Scar formation is a fundamental physiological
process through which the body repairs tissue damage following
injury (1–3). Although normal wound healing restores
tissue integrity, aberrant repair mechanisms often lead to abnormal
scarring, such as hypertrophic scars and keloids. Although scar
formation represents a physiological outcome of tissue repair,
excessive or dysregulated scarring can adversely affect the normal
wound healing process. Pathological scars are characterized by
persistent inflammation, excessive extracellular matrix (ECM)
deposition and prolonged myofibroblast activation. These
abnormalities disrupt the tightly regulated phases of wound
healing, particularly the transition from inflammation to tissue
remodeling, ultimately leading to abnormal tissue architecture and
impaired tissue regeneration (3,4).
These disruptions are closely associated with alterations in key
signaling pathways that regulate wound healing. For example,
sustained activation of the TGF-β/Smad pathway promotes persistent
myofibroblast differentiation and excessive ECM deposition, thereby
impairing the transition from the proliferative to the remodeling
phase. In addition, prolonged inflammatory signaling, mediated by
macrophage-derived cytokines such as IL-6 and platelet-derived
growth factor (PDGF), delays inflammation resolution. Furthermore,
an imbalance between matrix metalloproteinases (MMPs) and their
inhibitors (TIMPs) reduces ECM degradation, ultimately leading to
abnormal tissue remodeling and pathological scar formation. These
pathological scars, including hypertrophic scars and keloids,
impose a significant physical and psychological burden on patients
(3). These fibrotic conditions not
only cause aesthetic disfigurement and hinder social reintegration,
but they also lead to severe pruritus, pain and functional
impairment, leading to a significant deterioration in the patients'
quality of life (5). Despite the
availability of various therapeutic modalities, including
corticosteroid injections, laser therapy and surgical excision,
current treatments remain largely symptomatic, and are heavily
limited by unpredictable efficacies and high recurrence rates
(1). These difficulties highlight
an urgent clinical need to elucidate the precise molecular
mechanisms underlying scar pathogenesis, and also the need to
identify novel, targeted therapeutic strategies.
At the cellular level, tissue repair is
predominantly governed by the dynamic interplay between immune
cells and structural cells. The process of scar formation typically
occurs during wound healing when various tissues, such as skin,
muscle or internal organs, are damaged (6–8). In
response, the body activates a series of repair mechanisms, which
result in new tissue generation at an injury site (6–8).
Newly formed tissue is generally denser compared with the original
tissue, and often lacks its normal functionality (6–8).
Fibroblasts serve as the primary effector cells in the
proliferative and remodeling phases, and have the role of
synthesizing collagen and other ECM components to reconstruct the
damaged tissue (9–12). On the other hand, macrophages
orchestrate the inflammatory phase by clearing cellular debris and
pathogens, at the same time secreting cytokines that subsequently
modulate the wound microenvironment, thereby facilitating the
healing process (13–15). Studies have identified specific
cellular subpopulations that perform pivotal roles in these
processes (16–23). Secreted phosphoprotein 1 (SPP1), a
gene encoding the multifunctional matricellular protein osteopontin
(OPN), serves as a crucial marker for a distinct, highly active
macrophage subpopulation (16–19).
SPP1(+) macrophages are deeply involved in immune modulation, local
inflammation and the regulation of the tissue microenvironment
during repair (20–23). Similarly, periostin (POSTN), a
distinctive marker for fibroblasts, is a matricellular protein that
is extensively expressed in the ECM during tissue remodeling and
fibrosis (24–27). It acts as a key contributor to
structural repair and pathological scarring through excessively
secreting ECM components and driving fibrotic progression within
both the wound healing and the tumor microenvironments (28–31).
Emerging evidence has emphasized that the intricate
crosstalk between SPP1(+) macrophages and POSTN(+) fibroblasts
critically dictates the trajectories of both tissue repair and scar
formation (32–36). These two cellular subpopulations
are engaged in complex signaling networks that drive key biological
processes, including immune regulation and ECM remodeling (32); however, despite their recognized
importance, to date, research in this area has predominantly
focused on macrophages and fibroblasts as broad and heterogeneous
cell populations, typically examining macrophage polarization
states such as M1/M2 macrophages or describing fibroblasts as
general ECM-producing stromal cells (3,37,38).
Consequently, a significant knowledge gap remains regarding the
precise molecular mechanisms and synergistic interactions that
occur between distinct cellular subpopulations such as SPP1(+)
macrophages and POSTN(+) fibroblasts. Through synthesizing recent
findings on these specific cell types, the present study was
designed to bridge this critical gap. The present study aimed to
comprehensively clarify the specific roles, signaling pathways and
interaction mechanisms of SPP1(+) macrophages and POSTN(+)
fibroblasts during tissue scarring, with the goal of establishing a
robust theoretical framework for the development of targeted,
highly specific therapies for abnormal scar formation.
Several previous reviews have summarized the general
mechanisms of wound healing and scar formation, particularly
focusing on fibroblast activation, macrophage polarization and key
fibrotic signaling pathways (39).
However, most of these studies have primarily discussed macrophages
and fibroblasts as broad and heterogeneous populations, without
specifically addressing the functional roles of distinct cellular
subpopulations (40). These
previous studies have highlighted key pathways such as TGF-β/Smad
signaling, inflammatory cytokine-mediated pathways (such as IL-6
and PDGF), and ECM remodeling regulated by MMPs and TIMPs, which
collectively govern fibroblast activation, macrophage polarization
and scar formation.
Advances in single-cell transcriptomics have
revealed the presence of specialized macrophage and fibroblast
subsets (35), such as SPP1(+)
macrophages and POSTN(+) fibroblasts, which may play pivotal roles
in fibrosis progression. Nevertheless, systematic reviews
specifically examining the bidirectional crosstalk between SPP1(+)
macrophages and POSTN(+) fibroblasts during scar formation are
still lacking.
Therefore, the present review aimed to synthesize
current evidence regarding the roles, signaling mechanisms and
reciprocal interactions of SPP1(+) macrophages and POSTN(+)
fibroblasts in scar formation. By emphasizing the crosstalk between
these specific cellular subsets, it provides a more refined
perspective on the cellular mechanisms of fibrosis and highlights
potential therapeutic targets for abnormal scar formation.
To ensure a rigorous and comprehensive synthesis of
the current literature regarding the distinct roles of SPP1(+)
macrophages and POSTN(+) fibroblasts in scar formation, a
systematic literature search was executed. The identification,
screening and selection of relevant studies was carried out through
searching primary biomedical and scientific databases, including
the PubMed (https://pubmed.ncbi.nlm.nih.gov/), Web of Science
(https://clarivate.com/academia-government/scientific-and-academic-research/research-discovery-and-referencing/web-of-science/),
Embase (https://www.embase.com/) and Scopus
(https://www.elsevier.com/products/scopus). This
comprehensive literature retrieval process included publications
from database inception up to October 31, 2025. The search protocol
utilized a strategic combination of Medical Subject Headings (MeSH)
and free-text terms, linked by Boolean operators (‘AND’ and ‘OR’)
to maximize both search sensitivity and specificity. The primary
search terms, deployed both individually and in various
combinations, included ‘scar formation’, ‘hypertrophic scar’,
‘keloid’, ‘wound healing’, ‘SPP1’, ‘osteopontin’, ‘macrophage’,
‘POSTN’, ‘periostin’, ‘fibroblast’, ‘fibrosis’ and ‘single-cell
sequencing’. Subsequent to the initial retrieval, strict inclusion
criteria were applied to ensure high academic quality and relevance
to the objectives of the present study.
Selected studies were required to be peer-reviewed
original research articles, single-cell transcriptomic analyses,
meta-analyses or comprehensive foundational reviews. Furthermore,
eligible publications had to be written in English, and also had to
be directly concerned with investigating the cellular mechanisms,
molecular crosstalk or therapeutic targeting of the specific
SPP1(+) and POSTN(+) cellular subpopulations within the context of
tissue repair, fibrosis and scar pathogenesis.
To maintain a precise focus on the mechanisms of
scar formation, systematic exclusion criteria were concurrently
implemented. Publications were systematically excluded if: i) They
were non-English articles; ii) the abstract was lacking in
accessible full text; or iii) the study was devoid of specific
mechanistic insights into scarring. Additionally, research articles
that were primarily focused on unrelated oncological pathways or
other diseases without a distinct fibrotic or wound-healing context
were also omitted from this review, thereby ensuring that the
synthesized data directly contributed to an understanding of
abnormal scar formation.
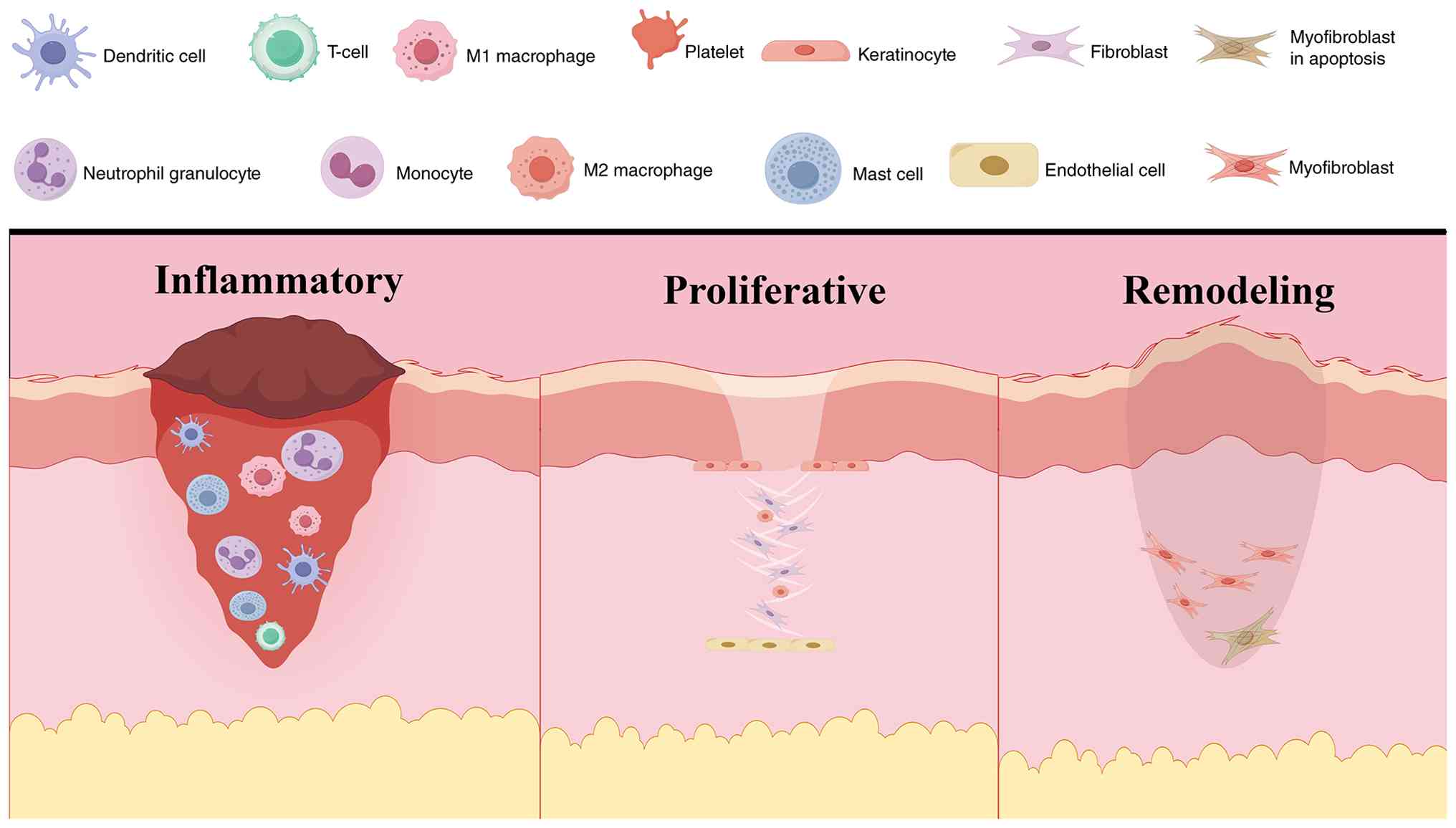
Scar formation is a physiological necessity during
tissue repair, culminating in the deposition of a collagen-rich ECM
to restore structural integrity following injury (Fig. 1) (41–43).
At the molecular level, macrophages regulate wound healing by
sensing DAMPs/PAMPs via Toll-like receptors and releasing
profibrotic mediators, including PDGF, IL-6, IL-11 and TGF-β1,
which drive fibroblast activation. TGF-β1 is a key regulator that
induces Smad2/3 phosphorylation, Smad4 complex formation and
nuclear translocation, thereby promoting transcription of collagen
and α-smooth muscle actin (α-SMA) and facilitating myofibroblast
differentiation. In parallel, excessive ECM accumulation is
reinforced by an imbalance between MMPs and TIMPs, leading to
reduced matrix degradation. These processes are normally restricted
by inhibitory Smads (Smad6/7) and ubiquitin-mediated degradation
via Smurf1/2. However, sustained activation of these pathways
results in persistent myofibroblast activity and pathological scar
formation. Although the process of normal wound healing follows a
highly coordinated spatiotemporal sequence that progresses through
hemostasis, inflammation, proliferation and remodeling, aberrant
repair mechanisms may cause a derailing of this progression
(44–46). Consequently, this dysregulation
leads to pathological scarring, including the development of
hypertrophic scars and keloids, which are characterized by
excessive ECM accumulation and distinct clinical presentations
(Table I) (4,47–49).
Rather than functioning as homogeneous populations, recent evidence
has demonstrated that specific cellular subsets orchestrate the
transition from normal repair to pathological fibrosis. During the
phases of normal scar formation, macrophages and fibroblasts
exhibit highly coordinated crosstalk (Table II) (50–52).
However, in pathological scarring, the prolonged activation of
specific subsets, most notably SPP1(+) macrophages and POSTN(+)
fibroblasts, creates a pro-fibrotic microenvironment. The
continuous infiltration of these specialized macrophages, coupled
with the hyperproliferation and impaired apoptosis of
myofibroblasts, drives the unabated ECM deposition that is
characteristic of hypertrophic scars and keloids (Fig. 2) (53–62).
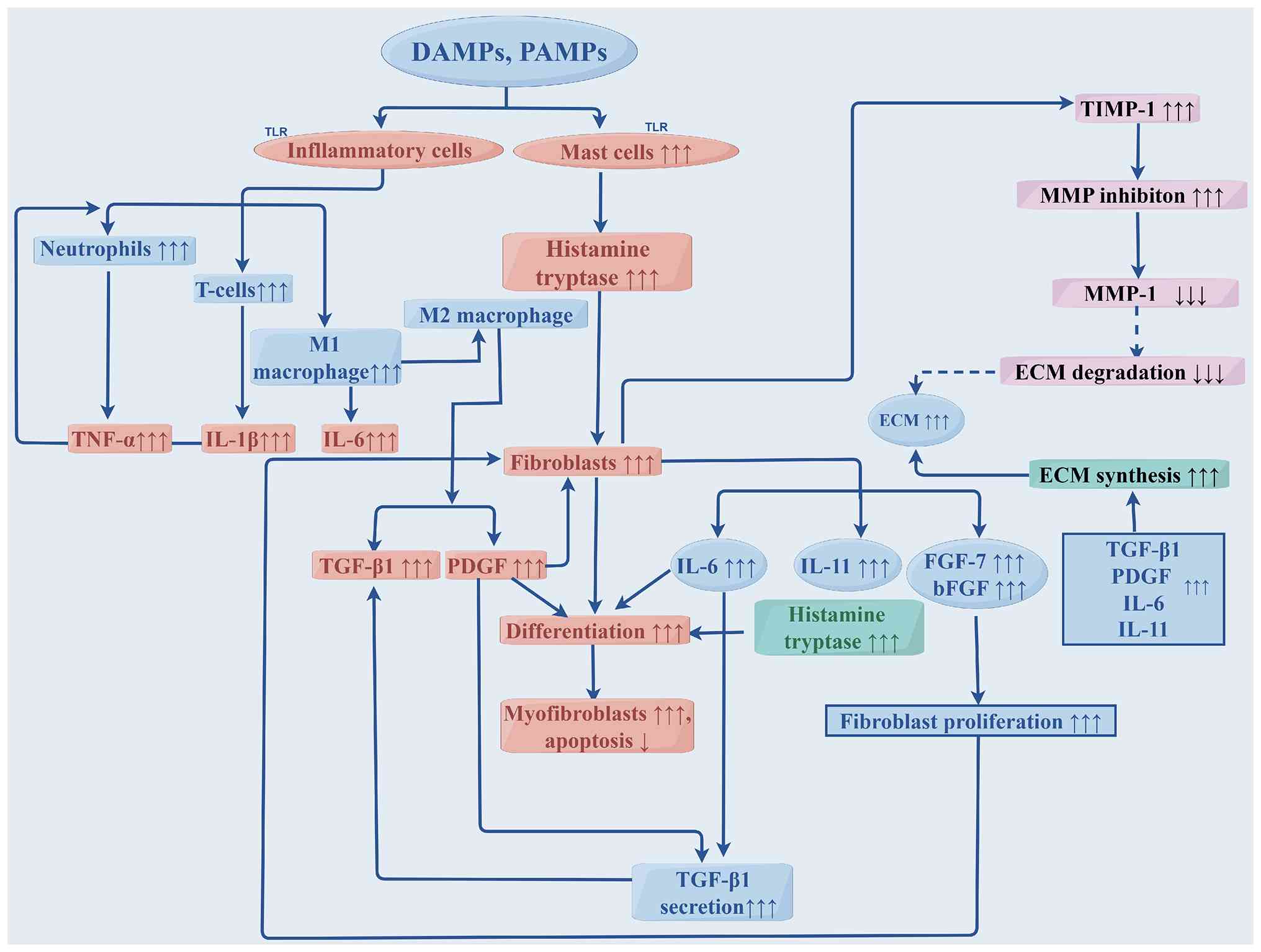
The initiation of this aberrant fibrotic response is
deeply rooted in prolonged innate immune activation.
Damage-associated molecular patterns (DAMPs) and
pathogen-associated molecular patterns (PAMPs) persistently
activate inflammatory cells via Toll-like receptors (63–65).
This sustained activation, in turn, triggers a cascade of
pro-fibrotic cytokines, including PDGF, IL-6 and TGF-β1.
Furthermore, the DAMP/PAMP-mediated stimulation of mast cells
induces the secretion of histamine and proteases, which leads to a
further amplification of the release of basic fibroblast growth
factor and fibroblast growth factor 7 (52,64,65).
Within this cytokine-rich milieu, PDGF, IL-6 and IL-11 act
synergistically with TGF-β1 to forcefully drive the differentiation
of fibroblasts into α-SMA-expressing myofibroblasts, which
especially enriches the highly active POSTN(+) subpopulation
(66). At the same time, elevated
levels of TIMPs, such as the protease TIMP-1, suppress MMP
activity, leading to the severe impairment of ECM degradation and
ensuring massive matrix accumulation (12,38,67).
Central to this pro-fibrotic signaling network is the TGF-β/Smad
axis, heavily secreted by highly active macrophage subsets,
including SPP1(+) macrophages (38,68,69).
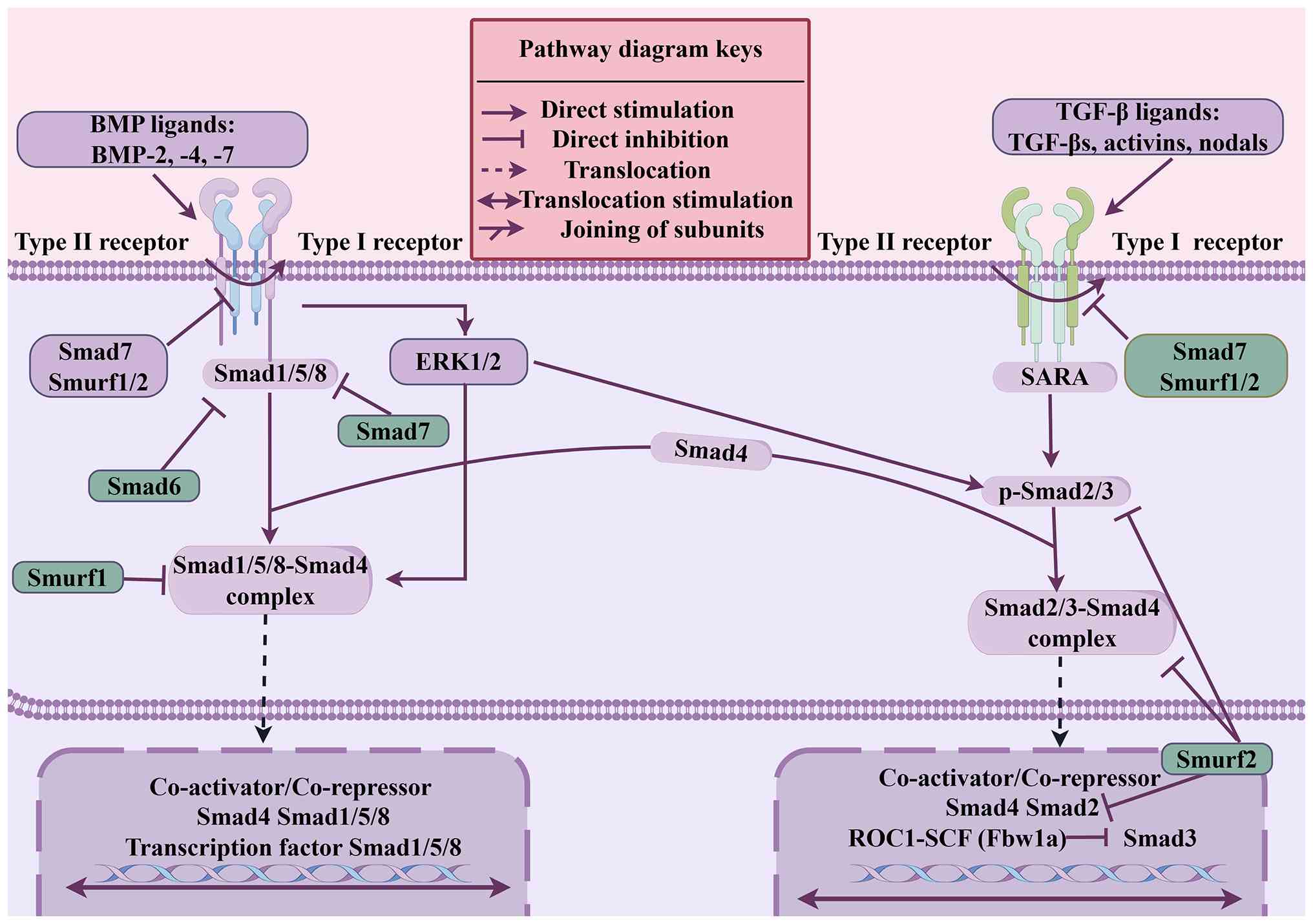
Specifically, TGF-β signaling initiates downstream
signal transduction by binding to type I and II TGF-β receptors on
the cell membrane (70). TGF-β1 is
the master regulator of the fibrotic response, binding to specific
membrane receptors to phosphorylate receptor-regulated Smads
(R-Smads), primarily Smad2 and Smad (71–73).
Once complexed with Co-Smad (Smad4), this transcriptional complex
is translocated to the nucleus, where it directly regulates gene
expression and stimulates the synthesis of α-SMA and collagen
(70–73).
This signaling pathway is crucial for the proper
functioning of fibroblasts and myofibroblasts, especially in the
context of fibrosis, where TGF-β signaling directly influences
collagen synthesis and deposition (74–76).
Moreover, the TGF-β signaling pathway is tightly regulated by
feedback mechanisms. Inhibitory SMADs (I-Smads), for example, Smad6
and Smad7, negatively modulate the pathway by preventing the
binding of R-Smads to the receptor, thereby reducing
phosphorylation and shutting down TGF-β signaling (77).
In normal tissue remodeling, this intense signaling
is strictly self-limiting, regulated by I-Smads such as Smad6 and
Smad7, as well as precise ubiquitination mechanisms (78–81).
Specifically, a number of E3 ubiquitin ligases, including Smad
ubiquitin-associated factors 1 (Smurf1) and Smad
ubiquitin-associated factors 2 (Smurf2), target R-Smads and the
TGF-β type I receptor for proteasomal degradation, thereby
effectively controlling the intensity and duration of TGF-β
signaling (Fig. 3) (78). Smurf1 specifically binds to Smads
1, 5 and 8 within the cytoplasm, facilitating the ubiquitination of
these proteins, whereas Smurf2 selectively targets phosphorylated
Smad2 within the cell nucleus, leading to its degradation (81–84).
Similarly, Smad3 ubiquitination is mediated by a ROC1-Skp1-cullin
1-F-box (SCF) E3 ubiquitin ligase complex via Fbw1a (84–86).
Ultimately, the complex of Smurf together with Smad7 facilitates
ubiquitination, as well as proteasomal degradation, targeting the
TGF-β type I receptor, ultimately leading to the cessation of
TGF-β/Smad signaling (87,88). Considered altogether, macrophages
and fibroblasts fulfill an essential role in pathological scar
formation, working in close association to facilitate a smooth
progression from the inflammatory phase to tissue repair.
Macrophages drive this process by regulating immune responses and
releasing growth factors, whereas fibroblasts contribute to the
process by synthesizing collagen and other ECM components, thereby
forming new tissue and ensuring both wound closure and scar
formation. Furthermore, the TGF-β signaling pathway is crucial for
regulating collagen synthesis and fibrosis during tissue repair via
Smad proteins, also ensuring that these processes are finely
controlled through feedback mechanisms.
The specific role and contribution of POSTN(+)
fibroblasts in scar formation have garnered significant attention.
During normal wound healing and pathological scarring, fibroblasts
undergo activation and transition into myofibroblasts, a process
that drives the massive secretion of collagen, and subsequent
structural remodeling (12,13,26,36,54).
Evidence has highlighted that distinct fibroblast subpopulations
dictate the intensity of this fibrotic response (89–96).
For example, in the study by Deng et al (89), the single-cell transcriptomic
profiling of human keloid tissues revealed a marked increase in the
expression of the POSTN gene in mesenchymal fibroblasts,
strongly correlating with enhanced ECM deposition. This finding
suggested that POSTN(+) fibroblasts possess a markedly amplified
collagen-secreting capacity compared with POSTN(−) fibroblasts.
Early foundational in vivo studies performed by Oka et
al (90) further established
this critical role. This research group also demonstrated that,
within the first 10 days post-myocardial infarction, mice lacking
the POSTN gene (Postn−/− mice) exhibited a
markedly higher susceptibility to ventricular rupture; however, the
surviving Postn−/− mice displayed markedly
attenuated fibrosis, with the preservation of ventricular function.
Furthermore, these knockout mice exhibited reduced levels of
fibrotic remodeling and hypertrophy under chronic pressure overload
conditions (90). In terms of the
underlying mechanism, the Postn−/− murine models
were shown to have profound molecular alterations as far as the
fibroblast functional program was concerned. Fibroblasts isolated
from these deficient hearts exhibited both severely compromised
adhesion to cardiomyocytes and notable transcriptional shifts,
suggesting that the Postn−/− fibroblasts are
incapable of secreting sufficient collagen to execute effective
structural repair (90). Expanding
on the origin of these highly active cells, Kanisicak et al
(91) demonstrated that
POSTN-expressing myofibroblasts in the fibrotic heart are derived
from tissue-resident fibroblasts of the Tcf21 lineage, rather than
from endothelial, myeloid or smooth muscle precursors.
Collectively, these foundational studies have provided robust
evidence that the POSTN(+) subpopulation is a primary driver of
collagen biosynthesis and fibrotic progression, a paradigm that has
been supported by extensive subsequent research studies across
various fibrotic diseases (92–96).
Despite the clear phenotypic evidence, elucidating
the precise intracellular mechanisms via which POSTN drives
myofibroblast hyperactivation remains a central focus. Recent
mechanotransduction studies have provided critical insights into
this process (89,97–103). Xu et al (97) demonstrated that Piezo-type
mechanosensitive ion channel component 1 (Piezo1) was highly
expressed in POSTN(+) myofibroblasts within a pulmonary fibrosis
model, serving as a pivotal node for mechanical activation.
Subsequently, the conditional deletion of Piezo1 in POSTN(+)
myofibroblasts led to a marked attenuation of pulmonary fibrosis by
inhibiting cellular activation and proliferation (97). At the molecular level, the loss of
Piezo1 both disrupted actin filament organization and blocked the
nuclear localization of the Yes-associated protein 1
(YAP)/WW-domain-containing transcription regulator 1 (WWTR1, or
TAZ) mechanotransducers (97). The
inhibition of these processes effectively shifted the
myofibroblasts from a highly proliferative state towards cellular
stress and apoptosis (97,99) Crucially, fibroblast-specific
deletion of YAP/TAZ replicated the protective phenotype observed in
conditional Piezo1 knockout models, thereby firmly establishing the
Piezo1/YAP/TAZ axis as being indispensable for POSTN(+) fibroblast
function (97,99).
Similar regulatory mechanisms have been identified
in the pathological cutaneous scarring process. Akita et al
(100) reported a positive
association between the density of POSTN(+) fibroblasts and the
expression of Piezo2 in human keloid tissues. Fibroblasts
exhibiting elevated levels of Piezo2 were found to have increased
levels of downstream signaling, leading in turn to increased ECM
production, suggesting a functional reliance of POSTN(+)
fibroblasts on Piezo2-mediated mechanotransduction, although
further functional validation of these findings is required
(100). Beyond mechanosensing,
POSTN(+) fibroblasts are heavily regulated by complex fibrotic
signaling cascades. Jiang et al (101) elucidated that POSTN performs a
vital role in keloid inflammation and fibrosis through activating
the JAK-STAT signaling pathway and dynamically modulating the
IL-4/IL-13 feedback loop within the T helper 2 cell immune
response. In this context, IL-4 actively upregulates POSTN
expression in fibroblasts, creating a pathogenic positive feedback
loop. Similarly, Maeda et al (102) confirmed that IL-4 induced POSTN
secretion subsequently triggers TGF-β1 signaling via the RhoA/ROCK
signaling pathway. The resulting elevation in TGF-β1 levels further
stimulates POSTN synthesis, thereby trapping the local
microenvironment in a continuous cycle of abnormal scar expansion
(102).
Crucially, the pathogenic influence of POSTN(+)
fibroblasts extends beyond autocrine collagen production to include
profound paracrine effects on the broader wound microenvironment.
For example, Zhang et al (103) discovered that POSTN(+)
fibroblasts directly activate the extracellular-regulated kinase
1/2 (ERK1/2) and focal adhesion kinase (FAK) signaling pathways in
adjacent endothelial cells. This paracrine cross-talk causes a
marked enhancement of local angiogenesis, thereby supplying the
metabolic demands of the expanding keloid lesion, and exacerbating
the fibrotic pathology (103).
Considered altogether, during pathological wound
healing, the highly active POSTN(+) fibroblast subpopulation
emerges as a central orchestrator of scar formation. Through
integrating profound mechanotransduction signals via Piezo channels
and participating in relentless cytokine feedback loops, these
cells not only drive massive autocrine collagen deposition, but
also facilitate complex paracrine networks, including the process
of angiogenesis, to sustain continuous and abnormal fibrosis.
The recognition of SPP1(+) macrophages as central
mediators of tissue repair dates back to the last century, where
high levels of SPP1 expression and secretion were detected in
macrophages that had infiltrated areas of the heart and brain that
had sustained ischemic-reperfusion injuries (104,105). Although the exact functional
significance of these cells has remained elusive since then, a
landmark study by Bevan et al (106) in 2020 established that
macrophage-derived SPP1 (also known as OPN) is indispensable for
fibrotic healing in cardiac injury. Subsequent studies have
transitioned from phenotypic observations to investigations aimed
at elucidating the associated mechanisms, and these have
characterized SPP1 as a critical paracrine regulator that modulates
the surrounding cellular microenvironment to facilitate tissue
repair and scar formation.
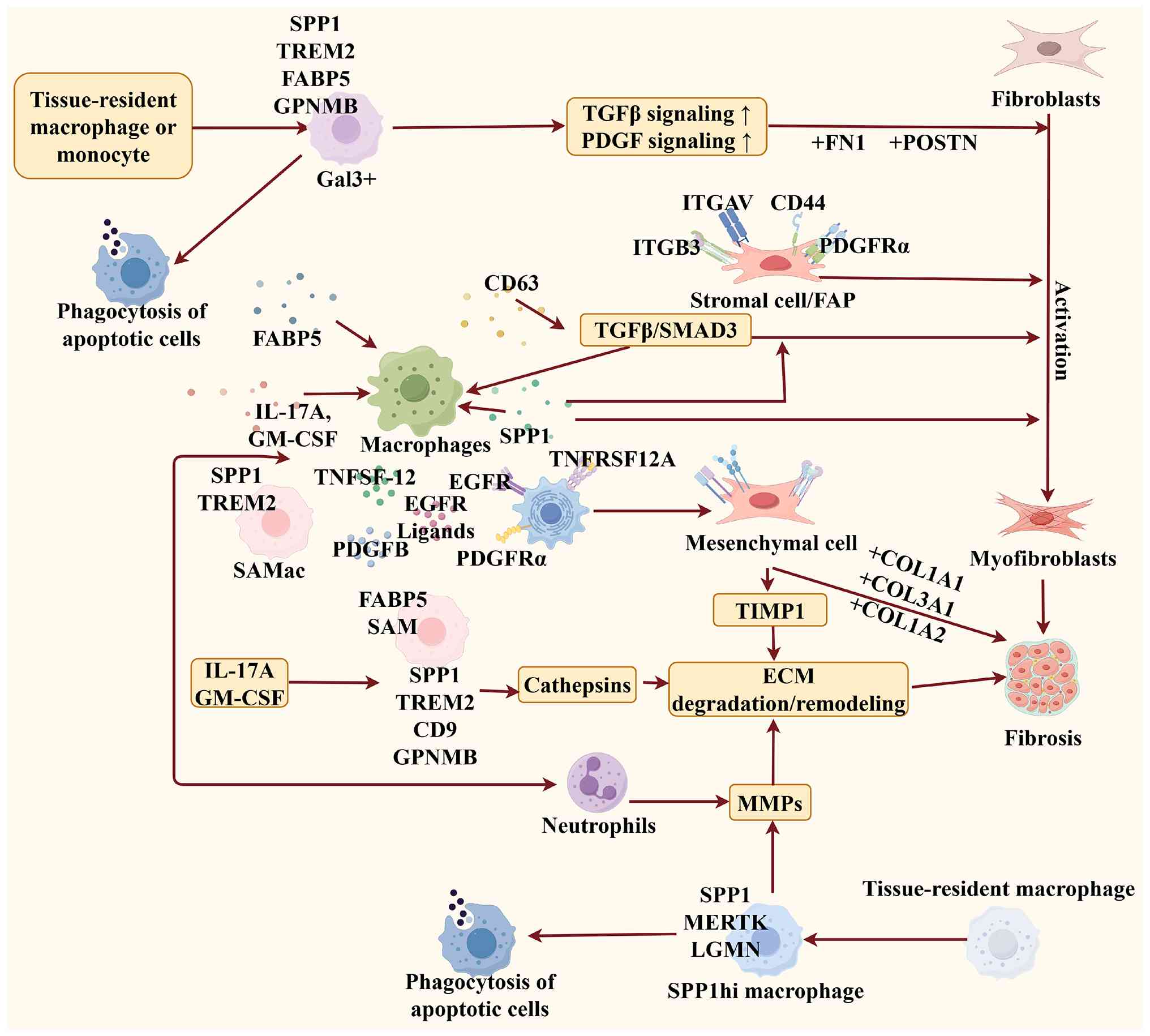
As a multifunctional secreted protein, SPP1 exerts
its regulatory effects by binding to various cell surface
receptors, thereby orchestrating diverse cellular responses across
different tissues (18,35,106–108). Gliem et al (107) demonstrate that SPP1 produced by
blood-derived macrophages induce astrocytes to extend their
processes towards the infarct boundaries, a morphological shift
that is essential for the structural repair of the ischemic
neurovascular unit. Similarly, in another study by Rotem et
al (108),
macrophage-secreted SPP1 was found to directly stimulate the
proliferation of myocardial cells, further supporting the repair of
damaged cardiac tissue. Beyond these reparative functions, a
growing body of evidence suggests that the primary contribution of
the SPP1(+) macrophage subpopulation to pathological scarring lies
in its ability to drive the transition of fibroblasts into
myofibroblasts (Fig. 4) (35,109–113).
Recent advances in single-cell transcriptomics have
further refined our understanding of the specific crosstalk between
immune and mesenchymal subpopulations. A pivotal study by Hong
et al (32) demonstrated
that SPP1(+) macrophages may exacerbate pathological scar formation
by establishing a specialized paracrine signaling axis with
POSTN(+) fibroblasts. Even though these findings were primarily
derived from computational bioinformatics analysis and necessitate
further experimental validation in vivo, they nonetheless
provide a compelling theoretical framework. This proposed
interaction highlights SPP1(+) macrophages as critical upstream
regulators that potentially dictate the fibrotic activity of
POSTN(+) fibroblasts, thereby positioning this specific cellular
axis as a key driver of aberrant tissue repair.
Emerging evidence suggests that SPP1(+) macrophages
and POSTN(+) fibroblasts may engage in functionally relevant
interactions during fibrotic processes (114–116); however, direct experimental
evidence of their crosstalk remains limited. To date, a direct
interaction between these two specific cellular subpopulations has
primarily been reported in a single-cell transcriptomic study of
acne keloidalis (32), which
identified ligand-receptor interactions indicative of active
communication. Most of the current mechanistic understanding is
derived from indirect evidence obtained from separate studies
focusing on either macrophages or fibroblasts in different fibrotic
contexts. For example, in pulmonary fibrosis and liver fibrosis
models, macrophage-derived SPP1 has been shown to bind to receptors
such as CD44 and integrins (such as αvβ3 and αvβ5) on fibroblasts,
thereby activating intracellular signaling pathways including FAK
and PI3K/AKT, which promote fibroblast activation and survival
(117–121). In addition, SPP1 has been
reported to facilitate the activation of latent TGF-β1, further
enhancing fibroblast-to-myofibroblast differentiation and ECM
production in fibrotic disease models (122,123). Conversely, studies investigating
fibroblast biology have demonstrated that POSTN can modulate the
local immune microenvironment in tumor and fibrotic settings,
including breast cancer and lung cancer models, by interacting with
integrins on macrophages, thereby promoting macrophage recruitment
and retention (24,25,28,124). Furthermore, increased ECM
stiffness driven by POSTN-expressing fibroblasts has been shown, in
mechanotransduction studies of fibrosis and tumor
microenvironments, to influence macrophage phenotype and sustain a
profibrotic state (97). Taken
together, while these findings collectively support the existence
of a potential bidirectional interaction between SPP1(+)
macrophages and POSTN(+) fibroblasts, most evidence remains
indirect and inferred from separate studies conducted in pulmonary
fibrosis, liver fibrosis and tumor-associated models. Further
experimental validation, particularly in disease-specific models of
pathological scarring, is required to definitively establish the
direct crosstalk between these two cellular subpopulations.
Disrupting this specific, localized communication network therefore
represents a crucial target in the development of definitive,
targeted therapies against hypertrophic scars and keloids, moving
beyond broad-spectrum, anti-inflammatory approaches.
The complex interplay between POSTN(+) fibroblasts
and SPP1(+) macrophages constitutes a core driving force in
pathological scar formation. Consequently, disrupting this specific
cellular crosstalk presents a highly promising therapeutic frontier
for managing keloids and hypertrophic scars. Since these cellular
subsets sustain the pro-fibrotic microenvironment through
continuous cytokine secretion and molecular feedback loops,
strategically targeting them could halt, or even reverse, the
progression of severe fibrosis. For SPP1(+) macrophages, the
primary pathogenic mechanism relies on the excessive local
secretion of the SPP1 protein (123,125). A highly viable clinical
intervention involves the deployment of engineered neutralizing
antibodies designed to specifically block the interaction between
SPP1 and both its own receptors and downstream receptors, including
CD44 and various integrins (for example, α5β1, αvβ3 and α4β1)
(18,108,119,123,125). Neutralizing antibodies targeting
OPN (SPP1) have been investigated in several fibrotic and
inflammatory disease models, including liver fibrosis, pulmonary
fibrosis and tumor-associated fibrosis, where inhibition of SPP1
signaling attenuates fibroblast activation and ECM deposition
(126–129). This targeted blockade directly
severs the pro-fibrotic signaling cascade, thereby preventing the
persistent activation of adjacent fibroblasts, and halting
subsequent ECM deposition (79,80).
Furthermore, the formation of immune complexes between these
therapeutic antibodies and SPP1 has been shown to enhance Fc
receptor-mediated phagocytosis by local macrophages (130). This process markedly accelerates
the clearance of SPP1 from the fibrotic microenvironment, massively
reducing its overall bioavailability. Even though the development
of direct anti-SPP1 therapies for cutaneous scarring is currently
in the preclinical stages, analogous SPP1-targeted monoclonal
antibodies are undergoing rigorous clinical evaluation for other
fibrotic and neoplastic conditions, providing a robust
translational framework for future scar treatments.
Concurrently, directly targeting the POSTN(+)
fibroblast subpopulation through genetic silencing affords an
exceptionally precise therapeutic avenue. Precision medicine
approaches utilizing RNA interference (RNAi) technologies,
specifically those involving small interfering RNA (siRNA) and
short hairpin RNA (shRNA), may be engineered to achieve the robust
knockdown of POSTN expression (131,132). In terms of the underlying
mechanism, these synthesized double-stranded RNA molecules are
processed into single strands that are incorporated into the
RNA-induced silencing complex, or RISC, within the fibroblast
cytoplasm (133,134). This complex subsequently binds to
the highly complementary POSTN mRNA sequence, inducing its precise
enzymatic cleavage, and also preventing the translation of the
POSTN protein (135). In addition
to synthetic RNAi, endogenous microRNAs (miRNAs), such as miR-876
and miR-577, have emerged as potent post-transcriptional regulators
(134).
Previous studies have demonstrated that these miRNAs
can directly regulate POSTN-related signaling pathways. For
example, miR-876 has been shown to suppress POSTN expression in
hepatocellular carcinoma-associated liver fibrosis models, where it
inhibits epithelial-mesenchymal transition by downregulating TGF-β
signaling and reducing the expression of fibrosis-related markers
such as α-SMA and collagen (132). Similarly, the miR-577/POSTN axis
has been investigated in breast cancer models, where miR-577
regulates POSTN expression and downstream ILK/AKT/mTOR signaling,
leading to reduced fibroblast proliferation and decreased ECM
production, including collagen deposition (131). In addition to miRNA-mediated
regulation, experimental strategies targeting POSTN have also been
explored. For instance, in in vitro fibroblast systems,
suppression of POSTN expression has been shown to reduce α-SMA
expression, inhibit fibroblast activation and decrease collagen
synthesis. In vivo studies using fibrotic disease models
have further demonstrated that genetic or functional inhibition of
POSTN attenuates fibrosis progression through modulation of TGF-β
signaling pathways and ECM remodeling (90,92).
However, direct evidence from siRNA/shRNA-based studies in fibrotic
models remains limited. Despite these promising molecular targets
and therapeutic strategies, however, several limitations must be
acknowledged within both the current research landscape and the
present study. However, most of these therapeutic strategies remain
at the preclinical stage and clinical trials specifically targeting
POSTN or SPP1 in scar formation are currently limited. At present,
no registered clinical trials specifically targeting POSTN or SPP1
for pathological scar formation have been reported, and most
available evidence remains limited to preclinical or early-phase
studies in other fibrotic or neoplastic conditions (136,137). A primary limitation in the
broader scientific field is that the majority of functional
validations heavily rely on murine models. These models
fundamentally lack the biological capacity to form true human
keloids, thereby limiting the direct clinical translatability of
the in vivo findings. Furthermore, although single-cell
transcriptomics has proven to be useful in terms of precisely
mapping the distinct presence of the SPP1(+) and POSTN(+)
subpopulations, numerous studies have been only observational and
descriptive in nature. More rigorous in vivo lineage tracing
and cell-specific knockout experiments are urgently required to
definitively prove direct causality in human tissue. Regarding the
limitations of the present study, the literature synthesis
deliberately focused on these two specific subpopulations and their
immediate molecular interactions. This highly targeted scope
inevitably omitted the potential contributions of other critical
microenvironmental factors, including regulatory T cells,
endothelial cell dysfunction and broader epigenetic modifications
that also dictate scar pathogenesis. Additionally, the rapid
evolution of single-cell sequencing technologies should lead to the
definitions of the transcriptomic markers for these cellular
subsets being further subdivided or refined in the near future.
In conclusion, POSTN(+) fibroblasts and SPP1(+)
macrophages are not merely passive participants in tissue repair,
but also act as central orchestrators of aberrant fibrosis and scar
formation. Through their continuous engagement in intricate
autocrine and paracrine signaling loops, they sustain the
inflammatory and proliferative phases indefinitely. Looking to the
future, treatment approaches targeting POSTN(+) fibroblasts and
SPP1(+) macrophages should offer new possibilities for treating
keloids and hypertrophic scars. The development of neutralizing
antibodies or other molecular strategies aimed at blocking the
binding of POSTN or SPP1 to their respective receptors may help to
slow, or even reverse, the scar formation process. Specifically,
neutralizing antibodies against SPP1 have been shown to reduce
fibroblast activation and collagen deposition, which, in turn, may
mitigate the progression of fibrosis. Additionally, silencing POSTN
expression in POSTN(+) fibroblasts prevents their excessive
activation, thereby reducing both scar formation and fibrosis.
Targeting the expression of these critical molecules through
techniques that employ siRNAs, shRNAs and miRNAs is likely to
provide effective strategies for managing abnormal scars. A primary
limitation in the current body of literature is that numerous
findings are derived from bioinformatics analyses and single-cell
transcriptomic studies, which require further experimental
validation. Although several studies have provided evidence from
in vitro experiments and animal models, large-scale in
vivo human clinical studies remain limited (136). Therefore, additional experimental
and clinical investigations are needed to confirm the causal roles
and therapeutic potential of these cellular subpopulations in scar
formation.
Therefore, POSTN(+) fibroblasts and SPP1(+)
macrophages serve not only as key regulators of scar formation, but
also as promising targets for future therapies. As research into
these cells and their molecular pathways continues to advance, it
will enable the development of novel therapeutic strategies for
treating a wide range of fibrotic diseases and improving scar
formation.
Funding: No funding was received.
Not applicable.
HW and XD designed the study and wrote the
manuscript. Data authentication is not applicable. Both authors
read and approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Waibel JS, Waibel H and Sedaghat E: Scar
therapy of skin. Facial Plast Surg Clin N Am. 31:453–462. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rosales Santillan M, Ozog D and Wu W:
Using Neuromodulators to Improve Scar Formation, Keloids, Rosacea,
and Antiaging. Dermatol Surg. 50 (Suppl 9):S91–S96. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Peña OA and Martin P: Cellular and
molecular mechanisms of skin wound healing. Nat Rev Mol Cell Biol.
25:599–616. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Knowles A and Glass DA: Keloids and
hypertrophic scars. Dermatol Clin. 41:509–517. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shen W, Chen L and Tian F: Research
progress of scar repair and its influence on physical and mental
health. Int J Burns Trauma. 11:442–446. 2021.PubMed/NCBI
|
|
6
|
Gong X, Zhao Q, Zhang H, Liu R, Wu J,
Zhang N, Zou Y, Zhao W, Huo R and Cui R: The effects of mesenchymal
stem cells-derived exosomes on metabolic reprogramming in scar
formation and wound healing. Int J Nanomed. 19:9871–9887. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lin X and Lai Y: Scarring skin: Mechanisms
and therapies. Int J Mol Sci. 25:14582024. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sun Q, Tang L and Zhang D: Molecular
mechanisms of uterine incision healing and scar formation. Eur J
Med Res. 28:4962023. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wen J, Li Z, Tan Y, Tey HL, Yu N and Wang
X: Endothelial dysfunction in keloid formation and therapeutic
insights. J Invest Dermatol. 145:2436–2448. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rennekampff HO, Tenenhaus M, Rennekampff I
and Alharbi Z: Roles of mechanosensitive channel Piezo1 in wound
healing and scar formation. Life (Basel). 14:3772024.PubMed/NCBI
|
|
11
|
Kim HJ and Kim YH: Comprehensive insights
into keloid pathogenesis and advanced therapeutic strategies. Int J
Mol Sci. 25:87762024. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schuster R, Younesi F, Ezzo M and Hinz B:
The role of myofibroblasts in physiological and pathological tissue
repair. Cold Spring Harbor Perspect Biol. 15:a0412312023.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hesketh M, Sahin KB, West ZE and Murray
RZ: Macrophage phenotypes regulate scar formation and chronic wound
healing. Int J Mol Sci. 18:15452017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ding JY, Chen MJ, Wu LF, Shu GF, Fang SJ,
Li ZY, Chu XR, Li XK, Wang ZG and Ji JS: Mesenchymal stem
cell-derived extracellular vesicles in skin wound healing: Roles,
opportunities and challenges. Mil Med Res. 10:362023.PubMed/NCBI
|
|
15
|
Wang X and Liu D: Macrophage polarization:
A novel target and strategy for pathological scarring. Tissue Eng
Regener Med. 21:1109–1124. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bill R, Wirapati P, Messemaker M, Roh W,
Zitti B, Duval F, Kiss M, Park JC, Saal TM, Hoelzl J, et al:
CXCL9:SPP1 macrophage polarity identifies a network of cellular
programs that control human cancers. Science. 381:515–524. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Han H, Ge X, Komakula SSB, Desert R, Das
S, Song Z, Chen W, Athavale D, Gaskell H, Lantvit D, et al:
Macrophage-derived osteopontin (SPP1) protects from nonalcoholic
steatohepatitis. Gastroenterology. 165:201–217. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fan G, Xie T, Li L, Tang L, Han X and Shi
Y: Single-cell and spatial analyses revealed the co-location of
cancer stem cells and SPP1+ macrophage in hypoxic region that
determines the poor prognosis in hepatocellular carcinoma. NPJ
Precis Oncol. 8:752024. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu X, Qin J, Nie J, Gao R, Hu S, Sun H,
Wang S and Pan Y: ANGPTL2+cancer-associated fibroblasts and
SPP1+macrophages are metastasis accelerators of colorectal cancer.
Front Immunol. 14:11852082023. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jie H, Wang B, Zhang J, Wang X, Song X,
Yang F, Fu C, Dong B and Yan F: Uncovering SPP1+ macrophage,
neutrophils and their related diagnostic biomarkers in intracranial
aneurysm and subarachnoid hemorrhage. J Inflammation Res.
17:8569–8587. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen H, Jin Z, Peng Y, Li Y, Li Z, Zhang
X, Xie Y, Dong J, Ma L and Ji Z: Malevolent alliance of MYBL2hi
cancer stem cell and SPP1+ macrophage confers resistance to
neoadjuvant immunotherapy in bladder cancer. J ImmunoTher Cancer.
13:e0113192025. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhao Y, Huang Z, Gao L, Ma H and Chang R:
Osteopontin/SPP1: A potential mediator between immune cells and
vascular calcification. Front Immunol. 15:13955962024. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ozato Y, Kojima Y, Kobayashi Y, Hisamatsu
Y, Toshima T, Yonemura Y, Masuda T, Kagawa K, Goto Y, Utou M, et
al: Spatial and single-cell transcriptomics decipher the cellular
environment containing HLA-G+ cancer cells and SPP1+ macrophages in
colorectal cancer. Cell Rep. 42:1119292023. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang H, Liang Y, Liu Z, Zhang R, Chao J,
Wang M, Liu M, Qiao L, Xuan Z, Zhao H and Lu L: POSTN+
cancer-associated fibroblasts determine the efficacy of
immunotherapy in hepatocellular carcinoma. J ImmunoTher Cancer.
12:e0087212024. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen C, Guo Q, Liu Y, Hou Q, Liao M, Guo
Y, Zang Y, Wang F, Liu H, Luan X, et al: Single-cell and spatial
transcriptomics reveal POSTN+ cancer-associated fibroblasts
correlated with immune suppression and tumour progression in
non-small cell lung cancer. Clin Transl Med. 13:e15152023.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Patel JR, Joel MZ, Lee KK, Kambala A,
Cornman H, Oladipo O, Taylor M, Imo BU, Ma EZ, Manjunath J, et al:
Single-cell RNA sequencing reveals dysregulated POSTN+WNT5A+
fibroblast subclusters in prurigo nodularis. J Invest Dermatol.
144:1568–1578.e5. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu S, Liu M, Zhang M, Ye X, Gu H, Jiang C,
Zhu H, Ye X, Li Q, Huang X and Cao M: The gene expression of CALD1,
CDH2, and POSTN in fibroblast are related to idiopathic pulmonary
fibrosis. Front Immunol. 15:12750642024. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lin SC, Liao YC, Chen PM, Yang YY, Wang
YH, Tung SL, Chuang CM, Sung YW, Jang TH, Chuang SE and Wang LH:
Periostin promotes ovarian cancer metastasis by enhancing M2
macrophages and cancer-associated fibroblasts via integrin-mediated
NF-κB and TGF-β2 signaling. J Biomed Sci. 29:1092022. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jin X, Deng Q, Ye S, Liu S, Fu Y, Liu Y,
Wu G, Ouyang G and Wu T: Cancer-associated fibroblast-derived
periostin promotes papillary thyroid tumor growth through
integrin-FAK-STAT3 signaling. Theranostics. 14:3014–3028. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Komura M, Wang C, Ito S, Kato S, Ueki A,
Ebi M, Ogasawara N, Tsuzuki T, Kasai K, Kasugai K, et al:
Simultaneous expression of CD70 and POSTN in cancer-associated
fibroblasts predicts worse survival of colorectal cancer patients.
Int J Mol Sci. 25:25372024. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ma H, Wang J, Zhao X, Wu T, Huang Z, Chen
D, Liu Y and Ouyang G: Periostin promotes colorectal tumorigenesis
through integrin-FAK-src pathway-mediated YAP/TAZ activation. Cell
Rep. 30:793–806.e6. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hong YK, Hwang DY, Yang CC, Cheng SM, Chen
PC, Aala WJ, I-Chen Harn H, Evans ST, Onoufriadis A, Liu SL, et al:
Profibrotic subsets of SPP1+ macrophages and POSTN+ fibroblasts
contribute to fibrotic scarring in acne keloidalis. J Invest
Dermatol. 144:1491–1504.e10. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ji X, Wu X, Sun W and Zhang H: Fibroblasts
in the tumor microenvironment: heterogeneity and dynamic
interactions in tumor progression revealed by spatial
transcriptomics. Cell Oncol (Dordr). 48:1615–1629. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xu Y, Ying L, Lang JK, Hinz B and Zhao R:
Modeling mechanical activation of macrophages during pulmonary
fibrogenesis for targeted anti-fibrosis therapy. Sci Adv.
10:eadj95592024. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Uhlig M, Billig S, Wienhold J and
Schumacher D: Pro-fibrotic macrophage subtypes: SPP1+ macrophages
as a key player and therapeutic target in cardiac fibrosis? Cells.
14:3452025. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gao Y, Li J, Cheng W, Diao T, Liu H, Bo Y,
Liu C, Zhou W, Chen M, Zhang Y, et al: Cross-tissue human
fibroblast atlas reveals myofibroblast subtypes with distinct roles
in immune modulation. Cancer Cell. 42:1764–1783.e10. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ji Y, Li X, Yao X, Sun J, Yi J, Shen Y,
Chen B and Sun H: Macrophage polarization: Molecular mechanisms,
disease implications, and targeted therapeutic strategies. Front
Immunol. 16:17327182025. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Moretti L, Stalfort J, Barker TH and
Abebayehu D: The interplay of fibroblasts, the extracellular
matrix, and inflammation in scar formation. J Biol Chem.
298:1015302022. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Roman J: Fibroblasts-Warriors at the
intersection of wound healing and disrepair. Biomolecules.
13:9452023. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Froom ZSCS, Callaghan NI and Davenport
Huyer L: Cellular crosstalk in fibrosis: Insights into macrophage
and fibroblast dynamics. J Biol Chem. 301:1102032025. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Feng Y, Li J, Mo X and Ju Q: Macrophages
in acne vulgaris: Mediating phagocytosis, inflammation, scar
formation, and therapeutic implications. Front Immunol.
15:13554552024. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Frech FS, Hernandez L, Urbonas R, Zaken
GA, Dreyfuss I and Nouri K: Hypertrophic scars and keloids:
advances in treatment and review of established therapies. Am J
Clin Dermatol. 24:225–245. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang Z, Zhao F, Xu C, Zhang Q, Ren H,
Huang X, He C, Ma J and Wang Z: Metabolic reprogramming in skin
wound healing. Burns Trauma. 12:tkad0472024. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhao W, Zhang H, Liu R and Cui R: Advances
in immunomodulatory mechanisms of mesenchymal stem cells-derived
exosome on immune cells in scar formation. Int J Nanomed.
18:3643–3662. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bhatt M, Sharma M and Das B: The role of
inflammatory cascade and reactive astrogliosis in glial scar
formation post-spinal cord injury. Cell Mol Neurobiol. 44:782024.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gardeazabal L and Izeta A: Elastin and
collagen fibres in cutaneous wound healing. Exp Dermatol.
33:e150522024. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bharadia SK, Burnett L and Gabriel V:
Hypertrophic scar. Phys Med Rehabil Clin N Am. 34:783–798. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Das RK, Al Kassis S and Thayer WP:
Hypertrophic scarring and scar revision in gender-affirming
mastectomy: A systematic review. Arch Dermatol Res. 316:5072024.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yuan B, Upton Z, Leavesley D, Fan C and
Wang XQ: Vascular and collagen target: A rational approach to
hypertrophic scar management. Adv Wound Care (New Rochelle).
12:38–55. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Shen L, Zhou Y, Gong J, Fan H and Liu L:
The role of macrophages in hypertrophic scarring: Molecular to
therapeutic insights. Front Immunol. 16:15039852025. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Cohen AJ, Nikbakht N and Uitto J: Keloid
disorder: Genetic basis, gene expression profiles, and
immunological modulation of the fibrotic processes in the skin.
Cold Spring Harbor Perspect Biol. 15:a0412452023. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zheng H, Cheng X, Jin L, Shan S, Yang J
and Zhou J: Recent advances in strategies to target the behavior of
macrophages in wound healing. Biomed Pharmacother. 165:1151992023.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Xu Y, Bian Q, Zhang Y, Zhang Y, Li D, Ma
X, Wang R, Hu W, Hu J, Ye Y, et al: Single-dose of integrated
bilayer microneedles for enhanced hypertrophic scar therapy with
rapid anti-inflammatory and sustained inhibition of myofibroblasts.
Biomaterials. 312:1227422025. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hong YK, Chang YH, Lin YC, Chen B, Guevara
BEK and Hsu CK: Inflammation in wound healing and pathological
scarring. Adv Wound Care (New Rochelle). 12:288–300. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Chen Y, Chen K, Zhong S, Wang J, Yu Z, Sun
X, Wang Y, Liu Y and Zhang Z: Transdermal transfersome nanogels
control hypertrophic scar formation via synergy of macrophage
phenotype-switching and anti-fibrosis effect. Adv Sci (Weinh).
11:e23054682024. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Li Z, Zhang L, Wang Y, Zhu Y, Shen H, Yuan
J, Li X, Yu Z and Song B: LA-peptide hydrogel-regulation of
macrophage and fibroblast fates and their crosstalk via attenuating
TGF-β to promote scarless wound healing. Bioact Mater. 47:417–431.
2025.PubMed/NCBI
|
|
57
|
Zhou Y, Hua T, Weng X, Ma D and Li X:
Calcitonin gene-related peptide alleviates hypertrophic scar
formation by inhibiting the inflammation. Arch Dermatol Res.
314:53–60. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Shi M, Zhang L, Bi F and Ma X: Exosomes
derived from M2 macrophages promote fibroblast autophagy to
contribute to hypertrophic scar formation via CXCL2/CXCR7/mTOR
pathway. Hum Exp Toxicol. 43:96032712413033202024. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Fei H, Qian Y, Pan T, Wei Y and Hu Y:
Curcumin alleviates hypertrophic scarring by inhibiting fibroblast
activation and regulating tissue inflammation. J Cosmet Dermatol.
23:227–235. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Yang L, Song Y, Wang T, Cui Z, Wu J, Shi
Y, Yu Z and Song B: Transcription factor c-maf drives macrophages
to promote hypertrophic scar formation. J Cosmet Dermatol.
23:639–647. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Honda A, Koike H, Dohi T, Toyohara E,
Hayakawa S, Tobe K, Manabe I, Ogawa R and Oishi Y: CD206+
macrophages facilitate wound healing through interactions with
gpnmbhi fibroblasts. EMBO Rep. 26:3679–3704. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Shang F, Qu Y, Li Y, Dong L, Liu D, Wang
Z, Li A, Li Y, Zhang D, Ming L and Jin R: Delivered baicalein
immunomodulatory hydrogel with dual properties of pH-responsive and
anti-infection orchestrates pro-regenerative response of
macrophages for enhanced hypertrophic scars therapy. Mater Today,
Bio. 34:1021602025. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Bhattacharyya S, Midwood KS and Varga J:
Tenascin-C in fibrosis in multiple organs: translational
implications. Semin Cell Dev Biol. 128:131–136. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Yan L, Wang J, Cai X, Liou YC, Shen HM,
Hao J, Huang C, Luo G and He W: Macrophage plasticity: Signaling
pathways, tissue repair, and regeneration. MedComm (2020).
5:e6582024. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kumar V and Stewart Iv JH:
Pattern-recognition receptors and immunometabolic reprogramming:
What we know and what to explore. J Innate Immun. 16:295–323. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Wang Z, An J, Zhu D, Chen H, Lin A, Kang
J, Liu W and Kang X: Periostin: An emerging activator of multiple
signaling pathways. J Cell Commun Signal. 16:515–530. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Urciuolo F, Passariello R, Imparato G,
Casale C and Netti PA: Bioengineered wound healing skin models: The
role of immune response and endogenous ECM to fully replicate the
dynamic of scar tissue formation in vitro. Bioengineering (Basel).
9:2332022. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Xiaojie W, Banda J, Qi H, Chang AK, Bwalya
C, Chao L and Li X: Scarless wound healing: Current insights from
the perspectives of TGF-beta, KGF-1, and KGF-2. Cytokine Growth
Factor Rev. 66:26–37. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Irma J, Kartasasmita AS, Kartiwa A, Irfani
I, Rizki SA and Onasis S: From growth factors to structure: PDGF
and TGF-β in granulation tissue formation. A literature review. J
Cell Mol Med. 29:e703742025. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Massagué J and Sheppard D: TGF-β signaling
in health and disease. Cell. 186:4007–4037. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Xin X, Cheng X, Zeng F, Xu Q and Hou L:
The role of TGF-β/SMAD signaling in hepatocellular carcinoma: from
mechanism to therapy and prognosis. Int J Biol Sci. 20:1436–1451.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Lee JH and Massagué J: TGF-β in
developmental and fibrogenic EMTs. Semin Cancer Biol. 86:136–145.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Aashaq S, Batool A, Mir SA, Beigh MA,
Andrabi KI and Shah ZA: TGF-β signaling: A recap of
SMAD-independent and SMAD-dependent pathways. J Cell Physiol.
237:59–85. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Yu XY, Sun Q, Zhang YM, Zou L and Zhao YY:
TGF-β/smad signaling pathway in tubulointerstitial fibrosis. Front
Pharmacol. 13:8605882022. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Li J, Zou Y, Kantapan J, Su H, Wang L and
Dechsupa N: TGF-β/smad signaling in chronic kidney disease:
Exploring post-translational regulatory perspectives (Review). Mol
Med Rep. 30:1432024. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Bakalenko N, Kuznetsova E and Malashicheva
A: The complex interplay of TGF-β and notch signaling in the
pathogenesis of fibrosis. Int J Mol Sci. 25:108032024. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Hayashi H, Abdollah S, Qiu Y, Cai J, Xu
YY, Grinnell BW, Richardson MA, Topper JN, Gimbrone MA Jr, Wrana JL
and Falb D: The MAD-related protein Smad7 associates with the
TGFbeta receptor and functions as an antagonist of TGFbeta
signaling. Cell. 89:1165–1173. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Zhu H, Kavsak P, Abdollah S, Wrana JL and
Thomsen GH: A SMAD ubiquitin ligase targets the BMP pathway and
affects embryonic pattern formation. Nature. 400:687–693. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Chen F, Lyu L, Xing C, Chen Y, Hu S, Wang
M and Ai Z: The pivotal role of TGF-beta/Smad pathway in fibrosis
pathogenesis and treatment. Front Oncol. 15:16491792025. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Feng F, Liu M, Pan L, Wu J, Wang C, Yang
L, Liu W, Xu W and Lei M: Biomechanical regulatory factors and
therapeutic targets in keloid fibrosis. Front Pharmacol.
13:9062122022. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Wang Q, Xiong F, Wu G, Wang D, Liu W, Chen
J, Qi Y, Wang B and Chen Y: SMAD proteins in TGF-β signalling
pathway in cancer: Regulatory mechanisms and clinical applications.
Diagnostics (Basel). 13:27692023. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
He H, Wang L, Xian B and Xia Y: Regulatory
roles of E3 ubiquitin ligases and deubiquitinases in bone.
Biomolecules. 15:6792025. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Souza-Costa LP, Andrade-Chaves JT, Andrade
JM, Costa VV and Franco LH: Uncovering new insights into the role
of the ubiquitin ligase Smurf1 on the regulation of innate immune
signaling and resistance to infection. Front Immunol.
14:11857412023. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Miyazawa K, Itoh Y, Fu H and Miyazono K:
Receptor-activated transcription factors and beyond: multiple modes
of Smad2/3-dependent transmission of TGF-β signaling. J Biol Chem.
300:1072562024. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Gu YY, Liu XS and Lan HY: Therapeutic
potential for renal fibrosis by targeting Smad3-dependent noncoding
RNAs. Mol Ther. 32:313–324. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Ke Y and Wang XJ: TGFβ signaling in
photoaging and UV-induced skin cancer. J Invest Dermatol.
141:1104–1110. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Laudisi F, Stolfi C, Monteleone I and
Monteleone G: TGF-β1 signaling and Smad7 control T-cell responses
in health and immune-mediated disorders. Eur J Immunol.
53:e23504602023. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Martin L, Gabbiani G and De Meyer GRY:
SMAD7: Riding on fibrosis-limiting routes and beyond. EMBO Mol Med.
17:2181–2190. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Deng CC, Hu YF, Zhu DH, Cheng Q, Gu JJ,
Feng QL, Zhang LX, Xu YP, Wang D, Rong Z and Yang B: Single-cell
RNA-seq reveals fibroblast heterogeneity and increased mesenchymal
fibroblasts in human fibrotic skin diseases. Nat Commun.
12:37092021. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Oka T, Xu J, Kaiser RA, Melendez J,
Hambleton M, Sargent MA, Lorts A, Brunskill EW, Dorn GW II, Conway
SJ, et al: Genetic manipulation of periostin expression reveals a
role in cardiac hypertrophy and ventricular remodeling. Circ Res.
101:313–321. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Kanisicak O, Khalil H, Ivey MJ, Karch J,
Maliken BD, Correll RN, Brody MJ, J Lin SC, Aronow BJ, Tallquist MD
and Molkentin JD: Genetic lineage tracing defines myofibroblast
origin and function in the injured heart. Nat Commun. 7:122602016.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Kaur H, Takefuji M, Ngai CY, Carvalho J,
Bayer J, Wietelmann A, Poetsch A, Hoelper S, Conway SJ, Möllmann H,
et al: Targeted ablation of periostin-expressing activated
fibroblasts prevents adverse cardiac remodeling in mice. Circ Res.
118:1906–1917. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Snider JC, Riley LA, Mallory NT, Bersi MR,
Umbarkar P, Gautam R, Zhang Q, Mahadevan-Jansen A, Hatzopoulos AK,
Maroteaux L, et al: Targeting 5-HT2B receptor signaling prevents
border zone expansion and improves microstructural remodeling after
myocardial infarction. Circulation. 143:1317–1330. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Crawford J, Nygard K, Gan BS and O'Gorman
DB: Periostin induces fibroblast proliferation and myofibroblast
persistence in hypertrophic scarring. Exp Dermatol. 24:121–126.
2015. View Article : Google Scholar
|
|
95
|
Ackerman JE, Muscat SN, Adjei-Sowah E,
Korcari A, Nichols AEC, Buckley MR and Loiselle AE: Identification
of periostin as a critical niche for myofibroblast dynamics and
fibrosis during tendon healing. Matrix Biol. 125:59–72. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Xu H, Wang Z, Yang H, Zhu J and Hu Z:
Bioinformatics analysis and identification of dysregulated POSTN in
the pathogenesis of keloid. Int Wound J. 20:1701–1711. 2023.
View Article : Google Scholar
|
|
97
|
Xu L, Li T, Cao Y, He Y, Shao Z, Liu S,
Wang B, Su A, Tian H, Li Y, et al: PIEZO1 mediates periostin+
myofibroblast activation and pulmonary fibrosis in mice. J Clin
Invest. 135:e1841582025. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Zeng X, Li J, Wang J, Zhang J, Wang Y,
Wang Y, Wang Y, Tian L and Zhu Z: Matrix stiffness promotes
DRP1-mediated myofibroblast senescence to drive silica-induced
pulmonary fibrosis. Aging Cell. 24:e702752025. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Tian J, Yang J, Fu W and Cheng B:
Platelet-rich plasma (PRP) bidirectionally modulates scar formation
via the Piezo1-YAP/TAZ axis: A novel mechanotransduction
hypothesis. Front Cell Dev Biol. 14:17342662026. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Akita S, Ikehara S, Kiuchi M, Kokubo K,
Azuma K, Ohki S, Matsuyama H, Kadarman JT, Hosokawa Y, Akimoto Y,
et al: Increased expression of the PIEZO2 mechanoreceptor in
fibroblasts and endothelial cells within the lymphatic and vascular
vessels of keloids. J Pathol. 267:105–119. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Jiang B, Zhuo F, Li X, Zhang K, Gu J, Wu
J, Zhong W, Zou Y, Yu B and Huang C: Regulatory role of POSTN in
keloid pathogenesis. Mol Med Rep. 32:3372025. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Maeda D, Kubo T, Kiya K, Kawai K,
Matsuzaki S, Kobayashi D, Fujiwara T, Katayama T and Hosokawa K:
Periostin is induced by IL-4/IL-13 in dermal fibroblasts and
promotes RhoA/ROCK pathway-mediated TGF-β1 secretion in abnormal
scar formation. J Plast Surg Hand Surg. 53:288–294. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Zhang Z, Nie F, Chen X, Qin Z, Kang C,
Chen B, Ma J, Pan B and Ma Y: Upregulated periostin promotes
angiogenesis in keloids through activation of the ERK 1/2 and focal
adhesion kinase pathways, as well as the upregulated expression of
VEGF and angiopoietin-1. Mol Med Rep. 11:857–864. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Ellison JA, Barone FC and Feuerstein GZ:
Matrix remodeling after stroke. De novo expression of matrix
proteins and integrin receptors. Ann N Y Acad Sci. 890:204–222.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Ellison JA, Velier JJ, Spera P, Jonak ZL,
Wang X, Barone FC and Feuerstein GZ: Osteopontin and its integrin
receptor alpha(v)beta3 are upregulated during formation of the
glial scar after focal stroke. Stroke. 29:1698–1707. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Bevan L, Lim ZW, Venkatesh B, Riley PR,
Martin P and Richardson RJ: Specific macrophage populations promote
both cardiac scar deposition and subsequent resolution in adult
zebrafish. Cardiovasc Res. 116:1357–1371. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Gliem M, Krammes K, Liaw L, van Rooijen N,
Hartung HP and Jander S: Macrophage-derived osteopontin induces
reactive astrocyte polarization and promotes re-establishment of
the blood brain barrier after ischemic stroke. Glia. 63:2198–2207.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Rotem I, Konfino T, Caller T, Schary Y,
Shaihov-Teper O, Palevski D, Lewis N, Lendengolts D, Naftali-Shani
N and Leor J: Osteopontin promotes infarct repair. Basic Res
Cardiol. 117:512022. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Fabre T, Barron AMS, Christensen SM, Asano
S, Bound K, Lech MP, Wadsworth MH II, Chen X, Wang C, Wang J, et
al: Identification of a broadly fibrogenic macrophage subset
induced by type 3 inflammation. Sci Immunol. 8:eadd89452023.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Dong E, Zhou Z, Chen T, Zhang B, Yin Y, Wu
X, Li X, Zhao J, He Y, Yang J, et al: Single-cell sequencing
uncovers disrupted stromal-macrophage communication as a driver of
intrauterine adhesion progression. Commun Biol. 8:11942025.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Coulis G, Jaime D, Guerrero-Juarez C,
Kastenschmidt JM, Farahat PK, Nguyen Q, Pervolarakis N, McLinden K,
Thurlow L, Movahedi S, et al: Single-cell and spatial
transcriptomics identify a macrophage population associated with
skeletal muscle fibrosis. Sci Adv. 9:eadd99842023. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Ramachandran P, Dobie R, Wilson-Kanamori
JR, Dora EF, Henderson BEP, Luu NT, Portman JR, Matchett KP, Brice
M, Marwick JA, et al: Resolving the fibrotic niche of human liver
cirrhosis at single-cell level. Nature. 575:512–518. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Morse C, Tabib T, Sembrat J, Buschur KL,
Bittar HT, Valenzi E, Jiang Y, Kass DJ, Gibson K, Chen W, et al:
Proliferating SPP1/MERTK-expressing macrophages in idiopathic
pulmonary fibrosis. Eur Respir J. 54:18024412019. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Zheng L, Li G, Cao J, Zhao Z, Wang H, Dong
Q and Hou Z: SPP1+macrophages promote fibroblast-to-myofibroblast
transformation during hypoxia in deep fascia of acute compartment
syndrome. Front Immunol. 16:15889262025. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Yu M and Fu J: The pro-fibrogenic role of
SPP1+ macrophages in medical implant fibrosis: Mechanisms and
therapeutic opportunities. Front Immunol. 17:17490982026.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Fu M, Shu S, Peng Z, Liu X, Chen X, Zeng
Z, Yang Y, Cui H, Zhao R, Wang X, et al: Single-Cell RNA sequencing
of coronary perivascular adipose tissue from end-stage heart
failure patients identifies SPP1+ macrophage
subpopulation as a target for alleviating fibrosis. Arterioscler
Thromb Vasc Biol. 43:2143–2164. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Pardo A, Gibson K, Cisneros J, Richards
TJ, Yang Y, Becerril C, Yousem S, Herrera I, Ruiz V, Selman M and
Kaminski N: Up-regulation and profibrotic role of osteopontin in
human idiopathic pulmonary fibrosis. PLoS Med. 2:e2512005.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Sun K, Yan C, Ji C, Han L, Sun J, Kang Z,
Sun J and Shi J: Integrin-mediated SPP1 signaling from macrophages
orchestrates extracellular matrix remodeling and chondrogenesis in
heterotopic ossification. J Transl Med. 24:1992025. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Tu Y, Xiong Y, Liu O and Peng Y: Secreted
phosphoprotein 1 promotes the activation of keloid fibroblasts via
the CD44/TGF-β1/Smad pathway. Exp Cell Res. 450:1146582025.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Cheng Y, Chen X, Feng L, Yang Z, Xiao L,
Xiang B, Wang X, Liu D, Lin P, Shi J, et al: Stromal architecture
and fibroblast subpopulations with opposing effects on outcomes in
hepatocellular carcinoma. Cell Discov. 11:12025. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
King EM, Zhao Y, Moore CM, Steinhart B,
Anderson KC, Vestal B, Moore PK, McManus SA, Evans CM, Mould KJ, et
al: Gpnmb and Spp1 mark a conserved macrophage injury response
masking fibrosis-specific programming in the lung. JCI Insight.
9:e1827002024. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
She Z, Chen H, Lin X, Li C and Su J: POSTN
regulates fibroblast proliferation and migration in laryngotracheal
stenosis through the TGF-β/RHOA pathway. Laryngoscope.
134:4078–4087. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Kramann R, Machado F, Wu H, Kusaba T,
Hoeft K, Schneider RK and Humphreys BD: Parabiosis and single-cell
RNA sequencing reveal a limited contribution of monocytes to
myofibroblasts in kidney fibrosis. JCI Insight. 3:e995612018.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Bao S, Ouyang G, Bai X, Huang Z, Ma C, Liu
M, Shao R, Anderson RM, Rich JN and Wang XF: Periostin potently
promotes metastatic growth of colon cancer by augmenting cell
survival via the Akt/PKB pathway. Cancer Cell. 5:329–339. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Lin SL, Kisseleva T, Brenner DA and
Duffield JS: Pericytes and perivascular fibroblasts are the primary
source of collagen-producing cells in obstructive fibrosis of the
kidney. Am J Pathol. 173:1617–1627. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Coombes JD, Swiderska-Syn M, Dollé L, Reid
D, Eksteen B, Claridge L, Briones-Orta MA, Shetty S, Oo YH, Riva A,
et al: Osteopontin neutralisation abrogates the liver progenitor
cell response and fibrogenesis in mice. Gut. 64:1120–1131. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Amer J, Salhab A, Hussini E, Shweiki R,
Zahran I and Far M: Osteopontin neutralization increases vitamin D
receptors on NKT cells and ameliorates liver fibrosis by promoting
their activity. Front Pharmacol. 15:14842782024. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Farrokhi V, Chabot JR, Neubert H and Yang
Z: Assessing the feasibility of neutralizing osteopontin with
various therapeutic antibody modalities. Sci Rep. 8:77812018.
View Article : Google Scholar : PubMed/NCBI
|
|
129
|
De Muynck K, Heyerick L, De Ponti FF,
Vanderborght B, Meese T, Van Campenhout S, Baudonck L, Gijbels E,
Rodrigues PM, Banales JM, et al: Osteopontin characterizes bile
duct-associated macrophages and correlates with liver fibrosis
severity in primary sclerosing cholangitis. Hepatology. 79:269–288.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Odell ID: Cross-tissue organization of
myeloid cells in scleroderma and related fibrotic diseases. Curr
Opin Rheumatol. 36:379–386. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Guo Y and Feng L:
N6-methyladenosine-mediated upregulation of LINC00520 accelerates
breast cancer progression via regulating miR-577/POSTN axis and
downstream ILK/AKT/mTOR signaling pathway. Arch Biochem Biophys.
729:1093812022. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Chen K, Li Z, Zhang M, Wang B, Peng T,
Shen Y, Zhang J, Ye J, Liu Y, Tang D, et al: miR-876 inhibits EMT
and liver fibrosis via POSTN to suppress metastasis in
hepatocellular carcinoma. Biomed Res Int. 2020:19642192020.
View Article : Google Scholar : PubMed/NCBI
|
|
133
|
MacFarlane LA, Gu Y, Casson AG and Murphy
PR: Regulation of fibroblast growth factor-2 by an endogenous
antisense RNA and by argonaute-2. Mol Endocrinol. 24:800–812. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Gao K, Cheng M, Zuo X, Lin J, Hoogewijs K,
Murphy MP, Fu XD and Zhang X: Active RNA interference in
mitochondria. Cell Res. 31:219–228. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Hu Y, Wang X, Ding F, Liu C, Wang S, Feng
T and Meng S: Periostin renders cardiomyocytes vulnerable to acute
myocardial infarction via pro-apoptosis. ESC Heart Fail. 9:977–987.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Yoshinami T, Sanada F, Ando Y, Kitano S,
Ueno T, Nakayama T, Kanemoto Y, Tokui R, Sato Y, Masunaga N, et al:
627TiP PT0101-CT1: A phase I/IIa, first-in-human, open-label,
multicenter trial of anti-periostin antibody (PT0101) in patients
with advanced solid cancers and HER2-negative recurrent or
metastatic breast cancer. Ann Oncol. 36 (Suppl 2):S442–S443. 2025.
View Article : Google Scholar
|
|
137
|
De Oliveira Macena Y, Cezar MEN, Lira CBF,
De Oliveira LBDM, Almeida TN, Costa ADAV, De Araujo BMD, de Almeida
Junior D, Dantas HM, De Mélo EC, et al: The roles of periostin
derived from cancer-associated fibroblasts in tumor progression and
treatment response. Cancer Metastasis Rev. 44:112025. View Article : Google Scholar : PubMed/NCBI
|