Introduction
Cardiovascular diseases (CVDs), including
atherosclerosis, myocardial infarction (MI), heart failure (HF),
myocardial hypertrophy, myocardial fibrosis, arrhythmia and
hypertension, remain the leading cause of global mortality. The
Global Burden of Cardiovascular Diseases Collaboration reported
that an estimated ~19.8 million individuals succumbed to CVDs in
2022 (1). These chronic conditions
arise from multiple risk factors, including diabetes, obesity,
inflammation, elevated blood pressure, poor lifestyle habits and
genetic predisposition (2–5). Achieving improvements in CVD
diagnosis and treatment is hindered by limited understanding of the
underlying pathogenesis, particularly the molecular mechanisms
driving the dysfunction of cardiovascular system cells, such as
smooth muscle cells (SMCs), endothelial cells (ECs),
cardiomyocytes, inflammatory cells and fibroblasts, as well as
abnormalities in signaling transduction and gene expression
(6,7).
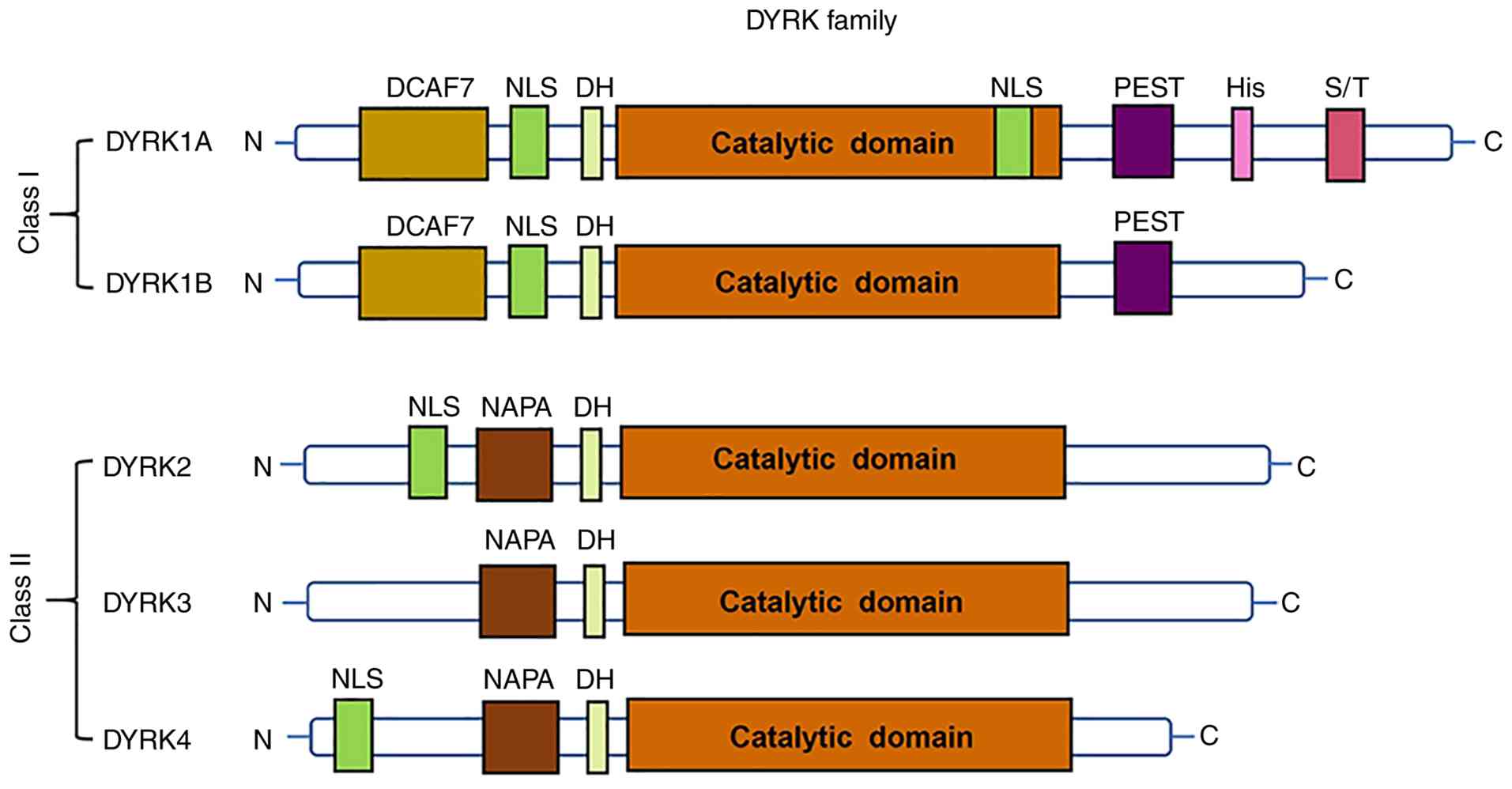
The dual-specificity tyrosine-regulated kinase
(DYRK) family, a highly conserved group of proline-directed kinases
within the cyclin-dependent kinase (CDK), mitogen-activated protein
kinase, glycogen synthase kinase 3 and CDC-like kinase superfamily,
plays important roles in neuronal development, cell cycle
regulation and mRNA splicing (8–12).
DYRK members are classified into two classes based on sequence
homology, as illustrated in Fig.
1. All DYRK proteins contain a conserved catalytic kinase
domain and a DYRK homology (DH) box located upstream of the kinase
domain (13). Among DYRK family
members, DYRK1A is the most extensively studied, whereas DYRK1B,
DYRK2, DYRK3 and DYRK4 have received less attention in
cardiovascular research. DYRK1B primarily regulates skeletal muscle
metabolism and tumor cell proliferation, with limited involvement
in cardiac or vascular pathology (9). DYRK2 functions mainly in DNA damage
repair and apoptosis; however, these roles have not been clearly
associated with major CVD-associated processes, such as
cardiomyocyte proliferation or pulmonary vascular remodeling
(14). By contrast, DYRK1A has
been demonstrated to be relevant to cardiovascular pathology.
Located within the Down syndrome critical region on chromosome 21,
both the upregulated and downregulated expression of DYRK1A have
been associated with multiple human diseases, including cancer,
Alzheimer's disease, diabetes, Down syndrome and CVDs (15–20).
Unlike other DYRK family members, DYRK1A is widely expressed in the
cardiovascular system, and its abnormal expression is consistently
observed in CVDs, highlighting its potential as a cardiovascular
therapeutic target.
A number of studies have demonstrated that DYRK1A is
involved in regulating the onset and progression of CVDs; however,
the underlying mechanisms in specific cardiovascular pathologies
are as yet not fully understood (21–25).
The present review summarizes recent findings regarding DYRK1A in
the cardiovascular system, focusing on its structural
characteristics, regulatory mechanisms and functional roles in
major CVDs. By integrating current evidence, the review aims to
clarify the relevance of DYRK1A to cardiovascular health and
provide insights into potential DYRK1A-targeted therapeutic
strategies.
A central concept addressed in this review is the
context-dependent duality of DYRK1A, whereby it exerts opposing
regulatory effects depending on disease context, cell type and
disease stage. For example, DYRK1A suppresses pathological
processes in cardiomyocyte hypertrophy but promotes disease
progression in pulmonary arterial hypertension (PAH). In MI, DYRK1A
appears to act as a detrimental regulator during the acute phase
but may exhibit protective effects during chronic cardiac
remodeling. This context-dependent behavior likely arises from its
ability to interact with diverse downstream signaling pathways,
respond to cell type-specific microenvironments and dynamically
adapt to pathological stimuli. Recognition of this duality is
essential for reconciling conflicting findings regarding DYRK1A
function across different CVDs, and for guiding the development of
precise, disease-specific therapeutic strategies targeting
DYRK1A.
Structural characteristics of DYRK1A and
brief regulatory insights
DYRK1A exerts diverse biological functions through
the phosphorylation of >30 substrates involved in transcription,
cell cycle progression, apoptosis, DNA damage repair and neuronal
migration (26–31). As a dual-specificity kinase, it
undergoes autophosphorylation, a property essential for its
maturation and subsequent phosphorylation of downstream substrates
to perform its regulatory functions (19,32,33).
The activity and functional specificity of DYRK1A
are largely determined by its structural characteristics, which
including several distinct structural regions. Consistent with all
members of the DYRK family, DYRK1A contains a conserved catalytic
kinase domain, with a DH box located immediately upstream. The
catalytic domain includes Lys188, a critical residue that serves as
the ATP-binding site and is indispensable for kinase activity
(34). DYRK1A also contains two
nuclear localization signals (NLSs), one upstream of the kinase
domain and the other within it. These NLSs facilitate the nuclear
translocation of DYRK1A, allowing it to phosphorylate nuclear
substrates, such as nuclear factor of activated T cells (NFAT), a
key regulator of myocardial hypertrophy, as well as transcription
factors involved in cell cycle regulation. This nuclear
localization is particularly important in cardiomyocytes, where
DYRK1A directly modulates cell cycle progression and hypertrophic
signaling (35).
In addition, DYRK1A contains a proline-, glutamic
acid-, serine- and threonine-rich (PEST) domain that mediates
protein degradation through the ubiquitin-proteasome system,
enabling the dynamic regulation of its own protein levels in
response to pathological stimuli, including myocardial ischemia and
hypoxic stress in pulmonary vascular cells (36,37).
The N-terminus of DYRK1A contains a 13-consecutive
histidine repeat region and serine/threonine-rich sequences. It has
been demonstrated that the histidine-rich region promotes phase
separation and the formation of liquid-like DYRK1A condensates
(38). This process may increase
the local concentration of DYRK1A and its substrates, thereby
promoting the phosphorylation of targets such as D-cyclins, which
regulate cardiomyocyte proliferation, and STAT3, which contributes
to pulmonary arterial SMC hyperproliferation.
The phase separation capacity of DYRK1A likely
contributes to its tissue-specific regulatory effects. In
cardiomyocytes, phase separation-mediated substrate phosphorylation
contributes to the inhibition of cell cycle progression, whereas in
pulmonary vascular cells, it promotes pathological proliferation
(39). These findings directly
associate the structural properties of DYRK1A with its
tissue-specific functional roles in cardiovascular pathologies.
The core enzymatic activation of DYRK1A is driven by
the intramolecular cis-autophosphorylation of a conserved tyrosine
residue, Tyr321, within the activation loop (A-loop) of its
catalytic kinase domain. This process occurs co-translationally
during ribosomal protein synthesis and folding, prior to the
complete release of the nascent DYRK1A polypeptide (40).
Under physiological conditions, constitutively
active DYRK1A is subjected to reversible inactivation and
functional silencing through several complementary mechanisms,
including reversible dephosphorylation of the A-loop,
post-translational modification-mediated subcellular sequestration
and PEST domain-dependent proteolytic degradation (41–43).
Collectively, these mechanisms form a tightly regulated network
that controls the spatiotemporal activity of DYRK1A in the
cardiovascular system.
While cis-autophosphorylation ensures the
constitutive enzymatic competence of DYRK1A, reversible
inactivation mechanisms including dephosphorylation, subcellular
sequestration and proteolytic degradation regulate its functional
activity. This is critical for the context-dependent regulatory
roles of DYRK1A in cardiovascular pathologies such as cardiomyocyte
hypertrophy and PAH.
DYRK1A in MI
MI is a life-threatening coronary disorder
characterized by sudden and prolonged ischemia of myocardial
tissue, leading to irreversible cardiomyocyte death (44,45).
The primary cause of MI is the rupture or erosion of
atherosclerotic plaques within the coronary arteries, which
triggers thrombus formation and the subsequent occlusion of blood
flow (46). This interruption of
perfusion initiates a cascade of pathological events, including the
release of pro-inflammatory cytokines, oxidative stress and
mitochondrial dysfunction (47).
Clinically, MI management prioritizes rapid reperfusion to restore
coronary blood flow, while complementary pharmacological therapies
are used to reduce myocardial oxygen demand (48). Despite advances in these
interventions, MI remains a major global cause of morbidity and
mortality. Therefore, the identification of molecular mechanisms
and potential therapeutic targets remains essential for the
development of improved treatment strategies.
A major limitation in post-MI cardiac recovery is
the restricted regenerative capacity of the adult mammalian
myocardium. Unlike lower vertebrates and neonatal mammals, adult
mammals cannot effectively replenish the cardiomyocytes lost
following MI-induced injury (49,50).
Persistent cardiomyocyte loss promotes progressive ventricular
remodeling and HF, significantly increasing mortality risk
(51). Notably, increased
cardiomyocyte proliferation has been found to be associated with
improved left ventricular function after MI, suggesting that the
stimulation of endogenous cardiomyocyte cell cycle activity
represents a potential therapeutic strategy (52–54).
DYRK1A was originally identified to act as a
regulator of tumor cell proliferation through the modulation of
cell cycle-related proteins (55).
However, it has since emerged as an important factor in cardiac
repair following myocardial injury (56,57).
A pivotal study using a mouse ischemia-reperfusion injury model
demonstrated that the pharmacological inhibition or
cardiomyocyte-specific deletion of DYRK1A improves cardiac function
and increases cardiomyocyte cell cycle activity. These findings
establish an association between DYRK1A and post-MI recovery, and
highlight its therapeutic potential (56). Notably, the role of DYRK1A in
post-MI repair is dynamic and phase-dependent, varying between the
acute injury and chronic remodeling phases. This temporal
specificity is critical for optimizing therapeutic benefit while
minimizing adverse effects.
During the acute injury phase, which typically
occurs within the first 72 h after MI, DYRK1A suppresses
cardiomyocyte cell cycle progression through two principal
mechanisms. First, DYRK1A directly phosphorylates core cell cycle
regulators, including cyclin D1 and p27Kip1, thereby inhibiting the
G1/S phase transition. Second, DYRK1A interacts with
chromatin remodeling complexes, such as histone deacetylase 4, to
suppress the transcription of proliferation-promoting genes,
including c-Myc and cyclin E1 (24,51,57,58).
Pharmacological inhibition or cardiomyocyte-specific deletion of
DYRK1A during this phase relieves cell cycle repression, promotes
the proliferation of surviving cardiomyocytes and preserves early
cardiac ejection fraction, demonstrating its therapeutic potential
for acute MI (59).
In the chronic remodeling phase, which typically
occurs ≥2 weeks after MI, DYRK1A participates in fibroblast
activation and extracellular matrix (ECM) regulation. Inhibition of
DYRK1A during this phase promotes transforming growth factor β
(TGF-β)/Smad3 signaling, thereby increasing the differentiation of
cardiac fibroblasts into myofibroblasts and increasing collagen
deposition (57,59,60).
This exacerbates ventricular stiffness and increases the risk of HF
with preserved ejection fraction, indicating that targeting DYRK1A
during the chronic phase may be detrimental and that phase-specific
therapeutic strategies are required.
DYRK1A suppresses cardiomyocyte proliferation
primarily through inhibition of the cell cycle. DYRK1A
phosphorylates WD repeat domain 82 and lysine acetyltransferase 6A,
thereby reducing the establishment of activating histone
modifications, including histone H3 lysine 4 trimethylation and
histone H3 lysine 27 acetylation, at the promoters of cell cycle
regulatory genes (51). In
addition, DYRK1A interacts with D-cyclin family members to reduce
their protein levels. For example, DYRK1A overexpression increases
the phosphorylation of cyclin D2, promoting its proteasomal
degradation (24,61). DYRK1A also induces the
hypophosphorylation of retinoblastoma protein 1 (Rb1), thereby
suppressing the Rb/E2f signaling pathway and the transcription of
E2f target genes. These mechanisms collectively result in
G1 phase arrest and the inhibition of cardiomyocyte cell
cycle progression (24).
DYRK1A in cardiomyocyte hypertrophy
Cardiomyocyte hypertrophy is an adaptive response to
diverse physiological or pathological stimuli, involving the
enlargement of individual cardiomyocytes. Physiological hypertrophy
is a beneficial adaptation to increased cardiac workload, such as
during regular exercise, which enhances cardiac contractile
function without compromising cardiac structure (59,62).
By contrast, pathological hypertrophy is primarily triggered by
chronic stressors, including hypertension, MI or genetic mutations,
leading to the abnormal growth of adult cardiomyocytes, impaired
contractility and pathological cardiac remodeling (63–65).
Pathological cardiomyocyte hypertrophy is closely associated with
reduced cardiac reserve, adverse clinical outcomes and an increased
risk of HF and sudden cardiac death; therefore, it is widely
recognized as a major predictor of cardiovascular morbidity and
mortality (62).
A complex network of molecular signals regulates the
development of cardiomyocyte hypertrophy. As a key component of the
renin-angiotensin-aldosterone system, angiotensin II (Ang II) binds
to the Ang II type 1 receptor, activating downstream pathways,
including mitogen-activated protein kinase, phosphatidylinositol
3-kinase/Akt and calcium signaling pathways (66,67).
These signaling cascades promote the synthesis of
hypertrophy-associated proteins and ultimately increase
cardiomyocyte size (68–70). In addition, catecholamines,
including norepinephrine and epinephrine, interact with
β-adrenergic receptors on cardiomyocytes, thereby activating G
protein-coupled receptor signaling and protein kinase A, which
promote the transcription of pro-hypertrophic genes, and contribute
to cardiomyocyte hypertrophy and subsequent HF (71–73).
Cytokine-mediated signaling pathways, including the TGF-β and tumor
necrosis factor α pathways, also play important roles in the
regulation of hypertrophic responses (74–76).
Epigenetic mechanisms, including DNA methylation, histone
modification and non-coding RNA-mediated regulation, have also
emerged as key modulators of cardiomyocyte hypertrophy, further
expanding understanding of its molecular basis (77,78).
Among the signaling pathways involved in
cardiomyocyte hypertrophy, the calcium/calmodulin-activated
phosphatase calcineurin and its downstream transcriptional
effector, NFAT, represent a central regulatory axis (63,79).
Under pro-hypertrophic conditions, calcineurin activation leads to
NFAT dephosphorylation, enabling NFAT to translocate to the nucleus
and facilitate the transcription of hypertrophy-associated genes
(79). Consistent with this,
genetic inhibition of calcineurin renders the heart unresponsive to
a broad range of hypertrophic stimuli and attenuates pathological
cardiac hypertrophy, highlighting the centrality of the
calcineurin-NFAT axis in this process (80).
DYRK1A functions as a negative regulator of the
calcineurin-NFAT signaling pathway through direct phosphorylation
of the conserved Ser-Pro repeat-3 motif of nuclear NFAT, promoting
its cytoplasmic re-localization and suppressing NFAT-mediated
pro-hypertrophic transcriptional activity (30,59).
However, conflicting findings have been reported regarding the
regulatory role of DYRK1A in pathological hypertrophy. In
vitro experiments using acute stimulation have demonstrated
that DYRK1A overexpression inhibits NFAT activation and attenuates
cardiomyocyte growth (18). By
contrast, transgenic mouse models have indicated that myocardial
DYRK1A overexpression does not suppress hypertrophy induced by
transverse aortic constriction (TAC) or constitutively active
calcineurin (25). These
contrasting observations highlight the complexity of the
DYRK1A-mediated regulation of cardiomyocyte hypertrophy, which may
depend on the duration of pathological stress and the nature of
hypertrophic stimuli.
DYRK1A-mediated acute signaling
inhibition in hypertrophy
During acute hypertrophic stimuli, such as
short-term Ang II exposure, DYRK1A exerts a rapid and direct
inhibitory effect on pro-hypertrophic signaling. By phosphorylating
nuclear NFAT, DYRK1A counteracts calcineurin-mediated NFAT
dephosphorylation, thereby preventing the nuclear retention of NFAT
and reducing the transcription of hypertrophic marker genes,
including atrial natriuretic peptide and brain natriuretic peptide
(18,59). This inhibition limits early
cardiomyocyte enlargement and represents a direct NFAT-dependent
regulatory role of DYRK1A. Such acute signaling inhibition has been
consistently observed in short-term in vitro and in
vivo experimental models.
Limited effect of DYRK1A on long-term
hypertrophic remodeling
By contrast to its acute signaling effects, DYRK1A
exhibits a diminished regulatory role during long-term pathological
remodeling driven by chronic stress, such as sustained pressure
overload induced by TAC. In chronic hypertrophy models, DYRK1A
overexpression fails to prevent pathological ventricular
remodeling, including cardiomyocyte hypertrophy, interstitial
fibrosis and impaired cardiac contractility (25). This reduced efficacy may be
attributed to the activation of DYRK1A-resistant pro-hypertrophic
pathways, which may function independently of the calcineurin-NFAT
axis or involve alternative NFAT isoforms that are not effectively
targeted by DYRK1A-mediated phosphorylation. In addition, chronic
pressure overload may alter the subcellular localization,
post-translational modification or protein-protein interactions of
DYRK1A, thereby disrupting its NFAT-mediated inhibitory function
(18). Collectively, these changes
contribute to the inability of DYRK1A to suppress long-term
hypertrophic remodeling despite sustained myocardial
expression.
To reconcile the differences between acute and
chronic effects, further research is necessary to identify
DYRK1A-resistant signaling pathways involved in chronic hypertrophy
and to determine whether chronic pathological stress induces
adaptive changes in the regulatory function of DYRK1A. Insights
obtained by such studies may aid in clarifying the
context-dependent role of DYRK1A and support the development of
therapeutic strategies that leverage its acute protective effects
while overcoming resistance during long-term pathological
remodeling.
DYRK1A in PAH
PAH is a progressive and heterogeneous disease
defined by a resting mean pulmonary arterial pressure of ≥20 mmHg
(81). Its pathogenesis is
characterized by the pathological remodeling of pulmonary arteries,
driven by abnormal proliferation of pulmonary artery SMCs (PASMCs),
pulmonary EC dysfunction and excessive ECM deposition. These
alterations increase vascular resistance, leading to chronic right
ventricular (RV) pressure overload and eventual RV failure, which
remains the primary cause of mortality in patients with PAH
(82,83).
PAH is classified into heritable, environmental and
comorbidity-associated subtypes. Heritable PAH is most commonly
caused by mutations in bone morphogenetic protein receptor type 2
(BMPR2), which disrupt anti-proliferative BMP signaling (84–86).
Environmental PAH is often triggered by chronic hypoxia, which
induces the phenotypic switching of PASMCs into a
hyperproliferative and apoptosis-resistant state (87–89).
Comorbidity-associated PAH occurs in conditions such as rheumatoid
arthritis, atrial septal defects, portal hypertension, acquired
immunodeficiency syndrome and Down syndrome (90). Notably, Down syndrome, caused by
trisomy 21, confers a significantly increased risk of PAH, largely
due to pulmonary endothelial dysfunction (91).
DYRK1A is a key contributor to PAH pathogenesis,
exerting cell-type-specific pathogenic effects through distinct
signaling pathways in PASMCs and pulmonary ECs. In hypoxia-induced
PAH, DYRK1A expression is upregulated in PASMCs and promotes
hyperproliferation through the STAT3/Pim-1/NFAT pathway. DYRK1A
phosphorylates STAT3, facilitating its nuclear translocation and
promoting the transcription of Pim-1. Pim-1 subsequently stabilizes
NFAT proteins, leading to the upregulated expression of genes
associated with cell cycle progression, including cyclin D1, as
well as anti-apoptotic factors, such as B cell lymphoma 2. These
effects sustain PASMC hyperproliferation and apoptosis resistance,
contributing to pulmonary vascular wall thickening (92).
In Down syndrome-associated PAH, DYRK1A
overexpression driven by trisomy 21 disrupts pulmonary EC function
through the DYRK1A/peroxisome proliferator-activated receptor γ
(PPARG)/early growth response protein 1 (EGR1) pathway. DYRK1A
phosphorylates PPARG, thereby impairing its DNA-binding ability and
downstream gene activation. Reduced PPARG activity leads to
increased EGR1 expression, which promotes the production of
pro-inflammatory cytokines and pro-remodeling proteins. These
molecular events impair endothelial angiogenesis, increase
apoptosis, elevate mitochondrial reactive oxygen species and
compromise endothelial integrity (93).
These cell-type-specific pathway activation effects
are influenced by differential cofactor expression and pathway
dependency. PASMCs exhibit hypoxia-induced upregulation of STAT3
and Pim-1, with limited EGR1 coactivator expression, whereas
pulmonary ECs exhibit high basal PPARG expression and relatively
limited NFAT signaling (92,93).
These differences render each cell type responsive to distinct
DYRK1A-mediated signaling pathways.
Harmine, a natural β-carboline alkaloid that targets
the ATP-binding pocket of DYRK1A, has emerged as a potential
therapeutic candidate for Down syndrome-associated PAH. Preclinical
experiments using isogenic trisomy 21 and corrected disomy 21
induced pluripotent stem cell-derived ECs have shown that harmine
reverses DYRK1A-mediated PPARG suppression, normalizes EGR1 levels
and restores endothelial function (93). However, harmine exhibits off-target
effects, including inhibition of DYRK1B, DYRK2 and CDK5, and may
cause adverse effects such as neurotoxicity and gastrointestinal
disturbances (94). In addition,
the metabolism of harmine via cytochrome P450 family 1 subfamily A
member 2 increases the risk of drug-drug interactions. These
limitations highlight the requirement for structural optimization
to improve DYRK1A selectivity, in addition to the development of
targeted delivery strategies, such as EC-specific nanoparticle
systems.
The cell-type specific role of DYRK1A in PASMCs and
ECs underscores its potential as a therapeutic target for PAH.
However, important gaps remain in understanding its interaction
with key PAH pathways, particularly BMPR2 signaling. BMPR2
mutations are the most common cause of heritable PAH, and BMPR2
loss-of-function produces phenotypes comparable to those associated
with DYRK1A overexpression, including PASMC hyperproliferation and
EC dysfunction (84,95). Future studies should investigate
whether DYRK1A acts upstream of BMPR2, for example through
phosphorylation-mediated inhibition, or whether both pathways
converge on shared downstream effectors, such as NFAT or EGR1.
Clarifying these mechanisms may identify novel regulatory nodes and
facilitate the development of therapeutic strategies targeting both
DYRK1A and BMPR2 pathways.
Although the investigation of DYRK1A in other CVDs,
such as atherosclerosis, may provide additional insights, it is
suggested that priority should be given to resolving key
PAH-specific challenges, such as optimizing DYRK1A inhibitors for
PASMC and EC targeting, and clarifying DYRK1A-BMPR2 crosstalk. Such
a focused approach may accelerate the translation of
DYRK1A-targeted therapies into effective and safe treatments for
PAH.
Future research directions for DYRK1A in
CVD
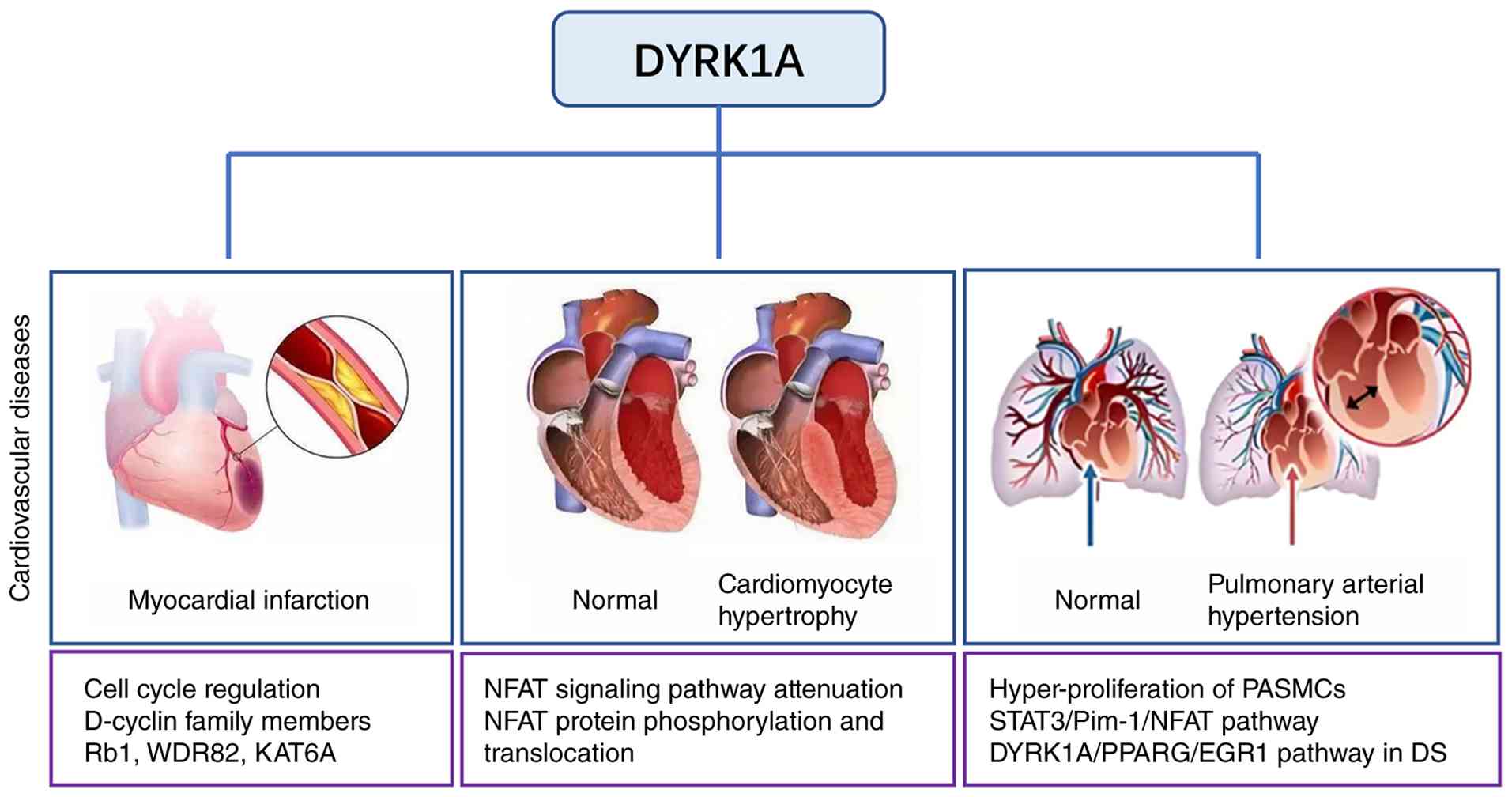
DYRK1A has been shown to function as an important
regulator in numerous CVDs, including MI, cardiomyocyte hypertrophy
and PAH (Fig. 2). In particular,
DYRK1A represses the cell cycle progression of cardiomyocytes and
limits cardiac repair following MI by phosphorylating transcription
factors involved in cell cycle regulation and interacting with
D-cyclins, regulates cardiomyocyte hypertrophy by modulating NFAT
signaling, and contributes to PAH by activating the
STAT3/Pim-1/NFAT pathway. In addition, evidence from harmine
treatment and transgenic mouse models suggest that DYRK1A is a
potential therapeutic target in CVDs. However, several aspects of
DYRK1A-mediated regulation remain unclear. For example, the
functional role of DYRK1A appears to differ between the acute phase
of MI and chronic remodeling, suggesting that stage-specific
therapeutic strategies may be required. Furthermore, the crosstalk
between DYRK1A and other cardiovascular regulators, including
AMP-activated protein kinase (AMPK), glycogen synthase kinase 3β
(GSK3β) and calcineurin, remains insufficiently characterized.
Elucidating these interactions will be critical for understanding
the broader regulatory network of DYRK1A in CVDs.
AMPK, a key energy-sensing kinase that regulates
cardiac metabolism and stress responses, may interact with DYRK1A
to coordinate cardiomyocyte cell cycle progression and metabolic
adaptation during post-MI repair. However, whether these proteins
directly phosphorylate one another or share downstream substrates
remains unclear. Similarly, GSK3β, which regulates cardiomyocyte
proliferation and hypertrophy (95), and calcineurin, an upstream
activator of NFAT signaling, may functionally interact with DYRK1A
to fine-tune pathological responses. Investigation of these
interactions may identify synergistic regulatory targets for
therapeutic intervention. In addition, the role of DYRK1A in other
CVDs, including atherosclerosis and doxorubicin-induced
cardiomyopathy, remains largely unexplored and merits further
investigation.
As maintaining balanced DYRK1A expression is
essential for physiological function, therapeutic strategies may be
required to differ according to disease context. Inhibition of
DYRK1A may protect cardiac function following acute MI or in PAH,
whereas activation of DYRK1A may be beneficial in cardiomyocyte
hypertrophy. From a translational perspective, the optimization of
DYRK1A modulators represents an important research priority.
Current DYRK1A inhibitors, such as harmine, have shown therapeutic
potential in preclinical models of Down syndrome-associated PAH;
however, off-target effects, including inhibition of DYRK1B, DYRK2
and CDK5, as well as adverse effects such as neurotoxicity and
gastrointestinal disturbances, limit their clinical applicability.
The development of harmine derivatives with improved selectivity
for the DYRK1A ATP-binding pocket may enhance therapeutic
specificity. In addition, cell-type-specific delivery systems, such
as pulmonary EC-targeted nanoparticles or cardiomyocyte-specific
lipid carriers, may reduce off-target exposure, particularly in
diseases with cell-type-specific DYRK1A functions.
Although the regulatory role of DYRK1A has been
partially elucidated in MI, PAH and cardiomyocyte hypertrophy, its
functions in the broader context of CVDs remain to be fully
defined. Therefore, further investigation of DYRK1A function and
mechanisms in additional cardiovascular conditions is warranted. To
further clarify DYRK1A roles across disease stages and cell types,
three specific research directions are proposed.
Single-cell transcriptomics
The application of single-cell RNA sequencing to
cardiac or pulmonary tissues from CVD models, such as MI or PAH
models, at different disease stages may reveal cell-type-specific
changes in DYRK1A expression and its downstream targets. This
approach could potentially identify rare cell populations, such as
proliferating cardiomyocytes or activated PASMCs, in which DYRK1A
exerts key regulatory effects, and aid the discovery of
stage-specific transcriptional programs mediated by DYRK1A,
including those associated with acute inflammation or chronic
fibrosis in MI.
Cardiac organoid modeling
Organoid technology, which closely mimics the
structure and function of in vivo organs, represents an
advanced in vitro platform for studying gene function,
modeling disease progression and evaluating therapeutic candidates.
Cardiac organoids can be used to support research advances in the
cardiovascular field (96). Human
induced pluripotent stem cell-derived cardiac organoids, which
recapitulate key components of human heart tissue, including
cardiomyocytes, fibroblasts and ECs, provide a physiologically
relevant system for investigating DYRK1A function. These models
enable the simulation of disease conditions such as hypoxic stress
for PAH and ischemic injury for MI, and facilitate the evaluation
of DYRK1A modulators in a human-relevant context. Such approaches
may reduce reliance on animal models and overcome species-specific
differences in cardiovascular biology.
Cell-type-specific knockout
studies
Generation of conditional knockout mouse models,
such as cardiomyocyte-specific and pulmonary SMC-specific DYRK1A
knockout models, represents an important strategy for dissecting
the cell-autonomous effects of DYRK1A. For example,
cardiomyocyte-specific knockout models may clarify whether the
DYRK1A-mediated regulation of post-MI cardiac repair occurs
directly within cardiomyocytes or indirectly through other cardiac
cell populations, such as fibroblasts. Similarly, pulmonary
EC-specific knockout models may help confirm the pathogenic
contribution of DYRK1A to Down syndrome-associated PAH.
Existing evidence suggests that DYRK1A is a
potential therapeutic target in CVDs and other pathological
conditions. However, translational studies investigating DYRK1A
modulators remain limited. Therefore, the development of highly
efficient and selective DYRK1A modulators, combined with advanced
mechanistic approaches using single-cell sequencing and
organoid-based models, is necessary to accelerate the clinical
translation of DYRK1A-targeted therapies for CVDs.
Conclusions
To facilitate understanding of the context-dependent
roles of DYRK1A and the corresponding therapeutic implications for
different cardiovascular pathologies, a summary of
cell-type-specific regulatory mechanisms, upstream triggers,
downstream signaling pathways and disease-specific therapeutic
strategies in MI, cardiomyocyte hypertrophy and PAH is presented in
Table I (18,24,44,46,48,56,57,59,62,63–66,81,84–89,92,93).
 | Table I.Summary of DYRK1A in major
cardiovascular diseases. |
Table I.
Summary of DYRK1A in major
cardiovascular diseases.
|
| Cardiovascular
disease |
|
|---|
|
|
|
|
|---|
| Feature | Myocardial
infarction | Cardiomyocyte
hypertrophy | Pulmonary arterial
hypertension | (Refs.) |
|---|
| Affected cell
type | Cardiomyocytes,
cardiac fibroblasts | Cardiomyocytes | PASMCs, ECs | (44,62,82) |
| Upstream
triggers | Myocardial
ischemia/reperfusion, hypoxia, inflammation | Chronic pressure
overload, Ang II/AT1R activation, catecholamine/β-AR signaling,
calcineurin activation | PASMCs: Hypoxia,
BMPR2 mutation; ECs: Trisomy 21, BMPR2 loss-of-function | (18,46,48,63,66,84–89,92,93) |
| Downstream
signaling pathways | Rb/E2f cell cycle,
histone modification (H3K4me3, H3K27ac) via WDR82/KAT6A,
TGF-β/Smad3 fibroblast activation | Calcineurin-NFAT
pro-hypertrophic signaling | PASMCs:
STAT3/Pim-1/NFAT; ECs: DYRK1A/ PPARG/EGR1 | (18,24,57,92,93) |
| Role of DYRK1A | Acute phase:
Inhibition of cardiomyocyte proliferation by blocking
G1/S phase transition; repression of
proliferation-promoting gene transcription via epigenetic
modification; chronic phase: Regulation of cardiac fibroblast
activation and extracellular matrix homeostasis to prevent
excessive fibrosis | Phosphorylation of
the Ser-Pro repeat-3 motif of NFAT, which promotes NFAT cytoplasmic
translocation and inhibits NFAT-driven pro-hypertrophic gene
transcription (inhibitory in pathological hypertrophy); partial
resistance under chronic stress | PASMCs: Drives
hyperproliferation and apoptosis resistance via STAT3/Pim-1/NFAT
activation, promoting vascular wall thickening; ECs: Suppresses
PPARG activity, upregulates EGR1, impairs EC homeostasis and
accelerates endothelial dysfunction and vascular remodeling
(pro-pathogenic in both cell types) | (24,59,92,93) |
| Therapeutic
strategies | Phase-specific
inhibition: In acute MI, DYRK1A should be inhibited to activate the
cardiomyocyte cycle and cardiac repair; DYRK1A inhibition may
reduce ischemia-reperfusion injury and infarct size; in chronic
remodeling, inhibition should be avoided to prevent excessive
myocardial fibrosis and ventricular stiffness | Activation/moderate
upregulation: Reinforcement of DYRK1A-mediated NFAT inhibition to
suppress pathological myocardial growth; DYRK1A activation may
prevent progression to heart failure; development of cell-specific
activators to overcome chronic stress resistance | Selective
inhibition: Pan-DYRK1A inhibition to block PASMC
hyperproliferation; EC-targeted DYRK1A inhibition (e.g.,
nanoparticle delivery) to restore PPARG/EGR1 balance; optimization
of inhibitors (e.g., harmine derivatives) to reduce off-target
effects; cell type-specific DYRK1A inhibition may reverse pulmonary
vascular remodeling | (18,59,84,95) |
The present review systematically summarizes the
context-dependent mechanistic roles of DYRK1A in three major CVDs.
In MI, DYRK1A inhibits cardiomyocyte proliferation by suppressing
cell cycle-associated signaling and through epigenetic mechanisms.
In cardiomyocyte hypertrophy, DYRK1A antagonizes pro-hypertrophic
NFAT signaling through the direct phosphorylation of NFAT proteins.
In PAH, DYRK1A promotes pathological vascular remodeling through
cell type-specific pathways, including STAT3/Pim-1/NFAT signaling
in PASMCs and DYRK1A/PPARG/EGR1 signaling in pulmonary ECs.
From a clinical perspective, the therapeutic
modulation of DYRK1A should be tailored to the specific disease
context and stage, as no universal strategy is applicable. DYRK1A
inhibition may be beneficial in acute MI by the promotion of
cardiac repair, and in PAH via the reduction of vascular
remodeling, particularly when combined with cell type-specific
targeting. By contrast, DYRK1A activation may be advantageous in
pathological cardiomyocyte hypertrophy Notably, the non-specific
inhibition of DYRK1A should be avoided in chronic MI and
hypertrophic conditions to prevent adverse outcomes, including
myocardial fibrosis and aggravated cardiac remodeling.
Despite these advances, important gaps remain,
including limited understanding of stage-specific regulatory shifts
of DYRK1A during chronic CVD progression, and of the crosstalk
between DYRK1A and other cardiovascular signaling pathways.
Addressing these gaps and developing highly selective DYRK1A
modulators will be critical for advancing the translational
application of DYRK1A-targeted therapies in CVDs.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant no. 82304578) and the Fujian Provincial
Natural Science Foundation (grant no. 2024J011419).
Availability of data and materials
Not applicable.
Authors' contributions
LY was responsible for writing the original draft,
and for reviewing and editing. QC was also responsible for writing
the original draft. JW was responsible for conceptualization and
supervision. Data authentication is not applicable. All authors
read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mensah GA, Fuster V and Roth GA: A
Heart-Healthy and Stroke-free world using data to inform global
action. J Am Coll Cardiol. 82:2343–2349. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sethwala AM, Goh I and Amerena JV:
Combating inflammation in cardiovascular disease. Heart Lung Circ.
30:197–206. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sebastian SA: Cardiovascular disease risk
communication: Strategies, impact, and future directions. Curr
Probl Cardiol. 49:1024902024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zuercher MD: Association between genetic
admixture and dietary inflammatory index with the risk of
cardiovascular disease and diabetes in postmenopausal hispanic
Women. 2021.UC Davis. Available from:. https://escholarship.org/uc/item/2r21303m
|
|
5
|
Zhao L, Wu S, Yu X, Huang Z, Liu L, Cheng
X, Yuan Z and Li Y: Chemical profiling and multi-dimensional
pharmacokinetic analysis of shengmaiyin oral liquid for cardiac
dysfunction. J Pharm Biomed Anal. 272:1173782026. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Akazawa H: Mechanisms of cardiovascular
homeostasis and pathophysiology-from gene expression, signal
transduction to cellular communication. Circ J. 79:2529–2536. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ghigo A, Laffargue M, Li M and Hirsch E:
PI3K and calcium signaling in cardiovascular disease. Circ Res.
121:282–292. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chu D, Lei L, Gu S, Liu F and Wu F:
Dual-specificity tyrosine phosphorylation-regulated kinase 1A
promotes the inclusion of amyloid precursor protein exon 7. Biochem
Pharmacol. 224:1162332024. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
de Souza MM, Cenci AR, Teixeira KF,
Machado V, Mendes Schuler MCG, Gonçalves AE, Paula Dalmagro A,
André Cazarin C, Gomes Ferreira LL, de Oliveira AS and Andricopulo
AD: DYRK1A inhibitors and perspectives for the treatment of
Alzheimer's Disease. Curr Med Chem. 30:669–688. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Johnson HK, Litovchick LL and Dickinson
AJG: The role of Goldilocks protein kinase DYRK1A in embryonic
development. Dev Biol. 525:216–228. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Soppa U, Schumacher J, Florencio Ortiz V,
Pasqualon T, Tejedor FJ and Becker W: The Down syndrome-related
protein kinase DYRK1A phosphorylates p27(Kip1) and Cyclin D1 and
induces cell cycle exit and neuronal differentiation. Cell Cycle.
13:2084–2100. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tejedor FJ and Hammerle B: MNB/DYRK1A as a
multiple regulator of neuronal development. FEBS J. 278:223–235.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sanchis-Juan A, Hasenahuer MA, Baker JA,
McTague A, Barwick K, Kurian MA, Duarte ST; NIHR BioResource, ;
Carss KJ, Thornton J and Raymond FL: Structural analysis of
pathogenic missense mutations in GABRA2 and identification of a
novel de novo variant in the desensitization gate. Mol Genet
Genomic Med. 8:e11062020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yoshida S and Yoshida K: Multiple
functions of DYRK2 in cancer and tissue development. FEBS Lett.
593:2953–2965. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Laham AJ, El-Awady R, Saber-Ayad M, Wang
N, Yan G, Boudreault J, Ali S and Lebrun JJ: Targeting the DYRK1A
kinase prevents cancer progression and metastasis and promotes
cancer cells response to G1/S targeting chemotherapy drugs. NPJ
Precis Oncol. 8:1282024. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gehlot P, Pathak R, Kumar S, Choudhary NK
and Vyas VK: A review on synthetic inhibitors of dual-specific
tyrosine phosphorylation-regulated kinase 1A (DYRK1A) for the
treatment of Alzheimer's disease (AD). Bioorg Med Chem.
113:1179252024. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rachdi L, Kariyawasam D, Guez F, Aïello V,
Arbonés ML, Janel N, Delabar JM, Polak M and Scharfmann R: Dyrk1a
haploinsufficiency induces diabetes in mice through decreased
pancreatic beta cell mass. Diabetologia. 57:960–969. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kuhn C, Frank D, Will R, Jaschinski C,
Frauen R, Katus HA and Frey N: DYRK1A is a novel negative regulator
of cardiomyocyte hypertrophy. J Biol Chem. 284:17320–17327. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang Y, Fan X and Liu Y, Ye D, Liu C, Yang
H, Su Z, Zhang Y and Liu Y: Function and inhibition of DYRK1A:
Emerging roles of treating multiple human diseases. Biochem
Pharmacol. 212:1155212023. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rozen EJ, Dowell RD and Allen MA: DYRK1A
in blood and immune function: Implications in leukemia,
inflammatory disorders, infection and Down syndrome. Front Cell
Deve Biol. 13:15870892025. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang J, Xu RM, Cao QM, Ma BC, Zhang H and
Hao HP: Mechanism of DYRK1a in myocardial ischemia-reperfusion
injury by regulating ferroptosis of cardiomyocytes. Kaohsiung J Med
Sci. 39:1190–1199. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lana-Elola E, Aoidi R, Llorian M, Gibbins
D, Buechsenschuetz C, Bussi C, Flynn H, Gilmore T, Watson-Scales S,
Haugsten Hansen M, et al: Increased dosage of DYRK1A leads to
congenital heart defects in a mouse model of Down syndrome. Sci
Transl Med. 16:eadd68832024. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cho HJ, Lee JG, Kim JH, Kim SY, Huh YH,
Kim HJ, Lee KS, Yu K and Lee JS: Vascular defects of DYRK1A
knockouts are ameliorated by modulating calcium signaling in
zebrafish. Dis Model Mech. 12:dmm0370442019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hille S, Dierck F, Kühl C, Sosna J,
Adam-Klages S, Adam D, Lüllmann-Rauch R, Frey N and Kuhn C: Dyrk1a
regulates the cardiomyocyte cell cycle via D-cyclin-dependent
Rb/E2f-signalling. Cardiovasc Res. 110:381–394. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Grebe C, Klingebiel TM, Grau SP, Toischer
K, Didié M, Jacobshagen C, Dullin C, Hasenfuss G and Seidler T:
Enhanced expression of DYRK1A in cardiomyocytes inhibits acute NFAT
activation but does not prevent hypertrophy in vivo. Cardiovasc
Res. 90:521–528. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Murakami N, Bolton DC, Kida E, Xie W and
Hwang YW: Phosphorylation by Dyrk1A of Clathrin Coated
Vesicle-Associated Proteins: Identification of the substrate
proteins and the effects of phosphorylation. PLoS One.
7:e348452012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Murakami N, Xie W, Lu RC, Chen-Hwang MC,
Wieraszko A and Hwang YW: Phosphorylation of amphiphysin I by
minibrain kinase/dual-specificity tyrosine
phosphorylation-regulated kinase, a kinase implicated in Down
syndrome. J Biol Chem. 281:23712–23724. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yoshida K: Role for DYRK family kinases on
regulation of apoptosis. Biochem Pharmacol. 76:1389–1394. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Boni J, Rubio-Perez C, Lopez-Bigas N,
Fillat C and de la Luna S: The DYRK family of kinases in cancer:
Molecular functions and therapeutic opportunities. Cancers (Basel).
12:21062020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Arron JR, Winslow MM, Polleri A, Chang CP,
Wu H, Gao X, Neilson JR, Chen L, Heit JJ, Kim SK, et al: NFAT
dysregulation by increased dosage of DSCR1 andDYRK1Aon chromosome
21. Nature. 441:595–600. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Park J, Oh Y, Yoo L, Jung MS, Song WJ, Lee
SH, Seo H and Chung KC: Dyrk1A phosphorylates p53 and inhibits
proliferation of embryonic neuronal cells. J Biol Chem.
285:31895–31906. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Adayev T, Chen-Hwang MC, Murakami N, Lee
E, Bolton DC and Hwang YW: Dual-specificity tyrosine
phosphorylation-regulated kinase 1A does not require tyrosine
phosphorylation for activity in vitro. Biochemistry. 46:7614–7624.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kentrup H, Becker W, Heukelbach J, Wilmes
A, Schürmann A, Huppertz C, Kainulainen H and Joost HG: Dyrk, a
dual specificity protein kinase with unique structural features
whose activity is dependent on tyrosine residues between subdomains
VII and VIII. J Biol Chem. 271:3488–3495. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Soundararajan M, Roos AK, Savitsky P,
Filippakopoulos P, Kettenbach AN, Olsen JV, Gerber SA, Eswaran J,
Knapp S and Elkins JM: Structures of down syndrome kinases, DYRKs,
reveal mechanisms of kinase activation and substrate recognition.
Structure. 21:986–996. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu T, Wang Y, Wang J, Ren C, Chen H and
Zhang J: DYRK1A inhibitors for disease therapy: Current status and
perspectives. Eur J Med Chem. 229:1140622022. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Galceran J, de Graaf K, Tejedor FJ and
Becker W: The MNB/DYRK1A protein kinase: Genetic and biochemical
properties. J Neural Transm Suppl. 67:139–148. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hammerle B, Elizalde C, Galceran J, Becker
W and Tejedor FJ: The MNB/DYRK1A protein kinase: Neurobiological
functions and Down syndrome implications. J Neural Transm Suppl.
67:129–137. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lu H, Yu D, Hansen AS, Ganguly S, Liu R,
Heckert A, Darzacq X and Zhou Q: Phase-separation mechanism for
C-terminal hyperphosphorylation of RNA polymerase II. Nature.
558:318–323. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cai Z, Mei S, Zhou L, Ma X, Wuyun Q, Yan J
and Ding H: Liquid-Liquid phase separation sheds new light upon
cardiovascular diseases. Int J Mol Sci. 24:154182023. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Quinones-Lombrana A and Blanco JG:
Comparative analysis of the DYRK1A-SRSF6-TNNT2 pathway in
myocardial tissue from individuals with and without Down syndrome.
Exp Mol Pathol. 110:1042682019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Marada A, Walter C, Suhm T, Shankar S,
Nandy A, Brummer T, Dhaouadi I, Vögtle FN and Meisinger C: DYRK1A
signalling synchronizes the mitochondrial import pathways for
metabolic rewiring. Nat Commun. 15:52652024. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lee YH, Im E, Hyun M, Park J and Chung KC:
Protein phosphatase PPM1B inhibits DYRK1A kinase through
dephosphorylation of pS258 and reduces toxic tau aggregation. J
Biol Chem. 296:1002452021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhang P, Zhang Z, Fu Y, Zhang Y, Washburn
MP, Florens L, Wu M, Huang C, Hou Z and Mohan M: K63-linked
ubiquitination of DYRK1A by TRAF2 alleviates Sprouty 2-mediated
degradation of EGFR. Cell Death Dis. 12:6082021. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ramanathan N, Babu HM, Nair A, Mohan D,
Rob MA, Dey RK, Jain R, Nallathambi N, Muthiah A, Lohakare T and
Rathi T: Clinical and coronary angiographic insights Into acute
ST-Elevation myocardial infarction in young patients: A study from
a tertiary care cardiac ICU. Cureus. 16:e684842024.PubMed/NCBI
|
|
45
|
Yeh RW, Sidney S, Chandra M, Sorel M,
Selby JV and Go AS: Population trends in the incidence and outcomes
of acute myocardial infarction. New Engl J Med. 362:2155–2165.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mlynarska E, Czarnik W, Fularski P, Hajdys
J, Majchrowicz G, Stabrawa M, Rysz J and Franczyk B: From
atherosclerotic plaque to myocardial Infarction-the leading cause
of coronary artery occlusion. Int J Mol Sci. 25:72952024.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wang KL, Balmforth C, Meah MN, Daghem M,
Moss AJ, Tzolos E, Kwiecinski J, Molek-Dziadosz P, Craig N, Bularga
A, et al: Coronary atherosclerotic plaque activity and risk of
myocardial infarction. J Am Coll Cardiol. 83:2135–2144. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Liu T, Hao Y, Zhang Z, Zhou H, Peng S,
Zhang D, Li K, Chen Y and Chen M: Advanced cardiac patches for the
treatment of myocardial infarction. Circulation. 149:2002–2020.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Porrello ER, Mahmoud AI, Simpson E, Hill
JA, Richardson JA, Olson EN and Sadek HA: Transient regenerative
potential of the neonatal mouse heart. Science. 331:1078–1080.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Jopling C, Sune G, Faucherre A, Fabregat C
and Belmonte JCI: Hypoxia induces myocardial regeneration in
zebrafish. Circulation. 126:3017–3027. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Bei Y, Wang H and Xiao J: DYRK1A: A
promising protein kinase target for cardiomyocyte cycling and
cardiac repair through epigenetic modifications. eBioMedicine.
82:1041682022. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Heallen T, Zhang M, Wang J,
Bonilla-Claudio M, Klysik E, Johnson RL and Martin JF: Hippo
pathway inhibits wnt signaling to restrain cardiomyocyte
proliferation and heart size. Science. 332:458–461. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Leach JP, Heallen T, Zhang M, Rahmani M,
Morikawa Y, Hill MC, Segura A, Willerson JT and Martin JF: Hippo
pathway deficiency reverses systolic heart failure after
infarction. Nature. 550:260–264. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Mohamed TM, Stone NR, Berry EC, Radzinsky
E, Huang Y, Pratt K, Ang YS, Yu P, Wang H, Tang S, et al: Chemical
enhancement of in vitro and in vivo direct cardiac reprogramming.
Circulation. 135:978–995. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Fernandez-Martinez P, Zahonero C and
Sanchez-Gomez P: DYRK1A: The double-edged kinase as a protagonist
in cell growth and tumorigenesis. Mol Cell Oncol. 2:e9700482015.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Young A, Bradley LA, Farrar E, Bilcheck
HO, Tkachenko S, Saucerman JJ, Bekiranov S and Wolf MJ: Inhibition
of DYRK1a enhances cardiomyocyte cycling after myocardial
infarction. Circ Res. 130:1345–1361. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lan C, Chen C, Qu S, Cao N, Luo H, Yu C,
Wang N, Xue Y, Xia X, Fan C, et al: Inhibition of DYRK1A, via
histone modification, promotes cardiomyocyte cell cycle activation
and cardiac repair after myocardial infarction. EBioMedicine.
82:1041392022. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Murillo BC, Young A, Wintruba KL, Eichert
AJ, Siejda K, Hoenig D, Bradley LA, Harris BN, Zhao C, Wu M, et al:
Network modeling predicts how DYRK1A inhibition promotes
cardiomyocyte cycling after Ischemic/Reperfusion Injury. bioRxiv.
Aug 25–2025.(Epub ahead of print). doi:
10.1101/2025.08.19.671147.
|
|
59
|
Gwack Y, Sharma S, Nardone J, Tanasa B,
Iuga A, Srikanth S, Okamura H, Bolton D, Feske S, Hogan PG and Rao
A: A genome-wide drosophila RNAi screen identifies DYRK-family
kinases as regulators of NFAT. Nature. 441:646–650. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Cao Y, Qian R, Yao R, Zheng Q, Yang C,
Yang X, Ji S, Zhang L, Zhan S, Wang Y, et al: DYRK1A-TGF-β
signaling axis determines sensitivity to OXPHOS inhibition in
hepatocellular carcinoma. Dev Cell. 60:1483–1497.e7. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Thompson BJ, Bhansali R, Diebold L, Cook
DE, Stolzenburg L, Casagrande AS, Besson T, Leblond B, Désiré L,
Malinge S and Crispino JD: DYRK1A controls the transition from
proliferation to quiescence during lymphoid development by
destabilizing Cyclin D3. J Exp Med. 212:723–740. 2015. View Article : Google Scholar
|
|
62
|
Nakamura M and Sadoshima J: Mechanisms of
physiological and pathological cardiac hypertrophy. Nat Rev
Cardiol. 15:387–407. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Frey N and Olson EN: Cardiac hypertrophy:
The good, the bad, and the ugly. Annu Rev Physiol. 65:45–79. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Frey N, Katus HA, Olson EN and Hill JA:
Hypertrophy of the heart-A new therapeutic target? Circulation.
109:1580–1589. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Hill JA and Olson EN: Cardiac plasticity.
N Engl J Med. 358:1370–1380. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Diniz GP, Carneiro-Ramos MS and
Barreto-Chaves ML: Angiotensin type 1 receptor mediates thyroid
hormone-induced cardiomyocyte hypertrophy through the
Akt/GSK-3beta/mTOR signaling pathway. Basic Res Cardiol.
104:653–667. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Bhullar SK and Dhalla NS: Angiotensin
II–Induced signal transduction mechanisms for cardiac hypertrophy.
Cells. 11:33362022. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Zucker IH, Schultz HD, Patel KP and Wang
H: Modulation of angiotensin II signaling following exercise
training in heart failure. Am J Physiol Heart Circ Physiol.
308:H781–H791. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Booz GW and Baker KM: Role of type 1 and
type 2 angiotensin receptors in angiotensin II-induced
cardiomyocyte hypertrophy. Hypertension. 28:635–640. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Palomeque J, Delbridge L and Petroff MV:
Angiotensin II: A regulator of cardiomyocyte function and survival.
Front Biosci (Landmark Ed). 14:5118–5133. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Simpson P: Norepinephrine-stimulated
hypertrophy of cultured rat myocardial cells is an alpha 1
adrenergic response. J Clin Invest. 72:732–738. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Koga Y, Itaya M and Toshima H: Increased
cardiovascular response to epinephrine in hypertrophic
cardiomyopathy. Jap Heart J. 26:727–740. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Glembotski CC: Classic studies of cultured
cardiac myocyte hypertrophy interview with a transformer. Circ Res.
113:1112–1116. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Ma Y, Zou H, Zhu XX, Pang J, Xu Q, Jin QY,
Ding YH, Zhou B and Huang DS: Transforming growth factor β: A
potential biomarker and therapeutic target of ventricular
remodeling. Oncotarget. 8:53780–53790. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Dobaczewski M, Chen W and Frangogiannis
NG: Transforming growth factor (TGF)-β signaling in cardiac
remodeling. J Mol Cell Cardiol. 51:600–606. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Hilfiker-Kleiner D, Hilfiker A, Schieffer
B, Engel D, Mann DL, Wollert KC and Drexler H: TNFα decreases α-MHC
expression by a NO mediated pathway: Role of E-box transcription
factors for cardiomyocyte specific gene regulation. Cardiovasc
Rese. 53:460–469. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Cheng MD, Li CL, Pei XY and Zhang YF, Jia
DD, Zuo YB, Cai SL, Li PF, Xin H and Zhang YF: Integrative analysis
of DNA methylome and transcriptome reveals epigenetic regulation of
bisphenols-induced cardiomyocyte hypertrophy. Ecotoxicol Environ
Saf. 263:1153912023. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Zheng L, Wang J, Zhang R, Zhang Y, Geng J,
Cao L, Zhao X, Geng J, Du X, Hu Y and Cong H: Angiotensin II
mediates cardiomyocyte hypertrophy in atrial cardiomyopathy via
epigenetic transcriptional regulation. Comput Math Methods Med.
2022:63121002022. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Molkentin JD, Lu JR, Antos CL, Markham B,
Richardson J, Robbins J, Grant SR and Olson EN: A
calcineurin-dependent transcriptional pathway for cardiac
hypertrophy. Cell. 93:215–228. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Zou Y, Hiroi Y, Uozumi H, Takimoto E, Toko
H, Zhu W, Kudoh S, Mizukami M, Shimoyama M, Shibasaki F, et al:
Calcineurin plays a critical role in the development of pressure
overload-induced cardiac hypertrophy. Circulation. 104:97–101.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Johnson S, Sommer N, Cox-Flaherty K,
Weissmann N, Ventetuolo CE and Maron BA: Pulmonary hypertension: A
contemporary review. Am J Respir Crit Care Med. 208:528–548. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Boucherat O, Agrawal V, Lawrie A and
Bonnet S: The latest in animal models of pulmonary hypertension and
right ventricular failure. Cir Res. 130:1466–1486. 2022. View Article : Google Scholar
|
|
83
|
Naeije R, Richter MJ and Rubin LJ: The
physiological basis of pulmonary arterial hypertension. Eur Respir
J. 59:21023342022. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Gu M, Shao NY, Sa S, Li D, Termglinchan V,
Ameen M, Karakikes I, Sosa G, Grubert F, Lee J, et al:
Patient-Specific iPSC-derived endothelial cells uncover pathways
that protect against pulmonary hypertension in BMPR2 mutation
Carriers. Cell Stem Cell. 20:490–504.e5. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Fan Y, Gu X, Zhang J, Sinn K, Klepetko W,
Wu N, Foris V, Solymosi P, Kwapiszewska G and Kuebler WM: TWIST1
drives smooth muscle cell proliferation in pulmonary hypertension
via loss of GATA-6 and BMPR2. Am J Respir Crit Care Med.
202:1283–1296. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Morrell NW, Yang X, Upton PD, Jourdan KB,
Morgan N, Sheares KK and Trembath RC: Altered growth responses of
pulmonary artery smooth muscle cells from patients with primary
pulmonary hypertension to transforming growth factor-beta(1) and
bone morphogenetic proteins. Circulation. 104:790–795. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Nathan SD, Barbera JA, Gaine SP, Harari S,
Martinez FJ, Olschewski H, Olsson KM, Peacock AJ, Pepke-Zaba J,
Provencher S, et al: Pulmonary hypertension in chronic lung disease
and hypoxia. Eur Respir J. 53:18019142019. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Wilkins MR, Ghofrani HA, Weissmann N,
Aldashev A and Zhao L: Pathophysiology and treatment of
high-altitude pulmonary vascular disease. Circulation. 131:582–590.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Calvier L, Boucher P, Herz J and Hansmann
G: LRP1 deficiency in vascular SMC leads to pulmonary arterial
hypertension that is reversed by PPARγ activation. Circu Res.
124:1778–1785. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Humbert M, Kovacs G, Hoeper MM,
Badagliacca R, Berger RMF, Brida M, Carlsen J, Coats AJS,
Escribano-Subias P, Ferrari P, et al: 2022 ESC/ERS Guidelines for
the diagnosis and treatment of pulmonary hypertension. G Ital
Cardiol (Rome). 24 (Suppl 1):e1–e116. 2023.(In Italian). PubMed/NCBI
|
|
91
|
Bush DS and Ivy DD: Pulmonary hypertension
in the population with down syndrome. Cardiol Ther. 11:33–47. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Lan C, Fang G, Qiu C, Li X, Yang F and
Yang Y: Inhibition of DYRK1A attenuates vascular remodeling in
pulmonary arterial hypertension via suppressing STAT3/Pim-1/NFAT
pathway. Clin Exp Hypertens. 46:22976422024. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Suginobe H, Ishida H, Ishii Y, Ueda K,
Yoshihara C, Ueyama A, Wang R, Tsuru H, Hashimoto K, Hirose M, et
al: Isogenic pairs of induced-pluripotent stem-derived endothelial
cells identify DYRK1A/PPARG/EGR1 pathway is responsible for Down
syndrome-associated pulmonary hypertension. Hum Mol Genet.
33:78–90. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Song Y, Kesuma D, Wang J, Deng Y, Duan J,
Wang JH and Qi RZ: Specific inhibition of cyclin-dependent kinases
and cell proliferation by harmine. Biochem Biophys Res Commun.
317:128–132. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Vainio L, Taponen S, Kinnunen SM,
Halmetoja E, Szabo Z, Alakoski T, Ulvila J, Junttila J, Lakkisto P,
Magga J, et al: GSK3β Serine 389 phosphorylation modulates
cardiomyocyte hypertrophy and ischemic injury. Int J Mol Sci.
22:135862021. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Liu S, Yang Y, Yang Y, Wei G, Zhou L, Lin
J, Yuan Z, Li Y, Wu Z, Liu T and Zhang G: A high-throughput
liver-kidney metabolic interaction chip for insights into the
nephrotoxicity mechanisms of triptolide. Lab Chip. 26:1830–1849.
2026. View Article : Google Scholar : PubMed/NCBI
|