Introduction
As a frequent and dangerous acute circulatory
condition, myocardial infarction (MI) is a significant burden
globally (1). The incidence of MI
is increasing worldwide, with certain high-income countries
reporting 50–100 cases per 100,000 individuals per year, whereas
low- and middle-income countries report 10–50 cases per 100,000
individuals per year (2). In
China, statistics show that the incidence of MI is increasing
annually, with an average growth rate of >20% in the last 10
years, and the mortality rate, which remains high despite a
decrease due to advancements in medical technology, even if a
patient survives an MI, the quality of life post-survival is
negatively affected (3,4). MI is now a major contributor to heart
failure and sudden cardiac death (5). Therefore, improving patient outcomes
requires a thorough examination of the pathophysiological
mechanisms underlying MI and novel therapeutic targets.
Impaired mitochondrial energy metabolism in
cardiomyocytes is hypothesized to be a key cause of the complex
pathological cascade that follows widespread cardiomyocyte
mortality after MI, in addition to directly resulting in loss of
cardiac function (6). The
functional integrity of mitochondria, as the ‘energy factories’ of
cardiomyocytes, is crucial for maintaining efficient cardiac
pumping (7). Increasingly, studies
have shown that, after MI, mitochondrial function in cardiomyocytes
is significantly impaired by persistent ischemia-hypoxia and
reperfusion injury, resulting in reduced energy production,
dysregulation of the redox balance and altered cell fate (8). For example, the tricarboxylic acid
(TCA) cycle and the oxidative phosphorylation (OXPHOS) process can
be disrupted by imbalances in mitochondrial dynamics (such as
Drp1-mediated hypersegmentation), abnormal opening of the
mitochondrial membrane permeability transition pore (mPTP) and
dysregulation of the sirtuin (SIRT)3 - AMP-activated protein kinase
(AMPK) - peroxisome proliferative activated receptor γ (PPARγ)
coactivator 1α (PGC-1α) metabolic axis. These disruptions can lead
to a notable reduction in ATP synthesis efficiency (9). In addition, there is a compensatory
shift in substrate utilization patterns from fatty acid oxidation
(FAO) to glycolysis, exacerbating the energy crisis of
cardiomyocytes; these metabolic disorders are also accompanied by
the release of a large quantities of reactive oxygen species (ROS),
mitochondrial DNA (mtDNA) and metabolites such as succinic acid,
which further amplifies impairment of mitochondrial function,
resulting in the widespread death of normal myocardium (9,10).
Mitochondria-derived metabolites can serve as
indicators of an energy imbalance and are crucial signals in the
control of innate immune responses (11). In a myocardial ischemic
environment, the accumulation of succinate activates
hypoxia-inducible factor-1α (HIF-1α) and enhances the expression of
pro-inflammatory factors (such as IL-1β and IL-6). Additionally,
ROS and mtDNA function as damage-associated molecular patterns
(DAMPs), activating the macrophage Toll-like receptor (TLR9)/NLR
family pyrin domain containing 3 (NLRP3) pathway, which
subsequently induces NLRP3 inflammasome activation and a surge in
the production and release of inflammatory factors (12). Subsequently, macrophages following
an MI are polarized towards an M1 or M2 phenotype by metabolic
reprogramming: M1-type macrophages primarily utilize glycolysis,
resulting in the secretion of numerous pro-inflammatory factors,
while M2-type macrophages depend on increased OXPHOS and FAO,
releasing reparative factors, such as IL-10, to facilitate tissue
healing and fibrotic remodeling, thereby establishing a closely
interconnected network of ‘metabolism-immunity-repair’ (13).
Consequently, following MI, compromised
mitochondrial energy metabolism is not only a pivotal factor in
cardiomyocyte damage but also a significant catalyst for
alterations in the immunological microenvironment. An increasing
body of evidence indicates that the ‘mitochondrial-immune axis’
plays bidirectional roles in regulating macrophage inflammatory
polarization and myocardial injury repair, offering novel avenues
for exploring metabolism-immunity-linked myocardial protection
strategies (14,15).
The present review examined mitochondrial energy
metabolism imbalance in cardiomyocytes, systematically elucidating
key molecular mechanisms and the role of metabolites in regulating
macrophage immune polarization. Additionally, the
‘mitochondria-immune axis’ is defined, highlighting the
bidirectional regulatory network that involves: i) Paracrine
metabolic signals released from damaged cardiomyocytes (such as
mtDNA, succinate and excessive ATP) that activate macrophage
inflammatory pathways, and ii) intrinsic mitochondrial metabolic
reprogramming within macrophages that determines their M1/M2
polarization phenotype. This review focuses on the bidirectional
interplay between metabolism and immunity in post-infarction repair
and injury, with the objective of providing a novel theoretical
foundation and potential targets for MI treatment, with a focus on
mitochondrial function and immune modulation.
The following sections describe the mechanisms of
mitochondrial metabolic impairment after MI, the regulation of
macrophage polarization by mitochondrial-derived metabolites and
the influence of metabolic-immune interactions on myocardial injury
and repair.
Molecular mechanisms of impaired
mitochondrial energy metabolism in cardiomyocytes
Mitochondrial kinetic dysregulation
and structural impairment
In typical physiological settings, the stable
preservation of mitochondrial morphology in cardiomyocytes relies
on a precise equilibrium between fusion and fission processes,
which is collaboratively controlled by fusion proteins [mitofusin
(Mfn)1/2 and OPA1 mitochondrial dynamin like GTPase (OPA1)] and
fission proteins [Drp1 and fission, mitochondrial 1 (Fis1)]. Mfn1
and Mfn2, dynamin-associated GTPases situated on the outer
mitochondrial membrane, facilitate the fusing of this membrane, and
when they form a trans-complex, they may approximate adjacent
mitochondria. OPA1 primarily facilitates mitochondrial inner
membrane fusion and cristae restructuring, with its long and short
isoforms working in concert to promote efficient inner membrane
fusion (16,17). Drp1 plays a central role in
mitochondrial division. Typically, Drp1 resides in the cytoplasm,
and several post-translational modifications are initiated upon
receipt of mitochondrial division signals (18). During mitosis, the
B1/cyclin-dependent kinase (Cdk)1 complex of the cell cycle
phosphorylates the Ser-585 site of Drp1, inducing its aggregation
in the outer mitochondrial membrane. Conversely, under healthy
conditions, CDK19/Cdk8 phosphorylates Drp1 at Ser616, facilitating
proper mitochondrial division (19). Furthermore, Rho-associated
coiled-coil protein kinase 1 can dephosphorylate the Ser637 site of
Drp1 by enhancing the activities of protein phosphatase 1 (PP1) and
protein phosphatase 2A(PP2A), therefore activating Drp1 and
facilitating its translocation to the mitochondria, where it
assembles into a helical shape (20). Fis1, an auxiliary protein involved
in mitochondrial division, interacts with Drp1 to recruit it to the
site of division. Subsequently, GTP hydrolysis by GTP-bound Drp1
induces a conformational change, generating a mechanical force that
facilitates mitochondrial membrane constriction and rupture,
thereby completing the division process (21).
However, in pathological situations like ischemia
and hypoxia, the balance of mitochondrial dynamics is disturbed in
the case of Drp1, ischemia and hypoxia can augment the activity of
calcium-modulated phosphatase (Calcineurin), which dephosphorylates
the Ser637 site of Drp1, thereby increasing its affinity for the
mitochondrial membrane and expediting its translocation to the
mitochondria (22). Du et
al (23) found that in the
cardiac ischemia-reperfusion model, Drp1-Ser616 phosphorylation was
markedly increased, accompanied by extensive Drp1 translocation to
the mitochondrial surface, resulting in excessive mitochondrial
division and fragmentation. In Sirt3-knockout mice, Sirt3
deficiency impaired deacetylation, thereby reducing activation of
downstream proteins, including kinases that regulate Drp1
phosphorylation. This resulted in reduced Drp1 phosphorylation
levels and heightened mitochondrial hypersegmentation, ultimately
causing a marked deterioration in cardiac function. The absence of
Mfn2 directly impaired mitochondrial fusion and disrupted calcium
signaling coupling between mitochondria and the endoplasmic
reticulum, owing to the role of Mfn2 in their linkage. Under
physiological conditions, calcium ions released from the
endoplasmic reticulum can be transported to mitochondria via
Mfn2-mediated channels, and moderate calcium signaling can activate
dehydrogenases and ATP synthase in the TCA cycle, thereby enhancing
ATP production (24) (Fig. 1).
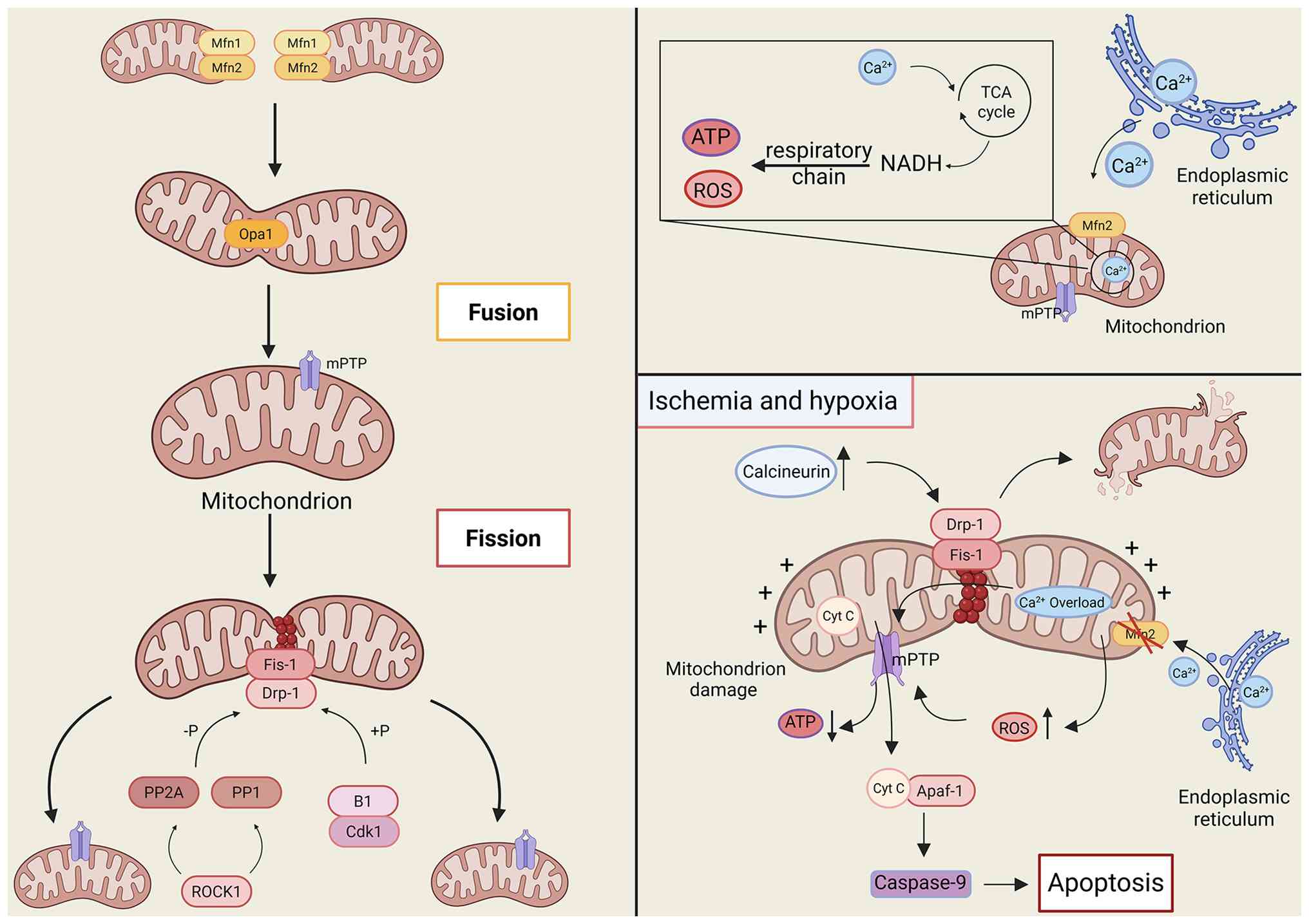 | Figure 1.Mechanisms regulating mitochondrial
morphologic stability in cardiomyocytes and the process of
imbalance under pathological conditions. Physiological
circumstances maintain a balance between mitochondrial fusion
(Mfn1/2 for outer membrane fusion, Opa1 for inner membrane and
cristae remodeling) and division (Drp1 in conjunction with Fis1,
regulated by Cdk1, ROCK1, etc.). Ischemia and hypoxia induce the
activation of calmodulin phosphatase, resulting in the
overactivation of Drp1 and excessive mitochondrial fragmentation.
Mfn2 aberrantly disrupts calcium signaling coupling, while
Ca2+ overload and ROS provoke the opening of the mPTP,
leading to the release of cytochrome C. This process activates
apoptosis mediated by caspase-9 through Apaf-1, thereby
exacerbating the injury. Mfn1, mitofusin 1; Opa1, optic atrophy 1;
Drp1, dynamin-related protein 1; Fis1, fission 1; Cdk1,
cyclin-dependent kinase 1; ROCK1, Rho-associated coiled-coil
containing protein kinase 1; mPTP, mitochondrial permeability
transition pore; Apaf-1, apoptotic protease activating factor 1;
Caspase-9, cysteinyl aspartate specific proteinase-9; ROS, reactive
oxygen species; Mfn2, mitofusin 2; PP1, protein phosphatase 1; TCA
cycle, tricarboxylic acid cycle; NADH, nicotinamide adenine
dinucleotide (reduced form); ATP, adenosine triphosphate; Cyt C,
cytochrome C. |
Following Mfn2 loss, mitochondrial calcium uptake
becomes disordered and susceptible to calcium excess. Excessive
calcium ions can activate calcium-dependent proteases and
phospholipases within the mitochondria, compromising mitochondrial
membrane integrity and inducing substantial ROS production, which
further impairs mitochondrial function. Mitochondrial morphological
impairment is closely associated with aberrant mPTP opening
(25). In compromised
cardiomyocytes, disrupted energy metabolism and diminished ATP
synthesis lead to impaired ion pump activity, which is essential
for maintaining intracellular ionic homeostasis. This leads to
excessive Ca2+ influx and intracellular accumulation,
while the mitochondrial respiratory chain is concurrently
dysregulated, hindering electron transfer and causing a significant
rise in ROS production (26).
Elevated Ca2+ and ROS levels can serve as cues to
initiate mPTP opening. mPTP is a protein complex situated between
the inner and outer mitochondrial membranes, which is typically in
a closed state. Upon opening of the mPTP, the mitochondrial
membrane potential rapidly dissipates; the proton electrochemical
gradient across the inner membrane is reduced; OXPHOS coupling is
disrupted; and ATP production decreases significantly.
Concurrently, mPTP opening facilitates the release of cytochrome c
from the mitochondrial intermembrane space into the cytosol, where
it associates with apoptotic protease-activating factor 1, thereby
recruiting and activating caspase-9, which subsequently initiates
the downstream caspase cascade and induces apoptosis (27). Drp1 activation has been shown to
promote excessive mitochondrial fission and to enhance mPTP opening
via interactions with mPTP-associated proteins, thereby
exacerbating mitochondrial injury. Conversely, the small molecule
mitochondrial division inhibitor 1 (Mdivi-1), functioning as a Drp1
inhibitor, selectively binds to Drp1, impedes its GTPase activity
and obstructs Drp1 from forming a helical shape with fission
activity on the mitochondrial membrane, hence postponing mPTP
opening (28). In the
MI-reperfusion injury model, Mdivi-1 treatment reduced
mitochondrial fragmentation, delayed mPTP opening and markedly
decreased the myocardial infarct area, suggesting that modulating
mitochondrial dynamics-related proteins may be an effective
intervention for myocardial ischemic injury (29).
Disorders of essential energy
metabolism pathways
The onset of MI involves the disruption of essential
mitochondrial energy metabolism pathways, significantly impairing
cardiomyocyte function, with the dysregulation of the
SIRT3-AMPK-PGC-1α axis being pivotal (30). SIRT3, a mitochondrial deacetylase,
is primarily localized in the mitochondrial matrix and modulates
the acetylation status of several mitochondrial proteins. Under
physiological conditions, SIRT3 preserves the proper function of
mitochondrial proteins through deacetylating action (31). Superoxide dismutase 2 (SOD2) is a
crucial mitochondrial antioxidant enzyme and its activity is
modulated by acetylation. Physiological expression of SIRT3
deacetylates Lys-68 and other residues of SOD2, thereby activating
SOD2, augmenting mitochondrial ROS scavenging capacity and
preserving mitochondrial redox homeostasis (32). Additionally, SIRT3 deacetylates
α-ketoglutarate dehydrogenase (OGDH), a pivotal enzyme in the TCA
cycle that catalyzes the conversion of α-ketoglutarate to succinyl
coenzyme A. Following deacetylation by SIRT3, OGDH activity is
increased to facilitate the uninterrupted progression of the TCA
cycle, allowing for the continuous supply of reducing equivalents
(NADH, FADH2) for OXPHOS (33). In MI, SIRT3 expression is markedly
downregulated by stressors such as ischemia and hypoxia. Research
indicates that in a mouse model of myocardial ischemia-reperfusion,
SIRT3 protein levels in cardiac tissues decreased by ~50% following
20 min of ischemia and 24 h of reperfusion. The absence of SIRT3
reduced mitochondrial protein deacetylation, resulting in
hyperacetylation (34). Excessive
acetylation of SOD2 markedly diminished its activity and impaired
its ability to scavenge ROS, leading to substantial ROS
accumulation within the cell. Excessive ROS not only directly
impaired the structural integrity of the mitochondrial membrane but
also altered critical enzymes in the TCA cycle, thereby inhibiting
their function (35).
Simultaneously, the catalytic activity of OGDH was significantly
diminished following hyperacetylation, thereby disrupting the TCA
cycle. Obstruction of the TCA cycle reduced NADH and
FADH2 generation, thereby impairing OXPHOS and
significantly reducing ATP synthesis efficiency, and resulting in a
failure to meet the energy requirements of cardiomyocytes (36).
Conversion of substrate
utilization
Under typical circumstances, cardiomyocytes
primarily obtain energy by FAO, with ~70% of ATP generated by fatty
acid β-oxidation, whereas glucose metabolism contributes
considerably less to energy production. The pivotal rate-limiting
step in the ingress of fatty acids into mitochondria for
β-oxidation is facilitated by carnitine palmitoyltransferase 1
(CPT-1), which is located in the outer mitochondrial membrane, and
catalyzes the conversion of lipoacyl-coenzyme A of long-chain fatty
acids to lipoacylcarnitine, thereby permitting their entry into the
mitochondrial matrix to ensure oxidation (37). At the onset of MI, the
intracellular environment undergoes considerable alterations,
resulting in inhibition of FAO and a gradual metabolic shift toward
glycolysis. During ischemia and hypoxia, intracellular ATP
concentrations decrease rapidly, whereas ADP and AMP levels rise,
thereby activating AMPK. Activated AMPK phosphorylates and inhibits
acetyl coenzyme A carboxylase, a crucial enzyme in fatty acid
synthesis, leading to decreased generation of malonyl coenzyme A
(38). Malonyl coenzyme A serves
as an endogenous inhibitor of CPT-1; thus, reduced malonyl coenzyme
A levels increase CPT-1 activity, presumably facilitating FAO. Of
note, poor mitochondrial function resulting from ischemia,
diminished activity of FAO-related enzymes and malfunction of fatty
acid transporters collectively limit FAO, despite elevated CPT-1
activity (39). Concurrently, the
cell redirects its metabolic flux toward glycolysis to meet its
fundamental energy requirements. On the one hand, HIF-1α expression
is upregulated and stabilized under ischemic hypoxic conditions.
HIF-1α binds to the promoter regions of key glycolytic genes, such
as glucose transporter 1 and hexokinase 2, thereby promoting their
transcription and enhancing glucose uptake and glycolytic
initiation. On the other hand, the activity of
phosphofructokinase-1 (PFK-1), a key rate-limiting enzyme in
glycolysis, was also increased (40,41).
Elevated intracellular AMP concentrations can allosterically
activate PFK-1 while concurrently increasing
fructose-2,6-bisphosphate levels, the most effective allosteric
activator of PFK-1. This interaction further enhances PFK-1
activity and accelerates glycolysis (42). The energy supply efficiency of
glycolysis is significantly lower than that of FAO in conjunction
with OXPHOS. One molecule of glucose can yield 30–32 molecules of
ATP via complete decomposition through aerobic oxidation, while
glycolysis results in the production of only 2 molecules of ATP.
This substrate utilization conversion serves as an emergency
strategy; however, prolonged dependence on glycolysis for energy
does not meet the substantial energy requirements of
cardiomyocytes. Additionally, the excessive lactate produced by
glycolysis further impairs cardiomyocyte function. This results in
a reduction of intracellular pH, subsequently compromising
cardiomyocyte function (43,44)
(Fig. 2).
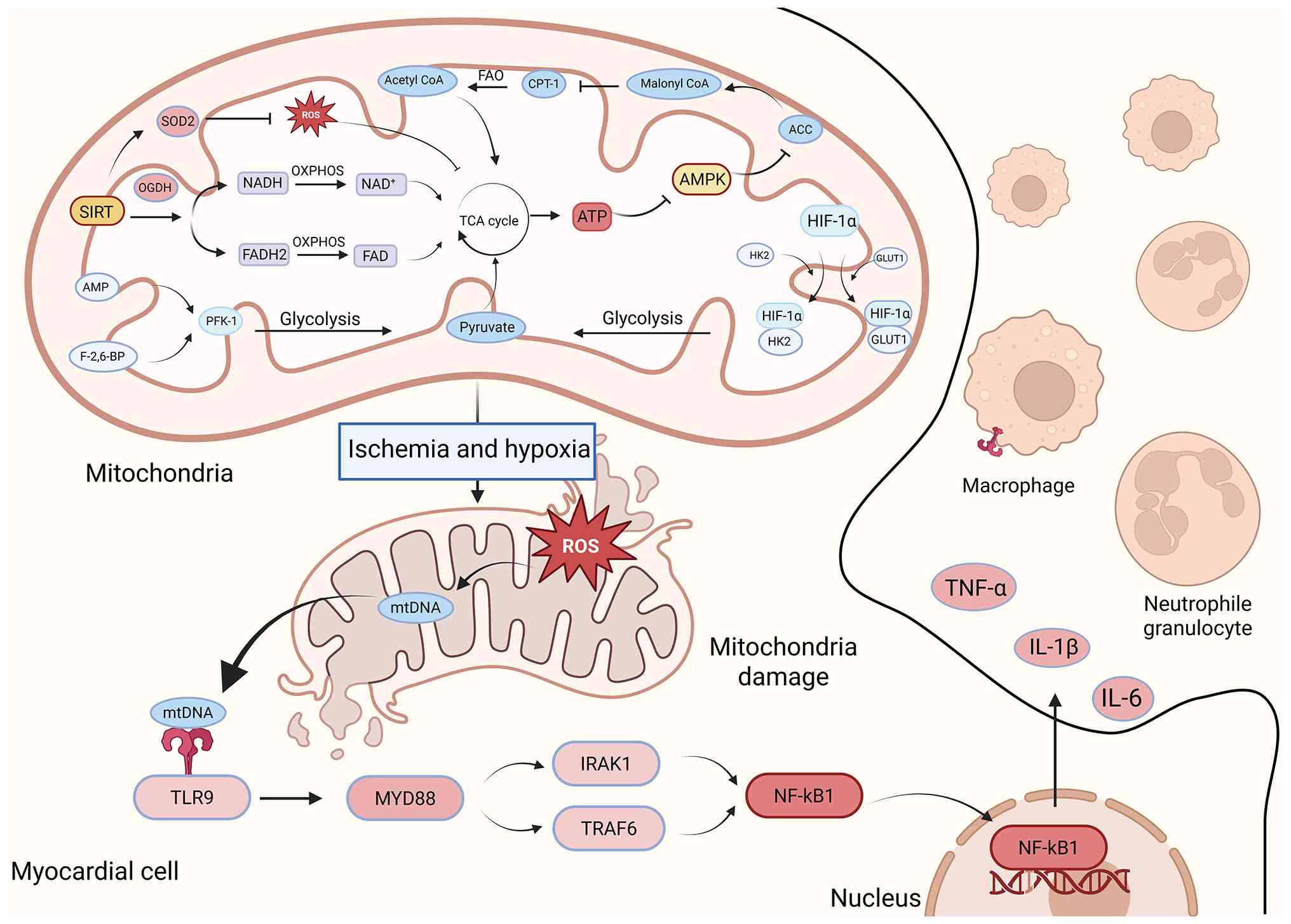 | Figure 2.Disturbed energy metabolism and
metabolic-immune cross-regulatory mechanisms in myocardial
infarction. Under physiological settings, SIRT3 activates SOD2 and
OGDH to sustain ROS scavenging and OXPHOS energy production,
whereas FAO is regulated by CPT-1 and ACC. In the context of
ischemia and hypoxia, the downregulation of SIRT3 results in the
accumulation of ROS and damage to the TCA cycle and OXPHOS.
Concurrently, AMPK and HIF-1α promote a metabolic shift towards
glycolysis. The release of ROS and mtDNA from mitochondria
activates the macrophage TLR9 pathway, leading to the release of
pro-inflammatory factors and establishing a cycle of injury. SIRT3,
sirtuin 3; SOD2, superoxide dismutase 2; OGDH, oxoglutarate
dehydrogenase; ROS, reactive oxygen species; OXPHOS, oxidative
phosphorylation; FAO, fatty acid oxidation; CPT-1, carnitine
palmitoyltransferase 1; ACC, acetyl-CoA carboxylase; AMPK,
adenosine 5′-monophosphate-activated protein kinase; HIF-1α,
hypoxia-inducible factor 1α; mtDNA, mitochondrial DNA; TLR9,
toll-like receptor 9; acetyl CoA, acetyl coenzyme A; NADH,
nicotinamide adenine dinucleotide (reduced form); NAD+,
nicotinamide adenine dinucleotide (oxidized form);
FADH2, flavin adenine dinucleotide (reduced form); FAD,
flavin adenine dinucleotide (oxidized form); ATP, adenosine
triphosphate; AMP, adenosine monophosphate; TCA cycle,
tricarboxylic acid cycle; PFK-1, phosphofructokinase-1; F-2,6-BP,
fructose-2,6-bisphosphate; HK2, hexokinase 2; GLUT1, glucose
transporter 1; MYD88, myeloid differentiation primary response 88;
IRAK1, interleukin-1 receptor-associated kinase 1; TRAF6, TNF
receptor-associated factor 6; NF-κB1, nuclear factor
kappa-light-chain-enhancer of activated B cells 1; TNF-α, tumor
necrosis factor-α; IL-1β, interleukin-1β. |
Metabolite release induces an
inflammatory response
Impaired mitochondrial energy metabolism affects
energy supply and leads to the abnormal release of various
metabolites. Notably, overproduction of ROS and mtDNA spillover are
significant factors that trigger aseptic inflammation (45). In the standard mitochondrial
respiratory chain, electrons are sent to oxygen through complexes
I, II, III and IV, while ADP is phosphorylated to generate ATP. In
MI, mitochondrial structural damage and respiratory chain
dysfunction impede electron transfer, leading to significant
electron leakage that reacts with oxygen to generate the superoxide
anion (O2·−). This ROS can subsequently
generate hydrogen peroxide (H2O2), hydroxyl
radicals (·OH) and other more reactive ROS (46). Excess ROS are potent oxidants that
can directly damage lipids, proteins and mtDNA within the
mitochondrial membrane. ROS in the mitochondrial membrane can
induce lipid peroxidation, thereby diminishing membrane fluidity,
increasing permeability and compromising the structural integrity
of the mitochondrial membrane. For example, ROS can oxidize
cardiolipin, a phospholipid unique to the inner mitochondrial
membrane, crucial for the stability and functionality of the
respiratory chain complex. The oxidation of cardiolipin impairs the
function of respiratory chain complexes, thereby exacerbating ROS
generation (47). Conversely, ROS
can affect mitochondrial proteins, hence changing their structure
and function. For example, ROS can induce oxidative alterations of
Drp1, thereby facilitating excessive mitochondrial division and
fragmentation (48). Concurrently,
ROS can also cause mtDNA damage, encompassing base mutations,
deletions and double-strand breaks (49).
In conclusion, impaired mitochondrial energy
metabolism in cardiomyocytes following MI leads to an injury
network characterized by kinetic imbalances, disruption of
metabolic pathways, alterations in substrate use and the release of
inflammatory signals. An imbalance in mitochondrial fusion and
fission disturbs energy production, dysregulation of the SIRT3 axis
impedes OXPHOS, a metabolic shift to glycolysis intensifies the
energy crisis and the activation of inflammation by ROS and mtDNA
deteriorates the microenvironment. Therefore, targeting the
mitochondrial metabolic network is a potential direction for
prevention and treatment; modulation of Drp1/Mfn2, activation of
the SIRT3 axis, optimization of substrate utilization and blockade
of the inflammatory cascade improve energy supply and promote
macrophage polarization toward the M2 phenotype by intervening in
the mitochondrial-immune axis, providing a novel direction for
clinical translation.
Dysfunctional mitochondrial metabolism
promotes immunological responses by macrophages
Metabolites as immunological
signals
Accumulation of succinate and activation of
inflammation
Under physiological conditions, the TCA cycle
operates in a coordinated manner, maintaining the equilibrium of
cellular energy metabolism. Nonetheless, during MI, mitochondrial
activity is compromised and the TCA cycle is disrupted (50). Succinate dehydrogenase activity is
suppressed, leading to intracellular accumulation of succinate due
to impaired conversion. Succinate, a crucial metabolic
intermediate, activates HIF-1α upon its buildup. HIF-1α is a
crucial transcription factor that is significant in hypoxic or
metabolic stress circumstances. In general, HIF-1α is expressed at
low levels in cells and undergoes hydroxylation by prolyl
hydroxylase (PHD) domains before being degraded by the
ubiquitin-proteasome system. Nonetheless, as succinate accumulates,
it inhibits PHD activity, thereby stabilizing HIF-1α, which permits
its substantial accumulation within the cell and subsequent
translocation to the nucleus. HIF-1α translocates to the nucleus,
attaches to the hypoxia response element in the promoter region of
the target gene and activates the transcription of numerous genes,
including the pro-inflammatory cytokines IL-1β and IL-6. IL-1β and
IL-6, significant inflammatory mediators, are released in
substantial amounts into the extracellular space, attracting immune
cells, such as neutrophils and lymphocytes, to the MI site and
initiating an inflammatory response. The inflammatory response
facilitates the removal of necrotic tissue in the early phases;
nevertheless, excessive or prolonged inflammation may cause further
damage to cardiomyocytes (51).
Activation of NLRP3 inflammasomes by
ATP/ROS
MI, resulting in mitochondrial impairment, leads to
succinate accumulation and the release of ATP and ROS into the
extracellular milieu, which function as DAMPs and can activate
NLRP3 inflammatory vesicles. Compromised mtDNA is less stable and
may be extruded from the mitochondria into the cytoplasm. mtDNA
functions as a DAMP detectable by TLR9 in the cytoplasm. TLR9 is
predominantly expressed on the surface of various cell types,
including immune cells (for example, macrophages and dendritic
cells) and cardiomyocytes. mtDNA binding to TLR9 initiates a
downstream signaling cascade dependent on myeloid differentiation
factor 88 (MyD88). MyD88 recruits IL-1 receptor-associated kinase
(IRAK) family members, which activate tumor necrosis factor
receptor-associated factor 6 (TRAF6), thereby activating nuclear
factor-κB (NF-κB). Activated NF-κB translocates to the nucleus and
associates with the promoter regions of pro-inflammatory cytokine
genes (for example, TNF-α, IL-1β and IL-6), thereby enhancing their
transcription and expression (52,53).
These pro-inflammatory cytokines are secreted extracellularly to
attract and activate immune cells, initiating a sterile
inflammatory response. While the inflammatory response serves as a
defensive mechanism, excessive inflammation can exacerbate
cardiomyocyte damage and worsen MI, creating a detrimental cycle
(54). The NLRP3 inflammasome is a
multiprotein complex that primarily comprises NLRP3,
apoptosis-associated speck-like protein (ASC) and caspase-1
(55). The construction and
activation of NLRP3 inflammatory vesicles are triggered by ATP
binding to the P2X7 receptor on the cell membrane or by ROS
stimulating redox-sensitive signaling pathways within the cell
(56). Specifically, ATP binding
to the P2X7 receptor induces channel opening, resulting in
intracellular potassium efflux that activates NLRP3. Conversely,
ROS can activate NLRP3 by oxidizing cysteine residues, thereby
inducing a conformational change. Activated NLRP3 inflammatory
vesicles assemble multimeric complexes by attracting ASC, which
then attracts and activates caspase-1 precursors, resulting in
their cleavage into active caspase-1. Activated caspase-1
preferentially cleaves pro-IL-1β, converting it into
physiologically active IL-1β, thereby facilitating the initiation
and progression of inflammatory responses (57). Additionally, caspase-1 cleaves the
Gasdermin D protein, releasing its N-terminal domain, which inserts
into the plasma membrane to form a pore. This process induces cell
death and the rapid release of inflammatory mediators, thereby
intensifying the inflammatory cascade. It causes osmotic imbalance
in cardiomyocytes, disrupts membrane integrity and impairs
organelle function, ultimately exacerbating structural damage and
functional loss in myocytes, while facilitating the pathological
progression of MI toward a reversible state and eventually
intensifying the structural impairment and functional decline of
cardiomyocytes, hence advancing the pathological progression of MI
irreversibly (58).
Metabolic reprogramming and
polarization of macrophages
In the context of MI, macrophage metabolic
reprogramming modulates inflammatory processes and tissue healing
through bidirectional polarization patterns (59). Pro-inflammatory M1 polarization is
induced by ischemia and a hypoxic milieu, in which increased HIF-1α
stability promotes macrophage polarization toward the
pro-inflammatory (M1) phenotype (60,61).
HIF-1α can modulate the glycolytic pathway by transcriptionally
activating pyruvate kinase M2 (PKM2), thereby steering glucose
metabolism toward the lactic acid and pentose phosphate pathways,
resulting in the accumulation of metabolic intermediates such as
succinate. Succinate not only establishes positive feedback by
stabilizing HIF-1α but also facilitates the maturation and release
of IL-1β via the mitochondrial ROS-NLRP3 inflammatory vesicle
pathway. In contrast, the glycolytic byproduct, lactic acid,
inhibits histone deacetylase (HDAC) activity, thereby alleviating
the transcriptional repression of NF-κB-mediated pro-inflammatory
genes, thereby enhancing the ‘metabolism-epigenetic’ cascade. In a
murine model of MI, PKM2 inhibition reduced the expression of the
M1 macrophage marker inducible nitric oxide synthase, significantly
decreased IL-1β levels and reduced infarct size (62).
Conversely, reparative M2 polarization is induced by
IL-4/IL-13 activation of the JAK1-STAT6 signaling pathway through
the IL-4Rα/IL-13Rα1 receptor (63). The phosphorylation of STAT6 within
the nucleus promotes the expression of PPAR-γ, which assembles a
transcriptional complex with retinoic acid X receptor (RXR) and
coactivator PGC-1α to collaboratively modulate mitochondrial
biosynthesis (for example, transcription factor A, mitochondrial
and nuclear respiratory factor 1 and essential enzymes of FAO
(e.g., CPT-1A and acyl-coa oxidase 1). PGC-1α facilitates repair
via two metabolic mechanisms: i) Promoting FAO of long-chain fatty
acids into β-oxidation, supplying acetyl coenzyme A for the TCA
cycle; and ii) upregulation of electron transport chain complex
expression to enhance OXPHOS efficiency. ATP generated by OXPHOS
facilitates the ubiquitin-mediated clearance of HIF-1α via the
AMPK/mTORC1 signaling pathway, thereby suppressing the
glycolysis-dependent pro-inflammatory response. Significantly, the
PPAR-γ agonist rosiglitazone augmented the percentage of CD206+
M2-type macrophages by 35% in the MI model and expedited
neovascularization by enhancing vascular endothelial growth factor
(VEGF) secretion. Conversely, PGC-1α knockdown led to a 40%
reduction in macrophage FAO, accompanied by a decrease in the
secretion of the anti-inflammatory factor IL-10, thereby supporting
the role of mitochondrial metabolism in promoting M2-type
polarization (64). This confirms
that mitochondrial metabolism facilitates M2-type polarization
(Fig. 3).
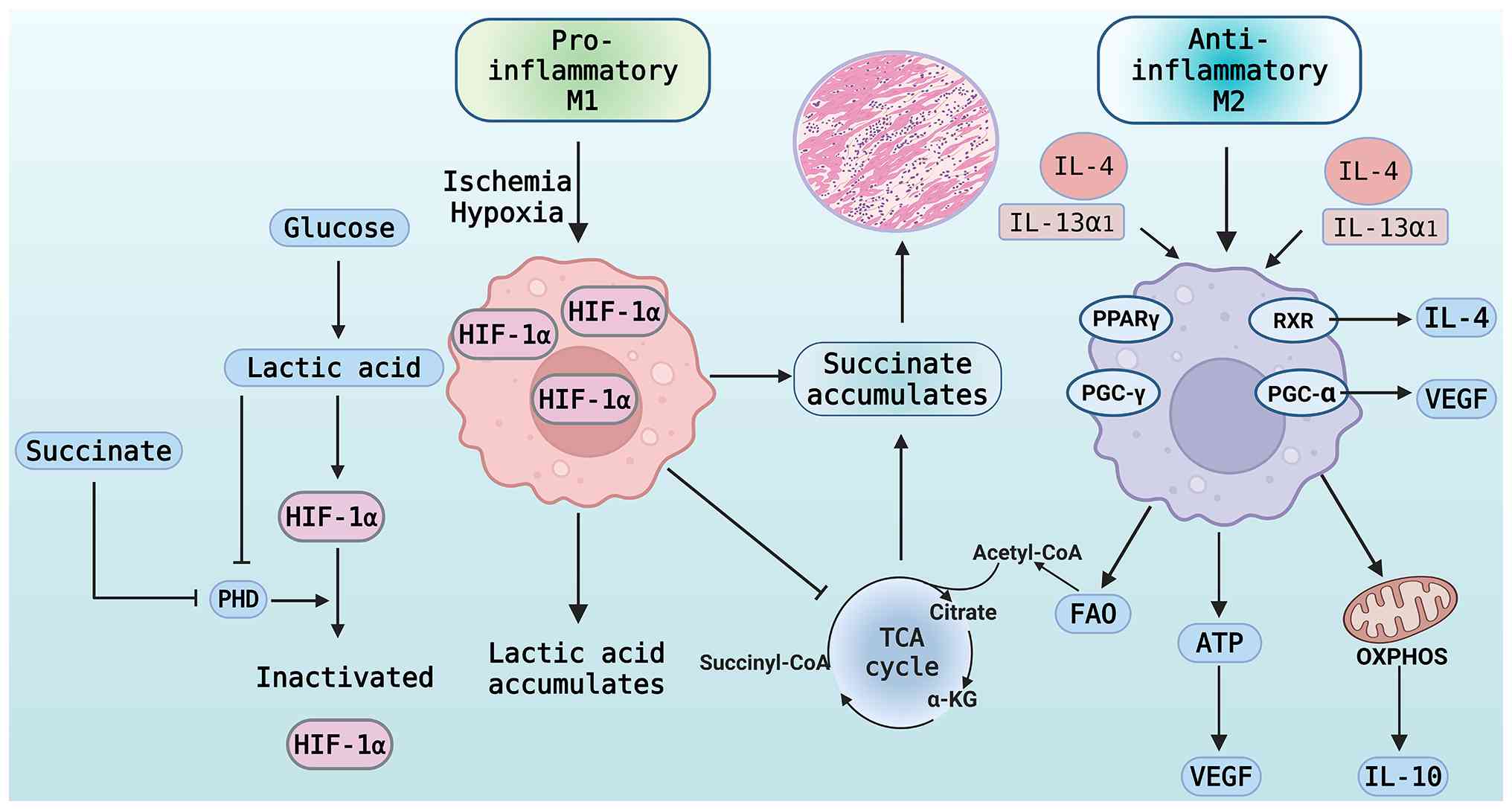 | Figure 3.Dynamic mechanisms by which
metabolites and cytokines synergistically regulate macrophage
polarization in the microenvironment of MI. In the microenvironment
of MI during the pro-inflammatory phase, mitochondrial dysfunction
disrupts the TCA cycle, resulting in Succinate accumulation that
inhibits PHD, stabilizes HIF-1α, activates the glycolytic pathway
and facilitates macrophage polarization towards the M1
pro-inflammatory phenotype, thereby establishing a ‘Succinate -
HIF-1α’ positive feedback loop. The repair phase involves IL-4
stimulating PPAR-γ to form a complex with RXR and PGC-1α, thereby
enhancing mitochondrial metabolism to inactivate HIF-1α. Acetyl
coenzyme A generated by the TCA cycle supports the
anti-inflammatory phenotype, induces the secretion of IL-10 and
VEGF, and promotes tissue repair, while lactate plays a regulatory
role, illustrating the interplay between metabolism and immunity.
MI, myocardial infarction; TCA, tricarboxylic acid cycle; HIF,
hypoxia-inducible factor; PHD, prolyl hydroxylase; IL-4,
interleukin-4; PPAR-γ, peroxisome proliferator-activated receptor
γ; RXR, retinoid X receptor; PGC-1α, peroxisome
proliferator-activated receptor gamma coactivator 1α; VEGF,
vascular endothelial growth factor; IL-13α1, IL-13 receptor α1;
PGC-γ, peroxisome proliferator-activated receptor gamma coactivator
γ; FAO, fatty acid oxidation; OXPHOS, oxidative phosphorylation;
IL-10, interleukin-10; α-KG, α-ketoglutarate; acetyl-CoA, acetyl
coenzyme A; succinyl-CoA, succinyl coenzyme A. |
In summary, compromised mitochondrial metabolism
during MI initiates a proinflammatory response through the release
of metabolites such as succinate and ATP/ROS, which activate the
HIF-1α/NF-κB and NLRP3 inflammatory vesicle pathways, thereby
promoting macrophage polarization toward the glycolysis-dominant M1
phenotype. Simultaneously, IL-4/IL-13 modifies mitochondrial
OXPHOS/FAO metabolism through the STAT6-PPARγ-PGC-1α pathway,
facilitating M2-type polarization to promote repair. Therefore,
focusing on the metabolism-immunity-linked interaction axis (for
example, modulating the SIRT3-AMPK pathway or targeting the
HIF-1α-PDK1 axis) may reshape macrophage polarization phenotype and
offer a novel approach for anti-inflammatory-restorative
combination therapy in MI. Future research should focus on the
spatial and temporal dynamics of transcellular metabolic
signaling.
Bidirectional influence of metabolic-immune
interactions in myocardial repair and damage
Initial injury phase
The immediate pathological response to MI is
characterized by a temporal sequence of metabolic homeostasis
disruption and inflammatory activation, which underlies early
damage (65). Within minutes to
hours following MI, the ischemia and hypoxic milieu swiftly induce
an imbalance in mitochondrial dynamics and metabolic signaling,
establishing a ‘metabolic-inflammatory’ positive feedback loop
(52). Specifically, enhanced
calcium-modulated phosphatase (Calcineurin) activity facilitates
dephosphorylation of Ser637 on Drp1, markedly increasing its
affinity for the mitochondrial membrane and leading to excessive
mitochondrial fragmentation. When the damaged inner mitochondrial
membrane ruptures during mitochondrial fission, mtDNA is released
into the cytoplasm. Macrophage TLR9 recognizes these molecules as
DAMPs. Through MyD88, TLR9 recruits IRAK, which, in turn, activates
TRAF6, thereby inducing NF-κB nuclear translocation and increasing
levels of pro-inflammatory cytokines such as IL-1β, TNF-α and IL-6
(66,67). These proinflammatory factors worsen
secondary myocardial injury through two mechanisms: TNF-α causes
cardiomyocytes to undergo apoptosis via the death receptor pathway,
increases vascular permeability and encourages neutrophil
infiltration; and IL-1β binds to IL-1R1 on cardiomyocytes,
activates the MAPK/p38 pathway, inhibits mitochondrial complex II
activity and further encourages succinate accumulation and ROS
generation. The key involvement of the mtDNA-TLR9 axis in early
inflammatory storms was confirmed by TLR9 silencing, which reduced
infarct size by 35% and IL-1β levels by 40% in a mouse model of
myocardial ischemia-reperfusion (68).
Repair stage
When the initial inflammatory surge reaches its
peak, the body activates a reparative process facilitated by
metabolic reprogramming, in which macrophage phenotypic switching
is pivotal (69). Between 24 and
72 h post-MI, macrophages commence phagocytosis by identifying
phosphatidylserine on the membranes of apoptotic cardiomyocytes, a
mechanism that activates metabolic reprogramming in macrophages.
Following phagocytosis of apoptotic cells, AMPK activity in
macrophages increases markedly, leading to phosphorylation and
activation of CPT-1, thereby facilitating FAO. Acetyl coenzyme A
generated by FAO enters the TCA cycle and increases OXPHOS
capability, supplying the energy required to sustain an
anti-inflammatory phenotype (70).
Concurrently, FAO intermediates, such as acetylcarnitine, stimulate
SIRT1, which deacetylates PPARγ and enhances its capacity to
heterodimerize with RXR. The PPARγ-RXR complex, in conjunction with
PGC-1α, stimulates transcription of the anti-inflammatory genes
IL-10, TGF-β and VEGF. IL-10 inhibits the release of
pro-inflammatory factors by binding to IL-10R1/IL-10R2 receptors on
macrophages, increasing JAK1/STAT3 signaling and suppressing NF-κB
activity (71,72). In a rat model of MI, PPARγ
overexpression led to a 2.3-fold increase in IL-10+ macrophages
within the infarcted region and a 40% decrease in myocardial
fibrosis area. Additionally, TGF-β facilitated organized type I
collagen deposition rather than disorganized fibrosis by inducing
fibroblast transformation into myofibroblasts, thereby preserving
cardiac structural integrity (73). Furthermore, Willenborg et al
(74) discovered that the
metabolic phenotype of repair-phase macrophages is dynamically
influenced by the oxygen partial pressure within the
microenvironment: In the initial hypoxic region (oxygen partial
pressure <10 mmHg), HIF-1α remained activated at a low level,
facilitating energy for phagocytosis by transiently sustaining
glycolysis through the induction of PDK1 expression; as
neovascularization occurred, the oxygen partial pressure increased
to 20–30 mmHg, leading to the dominance of the PPARγ/PGC-1α axis in
the metabolic transition. Neovascularization resulted in oxygen
partial pressure returning to 20–30 mmHg, leading to the
ubiquitination and degradation of HIF-1α, while the PPARγ/PGC-1α
axis governed the metabolic transition. This spatiotemporal
regulatory mechanism of metabolism-immunity-linked interaction
facilitated the timely transition of macrophages to a reparative
phenotype following the removal of necrotic tissues, thereby
preventing excessive inflammatory damage (74,75).
Molecular targeting methodologies for
metabolic therapies to promote repair
Based on the aforementioned regulatory mechanisms of
metabolism-immunity-linked interaction, targeted intervention in
the metabolic signaling axis has emerged as a novel paradigm for
enhancing cardiac healing. SIRT3-mediated modification of the
mitochondrial-immunological axis has considerable therapeutic
promise owing to its dual role in metabolic control and the
inhibition of inflammation (76).
Overexpression of SIRT3 ameliorated metabolic-immune disorders via
a dual mechanism: First, SIRT3 deacetylation activated liver kinase
B1, an upstream kinase of Drp1, thereby enhancing AMPK Thr172
phosphorylation, inhibiting Drp1 Ser637 dephosphorylation and
reducing mitochondrial fragmentation. In SIRT3 transgenic mice, MI
led to increased production of the mitochondrial fusion protein
Mfn2 and increased the recovery of the mitochondrial membrane
potential (77). Second, SIRT3
activated SOD2 via deacetylation, diminishing mitochondrial ROS
levels, mitigating mtDNA damage and release, and inhibiting the
TLR9/NF-κB inflammatory pathway (76,78).
Following the restoration of mitochondrial function, ATP synthesis
in cardiomyocytes increases, thereby influencing macrophage
phenotype via paracrine signaling. ATP is released by the Connexin
43 hemichannel, activating macrophage P2Y purinoceptor 12,
inhibiting adenylate cyclase via Gi proteins, reducing cyclic AMP
levels and facilitating PPARγ phosphorylation. Lactate
dehydrogenase A produced by cardiomyocytes can be taken up by
macrophages, facilitating PPARγ binding to DNA and augmenting
M2-type gene transcription via HDAC inhibition (79,80)
(Fig. 4).
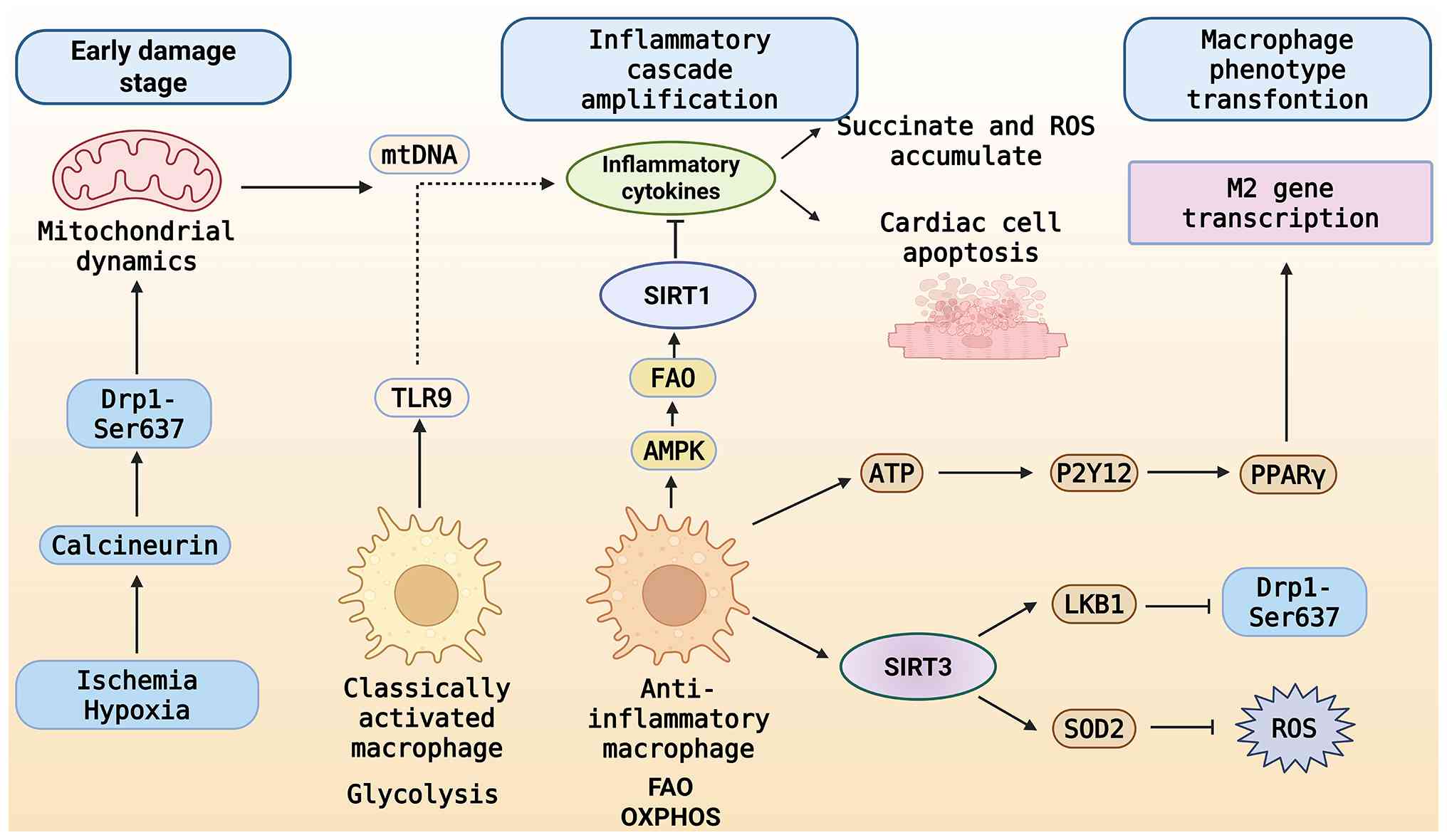 | Figure 4.Bidirectional mechanism of
metabolic-immune axis regulation of myocardial injury and repair
after myocardial infarction. During the initial phase of injury,
Ischemia and hypoxia stimulate calmodulin phosphatase, leading to
the dephosphorylation of Drp1 Ser637, resulting in excessive
mitochondrial fragmentation, the release of mtDNA, Succinate and
ROS, and the activation of the macrophage proinflammatory pathway
through TLR9, thereby polarizing it towards a proinflammatory
phenotype and intensifying myocardial apoptosis. During the repair
phase, SIRT3 enhances mitochondrial dynamics and diminishes ROS;
ATP and other factors propel macrophage metabolism towards FAO and
OXPHOS, facilitating a transition to an anti-inflammatory
phenotype, commencing M2 transformation and fostering repair. Drp1,
dynamin-related protein 1; mtDNA, mitochondrial DNA; ROS, reactive
oxygen species; TLR9, Toll-like receptor 9; SIRT3, Sirtuin 3; ATP,
adenosine triphosphate; FAO, fatty acid oxidation; OXPHOS,
oxidative phosphorylation; Ser637, serine 637; AMPK, adenosine
5′-monophosphate-activated protein kinase; P2Y12, purinergic
receptor P2Y12; PPAR-γ, peroxisome proliferator-activated receptor
γ; LKB1, liver kinase B1; SOD2, superoxide dismutase 2. |
Therefore, metabolic-immune interactions during MI
play a bidirectional regulatory function in both damage and
healing. During the initial injury phase, mitochondrial dysfunction
releases mtDNA, which activates the TLR9/NF-κB pathway, thereby
triggering a positive inflammatory feedback loop and exacerbating
myocardial injury. During the repair phase, macrophages initiate
the AMPK-CPT1-FAO axis via phagocytosis of apoptotic cells,
activate the PPARγ/PGC-1α pathway, shift to an anti-inflammatory M2
phenotype, and suppress inflammation and promote myocardial repair
through factors such as IL-10 and TGF-β. SIRT3 functions as a
metabolic-immune nexus that activates AMPK and SOD2, diminishes ROS
and mtDNA release and mitigates inflammation, while further
promoting macrophage reprogramming via ATP- and lactate-mediated
paracrine signaling. This mechanism reflects the coupled regulation
of metabolism and immunity across spatial and temporal scales,
providing theoretical support and potential targets for
interventions in myocardial injury.
Discussion
In recent years, the focus in myocardial healing and
injury research has been the metabolic-immune crossover between
mitochondrial failure and the macrophage immunological response
following MI (13). The molecular
mechanisms underlying impaired mitochondrial energy metabolism in
cardiomyocytes following MI are systematically reviewed in the
present article, with a focus on the critical roles of aberrant
substrate metabolism, imbalanced mitochondrial dynamics and
disruption of key signaling pathways in immunological activation
and cardiomyocyte death. Further investigation into functional
alterations in inflammatory polarization and the reprogramming of
macrophages in response to metabolites ultimately demonstrated the
reciprocal regulatory functions of immunity and metabolism in
myocardial damage and healing. Based on the above, this review
summarizes and provides an overview of the research focus and
challenges in this field across three in-depth aspects.
First, the inflammatory cascade reaction begins with
decreased mitochondrial metabolism in cardiomyocytes following MI.
Mitochondrial structural integrity and energy metabolism function
are severely compromised by imbalances in mitochondrial dynamics,
including Drp1-mediated hyperfission and downregulation of
Mfn2/OPA1 expression. When mitochondrial morphology is disrupted,
the mPTP opens abnormally and the membrane potential collapses,
triggering the mitochondrial-mediated apoptotic cascade.
Concurrently, the energy crisis in cardiomyocytes is exacerbated by
the buildup of metabolic intermediates like succinate in the TCA
cycle, SIRT3 deficiency-induced defective deacetylation of
important enzymes and the shift in substrate use from FAO to
glycolysis. In this context, a closed-loop disease known as
‘metabolic imbalance-immune activation’ is initiated when DAMPs,
including ROS and mtDNA, are released in significant amounts,
thereby triggering a sterile inflammatory response (18,21).
Second, mitochondrial metabolites play a major role
in regulating macrophage reprogramming and phenotypic change, which
are important effector cells in the immune response. While
substances such as ROS and ATP exacerbate secondary damage to
myocardial tissue by activating the NLRP3 inflammasome and
promoting caspase-1-dependent IL-1β maturation and cellular
pyroptosis, succinic acid is released from injured cells,
activating HIF-1α and driving pro-inflammatory polarization of
M1-type macrophages in the MI environment (57). M1-type macrophages' rapid
inflammatory response is supported by their glycolytic metabolic
advantage, whereas during the repair phase, FAO and
OXPHOS-dominated metabolism polarizes macrophages towards an M2
phenotype, which secretes VEGF and IL-10 to promote fibrosis and
neovascularization and finish tissue remodeling. Important
signaling axes, such as SIRT3, PGC-1α and PPARγ, control this
process, offering a biological foundation for the return of
metabolic-immune balance (64).
Third, the metabolic-immune crossover is a key
factor in the quality of myocardial repair and in the magnitude and
duration of the inflammatory response. In the initial stages of MI,
mtDNA leakage and mitochondrial damage induce Drp1 activation,
creating an inflammatory ‘signaling source’ that initiates the
macrophage TLR9-NF-κB pathway. This leads to the release of
numerous inflammatory factors, including TNF-α, IL-6 and IL-1β,
resulting in ‘secondary cell death’ and the development of MI
(68). Conversely, M2-type
macrophages regulate tissue healing throughout the repair phase by,
among other functions, eliminating apoptotic cells and promoting
collagen deposition and angiogenesis (75). Restoring mitochondrial fusion,
improving energy metabolism efficiency and lowering ROS load, all
of which promote the M2 polarization transition, by activating the
SIRT3 axis, may slow cardiac remodeling following MI (76).
In addition, it is important to acknowledge that
several regulatory mechanisms discussed above may exhibit
context-dependent or even contradictory effects. Although SIRT3 is
generally regarded as a protective metabolic regulator, recent
evidence suggests that its actions are not uniformly beneficial.
Excessive SIRT3-driven enhancement of FAO may increase metabolic
stress during acute ischemia, and its anti-inflammatory effects may
suppress the early M1-mediated clearance of damaged tissue, which
is essential for clearing necrotic cardiomyocytes. Similarly, while
prolonged M1 activation contributes to secondary injury, a timely
and transient M1 response is indispensable for proper removal of
debris and for establishing a microenvironment permissive to
subsequent M2-driven repair. Recognizing these complexities
highlights the need for stage-specific evaluation of metabolic and
immune interventions in MI, rather than assuming uniform
therapeutic benefits across all phases of injury and healing.
Several important questions remain unresolved,
despite research that has provided a clearer picture of the
molecular chain linking compromised mitochondrial metabolism,
immunological polarization and cardiac healing. For example, i)
whether mitochondrial structural alterations and metabolite release
are linked in a ‘lead event’ or feedback mechanism; ii) whether
macrophage phenotypes display more dynamic intermediate states; and
iii) the distinct functions of various cardiomyocyte subpopulations
in metabolic-immune crossover. Furthermore, current studies on
regulatory nodes, such as SIRT3 and PGC-1α, focus on cellular
models and animal experiments; comprehensive clinical translational
research is still required to confirm their expression profiles in
human cardiac tissues, to elucidate their dynamic patterns of
change and to assess their potential for pharmacological
regulation.
Taken together, these knowledge gaps highlight
several future perspectives and unresolved questions that warrant
further investigation. First, it remains unclear whether
mitochondrial fission and structural disruption serve as initiating
triggers that precede metabolite leakage, or whether they arise
secondarily as a consequence of sustained oxidative stress and DAMP
release. Time-resolved in vivo imaging and conditional
manipulation of Drp1/Mfn2 in cardiomyocytes will be essential to
disentangle causality along this axis. Second, the traditional
dichotomy of M1 vs. M2 macrophages is likely an oversimplification
in the evolving infarct microenvironment. Single-cell and spatial
multi-omics analyses suggest the presence of hybrid or transitional
states with mixed inflammatory and reparative signatures, yet their
temporal distribution, plasticity and functional contribution to
scar formation and ventricular remodeling remain poorly defined.
Third, cardiomyocytes are not a homogeneous population:
Subendocardial, subepicardial and conduction-system myocytes may
differ in mitochondrial density, substrate preference and
susceptibility to immune-mediated injury. How these distinct
cardiomyocyte subpopulations participate in, or shape, the
mitochondrial-immune crosstalk is largely unknown. Finally,
although SIRT3, PGC-1α and related metabolic checkpoints are
promising targets in preclinical models, their expression patterns,
pharmacodynamic responsiveness and safety profiles in human
myocardial tissue have not been systematically characterized.
Addressing these questions will be crucial for rationally designing
metabolism-based immunomodulatory therapies and for selecting the
right patients, timing and endpoints in future clinical trials.
From a therapeutic standpoint, targeting mitochondrial metabolic
remodeling has considerable potential. In animal studies,
Drp1-based inhibitors such as Mdivi-1, SIRT3 agonists such as
Honokiol, and AMPK-PGC-1α pathway activators have demonstrated
encouraging effects, improving energy metabolism and markedly
reducing myocardial infarct size and left ventricular remodeling.
In addition, immunometabolic modulators that have been shown to
enhance M2-type polarization and promote myocardial healing include
IL-10 carriers and FAO promoters. Future studies should investigate
the combined metabolic-immune intervention method in greater
detail, establish a more accurate treatment plan and timeline, and
recognize the shift from ‘symptomatic anti-inflammatory’ to ‘source
metabolic intervention’.
Conclusion
Myocardial infarction profoundly disrupts
mitochondrial energy metabolism, which not only compromises
cardiomyocyte survival but also shapes the inflammatory and
reparative responses by affecting macrophage activation and
polarization. Current evidence indicates that metabolic-immune
interactions, including mitochondrial dynamics, SIRT3-mediated
metabolic regulation, inflammasome activity and M1/M2 phenotypic
switching, collectively determine the balance between tissue injury
and healing.
These insights identify the mitochondria-immune axis
as a promising therapeutic target for coordinating both
inflammation control and myocardial repair. Future research should
focus on clarifying the temporal sequence of metabolic and immune
events, defining intermediate macrophage phenotypes and validating
metabolic regulators such as SIRT3 and Drp1 in human myocardial
tissue. Advancing this knowledge will facilitate the translation of
metabolism-based immunomodulatory strategies into clinically
applicable interventions for precise myocardial protection.
Acknowledgements
The figures were created with BioRender (https://app.biorender.com).
Funding
The present study was supported in part by research grants from
the ‘Project for Supporting Innovative Teams and Talents in
Traditional Chinese Medicine’ organized and implemented by the
National Administration of Traditional Chinese Medicine (grant no.
ZYYCXTD-C-202207) and Major and Key research Projects of
Traditional Chinese Medicine of Sichuan Provincial Administration
of Traditional Chinese Medicine in 2024 (grant no. 2024zd008).
Availability of data and materials
Not applicable.
Authors' contributions
YZ edited the manuscript. HF, QW and SY were
involved in writing - original draft. YJ and HX were involved in
the conception and design. LD and XL revised the manuscript. HC and
GL were responsible for conceptualization and supervised the study.
Data authentication is not applicable. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Reed GW, Rossi JE and Cannon CP: Acute
myocardial infarction. Lancet. 389:197–210. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Salari N, Morddarvanjoghi F, Abdolmaleki
A, Rasoulpoor S, Khaleghi AA, Hezarkhani LA, Shohaimi S and
Mohammadi M: The global prevalence of myocardial infarction: A
systematic review and meta-analysis. BMC Cardiovasc Disord.
23:2062023. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Song J, Murugiah K, Hu S, Gao Y, Li X,
Krumholz HM and Zheng X; China PEACE Collabortive Group, :
Incidence, predictors, and prognostic impact of recurrent acute
myocardial infarction in China. Heart. 107:313–318. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen H, Shi L, Xue M, Dong X, Wang N, Chen
J, Zhu W, Cai Y, Xu H and Meng Q: In-hospital mortality after acute
myocardial infarction in China: A nationwide cross-sectional
analysis. The Lancet. 390:S442017. View Article : Google Scholar
|
|
5
|
Curtain JP, Pfeffer MA, Braunwald E,
Claggett BL, Granger CB, Køber L, Lewis EF, Maggioni AP, Mann DL,
Rouleau JL, et al: Rates of sudden death after myocardial
infarction-insights from the VALIANT and PARADISE-MI trials. JAMA
Cardiol. 9:928–933. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhou H, Ren J, Toan S and Mui D: Role of
mitochondrial quality surveillance in myocardial infarction: From
bench to bedside. Ageing Res Rev. 66:1012502021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tahrir FG, Langford D, Amini S, Mohseni
Ahooyi T and Khalili K: Mitochondrial quality control in cardiac
cells: Mechanisms and role in cardiac cell injury and disease. J
Cell Physiol. 234:8122–8133. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhou M, Yu Y, Luo X, Wang J, Lan X, Liu P,
Feng Y and Jian W: Myocardial ischemia-reperfusion injury:
therapeutics from a mitochondria-centric perspective. Cardiology.
146:781–792. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Piamsiri C, Maneechote C, Jinawong K,
Arunsak B, Chunchai T, Nawara W, Kerdphoo S, Chattipakorn SC and
Chattipakorn N: Chronic mitochondrial dynamic-targeted therapy
alleviates left ventricular dysfunction by reducing multiple
programmed cell death in post-myocardial infarction rats. Eur J
Pharmacol. 977:1767362024. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Forte M, Schirone L, Ameri P, Basso C,
Catalucci D, Modica J, Chimenti C, Crotti L, Frati G, Rubattu S, et
al: The role of mitochondrial dynamics in cardiovascular diseases.
Br J Pharmacol. 178:2060–2076. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lyu Y, Wang T, Huang S and Zhang Z:
Mitochondrial damage-associated molecular patterns and metabolism
in the regulation of innate immunity. J Innate Immun. 15:665–679.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Francisco J and Del Re D: Inflammation in
myocardial ischemia/reperfusion injury: Underlying mechanisms and
therapeutic potential. Antioxidants (Basel). 12:19442023.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mouton AJ, DeLeon-Pennell KY, Rivera
Gonzalez OJ, Flynn ER, Freeman TC, Saucerman JJ, Garrett MR, Ma Y,
Harmancey R and Lindsey ML: Mapping macrophage polarization over
the myocardial infarction time continuum. Basic Res Cardiol.
113:262018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Weissman D and Maack C: Mitochondrial
function in macrophages controls cardiac repair after myocardial
infarction. J Clin Invest. 133:e1670792023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cai S, Zhao M, Zhou B, Yoshii A, Bugg D,
Villet O, Sahu A, Olson GS, Davis J and Tian R: Mitochondrial
dysfunction in macrophages promotes inflammation and suppresses
repair after myocardial infarction. J Clin Invest. 133:e1594982023.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Marín-García J and Akhmedov AT:
Mitochondrial dynamics and cell death in heart failure. Heart Fail
Rev. 21:123–136. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Spurlock BM, Xie Y, Song Y, Ricketts SN,
Hua JR, Chi HR, Nishtala M, Salmenov R, Liu J and Qian L:
Mitochondrial fusion and cristae reorganization facilitate
acquisition of cardiomyocyte identity during reprogramming of
murine fibroblasts. Cell Rep. 44:1153772025. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jin JY, Wei XX, Zhi XL, Wang XH and Meng
D: Drp1-dependent mitochondrial fission in cardiovascular disease.
Acta Pharmacol Sin. 42:655–664. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liao JZ, Chung HL, Shih C, Wong KKL, Dutta
D, Nil Z, Burns CG, Kanca O, Park YJ, Zuo Z, et al: Cdk8/CDK19
promotes mitochondrial fission through Drp1 phosphorylation and can
phenotypically suppress pink1 deficiency in Drosophila. Nat Commun.
15:33262024. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hu J, Zhang H, Li J, Jiang X, Zhang Y, Wu
Q, Shen L, Shi J and Gao N: ROCK1 activation-mediated mitochondrial
translocation of Drp1 and cofilin are required for arnidiol-induced
mitochondrial fission and apoptosis. J Exp Clin Cancer Res.
39:372020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nolden KA, Harwig MC and Hill RB: Human
Fis1 directly interacts with Drp1 in an evolutionarily conserved
manner to promote mitochondrial fission. J Biol Chem.
299:1053802023. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang J, Wang S, Zhang H, Yang X, Ren X,
Wang L, Yang Y, Yang Y and Wen Y: Drp1 acetylation mediated by
CDK5-AMPK-GCN5L1 axis promotes cerebral ischemic injury via
facilitating mitochondrial fission. Mol Med. 30:1732024. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Du J, Li H, Song J, Wang T, Dong Y, Zhan
A, Li Y and Liang G: AMPK activation alleviates myocardial
ischemia-reperfusion injury by regulating Drp1-mediated
mitochondrial dynamics. Front Pharmacol. 13:8622042022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
De Mario A, D'Angelo D, Zanotti G,
Raffaello A and Mammucari C: The mitochondrial calcium uniporter
complex-A play in five acts. Cell Calcium. 112:1027202023.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fu D, Luo J, Wu Y, Zhang L, Li L, Chen H,
Wen T, Fu Y and Xiong W: Angiotensin II-induced calcium overload
affects mitochondrial functions in cardiac hypertrophy by targeting
the USP2/MFN2 axis. Mol Cell Endocrinol. 571:1119382023. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chistiakov DA, Shkurat TP, Melnichenko AA,
Grechko AV and Orekhov AN: The role of mitochondrial dysfunction in
cardiovascular disease: A brief review. Ann Med. 50:121–127. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhou Y, Jing S, Liu S, Shen X, Cai L, Zhu
C, Zhao Y and Pang M: Double-activation of mitochondrial
permeability transition pore opening via calcium overload and
reactive oxygen species for cancer therapy. J Nanobiotechnology.
20:1882022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Su ZD, Li CQ, Wang HW, Zheng MM and Chen
QW: Inhibition of DRP1-dependent mitochondrial fission by Mdivi-1
alleviates atherosclerosis through the modulation of M1
polarization. J Transl Med. 21:4272023. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Maneechote C, Palee S, Kerdphoo S,
Jaiwongkam T, Chattipakorn SC and Chattipakorn N: Differential
temporal inhibition of mitochondrial fission by Mdivi-1 exerts
effective cardioprotection in cardiac ischemia/reperfusion injury.
Clin Sci (Lond). 132:1669–1683. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bugga P, Alam MJ, Kumar R, Pal S,
Chattopadyay N and Banerjee SK: Sirt3 ameliorates mitochondrial
dysfunction and oxidative stress through regulating mitochondrial
biogenesis and dynamics in cardiomyoblast. Cell Signal.
94:1103092022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang J, Xiang H, Liu J, Chen Y, He RR and
Liu B: Mitochondrial Sirtuin 3: New emerging biological function
and therapeutic target. Theranostics. 10:8315–8342. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
He J, Liu X, Su C, Wu F, Sun J, Zhang J,
Yang X, Zhang C, Zhou Z, Zhang X, et al: Inhibition of
mitochondrial oxidative damage improves reendothelialization
capacity of endothelial progenitor cell via SIRT3 (Sirtuin
3)-Enhanced SOD2 (Superoxide Dismutase) deacetylation in
hypertension. Arterioscler Thromb Vasc Biol. 39:1682–1698. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang Q, Siyuan Z, Xing C and Ruxiu L:
SIRT3 regulates mitochondrial function: A promising star target for
cardiovascular disease therapy. Biomed Pharmacother.
170:1160042024. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Porter GA, Urciuoli WR, Brookes PS and
Nadtochiy SM: SIRT3 deficiency exacerbates ischemia-reperfusion
injury: Implication for aged hearts. Am J Physiol Heart Circ
Physiol. 306:H1602–H1609. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang L, Chen CL, Kang PT, Jin Z and Chen
YR: Differential protein acetylation assists import of excess SOD2
into mitochondria and mediates SOD2 aggregation associated with
cardiac hypertrophy in the murine SOD2-tg heart. Free Radic Biol
Med. 108:595–609. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu X, Zhang Y, Deng Y, Yang L, Ou W, Xie
M, Ding L, Jiang C, Yu H, Li Q and Li T: Mitochondrial protein
hyperacetylation underpins heart failure with preserved ejection
fraction in mice. J Mol Cell Cardiol. 165:76–85. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Da Dalt L, Cabodevilla AG, Goldberg IJ and
Norata GD: Cardiac lipid metabolism, mitochondrial function and
heart failure. Cardiovasc Res. 119:1905–1914. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hopkins TA, JDyck JR and Lopaschuk GD:
AMP-activated protein kinase regulation of fatty acid oxidation in
the ischaemic heart. Biochem Soc Trans. 31(Pt 1): 207–212. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Edwards KS, Ashraf S, Lomax TM, Wiseman
JM, Hall ME, Gava FN, Hall JE, Hosler JP and Harmancey R:
Uncoupling protein 3 deficiency impairs myocardial fatty acid
oxidation and contractile recovery following ischemia/reperfusion.
Basic Res Cardiol. 113:472018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li H, Xiao F, Zhou C, Zhu T and Wang S:
Metabolic adaptations and therapies in cardiac hypoxia: Mechanisms
and Clinical Implications/Potential Strategies. JACC Basic Transl
Sci. 10:862–878. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sant'Ana PG, Tomasi LC, Murata GM,
Vileigas DF, Mota GAF, Souza SLB, Silva VL, Campos LP, Okoshi K,
Padovani CR and Cicogna AC: Hypoxia-inducible factor 1-alpha and
glucose metabolism during cardiac remodeling progression from
hypertrophy to heart failure. Int J Mol Sci. 24:62012023.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chauhan S, Lemaster M and England E: At
physiological concentrations, AMP increases phosphofructokinase-1
activity compared to fructose 2, 6-bisphosphate in postmortem
porcine skeletal muscle. Meat Sci. 172:1083322021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sun Q, Karwi QG, Wong N and Lopaschuk GD:
Advances in myocardial energy metabolism: Metabolic remodelling in
heart failure and beyond. Cardiovasc Res. 120:1996–2016. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li J, Duan X, Hu D, Liu P, Lin Y-W, Zhong
G, WenLLi S, Zhang H and Lin X: Abstract 4143725: Down-regulation
Of Cardiomyocyte Isocitrate Dehydrogenase 2 Mediates Metabolic
Reprogramming, Contributing To Heart Failure By Remodeling The
Lactate Microenvironment. Circulation. 150:2024. View Article : Google Scholar
|
|
45
|
Nakayama H and Otsu K: Mitochondrial DNA
as an inflammatory mediator in cardiovascular diseases. Biochem J.
475:839–852. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bugger H and Pfeil K: Mitochondrial ROS in
myocardial ischemia reperfusion and remodeling. Biochim Biophys
Acta Mol Basis Dis. 1866:1657682020. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Panov AV and Dikalov SI: Cardiolipin,
perhydroxyl radicals, and lipid peroxidation in mitochondrial
dysfunctions and aging. Oxid Med Cell Longev. 2020:13230282020.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Cho Ye and Kim YK: ROS-mediated
cytoplasmic localization of CARM1 induces mitochondrial fission
through DRP1 methylation. Redox Biol. 73:1032122024. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Quan Y, Xin Y, Tian G, Zhou J and Liu X:
Mitochondrial ROS-Modulated mtDNA: A potential target for cardiac
aging. Oxid Med Cell Longev. 2020:94235932020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ren J, Chen X, Wang T, Liu C and Wang K:
Regenerative therapies for myocardial infarction: Exploring the
critical role of energy metabolism in achieving cardiac repair.
Front Cardiovasc Med. 12:15331052025. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Pan J, Zhang L, Li D, Li Y, Lu M, Hu Y,
Sun B, Zhang Z and Li C: Hypoxia-inducible factor-1: Regulatory
mechanisms and drug therapy in myocardial infarction. Eur J
Pharmacol. 963. pp. 1762772024, View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Torp MK, Vaage J and Stensløkken KO:
Mitochondria-derived damage-associated molecular patterns and
inflammation in the ischemic-reperfused heart. Acta Physiol (Oxf).
237:e139202023. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Pagliaro P and Penna C: Inhibitors of
NLRP3 inflammasome in ischemic heart disease: Focus on functional
and redox aspects. Antioxidants (Basel). 12:13962023. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ong SB, Hernández-Reséndiz S,
Crespo-Avilan GE, Mukhametshina RT, Kwek XY, Cabrera-Fuentes HA and
Hausenloy DJ: Inflammation following acute myocardial infarction:
Multiple players, dynamic roles, and novel therapeutic
opportunities. Pharmacol Ther. 186:73–87. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Du Y, Duan C, Zhang X, Shi S, Zhu X, Lyu
M, Wei Y and Hu Y: Modulation of NLRP3 inflammasome: Advantages of
Chinese herbal medicine in treating myocardial ischemia/reperfusion
injury. Am J Chin Med. 53:737–769. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wang W, Hu D, Feng Y, Wu C, Song Y, Liu W,
Li A, Wang Y, Chen K, Tian M, et al: Paxillin mediates ATP-induced
activation of P2X7 receptor and NLRP3 inflammasome. BMC Biol.
18:1822020. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Drinkall S, Lawrence CB, Ossola B, Russell
S, Bender C, Brice NB, Dawson LA, Harte M and Brough D: The two
pore potassium channel THIK-1 regulates NLRP3 inflammasome
activation. Glia. 70:1301–1316. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Piamsiri C, Maneechote C, Chattipakorn SC
and Chattipakorn N: Therapeutic potential of gasdermin D-mediated
myocardial pyroptosis in ischaemic heart disease: Expanding the
paradigm from bench to clinical insights. J Cell Mol Med.
29:e703572025. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Kim Y, Nurakhayev S, Nurkesh A,
Zharkinbekov Z and Saparov A: Macrophage polarization in cardiac
tissue repair following myocardial infarction. Int J Mol Sci.
22:27152021. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
DeBerge M, Lantz C, Dehn S, Sullivan DP,
van der Laan AM, Niessen HWM, Flanagan ME, Brat DJ, Feinstein MJ,
Kaushal S, et al: Hypoxia-inducible factors individually facilitate
inflammatory myeloid metabolism and inefficient cardiac repair. J
Exp Med. 218:e202006672021. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
He H, Zhou Z, Zhang L, Lu Z, Li B and Li
X: HIF1α/MIF/CD74 signaling mediated OSA-induced atrial
fibrillation by promoting M1 macrophages polarization. Int
Immunopharmacol. 149:1142482025. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Lin ZJ, Dong X, He H, Jiang JL, Guan ZJ,
Li X, Lu L, Li H, Huang YS, Xian SX, et al: A simplified herbal
decoction attenuates myocardial infarction by regulating macrophage
metabolic reprogramming and phenotypic differentiation via
modulation of the HIF-1α/PDK1 axis. Chin Med. 19:752024. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Shiraishi M, Shintani Y, Shintani Y,
Ishida H, Saba R, Yamaguchi A, Adachi H, Yashiro K and Suzuki K:
Alternatively activated macrophages determine repair of the
infarcted adult murine heart. J Clin Invest. 126:2151–2166. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Liu S, Zhang H, Li Y, Zhang Y, Bian Y,
Zeng Y, Yao X, Wan J, Chen X, Li J, et al: S100A4 enhances protumor
macrophage polarization by control of PPAR-γ-dependent induction of
fatty acid oxidation. J Immunother Cancer. 9:e0025482021.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Wang X, Yang G, Li J, Meng C and Xue Z:
Dynamic molecular signatures of acute myocardial infarction based
on transcriptomics and metabolomics. Sci Rep. 14:101752024.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Sharp WW, Fang YH, Han M, Zhang HJ, Hong
Z, Banathy A, Morrow E, Ryan JJ and Archer SL: Dynamin-related
protein 1 (Drp1)-mediated diastolic dysfunction in myocardial
ischemia-reperfusion injury: Therapeutic benefits of Drp1
inhibition to reduce mitochondrial fission. FASEB J. 28:316–326.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Enzan N, Matsushima S, Ikeda S, Okabe K,
Ishikita A, Yamamoto T, Sada M, Miyake R, Tsutsui Y, Nishimura R,
et al: ZBP1 protects against mtDNA-induced myocardial inflammation
in failing hearts. Circ Res. 132:1110–1126. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Chu Y, Hua Y, He L, Chen Y, He J, Mckinney
K, Young M, Ballinger S, Hu H and Lal H: Abstract 4143940:
Circulating Mitochondrial DNA: Biomarker and Inflammation Mediator
in Cardiac Ischemia/Reperfusion Injury. Circulation. 150:2024.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Sun X, Li Y, Deng Q, Hu Y, Dong J, Wang W,
Wang Y and Li C: Macrophage polarization, metabolic reprogramming,
and inflammatory effects in ischemic heart disease. Front Immunol.
13:9340402022. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Ao-Di F, Han-Qing L, Xi-Zheng W, Ke Y,
Hong-Xin G, Hai-Xia Z, Guan-Wei F and Li-Lan: Advances in
macrophage metabolic reprogramming in myocardial
ischemia-reperfusion. Cell Signal. 123:1113702024. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Li W, Cao J, Wang X, Zhang Y, Sun Q, Jiang
Y, Yao J, Li C, Wang Y and Wang W: Ferruginol restores
SIRT1-PGC-1α-Mediated mitochondrial biogenesis and fatty acid
oxidation for the treatment of DOX–Induced cardiotoxicity. Front
Pharmacol. 12:7738342021. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Wilbers RHP, van Raaij DR, Westerhof LB,
Bakker J, Smant G and Schots A: Re-evaluation of IL-10 signaling
reveals novel insights on the contribution of the intracellular
domain of the IL-10R2 chain. PLoS One. 12:e01863172017. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Wang Y, Zhang Y, Li J, Li C, Zhao R, Shen
C, Liu W, Rong J, Wang Z, Ge J and Shi B: Hypoxia induces M2
macrophages to express VSIG4 and mediate cardiac fibrosis after
myocardial infarction. Theranostics. 13:2192–2209. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Willenborg S, Sanin DE, Jais A, Ding X,
Ulas T, Nüchel J, Popović M, MacVicar T, Langer T, Schultze JL, et
al: Mitochondrial metabolism coordinates stage-specific repair
processes in macrophages during wound healing. Cell Metab.
33:2398–2414.e9. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Semba H, Takeda N, Isagawa T, Sugiura Y,
Honda K, Wake M, Miyazawa H, Yamaguchi Y, Miura M, Jenkins DM, et
al: HIF-1α-PDK1 axis-induced active glycolysis plays an essential
role in macrophage migratory capacity. Nat Commun. 7:116352016.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Liu J, Yan W, Zhao X, Jia Q, Wang J, Zhang
H, Liu C, He K and Sun Z: Sirt3 attenuates post-infarction cardiac
injury via inhibiting mitochondrial fission and normalization of
AMPK-Drp1 pathways. Cell Signal. 53:1–13. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Liu M, Li X and Huang D: Mfn2
overexpression attenuates cardio-cerebrovascular
ischemia-reperfusion injury through mitochondrial fusion and
activation of the AMPK/Sirt3 signaling. Front Cell Dev Biol.
8:5980782020. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Xiong D, Wang X, Wang H, Chen X, Li H, Li
Y, Zhong M, Gao J, Zhao Z and Ren W: Quercetin inhibits
cardiomyocyte apoptosis via Sirt3/SOD2/mitochondrial reactive
oxygen species during myocardial ischemia-reperfusion injury.
Heliyon. 10:e390312024. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Chen Y, Liao Y, Bin J and Chen Y:
LDHA-mediated metabolic reprogramming promoted cardiomyocyte
proliferation by alleviating ROS and inducing M2 macrophage
polarization. Redox Biol. 56:1024462022. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Zhang S, Bories G, Lantz C, Emmons R,
Becker A, Liu E, Abecassis MM, Yvan-Charvet L and Thorp EB:
Immunometabolism of phagocytes and relationships to cardiac repair.
Front Cardiovasc Med. 6:422019. View Article : Google Scholar : PubMed/NCBI
|














