-1α has been shown
to inhibit its transcriptional activity (43). As such, SRT1720 may have enhanced
klotho expression and mitigated PCS-induced VIC calcification by
suppressing HIF-1α. The findings of the present study suggested
that klotho supplementation and SIRT1 activation attenuated the
pro-calcific effects of PCS on VICs. The present study also
demonstrated that SRT1720 treatment inhibited RUNX2 expression in
PCS-treated VICs, indicating that SIRT1 demonstrated a regulatory
role in valvular calcification. Therefore, SIRT1 signaling
activation may represent a promising therapeutic strategy for
mitigating PCS-enhanced CAVD.
Considering that the surgical or transcatheter
replacement of AVs remain the standard treatment strategies for
CAVD, the results of the present study support the feasibility of
the klotho/SIRT1 axis as a target for CAVD treatment, and suggest
that this pathway represents an important avenue for future
clinical translation. Therapeutic strategies aimed at restoring
klotho expression or enhancing SIRT1 activity therefore represent
potential approaches for mitigating CKD-associated CAVD.
Recombinant klotho protein, genetic or small-oligonucleotide-based
klotho upregulation strategies (44) and pharmacological SIRT1 activators,
for example resveratrol analogs and SRT1720 derivatives, are
currently under preclinical evaluation for use in CAVD-targeting
therapies; these strategies could, in principle, be combined with
PCS-lowering interventions, such as gut microbiota modulation,
protein-bound uremic toxin binders or optimized dialysis
modalities, to reduce valvular exposure to uremic toxins. The
findings of the present study offered mechanistic insights for
future translational studies targeting the klotho/SIRT1 axis for
decreasing PCS burden in patients with both CKD and CAVD.
In conclusion, the uremic toxin PCS induced VIC
calcification via HIF-1α/RUNX2 signaling pathway activation and
concurrent klotho downregulation. Activation of the klotho/SIRT1
pathway, via recombinant klotho or SRT1720, effectively attenuated
PCS-induced VIC calcification and may represent a potential
therapeutic strategy for mitigating CAVD progression in CKD.
Acknowledgements
Not applicable.
Funding
The present study was funded by grants from Wan Fang Hospital
(grant nos. 107TMU-WFH-01-1, 108TMU-WFH-01-3, 110-eva-24,
111-wf-eva-30, 112-wf-eva-17 and 113-wf-eva-38), Taipei Medical
University (grant no. TMU109-AE1-B09) and the National Science and
Technology Council, Taiwan, R.O.C. (grant nos.
MOST108-2314-B-038-120 and NSTC 112-2314-B-038-107).
Authors' contributions
SL, YK, NT, PC and YC were responsible for the
conceptualization of the present study. SL, WC, CC and TC analyzed
the data in the present study. SL, TC, WC and NT were responsible
for reviewing/investigation of existing research on the topics
discussed in the study. TC, NT, PC and YC were responsible for the
design of the methods used in the study. SL and TC were responsible
for the acquisition of materials used in the present study. SL, TC
and YC confirm the authenticity of all the raw data. SL and TC were
also responsible for writing the initial draft of the manuscript.
TC, YK, WC, CC, NT, PC and YC all contributed to reviewing and
editing the manuscript. All authors have read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Animal Care and Use Committee of Taipei Medical University
(approval nos. LAC 2020-0332 and LAC 2023-0087).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ikee R, Honda K, Ishioka K, Oka M, Maesato
K, Moriya H, Hidaka S, Ohtake T and Kobayashi S: Differences in
associated factors between aortic and mitral valve calcification in
hemodialysis. Hypertens Res. 33:622–626. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Guerraty MA, Chai B, Hsu JY, Ojo AO, Gao
Y, Yang W, Keane MG, Budoff MJ and Mohler ER III; CRIC Study
Investigators, : Relation of aortic valve calcium to chronic kidney
disease (from the Chronic Renal Insufficiency Cohort Study). Am J
Cardiol. 115:1281–1286. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Barreto FC, Barreto DV, Liabeuf S, Meert
N, Glorieux G, Temmar M, Choukroun G, Vanholder R and Massy ZA;
European Uremic Toxin Work Group (EUTox), : Serum indoxyl sulfate
is associated with vascular disease and mortality in chronic kidney
disease patients. Clin J Am Soc Nephrol. 4:1551–1558. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chiu CA, Lu LF, Yu TH, Hung WC, Chung FM,
Tsai IT, Yang CY, Hsu CC, Lu YC, Wang CP and Lee YJ: Increased
levels of total P-Cresylsulphate and indoxyl sulphate are
associated with coronary artery disease in patients with diabetic
nephropathy. Rev Diabet Stud. 7:275–284. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liabeuf S, Barreto DV, Barreto FC, Meert
N, Glorieux G, Schepers E, Temmar M, Choukroun G, Vanholder R and
Massy ZA; European Uraemic Toxin Work Group (EUTox), : Free
p-cresylsulphate is a predictor of mortality in patients at
different stages of chronic kidney disease. Nephrol Dial
Transplant. 2010.25:1183–1191. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Viaene L, Meijers BK, Bammens B,
Vanrenterghem Y and Evenepoel P: Serum concentrations of p-cresyl
sulfate and indoxyl sulfate, but not inflammatory markers, increase
in incident peritoneal dialysis patients in parallel with loss of
residual renal function. Perit Dial Int. 34:71–78. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Su H, Gao D, Chen Y and Zuo Z: The
relationship between klotho and SIRT1 expression in renal aging
related disease. Int J Gen Med. 15:7885–7893. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hu MC, Kuro-o M and Moe OW: Klotho and
chronic kidney disease. Contrib Nephrol. 180:47–63. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Giachelli CM: The emerging role of
phosphate in vascular calcification. Kidney Int. 75:890–897. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lindberg K, Amin R, Moe OW, Hu MC, Erben
RG, Ostman Wernerson A, Lanske B, Olauson H and Larsson TE: The
kidney is the principal organ mediating klotho effects. J Am Soc
Nephrol. 25:2169–2175. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hu MC, Shi M, Zhang J, Quinones H,
Griffith C, Kuro-o M and Moe OW: Klotho deficiency causes vascular
calcification in chronic kidney disease. J Am Soc Nephrol.
22:124–136. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yan J, Wang J, He JC and Zhong Y: Sirtuin
1 in chronic kidney disease and therapeutic potential of targeting
Sirtuin 1. Front Endocrinol (Lausanne). 13:9177732022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sun CY, Chang SC and Wu MS: Suppression of
Klotho expression by protein-bound uremic toxins is associated with
increased DNA methyltransferase expression and DNA
hypermethylation. Kidney Int. 81:640–650. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu H, Li Y and Xiong J: The role of
hypoxia-inducible factor-1 alpha in renal disease. Molecules.
27:73182022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Marxsen JH, Stengel P, Doege K, Heikkinen
P, Jokilehto T, Wagner T, Jelkmann W, Jaakkola P and Metzen E:
Hypoxia-inducible factor-1 (HIF-1) promotes its degradation by
induction of HIF-alpha-prolyl-4-hydroxylases. Biochem J. 381((Pt
3)): 761–767. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu TK, Wei CW, Pan YR, Hsu RJ, Wu CY and
Yu YL: The uremic toxin p-cresyl sulfate induces proliferation and
migration of clear cell renal cell carcinoma via microRNA-21/HIF-1α
axis signals. Sci Rep. 9:32072019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xie L, Wang Y, Li Q, Ji X, Tu Y, Du S, Lou
H, Zeng X, Zhu L, Zhang J and Zhu M: The
HIF-1α/p53/miRNA-34a/Klotho axis in retinal pigment epithelial
cells promotes subretinal fibrosis and exacerbates choroidal
neovascularization. J Cell Mol Med. 25:1700–1711. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen J, Lin Y and Sun Z: Deficiency in the
anti-aging gene Klotho promotes aortic valve fibrosis through
AMPKalpha-mediated activation of RUNX2. Aging Cell. 15:853–860.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li F, Yao Q, Ao L, Cleveland JC Jr, Dong
N, Fullerton DA and Meng X: Klotho suppresses high
phosphate-induced osteogenic responses in human aortic valve
interstitial cells through inhibition of Sox9. J Mol Med (Berl).
95:739–751. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li SJ, Kao YH, Chung CC, Chen WY, Cheng WL
and Chen YJ: Activated p300 acetyltransferase activity modulates
aortic valvular calcification with osteogenic transdifferentiation
and downregulation of Klotho. Int J Cardiol. 232:271–279. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gao D, Zuo Z, Tian J, Ali Q, Lin Y, Lei H
and Sun Z: Activation of SIRT1 attenuates klotho deficiency-induced
arterial stiffness and hypertension by enhancing AMP-activated
protein kinase activity. Hypertension. 68:1191–1199. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Carter S, Miard S, Roy-Bellavance C,
Boivin L, Li Z, Pibarot P, Mathieu P and Picard F: Sirt1 inhibits
resistin expression in aortic stenosis. PLoS One. 7:e351102012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cai Z, Wang J, Liu M, Yuan T and Li F:
Abstract 12300: Sirtuin 1 Prevents Age-associated Aortic Valve
Calcification in vivo. Circulation. 140 (Suppl 1):A123002019.
|
|
24
|
Lu C, Zhao H, Liu Y, Yang Z, Yao H, Liu T,
Gou T, Wang L, Zhang J, Tian Y, et al: Novel Role of the SIRT1 in
endocrine and metabolic diseases. Int J Biol Sci. 19:484–501. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu TF and McCall CE: Deacetylation by
SIRT1 reprograms inflammation and cancer. Genes Cancer. 4:135–147.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bartoli-Leonard F, Wilkinson FL, Schiro A,
Inglott FS, Alexander MY and Weston R: Suppression of SIRT1 in
diabetic conditions induces osteogenic differentiation of human
vascular smooth muscle cells via RUNX2 signalling. Sci Rep.
9:8782019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Poesen R, Viaene L, Verbeke K, Augustijns
P, Bammens B, Claes K, Kuypers D, Evenepoel P and Meijers B:
Cardiovascular disease relates to intestinal uptake of p-cresol in
patients with chronic kidney disease. BMC Nephrol. 15:872014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fu Q, Colgan SP and Shelley CS: Hypoxia:
The force that drives chronic kidney disease. Clin Med Res.
14:15–39. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hirakawa Y, Tanaka T and Nangaku M: Renal
hypoxia in CKD; Pathophysiology and detecting methods. Front
Physiol. 8:992017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu M, Ning X, Li R, Yang Z, Yang X, Sun S
and Qian Q: Signalling pathways involved in hypoxia-induced renal
fibrosis. J Cell Mol Med. 21:1248–1259. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chern YB, Chia KL, Liu CH, Lin YL, Tsai JP
and Hsu BG: Serum p-Cresyl sulfate is independently associated with
aortic stiffness in non-dialysis chronic kidney disease patients.
Life (Basel). 15:11162025.PubMed/NCBI
|
|
32
|
Zoccali C, Mallamaci F, Adamczak M, de
Oliveira RB, Massy ZA, Sarafidis P, Agarwal R, Mark PB, Kotanko P,
Ferro CJ, et al: Cardiovascular complications in chronic kidney
disease: A review from the European renal and cardiovascular
medicine working group of the European renal association.
Cardiovasc Res. 119:2017–2032. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xu Q, Liu Z, Guo L, Liu R, Li R, Chu X,
Yang J, Luo J, Chen F and Deng M: Hypoxia mediates runt-related
transcription factor 2 expression via induction of vascular
endothelial growth factor in periodontal ligament stem cells. Mol
Cells. 42:763–772. 2019.PubMed/NCBI
|
|
34
|
Li SJ, Kao YH, Chung CC, Cheng WL, Lin YK
and Chen YJ: Vascular endothelial growth factor on Runt-related
transcript factor-2 in aortic valve cells. Eur J Clin Invest.
51:e134702021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gryp T, Vanholder R, Vaneechoutte M and
Glorieux G: p-Cresyl Sulfate. Toxins (Basel). 9:522017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Watanabe H, Miyamoto Y, Enoki Y, Ishima Y,
Kadowaki D, Kotani S, Nakajima M, Tanaka M, Matsushita K, Mori Y,
et al: p-Cresyl sulfate, a uremic toxin, causes vascular
endothelial and smooth muscle cell damages by inducing oxidative
stress. Pharmacol Res Perspect. 3:e000922015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Afsar B, Kanbay M and Afsar RE:
Interconnections of fibroblast growth factor 23 and klotho with
erythropoietin and hypoxia-inducible factor. Mol Cell Biochem.
477:1973–1985. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
He T, Xiong J, Huang Y, Zheng C, Liu Y, Bi
X, Liu C, Han W, Yang K, Xiao T, et al: Klotho restrain RIG-1/NF-ĸB
signaling activation and monocyte inflammatory factor release under
uremic condition. Life Sci. 231:1165702019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu T, Zhang L, Joo D and Sun SC: NF-ĸB
signaling in inflammation. Signal Transduct Target Ther.
2:17023–2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhao Y, Banerjee S, Dey N, LeJeune WS,
Sarkar PS, Brobey R, Rosenblatt KP, Tilton RG and Choudhary S:
Klotho depletion contributes to increased inflammation in kidney of
the db/db mouse model of diabetes via RelA (serine)536
phosphorylation. Diabetes. 60:1907–1916. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Han W, Wang C, Yang Z, Mu L, Wu M, Chen N,
Du C, Duan H and Shi Y: SRT1720 retards renal fibrosis via
inhibition of HIF1A/GLUT1 in diabetic nephropathy. J Endocrinol.
241:85–98. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Szymanska M, Manthe S, Shrestha K, Girsh
E, Harlev A, Kisliouk T and Meidan R: Sirtuin-1 inhibits
endothelin-2 expression in human granulosa-lutein cells via hypoxia
inducible factor 1 alpha and epigenetic modifications †. Biol
Reprod. 104:387–398. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ryu DR, Yu MR, Kong KH, Kim H, Kwon SH,
Jeon JS, Han DC and Noh H: Sirt1-hypoxia-inducible factor-1α
interaction is a key mediator of tubulointerstitial damage in the
aged kidney. Aging Cell. 18:e129042019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Abraham CR, Chen C, Cuny GD, Glicksman MA
and Zeldich E: Small-molecule Klotho enhancers as novel treatment
of neurodegeneration. Future Med Chem. 4:1671–1679. 2012.
View Article : Google Scholar : PubMed/NCBI
|