Introduction
Humans are predisposed to low back pain (LBP) as an
evolutionary trade-off for a mechanically stressed spine that has
adapted from quadrupedal to bipedal locomotion. LBP is a prevalent
health problem. Statistical reports indicate that the global
incidence rate of LBP ranges between 13.1 and 28.5% (1). Intervertebral disc degeneration
(IVDD), a complex and progressive process, is the leading cause of
chronic LBP and imposes a substantial global health and economic
burden (2). The intervertebral
disc (IVD) is the largest avascular structure in the human body,
serving as the spine's primary mechanical and biological unit. Its
highly specialized architecture, comprising a proteoglycan-rich
nucleus pulposus (NP), a collagen-dense annulus fibrosus (AF) and
superior and inferior cartilage endplates (CEP), enables efficient
load distribution while preserving spinal flexibility, stability
and mobility (3–6).
Once regarded largely as a consequence of mechanical
wear and tear, IVDD is now recognized as a complex, age-associated
degenerative process shaped by the interplay between biomechanical
forces, systemic metabolic stressors and tightly regulated local
biological signals (7). At the
cellular level, progressive loss and dysfunction of NP cells
(NPCs), including chondrocyte-like, notochordal and progenitor
populations, occur through senescence and apoptosis, fundamentally
undermining disc homeostasis (8).
Increasing evidence suggests that metabolic dysregulation exerts a
stronger influence on IVDD progression than biomechanical
alterations alone (9). Mendelian
randomization analyses have identified type 2 diabetes,
hypertriglyceridemia, elevated fasting glucose and increased HbA1c
as notable causal risk factors for IVDD, whereas the contribution
of obesity-related traits remains less certain (10).
Pathologically, IVDD unfolds through a cascade
initiated by disruption of the balance between anabolic and
catabolic activities within the disc (2,7,8,11).
Hallmark features include extracellular matrix (ECM) degradation,
chronic inflammation, oxidative stress, cellular senescence and
dysregulated cell death modalities, such as apoptosis, pyroptosis
and ferroptosis, accompanied by aberrant neoinnervation and
neovascularization. Anabolic synthesis of type I/II collagens,
glycosaminoglycans (GAGs) and proteoglycans such as aggrecan (ACAN)
and versican progressively declines, while catabolic mediators,
notably matrix metalloproteinases (MMPs) and a disintegrin and
metalloproteinase with thrombospondin motifs (ADAMTS) family
members, are upregulated, accelerating ECM breakdown and loss of
disc hydration and structural integrity (11,12).
These molecular and structural derangements culminate in disc
bulging, reduced NP cellularity and water content, disc height loss
and impaired biomechanical competence. In combination with disc
protrusion and local inflammatory responses, such changes may
compress adjacent nerve roots and manifest clinically as lumbar
disc herniation (13). Compounding
these challenges, the avascularity and hostile microenvironment of
mature IVDs, characterized by hypoxia, acidosis and restricted
nutrient diffusion, severely limit intrinsic regenerative capacity
(5,14–17).
Aging and accelerated cellular senescence are
central drivers of IVDD and are tightly linked to epigenetic
dysregulation. Epigenetic alterations fuel metabolic imbalance,
persistent inflammation, oxidative damage, genomic instability and
progressive ECM degradation. Key regulatory layers include DNA
methylation, histone modifications, diverse classes of non-coding
RNAs (ncRNAs) and RNA-specific modifications such as
N6-methyladenosine (m6A) methylation. Specifically, DNA
methylation participates in the degenerative process by altering
the methylation status of cytosine bases and regulating the
expression of genes associated with ECM metabolism, inflammatory
responses and ion channel function. Mechanistically, this
epigenetic modification involves the addition of methyl groups to
cytosine residues, typically silencing gene expression when
occurring at promoter regions (18). Histone modifications (such as
methylation and acetylation) modulate cellular senescence and
inflammatory responses by remodeling chromatin structure and
influencing the accessibility of downstream pro-degenerative or
protective genes (19). Meanwhile,
microRNAs (miRNAs), long non-coding RNAs (lncRNAs) and circular
RNAs (circRNAs) function as powerful epigenetic regulators by
shaping chromatin landscapes, recruiting chromatin-modifying
complexes, and controlling mRNA stability and translation (20). Beyond these classical mechanisms,
RNA-specific modifications such as m6A methylation
represent a rapidly emerging layer of epigenetic control that
influences RNA stability, splicing and translation, and has been
implicated in regulating cellular senescence and matrix metabolism
in degenerative conditions such as IVDD (21). Through extensive crosstalk with
signaling pathways, these mechanisms orchestrate nearly every stage
of IVDD pathogenesis.
In the context of aging, epigenetic changes mainly
exhibit two patterns: One is a stochastic and cumulative
‘epigenetic drift’ and the other is a highly programmed ‘epigenetic
clock’. Epigenetic drift refers to the random and disordered
alterations in the epigenome (especially DNA methylation patterns)
that occur with advancing age, reflecting the gradual failure of
epigenetic maintenance systems, increased genomic instability and
exacerbated interindividual heterogeneity (22). By contrast, the epigenetic clock is
an algorithmic model constructed by integrating age-dependent DNA
methylation profiles at specific CpG sites, which can accurately
estimate an individual's biological age (23). Notably, epigenetic drift forms the
basis of epigenetic clock signals, yet the two display markedly
distinct response patterns to lifespan interventions, developmental
processes and cellular dedifferentiation (24). Therefore, deciphering how
physiological epigenetic drift transitions into pathological
degeneration, how epigenetic signatures evolve across disease
stages and populations, and whether targeted epigenetic
reprogramming can halt or reverse IVDD represents a critical
frontier with notable therapeutic promise (2,13,18,25).
Crucially, the translation of epigenetic discoveries
into clinical relevance depends on the choice of appropriate
experimental models. Although murine needle-puncture models are
widely used, their capacity to mimic the slow, cumulative
epigenetic drift characteristic of human disc aging is increasingly
questioned due to evolutionary, anatomical and biomechanical
disparities. In the present review, current epigenetic research
methodologies applied to IVDD are evaluated, highlighting their
strengths and limitations in resolving complex regulatory networks.
Commonly used animal models are further critically evaluated,
focusing on their translational relevance for epigenetic studies,
to provide a framework for more precise and clinically meaningful
disc degeneration research.
Epigenetic research methods in IVDD
DNA methylation
DNA methylation status reflects a dynamic
equilibrium between DNA methyltransferases (DNMTs) and
demethylating ten-eleven translocation (TET) enzymes. DNMTs add
methyl groups to the fifth carbon of cytosine residues, forming
5-methylcytosine (5-mC) within CpG dinucleotides. The DNMT family
orchestrates this landscape: DNMT1 functions as the maintenance
methyltransferase during replication, whereas DNMT3A and DNMT3B are
responsible for de novo methylation. Conversely, these
methylation footprints can be removed by TETs, which oxidize 5-mC
to 5-hydroxymethylcytosine (5-hmC) and further to formyl and
carboxyl-cytosine derivatives (18,26,27).
The central role of these enzymes in disease progression and their
therapeutic potential has been highlighted.
Mechanistic drivers in IVDD
pathogenesis
DNA methylation acts as a pivotal switch in distinct
pathological cascades, such as senescence and ferroptosis (DNMT3B
axis), and inflammation and matrix catabolism (DNMT3A axis).
Upregulation of DNMT3B can be a dual driver of NPC
senescence and ferroptosis. In one pathway, excessive DNMT3B
activity, triggered by loss of m6A modification due to
elevated activity of AlkB homolog 5, RNA demethylase (ALKBH5), an
m6A demethylase, induces hypermethylation of the
transcription factor E4F1, promoting NPC senescence and IVDD.
Silencing ALKBH5 or DNMT3B in IVDD rats confers protection against
degeneration in vivo (28).
In parallel, inside the tert-butyl hydroperoxide-induced
degenerative human NPCs, DNMT3B is upregulated and exacerbates
ferroptosis and oxidative stress by downregulating the ferroptosis
suppressor SLC40A1. Therapeutic intervention with the pan-DNMT
inhibitor 5-azacytidine (5-Aza) restores SLC40A1 and alleviates
ferroptosis in rat models (29).
DNMT3A abundance and activity increase with disease
severity, promoting hypermethylation and suppression of the
peroxisome proliferator-activated receptor γ (PPARγ) promoter. This
triggers a chain reaction leading to NF-κB activation, exacerbating
apoptosis and matrix imbalance. Suppression of DNMT activity
effectively preserves NPC viability and matrix homeostasis.
Specifically, silencing DNMT3A reduces IL-1β-induced apoptosis and
ECM degradation, while 5-Aza treatment counters endoplasmic
reticulum stress by inhibiting DNMT1/3A, thereby demethylating the
PPARγ promoter to restore its expression in vitro.
Intradiscal delivery of short hairpin RNA-DNMT3A in rat IVDD models
restored ECM anabolism and reduced NPC apoptosis (30,31).
The epigenetic landscape may also differ between
symptomatic and asymptomatic IVDD. A study using DNA methylation
arrays comparing painful vs. non-painful IVDD discovered
differential promoter CpG methylation in equally degenerated discs.
Genes related to matrix catabolism, such as MMP2 and MMP28, were
less methylated and upregulated in the painful discs, whereas genes
associated with matrix anabolism, including CHSY1 and
chondroadherin, show an opposite pattern (32). In addition, the global DNA
methylation status in rat AF following puncture-induced IVDD has
been assessed by measuring epigenetic markers, including 5-mC,
5-hmC, DNMTs and TETs. Elevated global hypermethylation and
increased DNMT1 expression are notably associated with abnormal
expression of the pain-related ion channel transient receptor
potential cation channel subfamily V member 1, suggesting a key
role for these epigenetic changes in ectopic nerve innervation of
the IVD and pain development (33). However, few studies have directly
or systematically stratified epigenetic signatures in IVDD by
etiology, sex or other demographic characteristics (34).
Evolution of detection techniques
A plethora of high-throughput detection techniques
have been applied to DNA methylation analysis in IVDD, ranging from
single-locus (site-specific DNA methylation typing) to global
methylome profiling, as well as cutting-edge epigenome editing
tools such as CRISPR-dCas9-Tet1-mediated DNA methylation editing.
Locus-specific approaches focus on individual CpG sites or regions
and employ techniques such as methylation-specific PCR (MSP) and
bisulfite sequencing PCR (BSP), sometimes combined with restriction
analysis or matrix-assisted laser desorption ionization
time-of-flight mass spectrometry for quantitative assessment. Using
MSP and BSP, hypermethylation of the secreted protein acidic and
rich in cysteine (SPARC) promoter was observed in NP tissues from
middle-aged (6 months) mice, increasing further in older (15
months) mice, whereas young (3 months) mice showed minimal
methylation. This age-related SPARC silencing was associated with
anatomical signs of intervertebral disc degeneration (e.g., disc
height loss in mice and imaging evidence of degeneration in humans)
and behavioral signs of chronic low back pain (e.g.,
movement-evoked axial discomfort and cold hypersensitivity in mice,
and self-reported chronic pain in humans) in both species (35).
By comparison, global DNA methylome profiling
provides genome-wide quantitative information on DNA methylation
patterns. These approaches include whole-genome bisulfite
sequencing (WGBS), methylation-sensitive restriction enzyme-based
sequencing and methylated DNA immunoprecipitation sequencing
(36). Human NP studies using WGBS
have identified 220 differentially methylated loci in NF-κB, MAPK
and Wnt signaling pathways between early and advanced stages, with
late-stage samples showing predominant hypermethylation (216 loci).
This hypermethylation is associated with silencing of regulatory
genes and dysregulation of cell adhesion, contributing to the
progression of disc degeneration (37).
Histone modification
Histone acetylation
Post-translational modifications (PTMs) of histones,
including methylation, acetylation, phosphorylation, ubiquitination
and ADP-ribosylation, modulate chromatin, thereby controlling
genome accessibility architecture. Central to this regulation is
the dynamic equilibrium of histone acetylation. The dysregulation
of histone acetylation at the N-terminal tail and core domains is
catalyzed by histone acetyltransferases (HATs) and histone
deacetylases (HDACs) (38). HATs
neutralize the positive charge of lysine residues, relaxing
chromatin to promote transcription, whereas HDACs restore this
charge, tightening histone-DNA interaction and inducing chromatin
condensation and transcriptional repression (39). In IVDD, the dysregulation of the
HAT/HDAC balance reflects broader epigenetic regulatory network
(ERN) instability, with specific HDACs serving opposing roles.
Elevated expression of HDAC7 and HDAC4, which act as
detrimental regulators, is associated with disease progression
(40–42). Conversely, broad inhibition using
HDAC inhibitors (such as SAHA) can shift cytokine-stimulated
chondrocytes from a catabolic (MMP-2/3/9, ADAMTS-5) to an anabolic
(COL2A1, aggrecan) phenotype (43). However, not all deacetylases are
deleterious; HDAC9 and SIRT6 appear protective in both aging and
injury models: HDAC9 preserves nucleus pulposus cell viability and
suppresses apoptosis by deacetylating RUNX3 and promoting its
ubiquitin-proteasomal degradation, while SIRT6 maintains disc
health by reducing DNA damage, enhancing autophagy, and inhibiting
cellular senescence and the SASP (44,45).
Modulating this landscape offers tangible therapeutic avenues,
exemplified by the plasmid-mediated delivery of pluripotency
factors (Oct4, Krüppel-like factor 4 and Sox2), which reduces
repressive H4K20me3 levels to alleviate low back pain and
degeneration in rat models (46).
Canonical histone methylation and
inflammation
While acetylation acts as a rapid switch, histone
methylation provides stable, long-term regulation of gene
expression. Previous high-impact evidence highlights the enhancer
of zeste homolog 2 (EZH2)-H3K27me3 axis as a critical gatekeeper of
inflammation in IVDD. EZH2 is the methyltransferase responsible for
placing the repressive H3K27me3 mark. In degenerative NPCs, EZH2
expression is markedly downregulated. This loss leads to the
erasure of H3K27me3 at the promoters of key pro-inflammatory and
pro-pyroptotic genes, such as Dickkopf Wnt signaling pathway
inhibitor 1 (DKK1) and MAPK1. The consequent de-repression of these
genes activates the NLRP3 inflammasome and drives pyroptosis
(inflammatory cell death). Mechanistically, restoring EZH2 function
or inhibiting its downstream targets (such as DKK1) has been
demonstrated to re-establish the repressive chromatin state,
thereby attenuating the inflammatory cascade and preserving the
disc matrix (47,48).
Histone lactylation
Lactate-derived histone lysine lactylation (Kla) has
emerged as a novel mechanism linking cellular metabolism directly
to gene transcription. Catalyzed by lactyltransferases (such as
p300) and removed by delactylases, this modification involves
attaching lactyl groups from glycolysis-derived lactate to histone
lysines (49).
In IVDD, a metabolic shift toward hyperglycolysis
leads to intracellular lactate accumulation. Single-cell RNA
sequencing (scRNA-seq) confirms that this excess lactate drives
H3K18la (lactylation of H3 lysine 18) specifically at the promoter
of acyl-CoA synthetase long-chain family member 4 (ACSL4). The
transcriptional activation of ACSL4 primes NPCs for lipid
peroxidation and ferroptosis. Consequently, targeting this axis,
either by silencing lactate dehydrogenase A [LDHA; via AAV9-small
interfering (si)RNA-Ldha] or inhibiting glycolysis with
2-deoxy-D-glucose, suppresses H3K18la levels and ameliorates
degeneration. Clinical relevance is further supported by integrated
bulk and single-cell analyses showing that histone lactylation
levels, and lactylation-related genes such as chromobox 3, are
strongly associated with IVDD severity in human tissues (50,51).
High-resolution profiling
Deciphering these complex modifications requires a
transition from bulk assays to high-resolution genomic mapping. By
combining chromatin immunoprecipitation-sequencing (ChIP-seq) with
RNA-seq, researchers have mapped genomic interactions, identifying
nuclear receptor 4A3 as a direct downstream target of the regulator
early growth response factor 1, whose binding promotes NPC
apoptosis and ECM breakdown (52).
To understand chromatin accessibility, transposase accessible
chromatin sequencing has been integrated with transcriptional
profiling, revealing that activating protein-1 transcription
factors, particularly c-Fos, increase chromatin accessibility to
upregulate cell migration-inducing and hyaluronan-binding protein,
a hyaluronidase that disrupts matrix homeostasis. Emerging
techniques such as Cleavage Under Targets and Tagmentation
(CUT&Tag) now offer higher signal-to-noise ratios than
traditional ChIP, revealing that H3K18la is specifically enriched
at the promoters of pro-inflammatory genes such as thrombospondin 1
in degenerative NPCs. Furthermore, advanced multi-omics integration
has uncovered non-canonical mechanisms, such as the
methyltransferase EZH2 monomethylating the RNA-binding protein
DDX1. Validated by RNA-seq and RNA immunoprecipitation (RIP)-seq,
this modification triggers exon skipping in matrin 3 (MATR3),
generating a short isoform (MATR3-S) that disrupts nuclear
architecture and activates Wnt signaling (53–56).
m6A methylation
RNA methylation constitutes the most abundant
internal RNA modification. Particularly, m6A methylation
has drawn great attention for its role in fine-tuning gene
expression in IVDD. This reversible modification involves adding a
methyl group to the nitrogen-6 position of adenosine in RNA,
regulating RNA stability, splicing, decay and translation.
Imbalances in m6A writers [methyltransferase 3,
N6-adenosine-methyltransferase complex catalytic subunit (METTL3),
methyltransferase 14, N6-adenosine-methyltransferase non-catalytic
subunit and WT1-associating protein], erasers
[α-ketoglutarate-dependent dioxygenase FTO (FTO) and ALKBH5] and
reader proteins have been implicated in disease progression.
Advances in profiling the m6A
epitranscriptome are flourishing, driven by a wide spectrum of
high-throughput techniques. The current toolkit encompasses three
major categories based on detection principles: Antibody-based
methods use anti-m6A antibodies to immunoprecipitate
methylated RNA fragments followed by sequencing. Owing to their
relative maturity and ease of use, such approaches are the
preferred choice for early exploratory research and large-scale
sample screening. m6A-seq/m6A RNA
immunoprecipitation sequencing (MeRIP-seq) represents the
foundational approach, enriching m6A-containing RNA
fragments through immunoprecipitation and identifying methylated
regions at 100–200 nucleotide resolution; this method has been
widely employed for mapping m6A distribution
transcriptome-wide in diverse biological systems.
m6A-seq2 improves upon this with optimized fragmentation
and library preparation protocols. m6A
individual-nucleotide resolution crosslinking and
immunoprecipitation sequencing employs UV crosslinking to
covalently link antibodies to m6A sites, enabling
single-nucleotide resolution mapping through characteristic
mutation signatures at crosslink sites. m6A-level and
isoform-characterization sequencing combines immunoprecipitation
with full-length transcript sequencing to quantify m6A
levels on individual transcript isoforms (57–59).
While antibody-based methods are widely adopted for
transcriptome-wide profiling, they require substantial RNA input
(typically 300 µg for standard protocols, although optimized
versions can work with as little as 500 ng) and may exhibit
cross-reactivity with other modifications such as
N6,2′-O-dimethyladenosine (57,60).
Refined protocols using alternative antibodies (such as the highly
specific, commercially available monoclonal anti-m6A antibody
produced by Cell Signaling Technology, Inc.) have markedly reduced
input requirements and costs while maintaining comparable
sensitivity (61).
Chemical-based approaches achieve antibody-free
detection through selective chemical reactions that distinguish
methylated from unmethylated adenosines. By directly labeling or
differentiating modified bases via chemical reactions, these
approaches enable more accurate quantification and absolute
detection. m6A-selective electrophilic affinity labeling
sequencing (m6A-SEAL-seq, a chemical strategy that
covalently captures oxidized m6A electrophilic
intermediates using specific probes) uses FTO-assisted chemical
labeling to tag m6A sites, enabling enrichment, imaging
and sequencing applications with good sensitivity and specificity
(62). m6A-selective
allyl chemical labeling-seq employs chemical derivatization to
enable base-resolution mapping with quantification capabilities.
m6A-label-seq utilizes metabolic labeling combined with
chemical reactions.
m6A-glyoxal and nitrite-mediated
deamination of unmethylated adenosines-seq represents a
breakthrough method that deaminates unmethylated adenosines to
inosines while leaving m6A residues intact, enabling
absolute quantification of m6A stoichiometry at
single-base resolution, conceptually analogous to bisulfite
sequencing for DNA methylation (63,64).
This unbiased approach reveals clustered m6A
modifications with differential distribution and stoichiometry, and
characterizes m6A dynamics under stress conditions
(64). Updated versions (GLORI 2.0
and 3.0) have dramatically reduced RNA input requirements to as low
as 500–1,000 cells while accelerating reaction times and minimizing
RNA degradation, greatly expanding applicability for low-input
samples (65). Chemical methods
offer unbiased, quantitative m6A detection without
antibody-related artifacts.
Enzyme-based methods exploit enzymatic activities
that discriminate between m6A and unmodified adenosine.
By utilizing the specific cleavage or editing activities of
enzymes, these approaches offer an antibody-independent
alternative, which is particularly suitable for studies requiring
high sensitivity or aiming to avoid antibody-related biases.
m6A-m6A-sensitive
RNA-endoribonuclease-facilitated (MATZER)-seq uses the bacterial
endoribonuclease MazF, which cleaves unmethylated ACA motifs but
not m6A-containing ACA sequences, generating
characteristic cleavage patterns that reveal m6A
positions at single-nucleotide resolution. This method permits
systematic quantitative profiling of m6A at 16–25% of
expressed sites and enables de novo discovery of
m6A sites, calibration of antibody-based approaches and
quantitative tracking of m6A dynamics during
differentiation. m6A-sensitive
RNA-endoribonuclease-facilitated sequencing with refinement
improves upon MAZTER-seq with enhanced computational analysis
(66).
m6A-deamination adjacent to RNA
modification targets (DART)-seq employs a fusion protein combining
the cytidine deaminase apolipoprotein B mRNA editing enzyme
catalytic subunit 1 with the m6A-binding YTH domain,
which induces C-to-U deamination at sites adjacent to
m6A that are then detected by standard RNA-seq (60,67,68).
DART-seq requires minimal RNA input (as little as 10–50 ng of total
RNA) and can track m6A accumulation in cells over time;
long-read DART-seq provides insights into m6A
distribution along individual transcripts. However, DART-seq
depends on the presence of cytidines near m6A sites and
may have variable efficiency across different sequence contexts
(68,69).
Each methodological category offers distinct
advantages: Antibody-based methods provide established workflows
for transcriptome-wide profiling with extensive bioinformatics
support; chemical-based approaches enable absolute quantification
without antibody bias; and enzyme-based methods offer low-input,
antibody-free alternatives with unique detection mechanisms. The
choice of method depends on research goals, available sample
quantity, desired resolution (region-level vs. single-nucleotide)
and whether absolute or relative quantification is needed. For IVDD
research involving limited clinical specimens, low-input methods
such as GLORI 3.0 or DART-seq may be particularly advantageous.
Mapping the degenerative epitranscriptome:
Inflammation and autophagy
Global and focal m6A epitranscriptomic
changes in IVDD have been mapped both in human and rodent tissues.
MeRIP-seq analysis [validated by reverse transcription-quantitative
PCR (RT-qPCR)] identified distinct signatures: Annexin A2
transcripts show increased m6A levels in degenerative
discs, whereas SLC3A2 and pre-B-cell leukemia transcription factor
3 show decreased methylation. These differentially methylated
transcripts were enriched in pathways highly relevant to disc
pathology, such as NF-κB signaling and ECM-associated processes,
suggesting that m6A modifications are central to
influencing inflammation and maintaining matrix homeostasis to IVDD
progression (70). Dynamic
m6A profiling of aging rat NP tissue using MeRIP-seq
combined with RNA-seq further revealed a progressive, age-dependent
increase in global m6A levels. This accumulation is
associated with gene expression shifts in pathways linked to
inflammation, cellular stress responses and ECM metabolism
(71).
Beyond these foundational findings, recent evidence
highlights the pivotal role of m6A in regulating
autophagy and pyroptosis, two critical fate determinants in IVDD.
New studies indicate that METTL3-mediated m6A
modification stabilizes autophagy related 7 mRNA, a core autophagy
gene. In degenerative NPCs, elevated METTL3 promotes senescence by
disrupting the autophagic flux (72,73).
Conversely, the eraser FTO has been shown to demethylate NLRP3
mRNA. Downregulation of FTO during degeneration leads to
m6A-dependent stability of NLRP3, thereby activating the
inflammasome and driving pyroptosis in NPCs (74). These findings suggest that the
m6A machinery acts as a ‘molecular switch’ between cell
survival (autophagy) and inflammatory death (pyroptosis).
Diagnostic stratification via
regulator variability
Beyond pathogenesis, m6A regulator
expression may serve as a sensitive indicator of inter-individual
epigenetic variability. A comprehensive analysis using Gene
Expression Omnibus datasets, assisted by machine learning and LASSO
regression, screened for differentially expressed m6A
regulators and analyzed associated immune infiltration
characteristics in IVDD. The study generated a predictive model
based on five key regulators (RBM15, YTHDC1, YTHDF3, HNRNPA2B1 and
ALKBH5). Validated in rat models, this signature stratifies
patients into eight distinct clusters, each associated with unique
immune infiltration characteristics. These clusters may underlie
subpopulation-specific IVDD trajectories and immune responses,
offering a roadmap for personalized epigenetic diagnostics
(75).
ncRNAs
Regulatory triad: miRNAs, lncRNAs and
circRNAs
ncRNAs orchestrate gene expression at
transcriptional or post-transcriptional levels without encoding
proteins. In IVDD, three major classes interact synergistically to
modulate ECM metabolism, apoptosis, senescence, inflammation and
autophagy (20,76–78).
miRNAs, which are 18–22 nucleotides in length, act as endogenous
silencers by binding to the 3′ untranslated regions of target
mRNAs, leading to mRNA degradation or translational inhibition.
Their effects frequently converge on the NF-κB signaling hub,
thereby driving matrix degradation and cell death. These miRNAs are
themselves regulated by longer transcripts, including lncRNAs and
circRNAs. lncRNAs, typically exceeding 200 nucleotides, modulate
gene expression via complex secondary structures that influence
chromatin remodeling and mRNA stability. Meanwhile, circRNAs are
characterized by a covalent closed-loop structure that renders them
resistant to exonuclease degradation, and also serve as protein
decoys or scaffolds, influencing diverse cellular processes. A
dominant regulatory mechanism in disc homeostasis is the competing
endogenous RNA (ceRNA) hypothesis, wherein lncRNAs and circRNAs
contain miRNA response elements that sponge specific miRNAs,
thereby lifting the repression of downstream target genes.
IVDD-associated ncRNAs can be protective or detrimental; what
matters most is the perturbation of the regulatory network
(20,76–78).
High-resolution research
methodologies
Research on ncRNAs employs an integrated workflow
combining high-throughput discovery with mechanistic validation.
The process typically begins with omics-based profiling, using
RNA-seq, microarray hybridization or single-cell transcriptomics to
map dysregulated profiles (79–81).
For instance, microarray analysis has identified the lncRNA
RP11-296A18.3 and its neighboring gene Fas associated factor 1 as
pro-apoptotic drivers in degeneration (82). Once candidates are identified,
their spatial expression is defined using RT-qPCR, FISH or in
situ hybridization. To confirm the physical interaction between
ncRNAs, researchers utilize luciferase reporter assays to verify
binding sites and RIP to identify protein partners. Specifically,
Argonaute 2 (AGO2)-RIP is crucial for validating the sponging
mechanism; since AGO2 is the core component of the RNA-induced
silencing complex, enriching for AGO2-bound transcripts confirms
the regulatory roles of key molecules such as circVMA21, circFOXO3,
circEYA3 and circATXN1 (83–86).
Finally, functional roles are assessed via gain- and
loss-of-function strategies (using mimics, inhibitors or CRISPR)
paired with phenotypic readouts such as TUNEL staining for
apoptosis, EdU incorporation for proliferation and Alcian blue
staining for matrix synthesis (87–90).
Beyond cytoplasmic sponging, advanced techniques are
uncovering novel ncRNA functions in the nucleus and extracellular
space. Emerging methods such as CUT&Tag and CUT & Release
Using Nuclease bridge RNA-protein binding with genomic
localization, revealing that some ncRNAs recruit transcriptional
regulators directly to chromatin (91). For example, the nuclear circFUNDC1
was found to recruit CDK9 to the FUNDC1 promoter, boosting
transcription and promoting mitophagy to protect against oxidative
stress (92). Furthermore,
previous evidence highlights that exosomes serve as critical
vehicles for intercellular rejuvenation. Urine-derived stem cell
exosomes have been shown to deliver the matrilin-3 protein to NPCs,
where it activates the TGF-β signaling pathway to promote matrix
synthesis and suppress senescence (93). These findings establish exosomal
ncRNA tracking and delivery as a promising non-viral strategy for
disc repair.
Notably, the plasticity and complexity of ncRNA
networks are best illustrated by the conflicting roles of the
lncRNA HOTAIR, which suggests a biphasic function driven by disease
stage or stress type. Although some studies report that HOTAIR is
downregulated in patients, where its restoration exerts a
protective effect by suppressing MMP-13 via the sponging of
miR-642a-5p (94) or preventing
apoptosis by sequestering miR-34a (95,96),
more evidence suggests that HOTAIR upregulation is associated with
IVDD severity by activating the Wnt/β-catenin pathway and
AMPK/mTOR/ULK1 pathway to drive the NPC degenerative changes.
Moreover, targeted inhibition via siHOTAIR effectively improved ECM
composition and attenuated IVDD symptoms in rat models (97,98).
This discrepancy likely stems from variations in sample source, as
‘control’ samples often differ (such as idiopathic scoliosis vs.
trauma) and ‘disease’ samples represent different pathological
stages (acute inflammation vs. chronic oxidative stress). It
remains an intriguing research avenue to discern how regulators
such as HOTAIR may transition from an anti-apoptotic factor under
inflammatory cues to a pro-degenerative factor under chronic stress
conditions.
Advances in mitochondrial epigenetics
(mitoepigenetics) in IVDD
Mitoepigenetics represents an emerging regulatory
layer distinct from nuclear epigenetics, with critical implications
for IVDD. Epigenetic mechanisms regulate mitochondrial quality
control (MQC) through interconnected pathways involving DNA
methylation, histone modifications and m6A RNA
methylation, directly influencing mitochondrial biogenesis,
mitophagy, dynamics and oxidative stress responses in NPCs
(14,99,100).
Distinguishing mitochondrial from
nuclear epigenetics
Mitoepigenetics differs fundamentally from nuclear
epigenetics in DNA packaging, modification patterns and functional
consequences. Nuclear DNA is packaged with histone proteins into
nucleosomes, allowing extensive histone modifications (acetylation,
methylation and phosphorylation) that regulate gene expression
(99,101). By contrast, mitochondrial DNA
(mtDNA) exists in nucleoids, DNA-protein complexes lacking histones
that instead contain mitochondrial transcription factor A and other
nucleoid-associated proteins (99,102). Epigenetic regulation in
mitochondria therefore relies on PTMs of these nucleoid proteins
rather than histone modifications (103).
Although DNA methylation occurs in both genomes,
critical differences exist. Nuclear DNA methylation primarily
occurs at CpG dinucleotides and typically represses gene
transcription when present at promoters (104). mtDNA methylation involves
multiple patterns including 5-mC, 5-hmC, and N6-methyladenine
(105). Notably, experimental
evidence suggests that GpC methylation, not CpG methylation, may be
the primary functional regulator of mitochondrial gene expression,
and mtDNA methylation at promoters appears to activate rather than
repress transcription; diametrically opposed to nuclear DNA
methylation effects (105,106).
Mitoepigenetic modifications utilize mitochondrial
isoforms of nuclear enzymes. DNMTs (mtDNMT1 and DNMT3B) localize to
mitochondria and methylate mtDNA, while TET-like hydroxymethylase
activity has been detected in mitochondria for demethylation
(106). A unique aspect of
mitochondrial epigenetics is its bidirectional relationship with
nuclear epigenetics: Mitochondria produce metabolites (acetyl-CoA,
α-ketoglutarate and S-adenosylmethionine) that serve as substrates
for nuclear epigenetic enzymes, while nuclear-encoded factors
regulate mitochondrial epigenetic machinery through anterograde
signaling (101,104,107).
mtDNA D-loop hypermethylation in
IVDD
Recent studies indicate that hypermethylation of the
mtDNA displacement loop (D-loop), the control region for
mitochondrial replication and transcription, is markedly elevated
in degenerative human NP tissues. This D-loop hypermethylation,
mediated by DNMT1 translocation to mitochondria, represses
mitochondrial gene expression (such as ND1 and COX1), driving
metabolic reprogramming and oxidative stress (108–110). In other disease models, D-loop
methylation levels are inversely associated with mtDNA copy number,
suggesting compensatory mechanisms in response to mitochondrial
dysfunction (109,111).
Single-cell approaches to
mitochondrial dysfunction
scRNA-seq has proven particularly valuable for
dissecting cellular heterogeneity and identifying mitochondrial
dysfunction signatures in degenerative disc tissues (112,113). Recent scRNA-seq-guided studies
revealed critical roles for mtDNA/SPARC-STING signaling pathways in
fibrotic phenotype polarization of NP cells during IVDD
progression. These findings have informed novel therapeutic
strategies, including engineered mitochondrial transplantation that
improves MQC in NP cells under pathological conditions (114–116).
Nuclear epigenetic regulation of
MQC
DNA methylation and mitochondrial oxidative damage.
DNMT3B-mediated DNA hypermethylation represents a key epigenetic
driver of mitochondrial dysfunction in IVDD. DNMT3B is highly
expressed in degenerated NP tissue and promotes hypermethylation of
genes enriched in oxidative stress and ferroptosis pathways.
Specifically, DNMT3B-mediated hypermethylation silences the
ferroptosis suppressor gene SLC40A1 (ferroportin), leading to iron
accumulation, lipid peroxidation and mitochondrial oxidative
damage. Treatment with the DNA methylation inhibitor 5-azacytidine
(5-Aza) alleviates IVDD in rat models by restoring SLC40A1
expression and reducing ferroptosis-associated mitochondrial
dysfunction (29,100).
Histone modifications and metabolic homeostasis.
H3K4me3 catalyzed by SET domain containing protein 1A (SETD1A)
serves a protective role in maintaining mitochondrial metabolic
function. H3K4me3 levels are markedly decreased in degenerated NP
tissues (117). SETD1A regulates
glycolytic metabolism, the primary energy pathway in the avascular
intervertebral disc., through the H3K4me3-HELZ2/PPARα-hypoxia
inducible factor 1α (HIF1α) axis. Loss of SETD1A reduces H3K4me3
enrichment at the HELZ2 promoter, suppressing the HELZ2/PPARα
complex and downregulating HIF1α, which impairs glycolytic capacity
and induces cellular senescence (117). Since mitochondria participate in
metabolic regulation beyond glycolysis in disc cells, this histone
modification pathway indirectly affects mitochondrial quality by
altering cellular metabolic homeostasis.
m6A methylation and mitophagy regulation.
m6A methylation represents a critical epitranscriptomic
mechanism linking metabolic intermediates to MQC. METTL3-mediated
m6A modification regulates α-ketoglutarate
(α-KG)-dependent mitophagy through the MALAT1/miR-23c/isocitrate
dehydrogenase 1 (IDH1) axis. α-KG, a tricarboxylic acid cycle
intermediate which is markedly decreased in degenerated NP tissues,
serves dual functions: As a metabolic substrate and as a cofactor
for TET DNA demethylases and Jumonji-C histone demethylases.
Mechanistically, METTL3-mediated m6A modification
destabilizes the lncRNA MALAT1, releasing miR-23c to suppress IDH1,
the enzyme responsible for α-KG production. Reduced α-KG levels
impair mitophagy, leading to accumulation of dysfunctional
mitochondria, increased reactive oxygen species (ROS) production
and enhanced apoptosis. Supplementation with α-KG restores NP cell
proliferation, reduces apoptosis and reestablishes ECM homeostasis
(100,118).
m6A modifications also regulate
autophagy-related genes under stress conditions, providing
additional control over mitochondrial quality (21). Multiple m6A regulators
including RBM15, YTHDC1, YTHDF3, HNRNPA2B1 and ALKBH5 show
differential expression in IVDD, with METTL3/YTHDC1 co-regulation
mediating m6A hypermethylation of RNF41 mRNA to impair
autophagy and ECM integrity (119,120).
Bidirectional metabolic-epigenetic
crosstalk
A bidirectional relationship exists between
mitochondrial metabolism and epigenetic regulation in IVDD.
Mitochondria-derived metabolites serve as essential cofactors for
epigenetic enzymes: NAD+ for sirtuin deacetylases, α-KG
for TET demethylases and Jumonji-C demethylases, and
S-adenosylmethionine for methyltransferases (100,107). Conversely, epigenetic
modifications regulate expression of genes encoding MQC machinery,
including those governing mitochondrial dynamics (fusion/fission),
mitophagy pathways (PINK1-PRKN) and mitochondrial biogenesis
(PPARGC1A/PGC-1α) (121,122).
Protein-level integration with
epigenetic control
The NOD-like receptor X1 (NLRX1)-SLC39A7 complex
exemplifies how protein-level regulation integrates with epigenetic
control to maintain mitochondrial quality. This complex
orchestrates mitochondrial dynamics and mitophagy by modulating
mitochondrial zinc trafficking, coupling mitochondrial fission
factors (dynamin-1-like protein), fusion regulators (OPA1
mitochondrial dynamin like GTPase) and mitophagy activity. Loss of
NLRX1 triggers compensatory activation of the PINK1-PRKN pathway,
leading to excessive mitophagy and accelerated NP cell senescence
(123,124). NLRX1 is the only known
mitochondria-localized NLR family member and functions as a
negative regulator of inflammation while modulating mitochondrial
metabolism and autophagy (124,125).
Targeted epigenetic editing
To establish a definitive causal link between
epigenetic modulation and IVDD, and to facilitate the transition
from observation to therapy, the field is increasingly adopting
targeted epigenetic editing. Unlike traditional gene therapy that
alters the genomic sequence, platforms such as CRISPR/dCas9,
transcription activator-like effector nucleases or zinc-finger
proteins fused to chromatin-modifying enzymes (such as DNMT3A,
TET1, KRAB and p300) allow for the precise, reversible regulation
of gene expression. These approaches are currently in various
stages of in vivo investigation across a spectrum of
metabolic, ophthalmic, neurobiological and musculoskeletal
disorders (126,127). The translational feasibility of
this technology in complex tissue environments has been
demonstrated in non-spinal models; for instance, mice receiving
lipid nanoparticle-derived mRNA encoding engineered zinc-finger
repressors for PCSK9 achieved stable transcriptional repression
through repressive chromatin remodeling, offering a potential
treatment for hypercholesterolemia (128). Similarly, AAV-mediated delivery
of dSaCas9 fused to transcriptional repression domains derived from
KRAB and MeCP2 successfully induced localized chromatin repression
of ApoE in the mouse brain, mitigating the most prominent genetic
risk factor for Alzheimer's disease (129).
In the specific context of IVDD and discogenic
pain, research has progressed from germline-edited models [such as
zygote microinjection with dCas9-DNMT3A (130) or scaffold-based recruitment
systems (131)] to somatic,
therapeutically relevant interventions. A primary focus has been
the modulation of the ‘gut-disc-nerve’ or inflammatory axes.
Utilizing in vitro and ex vivo models, researchers
have employed lentiviral dCas9-KRAB constructs to target the
promoters of inflammatory receptors IL6st, TNFR1 and IL1R1 in rat
dorsal root ganglion neurons. This multiplex epigenome editing
effectively silenced cytokine signaling cascades, thereby
desensitizing nociceptive neurons to the inflammatory milieu of the
degenerative disc (132,133). Building on this, the same group
demonstrated the in vivo therapeutic efficacy of CRISPR
epigenome editing of TNFR1 in a needle puncture-induced rat model,
demonstrated that targeted repression of inflammation can alleviate
behavioral signs of pain (134).
However, halting inflammation addresses only one
side of the degenerative coin; restoring the matrix requires
reactivating silenced anabolic genes. Complementing repressive
strategies, recent ‘CRISPR activation’ (CRISPRa) approaches,
utilizing dCas9 fused to transcriptional activators such as VPR or
p300, are now being explored to rejuvenate the disc matrix.
Emerging evidence suggests that targeting the promoters of core
anabolic genes, such as ACAN and COL2A1, can reverse the
age-related epigenetic silencing of these loci. By depositing
active histone marks (such as H3K27ac) at these promoters, CRISPRa
systems can restore physiological gene expression levels and
promote functional matrix repair in degenerative NPCs, offering a
dual-pronged strategy when combined with inflammatory repression
(Table I) (28–30,33,35,37,50,52,54–56,70,71,79–86,92,133–135).
 | Table I.Core epigenetic techniques in IVDD
research. |
Table I.
Core epigenetic techniques in IVDD
research.
| Epigenetic
category | Methodological
approach | Specific
techniques/tools | Key
applications& insights in IVDD | (Refs.) |
|---|
| DNA
methylation | Locus-specific
methylation typing | MSP/BSP,
COBRA/MALDI-TOF MS | Observed
age-related hypermethylation of the SPARC promoter associated with
chronic low back pain. | (35) |
|
|
| BSP + dot blot | Revealed global
hypermethylation and DNMT1 expression associated with the
upregulation of the pain-related channel TRPV1 in rat AF
tissue. | (33) |
|
|
| MSP/western
blot | 5-Azacytidine
inhibits ER stress and apoptosis in NPCs by preser ving
PPARγexpression via promoter demethylation. | (30) |
|
|
| BSP/ChIP/western
blot | Oxidative stress
upregulates DNMT3B, causing hypermethylation of SLC40A1
(ferroptosis inhibitor), leading to ferroptosis; reversed by
5-Azacytidine. | (29) |
|
| Global methylome
profiling | WGBS | Identified 220
differentially methylated loci distinguishing early vs. late-stage
IVDD. | (37) |
| m6A
methylation | High-throughput
profiling | MeRIP-seq | Identified altered
m6A methylation in ANXA2, SLC3A2 and PBX3, which are
enriched in NF-κB and ECM degradation pathways. | (70) |
|
|
|
RNA-seq/LC-MS/MS | Characterized
age-dependent increases in global m6A levels in rat NP
tissue that regulate ECM metabolism and inflammation. | (71) |
|
| Mechanistic
validation | MeRIP-qPCR/RIP | Demonstrated that
ALKBH5-mediated m6A hypomethylation of DNMT3B
transcripts promotes IVDD via E4F1 deficiency. | (28) |
| Histone
modifications | Chromatin
profiling | ChIP-seq +
RNA-seq | Identified NR4A3 as
a direct downstream target of the transcription regulator EGR1,
promoting oxidative stress-induced apoptosis. | (52) |
|
|
| CUT&Tag | Revealed that
mitophagy reduces H3K18la enrichment at the THBS1 promoter, linking
mitophagy and lactate metabolism. | (56) |
|
| Chromatin
accessibility | ATAC-seq | Demonstrated that
upregulated CEMIP promotes IVDD by altering chromatin accessibility
via the AP-1 transcription factor. | (54) |
|
| Proteomics and
chromatin | LC-MS/MS | Discovered that
EZH2-mediated methylation of DDX1 alters MATR3 splicing, initiating
chromatin reprogramming and degeneration. | (55) |
|
|
Metabolic-epigenetic link | scRNA-seq +
ChIP-seq | Detected metabolic
shifts (glycolysis) driving H3K18la enrichment at the ACSL4
promoter, which activates ferroptosis in NPCs. | (50) |
| Non-coding RNA | Expression
profiling |
RNA-seq/microarray | Systematic
identification of dysregulated miRNA, lncRNA and circRNA expression
profiles in degenerative human disc tissues. | (79–82) |
|
| Functional
interaction | Luciferase/RIP/RNA
pull-down | Validated
circRNA-miRNA-mRNA axes (such as circVMA21/miR-200c/XIAP,
circEYA3/miR-196a-5p/EBF1) that regulate apoptosis and ECM
balance. | (83–86) |
|
|
|
CUT&Tag/ChIP | Demonstrated that
circFUNDC1 recruits CDK9 to the FUNDC1 promoter to enhance
mitophagy under oxidative stress. | (92) |
| Targeted epigenetic
editing | Multiplex
repression | Lentiviral
dCas9-KRAB | Simultaneous
repression of IL6st, TNFR1 and IL1R1 in DRG neurons completely
abolished IVDD-induced mechanical sensitivity. | (133) |
|
| In vivo
epigenome editing |
AAV-CRISPR-dCas9 | Successful
therapeutic epigenome editing of TNFR1 in rat IVDD models
significantly reduced inflammation and pain behavior. | (134) |
Animal models used in IVDD epigenetics
research
Common models and translational
challenges
Animal models remain indispensable for IVDD
epigenetics research because chromatin remodeling is dynamically
regulated by systemic cues (such as vascularization, immune
surveillance and neural interaction) that are notably absent in
static in vitro or organ-culture systems (136). The experimental repertoire is
extensive, utilizing both small animals (murine, rabbit or canine)
for mechanistic screening and large animals (ovine, porcine or
non-human primates) for pre-clinical validation (137). Recently, weight-bearing species
such as camelids and kangaroos have been introduced to improve
approximation of human spinal loading. These models are generally
classified into three distinct categories: Spontaneous, induced and
genetically engineered. Spontaneous models, such as the sand rat
(Psammomys obesus) or chondrodystrophoid (ChD) dogs,
naturally replicate age-related degeneration, with porcine models
offering anatomical dimensions that closely mirror human disc aging
(138,139). Conversely, induced models utilize
surgical needle puncture, enzymatic injection or controlled
mechanical vibration to trigger rapid, reproducible degeneration,
making them ideal for isolating specific mechanobiological
pathways. Bridging these approaches, genetically engineered models
introduce targeted mutations to probe specific signaling or
metabolic mediators; notably, the lamin A/C G609G/G609G knock-in
mouse, which models Hutchinson-Gilford progeria, develops
spontaneous IVDD that closely mimics the accelerated epigenetic
aging seen in humans (140).
Despite this diversity, species-specific variations
impose notable constraints on translational relevance. The primary
limitation is biomechanical; the quadrupedal posture of most models
fails to replicate the axial loading forces of the human spine.
While primates and surgically bipedal rodents offer partial
solutions, their use is limited by ethical concerns and high costs
(135,141,142). To address this, a novel
non-surgical bipedal rat model has been developed by encouraging
upright posture via specially designed cages; this approach induces
progressive degeneration over months, improving the mimicking of
chronic mechanical stress compared with acute injury models,
although it still incompletely replicates human kinematics
(143). Furthermore, the
persistence of notochordal cells into adulthood in rodents
contrasts with their disappearance in humans, obscuring the study
of the ‘chondrocyte-like’ cells that populate the adult human
NP.
From an epigenetic perspective, the ‘time gap’
presents a formidable barrier. The short lifespan of rodents
prevents the modeling of gradual, accumulative ‘epigenetic drift’,
the loss of stable methylation patterns over decades, that
characterizes human disease. Moreover, previous comparative
epigenomics data from the Encyclopedia of DNA Elements (ENCODE)
project highlights that while DNA sequences may be conserved, the
functional activity of regulatory elements (enhancers/promoters)
often diverges significantly between mice and humans, meaning an
epigenetic therapy valid in mice may lack a functional target in
humans (144). Consequently, the
field is increasingly integrating human pluripotent stem
cell-derived organoids alongside animal models. These
‘disc-on-a-chip’ systems provide a human genetic background to
validate epigenetic mechanisms found in animals, offering a bridge
to overcome species-specific regulatory divergence while adhering
to the 3R principles of animal research (145).
Needle-puncture models used in IVDD
epigenetics
Model utility and biological relevance
For epigenetic studies, murine needle puncture
remains the overwhelmingly dominant method to induce controlled AF
or NP disruption. The protocol allows for precise modulation
through variables such as the insertion site (lumbar vs. coccygeal
disc), needle gauge (21G to 32G), needle manipulation (retained or
rotated) and intradiscal reagent injections (135,146). Despite the biomechanical
differences inherent to quadrupeds, the murine caudal disc serves
as a surprisingly robust surrogate for the human lumbar spine; it
mirrors the human phenotype in disc height, anteroposterior width,
torsional resistance, axial compressive loading and biochemical
composition, particularly in glycosaminoglycan content.
Consequently, the model rapidly recapitulates a spectrum of
pathological features strikingly similar to human IVDD, including
radial/concentric annular tears, endplate trabecular bone
deposition, profound NP matrix degradation, cell apoptosis and
inflammatory responses. These extensive biological parallels,
combined with low cost, short life cycles and amenability to
genetic manipulation, establish the murine tail disc as a
clinically relevant proxy for studying structural degeneration
(147).
Epigenetic limitations: Acute injury
and evolutionary divergence
However, the application of this model to
epigenetics requires rigorous scrutiny due to two fundamental
disconnects. First, the temporal nature of the epigenetic
modifications differs; because the alterations detected in puncture
models are triggered by acute mechanical injury, they generate an
‘acute inflammatory epigenetic spike’ that may obscure or
completely miss the gradual, wear-and-tear-related epigenetic drift
that cumulatively drives IVDD onset in humans (148). Second, epigenetic findings from
murine models must be interpreted with caution due to evolutionary
divergence in regulatory elements (REs). Data from the latest phase
of the ENCODE project reveals a notable discordance between
sequence conservation and functional activity. While a substantial
proportion of epigenome REs appear orthologous at the sequence
level (~56% of human REs and 72% of mouse REs), only a minority
retain conserved regulatory activity. This functional drop-off is
particularly severe when translating from humans to mice: Only 18%
of human REs display analogous activity in mice, whereas 46% of
mouse REs have functional counterparts in humans (149,150). This disparity, attributed to
species-specific regulatory evolution, context-dependent enhancer
usage and heterogeneity in bulk-tissue profiling, implies that an
epigenetic target validated in a murine puncture model has a high
probability of lacking a functional equivalent in the human
genome.
Non-puncture models used in IVDD
epigenetics
Genetic engineering: Dissecting molecular
homeostasis
In contrast to acute injury models, only a few
non-puncture IVDD animal models have been used in IVDD epigenetic
research, including natural aging, gene editing (alone or combined
with aging) and models based on advanced glycation end products
(AGEs) or more systemic conditions. These models capture distinct
facets of epigenetic modifications. For example, age-related SPARC
downregulation due to hypermethylation is linked to disc
degeneration, as aged and SPARC-null mice exhibit highly similar
pain phenotypes (35). HDAC9
knockout mice at different ages exhibit accelerated IVDD due to
impaired NPC vitality and increased apoptosis. HDAC9 enhances
acetylation and ubiquitin-proteasomal degradation of RUNX3, and its
overexpression restores NPC viability in vivo (44). miR-141 knockout prevents both
spontaneous IVDD in aged mice and IVDD progression in a
puncture-induced mouse model, acting through the SIRT1/NF-κB
pathway. Local injection of nanoparticle-coupled miR-141 inhibitors
demonstrated therapeutic benefit (87). The epigenetic regulator SIRT6, a
nuclear NAD+-dependent HDAC, maintains disc homeostasis
by supporting anabolic and proliferative responses via IGF-1 and
MYC. Disc-specific Sirt6-knockout causes global H3K9
hyperacetylation, increased H3K27 and H3K36 methylation, altered
chromatin accessibility and dysregulation of DNA-repair pathways.
Together these changes result in accumulated DNA damage (γH2AX
foci), reduced autophagy, increased cellular senescence with a
heightened senescence-associated secretory phenotype burden in NP
and AF cells, and accelerated structural disc degeneration in an
age-dependent manner (45). Disc
cell-specific Foxo3 knockout induces IVDD with pronounced type II
collagen and ECM loss, revealing a protective mechanism whereby
FOXO3 activates the lncRNA HOTTIP, which sequesters miR-615-3p to
preserve COL2A1 expression (151).
Chemical and metabolic induction: The
‘slow-burn’ of aging
Using a rat IVDD model induced by AGEs, researchers
show that lentiviral overexpression of lncRNA FAM83H-AS1
competitively binds miR-22-3p via a ceRNA mechanism, reducing the
inflammatory mediators IL-1β and TNF-α (152). Unlike mechanical injury models,
AGE administration-based models reproduce a gradual accumulation of
crosslinked matrix proteins, oxidative stress, inflammation and
activation of the receptor for AGE, the ‘slow-burn’ aging chemistry
that contributes to IVDD. A similar AGE-based rat model has also
been used to evaluate the therapeutic effects of palladium
nanoparticles in IVDD, which facilitates autophagic degradation of
AGEs (153). However, exogenous
AGE delivery may not fully reflect the temporal or tissue-specific
distribution of AGE accumulation seen in natural aging, and it
underrepresents other contributors such as mechanical loading and
vascular or immune responses.
Systemic regulators: Exercise and
circadian rhythms
Animal studies have also explored how systemic
conditions may have a strong impact on epigenetic changes and
disease development. Although lumbar IVDs from SPARC-null and
age-matched wild-type mice show comparable global DNA methylation,
as assessed via 5-mC ELISA, long-term exercise lowers global
methylation in both groups and attenuates LBP-related symptoms
manifested in a sex-specific manner. In males, exercise reduces
Dnmt3a, Mecp2 and Tet1 mRNA expression, while in females it reduces
Dnmt3b and Mecp2 but increases Mbd2a/b, with Mecp2a reduction
shared across sexes, especially in SPARC-null mice. Because the
exercise intervention began at 8 months (middle-aged) and ended at
14 months, this comparison reflects the function of SPARC in
age-associated methylation (154). In a streptozotocin (STZ)-induced
diabetic rat model, hyperglycemia markedly suppresses SIRT1
expression and activity in NPCs. As a class III HDAC, SIRT1
deacetylates multiple substrates, including the tumor suppressor
protein p53. Its reduction increases p53 acetylation, enhancing its
transcriptional activity that favors apoptosis and cellular
senescence. Pharmacological or gene-based reactivation of SIRT1
reverses these effects and protects NPCs from diabetes-associated
IVDD (155).
Expanding on systemic regulation, recent work has
established circadian rhythm disruption models (such as
environmental light-cycle shifting or Bmal1 knockout) as a novel
non-puncture paradigm. These models reveal that the core circadian
clock protein BMAL1 directly binds to the promoters of anabolic
genes (such as aggrecan and Col2a1) to maintain their rhythmic
expression, a mechanism critical for cartilaginous tissue
integrity. Furthermore, in the specific context of the
intervertebral disc, disrupting this rhythm leads to degeneration
through an autophagy-dependent axis; the loss of BMAL1 impairs the
rhythmic expression of nuclear receptors that govern autophagic
flux, thereby compromising NPC survival (156). Collectively, these models
underscore that the IVDD epigenome is not a static entity but a
dynamic record of genetic, metabolic and lifestyle inputs (156,157).
Common pitfalls and misinterpretations
in IVDD epigenetics research
Progress in IVDD epigenetics research into clinical
trials is limited by several obstacles, including over-reliance on
single epigenetic marks, neglect of disc zonal heterogeneity,
conflation of correlation with causation, and mismatches between
models and methods.
A persistent limitation in IVDD epigenetic studies
is the tendency to focus on isolated epigenetic features, such as
DNA methylation or individual histone marks, rather than the
coordinated behavior of the ERN. Age-associated epigenetic
alterations accumulate in a non-random manner, preferentially
affecting genes involved in development and transcriptional
regulation, indicating that degeneration reflects network-level
erosion rather than discrete molecular events. Studies that fail to
account for this functional redundancy risk oversimplifying disc
aging as a sum of independent alterations rather than a systemic
disruption of regulatory control.
The IVD exhibits pronounced spatial heterogeneity,
with distinct epigenetic and transcriptional programs across the
NP, AF and CEP. Failure to resolve these zonal differences,
particularly in bulk epigenomic assays, can obscure focal chromatin
architectures that drive localized degeneration. Advanced spatial
and single-cell epigenomic approaches are therefore critical for
accurately interpreting disc-specific regulatory states and
avoiding the conflation of global aging signatures with
region-specific pathology.
Early machine learning studies successfully
identified senescence-associated hub genes, such as TATA-box
binding protein associated factor 13 (158), and predictive mRNA triads (BTG
anti-proliferation factor 2, MDM4 regulator of p53, acyl-CoA
oxidase 1) using approaches including weighted gene co-expression
network analysis, random forest and support vector
machine-recursive feature elimination (159). While these correlation-based
frameworks are valuable for hypothesis generation, they cannot
establish causality. Without functional perturbation or
longitudinal validation, there remains a risk of misinterpreting
downstream epigenetic consequences as primary drivers of
degeneration.
A critical and often overlooked pitfall lies in
mismatching experimental models with the biological timescale of
human IVDD. Selecting an appropriate animal model requires
consideration of its age-related characteristics relative to human
IVDD, as different species have distinct life spans and aging rates
that shift the timing of degeneration (160). Across mammals, shorter-lived
species have a higher rate of epigenetic drift than longer-lived
species, and this rate scales with maximum lifespan. Age-associated
epigenetic drift accumulates faster in genome regions with low CpG
density, which are more common in shorter-lived species, whereas
longer-lived species tend to possess CpG-dense regulatory elements
in shared genes and appear to maintain more robust protective
mechanisms, such as sirtuin efficiency and DNA repair capacity.
This discrepancy is detectable even within murine rodents. When
measured per unit chronological time, epigenetic disorder occurs
more rapidly in rats than in mice (22). Such mismatches undermine
translational relevance when acute injury models are used to infer
mechanisms of chronic degeneration.
Future directions
Methodological innovations needed
The avascular nature of disc tissue and limited
cell yields necessitate low-input and single-cell epigenomic
technologies. Integrating single-cell multi-omics, spatial
transcriptomics and long-read epigenomics will enable
high-resolution mapping of chromatin states while preserving zonal
and cellular context. Human IVDD is defined by slow, progressive
epigenetic drift rather than abrupt molecular change. Longitudinal
sampling strategies, particularly in aging animal models, are
essential to distinguish stochastic drift from pathological
transitions and to identify early epigenetic states that predispose
discs to failure. Establishing standardized, disc-specific
epigenetic reference maps across age, degeneration stage and disc
region will provide a foundational framework for interpreting
disease-associated changes. Such atlases are critical for
cross-study comparability and for anchoring artificial intelligence
(AI)-driven analyses in biologically meaningful ground truth.
Challenges in therapeutic delivery remain stubborn.
The NP tissue presents formidable barriers, creating a dilemma
between achieving tissue penetration and maintaining therapeutic
retention. The avascular nature and dense ECM make it exceptionally
challenging for systemically administered drugs to reach target
cells, necessitating local intradiscal injection as the clinically
relevant delivery approach (161). However, direct injection suffers
from rapid diffusion outside the injection site, resulting in
short-lived benefits while potentially causing systemic toxicity
(162). The high negative fixed
charge density of the NP, driven by aggrecan glycosaminoglycans,
can be exploited to enhance intra-NP residence time through
positively charged delivery vehicles that utilize long-range
electrostatic interactions (163).
Additionally, degenerative disc tissues develop
fibrotic barriers that further impede drug penetration, further
complicating therapeutic delivery (164). Advanced nanocarrier systems have
emerged as promising solutions to overcome these dual challenges.
Injectable hydrogel gene delivery systems incorporating
functionalized nanoparticles enable sustained, on-demand release of
therapeutic agents directly into the NP, often utilizing
MMP-responsive mechanisms (165).
Tannic acid-based nanoparticles provide both antioxidant and
anti-inflammatory effects while serving as effective gene delivery
vectors (166). Multiple
engineering strategies can enhance nanocarrier performance.
Stimuli-responsive nanoparticles offer precise targeting and
controlled therapeutic release, improving drug localization and
enabling sustained delivery (167). Inflammation-responsive drug
release systems can exploit the pathological microenvironment, with
nanoscaffolds demonstrating disc-mimetic stiffness, excellent
biodegradability and robust scavenging of ROS and cell-free nucleic
acids (168). Phenylboronic
acid-functionalized microspheres create microenvironment-responsive
systems enabling sustained gene release while modulating
inflammation and alleviating apoptosis (169). Charge-reversal and
self-propelling nanocarriers represent cutting-edge approaches to
the penetration-retention paradox. Avidin-grafted dextran
nanostructures utilize electrostatic interactions to achieve
month-long intra-discal retention, with binding strong enough to
prevent rapid clearance yet reversible enough to allow movement
throughout the tissue (163).
Self-enriching nanocarriers catalyze hydrogen peroxide to generate
asymmetric bubble propulsion, enabling selective penetration into
degenerated tissues without accumulation in healthy tissues,
overcoming fibrotic barriers that impede conventional passive
diffusion (164). Surface
modification further enhances functionality. NP-targeting
nanocarriers facilitate miRNA-based therapeutics by enhancing
transportation to target cells, which enables effective in
vivo inhibition of pathological miRNAs (166). Multifunctional nanocarriers can
simultaneously deliver multiple therapeutic agents, such as
combining gene therapy with mitochondrial-targeted peptides, to
address both ECM metabolism and mitochondrial dysfunction (170).
Despite these challenges, nanocarrier-based
delivery systems have demonstrated the capacity to enhance targeted
delivery, improve local drug concentration and sustain drug
retention. This provides disease-modifying therapeutic strategies
that address fundamental pathophysiological mechanisms of IVDD
(Fig. 1) (162,171,172).
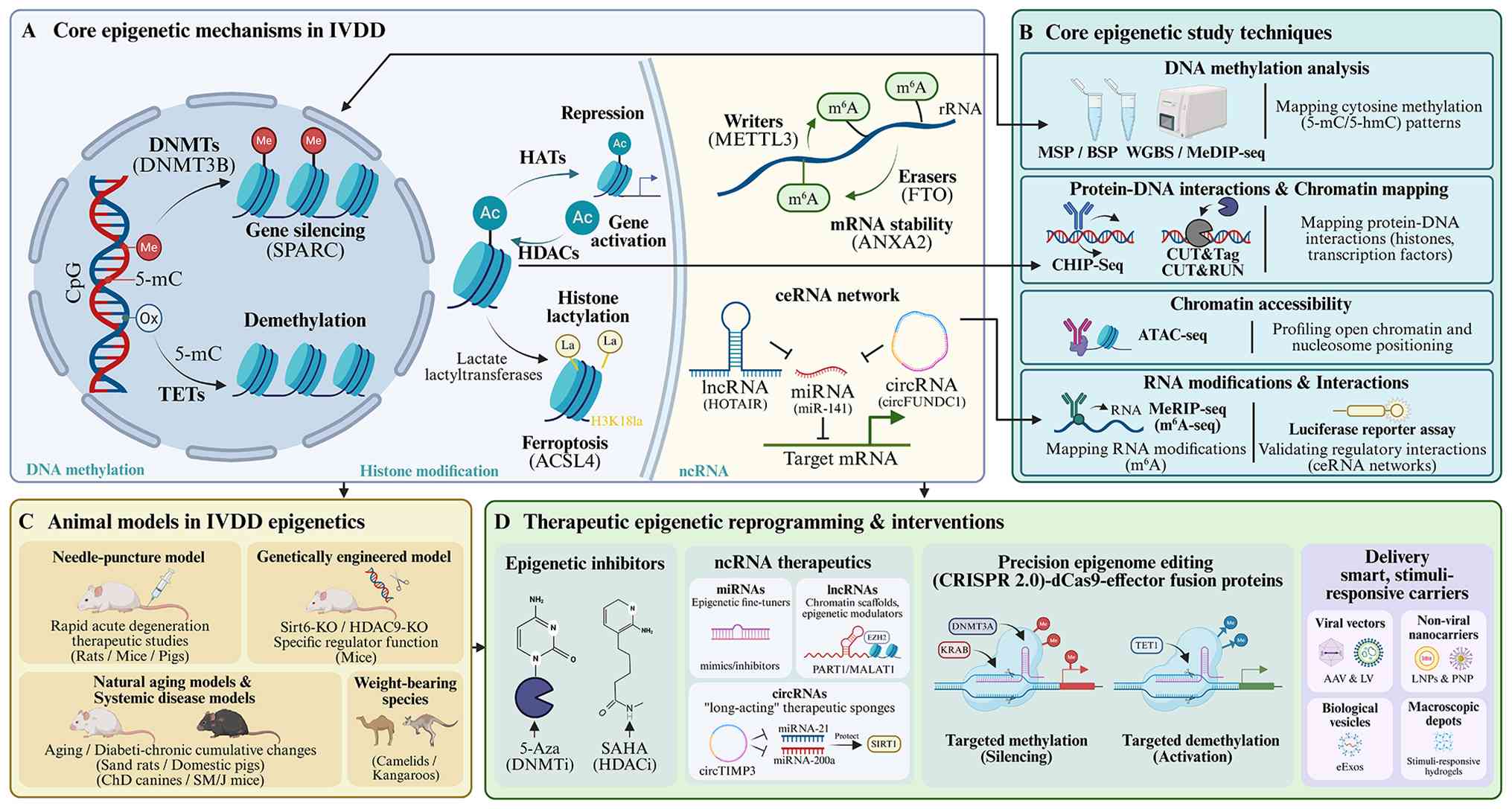 | Figure 1.Epigenetic toolkit in IVDD:
Mechanisms, techniques, models and translational applications. (A)
Core epigenetic mechanisms in IVDD. (B) Core epigenetic study
techniques (C) Animal models in IVDD epigenetics. (D) Therapeutic
epigenetic reprogramming and interventions. 5-mc, 5-methylcytosine;
5-hmc, 5-hydroxymethylcytosine; DNMTs, DNA methyltransferases;
TETs, Ten-eleven translocation enzymes; HATs, histone
acetyltransferases; MSP, methylation-specific PCR; BSP, bisulfite
sequencing PCR; WGBS, whole-genome bisulfite sequencing; MeDIP-seq,
methylated DNA immunoprecipitation sequencing; CHIP-Seq, chromatin
immunoprecipitation sequencing; CUT&Tag, Cleavage Under Targets
and Tagmentation; CUT&RUN, Cleavage Under Targets and Release
Using Nuclease; ATAC-seq, assay for transposase-accessible
chromatin using sequencing; MeRIP-seq, methylated RNA
immunoprecipitation sequencing; m6A-seq,
N6-methyladenosine sequencing; AAV, adeno-associated virus; LV,
lentivirus; LNPs, lipid nanoparticles; PNP, polymeric
nanoparticles; eExos, engineered exosomes; miRNA, microRNA, HDAC,
histone deacetylase. |
Model innovation
Non-human primates such as Macaca mulatta
live to about 27 years and age at about three times faster than
humans. This degeneration, similar to that of middle-aged and
elderly humans (40–60 years old), appears at 8–12 years in monkeys
(173). ChD dogs exhibit clinical
IVDD between 3 and 7 years of age (174), whereas gerbils develop early
degeneration at 4 months, progressing extensively by 9 months,
which helps model the continuum from youth to old age in humans
(175). Future research must
prioritize such longer-lived models to capture the slow, cumulative
epigenetic drift that characterizes human pathology, while
permitting controlled mechanical perturbation. Humanized and
genetically engineered progeroid models represent a promising
direction for aligning epigenetic timescales with human pathology.
These systems allow interrogation of pre-existing epigenetic
vulnerabilities that are invisible in acute injury paradigms yet
decisive for clinical translation.
From descriptive epigenetics to causal
biology
Recent deep learning models published between 2024
and 2025 have pushed analysis far beyond correlation-based methods,
enabling the end-to-end prediction of complex regulatory
interactions directly from DNA sequence. Key advances include
DeepEPI (176) and EPI-Trans
(177), which combine
convolutional neural networks to capture local motifs with
transformers to model long-range 3D chromatin dependencies.
Building on this, DeepMethyGene integrates multi-scale methylation
features (CpG islands, shores and shelves) to predict gene
expression with high interpretability (178). These models enable
hypothesis-driven perturbation experiments that test causality
rather than correlation. Advanced architectures incorporating
transformers and Kolmogorov-Arnold Networks (such as KansformerEPI)
facilitate interpretable modeling of long-range chromatin
dependencies. By learning the non-linear regulatory grammar of the
disc, these tools support experimental validation of cooperative
network behaviors underlying senescence. More sophisticated
architectures add biological flexibility; EPI-DynFusion introduces
dynamic fusion gates to adaptively weigh sequence representations
based on cell context (179),
while KansformerEPI replaces traditional perceptrons with
Kolmogorov-Arnold Networks, combining functional expressiveness
with interpretability (180).
Together, these tools are reshaping the paradigm from studying
isolated epigenetic marks towards learning the non-linear,
context-dependent regulatory grammar governing disc senescence.
Conclusions
IVDD represents a systemic collapse of the ERN,
rather than isolated molecular failures. Future research must
integrate single-cell multi-omics, spatial transcriptomics and AI
to decode the cumulative erosion of cooperative network behaviors
and the complex regulatory mechanisms of disc senescence,
prioritizing physiologically relevant aging models to identify
epigenetic states predisposing to degeneration. Successful clinical
translation depends on coupling these insights with advanced
delivery strategies that navigate the disc's avascular environment,
shifting the paradigm from symptom preservation toward active
epigenetic reprogramming and rejuvenation of the aging spine.
Acknowledgements
Fig. 1. was
created in BioRender. c, C. (2026; http://BioRender.com/hzv1ofn) with a publication
license.
Funding
This work was supported by the Research and Innovation Team
Project for Scientific Breakthroughs at Shanxi Bethune Hospital
(grant no. 2024ZHANCHI10).
Availability of data and materials
Not applicable.
Authors' contributions
XLC contributed to the conceptualization, writing
and figures and tables creation. LZ contributed to the
conceptualization and writing. HJ contributed to the
conceptualization and reviewing and editing the manuscript. WZ
contributed to the writing. LX and DJ contributed to the review,
editing and language checking. RW and S contributed to the
conceptualization. HF contributed to the review and editing. All
authors read and approved the final version of the manuscript. Data
authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ACAN
|
aggrecan
|
|
ADAMTS
|
a disintegrin and metalloproteinase
with thrombospondin motifs
|
|
AGEs
|
advanced glycation end products
|
|
AF
|
annulus fibrosus
|
|
ATAC-seq
|
transposase accessible chromatin
sequencing
|
|
CEP
|
cartilage endplates
|
|
ceRNAs
|
competitive endogenous RNAs
|
|
ChIP-Seq
|
chromatin immunoprecipitation
sequencing
|
|
circRNAs
|
circular RNAs
|
|
COL2A1
|
type II collagen
|
|
DNMTs
|
DNA methyltransferases
|
|
ECM
|
extracellular matrix
|
|
ERN
|
epigenetic regulatory network
|
|
GAGs
|
glycosaminoglycans
|
|
HATs
|
histone acetyltransferases
|
|
HDACs
|
histone deacetylases
|
|
IVD
|
intervertebral disc
|
|
IVDD
|
intervertebral disc degeneration
|
|
LBP
|
low back pain
|
|
lncRNAs
|
long non-coding RNAs
|
|
miRNAs
|
microRNAs
|
|
MMPs
|
matrix metalloproteinases
|
|
MNase-seq
|
micrococcal nuclease sequencing
|
|
MSP
|
methylation-specific PCR
|
|
mtDNA
|
mitochondrial DNA
|
|
m6A
|
N6-methyladenosine
|
|
ncRNA
|
non-coding RNA
|
|
NP
|
nucleus pulposus
|
|
NPCs
|
nucleus pulposus cells
|
|
PPARγ
|
peroxisome proliferator-activated
receptor γ
|
|
PTM
|
post-translational modification
|
|
ROS
|
reactive oxygen species
|
|
TETs
|
ten-eleven translocation enzymes
|
|
WGBS
|
whole-genome bisulfite sequencing
|
|
3′UTR
|
3' untranslated region
|
|
5-Aza
|
5-azacytidine
|
|
5-hmC
|
5-hydroxymethylcytosine
|
|
5-mC
|
5-methylcytosine
|
References
|
1
|
Maher C, Underwood M and Buchbinder R:
Non-specific low back pain. Lancet. 389:736–747. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Samanta A, Lufkin T and Kraus P:
Intervertebral disc degeneration-current therapeutic options and
challenges. Front Public Health. 11:11567492023. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Taylor W and Erwin WM: Intervertebral disc
degeneration and regeneration: New molecular mechanisms and
therapeutics: Obstacles and potential breakthrough technologies.
Cells. 13:21032024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mohd Isa IL, Teoh SL, Mohd Nor NH and
Mokhtar SA: Discogenic low back pain: Anatomy, pathophysiology and
treatments of intervertebral disc degeneration. Int J Mol Sci.
24:2082022. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kirnaz S, Capadona C, Wong T, Goldberg JL,
Medary B, Sommer F, McGrath LB Jr and Härtl R: Fundamentals of
intervertebral disc degeneration. World Neurosurg. 157:264–273.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Du J: Global, regional, and national
burden of low back pain: Findings from the global burden of disease
study 2021 and projections to 2050. Spine (Phila Pa 1976).
50:E3892025. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Oichi T, Taniguchi Y, Oshima Y, Tanaka S
and Saito T: Pathomechanism of intervertebral disc degeneration.
JOR spine. 3:e10762020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ferreira JR, Moura ML, Pilão S, Castro AL,
Fiordalisi M, Leite Pereira C, Caldeira J and Gonçalves RM:
Decoding intervertebral disc cell populations: Challenges in
isolation and phenotype definition. Tissue Eng Part B Rev.
tenteb20240350. 2025.(Epub ahead of print). PubMed/NCBI
|
|
9
|
Urban JPG and Fairbank JCT: Current
perspectives on the role of biomechanical loading and genetics in
development of disc degeneration and low back pain; a narrative
review. J Biomech. 102:1095732020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Guo W, Li BL, Zhao JY, Li XM and Wang LF:
Causal associations between modifiable risk factors and
intervertebral disc degeneration. Spine J. 24:195–209. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vo NV, Hartman RA, Yurube T, Jacobs LJ,
Sowa GA and Kang JD: Expression and regulation of
metalloproteinases and their inhibitors in intervertebral disc
aging and degeneration. Spine J. 13:331–341. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tseranidou S, Segarra-Queralt M, Chemorion
FK, Le Maitre CL, Piñero J and Noailly J: Nucleus pulposus cell
network modelling in the intervertebral disc. NPJ Syst Biol Appl.
11:132025. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kaneda G, Zila L, Wechsler JT, Shafi K,
Cheema K, Bae H, Kim SD, Tuchman A, Li D and Sheyn D: What a pain
in the back: Etiology, diagnosis and future treatment directions
for discogenic low back pain. Bone Res. 13:892025. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kang L, Zhang H, Jia C, Zhang R and Shen
C: Epigenetic modifications of inflammation in intervertebral disc
degeneration. Ageing Res Rev. 87:1019022023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ząbek Z, Wyczałkowska-Tomasik A, Poboży K,
Adamus JP, Turek G, Ząbek M and Pączek L: Understanding the
microenvironment of intervertebral disc degeneration: A
comprehensive review of pathophysiological insights and therapeutic
implications. Int J Mol Sci. 26:99382025. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xu J, Shao T, Lou J, Zhang J and Xia C:
Aging, cell senescence, the pathogenesis and targeted therapies of
intervertebral disc degeneration. Front Pharmacol. 14:11729202023.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hickman TT, Rathan-Kumar S and Peck SH:
Development, pathogenesis, and regeneration of the intervertebral
disc: Current and future insights spanning traditional to omics
methods. Front Cell Dev Biol. 10:8418312022. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li G, Zhang W, Liang H and Yang C:
Epigenetic regulation in intervertebral disc degeneration. Trends
Mol Med. 28:803–805. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang Y, Wang J, Hua D, Fan C, He W, Deng
Y, Tang M, Geng D, Wu X and Mao H: Histone modifications: Unveiling
the epigenetic enigma of degenerative skeletal diseases. J Orthop
Translat. 55:245–266. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jiang C, Chen Z, Wang X, Zhang Y, Guo X,
Xu Z, Yang H and Hao D: The potential mechanisms and application
prospects of non-coding RNAs in intervertebral disc degeneration.
Front Endocrinol (Lausanne). 13:10811852022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tao Y, Yu X, Li X, Xu Y, Wang H, Zhang L,
Lin R, Wang Y and Fan P: M6A methylation-regulated autophagy may be
a new therapeutic target for intervertebral disc degeneration. Cell
Biol Int. 48:389–403. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bertucci-Richter EM and Parrott BB: The
rate of epigenetic drift scales with maximum lifespan across
mammals. Nat Commun. 14:77312023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Moqri M, Poganik JR, Horvath S and
Gladyshev VN: What makes biological age epigenetic clocks tick. Nat
Aging. 5:335–336. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bertucci-Richter EM, Shealy EP and Parrott
BB: Epigenetic drift underlies epigenetic clock signals, but
displays distinct responses to lifespan interventions, development,
and cellular dedifferentiation. Aging (Albany NY). 16:1002–1020.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Diwan AD and Melrose J: Intervertebral
disc degeneration and how it leads to low back pain. JOR Spine.
6:e12312022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Younesian S, Mohammadi MH, Younesian O,
Momeny M, Ghaffari SH and Bashash D: DNA methylation in human
diseases. Heliyon. 10:e323662024. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu X and Zhang Y: TET-mediated active DNA
demethylation: Mechanism, function and beyond. Nat Rev Genet.
18:517–534. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li G, Luo R, Zhang W, He S, Wang B, Liang
H, Song Y, Ke W, Shi Y, Feng X, et al: m6A hypomethylation of
DNMT3B regulated by ALKBH5 promotes intervertebral disc
degeneration via E4F1 deficiency. Clin Transl Med. 12:e7652022.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen J, Yang X, Li Q, Ma J, Li H, Wang L,
Chen Z and Quan Z: Inhibiting DNA methyltransferase DNMT3B confers
protection against ferroptosis in nucleus pulposus and ameliorates
intervertebral disc degeneration via upregulating SLC40A1. Free
Radic Biol Med. 220:139–153. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cheng P, Li H, Chen HW, Wang ZQ, Li PW and
Zhang HH: 5-Azacytidine inhibits endoplasmic reticulum stress and
apoptosis of nucleus pulposus cells by preserving PPARγ via
promoter demethylation. In Vitro Cell Dev Biol Anim. 61:288–297.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cheng P, Wei HZ, Chen HW, Wang ZQ, Mao P
and Zhang HH: DNMT3a-mediated methylation of PPARγ promote
intervertebral disc degeneration by regulating the NF-κB pathway. J
Cell Mol Med. 28:e180482024. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yeater TD, Kawarai Y, Lee S, Belani KG,
Beebe DS, Sheyn D, Pinto MR and Stone LS: Investigating the
epigenetic landscape of symptomatic disk degeneration: A case
study. Pain Rep. 10:e12372025. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hong JY, Kim H, Jeon WJ, Lee J, Yeo C, Lee
YJ and Ha IH: Epigenetic changes within the annulus fibrosus by DNA
methylation in rat intervertebral disc degeneration model. Cells.
11:35472022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Karchevskaya AE, Poluektov YM and
Korolishin VA: Understanding intervertebral disc degeneration:
Background factors and the role of initial injury. Biomedicines.
11:27142023. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tajerian M, Alvarado S, Millecamps M,
Dashwood T, Anderson KM, Haglund L, Ouellet J, Szyf M and Stone LS:
DNA methylation of SPARC and chronic low back pain. Mol Pain.
7:652011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li S and Tollefsbol TO: DNA methylation
methods: Global DNA methylation and methylomic analyses. Methods.
187:28–43. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ikuno A, Akeda K, Takebayashi SI, Shimaoka
M, Okumura K and Sudo A: Genome-wide analysis of DNA methylation
profile identifies differentially methylated loci associated with
human intervertebral disc degeneration. PLoS One. 14:e02221882019.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sun P, Huang T, Huang C, Wang Y and Tang
D: Role of histone modification in the occurrence and development
of osteoporosis. Front Endocrinol (Lausanne). 13:9641032022.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dueva R, Akopyan K, Pederiva C, Trevisan
D, Dhanjal S, Lindqvist A and Farnebo M: Neutralization of the
positive charges on histone tails by RNA promotes an open chromatin
structure. Cell Chem Biol. 26:1436–1449.e5. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Higashiyama R, Miyaki S, Yamashita S,
Yoshitaka T, Lindman G, Ito Y, Sasho T, Takahashi K, Lotz M and
Asahara H: Correlation between MMP-13 and HDAC7 expression in human
knee osteoarthritis. Mod Rheumatol. 20:11–17. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zheng Q, Li XX, Xiao L, Shao S, Jiang H,
Zhang XL, Sun LY and Xu HG: MicroRNA-365 functions as a
mechanosensitive microRNA to inhibit end plate chondrocyte
degeneration by targeting histone deacetylase 4. Bone.
128:1150522019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Xiao L, Gong D, Liang L, Liang A, Liang H,
Xu X and Teng H: Inhibition of HDAC4 by GSK3β leads to
downregulation of KLF5 and ASK1 and prevents the progression of
intravertebral disc degeneration. Clin Epigenetics. 13:532021.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Makki MS and Haqqi TM: Histone deacetylase
inhibitor vorinostat (SAHA) suppresses IL-1β-induced matrix
metallopeptidase-13 expression by inhibiting IL-6 in osteoarthritis
chondrocyte. Am J Pathol. 186:2701–2708. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lei M, Lin H, Shi D, Hong P, Song H,
Herman B, Liao Z and Yang C: Molecular mechanism and therapeutic
potential of HDAC9 in intervertebral disc degeneration. Cell Mol
Biol Lett. 28:1042023. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ramteke P, Watson B, Toci M, Tran VA,
Johnston S, Tsingas M, Barve RA, Mitra R, Loeser RF, Collins JA and
Risbud MV: SIRT6 loss causes intervertebral disc degeneration in
mice by promoting senescence and SASP status. bioRxiv [Preprint].
2024.09.09.612072. 2024.PubMed/NCBI
|
|
46
|
Ma W, Wang W, Zhao L, Fan J, Liu L, Huang
L, Peng B, Wang J, Xu B, Liu H, et al: Reprogramming to restore
youthful epigenetics of senescent nucleus pulposus cells for
mitigating intervertebral disc degeneration and alleviating low
back pain. Bone Res. 13:352025. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yao Q, Lei Y, Zhang Y, Chen H, Dong X, Ye
Z and Liang H: EZH2-H3K27me3-mediated epigenetic silencing of DKK1
induces nucleus pulposus cell pyroptosis in intervertebral disc
degeneration by activating NLRP3 and NAIP/NLRC4. Inflammation.
48:902–918. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhou M, He SJ, Liu W, Yang MJ, Hou ZY,
Meng Q and Qian ZL: EZH2 upregulates the expression of MAPK1 to
promote intervertebral disc degeneration via suppression of
miR-129-5p. J Gene Med. 24:e33952022. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhang D, Tang Z, Huang H, Zhou G, Cui C,
Weng Y, Liu W, Kim S, Lee S, Perez-Neut M, et al: Metabolic
regulation of gene expression by histone lactylation. Nature.
574:575–580. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sun K, Shi Y, Yan C, Wang S, Han L, Li F,
Xu X, Wang Y, Sun J, Kang Z and Shi J: Glycolysis-derived lactate
induces ACSL4 expression and lactylation to activate ferroptosis
during intervertebral disc degeneration. Adv Sci (Weinh).
12:e24161492025. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Shi Y, Li F, Lin W, Han L, Wang J, Yan C,
Sun J, Ji C, Shi J and Sun K: Integrating bulk RNA and single-cell
RNA sequencing identifies and validates lactylation-related
signatures for intervertebral disc degeneration. J Cell Mol Med.
28:e702622024. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zheng SK, Zhao XK, Wu H, He DW, Xiong L
and Cheng XG: Oxidative stress-induced EGR1 upregulation promotes
NR4A3-mediated nucleus pulposus cells apoptosis in intervertebral
disc degeneration. Aging (Albany NY). 16:10216–10238. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Saotome M, Goodman J and Takaku M:
Capturing chromatin organization by MNase-seq and ATAC-seq. Methods
Mol Biol. 2889:167–180. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Shu S, Zhang X, Feng Z, Liu Z, Wang K, Li
F, Wu Y, Shi B, Qiu Y, Zhu Z and Bao H: Upregulated CEMIP promotes
intervertebral disc degeneration via AP-1-mediated change in
chromatin accessibility. Clin Transl Med. 15:e703222025. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhu D, Liang H, Tong B, Du Z, Li G, Zhang
W, Wu D, Zhou X, Lei J, Zhang X, et al: DDX1 methylation mediated
MATR3 splicing regulates intervertebral disc degeneration by
initiating chromatin reprogramming. Nat Commun. 16:61532025.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wang K, Wu X, Li H, Xu X, Cheng F, Chen J,
Zhou H, Xu J, Yu C, Li Y, et al: Mitophagy reprograms lactate
metabolism to suppress THBS1 via H3K18la reduction, alleviating
intervertebral disc degeneration. Research (Wash D C).
8:09572025.PubMed/NCBI
|
|
57
|
Zeng Y, Wang S, Gao S, Soares F, Ahmed M,
Guo H, Wang M, Hua JT, Guan J, Moran MF, et al: Refined RIP-seq
protocol for epitranscriptome analysis with low input materials.
PLoS Biol. 16:e20060922018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Dominissini D, Moshitch-Moshkovitz S,
Amariglio N and Rechavi G: Transcriptome-wide mapping of
N6-methyladenosine by m6A-Seq. Methods Enzymol. 560:131–147. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Körtel N, Rücklé C, Zhou Y, Busch A,
Hoch-Kraft P, Sutandy FXR, Haase J, Pradhan M, Musheev M, Ostareck
D, et al: Deep and accurate detection of m6A RNA modifications
using miCLIP2 and m6Aboost machine learning. Nucleic Acids Res.
49:e922021. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Yang Y, Lu Y, Wang Y, Wen X, Qi C, Piao W
and Jin H: Current progress in strategies to profile transcriptomic
m6A modifications. Front Cell Dev Biol. 12:13921592024. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Xia W, Guo L, Su H, Li J, Lu J, Li H and
Huang B: A low-cost, low-input method establishment for m6A
MeRIP-seq. Biosci Rep. 44:BSR202314302024. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Wang Y, Xiao Y, Dong S, Yu Q and Jia G:
Antibody-free enzyme-assisted chemical approach for detection of
N6-methyladenosine. Nat Chem Biol. 16:896–903. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Zhang LS, Dai Q and He C: Base-resolution
sequencing methods for whole-transcriptome quantification of mRNA
modifications. Acc Chem Res. 57:47–58. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Liu C, Sun H, Yi Y, Shen W, Li K, Xiao Y,
Li F, Li Y, Hou Y, Lu B, et al: Absolute quantification of
single-base m6A methylation in the mammalian
transcriptome using GLORI. Nat Biotechnol. 41:355–366. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Sun H, Lu B, Zhang Z, Xiao Y, Zhou Z, Xi
L, Li Z, Jiang Z, Zhang J, Wang M, et al: Mild and ultrafast GLORI
enables absolute quantification of m6A methylome from
low-input samples. Nat Methods. 22:1226–1236. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Garcia-Campos MA, Edelheit S, Toth U,
Safra M, Shachar R, Viukov S, Winkler R, Nir R, Lasman L, Brandis
A, et al: Deciphering the ‘m6A Code’ via
antibody-independent quantitative profiling. Cell. 178:731–747.e16.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Capitanchik C, Toolan-Kerr P, Luscombe NM
and Ule J: How do you identify m6 A methylation in
transcriptomes at high resolution? A comparison of recent datasets.
Front Genet. 11:3982020. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Meyer KD: DART-seq: An antibody-free
method for global m6A detection. Nat Methods.
16:1275–1280. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Tegowski M, Zhu H and Meyer KD: Detecting
m6A with in vitro DART-Seq. Methods Mol Biol.
2404:363–374. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Shen J, Zhang Q, Lan Y, Peng Q, Ji Z, Wu Y
and Liu H: Identification and characterisation of potential targets
for N6-methyladenosine (m6A) modification during intervertebral
disc degeneration. Front Biosci (Landmark Ed). 29:4052024.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Liu L, Sun H, Zhang Y, Liu C, Zhuang Y,
Liu M, Ai X, Long D, Huang B, Li C, et al: Dynamics of
N6-methyladenosine modification during aging and their potential
roles in the degeneration of intervertebral disc. JOR Spine.
7:e13162024. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Chen X, Gong W, Shao X, Shi T, Zhang L,
Dong J, Shi Y, Shen S, Qin J, Jiang Q and Guo B: METTL3-mediated
m6A modification of ATG7 regulates autophagy-GATA4 axis
to promote cellular senescence and osteoarthritis progression. Ann
Rheum Dis. 81:87–99. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Wu O, Jin Y, Zhang Z, Zhou H, Xu W, Chen
L, Jones M, Kwan KYH, Gao J, Zhang K, et al: KMT2A regulates the
autophagy-GATA4 axis through METTL3-mediated m6A
modification of ATG4a to promote NPCs senescence and IVDD
progression. Bone Res. 12:672024. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Sun R, Wu XT, Shi H, Wang F, Gao JW, Wang
PY, Xu ZY, Gan WW, Wang YT and Zhang C: Mechanism of FTO-mediated
m6A demethylation regulation of YAP1 in nucleus pulposus cell
senescence. Mech Ageing Dev. 227:1121012025. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Xu X, Shen L, Qu Y, Li D, Zhao X, Wei H
and Yue S: Experimental validation and comprehensive analysis of
m6A methylation regulators in intervertebral disc degeneration
subpopulation classification. Sci Rep. 14:84172024. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Guo C, Chen Y, Wang Y and Hao Y:
Regulatory roles of noncoding RNAs in intervertebral disc
degeneration as potential therapeutic targets (Review). Exp Ther
Med. 25:442022. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Jiang J, Sun Y, Xu G, Wang H and Wang L:
The role of miRNA, lncRNA and circRNA in the development of
intervertebral disk degeneration (Review). Exp Ther Med.
21:5552021. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Wang C, Cui L, Gu Q, Guo S, Zhu B, Liu X,
Li Y, Liu X, Wang D and Li S: The mechanism and function of miRNA
in intervertebral disc degeneration. Orthop Surg. 14:463–471. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Li Y, Wang B, Sun W, Kong C, Ding J, Hu F,
Li J, Chen X and Lu S: Construction of circ_0071922-miR-15a-5p-mRNA
network in intervertebral disc degeneration by RNA-sequencing. JOR
Spine. 7:e12752023. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Zhao B, Lu M, Wang D, Li H and He X:
Genome-wide identification of long noncoding RNAs in human
intervertebral disc degeneration by RNA sequencing. Biomed Res Int.
2016:36848752016. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Guo ZL, Liu YY, Gao Y, Guan XM, Li H and
Cheng M: Circ-RNA expression pattern and circ-RNA-miRNA-mRNA
network in the pathogenesis of human intervertebral disc
degeneration. Cell J. 23:218–224. 2021.PubMed/NCBI
|
|
82
|
Wan ZY, Song F, Sun Z, Chen YF, Zhang WL,
Samartzis D, Ma CJ, Che L, Liu X, Ali MA, et al: Aberrantly
expressed long noncoding RNAs in human intervertebral disc
degeneration: A microarray related study. Arthritis Res Ther.
16:4652014. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Cheng X, Zhang L, Zhang K, Zhang G, Hu Y,
Sun X, Zhao C, Li H, Li YM and Zhao J: Circular RNA VMA21 protects
against intervertebral disc degeneration through targeting miR-200c
and X linked inhibitor-of-apoptosis protein. Ann Rheum Dis.
77:770–779. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Yu C, Zhao J, Cheng F, Chen J, Chen J, Xu
H, Shi K, Xia K, Ding S, Wang K, et al: Silencing circATXN1 in
aging nucleus pulposus cell alleviates intervertebral disc
degeneration via correcting progerin mislocalization. Research
(Wash D C). 7:03362024.PubMed/NCBI
|
|
85
|
Wang T, Yan X, Song D, Li Y, Li Z and Feng
D: CircEYA3 aggravates intervertebral disc degeneration through the
miR-196a-5p/EBF1 axis and NF-κB signaling. Commun Biol. 7:3902024.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Chen X, Song Y, Chen G, Zhang B, Bai Y,
Sun C, Fan D and Chen Z: Circular RNA CircFOXO3 functions as a
competitive endogenous RNA for acid-sensing ion channel subunit 1
mediating oxeiptosis in nucleus pulposus. Biomedicines. 12:6782024.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Ji ML, Jiang H, Zhang XJ, Shi PL, Li C, Wu
H, Wu XT, Wang YT, Wang C and Lu J: Preclinical development of a
microRNA-based therapy for intervertebral disc degeneration. Nat
Commun. 9:50512018. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Huang Y, Zhang Z, Wang J, Shen S, Yao T,
Xu Y, Chen Z, Fang B and Ma J: circSPG21 protects against
intervertebral disc disease by targeting miR-1197/ATP1B3. Exp Mol
Med. 53:1547–1558. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Du X, Chen S, Cui H, Huang Y, Wang J, Liu
H, Li Z, Liang C, Zheng Z and Wang H: Circular RNA hsa_circ_0083756
promotes intervertebral disc degeneration by sponging miR-558 and
regulating TREM1 expression. Cell Prolif. 55:e132052022. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Xie L, Huang W, Fang Z, Ding F, Zou F, Ma
X, Tao J, Guo J, Xia X, Wang H, et al: CircERCC2 ameliorated
intervertebral disc degeneration by regulating mitophagy and
apoptosis through miR-182-5p/SIRT1 axis. Cell Death Dis.
10:7512019. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Yao W, Hu X and Wang X: Crossing
epigenetic frontiers: The intersection of novel histone
modifications and diseases. Signal Transduct Target Ther.
9:2322024. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Gu T, He Y, Zhou J, Qiu X, Yang W, Zhu Q,
Liang Y, Zheng Y, Yik JHN, Haudenschild DR, et al: CircFUNDC1
interacts with CDK9 to promote mitophagy in nucleus pulposus cells
under oxidative stress and ameliorates intervertebral disc
degeneration. Cell Death Dis. 16:942025. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Guo Z, Su W, Zhou R, Zhang G, Yang S, Wu
X, Qiu C, Cong W, Shen N, Guo J, et al: Exosomal MATN3 of
urine-derived stem cells ameliorates intervertebral disc
degeneration by antisenescence effects and promotes NPC
proliferation and ECM synthesis by activating TGF-β. Oxid Med Cell
Longev. 2021:55422412021. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Wang Z, Zhang J, Zheng W and He Y: Long
non-coding RNAs H19 and HOTAIR implicated in intervertebral disc
degeneration. Front Genet. 13:8435992022. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Yu Y, Zhang X, Li Z, Kong L and Huang Y:
LncRNA HOTAIR suppresses TNF-α induced apoptosis of nucleus
pulposus cells by regulating miR-34a/Bcl-2 axis. Biomed
Pharmacother. 107:729–737. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Shao T, Hu Y, Tang W, Shen H, Yu Z and Gu
J: The long noncoding RNA HOTAIR serves as a microRNA-34a-5p sponge
to reduce nucleus pulposus cell apoptosis via a NOTCH1-mediated
mechanism. Gene. 715:1440292019. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Zhang S, Song S, Cui W, Liu X and Sun Z:
Mechanism of long noncoding RNA HOTAIR in nucleus pulposus cell
autophagy and apoptosis in intervertebral disc degeneration. Evid
Based Complement Alternat Med. 2022:85046012022.PubMed/NCBI
|
|
98
|
Zhan S, Wang K, Song Y, Li S, Yin H, Luo
R, Liao Z, Wu X, Zhang Y and Yang C: Long non-coding RNA HOTAIR
modulates intervertebral disc degenerative changes via
Wnt/β-catenin pathway. Arthritis Res Ther. 21:2012019. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Donato L, Mordà D, Scimone C, Alibrandi S,
D'Angelo R and Sidoti A: From powerhouse to regulator: The role of
mitoepigenetics in mitochondrion-related cellular functions and
human diseases. Free Radic Biol Med. 218:105–119. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Hou Y, Liu L, Guo Y and Shi J: Epigenetic
crossroads in intervertebral disc degeneration: Unlocking novel
therapeutic avenues (Review). Mol Med Rep. 33:1132026. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Wiese M and Bannister AJ: Two genomes, one
cell: Mitochondrial-nuclear coordination via epigenetic pathways.
Mol Metab. 38:1009422020. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Cao K, Feng Z, Gao F, Zang W and Liu J:
Mitoepigenetics: An intriguing regulatory layer in aging and
metabolic-related diseases. Free Radic Biol Med. 177:337–346. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Sharma J, Kumari R, Bhargava A, Tiwari R
and Mishra PK: Mitochondrial-induced epigenetic modifications: From
biology to clinical translation. Curr Pharm Des. 27:159–176. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Matilainen O, Quirós PM and Auwerx J:
Mitochondria and epigenetics-crosstalk in homeostasis and stress.
Trends Cell Biol. 27:453–463. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Yin MT and Guo L: Mitochondrial DNA
methylation: State-of-the-art in molecular mechanisms and disease
implications. J Adv Res. 83:455–473. 2026. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Shock LS, Thakkar PV, Robinson JM and
Taylor SM: Cytosine methylase and hydroxymethylase activity in
mammalian mitochondria. Front Cell Dev Biol. 13:16774022025.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Santos JH: Mitochondria signaling to the
epigenome: A novel role for an old organelle. Free Radic Biol Med.
170:59–69. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Liu YF, Zhu JJ, Yu Tian X, Liu H, Zhang T,
Zhang YP, Xie SA, Zheng M, Kong W, Yao WJ, et al: Hypermethylation
of mitochondrial DNA in vascular smooth muscle cells impairs cell
contractility. Cell Death Dis. 11:352020. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Coppedè F and Stoccoro A: Mitoepigenetics
and neurodegenerative diseases. Front Endocrinol (Lausanne).
10:862019. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Mishra M and Kowluru RA: Epigenetic
modification of mitochondrial DNA in the development of diabetic
retinopathy. Invest Ophthalmol Vis Sci. 56:5133–5142. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Zhang J, Shang J, Wang F, Huo X, Sun R,
Ren Z, Wang W, Yang M, Li G, Gao D, et al: Decreased mitochondrial
D-loop region methylation mediates an increase in mitochondrial DNA
copy number in CADASIL. Clin Epigenetics. 14:22022. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Hu X, Wang Z, Zhang H, Cui P, Li Y, Chen
X, Kong C, Wang W and Lu S: Single-cell sequencing: New insights
for intervertebral disc degeneration. Biomed Pharmacother.
165:1152242023. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Yu C, Yang W, Wang Y and Zhang Y:
Single-cell sequencing reveals key biological insights into
intervertebral disc development and degeneration. Curr Mol Med. Jan
13–2026.(Epub ahead of print). View Article : Google Scholar
|
|
114
|
Yang G, Dong C, Wu Z, Wu P, Yang C, Li L,
Zhang J and Wu X: Single-cell RNA sequencing-guided engineering of
mitochondrial therapies for intervertebral disc degeneration by
regulating mtDNA/SPARC-STING signaling. Bioact Mater. 48:564–582.
2025.PubMed/NCBI
|
|
115
|
Zhao L, Liang K, Cheng W, Zhou X, Yang L,
Mao X, Qi W, Jin H, Zhang W, Pan H and Wang D: A multifaceted
strategy for intra- and extracellular nucleic acid regulation to
alleviate intervertebral disc degeneration. Nat Commun.
16:79362025. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Zhou H, Liang C, Cheng F, Wang K, Zhang Y,
Chen J, Xu H, Yu C, Xia K, Li Y, et al: Mitochondria-targeting
polymeric micelles for intervertebral disc degeneration alleviation
via coordinated cascade energetic intervention. ACS Nano.
19:41121–41135. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Fu J, Leng X, Long J, Liao Z, An X, Ai X,
Long D, Li C, Huang B, Zhou Y, et al: SETD1A regulates glycolysis
and senescence of nucleus pulposus cells via
H3K4me3-HELZ2/PPARα-HIF1α axis to drive intervertebral disc
degeneration. Adv Sci (Weinh). e751052026.(Epub ahead of print).
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Wu O, Jin Y, Weng S, Zhang Z, Tu K, Sun J,
Chen L, Chen Q, Chen Z, Jones M, et al: METTL3 regulates
α-KG-dependent mitophagy and apoptosis in nucleus pulposus cells
through the MALAT1/miR-23c/IDH1 axis. Cell Rep. 45:1167932026.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Yao B, Wan L, Deng J, Chen Z, Zhao L, Wang
W and Han Z: m6A-mediated silencing of RNF41 by METTL3/YTHDC1
disrupts autophagy to drive intervertebral disc degeneration. Cell
Biol Toxicol. 41:1472025. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Yang Q, Huang F, Wang C, Liang X, Huang L,
Xu H, Liu J, Wei Q and Jiang H: METTL3-mediated m6A
modification promotes intervertebral disc degeneration. Ann Med.
57:25466702025. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Song C, Xu Y, Peng Q, Chen R, Zhou D,
Cheng K, Cai W, Liu T, Huang C, Fu Z, et al: Mitochondrial
dysfunction: A new molecular mechanism of intervertebral disc
degeneration. Inflamm Res. 72:2249–2260. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Qiu B, Xie X and Xi Y: Mitochondrial
quality control: The real dawn of intervertebral disc degeneration?
J Transl Med. 22:11262024. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Song Y, Liang H, Li G, Ma L, Zhu D, Zhang
W, Tong B, Li S, Gao Y, Wu X, et al: The NLRX1-SLC39A7 complex
orchestrates mitochondrial dynamics and mitophagy to rejuvenate
intervertebral disc by modulating mitochondrial Zn2+
trafficking. Autophagy. 20:809–829. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Liu M, Liu K, Cheng D, Zheng B, Li S and
Mo Z: The regulatory role of NLRX1 in innate immunity and human
disease. Cytokine. 160:1560552022. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Stokman G, Kors L, Bakker PJ, Rampanelli
E, Claessen N, Teske GJD, Butter L, van Andel H, van den Bergh
Weerman MA, Larsen PWB, et al: NLRX1 dampens oxidative stress and
apoptosis in tissue injury via control of mitochondrial activity. J
Exp Med. 214:2405–2420. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Roth GV, Gengaro IR and Qi LS: Precision
epigenetic editing: Technological advances, enduring challenges,
and therapeutic applications. Cell Chem Biol.
S2451-9456(24)00309-X. 2024.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Yuan L, Xiong Y, Zhang Y, Gu S and Lei Y:
Epigenome editing based treatment: Progresses and challenges. Mol
Ther. 34:46–67. 2026. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Cappelluti MA, Mollica Poeta V, Valsoni S,
Quarato P, Merlin S, Merelli I and Lombardo A: Durable and
efficient gene silencing in vivo by hit-and-run epigenome editing.
Nature. 627:416–423. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Kantor B, O'Donovan B, Rittiner J, Hodgson
D, Lindner N, Guerrero S, Dong W, Zhang A and Chiba-Falek O: The
therapeutic implications of all-in-one AAV-delivered
epigenome-editing platform in neurodegenerative disorders. Nat
Commun. 15:72592024. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Lei Y, Zhang X, Su J, Jeong M, Gundry MC,
Huang YH, Zhou Y, Li W and Goodell MA: Targeted DNA methylation in
vivo using an engineered dCas9-MQ1 fusion protein. Nat Commun.
8:160262017. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Horii T, Morita S, Hino S, Kimura M, Hino
Y, Kogo H, Nakao M and Hatada I: Successful generation of
epigenetic disease model mice by targeted demethylation of the
epigenome. Genome Biol. 21:772020. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Stover JD, Trone MA, Lawrence B and Bowles
RD: Multiplex epigenome editing of ion channel expression in
nociceptive neurons abolished degenerative IVD-conditioned
media-induced mechanical sensitivity. JOR Spine. 6:e12532023.
View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Stover JD, Farhang N, Lawrence B and
Bowles RD: Multiplex epigenome editing of dorsal root ganglion
neuron receptors abolishes redundant interleukin 6, tumor necrosis
factor alpha, and interleukin 1β signaling by the degenerative
intervertebral disc. Hum Gene Ther. 30:1147–1160. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Stover JD, Trone MAR, Weston J, Lewis C,
Levis H, Farhang N, Philippi M, Zeidan M, Lawrence B and Bowles RD:
Therapeutic CRISPR epigenome editing of inflammatory receptors in
the intervertebral disc. Mol Ther. 32:3955–3973. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Liang T, Gao B, Zhou J, Qiu X, Qiu J, Chen
T, Liang Y, Gao W, Qiu X and Lin Y: Constructing intervertebral
disc degeneration animal model: A review of current models. Front
Surg. 9:10892442023. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Tang SN, Bonilla AF, Chahine NO, Colbath
AC, Easley JT, Grad S, Haglund L, Le Maitre CL, Leung V, McCoy AM,
et al: Controversies in spine research: Organ culture versus in
vivo models for studies of the intervertebral disc. JOR Spine.
5:e12352022. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Daly C, Ghosh P, Jenkin G, Oehme D and
Goldschlager T: A review of animal models of intervertebral disc
degeneration: Pathophysiology, regeneration, and translation to the
clinic. Biomed Res Int. 2016:59521652016. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Choi H, Tessier S, Silagi ES, Kyada R,
Yousefi F, Pleshko N, Shapiro IM and Risbud MV: A novel mouse model
of intervertebral disc degeneration shows altered cell fate and
matrix homeostasis. Matrix Biol. 70:102–122. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Bergknut N, Rutges JPHJ, Kranenburg HJC,
Smolders LA, Hagman R, Smidt HJ, Lagerstedt AS, Penning LC,
Voorhout G, Hazewinkel HAW, et al: The dog as an animal model for
intervertebral disc degeneration? Spine (Phila Pa 1976).
37:351–358. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Xu X, Wang D, Zheng C, Gao B, Fan J, Cheng
P, Liu B, Yang L and Luo Z: Progerin accumulation in nucleus
pulposus cells impairs mitochondrial function and induces
intervertebral disc degeneration and therapeutic effects of
sulforaphane. Theranostics. 9:2252–2267. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Castillo ER and Lieberman DE: Lower back
pain. Evol Med Public Health. 2015:2–3. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Alini M, Diwan AD, Erwin WM, Little CB and
Melrose J: An update on animal models of intervertebral disc
degeneration and low back pain: Exploring the potential of
artificial intelligence to improve research analysis and
development of prospective therapeutics. JOR Spine. 6:e12302023.
View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Kong M, Gao C, Han S, Jin C, Hao M, Zhao
J, Luan J, Lin Y, Li Q and Ma X: Establishing an intervertebral
disc degeneration model in bipedal rats via a modified feeding
approach. Sci Rep. 15:353192025. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Cheng Y, Ma Z, Kim BH, Wu W, Cayting P,
Boyle AP, Sundaram V, Xing X, Dogan N, Li J, et al: Principles of
regulatory information conservation between mouse and human.
Nature. 515:371–375. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Mainardi A, Cambria E, Occhetta P, Martin
I, Barbero A, Schären S, Mehrkens A and Krupkova O: Intervertebral
disc-on-a-chip as advanced in vitro model for mechanobiology
research and drug testing: A review and perspective. Front Bioeng
Biotechnol. 9:8268672022. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Poletto DL, Crowley JD, Tanglay O, Walsh
WR and Pelletier MH: Preclinical in vivo animal models of
intervertebral disc degeneration. Part 1: A systematic review. JOR
Spine. 6:e12342022. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Romaniyanto F, Mahyudin F, Utomo DN,
Suroto H, Sari WA, Fachreza MS, Sadewa D, Dzikri DN and Nofaldi F:
Effectivity of puncture method for intervertebral disc degeneration
animal models: Review article. Ann Med Surg (Lond). 85:3501–3505.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Sakai D and Grad S: Advancing the cellular
and molecular therapy for intervertebral disc disease. Adv Drug
Deliv Rev. 84:159–171. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
ENCODE Project Consortium, . Moore JE,
Purcaro MJ, Pratt HE, Epstein CB, Shoresh N, Adrian J, Kawli T,
Davis CA, Dobin A, et al: Expanded encyclopaedias of DNA elements
in the human and mouse genomes. Nature. 583:699–710. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Zhou H, Clark E, Guan D, Lagarrigue S,
Fang L, Cheng H, Tuggle CK, Kapoor M, Wang Y, Giuffra E and Egidy
G: Comparative genomics and epigenomics of transcriptional
regulation. Annu Rev Anim Biosci. 13:73–98. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Hao Y, Zhu G, Yu L, Ren Z, Zhou W, Zhang P
and Lian X: FOXO3-activated HOTTIP sequesters miR-615-3p away from
COL2A1 to mitigate intervertebral disc degeneration. Am J Pathol.
194:280–295. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Jiang X and Chen D: LncRNA FAM83H-AS1
maintains intervertebral disc tissue homeostasis and attenuates
inflammation-related pain via promoting nucleus pulposus cell
growth through miR-22-3p inhibition. Ann Transl Med. 8:15182020.
View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Yang X, Cao X, Wang X, Guo J, Yang Y, Lu
L, Zhang P, Yang H, Rong K, Zhou T, et al: Palladium nanoparticles
degrade advanced glycation end products via valosin-containing
protein mediated autophagy to attenuate
high-glucose/high-fat-induced intervertebral disc degeneration.
Exploration (Beijing). 5:202301742025. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Kawarai Y, Jang SH, Lee S, Millecamps M,
Kang H, Gregoire S, Suzuki-Narita M, Ohtori S and Stone LS:
Exercise attenuates low back pain and alters epigenetic regulation
in intervertebral discs in a mouse model. Spine J. 21:1938–1949.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Zhang Z, Lin J, Nisar M, Chen T, Xu T,
Zheng G, Wang C, Jin H, Chen J, Gao W, et al: The Sirt1/P53 axis in
diabetic intervertebral disc degeneration pathogenesis and
therapeutics. Oxid Med Cell Longev. 2019:79595732019.PubMed/NCBI
|
|
156
|
Zhang TW, Li ZF, Dong J and Jiang LB: The
circadian rhythm in intervertebral disc degeneration: An autophagy
connection. Exp Mol Med. 52:31–40. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Dudek M, Gossan N, Yang N, Im HJ,
Ruckshanthi JP, Yoshitane H, Li X, Jin D, Wang P, Boudiffa M, et
al: The chondrocyte clock gene Bmal1 controls cartilage homeostasis
and integrity. J Clin Invest. 126:365–376. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Xiang M, Peng Q, Dai F, Wu Y, Liu L and
Liu H: Machine learning-enabled identification of nucleus pulposus
senescence-associated genes as potential biomarkers for
intervertebral disc degeneration. Eur J Med Res. 30:8872025.
View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Huang K, Shen L, Guan H, Dai L, Huang X,
Zhang X, Xu X and Liu C: Construction of a lncRNA-miRNA-mRNA
network for biomarker identification in intervertebral disc
degeneration. J Musculoskelet Neuronal Interact. 25:316–327. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Murphy K, Lufkin T and Kraus P:
Development and degeneration of the intervertebral disc-insights
from across species. Vet Sci. 10:5402023.PubMed/NCBI
|
|
161
|
Hu Y, Yang R, Liu S, Song Z and Wang H:
The emerging roles of nanocarrier drug delivery system in treatment
of intervertebral disc degeneration-current knowledge, hot spots,
challenges and future perspectives. Drug Des Devel Ther.
18:1007–1022. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Ding Y, Li F, Wang Y, Pan W, Fu X and Tan
S: Nanomedicine approaches for intervertebral disc regeneration:
From bench to bedside. Pharmaceutics. 17:3132025. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Wagner EK, Vedadghavami A, Jacobsen TD,
Goel SA, Chahine NO and Bajpayee AG: Avidin grafted dextran
nanostructure enables a month-long intra-discal retention. Sci Rep.
10:120172020. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Zhou H, Ning H, Liu Q, Tan Q, Zeng D, Feng
X, Wang D, Zhao Q, Wu X, He H, et al: A single-cell-inspired
self-enrichment therapeutic strategy delays intervertebral disc
degeneration by inhibiting pyroptosis. Adv Mater. 38:e164052026.
View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Wang Y, Wu Y, Zhang B, Zheng C, Hu C, Guo
C, Kong Q and Wang Y: Repair of degenerative nucleus pulposus by
polyphenol nanosphere-encapsulated hydrogel gene delivery system.
Biomaterials. 298:1221322023. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Wang Y, Deng M, Wu Y, Zheng C, Zhang F,
Guo C, Zhang B, Hu C, Kong Q and Wang Y: A multifunctional
mitochondria-protective gene delivery platform promote
intervertebral disc regeneration. Biomaterials. 317:1230672025.
View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Shi S, Ou X, Liu C, Li R, Zheng Q and Hu
L: Nanotechnology-enhanced pharmacotherapy for intervertebral disc
degeneration treatment. Int J Nanomedicine. 19:14043–14058. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Yang L, Bhujel B, Hou Y, Luo J, An SB, Han
I and Lee KB: Effective modulation of inflammation and oxidative
stress for enhanced regeneration of intervertebral discs using 3D
porous hybrid protein nanoscaffold. Adv Mater. 35:e23030212023.
View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Guo C, Liu Y, Zhao Z, Wu Y, Kong Q and
Wang Y: Regulating inflammation and apoptosis: A smart microgel
gene delivery system for repairing degenerative nucleus pulposus. J
Control Release. 365:1004–1018. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Chen Z, Liao Z, Liu M, Lin F, Chen S, Wang
G, Zheng Z, Liu B, Li C, Wang Z, et al: Nucleus pulposus-targeting
nanocarriers facilitate mirna-based therapeutics for intervertebral
disc degeneration. Adv Healthc Mater. 12:e23013372023. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Song T, Chen H, Huang J, Tian Y, Zhang X,
Chen H, Xie Y and Chen L: Engineering nanomaterials for the
treatment of intervertebral disc degeneration: From application to
therapeutic mechanisms. Small. 21:e098062025. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Liu W, Ma Z, Wang Y and Yang J: Multiple
nano-drug delivery systems for intervertebral disc degeneration:
Current status and future perspectives. Bioact Mater. 23:274–299.
2022.PubMed/NCBI
|
|
173
|
Bailey JF, Fields AJ, Liebenberg E,
Mattison JA, Lotz JC and Kramer PA: Comparison of vertebral and
intervertebral disc lesions in aging humans and rhesus monkeys.
Osteoarthritis Cartilage. 22:980–985. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Thompson K, Moore S, Tang S, Wiet M and
Purmessur D: The chondrodystrophic dog: A clinically relevant
intermediate-sized animal model for the study of intervertebral
disc-associated spinal pain. JOR Spine. 1:e10112018. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Gruber HE and Hanley EN Jr: Morphologic
features of spontaneous annular tears and disc degeneration in the
aging sand rat (Psammomys obesus obesus). Biotech Histochem.
92:402–410. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Tabatabaei SF, Akbari Roknabadi S and
Koohi S: DeepEPI: CNN-transformer-based model for extracting TF
interactions through predicting enhancer-promoter interactions.
Bioinform Adv. 5:vbaf2212025. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Ahmed FS, Aly S and Liu X: EPI-Trans: An
effective transformer-based deep learning model for enhancer
promoter interaction prediction. BMC Bioinformatics. 25:2162024.
View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Yan Y, Chai X, Liu J, Wang S, Li W and
Huang T: DeepMethyGene: A deep-learning model to predict gene
expression using DNA methylations. BMC Bioinformatics. 26:992025.
View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Zhang A, Jia J, Sun M and Wei X:
EPI-DynFusion: Enhancer-promoter interaction prediction model based
on sequence features and dynamic fusion mechanisms. Front Genet.
16:16142222025. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Zhang T, Shao S, Zhang H, Zhao Z, Zhao X,
Zhang X, Wang Z and Wang G: KansformerEPI: A deep learning
framework integrating KAN and transformer for predicting
enhancer-promoter interactions. Brief Bioinform. 26:bbaf2722025.
View Article : Google Scholar : PubMed/NCBI
|














