Thyroid cancer (TC) is currently the most common
malignancy of the endocrine system and its incidence has steadily
increased worldwide over the past 20 years (1,2). TC
exhibits considerable heterogeneity and shares typical
histopathological features with other tumors (3). The two varieties of epithelial cells
that constitute the thyroid gland are follicular and parafollicular
cells. Follicular cells convert iodine into T4 and T3, and the
hormones T3 and T4 (tyrosine) regulate metabolism (4). By contrast, parafollicular cells are
epithelial cells that produce calcitonin. Primary TC predominantly
originates from thyroid follicular cells and is classified as an
epithelial tumor, which can be categorized into three primary
pathological types. The most prevalent type is papillary TC (PTC),
followed by follicular TC (FTC), with the most aggressive form
being anaplastic TC (ATC). Furthermore, medullary TC (MTC) arises
from parafollicular cells (5).
Tumorigenesis is mainly driven by somatic mutations
that occur during the initial stages of transformation. According
to various genetic studies on TC, several oncogenes and tumor
suppressor genes have been identified that can be used as
diagnostic markers and therapeutic targets (6–8). For
example, pathogenic loss-of-function mutations in genes responsible
for tumor suppression constitute a common genetic event involved in
TC. Crucial examples include phosphatase and tensin homolog and
tumor protein 53, which are frequently inactivated via such
mutations. These alterations are highly prevalent and
well-characterized events driving the initiation and progression of
TC. The use of targeted therapy in affected patients has become
possible due to the development of tyrosine kinase inhibitors
(TKIs) (9–11). The major driver mutations involved
in TC mainly include B-Raf proto-oncogene (BRAF), RET
(mutations/fusions) and RAS mutations. In addition, gene mutations
related to the VEGFR and PI3K/AKT/mTOR signaling pathways, as well
as mutations in drug resistance-associated genes such as
mesenchymal epithelial transition and neurofibromin 2, are also
closely connected with the regulation of TKI efficacy and the
mechanisms of drug resistance. TKIs function by competing with
adenosine triphosphate (ATP) for binding sites, thus inhibiting
phosphorylation by kinases, and ultimately preventing the signaling
and proliferation of tumor cells (12). Novel and state-of-the-art genetic
testing based on advanced next-generation sequencing has promoted
the development of tumor-targeted therapies, in which the molecular
drivers of tumorigenesis act as the therapeutic targets. Previous
studies (13,14) have compared targeted therapies with
non-targeted agents and immunotherapeutic agents lacking
well-defined molecular targets, including chemotherapeutic drugs
for advanced anaplastic thyroid carcinoma (ATC) and
radioiodine-refractory differentiated thyroid carcinoma (RAIR-DTC),
such as taxanes (paclitaxel, docetaxel), anthracyclines
(doxorubicin), platinum-based agents (cisplatin, carboplatin,
etc.), and PD-1/PD-L1 inhibitors (pembrolizumab, nivolumab). The
results indicate that targeted therapies achieve superior efficacy
and a lower incidence of off-target adverse events. The aim of the
present study was to examine and summarize the role and
organization of the rearranged during transfection (RET)
proto-oncogene, along with its disease-causing mechanisms, in
different variants of TC. In addition, the present revied aimed to
investigate small-molecule inhibitors of tyrosine kinases that
target RET mutations, and present a summary of advances in research
on the management and treatment of TC linked to alterations in the
RET gene.
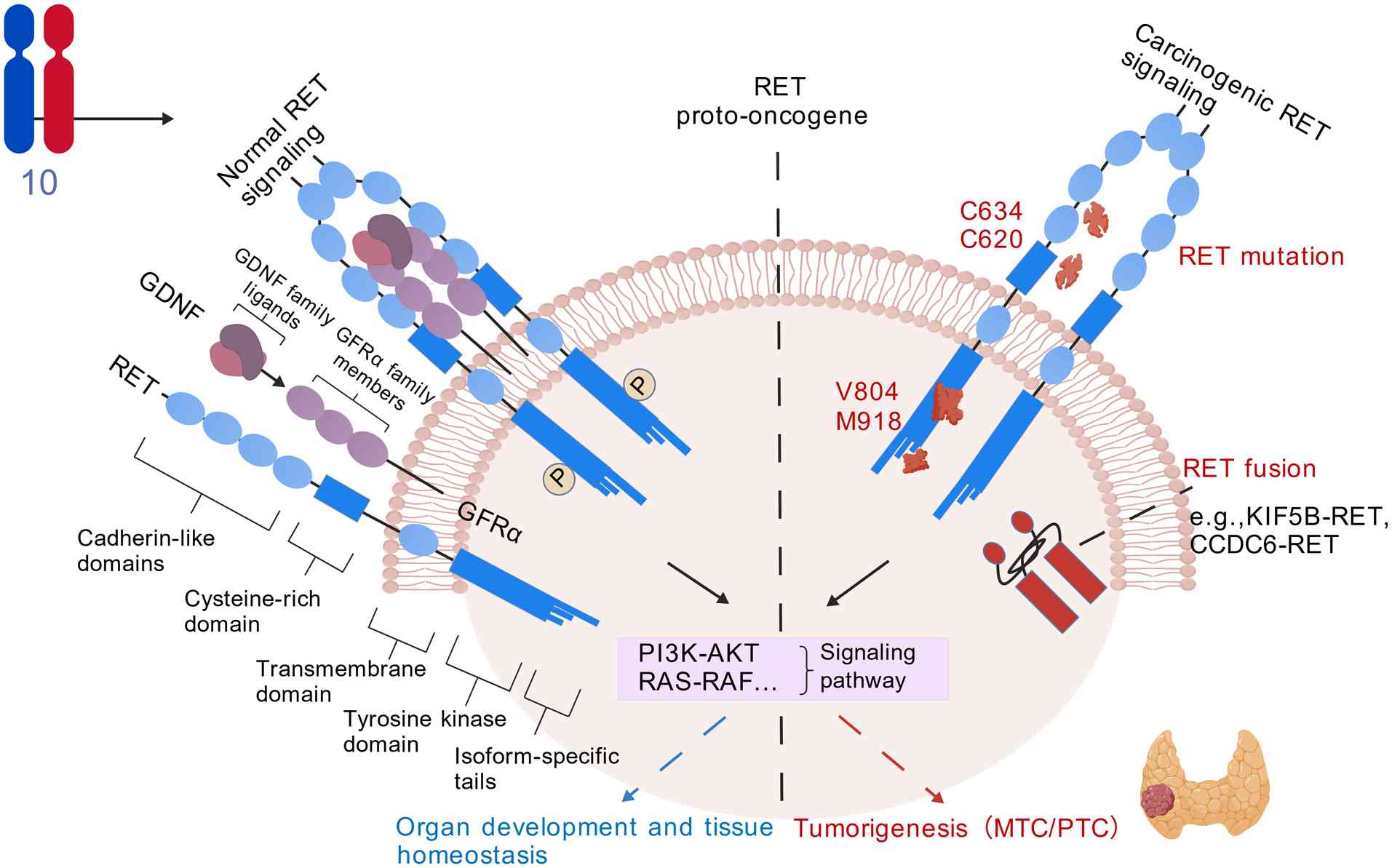
The RET gene, which is located on chromosome 10q11.2
and encodes a 170 kDa transmembrane receptor tyrosine kinase, was
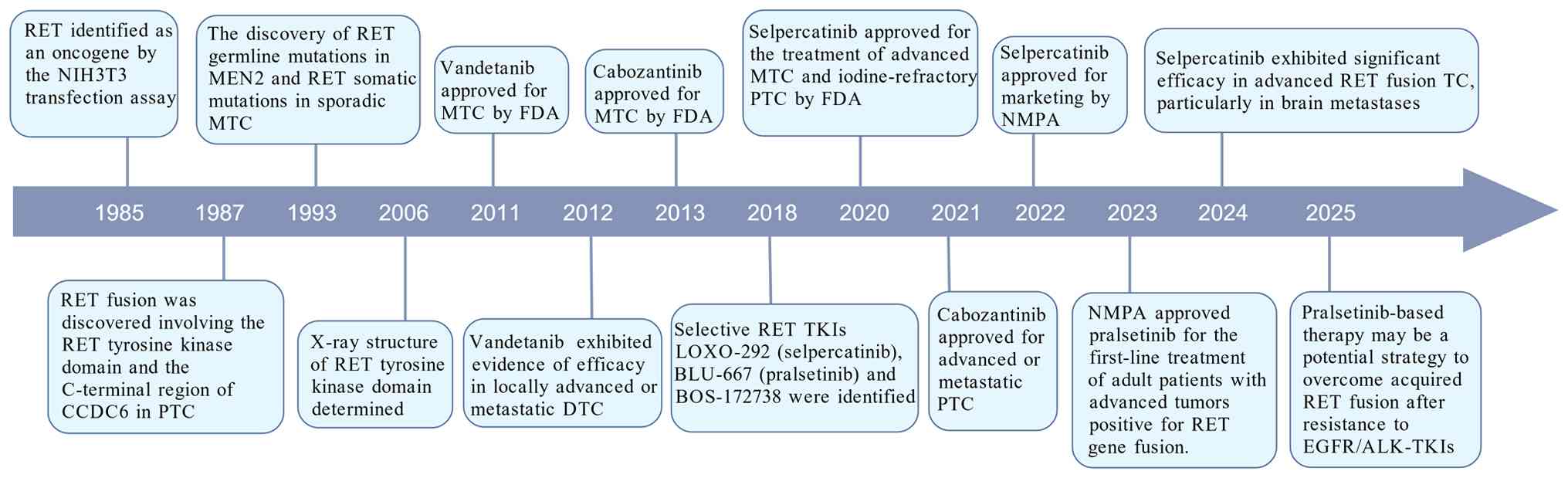
first discovered by researchers in 1985 (15). Structurally, the RET protein
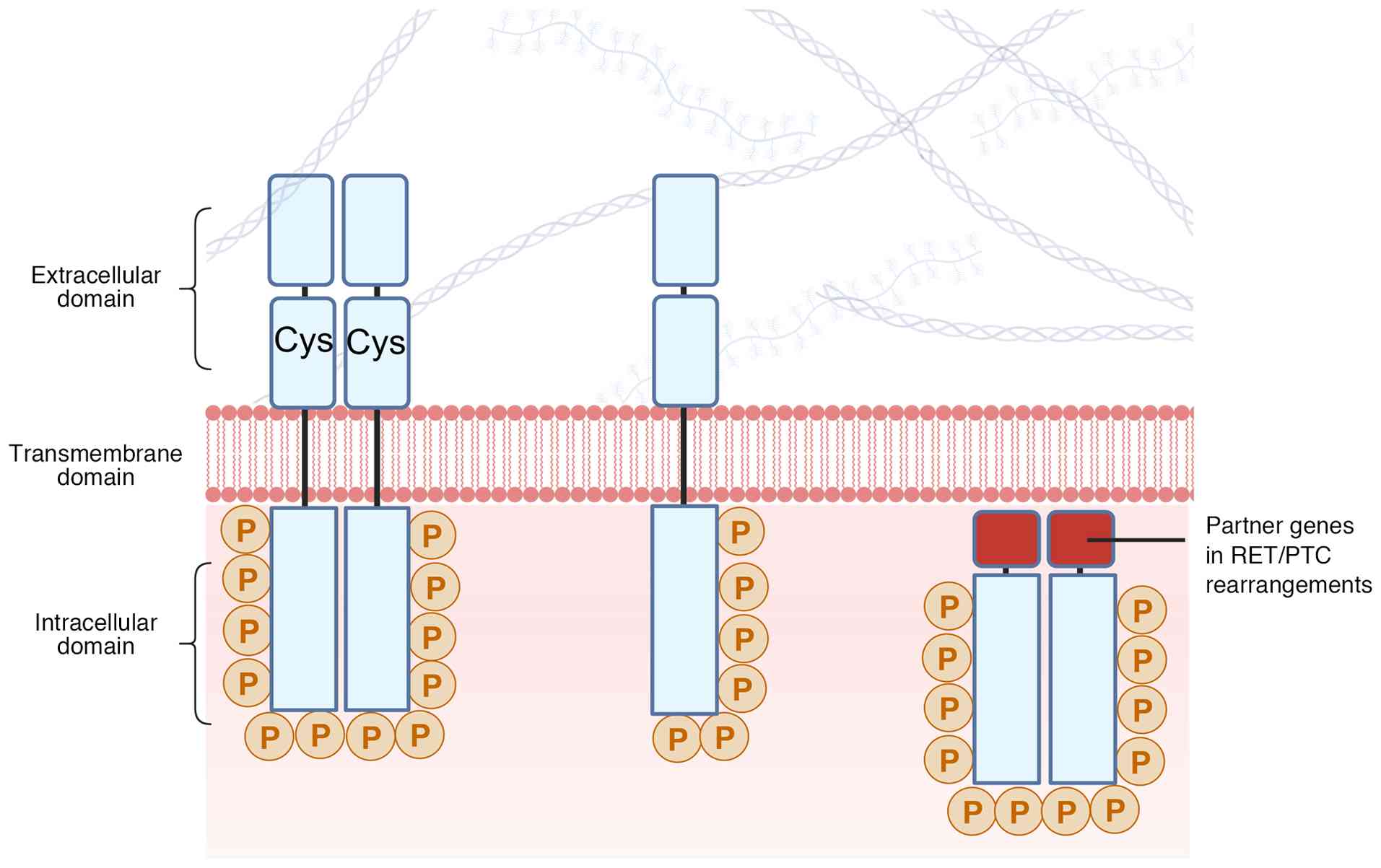
comprises an extracellular region featuring four cadherin-like
domains (CLD1-4), a calcium-binding site and a cysteine-rich domain
(CRD), a transmembrane region, and an intracellular region
containing the juxtamembrane domain and a bilobed tyrosine kinase
domain (TKD). Under normal physiological conditions, RET activation
is ligand-dependent. The binding of the glial cell line-derived
neurotrophic factor (GDNF) family ligands, along with their
co-receptor GDNF family receptor α, induces RET dimerization. This
dimerization triggers the trans-autophosphorylation of specific
tyrosine residues within the TKD (16–18).
Subsequently, these phosphorylated residues serve as docking sites
for downstream adaptor proteins, initiating crucial signaling
cascades such as the RAS/MAPK and PI3K/AKT pathways, which govern
essential cellular processes, including proliferation,
differentiation and survival (Fig.
1) (19).
The constitutive, ligand-independent activation of
RET, primarily driven by specific mutations, is central to the
pathogenesis of MTC (20–22). These activating mutations occur
either in the germline (hereditary, 95–98%) or somatically
(sporadic, 25–50%) (23–25). The 95–98% refers to germline
mutations accounting for the majority of mutations in hereditary TC
cases, while the 25–50% refers to somatic mutations in all TC cases
(including sporadic ones); these two proportions describe different
mutation subsets and are not mutually exclusive. The global
age-adjusted incidence rate of germline RET mutations is estimated
to be 0.06 cases per 100,000 persons/year, and the corresponding
prevalence is 1.3 cases per 100,000 individuals (26,27).
Germline RET mutations are responsible for virtually
all cases of hereditary MTC, which is categorized as a type of MEN2
syndrome. MEN2 is stratified into two primary clinical subtypes
based on distinct genotype-phenotype associations (28,29).
MEN2A (~95% of cases): This subtype is characterized
by MTC, a high frequency (~50%) of pheochromocytoma and primary
hyperparathyroidism in 20–30% of patients (30). Approximately 95% of MEN2A mutations
(all mutations described in the MEN2A subtype involve cysteine
residues in the extracellular CRD and codon 634 in exon 11) affect
cysteine residues within the extracellular CRD, with codon 634 in
exon 11 being the most prevalent site (85%). The substitution of
cysteine disrupts normal disulfide bonding, leading to aberrant,
ligand-independent receptor dimerization and constitutive kinase
activation (31–33).
MEN2B (~5% of cases): Recognized as the most
aggressive form, MEN2B presents with early-onset, metastatic MTC,
frequent pheochromocytoma, ganglioneuromatosis and marfanoid
habitus (34,35). Over 95% of MEN2B cases are caused
by a specific methionine-to-threonine substitution at codon 918
(M918T) in the TKD activation loop (36). This mutation fundamentally alters
the conformation of the kinase, markedly enhancing its catalytic
activity and enabling robust signaling even in a monomeric or
dimerized state, independent of ligand binding (37,38).
In addition, in <10% of MEN2B cases, A883F mutations or other
combination mutations, such as valine-to-methionine substitution at
codon 804 of RET (V804M), are involved (Fig. 2) (39,40).
These mutations all occur in the RET gene.
Sporadic MTC: Somatic RET mutations are identified
in ~55% of non-hereditary MTC cases (41). The prevalence of these mutations is
associated with tumor burden, markedly rising in advanced or
metastatic disease (42). The
M918T mutation is again the most common somatic variant (~40%),
underscoring its potent oncogenic potential. Other recurrent
somatic mutation sites include codons 611, 618, 620, 630, 634, 768,
883 and 891 (43–47).
RET-activated tumors may arise from chromosomal
rearrangements, which involve the fusion of the RET kinase domain
with various protein partners that possess dimerization domains
(48–50). This phenomenon occurs along with
germline and somatic mutations that activate RET. Previous studies
have proposed that the erroneous repair of DNA double-strand breaks
serves a critical role in the molecular mechanisms that lead to RET
fusion. Specifically, these chromosomal breaks facilitate the
fusion of the 3′ sequence that encodes the RET mRNA kinase domain
with the 5′ sequence that encodes domains responsible for both the
dimerization and localization of an upstream partner gene. This
fusion ultimately results in the production of active RET fusion
proteins, which contribute to tumorigenesis (51–53).
The RET fusion protein can cause cancer through two pathways:
First, by sending a message that promotes the continued
proliferation of cancerous cells due to the ability to send such
messages without a ligand; and secondly, due to the unique
endocytosis process, which prevents RET from being inhibited by
blocking the attachment of ubiquitin to the protein receptor
(54).
The rate at which RET fusion occurs is even higher
in radiation-induced TC. A previous study was performed to analyze
molecular genetic aberrations and associated phenotypes in the
pathological tissues of 191 patients with PTC who were exposed to
radioactive iodine from the Chernobyl reactor as young children.
The results revealed that the frequency of RET gene rearrangements
increased to 62.3% in the first decade following exposure to
radiation. In addition, ELE1/RET (PTC3) rearrangements were
markedly more common than H4/RET (PTC1) rearrangements (75). Note that NCOA4 is the official
symbol for the nuclear receptor coactivator gene involved in this
rearrangement, which is also widely known by the aliases ELE1 and
ARA70. RET/PTC1 is defined as the CCDC6 (formerly H4)-RET gene
fusion, whereas RET/PTC3 corresponds to the NCOA4 (ELE1/ARA70)-RET
gene fusion. An additional investigation of the survivors of the
atomic bombings that took place in Hiroshima and Nagasaki (Japan),
revealed that among 50 patients with PTC who were exposed to
nuclear radiation, 11 (22%) exhibited RET rearrangements, whereas
only 5% of the 21 unexposed patients exhibited the same RET
rearrangements. To the best of our knowledge, this study is the
first to clearly demonstrate a statistically significant positive
association between radiation exposure dose and the incidence rate
of RET/PTC rearrangement in a large-scale human sample (analyzing
249 PTC tumor samples from atomic bomb survivors). Specifically,
the higher the radiation dose received by the survivors, the higher
the proportion of RET/PTC rearrangement in the PTC tumors they
later developed (76).
The specific phosphotyrosine residues on the
activated RET intracellular tail recruit distinct adaptor and
effector proteins, thereby activating multiple downstream signaling
pathways critical for both normal development and oncogenesis
(77–79). The RAS/RAF/MEK/ERK pathway
(80–84) is primarily initiated through
phosphorylated-Y1062 and -Y1096 (these phosphorylated tyrosine
residues are located on the RET protein. Specifically,
phosphorylated-Y1062 and phosphorylated-Y1096 are tyrosine
phosphorylation sites on the intracellular domain of the activated
RET receptor tyrosine kinase), and this pathway is a key driver of
cellular proliferation and differentiation. The PI3K/AKT pathway
(85–87) is a pro-survival pathway that is
activated through several mechanisms, including the recruitment of
SHP2 to phosphorylated-Y687 (phosphorylated-Y687 is also a tyrosine
phosphorylation site on the intracellular domain of the activated
RET receptor tyrosine kinase). The JAK2/STAT3 pathway (88,89)
is activated following the binding of STAT3 to phosphorylated-Y752
and -Y928 (phosphorylated-Y752 and -Y928 are tyrosine
phosphorylation sites of the RET protein), leading to nuclear
translocation and the regulation of target gene expression. The
phospholipase (PLC)γ/protein kinase C pathway (90) is triggered by PLCγ binding to
phosphorylated-Y1015 (phosphorylated-Y1015 is a tyrosine
phosphorylation site on the intracellular domain of the activated
RET receptor tyrosine kinase); this pathway contributes to various
cellular responses.
In cancer, genetic alterations subvert this tightly
regulated system. In MTC, extracellular cysteine mutations (C609,
C611, C618, C620, C630 and C634) cause constitutive dimerization
(91,92), whereas intracellular kinase domain
mutations (for example, M918T and V804M) stabilize the active
conformation, enhance ATP binding or impair autoinhibitory
mechanisms. In PTC, chromosomal rearrangements create RET fusion
genes (93–96). These chimeric proteins, often
lacking the transmembrane domain and fused to dimerization
partners, exhibit ligand-independent, constitutive kinase activity
localized in the cytoplasm. Collectively, these diverse genetic
events lead to the persistent and unregulated activation of
RET-driven oncogenic signaling, fueling tumor initiation, growth
and progression.
Numerous TKI medications have advanced to the stages
of preclinical and clinical development (97–99).
For example, the 2025 National Comprehensive Cancer Network
Guidelines recommendations on TKI inhibitors for TC are as follows
(100): For iodine-refractory PTC
and FTC, both lenvatinib and sorafenib are Category 1
recommendations, with lenvatinib as the preferred first-line option
for locally recurrent/metastatic, progressive radioactive
iodine-refractory PTC and FTC. For MTC, first-line agents include
vandetanib and cabozantinib (Category 1 recommendations). For MTC
with RET mutations, selpercatinib and pralsetinib (highly selective
RET inhibitors) are preferred. For ATC, treatment is mainly
chemotherapy combined with immunotherapy, whereas TKIs may be used
in the setting of positive specific targets. For example,
dabrafenib plus trametinib can be used for tumors with the BRAF
V600E mutation.
The recommendations on TKI inhibitors for TC in the
2025 American Thyroid Association Guidelines (101) include: For TC with RET fusions,
highly selective TKIs, such as selpercatinib (for RET) and
larotrectinib (for neurotrophic tyrosine receptor kinase), are
preferred first-line treatments. These agents demonstrate superior
efficacy and safety compared with conventional multikinase
inhibitors, such as lenvatinib and sorafenib. In the absence of
specific targets, lenvatinib is the first-line choice, and
sorafenib is an alternative. For TC with the BRAF V600E mutation,
dabrafenib plus trametinib may be used as first-line in patients
intolerant to multi-target TKIs; in those who are tolerant, this
combination is reserved for later-line therapy. These kinase
inhibitors are organic compounds with small molecular structures
that interact with the nucleotide-binding site within the kinase
domain, either entirely or in part, and block kinase function
(102,103). Depending on the orientation of
the activation loop, kinases can adopt either an active or an
inactive conformation. A kinase achieves its active state when the
aspartate-phenylalanine-glycine (DFG) motif is located at the
N-terminus of the activation loop; this configuration is referred
to as DFG-in. By contrast, when the DFG motif is found at the
C-terminus of the activation loop, the kinase is considered to be
in an inactive conformation, referred to as DFG-out (104–106). TKIs are divided into two primary
categories based on their preference for DFG-in (Type I) or DFG-out
(Type II) conformations. Type I TKIs function by competing with ATP
at the active site. By contrast, Type II inhibitors promote the
inactive state of kinases by binding both to the ATP-binding pocket
and the nearby allosteric site, which can only be accessed in the
DFG-out conformation (107,108).
Several multikinase inhibitors (MKIs) exhibit
anti-RET activity and are approved for the treatment of TC,
including vandetanib (MTC), cabozantinib (MTC), regorafenib and
sorafenib (differentiated TC). A variety of highly selective RET
inhibitors have been developed. These agents target specific RET
mutations and are associated with lower toxicity, lower required
doses, and lower discontinuation rates compared with MKIs, such as
vandetanib, cabozantinib, regorafenib and sorafenib (109–112).
The LIBRETTO-001 (NCT03157128) trial was an
international, multicenter, open-label, phase I/II trial assessing
the safety and efficacy of selpercatinib (49). A total of 531 patients aged 12
years were enrolled in the study. All of the patients had locally
advanced or metastatic solid tumors of various types. This occurs
with the activation of RET alterations. Of the 531 patients, 162
had RET-altered TC. In the TC subgroup, 55 patients had RET-mutated
medullary TC (MTC) previously treated with cabozantinib and
vandetanib, 88 patients had RET-mutated MTC without prior
cabozantinib or vandetanib treatment, and 19 patients had RET
fusion-positive non-MTC. Notably, these 19 patients had been
previously treated for fusion-positive non-MTC, not for RET-altered
MTC. Based on the findings from the clinical trial, RET-mutant
patients with MTC who were not previously treated with cabozantinib
and vandetanib exhibited an overall response rate (ORR) of 69% and
progression-free survival (PFS) rate of 82% after 1 year. By
contrast, patients with RET-mutant MTC who had not previously
received these treatments had a 73% ORR and a 1-year PFS rate of
92%. For patients with RET fusion-positive non-MTC, the ORR reached
79%, with treatment effectiveness noted across various histological
subtypes and a corresponding 1-year progression-free survival (PFS)
rate. Furthermore, selpercatinib demonstrated overall treatment
effectiveness in all patients with MTC. All patients enrolled in
the LIBRETTO-001 trial, including the 162 patients with RET-altered
TC described in the text, were treated with selpercatinib,
regardless of their previous exposure to MKIs, radioactive iodine,
or the specific type of RET mutation or fusion.
Based on the current research status and clinical
practice, it is proposed that future studies on TKIs targeting RET
alterations should focus on the following aspects: i) The
development of more efficient selective RET inhibitors to overcome
existing resistance mechanisms; ii) the exploration of combinations
of RET inhibitors with other targeted therapies or immunotherapies
to enhance treatment efficacy; and iii) the utilization of
technologies such as liquid biopsy to monitor RET mutations and
resistance in real time, thereby providing a foundation for
personalized treatment.
In conclusion, TC is an increasingly common
endocrine malignancy with genetic drivers, particularly RET
mutations and rearrangements, which serve key roles in MTC and PTC
subtypes, respectively. While targeted RET inhibitors represent a
promising therapeutic advance, future research should prioritize
deeper molecular studies of RET signaling, the interplay between
genetic and environmental risk factors such as radiation, and the
development of improved strategies for early detection and
precision treatment. Advancing these areas will be critical to
enhancing patient outcomes and understanding TC pathogenesis.
Not applicable.
This research was funded by the Anhui Provincial New Era
Education Quality Engineering Project (Graduate Education) (grant
no. 2024qyw/sysfkc017) and the Anhui Medical University Quality
Engineering Project (grant no. 2024×jxm72).
Not applicable.
MW and RW wrote the manuscript. JQ made substantial
contributions to the conception and design of the review's visual
data presentation, created the figures and verified that the visual
content accurately reflected the core research findings and
academic conclusions of the review. JT and QF supervised the
research, revised the manuscript, obtained financial support,
conceptualized the review and performed the literature search. Data
authentication is not applicable. All authors read and approved the
final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Seib CD and Sosa JA: Evolving
understanding of the epidemiology of thyroid cancer. Endocrinol
Metab Clin North Am. 48:23–35. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Miranda-Filho A, Lortet-Tieulent J, Bray
F, Cao B, Franceschi S, Vaccarella S and Dal Maso L: Thyroid cancer
incidence trends by histology in 25 countries: A population-based
study. Lancet Diabetes Endocrinol. 9:225–234. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ni Z, Cong S, Li H, Liu J, Zhang Q, Wei C,
Pan G, He H, Liu W and Mao A: Integration of scRNA and bulk
RNA-sequence to construct the 5-gene molecular prognostic model
based on the heterogeneity of thyroid carcinoma endothelial cell:
Five-gene TC prognostic model. Acta Biochim Biophys Sin (Shanghai).
56:255–269. 2024.PubMed/NCBI
|
|
4
|
Dahiya V, Vasudeva N, Sharma S and Kumar
A: Role of dietary supplements in thyroid diseases. Endocr Metab
Immune Disord Drug Targets. 22:985–996. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Elisei R and Romei C: Looking for RET
alterations in thyroid cancer: Clinical relevance, methodology and
timing. Endocrine. 81:206–215. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Agrawal N, Akbani R, Aksoy BA, Ally A,
Arachchi H, Asa SL, Auman JT, Balasundaram M, Balu S, Baylin SB, et
al: Integrated genomic characterization of papillary thyroid
carcinoma. Cell. 159:676–690. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Simbolo M, Mian C, Barollo S, Fassan M,
Mafficini A, Neves D, Scardoni M, Pennelli G, Rugge M, Pelizzo MR,
et al: High-throughput mutation profiling improves diagnostic
stratification of sporadic medullary thyroid carcinomas. Virchows
Arch. 465:73–78. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xiao X, Chen M, Sang Y, Xue J, Jiang K,
Chen Y, Zhang L, Yu S, Lv W, Li Y, et al: Methylation-mediated
silencing of ATF3 promotes thyroid cancer progression by regulating
prognostic genes in the MAPK and PI3K/AKT pathways. Thyroid.
33:1441–1454. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fallahi P, Ferrari SM, Galdiero MR,
Varricchi G, Elia G, Ragusa F, Paparo SR, Benvenga S and Antonelli
A: Molecular targets of tyrosine kinase inhibitors in thyroid
cancer. Semin Cancer Biol. 79:180–196. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zago E, Galluzzo A, Pradella S, Antonuzzo
L, Maggi M, Petrone L and Sparano C: Cabozantinib for different
endocrine tumours: Killing two birds with one stone. A systematic
review of the literature. Endocrine. 83:26–40. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Saltiki K, Simeakis G, Karapanou O,
Paschou SA and Alevizaki M: Metastatic medullary thyroid carcinoma
(MTC): Disease course, treatment modalities and factors
predisposing for drug resistance. Endocrine. 80:570–579. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jiao Q, Bi L, Ren Y, Song S, Wang Q and
Wang YS: Advances in studies of tyrosine kinase inhibitors and
their acquired resistance. Mol Cancer. 17:362018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cabanillas ME, Ryder M and Jimenez C:
Targeted Therapy for Advanced Thyroid Cancer: Kinase Inhibitors and
Beyond. Endocr Rev. 40:1573–1604. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
De Leo S, Trevisan M and Fugazzola L:
Recent advances in the management of anaplastic thyroid cancer.
Thyroid Res. 13:172020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Takahashi M, Ritz J and Cooper GM:
Activation of a novel human transforming gene, ret, by DNA
rearrangement. Cell. 42:581–588. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Takahashi M: RET receptor signaling:
Function in development, metabolic disease, and cancer. Proc Jpn
Acad Ser B Phys Biol Sci. 98:112–125. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Knowles PP, Murray-Rust J, Kjaer S, Scott
RP, Hanrahan S, Santoro M, Ibáñez CF and McDonald NQ: Structure and
chemical inhibition of the RET tyrosine kinase domain. J Biol Chem.
281:33577–33587. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Treanor JJ, Goodman L, de Sauvage F, Stone
DM, Poulsen KT, Beck CD, Gray C, Armanini MP, Pollock RA, Hefti F,
et al: Characterization of a multicomponent receptor for GDNF.
Nature. 382:80–83. 1996. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Arighi E, Borrello MG and Sariola H: RET
tyrosine kinase signaling in development and cancer. Cytokine
Growth Factor Rev. 16:441–467. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Melillo RM, Cirafici AM, De Falco V,
Bellantoni M, Chiappetta G, Fusco A, Carlomagno F, Picascia A,
Tramontano D, Tallini G and Santoro M: The oncogenic activity of
RET point mutants for follicular thyroid cells may account for the
occurrence of papillary thyroid carcinoma in patients affected by
familial medullary thyroid carcinoma. Am J Pathol. 165:511–521.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ciampi R, Romei C, Pieruzzi L, Tacito A,
Molinaro E, Agate L, Bottici V, Casella F, Ugolini C, Materazzi G,
et al: Classical point mutations of RET, BRAF and RAS oncogenes are
not shared in papillary and medullary thyroid cancer occurring
simultaneously in the same gland. J Endocrinol Invest. 40:55–62.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pham VH, Pham QT, Nguyen M, Ngo HN, Luu
TTT, Minh NDT, Đặng T, Thai AT, Vu HA and Ngo DQ: Characteristics
of RET gene mutations in Vietnamese medullary thyroid carcinoma
patients: A single-center analysis. J Pathol Transl Med.
59:125–132. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hofstra RM, Landsvater RM, Ceccherini I,
Stulp RP, Stelwagen T, Luo Y, Pasini B, Höppener JW, van Amstel HK,
Romeo G, et al: A mutation in the RET proto-oncogene associated
with multiple endocrine neoplasia type 2B and sporadic medullary
thyroid carcinoma. Nature. 367:375–376. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Barletta JA, Nosé V and Sadow PM: Genomics
and epigenomics of medullary thyroid carcinoma: From sporadic
disease to familial manifestations. Endocr Pathol. 32:35–43. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Prinzi A, Vella V, Bosco A, Mirone A,
Russo M, Piticchio T, Di Benedetto G, Bartoloni G, Frasca F and
Malandrino P: Sporadic and familial medullary thyroid carcinoma: A
retrospective single center study on presentation and outcome.
Endocr Res. 49:179–185. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mathiesen JS, Kroustrup JP, Vestergaard P,
Stochholm K, Poulsen PL, Rasmussen ÅK, Feldt-Rasmussen U, Schytte
S, Londero SC, Pedersen HB, et al: Incidence and prevalence of
sporadic and hereditary MTC in Denmark 1960–2014: A nationwide
study. Endocr Connect. 7:829–839. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Machens A and Dralle H: Long-term outcome
after DNA-based prophylactic neck surgery in children at risk of
hereditary medullary thyroid cancer. Best Pract Res Clin Endocrinol
Metab. 33:1012742019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yasir M, Mulji NJ and Kasi A: Multiple
endocrine neoplasia type 2. StatPearls [Internet] Treasure Island
(FL): StatPearls Publishing; 2025
|
|
29
|
Greenberg LA: Multiple endocrine neoplasia
type 1, type 2A, and type 2B. Prim Care. 51:483–494. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wells SA Jr: Advances in the management of
MEN2: From improved surgical and medical treatment to novel kinase
inhibitors. Endocr Relat Cancer. 25:T1–T13. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Eng C, Clayton D, Schuffenecker I, Lenoir
G, Cote G, Gagel RF, van Amstel HK, Lips CJ, Nishisho I, Takai SI,
et al: The relationship between specific RET proto-oncogene
mutations and disease phenotype in multiple endocrine neoplasia
type 2: International RET mutation consortium analysis. JAMA.
276:1575–1579. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Santoro M, Carlomagno F, Romano A, Bottaro
DP, Dathan NA, Grieco M, Fusco A, Vecchio G, Matoskova B, Kraus MH,
et al: Activation of RET as a dominant transforming gene by
germline mutations of MEN2A and MEN2B. Science. 267:381–383. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Asai N, Iwashita T, Matsuyama M and
Takahashi M: Mechanism of activation of the ret protooncogene by
multiple endocrine neoplasia type 2A mutations. Mol Cell Biol.
15:1613–1619. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Castinetti F, Moley J, Mulligan L and
Waguespack SG: A comprehensive review on MEN2B. Endocr Relat
Cancer. 25:T29–T39. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Frank-Raue K and Raue F: Hereditary
Medullary Thyroid Cancer: Genotype-Phenotype Correlation. Recent
Results Cancer Res. 223:183–209. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang ZW, Guo X and Qi XP: Molecular
diagnosis and treatment of multiple endocrine neoplasia type 2b in
ethnic Han Chinese. Endocr Metab Immune Disord Drug Targets.
21:534–543. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Plaza-Menacho I, Barnouin K, Goodman K,
Martínez-Torres RJ, Borg A, Murray-Rust J, Mouilleron S, Knowles P
and McDonald NQ: Oncogenic RET kinase domain mutations perturb the
autophosphorylation trajectory by enhancing substrate presentation
in trans. Mol Cell. 53:738–751. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gujral TS, Singh VK, Jia Z and Mulligan
LM: Molecular mechanisms of RET receptor-mediated oncogenesis in
multiple endocrine neoplasia 2B. Cancer Res. 66:10741–10749. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Menko FH, van der Luijt RB, de Valk IA,
Toorians AW, Sepers JM, van Diest PJ and Lips CJ: Atypical MEN type
2B associated with two germline RET mutations on the same allele
not involving codon 918. J Clin Endocrinol Metab. 87:393–397. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cranston AN, Carniti C, Oakhill K,
Radzio-Andzelm E, Stone EA, McCallion AS, Hodgson S, Clarke S,
Mondellini P, Leyland J, et al: RET is constitutively activated by
novel tandem mutations that alter the active site resulting in
multiple endocrine neoplasia type 2B. Cancer Res. 66:10179–10187.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Censi S, Galuppini F, Clausi C, Battheu F,
Manso J, Piva I, Corvaglia S, Pedron MC, Mondin A, Iacobone M, et
al: Tumor grade and molecular characteristics associated with
survival in sporadic medullary thyroid carcinoma. Thyroid.
34:177–185. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Heilmann AM, Subbiah V, Wang K, Sun JX,
Elvin JA, Chmielecki J, Sherman SI, Murthy R, Busaidy NL, Subbiah
I, et al: Comprehensive genomic profiling of clinically advanced
medullary thyroid carcinoma. Oncology. 90:339–346. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Keskin Ç, Canpolat AG, Canlar Ş,
Bahçecioğlu Mutlu AB and Erdoğan MF: Men 2B cases with atypical
presentation, unusual clinical course and a literature review. Acta
Endocrinol (Buchar). 19:260–266. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Erickson TA, Shih YP, Fass J, Jang M and
Tran E: T cells engineered to express immunoreceptors targeting the
frequently expressed medullary thyroid cancer antigens calcitonin,
CEA, and RET M918T. Thyroid. 32:789–798. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Qi XP, Lin GB, Chen B, Li F, Cao ZL, Zheng
WH and Zhao JQ: Multiple endocrine neoplasia type 2B associated
mixed medullary and follicular thyroid carcinoma in a Chinese
patient with RET M918T germline mutation. Endocr Metab Immune
Disord Drug Targets. 21:554–560. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ciampi R, Romei C, Ramone T, Prete A,
Tacito A, Cappagli V, Bottici V, Viola D, Torregrossa L, Ugolini C,
et al: Genetic landscape of somatic mutations in a large cohort of
sporadic medullary thyroid carcinomas studied by next-generation
targeted sequencing. iScience. 20:324–336. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Romei C, Ciampi R and Elisei R: A
comprehensive overview of the role of the RET proto-oncogene in
thyroid carcinoma. Nat Rev Endocrinol. 12:192–202. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Thomas GA, Bunnell H, Cook HA, Williams
ED, Nerovnya A, Cherstvoy ED, Tronko ND, Bogdanova TI, Chiappetta
G, Viglietto G, et al: High prevalence of RET/PTC rearrangements in
Ukrainian and Belarussian post-Chernobyl thyroid papillary
carcinomas: A strong correlation between RET/PTC3 and the
solid-follicular variant. J Clin Endocrinol Metab. 84:4232–4238.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wirth LJ, Sherman E, Robinson B, Solomon
B, Kang H, Lorch J, Worden F, Brose M, Patel J, Leboulleux S, et
al: Efficacy of selpercatinib in RET-altered thyroid cancers. N
Engl J Med. 383:825–835. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Nacchio M, Pisapia P, Pepe F, Russo G,
Vigliar E, Porcelli T, Luongo C, Iaccarino A, Pagni F, Salvatore D,
et al: Predictive molecular pathology in metastatic thyroid cancer:
The role of RET fusions. Expert Rev Endocrinol Metab. 17:167–178.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Subbiah V, Yang D, Velcheti V, Drilon A
and Meric-Bernstam F: State-of-the-art strategies for targeting
RET-dependent cancers. J Clin Oncol. 38:1209–1221. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Yao Y, Yu Z, Ma Y, Ou Q, Wu X, Lu D and Li
X: Characterizing kinase intergenic-breakpoint rearrangements in a
large-scale lung cancer population and real-world clinical
outcomes. ESMO Open. 7:1004052022. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sorokin M, Rabushko E, Rozenberg JM,
Mohammad T, Seryakov A, Sekacheva M and Buzdin A: Clinically
relevant fusion oncogenes: Detection and practical implications.
Ther Adv Med Oncol. 14:175883592211441082022. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Mahato AK and Sidorova YA: RET receptor
tyrosine kinase: Role in neurodegeneration, obesity, and cancer.
Int J Mol Sci. 21:71082020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Grieco M, Santoro M, Berlingieri MT,
Melillo RM, Donghi R, Bongarzone I, Pierotti MA, Della Porta G,
Fusco A and Vecchio G: PTC is a novel rearranged form of the ret
proto-oncogene and is frequently detected in vivo in human thyroid
papillary carcinomas. Cell. 60:557–563. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Saenko V, Rogounovitch T, Shimizu-Yoshida
Y, Abrosimov A, Lushnikov E, Roumiantsev P, Matsumoto N, Nakashima
M, Meirmanov S, Ohtsuru A, et al: Novel tumorigenic rearrangement,
delta rfp/ret, in a papillary thyroid carcinoma from externally
irradiated patient. Mutat Res. 527:81–90. 2003.PubMed/NCBI
|
|
57
|
Zhao Y, Du R, Chen M and Chen Z: The
fusion characteristics of RET fusion in pan-cancer among the
Chinese population: A comprehensive genomic analysis. Transl Oncol.
55:1023842025. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Nakazawa T, Kondo T, Kobayashi Y, Takamura
N, Murata S, Kameyama K, Muramatsu A, Ito K, Kobayashi M and Katoh
R: RET gene rearrangements (RET/PTC1 and RET/PTC3) in papillary
thyroid carcinomas from an iodine-rich country (Japan). Cancer.
104:943–951. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Demin DE, Murashko MM, Uvarova AN,
Stasevich EM, Shyrokova EY, Gorlachev GE, Zaretsky AR, Korneev KV,
Ustiugova AS, Tkachenko EA, et al: Adversary of DNA integrity: A
long non-coding RNA stimulates driver oncogenic chromosomal
rearrangement in human thyroid cells. Int J Cancer. 152:1452–1462.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Alswailem M, Alghamdi B, Alotaibi A,
Aljomiah A, Al-Hindi H, Murugan AK, Abouelhoda M, Shi Y and
Alzahrani AS: Molecular genetics of diffuse sclerosing papillary
thyroid cancer. J Clin Endocrinol Metab. 108:e704–e711. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Yokota K, Sasaki H, Okuda K, Shimizu S,
Shitara M, Hikosaka Y, Moriyama S, Yano M and Fujii Y: KIF5B/RET
fusion gene in surgically-treated adenocarcinoma of the lung. Oncol
Rep. 28:1187–1192. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Lee MR, Shin JY, Kim MY, Kim JO, Jung CK
and Kang J: FOXA2 and STAT5A regulate oncogenic activity of
KIF5B-RET fusion. Am J Cancer Res. 13:638–653. 2023.PubMed/NCBI
|
|
63
|
Santoro M, Moccia M, Federico G and
Carlomagno F: RET gene fusions in malignancies of the thyroid and
other tissues. Genes (Basel). 11:4242020. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Ju YS, Lee WC, Shin JY, Lee S, Bleazard T,
Won JK, Kim YT, Kim JI, Kang JH and Seo JS: A transforming KIF5B
and RET gene fusion in lung adenocarcinoma revealed from
whole-genome and transcriptome sequencing. Genome Res. 22:436–445.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Tanaka A, Okita R, Morishige T, Okada M,
Inokawa H, Hirazawa K, Kameyama K, Ikeda A and Ikeda E: A case of
primary lung adenocarcinoma mimicking metastatic papillary thyroid
carcinoma. Thorac Cancer. 15:353–357. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Zhao M, Yin X, He H, Xia Q and Ru G:
Recurrent RET fusions in fibrosarcoma-like neoplasms in adult
viscera: Expanding the clinicopathological and genetic spectrum.
Histopathology. 82:633–645. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Salassidis K, Bruch J, Zitzelsberger H,
Lengfelder E, Kellerer AM and Bauchinger M: Translocation
t(10;14)(q11.2:q22.1) fusing the kinetin to the RET gene creates a
novel rearranged form (PTC8) of the RET proto-oncogene in
radiation-induced childhood papillary thyroid carcinoma. Cancer
Res. 60:2786–2789. 2000.PubMed/NCBI
|
|
68
|
Mizukami T, Shiraishi K, Shimada Y,
Ogiwara H, Tsuta K, Ichikawa H, Sakamoto H, Kato M, Shibata T,
Nakano T and Kohno T: Molecular mechanisms underlying oncogenic RET
fusion in lung adenocarcinoma. J Thorac Oncol. 9:622–630. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Duque CS, Vélez A, Cuartas J, Jaimes F,
Dueñas JP, Agudelo M, Nikiforova MN, Nikiforov YE and Condello V:
Molecular profiling of papillary thyroid carcinomas in healthcare
workers exposed to low dose radiation at the workplace. Endocrine.
76:95–100. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Ameziane-El-Hassani R, Boufraqech M,
Lagente-Chevallier O, Weyemi U, Talbot M, Métivier D, Courtin F,
Bidart JM, El Mzibri M, Schlumberger M and Dupuy C: Role of H2O2 in
RET/PTC1 chromosomal rearrangement produced by ionizing radiation
in human thyroid cells. Cancer Res. 70:4123–4132. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Djerir B and Maréchal A: Detection of
γ-H2A.X for rapid assessment of genotoxic agent-induced
double-strand DNA breaks by immunofluorescence. Methods Mol Biol.
2019:83–89. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Su X, Li Z, He C, Chen W, Fu X and Yang A:
Radiation exposure, young age, and female gender are associated
with high prevalence of RET/PTC1 and RET/PTC3 in papillary thyroid
cancer: A meta-analysis. Oncotarget. 7:16716–16730. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Vanden Borre P, Schrock AB, Anderson PM,
Morris JC III, Heilmann AM, Holmes O, Wang K, Johnson A, Waguespack
SG, Ou SI, et al: Pediatric, adolescent, and young adult thyroid
carcinoma harbors frequent and diverse targetable genomic
alterations, including kinase fusions. Oncologist. 22:255–263.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Tian T, Huang S, Dai H, Qi M, Liu B and
Huang R: Radioactive iodine-refractory pulmonary metastases of
papillary thyroid cancer in children, adolescents, and young
adults. J Clin Endocrinol Metab. 108:306–314. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Rabes HM, Demidchik EP, Sidorow JD,
Lengfelder E, Beimfohr C, Hoelzel D and Klugbauer S: Pattern of
radiation-induced RET and NTRK1 rearrangements in 191
post-chernobyl papillary thyroid carcinomas: Biological,
phenotypic, and clinical implications. Clin Cancer Res.
6:1093–1103. 2000.PubMed/NCBI
|
|
76
|
Hamatani K, Eguchi H, Ito R, Mukai M,
Takahashi K, Taga M, Imai K, Cologne J, Soda M, Arihiro K, et al:
RET/PTC rearrangements preferentially occurred in papillary thyroid
cancer among atomic bomb survivors exposed to high radiation dose.
Cancer Res. 68:7176–7182. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Ibáñez CF: Structure and physiology of the
RET receptor tyrosine kinase. Cold Spring Harb Perspect Biol.
5:a0091342013. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Krampitz GW and Norton JA: RET gene
mutations (genotype and phenotype) of multiple endocrine neoplasia
type 2 and familial medullary thyroid carcinoma. Cancer.
120:1920–1931. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Li J, Shang G, Chen YJ, Brautigam CA, Liou
J, Zhang X and Bai XC: Cryo-EM analyses reveal the common mechanism
and diversification in the activation of RET by different ligands.
Elife. 8:e476502019. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Zhang Y, Liu H, Wang K, Zheng J, Luan H
and Xin M: RET inhibitor SPP86 triggers apoptosis and activates the
DNA damage response through the suppression of autophagy and the
PI3K/AKT signaling pathway in melanoma cells. Drug Des Devel Ther.
19:67–82. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Asai N, Murakami H, Iwashita T and
Takahashi M: A mutation at tyrosine 1062 in MEN2A-Ret and MEN2B-Ret
impairs their transforming activity and association with shc
adaptor proteins. J Biol Chem. 271:17644–17649. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Alberti L, Borrello MG, Ghizzoni S,
Torriti F, Rizzetti MG and Pierotti MA: Grb2 binding to the
different isoforms of Ret tyrosine kinase. Oncogene. 17:1079–1087.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Srivastava A, Tommasi C, Sessions D, Mah
A, Bencomo T, Garcia JM, Jiang T, Lee M, Shen JY, Seow LW, et al:
MAB21L4 deficiency drives squamous cell carcinoma via activation of
RET. Cancer Res. 82:3143–3157. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Pu X, Xu C, Wang Q, Wang W, Wu F, Cai X,
Song Z, Yu J, Zhong W, Wang Z, et al: Expert consensus on the
diagnosis and treatment of RET gene fusion non-small cell lung
cancer in China. Thorac Cancer. 14:3166–3177. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Hong DS, Cabanillas ME, Wheler J, Naing A,
Tsimberidou AM, Ye L, Busaidy NL, Waguespack SG, Hernandez M, El
Naggar AK, et al: Inhibition of the Ras/Raf/MEK/ERK and RET kinase
pathways with the combination of the multikinase inhibitor
sorafenib and the farnesyltransferase inhibitor tipifarnib in
medullary and differentiated thyroid malignancies. J Clin
Endocrinol Metab. 96:997–1005. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Perrinjaquet M, Vilar M and Ibáñez CF:
Protein-tyrosine phosphatase SHP2 contributes to GDNF neurotrophic
activity through direct binding to phospho-Tyr687 in the RET
receptor tyrosine kinase. J Biol Chem. 285:31867–31875. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Wang Z, Chen Y, Zhang S, et al: SHP2 is
required for the oncogenic activity of the EML4-ALK fusion protein
in non-small cell lung cancer. Oncogene. 39:3281–3295. 2020.
|
|
88
|
Plaza Menacho I, Koster R, van der Sloot
AM, Quax WJ, Osinga J, van der Sluis T, Hollema H, Burzynski GM,
Gimm O, Buys CH, et al: RET-familial medullary thyroid carcinoma
mutants Y791F and S891A activate a Src/JAK/STAT3 pathway,
independent of glial cell line-derived neurotrophic factor. Cancer
Res. 65:1729–1737. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Schuringa JJ, Wojtachnio K, Hagens W,
Vellenga E, Buys CH, Hofstra R and Kruijer W: MEN2A-RET-induced
cellular transformation by activation of STAT3. Oncogene.
20:5350–5358. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Borrello MG, Alberti L, Arighi E,
Bongarzone I, Battistini C, Bardelli A, Pasini B, Piutti C,
Rizzetti MG, Mondellini P, et al: The full oncogenic activity of
Ret/ptc2 depends on tyrosine 539, a docking site for phospholipase
Cgamma. Mol Cell Biol. 16:2151–2163. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Arlt D, Baur B, Wagner B and Höppner W: A
novel type of mutation in the cysteine rich domain of the RET
receptor causes ligand independent activation. Oncogene.
19:3445–3448. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Tabata J, Nakaoku T, Araki M, Yoshino R,
Kohsaka S, Otsuka A, Ikegami M, Ui A, Kanno SI, Miyoshi K, et al:
Novel calcium-binding ablating mutations induce constitutive RET
activity and drive tumorigenesis. Cancer Res. 82:3751–3762. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Carlomagno F, Guida T, Anaganti S,
Provitera L, Kjaer S, McDonald NQ, Ryan AJ and Santoro M:
Identification of tyrosine 806 as a molecular determinant of RET
kinase sensitivity to ZD6474. Endocr Relat Cancer. 16:233–241.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Carlomagno F, Guida T, Anaganti S, Vecchio
G, Fusco A, Ryan AJ, Billaud M and Santoro M: Disease associated
mutations at valine 804 in the RET receptor tyrosine kinase confer
resistance to selective kinase inhibitors. Oncogene. 23:6056–6063.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Gimm O, Marsh DJ, Andrew SD, Frilling A,
Dahia PL, Mulligan LM, Zajac JD, Robinson BG and Eng C: Germline
dinucleotide mutation in codon 883 of the RET proto-oncogene in
multiple endocrine neoplasia type 2B without codon 918 mutation. J
Clin Endocrinol Metab. 82:3902–3904. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Carlson KM, Dou S, Chi D, Scavarda N,
Toshima K, Jackson CE, Wells SA Jr, Goodfellow PJ and Donis-Keller
H: Single missense mutation in the tyrosine kinase catalytic domain
of the RET protooncogene is associated with multiple endocrine
neoplasia type 2B. Proc Natl Acad Sci USA. 91:1579–1583. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Zhang J, Yang PL and Gray NS: Targeting
cancer with small molecule kinase inhibitors. Nat Rev Cancer.
9:28–39. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Gross S, Rahal R, Stransky N, Lengauer C
and Hoeflich KP: Targeting cancer with kinase inhibitors. J Clin
Invest. 125:1780–1789. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Mok T, Jänne PA, Nishio M, Novello S, Reck
M, Steuer C, Wu YL, Fougeray R, Fan PD, Meng J, et al:
HERTHENA-Lung02: Phase III study of patritumab deruxtecan in
advanced EGFR-mutated NSCLC after a third-generation EGFR TKI.
Future Oncol. 20:969–980. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
National Comprehensive Cancer Network
(NCCN), . NCCN clinical practice guidelines in oncology: Thyroid
carcinoma. Version 1.2025. NCCN; Washington, PA: 2025
|
|
101
|
Haugen BR, Alexander EK, Doherty GM, et
al: 2025 American Thyroid Association (ATA) Guidelines for Adult
Patients with Differentiated Thyroid Cancer. American Thyroid
Association; 2025
|
|
102
|
Wang Z, Wang H, Bu C, Meng B, Mu Y, Gao S,
Chen W and Tao X: Tyrosine kinase inhibitor-induced hypothyroidism:
Mechanism and clinical implications. Eur J Clin Pharmacol.
80:827–838. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Fu M, Zhao J, Zhang L, Sheng Z, Li X, Qiu
F, Feng Y, You M, Xu H, Zhang J, et al: Overcoming tyrosine kinase
inhibitor resistance in lung cancer brain metastasis with CTLA4
blockade. Cancer Cell. 42:1882–1897.e7. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Zhao Z, Wu H, Wang L, Liu Y, Knapp S, Liu
Q and Gray NS: Exploration of type II binding mode: A privileged
approach for kinase inhibitor focused drug discovery? ACS Chem
Biol. 9:1230–1241. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Modi SJ and Kulkarni VM: Exploration of
structural requirements for the inhibition of VEGFR-2 tyrosine
kinase: Binding site analysis of type II, ‘DFG-out’ inhibitors. J
Biomol Struct Dyn. 40:5712–5727. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Xu G, Zhang W, Du J, Cong J, Wang P, Li X,
Si X and Wei B: Binding mechanism of inhibitors to DFG-in and
DFG-out P38α deciphered using multiple independent Gaussian
accelerated molecular dynamics simulations and deep learning. SAR
QSAR Environ Res. 36:101–126. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Vijayan R, He P, Modi V, Duong-Ly KC, Ma
H, Peterson JR, Dunbrack RL Jr and Levy RM: Conformational analysis
of the DFG-out kinase motif and biochemical profiling of
structurally validated type II inhibitors. J Med Chem. 58:466–479.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Peng YH, Shiao HY, Tu CH, Liu PM, Hsu JT,
Amancha PK, Wu JS, Coumar MS, Chen CH, Wang SY, et al: Protein
kinase inhibitor design by targeting the Asp-Phe-Gly (DFG) motif:
The role of the DFG motif in the design of epidermal growth factor
receptor inhibitors. J Med Chem. 56:3889–3903. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Buffet C, Leboulleux S, Kraeber-Bodéré F,
Bodet-Milin C, Cabanes L, Dohan A, Leprince P, Schlumberger M,
Huillard O and Groussin L: Cardiac metastasis from medullary
thyroid cancers with long-term survival under vandetanib. Eur
Thyroid J. 10:517–522. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Ly NS, Li J, Faggioni R, Roskos LK and
Brose MS: Population pharmacokinetics and exposure-response
analysis for the phase 3 COSMIC-311 trial of cabozantinib for
radioiodine-refractory differentiated thyroid cancer. Clin
Pharmacokinet. 62:587–598. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Sherman EJ, Dunn LA, Ho AL, Baxi SS,
Ghossein RA, Fury MG, Haque S, Sima CS, Cullen G, Fagin JA and
Pfister DG: Phase 2 study evaluating the combination of sorafenib
and temsirolimus in the treatment of radioactive iodine-refractory
thyroid cancer. Cancer. 123:4114–4121. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Hadoux J, Elisei R, Brose MS, Hoff AO,
Robinson BG, Gao M, Jarzab B, Isaev P, Kopeckova K, Wadsley J, et
al: Phase 3 trial of selpercatinib in advanced RET-mutant medullary
thyroid cancer. N Engl J Med. 389:1851–1861. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Kassir N, McDougall D, Kuruvilla D, Kim S,
Kumar S, Rahman A, Ruf T, Cheeti S and Ankrom W: Exposure-response
relationships for pralsetinib in patients with RET-altered thyroid
cancer or RET fusion-positive nonsmall cell lung cancer. J Clin
Pharmacol. 64:685–696. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Subbiah V, Hu MI, Wirth LJ, Schuler M,
Mansfield AS, Curigliano G, Brose MS, Zhu VW, Leboulleux S, Bowles
DW, et al: Pralsetinib for patients with advanced or metastatic
RET-altered thyroid cancer (ARROW): A multi-cohort, open-label,
registrational, phase 1/2 study. Lancet Diabetes Endocrinol.
9:491–501. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Subbiah V, Hu MI, Mansfield AS, Taylor MH,
Schuler M, Zhu VW, Hadoux J, Curigliano G, Wirth L, Gainor JF, et
al: Pralsetinib in patients with advanced/metastatic rearranged
during transfection (RET)-altered thyroid cancer: Updated efficacy
and safety data from the ARROW study. Thyroid. 34:26–40. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Subbiah V, Cassier PA, Siena S, Garralda
E, Paz-Ares L, Garrido P, Nadal E, Vuky J, Lopes G, Kalemkerian GP,
et al: Pan-cancer efficacy of pralsetinib in patients with RET
fusion-positive solid tumors from the phase 1/2 ARROW trial. Nat
Med. 28:1640–1645. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Griesinger F, Curigliano G, Thomas M,
Subbiah V, Baik CS, Tan DSW, Lee DH, Misch D, Garralda E, Kim DW,
et al: Safety and efficacy of pralsetinib in RET fusion-positive
non-small-cell lung cancer including as first-line therapy: update
from the ARROW trial. Ann Oncol. 33:1168–1178. 2022. View Article : Google Scholar : PubMed/NCBI
|