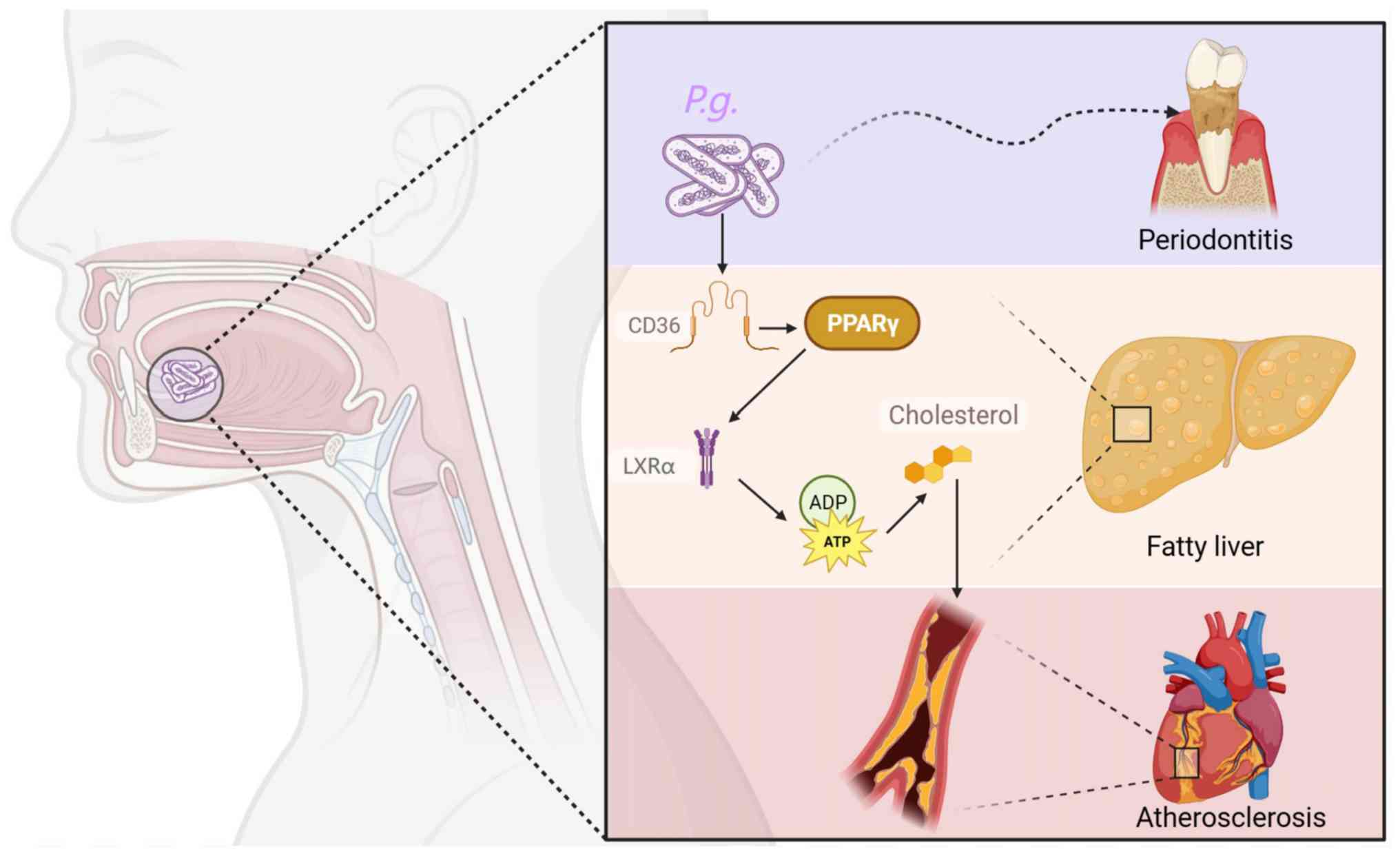
Introduction
Atherosclerotic cardiovascular disease is
characterized by the thickening and hardening of the arterial wall,
the formation or rupture of subendothelial plaques, and a reduction
in the arterial blood supply due to stenosis or occlusion of the
lumen, and is reported as the main cause of mortality worldwide
(1). Periodontitis is an
independent risk factor for atherosclerosis, and its molecular
mechanism involves a number of inflammatory cytokines and signaling
pathways. Lipoplysaccharide (LPS) from Porphyromonas
gingivalis activates Toll-like receptor (TLR)4, inducing IL-6
release and stimulating hepatic C-reactive protein production,
which subsequently upregulates vascular endothelial adhesion
molecule expression; this promotes monocyte adhesion and foam cell
formation, accelerating the progression of atherosclerosis
(2). P. gingivalis is a
gram-negative bacterium commonly found at sites with severe loss of
periodontal tissue attachment and in deep periodontal pockets, and
is associated with the progression of periodontal disease (3). P. gingivalis is closely
related to the occurrence and development of periodontitis, as well
as coronary atherosclerotic heart disease. Infection with this
bacterium can, for example, lead to lipid metabolism disorders,
resulting in elevated total cholesterol (TC), triglyceride (TG) and
low-density lipoprotein (LDL) levels, and decreased high-density
lipoprotein (HDL) levels (4,5).
P. gingivalis promotes atherosclerosis by affecting lipid
metabolism. Experimental studies have suggested that P.
gingivalis can invade endothelial and phagocytic cells within
the atheroma, leading to pathogenic changes and progression of the
atheroma lesion (6,7). As the primary pathogens of chronic
periodontitis, P. gingivalis and related toxins can enter
the bloodstream through the gingival epithelium, which causes
vascular endothelial dysfunction, activates blood inflammatory
factors and ultimately stimulates intravascular plaque formation
(8).
Peroxisome proliferator-activated receptor γ (PPARγ)
has been reported to be highly expressed in the early and middle
stages of human atherosclerotic lesions. In a previous study, in
response to administration of 5 g/l garlic allicin, the expression
levels of PPARγ in the atherosclerotic lesions of the aortic root
in ApoE−/− mice were reported to be increased
(9). In addition, PPARγ has been
revealed to be highly expressed in RAW-derived foam cells, whereas
its expression may be reduced after Dansameum extract (DSE)
treatment. PPARγ is also highly expressed in the atherosclerotic
lesion area of apolipoprotein E-knockout
(ApoE−/−) mice, with its expression further
increased in the aortic plaque of mice fed a high-fat diet; by
contrast, its expression can be reduced by DSE treatment (10). ATP-binding cassette transporter
(ABC)A1 is the key initiator protein that mediates reverse
cholesterol transport (RCT) and it has been shown that
macrophage-specific overexpression of ABCA1 can reduce
atherosclerotic plaque area (11).
ABCG1 is a key member of the ABC superfamily, which serves a
critical role in lipid metabolism. High expression of ABCG1 in
macrophages reportedly reduces cholesterol accumulation and
inhibits foam cell formation, thereby decelerating the progression
of atherosclerotic plaques (12).
Liver X receptor α (LXRα) is a key member of the nuclear receptor
superfamily and is involved primarily in the regulation of
cholesterol metabolism (for example, promoting the expression of
ABCA1 and ABCG1 to mediate RCT) and bile acid synthesis (13). The PPARγ-LXRα-ABCA1/ABCG1 pathway
regulates cholesterol efflux, whereas CD36 regulates cholesterol
uptake during lipid metabolism (14). In the present study, a model of
atherosclerosis and P. gingivalis infection was established,
and the changes in lipid metabolism and inflammatory responses were
observed. In addition, whether the expression of
PPARγ-LXRα-ABCA1/ABCG1 and CD36 served as the mechanism of lipid
metabolism disorders caused by P. gingivalis was
investigated. As illustrated in Fig.
1, P. gingivalis infection may disrupt lipid homeostasis
by promoting cholesterol uptake via CD36 while simultaneously
inhibiting cholesterol efflux through the downregulation of the
PPARγ-LXRα-ABCA1/ABCG1 pathway. This imbalance leads to lipid
accumulation in macrophages and hepatocytes, thereby accelerating
the progression of atherosclerosis and hepatic steatosis.
Septin 4 is a member of the GTP-binding protein
family that serves an important role in preventing foam cell
formation through the activation of PPARγ/LXRα signaling and
subsequent enhancement of ABCA1/ABCG1 expression (15). ABCA1 expression in endothelial
cells protects against atherosclerosis, and this atheroprotective
effect partially contributes to enhancing ApoAI-mediated
cholesterol efflux. Notably, ABCA1 is a target gene for LXR and RXR
(16); therefore, treating
endothelial cells with LXR and/or RXR agonists may increase ABCA1
expression (17).
However, whether P. gingivalis is involved in
cholesterol metabolism and its mechanism remains unclear. The
present study examined the effects of long-term, low-dose intraoral
infection of ApoE−/− mice with P.
gingivalis on lipid metabolism disorders, with the aim of
providing further experimental evidence for the association between
periodontitis and atherosclerosis.
Materials and methods
Bacterial culture
P. gingivalis (cat. no. 33277; American Type
Culture Collection) was cultured in 30 g/l trypsin soybean broth
supplemented with 1 g/l yeast extract, 50 mg/l heme chloride and 10
mg/l vitamin K3, inside an anaerobic tank, at 37°C for 3 days.
Subsequently, a PBS bacterial mixture with a P. gingivalis
concentration of 109/ml (containing 20 g/l sodium
carboxymethyl cellulose) was obtained for further use.
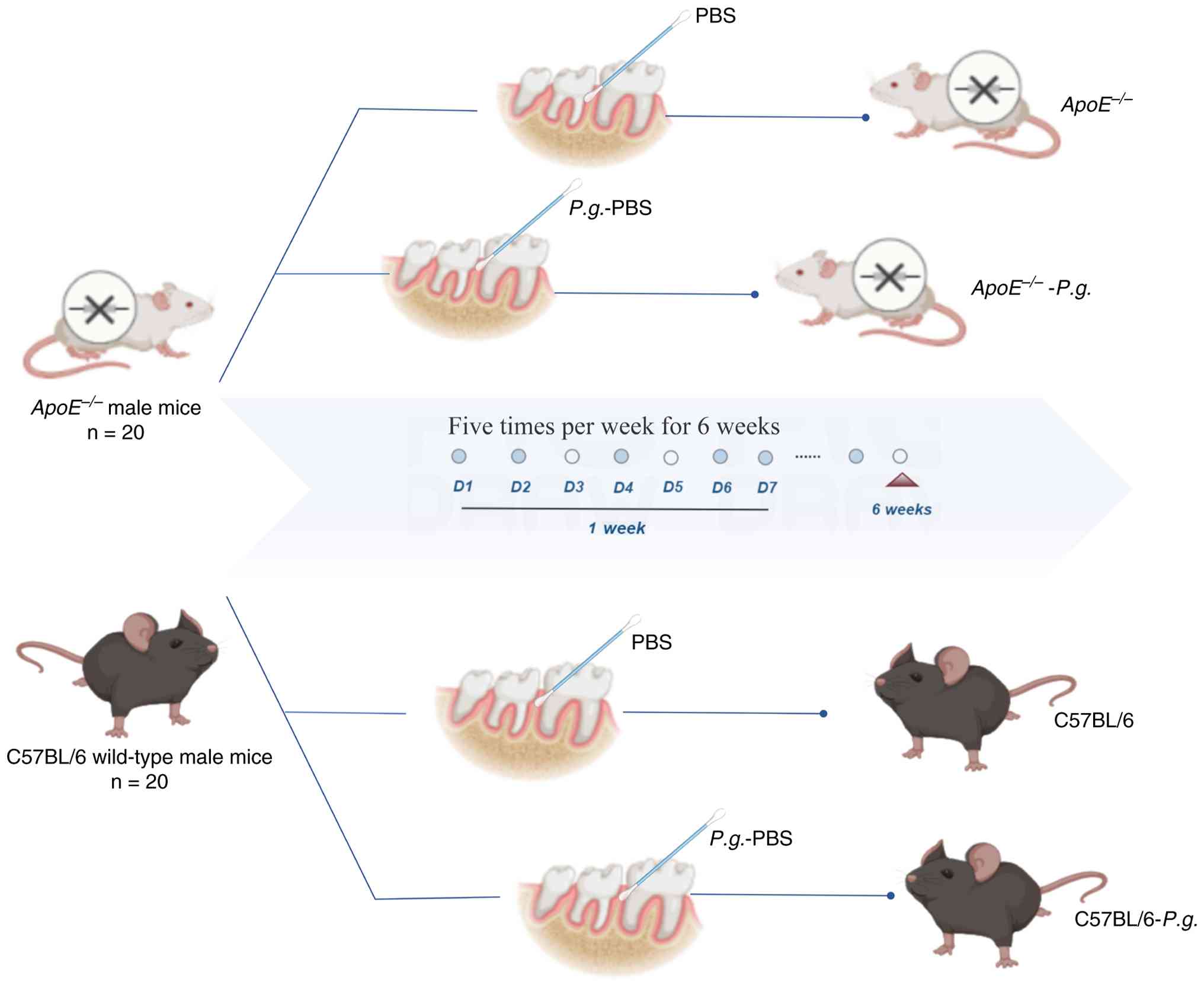
Mouse feeding
A total of 20 specific pathogen-free
ApoE−/− male mice and 20 C57BL/6 wild-type male
mice (both acquired from the Model Animal Research Center of
Nanjing University, Nanjing China) were randomly selected and fed a
high-fat diet (cat. no. 88137; Inotiv Inc.; 21% fat, 0.15%
cholesterol, no cholate) from the age of 4 weeks. The initial body
weight of ApoE−/− mice was 14.5±1.5 g, whereas
that of C57BL/6 mice was 15.0±1.5 g. The mice were maintained under
specific pathogen-free conditions, under a constant temperature
(22±2°C) and humidity (40–70% relative humidity), with a 12-h
light/dark cycle. Both the high-fat diet and autoclaved drinking
water were provided ad libitum. At the age of 5 weeks, each
mouse was intragastrically administered ampicillin and kanamycin (2
mg each per mouse) daily for 4 days. The present study received
complete ethical clearance from the ethics committee of Hainan
Medical University (approval no. HYLL-2021-121; Haikou, China) and
was conducted rigorously following the relevant guidelines
established by the National Institutes of Health (18). Every effort was made to minimize
the number of utilized mice and to mitigate any distress caused to
the mice during experiments.
Experimental grouping and oral
administration
ApoE−/− mice were randomly divided
into the following two groups (n=10/group): The infection group and
the control group. The wild-type mice were also randomly divided
into an infection group and a control group (n=10/group). The
sample size is consistent with that used in prior studies
investigating P. gingivalis-induced atherosclerosis in
ApoE−/− mice (19,20).
From the age of 6 weeks, the abdominal area of each mouse was
aseptically prepared with iodine, after which the mouse was
subjected to anesthesia through an intraperitoneal (IP) injection
of 1% pentobarbital sodium solution (60 mg/kg). The PBS bacterial
mixture with a concentration of 109/ml P.
gingivalis (containing 20 g/l sodium carboxymethyl cellulose)
was smeared on the gingival region of each mouse in the infection
group. The uninfected control mice received 0.1 ml PBS with 20 g/l
sodium carboxymethyl cellulose in the same manner. Oral inoculation
with P. gingivalis was performed five times per week for 6
weeks (Fig. 2). At the end of week
6, the mice were euthanized using pentobarbital overdose (200 mg/kg
IP), followed by cervical dislocation and collection of the
mandibles. The remaining experimental mice (10
ApoE−/− type and 10 wild-type) were sacrificed at
the 12th week for sample collection.
Microcomputed tomography (micro-CT)
analysis
The mandibles were fixed in 10% buffered formalin
for 24 h at 4°C, followed by storage in PBS at 4°C prior to
micro-CT (USPECT-II/CT; MILabs B.V.) analysis. The mandibles of
mice were imaged using a voxel size of 25 µm, scan settings of 70
kVp, 114 µA and 0.5 mm aluminum filter, and an integration time of
500 msec. A 3D construction was created using Avizo software
(version 9.1; Thermo Fisher Scientific, Inc.).
Determination of serum lipid and
inflammatory factor levels
At the ages of 6 and 12 weeks, the weights of all
mice were recorded, and each group of mice was subjected to
pentobarbital overdose (200 mg/kg IP), followed by cervical
dislocation for euthanasia. In week 6, 10 ApoE−/−
male mice and 10 C57BL/6 wild-type male mice were sacrificed, and
in week 12, 10 ApoE−/− male mice and 10 C57BL/6
wild-type male mice were sacrificed; within each group of mice,
five were from the infection group and five were from the control
group. After euthanasia, the height of the alveolar bone of each
mouse was measured and blood samples (0.4 ml) were collected from
the mouse eyeballs via the retro-orbital vein and anticoagulated
using heparin. After centrifugation at 4°C for 10 min at 1,960 × g,
the serum from the blood samples was collected and stored at −70°C
for further use in the measurement of the serum levels of TC (cat.
no. 100051013), TG (cat. no. 100051014), HDL (cat. no. 100020235)
and LDL (cat. no. 100020245) (all from Zhongsheng Beikong
Biotechnology Co., Ltd.) using blood biochemical methods. In
addition, the serum levels of oxidized LDL (ox-LDL; cat. no.
CSB-E07933m), IL-6 (cat. no. CSB-E04639m) and monocyte chemotactic
protein-1 (MCP-1; cat. no. CSB- E07430m) (all from Cusabio
Technology, LLC) were determined using ELISA.
Preparation of periodontal tissues,
liver and aorta samples
Collection of atherosclerotic plaques: The heart and
aorta were perfused slowly with normal saline at 4°C through the
left ventricle for 10 min, and the aorta (including the aortic
arch, thoracic aorta and abdominal aorta) was identified. The
connective tissue around the artery was stripped, and the integrity
of its bifurcation to the iliac bone was ensured. The isolated
mouse aorta was washed with normal saline at 4°C, and the excess
liquid was dried using filter paper and immediately placed into
liquid nitrogen for freezing, and preservation at −80°C. Meanwhile,
the liver was removed for further use. The levels of PPARγ (cat.
no. CSB-E08625m; Cusabio Technology, LLC), ABCA1 (cat. no.
LS-F21311; LS Bio; Vector Laboratories, Inc.) and ABCG1 (cat. no.
ASET-2063; Ace Therapeutics), and LXRα (cat. no. MBS2020587;
MyBioSource, Inc.), in periodontal tissues and plaque were detected
by ELISA. The specific procedure was performed according to the
corresponding kit instructions. After collection, the liver samples
were washed with normal saline and fixed with 40 g/l neutral
polycarboxylic acid at 4°C for 24 h. Paraffin-embedded sections
(4–5 µm) were subjected to hematoxylin and eosin (H&E) staining
at room temperature (20–25°C) and observed under a bright-field
microscope. Collection of periodontal tissues: After the mice were
sacrificed and fully perfused, the gingival and periodontal
ligament soft tissues surrounding the maxillary molars were cut
off, washed, frozen quickly and stored at −80°C for later use in
ELISA.
Oil red O staining of atherosclerotic
plaques
The aortic tissues were first embedded with frozen
embedding agent OCT (Thermo Fisher Scientific, Inc.) and were then
sliced across the aorta at a thickness of 5 µm from the proximal
end to the distal end. The sections were dried at room temperature
and soaked in 60% isopropanol for 5 min. Dip dyeing with oil red O
dye solution was performed for 30 min in the dark at room
temperature. Under a light microscope, the area stained with oil
red O was defined as the atherosclerotic lesion area.
Detection of the mRNA expression
levels of ABCA1, ABCG1, PPARγ, LXRα and CD36 in the liver
Total RNA was extracted from mouse liver tissue
using TRIzol® reagent (cat. no. 15596026; Invitrogen;
Thermo Fisher Scientific, Inc.), purified and reverse transcribed
into cDNA using the PrimeScript™ RT reagent kit (cat. no. RR037A;
Takara Bio, Inc.) according to the manufacturer's protocol. The RT
reaction was performed at 37°C for 15 min, followed by heat
inactivation at 85°C for 5 sec. The cDNA was stored at −20°C until
further use. The mRNA expression levels of CD36, ABCA1, ABCG1,
PPARγ and LXRα were determined using qPCR (StepOne; Applied
Biosystems; Thermo Fisher Scientific, Inc.) with TB Green™ Premix
Ex Taq™ II (Tli RNaseH Plus) (cat. no. RR820A; Takara Bio, Inc.)
and were normalized to the levels of GAPDH. Primer sequence
information is presented in Table
I. The qPCR thermocycling conditions were as follows:
Pre-denaturation at 95°C for 10 sec, followed by 40 cycles of
heating at 95°C for 5 sec and 62°C for 20 sec. The product was
slowly and evenly heated from 50°C to 95°C, and the melting product
curve was automatically drawn using the qPCR equipment. The Cq
value was determined using a computer program, the
2−ΔΔCq value serves as the expression level of the
target gene mRNA, and the software automatically generated the
amplification and melting curves and calculated the relative mRNA
expression levels (21,22).
 | Table I.Sequences of the primers used for
quantitative PCR. |
Table I.
Sequences of the primers used for
quantitative PCR.
| Gene | Primer sequence,
5′-3′ |
|---|
| CD36 | F:
TGATTAACGGGACAGACGGAGAC |
|
| R:
ACGTTCTCAAAGCTGCTGAAAGTG |
| ABCA1 | F:
AAAACCGCAGACATCCTTCAG |
|
| R:
CATACCGAAACTCGTTCACCC |
| ABCG1 | F:
ATACAGGGGAAAGGTCTCCAA |
|
| R:
CCCCCGAGGTCTCTCTTATAGT |
| PPARγ | F:
GTACTGTCGGTTTCAGAAGTGCC |
|
| R:
ATCTCCGCCAACAGCTTCTCCT |
| LXRα | F:
GTTATAACCGGGAAGACTTTGCCA |
|
| R:
GCCTCTCTACCTGGAGCTGGT |
| GAPDH | F:
CATCACTGCCACCCAGAAGACTG |
|
| R:
ATGCCAGTGAGCTTCCCGTTCAG |
Detection of oxidative stress
molecules
Malondialdehyde (MDA) levels were determined as
follows: The liver was placed into a mortar in an ice bath, and 2
ml 10% trichloroacetic acid (TCA; cat. no. T0699; Sigma-Aldrich;
Merck KGaA) and a small amount of quartz sand were added to it. The
mixture was then crushed until it was homogenized and was
subsequently centrifuged at 12,000 × g at 4°C for 15 min, after
which the supernatants were collected. The supernatant was further
centrifuged at 5,000 × g for 10 min at 4°C, and then adjusted to a
volume of 1.5 ml with 10% TCA; the control group consisted of 1.5
ml 10% TCA in the absence of a tissue sample. An equal volume of
0.5% thiobarbituric acid (cat. no. T5500; Sigma-Aldrich; Merck
KGaA) solution was added, and the mixture was incubated in a
boiling water bath for 30 min. After rapid cooling, the mixture was
centrifuged at 3,000 × g for 10 min at room temperature. Again, the
mixture was allowed to react in a boiling water bath for 30 min,
and then cooled quickly and centrifuged at 3,000 × g at room
temperature for 10 min. The absorbance of the supernatant was
measured at 532, 450 and 600 nm.
Superoxide dismutase (SOD) was detected using a kit
(cat. no. 706002; Cayman Chemical Company), as follows: Organ
homogenates from the liver were prepared in cold Tris buffer (5
mmol/l, containing 2 mmol/l EDTA, pH 7.4), utilizing a homogenizer
with a 1,500 rotatory speed of piston/min. The homogenates were
then centrifuged at 10,000 × g for 10 min at 4°C, and the
supernatants were collected. A total of 0.2 ml supernatant obtained
after centrifugation (1,500 × g at 4°C for 10 min, followed by
10,000 × g at 4°C for 15 min) of 10% liver homogenate was added to
the reaction system containing xanthine oxidase, nitroblue
tetrazolium and xanthine in phosphate buffer (pH 7.8) to form the
reaction mixture. The enzyme reaction was initiated by adding 0.2
ml NADH (780 µmol/l) and stopped precisely after 1 min by adding 1
ml glacial acetic acid. The amount of chromogen formed was measured
at 560 nm. SOD activity was calculated from the percentage
inhibition of formazan formation, with one unit defined as the
amount of enzyme causing 50% inhibition of the color
development.
To determine the glutathione (GSH) content, a kit
(cat. no. 703002; Cayman Chemical Company) was used. Briefly, 1 ml
liver homogenate supernatant was mixed with 3 ml 0.25 mol/l
Tris-HCl buffer (pH 8.0), and the mixture was shaken well.
Subsequently, 1 ml 3% formaldehyde was added, and the mixture was
shaken again. After being incubated at room temperature for 2 and
60 min, 1 ml was immediately collected, and 5 ml
5,5′-Dithiobis(2-nitrobenzoic acid) analytical solution was added
to it in a water bath at a constant temperature of 25°C. After
shaking well, the samples were allowed to stand for 5 min, after
which the absorbance was measured immediately at a wavelength of
412 nm. The difference between absorbance value at the 2-min time
point and the absorbance value at the 60-min time point was
calculated and substituted into the regression equation to
calculate the corresponding GSH concentration; the maximum error
did not exceed 6%.
Statistical analysis
Data are presented as the mean ± SEM and each
experimental procedure was conducted at least three times.
Statistical analyses were carried out using SPSS software (version
22.0; IBM Corp.). Data were analyzed using one-way ANOVA followed
by Tukey's or Dunnett's post hoc tests for normally distributed
data, or Kruskal-Wallis test followed by Dunn's post hoc test for
non-normally distributed data. Normality was assessed using the
Shapiro-Wilk test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Comparison of the weights and alveolar
bone heights of mice
There was no significant difference in weight
between the ApoE−/− infected group and the
ApoE−/− control group at the ages of 6 and 12
weeks (P>0.05; Table II). In
addition, no significant difference in weight was observed between
the wild-type infected group and the wild-type control group at the
ages of 6 and 12 weeks (P>0.05; Table II). Alveolar bone loss in response
to P. gingivalis oral infection in both groups was
determined using micro-CT. Micro-CT analysis of the mandibles
revealed obvious alveolar bone resorption in both the P.
gingivalis-infected C57BL/6 group (Fig. 3B) and the P.
gingivalis-infected ApoE−/− group (Fig. 3D) compared with in their respective
uninfected controls (Fig. 3A and
C). In addition, the levels of PPARγ, LXRα, ABCA1 and ABCG1 in
the periodontal tissues of the ApoE−/− infected
group were lower than those in the ApoE−/−
control group and the C57BL/6-P.g. group (P<0.05;
Fig. 3E-H).
 | Figure 3.Effects of P.g. infection on
the periodontium in mice. (A) C57B6/L: No notable alveolar bone
resorption was observed. (B) C57B6/L-P.g.: Compared with the
C57B6/L group, there was marked alveolar bone resorption. (C)
ApoE−/− control group: Slight alveolar bone
resorption was detected. (D) ApoE−/−-P.g.
infection group: Compared with the ApoE−/−
control group, alveolar bone resorption was markedly aggravated.
Scale bars: 700 µm. The protein levels of (E) PPARγ, (F) LXRα, (G)
ABCA1 and (H) ABCG1 in the periodontal tissues in the
ApoE−/− infected group were lower than those in
the ApoE−/− control group and the wild-type
infected group. Data are presented as the mean ± SEM (n=5).
*P<0.05, **P<0.01 vs. ApoE−/−;
†P<0.05 vs. C57B6/L-P.g. Data for (E-H) were
not normally distributed (Shapiro-Wilk test, P<0.05) and were
analyzed using the Kruskal-Wallis test followed by Dunn's post hoc
test. ABC, ATP-binding cassette transporter;
ApoE−/−, apolipoprotein E-knockout; LXRα, liver X
receptor α; P.g., Porphyromonas gingivalis; PPARγ,
peroxisome proliferator-activated receptor γ. |
| Table II.Weights of C57B6/L and
ApoE−/− mice at 6 and 12 weeks of age
(n=5/group). |
Table II.
Weights of C57B6/L and
ApoE−/− mice at 6 and 12 weeks of age
(n=5/group).
|
| Weight, g |
|---|
|
|
|
|---|
| Age |
ApoE−/− |
ApoE−/−-P.g. | C57B6/L |
C57B6/L-P.g. |
|---|
| 6 weeks | 23.32±2.23 | 23.48±0.96 | 26.27±0.60 | 26.97±0.63 |
| 12 weeks | 24.67±2.66 | 23.50±1.33 | 28.23±1.19 | 27.05±1.46 |
Serum levels of MCP-1 and IL-6
IL-6 has been shown to upregulate the expression of
cell adhesion molecules (such as CD54, CD102 and CD31) and
potentiate vascular permeability, whereas MCP-1 can induce
monocytes to penetrate the arterial wall, which leads to sustained
loss of endothelial barrier function (23,24).
In order to confirm whether P. gingivalis infection could
induce an inflammatory reaction, serum MCP-1 and IL-6 levels in
ApoE−/− and wild-type mice were measured. As
shown in Fig. 4C and D, the serum
MCP-1 and IL-6 levels in the ApoE−/− and
wild-type infected groups were significantly greater than those in
the comparable control groups, indicating that P. gingivalis
infection may trigger an effective systemic inflammatory response
in the mice. Therefore, the effects of PPARγ-LXRα-ABCA1/ABCG1 and
CD36 signaling on the development of atherosclerosis induced by
P. gingivalis were investigated.
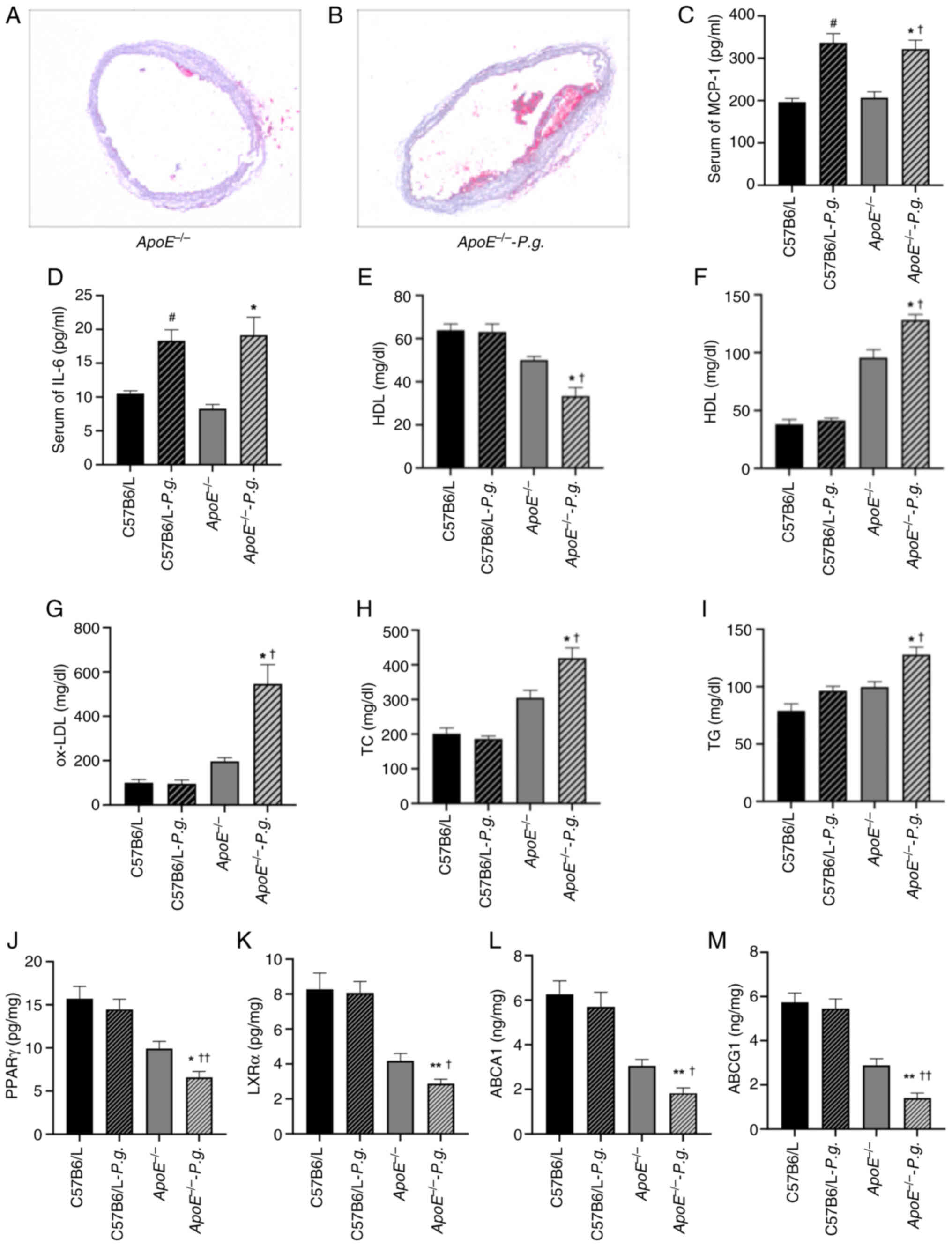 | Figure 4.Effects of P.g. on blood lipid
metabolism and atherosclerotic plaques in mice. (A) Oil red O
staining of aortic root cross-sections from
ApoE−/− mice (magnification, ×200). The image
shows scattered lipid deposits (red-stained areas) in the aortic
wall in the absence of P.G. infection, characteristic of
early atherosclerotic lesions. (B) Oil red O staining of aortic
root cross-sections from ApoE−/− mice following
P.g. infection (magnification, ×200). Compared with in the
control, the atherosclerotic plaque area was markedly increased,
with more extensive and densely distributed red-stained regions
indicating enhanced lipid deposition. Serum levels of (C) MCP-1 and
(D) IL-6 were determined using ELISA. Levels of (E) HDL, (F) LDL,
(H) TC and (I) TG were determined using blood biochemical methods.
(G) Serum levels of ox-LDL were determined using ELISA. Levels of
(J) PPARγ, (K) LXRα, (L) ABCA1 and (M) ABCG1 were determined using
ELISA. Data are presented as the mean ± SEM (n=5). *P<0.05,
**P<0.01 vs. ApoE−/−; †P<0.05,
††P<0.01 vs. C57B6/L-P.g;
#P<0.05 vs. C57B6/L. Data for (C, D, G and J-M) were
not normally distributed (Shapiro-Wilk test, P<0.05) and were
analyzed using the Kruskal-Wallis test followed by Dunn's post hoc
test. Data for (E, F, H and I) were normally distributed and
analyzed using one-way ANOVA followed by Tukey's or Dunnett's post
hoc test. ABC, ATP-binding cassette transporter;
ApoE−/−, apolipoprotein E-knockout; HDL,
high-density lipoprotein; LDL, low-density lipoprotein; LXRα, liver
X receptor α; MCP-1, monocyte chemotactic protein-1; ox-LDL,
oxidized LDL; P.g., Porphyromonas gingivalis; PPARγ,
peroxisome proliferator-activated receptor γ; TC, total
cholesterol; TG, triglyceride. |
Effects of P. gingivalis on blood
lipid metabolism in atherosclerotic plaques in mice
As shown in Fig. 4A and
B, the results of the histological analysis of the Oil Red
O-stained sections revealed irregularly elevated atherosclerotic
plaques along the intimal surface in both ApoE−/−
control and P. gingivalis-infected groups. Notably, the
plaques in the infected group were larger, exhibited greater
protrusion into the intima, and led to a more pronounced
compression of the underlying medial layer. The plaque was further
elevated by the rupturing of new blood vessels within the plaque,
leading to hematoma formation. In addition, P. gingivalis
infection inhibited the levels of PPARγ, LXRα, ABCA1 and ABCG1 in
plaque; this was manifested by the lower levels of PPARγ, LXRα,
ABCA1 and ABCG1 in the ApoE−/− infected group
compared with in the ApoE−/− control group, and
the levels in the ApoE−/− infected group also
lower than those in the wild-type infected group (P<0.05)
(Fig. 4J-M). Next, the effects of
P. gingivalis on lipid metabolism were evaluated by
determining the levels of HDL, LDL, ox-LDL, TG and TC. As shown in
Fig. 4E-I, there was no
significant difference in these five blood lipid indices between
the wild-type control group and the wild-type infected group,
indicating that P. gingivalis infection cannot directly
cause lipid metabolism disorders. Notably, the levels of LDL,
ox-LDL, TC and TG in the serum of the ApoE−/−
infected group were significantly greater compared with those in
the serum of the ApoE−/− control group, whereas
the HDL levels were significantly lower (P<0.05). These results
revealed that P. gingivalis could exacerbate lipid
metabolism disorders.
Effects of oral infection with P.
gingivalis on liver hepatocytes
As shown in Fig.
5A-D, the images of the H&E-stained sections revealed that
compared with in the C57B6/L group, P. gingivalis infection
in the wild-type group resulted in a mild increase in hepatocyte
volume with a small number of lipid droplets. Furthermore, compared
with in the P. gingivalis-infected C57B6/L group, the
ApoE−/− group showed moderately increased
hepatocyte volume with increased lipid droplets, whereas the
ApoE−/− infection group displayed the most
notable increase in hepatocyte volume, with numerous lipid droplets
fusing into large vacuoles and nuclei displaced to the cell
periphery, presenting obvious macrovesicular steatosis. These
results indicated that P. gingivalis infection may
exacerbate hepatocyte steatosis in ApoE−/− mice.
Furthermore, the effects of P. gingivalis infection on the
expression of PPARγ, LXRα, ABCA1, ABCG1 and CD36 in
the liver were measured using RT-qPCR. As shown in Fig. 5E-H, compared with in the
ApoE−/− control group, there was no significant
difference in the mRNA levels of ABCG1 in the livers of mice in the
ApoE−/− P. gingivalis infection group
(P>0.05), whereas the levels of ABCA1, PPARγ and LXRα were
significantly decreased (P<0.05). In addition, the mRNA levels
of CD36 were significantly increased in the
ApoE−/− infected group compared with those in the
ApoE−/− control group (P<0.05; Fig. 5I). Compared with in the wild-type
control group, the levels of ABCA1 in the wild-type infected group
were decreased, whereas the levels of ABCG1 and PPARγ were
increased (P<0.05), and there was no significant difference in
the levels of CD36 and LXRα between these two groups
(P>0.05).
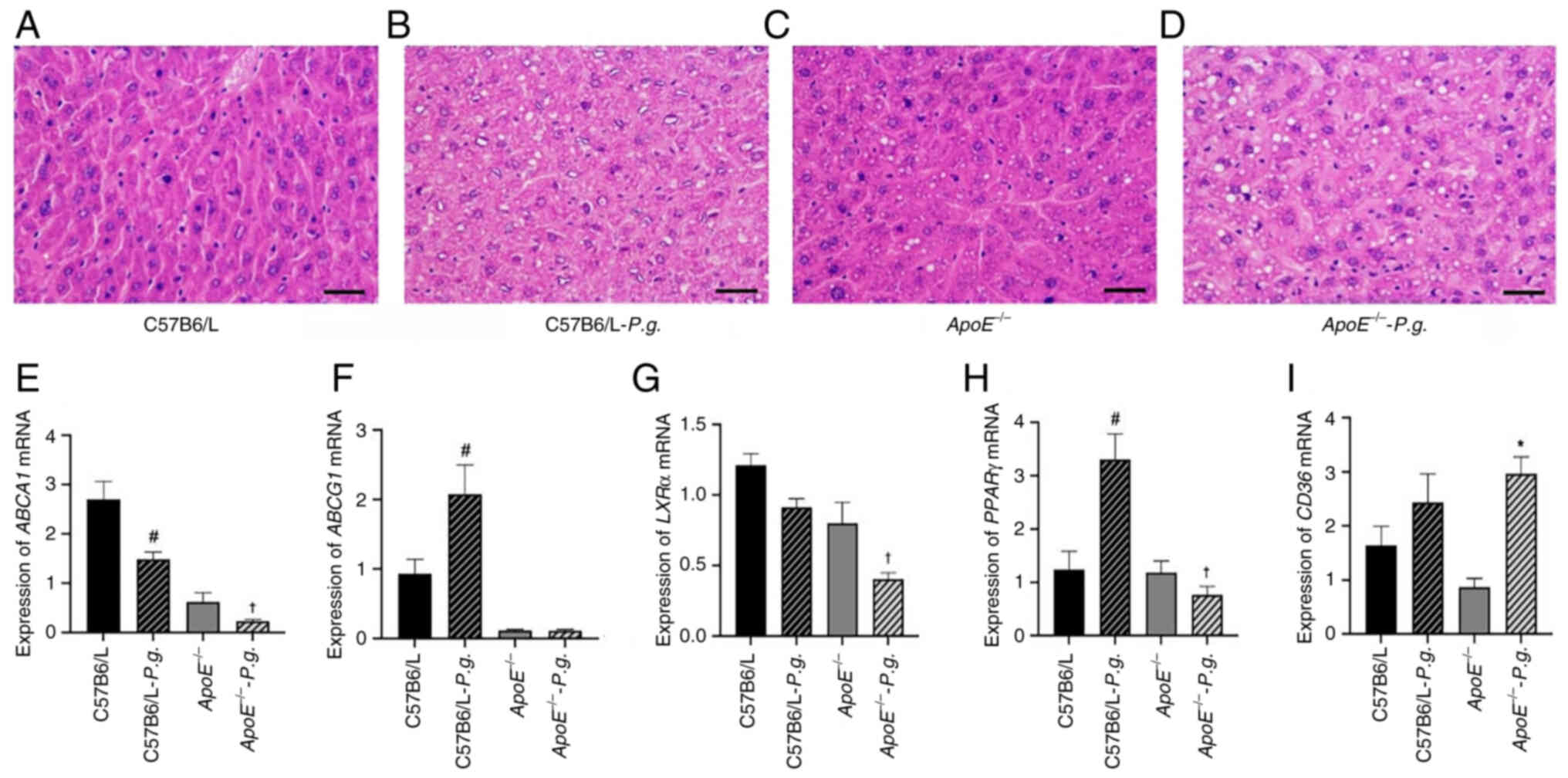 | Figure 5.Effects of P.g. infection on
lipid metabolism disorders and liver cells. (A-D) Representative
images of hematoxylin and eosin staining in liver hepatocytes
(magnification, ×400). (A) Liver cells in the C57B6/L group had a
regular shape, uniform size and uniform cytoplasm. There was no
obvious lipid droplet deposition. The liver lobule structure was
intact, and no hepatic cell fatty degeneration or morphological
abnormalities were observed. (B) Compared with in the C57B6/L
group, the liver cell volume in the C57B6/L-P.g. group was
slightly enlarged. A few scattered small lipid droplets can be seen
in the cytoplasm, without obvious lipid droplet fusion or nuclear
displacement. There was a slight change of hepatic cell fatty
degeneration. (C) Liver cell volume in the
ApoE−/− group was moderately enlarged. The number
of lipid droplets in the cytoplasm was more than that in the
C57B6/L-P.g. group. Some lipid droplets showed small-scale
fusion, and a few liver cells showed a slight displacement of the
nucleus, presenting moderate characteristics of hepatic cell fatty
degeneration. (D) Compared with in the ApoE−/−
control group, the liver cell volume in the
ApoE−/−-P.g. group was markedly enlarged.
A large number of lipid droplets in the cytoplasm fused to form
large vacuoles, occupying most of the cytoplasm. The nucleus was
compressed to the edge of the cell, presenting typical large
vacuolar fatty degeneration. The degree of hepatic cell fatty
degeneration was severe. Effects of P.g. on the mRNA
expression levels of (E) ABCA1, (F) ABCG1, (G)
LXRα, (H) PPARγ and (I) CD36 in the liver were
determined using reverse transcription-quantitative PCR. Data for
(E-I) were not normally distributed (Shapiro-Wilk test, P<0.05)
and were analyzed using the Kruskal-Wallis test followed by Dunn's
post hoc test. Data are presented as the mean ± SEM (n=5).
#P<0.05 vs. C57B6/L; *P<0.05 vs.
ApoE−/−; †P<0.05 vs.
C57B6/L-P.g. ABC, ATP-binding cassette transporter;
ApoE−/−, apolipoprotein E-knockout; LXRα, liver X
receptor α; P.g., Porphyromonas gingivalis; PPARγ,
peroxisome proliferator-activated receptor γ. |
Effects of P. gingivalis on the levels
of oxidative stress molecules
Compared with those in the ApoE−/−
control group, the levels of GSH and SOD were significantly lower
in the livers of the ApoE−/− infected group mice,
whereas the levels of MDA were significantly greater (P<0.05;
Table III). By contrast, there
was no significant difference in the levels of GSH, SOD and MDA
between the two groups of C57BL/6 wild-type mice (P>0.05;
Table III).
| Table III.Oxidative stress molecules in the
livers of C57BL6/6 and ApoE−/− mice
(n=5/group). |
Table III.
Oxidative stress molecules in the
livers of C57BL6/6 and ApoE−/− mice
(n=5/group).
| Parameter |
ApoE−/− |
ApoE−/−-P.g. | C57B6/L |
C57B6/L-P.g. |
|---|
| GSH,
µmol/gprot | 6.74±0.35 |
5.21±0.33a | 8.62±0.41 | 8.33±0.25 |
| SOD, µ/mgprot | 3.12±0.21 |
2.30±0.36b | 3.92±0.20 | 3.62±0.36 |
| MDA,
µmol/gprot | 2.76±0.44 |
3.55±0.39b | 1.86±0.20 | 1.90±0.19 |
Discussion
Atherosclerotic plaque initiation and progression
are characterized by massive deposition and accumulation of
lipid-loaded macrophages within arterial walls (12). Numerous studies have shown that the
pathogenesis of atherosclerosis mainly involves inflammation, with
bacteria serving the role of inflammatory initiators in this
process (25,26). Periodontal pathogens and their
products have been reported to interfere with lipid distribution
and metabolism in macrophages (27,28).
Any disturbance in the cholesterol-handling machinery in
macrophages, particularly the impairment of their cholesterol
efflux capacity, is closely associated with foam cell formation, an
aberrant serum lipid profile and reduced RCT efficiency, all of
which contribute to the development of atherogenesis (29). In the present study, the effects of
P. gingivalis infection on atheromatous plaque formation in
ApoE−/− mice were investigated, and the
involvement of the PPARγ-LXRα-ABCA1/ABCG1 and CD36 signaling
pathways as potential mechanisms underlying the P.
gingivalis-induced lipid metabolism disorders was explored. For
this, male ApoE−/− mice were fed a high-fat diet
and were then injected with ampicillin and kanamycin at the age of
5 weeks to control for biological variables that may interfere with
the establishment of a P. gingivalis infection model.
According to previous studies, mice were treated with ampicillin
and kanamycin (2 mg/mouse) daily for 4 days to suppress the native
oral flora and promote subsequent colonization of P.
gingivalis (30). An oral
bacterial mixture was used to establish an atherosclerosis model of
P. gingivalis infection. A comparison of the alveolar bone
height and aortic plaque area between the infected and control
groups confirmed that P. gingivalis infection promoted the
formation of atherosclerotic plaques and decreased the alveolar
bone height. In addition, P. gingivalis infection led to an
imbalance of oxidation and antioxidation. Finally, it was revealed
that PPARγ-LXRα-ABCA1/ABCG1 and CD36 signaling pathways may be
involved in P. gingivalis-induced lipid metabolism disorders
in ApoE−/− mice, which further exacerbated
atherosclerosis progression. The results indicated that in the
liver samples of ApoE−/− mice infected with P.
gingivalis, the levels of the core molecules of the
PPARγ-LXRα-ABCA1/ABCG1 pathway were abnormally downregulated,
whereas the levels of the key molecule for lipid uptake, CD36, were
significantly upregulated. Together, CD36 upregulation and
downregulation of the PPARγ-LXRα-ABCA1/ABCG1 pathway may mediate an
imbalance between excessive lipid uptake and impaired cholesterol
efflux in mice, leading to severe lipid metabolism disorders
characterized by elevated serum TC, TG, LDL and ox-LDL, and
decreased HDL. This could further aggravate the pathological damage
of atherosclerotic plaques in the mice, and simultaneously cause
foamy fatty degeneration in the liver, resulting in severe damage
to liver lipid metabolism.
In chronic periodontitis, virulence factors,
including LPS, fimbriae and outer membrane vesicles, continue to
enter the circulatory system, increasing the number of inflammatory
cells and the released cytokines. Notably, IL-6 is abundantly
released during the development of atherosclerosis. Moreover, an
established connection exists among IL-6 levels, endothelial
dysfunction and subclinical atherosclerosis, and multiple studies
have reported that IL-6 signaling serves a role in atherothrombosis
(23,31). Smooth muscle cells in plaques
induce MCP-1, which promotes the inflammatory response to blood
lipids (32). MCP-1 has an
important role in the process of monocyte accumulation at the
lesion site of atherosclerosis and entry into the vascular wall
through endothelial cells. Other studies have reported similar
results: P. gingivalis infection has been shown to
accelerate atherosclerotic plaque development in
ApoE−/− mice, which was revealed to be associated
with increased serum levels of the atherosclerotic factors MCP-1
and IL-6 (33). The micro-CT
results in the current study showed that P. gingivalis
infection exacerbated alveolar bone resorption, and the ELISA
results revealed that the levels of IL-6, MCP-1, TC, TG and LDL
were significantly increased, whereas the levels of HDL were
decreased in the serum of ApoE−/− mice after
stimulation with P. gingivalis, which aggravated the
inflammatory response. These findings further confirmed that P.
gingivalis infection stimulated the levels of IL-6 and MCP-1,
and aggravated the atherosclerotic process in
ApoE−/− mice.
The accumulation of free cholesterol in the liver
mitochondria causes mitochondrial dysfunction, activating the
unfolded protein response of the endoplasmic reticulum, and leading
to endoplasmic reticulum stress and hepatocyte apoptosis (34). H&E staining of the liver tissue
samples from mice in the present study revealed an increase in
hepatocyte volume and steatosis in the ApoE−/−
mice infected with P. gingivalis. Lipid deposition can also
induce oxidative stress injury, and these two together accelerate
the formation of lipid peroxidation products (35). By contrast, SOD is considered to
have anti-atherosclerosis effects. Studies have shown that carotid
intimal-medial thickness values are positively associated serum
small dense LDL (sdLDL) and MDA levels, and are negatively
associated with SOD activity, which is also in line with the
protective mechanism of oxidative damage and antioxidation
(36,37). In the present study, the levels of
GSH and SOD in the livers of ApoE−/− mice
infected with P. gingivalis were significantly lower than
those in non-infected ApoE−/− mice, whereas the
levels of MDA were significantly increased. Some studies have shown
that changes in the protein levels of SOD are not significant, but
SOD activity still varies significantly across disease states
(38,39). LDL is transformed into modified LDL
under oxidative stress. Therefore, a study of the oxidative stress
response in infected mice can further reveal the possible mechanism
of P. gingivalis-induced atherosclerosis.
The levels of TC, TG, LDL and HDL are commonly used
to evaluate the blood lipid levels of patients. TC is the sum of
free cholesterol and cholesterol esters in the blood, which are
synthesized and stored in the liver. Serum TC, therefore, serves as
an indicator of lipid metabolism in patients. TG primarily serves
as an energy storage molecule. LDL is positively associated with
the incidence of atherosclerosis and is easily modified by reactive
oxygen species to form ox-LDL, which can activate endothelial
cells. HDL is an anti-atherosclerosis lipoprotein. With a decrease
in plasma HDL levels, cholesterol metabolism becomes dysregulated;
HDL can accept excessive cholesterol in the body and transport
cholesterol from damaged plaques to the liver (a process known as
RCT) (1,11). The liver inflammatory response can
increase the levels of proinflammatory substances in systemic
blood, which then exacerbates the development of atherosclerosis
(40). Few studies have explored
the effects of metabolic inflammation on lipid metabolism. For
example, some experiments have shown that the endothelial
protective function of HDL deteriorates sharply in patients with
periodontitis (41). The
inflammatory response also promotes the oxidation of LDL to ox-LDL
and the formation of atherosclerotic plaques (42). In the present study, the serum
levels of LDL, ox-LDL, TG and TC were significantly increased,
whereas HDL levels were significantly decreased in
ApoE−/− mice infected with P. gingivalis
compared with those in the uninfected ApoE−/−
group. Additionally, the plaques that appeared on the intimal
surface in the P. gingivalis-infected
ApoE−/− group were larger than those in the
uninfected ApoE−/− group and protruded into the
intimal surface, more deeply compressing the medial membrane.
Different P. gingivalis strains vary in their virulence,
adherence ability, invasion and survivability. Therefore, the
variability in the effects of P. gingivalis on lipid
metabolism needs to be confirmed by further studies. The present
study indicated that inflammatory reactions and lipid metabolism
disorders are important pathogenic mechanisms underlying
atherosclerosis. The interrelationship between these two factors
and the order of their effects after P. gingivalis infection
need to be further investigated and confirmed in future studies
investigating the progression of atherosclerosis.
The intake of modified LDL mediated by CD36 is an
important mechanism of atherosclerosis induction by P.
gingivalis. CD36 is a pattern recognition receptor that binds
to multiple ionic ligands on both pathogens and host cells
(43). CD36 effectively regulates
lipid metabolism by promoting HDL synthesis in macrophages,
participating in cholesterol efflux, and upregulating ABCA1
expression (44). When CD36 uptake
of ox-LDL is unrestricted, macrophages transform into foam cells
when intracellular lipid accumulation exceeds the ability of ABCA1
and ABCG1 to excrete excess cholesterol. The RT-qPCR results of the
present study indicated that the expression levels of CD36 were
increased in ApoE−/− infected group, whereas no
significant change was observed in wild-type mice following
infection. This finding is consistent with those reported in
previous studies. In a previous study, after periodontal
inflammation was induced through periodontal ligation with P.
gingivalis, periodontitis could promote CD36-mediated fat
deposition in the liver (45).
Additionally, it has been shown that oral P. gingivalis
infection can lead to notable TLR2-CD36/Sr-B2-dependent IL-1β
release, resulting in increased foam cell formation and plaque
development (46). Taken together,
these findings suggested that oral P. gingivalis infection
can promote CD36 fat deposition in the liver, affect lipid
metabolism, increase the release of inflammatory factors and
promote plaque enlargement. Further investigation suggested that
targeting CD36 may be a feasible strategy for reducing the
additional risk of atherosclerosis through factors that contribute
to systemic inflammation, such as periodontal disease (47).
PPARs serve as fatty acid sensors, which regulate a
number of pathways involved in lipid and glucose metabolism, as
well as overall energy metabolism. LXRs are sterol sensors, which
predominantly regulate cholesterol, fatty acid and glucose
homeostasis; these receptors can also inhibit the development of
atherosclerosis (48). PPARγ,
which is a key regulator of CD36, regulates lipid metabolism by
activating LXRα and upregulating ABCA1 expression (49). However, the genes involved in lipid
efflux reportedly downregulate PPARγ and LXRα during P.
gingivalis-induced macrophage formation in foam cells. In the
presence of LDL, this downregulation is dose-dependent (50). In the current study, the levels of
PPARγ and LXRα in P. gingivalis-infected mice were
determined, and it was revealed that LXRα levels were significantly
lower in ApoE−/− infected group mice than those
in in wild-type infected mice. Similarly, PPARγ levels were
significantly lower in the ApoE−/− infected group
than in the wild-type infected group. However, no significant
difference in PPARγ levels was observed between the
ApoE−/− control group and the
ApoE−/− infected group. These findings suggested
that P. gingivalis infection may further aggravate
atherosclerosis by affecting the levels of PPARγ and LXRα. In
addition, TLR4 signaling contributes to P.
gingivalis-induced reduction in LXRα gene expression via the
TRIF branch, whereas the IRF3 status affects P.
gingivalis-induced LXRα gene expression in macrophages
(51). These findings further
confirmed that LXRα expression is downregulated in the inflammatory
state. ABCA1 and ABCG1 are considered key proteins that mediate
liver cholesterol output. The expression of these transporters is
predominantly dependent on the activation of the PPARγ and LXRα
transcription factors (52). In
order to determine whether the expression levels of ABCA1/G1,
downstream genes of PPARγ and LXRα, were also affected by P.
gingivalis, the expression levels of ABCA1 and ABCG1 in mice
were determined in the current study. It was revealed that,
compared with those in the wild-type mice, the expression levels of
ABCG1 in the liver of P. gingivalis-infected wild-type mice
were increased; this upregulation may be a protective reaction of
wild-type mice to a mild infection by P. gingivalis. High
levels of PPARγ have been suggested to directly promote the
expression of ABCG1, thereby enhancing the excretion of cholesterol
in the liver (11). This finding
was consistent with previous studies, which showed that treatment
of THP-1-derived macrophages co-cultured with LDL with LPS from
P. gingivalis could inhibit ABCG1 expression in a time- and
concentration-dependent manner (26,53).
Reduced ABCA1 and ABCG1 expression can mediate lipid accumulation.
It has also been suggested that the activation of PPARγ facilitates
cholesterol efflux mainly through ABCG1 but not through ABCA1
(54). However, compared with
those in the uninfected ApoE−/− group, the
expression levels of ABCA1 and ABCG1 were not significantly
decreased in the ApoE−/− infection group. This
may be related to the regulation of ABCA1 and ABCG1 by ILs in the
inflammatory state; however, further experimental validation of
this finding and inference is needed.
In conclusion, the present study revealed that oral
P. gingivalis infection can exacerbate inflammatory
responses and lipid metabolism disorders, further aggravating
atherosclerosis. The PPARγ-LXRα-ABCA1/ABCG1 signaling pathway may
serve a role in this process, although the specific interaction
between the inflammatory response and lipid metabolism remains to
be explored further.
Acknowledgements
Not applicable.
Funding
This research was funded by the National Natural Science
Foundation of China (grant no. 82201080), the Hainan S&T
Program (grant no. KJTP202561) and the Academic Enhancement Support
Program of Hainan Medical University (grant no. XSTS2025027).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
XJL and MY participated in the conception and
design of the study, acquisition and analysis of data, and drafting
of the manuscript. ZLG participated in the conceptualization and
supervision of the study. WYL, QY, JZ, LHW and DS were involved in
data collection and analysis. MZUS, WXX, YLD, MMZ and MWC
participated in the experimental design and methodological
optimization. XJL, MY, ZLG, WYL, QY, JZ, LHW, DS, MZUS, WXX, YLD,
MMZ and MWC confirm the authenticity of all the raw data. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
This research protocol obtained full ethical
clearance from the ethics committee of Hainan Medical University
(approval no. HYLL-2021-121) and rigorously adhered to the relevant
guidelines established by the National Institutes of Health.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hussain A, Ballantyne CM, Saeed A and
Virani SS: Triglycerides and ASCVD risk reduction: Recent insights
and future directions. Curr Atheroscler Rep. 22:252020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mendoza MF, Anzelmo MA, Suan NM, Cuccia CS
and Lavie CJ: More than just a toothache: Inflammatory mechanisms
linking periodontal disease to cardiovascular disease and the
protective impact of cardiorespiratory fitness. Biomedicines.
13:15122025. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tang Y, Qi Y, Chen Y, Wang YQ, Zhang C,
Sun Y, Huang C and Zhang XZ: Erythrocyte-mimicking nanovesicle
targeting porphyromonas gingivalis for periodontitis. ACS Nano.
18:21077–21090. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ding C, Shen Z, Xu R, Liu Y, Xu M, Fan C,
Hu D and Xing T: Exosomes derived from periodontitis induce hepatic
steatosis through the SCD-1/AMPK signaling pathway. Biochim Biophys
Acta Mol Basis Dis. 1870:1673432024. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yuan Q, Dong YL and Guo ZL: Research
progress on the intervention of Porphyromonas gingivalis in chronic
obstructive pulmonary disease. J Hainan Med Univ. 31:229–240.
2025.(In Chinese).
|
|
6
|
Wu Q, Li Z, Zhang Y, Luo K, Xu X, Li J,
Peng X and Zhou X: Cyclic di-AMP rescues porphyromonas
gingivalis-aggravated atherosclerosis. J Dent Res. 102:785–794.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schenkein HA, Papapanou PN, Genco R and
Sanz M: Mechanisms underlying the association between periodontitis
and atherosclerotic disease. Periodontol 2020. 83:90–106.
2000.PubMed/NCBI
|
|
8
|
Kim Y, Lee H, Park HJ, Kim MK, Kim YI, Kim
HJ, Bae SK, Kim YJ and Bae MK: Hispidulin inhibits the vascular
inflammation triggered by porphyromonas gingivalis
lipopolysaccharide. Molecules. 28:67172023. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
He XW, Yu D, Li WL, Zheng Z, Lv CL, Li C,
Liu P, Xu CQ, Hu XF and Jin XP: Anti-atherosclerotic potential of
baicalin mediated by promoting cholesterol efflux from macrophages
via the PPARγ-LXRα-ABCA1/ABCG1 pathway. Biomed Pharmacother.
83:257–264. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen H, Tan H, Wan J, Zeng Y, Wang J, Wang
H and Lu X: PPAR-γ signaling in nonalcoholic fatty liver disease:
Pathogenesis and therapeutic targets. Pharmacol Ther.
245:1083912023. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Matsuo M: ABCA1 and ABCG1 as potential
therapeutic targets for the prevention of atherosclerosis. J
Pharmacol Sci. 148:197–203. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang W, Zhang Y, Wang Z, Zhang J and Jia
L: Ganoderma lucidum polysaccharides improve lipid metabolism
against high-fat diet-induced dyslipidemia. J Ethnopharmacol.
309:1163212023. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lockhart SM, Muso M, Zvetkova I, Lam BYH,
Ferrari A, Schoenmakers E, Duckett K, Leslie J, Collins A,
Romartínez-Alonso B, et al: Damaging mutations in liver X
receptor-α are hepatotoxic and implicate cholesterol sensing in
liver health. Nat Metab. 6:1922–1938. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang H, Yang Y, Sun X, Tian F, Guo S, Wang
W, Tian Z, Jin H, Zhang Z and Tian Y: Sonodynamic therapy-induced
foam cells apoptosis activates the phagocytic
PPARγ-LXRα-ABCA1/ABCG1 pathway and promotes cholesterol efflux in
advanced plaque. Theranostics. 8:4969–4984. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Song X, Yan G, Wang H and Lou D: Septin 4
activates PPARγ/LXRα signaling by upregulating ABCA1 and ABCG1
expression to inhibit the formation of THP-1 macrophage-derived
foam cells. Exp Ther Med. 22:7632021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mahdavi-Roshan M, Salari A, Kheirkhah J
and Ghorbani Z: The effects of probiotics on inflammation,
endothelial dysfunction, and atherosclerosis progression: A
mechanistic overview. Heart Lung Circ. 31:e45–e71. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Litvinchuk A, Suh JH, Guo JL, Lin K, Davis
SS, Bien-Ly N, Tycksen E, Tabor GT, Remolina Serrano J, Manis M, et
al: Amelioration of Tau and ApoE4-linked glial lipid accumulation
and neurodegeneration with an LXR agonist. Neuron. 112:384–403.e8.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hofseth LJ: Getting rigorous with
scientific rigor. Carcinogenesis. 39:21–25. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xuan Y, Cai Y, Wang XX, Shi Q, Qiu LX and
Luan QX: Effect of Porphyromonas gingivalis infection on
atherosclerosis in apolipoprotein-E knockout mice. Beijing Da Xue
Xue Bao Yi Xue Ban. 52:743–749. 2020.PubMed/NCBI
|
|
20
|
Miyamoto T, Yumoto H, Takahashi Y, Davey
M, Gibson FC III and Genco CA: Pathogen-accelerated atherosclerosis
occurs early after exposure and can be prevented via immunization.
Infect Immun. 74:1376–1380. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chatterjee S: Endothelial
Mechanotransduction, redox signaling and the regulation of vascular
inflammatory pathways. Front Physiol. 9:5242018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ridker PM and Rane M: Interleukin-6
signaling and anti-interleukin-6 therapeutics in cardiovascular
disease. Circ Res. 128:1728–1746. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nakagawa K and Nakashima Y: Pathologic
intimal thickening in human atherosclerosis is formed by
extracellular accumulation of plasma-derived lipids and dispersion
of intimal smooth muscle cells. Atherosclerosis. 274:235–242. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xie M, Tang Q, Nie J, Zhang C, Zhou X, Yu
S, Sun J, Cheng X, Dong N, Hu Y and Chen L: BMAL1-Downregulation
aggravates porphyromonas gingivalis-induced atherosclerosis by
encouraging oxidative stress. Circ Res. 126:e15–e29. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zou Y, Huang Y, Liu S, Yang J, Zheng W,
Deng Y, Zhang M, Yan Z and Xie H: Periodontopathic microbiota and
atherosclerosis: Roles of TLR-mediated inflammation response. Oxid
Med Cell Longev. 2022:96113622022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huang X, Xie M, Lu X, Mei F, Song W, Liu Y
and Chen L: The roles of periodontal bacteria in atherosclerosis.
Int J Mol Sci. 24:128612023. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lin XJ, Yuan Q, Zhou J, Dong YL, Sunchuri
D and Guo ZL: Cellular senescence: A new perspective on the
suppression of periodontitis (Review). Mol Med Rep. 30:2382024.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hou L, Feng X, Zhu Z, Mi Y, He Q, Yin K
and Zhao G: IGFBPL1 inhibits macrophage lipid accumulation by
enhancing the activation of IGR1R/LXRα/ABCG1 pathway. Aging (Albany
NY). 15:14791–14802. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kuula H, Salo T, Pirilä E, Tuomainen AM,
Jauhiainen M, Uitto VJ, Tjäderhane L, Pussinen PJ and Sorsa T:
Local and systemic responses in matrix metalloproteinase
8-deficient mice during Porphyromonas gingivalis-induced
periodontitis. Infect Immun. 77:850–859. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wacker M, Ball A, Beer HD, Schmitz I,
Borucki K, Azizzadeh F, Scherner M, Awad G, Wippermann J and
Veluswamy P: Immunophenotyping of monocyte migration markers and
therapeutic effects of selenium on IL-6 and IL-1β cytokine axes of
blood mononuclear cells in preoperative and postoperative coronary
artery disease patients. Int J Mol Sci. 24:71982023. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ledard N, Liboz A, Blondeau B, Babiak M,
Moulin C, Vallin B, Guillas I, Mateo V, Jumeau C, Blirando K, et
al: Slug, a cancer-related transcription factor, is involved in
vascular smooth muscle cell transdifferentiation induced by
platelet-derived growth factor-BB during atherosclerosis. J Am
Heart Assoc. 9:e0142762020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ruan Q, Guan P, Qi W, Li J, Xi M, Xiao L,
Zhong S, Ma D and Ni J: Porphyromonas gingivalis regulates
atherosclerosis through an immune pathway. Front Immunol.
14:11035922023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mendez-Sanchez N, Cruz-Ramon VC,
Ramirez-Perez OL, Hwang JP, Barranco-Fragoso B and Cordova-Gallardo
J: New aspects of lipotoxicity in nonalcoholic steatohepatitis. Int
J Mol Sci. 19:20342018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu C, Schönke M, Zhou E, Li Z, Kooijman
S, Boon MR, Larsson M, Wallenius K, Dekker N, Barlind L, et al:
Pharmacological treatment with FGF21 strongly improves plasma
cholesterol metabolism to reduce atherosclerosis. Cardiovasc Res.
118:489–502. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhou P, Shen Y, Wang L, Cao Z, Feng W, Liu
J, Wang L, Meng P, Yang J, Xu WY and Gao P: Association between
carotid intima media thickness and small dense low-density
lipoprotein cholesterol in acute ischaemic stroke. Lipids Health
Dis. 19:1772020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Canbay E, Canda E, Yazıcı H, Kasıkcı GK,
Durmaz B, Copur O, Tahhan B, Düzgün D, Koru ZE, Sezer E, et al:
Determination of selected oxysterol levels, oxidative stress, and
macrophage activation indicators in children and adolescents with
familial hypercholesterolemia. Lipids Health Dis. 23:3742024.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Robinett NG, Culbertson EM, Peterson RL,
Sanchez H, Andes DR, Nett JE and Culotta VC: Exploiting the
vulnerable active site of a copper-only superoxide dismutase to
disrupt fungal pathogenesis. J Biol Chem. 294:2700–2713. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Schatzman SS, Peterson RL, Teka M, He B,
Cabelli DE, Cormack BP and Culotta VC: Copper-only superoxide
dismutase enzymes and iron starvation stress in Candida fungal
pathogens. J Biol Chem. 295:570–583. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Calcaterra V, Verduci E, Cena H, Magenes
VC, Todisco CF, Tenuta E, Gregorio C, De Giuseppe R, Bosetti A, Di
Profio E and Zuccotti G: Polycystic ovary syndrome in
insulin-resistant adolescents with obesity: The role of nutrition
therapy and food supplements as a strategy to protect fertility.
Nutrients. 13:18482021. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Al-Ahmadi W, Webberley TS, Joseph A,
Harris F, Chan YH, Alotibi R, Williams JO, Alahmadi A, Decker T,
Hughes TR and Ramji DP: Pro-atherogenic actions of signal
transducer and activator of transcription 1 serine 727
phosphorylation in LDL receptor deficient mice via modulation of
plaque inflammation. FASEB J. 35:e218922021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang F, Liu P, He Z, Zhang L, He X, Liu F
and Qi J: Crocin ameliorates atherosclerosis by promoting the
reverse cholesterol transport and inhibiting the foam cell
formation via regulating PPARγ/LXR-α. Cell Cycle. 21:202–218. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Huang R, Pang Q, Zheng L, Lin J, Li H, Wan
L and Wang T: Cholesterol metabolism: physiological versus
pathological aspects in intracerebral hemorrhage. Neural Regen Res.
20:1015–1030. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang H, Wang L, Liu Y, Men W, Hao W, Fang
C, Li C and Zhang L: Plasma levels of CD36 and glutathione as
biomarkers for ruptured intracranial aneurysm. Open Life Sci.
18:202207572023. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ahn JS, Yang JW, Oh SJ, Shin YY, Kang MJ,
Park HR, Seo Y and Kim HS: Porphyromonas gingivalis exacerbates the
progression of fatty liver disease via CD36-PPARgamma pathway. BMB
Rep. 54:323–328. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kim D, Choi H, Oh H, Lee J, Hwang Y and
Kang SS: Mutanolysin-Digested Peptidoglycan of lactobacillus
reuteri promotes the inhibition of porphyromonas gingivalis
lipopolysaccharide-induced inflammatory responses through the
regulation of signaling cascades via TLR4 suppression. Int J Mol
Sci. 25:422023. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Rekhi UR, Catunda RQ, Alexiou M, Sharma M,
Fong A and Febbraio M: Impact of a CD36 inhibitor on Porphyromonas
gingivalis mediated atherosclerosis. Arch Oral Biol.
126:1051292021. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Cariello M, Piccinin E and Moschetta A:
Transcriptional regulation of metabolic pathways via lipid-sensing
nuclear receptors PPARs, FXR, and LXR in NASH. Cell Mol
Gastroenterol Hepatol. 11:1519–1539. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Afzoon S, Amiri MA, Mohebbi M, Hamedani S
and Farshidfar N: A systematic review of the impact of
Porphyromonas gingivalis on foam cell formation: Implications for
the role of periodontitis in atherosclerosis. BMC Oral Health.
23:4812023. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Huang N, Shaik-Dasthagirisaheb YB,
LaValley MP and Gibson FC III: Liver X receptors contribute to
periodontal pathogen-elicited inflammation and oral bone loss. Mol
Oral Microbiol. 30:438–450. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Mao M, Deng Y, Wang L, Zhao G, Qi R, Gong
H, Shen T, Xu Y, Liu D and Chen B: Chronic unpredictable mild
stress promotes atherosclerosis via adipose tissue dysfunction in
ApoE(−/-) mice. PeerJ. 11:e160292023. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Javadifar A, Rastgoo S, Banach M,
Jamialahmadi T, Johnston TP and Sahebkar A: Foam cells as
therapeutic targets in atherosclerosis with a focus on the
regulatory roles of non-coding RNAs. Int J Mol Sci. 22:25292021.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Li XY, Wang C, Xiang XR, Chen FC, Yang CM
and Wu J: Porphyromonas gingivalis lipopolysaccharide increases
lipid accumulation by affecting CD36 and ATP-binding cassette
transporter A1 in macrophages. Oncol Rep. 30:1329–1336. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Shen X, Zhang S, Guo Z, Xing D and Chen W:
The crosstalk of ABCA1 and ANXA1: A potential mechanism for
protection against atherosclerosis. Mol Med. 26:842020. View Article : Google Scholar : PubMed/NCBI
|