Introduction
The progression of liver disease from initial
steatosis through chronic hepatitis and fibrosis to hepatocellular
carcinoma (HCC) constitutes a major global health burden (1), accounting for ~2 million deaths
annually worldwide, with HCC alone responsible for >800,000
deaths each year (2). While
traditionally viewed as a linear sequence triggered by diverse
etiologies, emerging evidence reveals a shared, deeper molecular
foundation, including the systematic disruption and reprogramming
of the ubiquitin code, which is a central post-translational
regulatory system governing hepatic proteostasis. The ubiquitin
system was first discovered in the late 1970s and early 1980s as a
pathway responsible for ATP-dependent protein degradation, a
finding that was later recognized with the Nobel Prize in Chemistry
in 2004 (3). Subsequent studies
have revealed that beyond its canonical role in proteolysis, the
ubiquitin code governs a wide array of cellular processes,
including cell cycle control, DNA repair and immune signaling
(4–6). The implication of ubiquitin signaling
in tumorigenesis emerged from seminal observations that aberrant
expression or mutation of E3 ubiquitin ligases (E3 ligases) and
deubiquitinases (DUBs) frequently occurs in human cancers, leading
to dysregulation of oncoproteins and tumor suppressors. This
historical trajectory underscores the relevance of the ubiquitin
system as a critical node in cancer biology (7,8).
In physiological conditions, the ubiquitin system,
coordinately executed by E3 ligases and DUBs, functions as a
precise molecular operating system. It maintains immune tolerance,
metabolic equilibrium and cell survival by decoding ubiquitin
signals to determine the fate of key signaling proteins, thereby
preserving hepatic homeostasis despite continuous antigen exposure.
Under persistent pathological stress, including metabolic
lipotoxicity, chronic inflammatory signals and cell death, this
regulatory network becomes fundamentally corrupted. Key E3 ligases
and DUBs are functionally subverted, transitioning from homeostatic
guardians to pathogenic drivers. For instance, the
immune-regulatory functions of A20 and casitas B-lineage lymphoma-b
(Cbl-b) are compromised, whereas destructive pathways, such as
NOD-like receptor family pyrin domain-containing 3 (NLRP3)
inflammasome activation [which has been linked to
BRCA1-BRCA2-containing complex subunit 3 (BRCC3) in certain
contexts] and cylindromatosis (CYLD)-dependent death, are
aberrantly engaged (9,10). This reprogramming converts
protective responses into a chronic engine of tissue damage,
fibrosis and genomic instability.
Ultimately, during cirrhosis and carcinogenesis, the
ubiquitin code is comprehensively hijacked to establish a
pro-tumorigenic state. Oncoproteins including β-catenin and c-Myc
evade degradation, while tumor suppressors such as p53 are targeted
by E3 ligases such as MDM2. Concurrently, immune checkpoint
molecules such as programmed death-ligand 1 (PD-L1) are stabilized
by specific DUBs, reinforcing an immunosuppressive microenvironment
(11).
The present review hypothesized that the spectrum of
liver disease progression reflects the systematic corrosion and
reprogramming of the hepatic ubiquitin code by a pathological
microenvironment. The functional metamorphosis of E3 ligases and
DUBs represents a unifying molecular axis linking steatosis,
inflammation, fibrosis and carcinogenesis. In addition, this
paradigm was systematically deciphered and novel therapeutic
strategies, including targeted inhibitors and proteolysis-targeting
chimeras, designed to reset hepatic homeostasis and intercept
disease progression were explored.
Regulatory mechanisms of the ubiquitin
system in hepatic immunity
Composition and function of the
ubiquitin system
Ubiquitination is a post-translational modification
wherein ubiquitin is covalently attached to substrate proteins. The
functional outcome is determined by polyubiquitin chain topology,
which can be linked via any of the seven lysine residues of
ubiquitin (K6, K11, K27, K29, K33, K48 and K63) or its N-terminal
methionine (Met1). K48-linked chains primarily target substrates
for proteasomal degradation, whereas K63-linked chains regulate
non-proteolytic processes including signal transduction and DNA
repair. Linear (Met1-linked) chains play specialized roles in
nuclear factor κB (NF-κB) signaling and cell death regulation.
Ubiquitination machinery: A tripartite
enzymatic cascade
Ubiquitination proceeds via a three-step enzymatic
cascade involving E1 (activating), E2 (conjugating) and E3
(ligating) enzymes (12–14). E3 ligases confer substrate
specificity, with >600 members encoded in the human genome
classified into three families based on catalytic mechanism
(15,16). Fig.
1 describes the mechanisms of each family, demonstrating that
each family uses a distinct strategy to transfer ubiquitin to the
substrate.
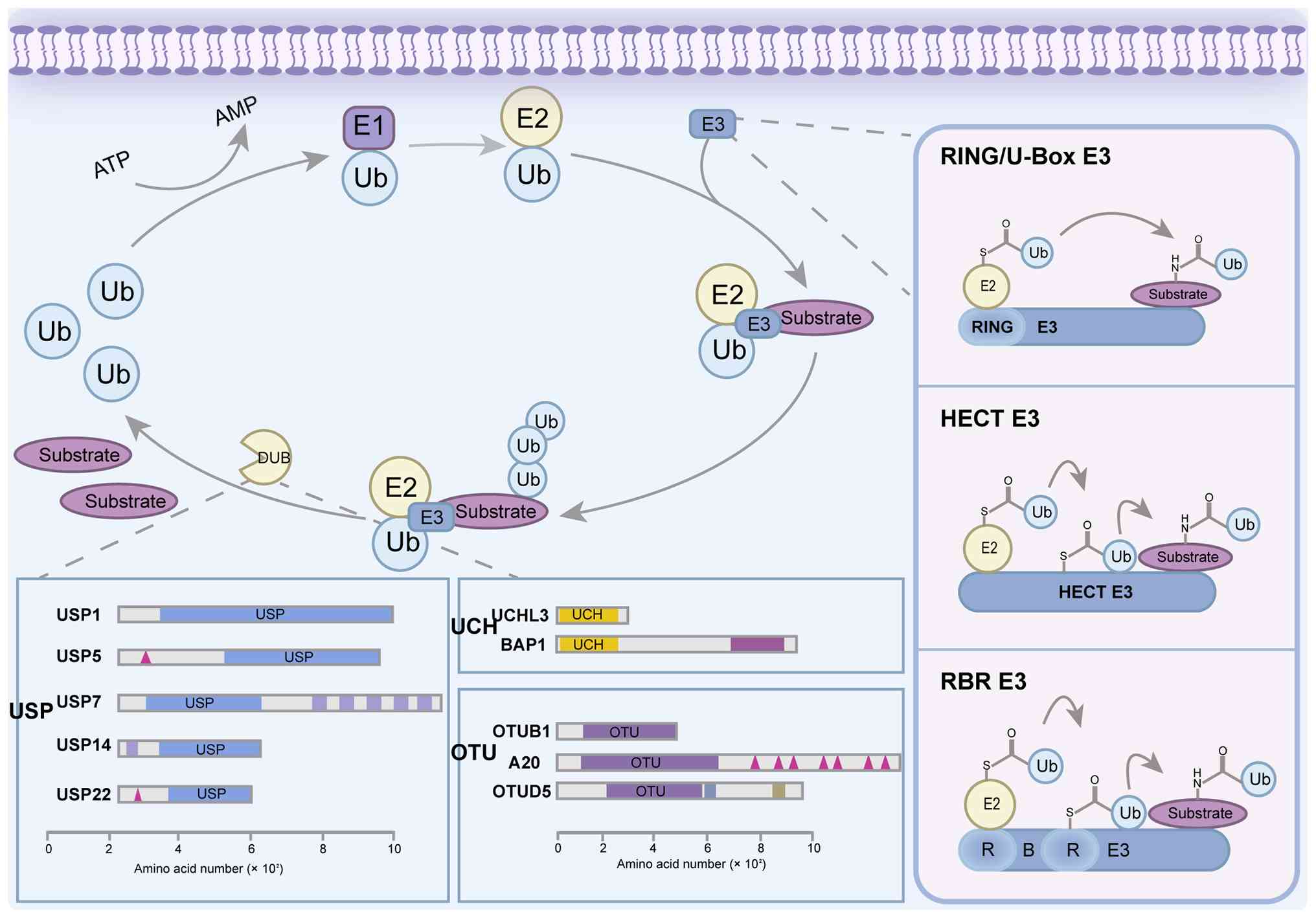 | Figure 1.Main families of Ub ligases and DUBs
and their core features. E3 Ub ligases (key subtypes) mainly
include HECT (with C-terminal HECT domain), RING (with N-terminal
zinc-binding RING domain), U-box (with C-terminal U-box domain) and
RBR (with RING1/IBR/RING2 domains). DUBs (main families) mainly
include USP, UCHL and OTU families, and the domain composition of
these families (such as catalytic domains and accessory domains of
USP) is described. Ub, ubiquitin; E1, ubiquitin-activating enzyme;
E2, ubiquitin-conjugating enzyme; E3, ubiquitin ligase; RING,
really interesting new gene; HECT, homologous to E6-AP C-terminus;
RBR, RING-between-RING; DUBs, deubiquitinating enzymes; USP,
ubiquitin-specific protease; UCH, ubiquitin C-terminal hydrolase;
OTU, ovarian tumor protease; UCHL3, ubiquitin C-terminal hydrolase
L3; BAP1, BRCA1-associated protein 1; A20, TNF-α-induced protein 3;
OTUB1, OTU domain-containing ubiquitin aldehyde-binding protein 1;
OTUD5, OTU deubiquitinase 5. |
Really interesting new gene (RING)-type E3s act as
scaffolds that facilitate direct ubiquitin transfer from E2 to the
substrate (17–21). Homologous to E6-AP carboxyl
terminus (HECT)-type E3s form a catalytic intermediate, accepting
ubiquitin from E2 via a thioester bond before transferring to
substrate (19–21). RING-between-RING (RBR)-type E3s
utilize a hybrid RING-HECT mechanism involving a transient E3
ubiquitin intermediate (16,20).
Deubiquitination: Reversal and
regulation
Ubiquitination is reversed by DUBs (22–24),
whereby the 100 human DUBs cleave ubiquitin-substrate bonds to
terminate signaling, rescue proteins from degradation and recycle
ubiquitin monomers to maintain cellular homeostasis (23). As shown in Fig. 1, major DUB families include: i)
Ubiquitin-specific proteases (USPs); ii) ovarian tumor proteases
(OTUs); and iii) ubiquitin C-terminal hydrolases (24).
Central role of ubiquitination in
cellular processes
The ubiquitin system regulates numerous fundamental
cellular processes (24). It
modulates the intensity and duration of signal transduction by
controlling the stability of receptors, adaptors and kinases. In
inflammatory responses, ubiquitination exerts precise control over
inflammasome components and key molecules within the NF-κB pathway,
thereby orchestrating the initiation and resolution of
inflammation. Furthermore, the system directly regulates apoptotic
and necroptotic pathways by influencing caspase activity and the
stability of critical signaling molecules such as
receptor-interacting serine/threonine-protein kinase 1 (RIPK1)
(25). During autophagy, ubiquitin
acts as a degradation signal that labels damaged organelles for
clearance (26). Within metabolic
pathways, ubiquitination controls the stability of metabolic
enzymes and transcription factors, including sterol regulatory
element-binding proteins, thereby coordinating glucose and lipid
homeostasis.
Hepatic immune milieu and the role of
ubiquitination
The liver constitutes an immunologically-privileged
organ, continuously exposed to gut-derived antigens and metabolites
via the portal venous circulation (27). This results in a microenvironment
characterized by a high antigenic load (28). To maintain homeostasis under such
conditions, the liver has developed robust tolerogenic mechanisms
that enable distinction between harmless substances and genuine
threats, thereby preventing excessive immune damage.
Within this unique immunological setting, the
ubiquitin system serves as a pivotal regulator of hepatic immune
homeostasis (29). It establishes
and sustains immune tolerance by precisely controlling the
initiation and resolution of innate immune signaling, such as the
Toll-like receptor (TLR) and NF-κB pathway, and by setting
stringent activation thresholds for adaptive immune cells, such as
both T and B lymphocytes. This regulatory function is exemplified
by E3 ligases such as Cbl-b and itchy E3 ubiquitin ligase (Itch)
(30,31). Consequently, the stability of the
hepatic microenvironment is highly dependent on the precise,
dynamic and context-dependent regulation of immune pathways by the
ubiquitin system.
Central role of the ubiquitin system in
maintaining hepatic homeostasis
Ubiquitin-mediated regulation of
immune tolerance
The liver is a central organ for systemic metabolism
and immune regulation. Due to its unique anatomical location and
continuous exposure to gut-derived antigens via the portal
circulation, the liver requires sophisticated regulatory mechanisms
to distinguish self from non-self, balance immune clearance with
tolerance and maintain metabolic stability. The ubiquitin system,
comprising E1 activating enzymes, E2 conjugating enzymes, E3
ligases, DUBs and downstream degradation pathways, plays a central
regulatory role in this context. Through reversible ubiquitin-chain
modifications, this system precisely governs protein activity,
subcellular localization, protein-protein interactions and
stability (32,33). Under physiological conditions, a
coordinated network of E3 ligases and DUBs sustains hepatic
homeostasis by regulating immune responses, metabolic equilibrium
and cellular quality control.
Hepatic immune homeostasis depends on the precise
negative regulation of innate immune signaling. In the TLR4-NF-κB
pathway, the E3 ligase tumor necrosis factor (TNF)
receptor-associated factor 6 (TRAF6) activates the IκB kinase
(IKK)-NF-κB axis via K63-linked polyubiquitination, initiating
pro-inflammatory gene expression (34). To prevent excessive activation, a
built-in negative feedback mechanism operates through the
ubiquitin-editing enzyme A20 (TNFAIP3) (35,36).
A20 terminates signaling by removing K63-linked chains from TRAF6
and RIPK1 via its OTU domain, while simultaneously targeting these
substrates for proteasomal degradation through K48-linked
ubiquitination mediated by its zinc-finger domains (Fig. 2) (37). This coordinated activity ensures
the self-limiting nature of inflammatory responses.
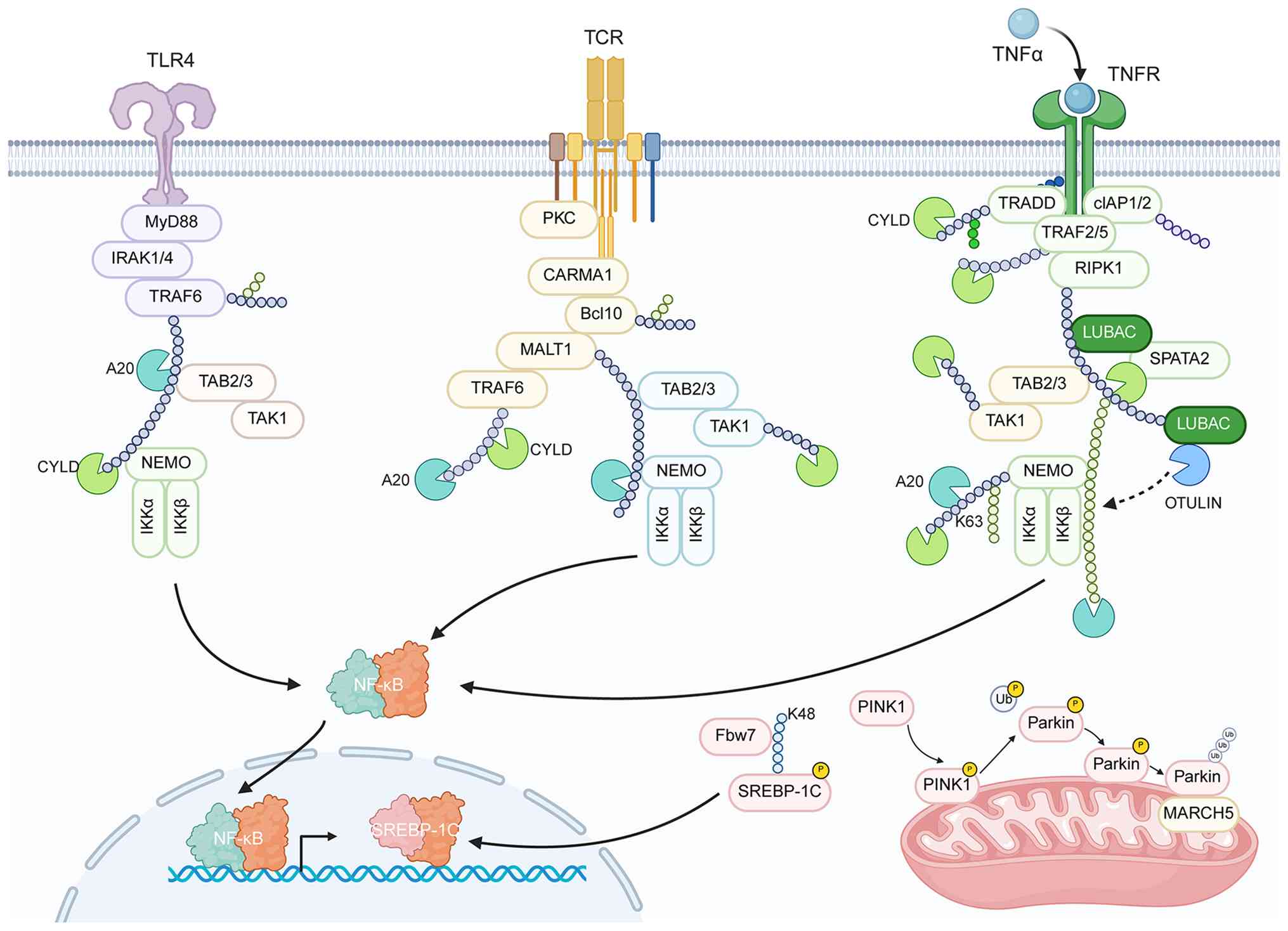 | Figure 2.Hepatic ubiquitin system-mediated
homeostatic regulatory networks under physiological conditions. Key
components (such as A20, CYLD, Fbw7, PINK1-PARKIN and MARCH5)
acting across pathways: i) Fine-tuning immune responses downstream
of TLR4, TCR and TNFR signaling; ii) governing metabolic pathways
via regulators such as Fbw7; and iii) executing protein quality
control through the PINK1-PARKIN axis and MARCH5. The coordinated,
context-dependent actions of these components underpin hepatic
functional homeostasis. TLR4, Toll-like receptor 4; MyD88, myeloid
differentiation primary response 88; IRAK1/4, interleukin-1
receptor-associated kinase 1/4; TRAF6, TNF receptor-associated
factor 6; A20, TNF-α-induced protein 3; TAB2/3, TAK1-binding
protein 2/3; TAK1, TGF-β-activated kinase 1; CYLD, cylindromatosis;
NEMO, NF-κB essential modulator; IKKα/β, IκB kinase α/β; NF-κB,
nuclear factor κB; TCR, T-cell receptor; Cbl-b, casitas B-lineage
lymphoma-b; Itch, itchy E3 ubiquitin ligase; PKCθ, protein kinase C
θ; PLCγ1, phospholipase C γ1; Fbw7, F-box/WD repeat-containing
protein 7; SREBP-1c, sterol regulatory element-binding protein 1c;
PINK1, PTEN-induced kinase 1; PARKIN, Parkin RBR E3 ubiquitin
ligase; MARCH5, membrane-associated RING-CH 5; NLRP3, NOD-like
receptor family pyrin domain-containing 3; Ub, ubiquitin; K48,
lysine 48; K63, lysine 63. |
The ubiquitin system also establishes stringent
activation thresholds for adaptive immunity, particularly in T-cell
activation. Full T-cell activation requires signaling through both
the T-cell receptor (TCR) and the CD28 costimulatory pathway
(38). As illustrated in Fig. 2, this threshold is enforced by a
network of E3 ligases, including Cbl-b, Itch and GRAIL, that
collectively restrain TCR and costimulatory signaling. The figure
visually organizes the mechanism by which the ligases intersect
distinct signaling nodes to cooperatively maintain immune
tolerance. In the absence of CD28 co-stimulation, activated Cbl-b
catalyzes K48-linked ubiquitination and degradation of key
TCR-signaling components, including protein kinase C θ,
phospholipase C γ1 and CD28 itself, thereby attenuating signal
transduction (39). Cbl-b further
impairs immunological-synapse formation by targeting the adaptor
protein Crk-L for degradation (40). Additional E3 ligases contribute to
this layered control, including Itch, which, in concert with the E2
enzyme ubiquitin-conjugating enzyme H7, mediates degradation of the
TCRζ chain, whereas GRAIL restricts T-cell activation and clonal
expansion by promoting the endocytosis and degradation of membrane
proteins such as CD154. Collectively, these E3 ligases constitute a
multilayered regulatory network that actively maintains the
immune-tolerant state of the liver.
Ubiquitin-dependent regulation of
metabolic pathways
The metabolic function of the liver is critically
regulated by the ubiquitin system, which controls the stability of
key metabolic transcription factors. This regulation occurs through
targeted ubiquitination and degradation of key metabolic
transcription factors, a mechanism that integrates hepatic
metabolic control with the broader ubiquitin-mediated regulatory
framework (Fig. 2). Lipid
homeostasis exemplifies this regulation. The F-box/WD
repeat-containing protein 7 (Fbw7), a substrate-recognition
component of the Skp1-cullin-F-box E3 ligase complex, targets
phosphorylated nuclear sterol regulatory element-binding protein 1c
for K48-linked ubiquitination and proteasomal degradation. This
process establishes a negative feedback loop that limits excessive
hepatic lipid accumulation (41).
Furthermore, the typically low expression of peroxisome
proliferator-activated receptor γ in hepatocytes is maintained
partly through constitutive ubiquitination and degradation mediated
by E3 ligases, such as neural precursor cell expressed
developmentally downregulated protein 4, thereby suppressing
hepatocyte transdifferentiation toward an adipocyte-like phenotype
(42).
Ubiquitin-dependent quality control in
organelle stress
The ubiquitin system plays a critical role in
quality control mechanisms that counteract organelle stress induced
by high metabolic activity. For the clearance of damaged
mitochondria, the phosphatase and tensin homolog-induced kinase 1
(PINK1) - parkin RBR E3 ubiquitin ligase (PARKIN) pathway serves as
a central regulatory axis. As shown in Fig. 2, two key stress-resisting
mechanisms mediated by the ubiquitin system, mitochondrial quality
control through mitophagy and constrained NLRP3 inflammasome
activation, are presented side by side to illustrate their
coordinated roles in preserving cellular homeostasis. PINK1
stabilizes on the outer mitochondrial membrane and phosphorylates
ubiquitin, leading to recruitment and activation of the cytosolic
E3 ligase PARKIN (43). Activated
PARKIN deposits ubiquitin chains on mitochondrial surface proteins,
thereby tagging damaged organelles for elimination via
mitophagy.
Similarly, the activation of the NLRP3 inflammasome
is tightly regulated by ubiquitination. Under basal conditions,
constitutive ubiquitination of NLRP3 suppresses its oligomerization
and activation. This inhibitory state is maintained by the
mitochondria-localized E3 ligase membrane-associated RING-CH 5
(MARCH5), which mediates K48-linked ubiquitination and degradation
of NLRP3. Additionally, tripartite motif containing 31 (TRIM31)
promotes NLRP3 clearance under cellular stress, further
highlighting the multilayered ubiquitin-dependent regulation of
inflammasome activity (44).
Summary
Under physiological conditions, the hepatic
ubiquitin system operates as an integrated and dynamically
regulated network that sustains homeostasis. Key components,
including A20, Cbl-b and Itch, precisely modulate immune responses;
regulators such as Fbw7 govern metabolic pathways; and mediators
such as the PINK1-PARKIN axis and MARCH5 execute quality control.
The precision and context-dependency of this system, supported by
multi-layered feedback, are essential for maintaining hepatic
function. However, under sustained metabolic, toxic or
immunological stress, core elements of this network can be
subverted or impaired. This functional conversion transforms
regulatory components from homeostatic regulators into pathological
progression.
Mechanisms of hepatic ubiquitin homeostasis
disruption by metabolic stress
Metabolic stress as a pathogenic
initiator in liver disease
In liver disease pathogenesis, metabolic
dysregulation represents a key initiating event (45). Within the shared multiple-hit model
of non-alcoholic and alcoholic fatty liver disease, persistent
metabolic stressors, such as lipotoxicity and oxidative stress, act
as primary inducers (1,46,47).
These stressors not only disturb metabolic pathways but also
disrupt the hepatic ubiquitin system. This disruption involves the
functional conversion of core E3 ligases and DUBs from homeostatic
regulators into promoters of pathological efforts (48). The process is characterized by
aberrant activation of innate immune and inflammatory pathways
alongside impaired cellular quality control.
Aberrant activation of the NLRP3
inflammasome: Dysregulation of ubiquitin-dependent control
Under physiological conditions, the activation of
innate immune signaling pathways, such as the NLRP3 inflammasome,
is tightly regulated by ubiquitination (49). However, in the pathological
contexts of non-alcoholic fatty liver disease (NAFLD) and alcoholic
steatohepatitis, metabolic danger signals, including excess
saturated fatty acids, free cholesterol and its crystals, and lipid
peroxidation products, disrupt this ubiquitin-mediated control,
leading to aberrant activation of these pathways (50).
NLRP3 inflammasome activation constitutes a crucial
checkpoint for the proteolytic maturation of IL-1β and IL-18,
dependent on caspase-1, thereby orchestrating the transition from
simple fatty liver to steatohepatitis (51). Under basal conditions, NLRP3 is
tonically inhibited by constitutive ubiquitin modifications
(52). A two-step process,
involving priming and activation signals, is necessary for its
complete engagement (53).
Metabolic stress fulfills this dual requirement by acting as a
priming stimulus and, through the direct perturbation of NLRP3
ubiquitination, serving as an activation trigger (54,55).
The DUB BRCC3 has been implicated as a key mediator
in this pathway (56).
Specifically, BRCC3 possesses K63-specific DUB activity and
directly binds to NLRP3, removing the K63-linked ubiquitin chains
from it. Exposure to saturated fatty acids (such as palmitate) and
cholesterol crystals in macrophages and Kupffer cells potentiates
this interaction and augments the catalytic function of BRCC3
(57). This deubiquitination is
proposed to serve as a molecular prerequisite for the
oligomerization of NLRP3 and its subsequent assembly into an active
inflammasome. Consistently, attenuating BRCC3 expression blunts the
assembly of the NLRP3 inflammasome and the secretion of IL-1β
triggered by metabolic danger signals. Consistent with this,
preclinical studies have established that BRCC3-mediated
deubiquitination is a critical regulatory step for NLRP3
inflammasome activation (52,58).
Given the central role of NLRP3-driven inflammation in NASH
pathogenesis, it is plausible that similar dysregulation of the
BRCC3-NLRP3 axis may contribute to the hepatic inflammatory milieu
in NASH, although direct evidence from NASH-specific models remains
to be fully elucidated.
However, it is worth noting that the role of DUBs in
NLRP3 inflammasome regulation is highly context-dependent. While
BRCC3 has been shown to promote NLRP3 activation in myeloid cells
under lipotoxic conditions, other DUBs such as USP7 and USP47 have
been reported to enhance NLRP3 activation through facilitating ASC
oligomerization, and USP50 by removing K48-linked ubiquitin chains
to stabilize NLRP3 (59,60). By contrast, the DUB A20 has been
shown to suppress NLRP3 inflammasome activation by competitively
binding to NIMA-related kinase 7 (61), highlighting that different DUBs can
exert opposing effects on NLRP3 depending on the cellular context
and specific protein-protein interactions. Furthermore, the current
evidence linking BRCC3 to NLRP3 activation in the liver remains
largely correlative. The majority of studies, including those cited
in the present review, are derived from macrophage or Kupffer cell
models; whether BRCC3 exerts a similar function in other hepatic
cell types (such as hepatocytes or hepatic stellate cells) under
lipotoxic stress is less well-established. Additionally, direct
evidence for BRCC3-mediated NLRP3 deubiquitination in vivo,
such as from conditional knockout models or in situ
enzymatic assays, is still limited.
Fig. 3 illustrates
the proposed mechanism by which metabolic stress triggers NLRP3
inflammasome assembly through BRCC3-driven deubiquitination. The
figure also highlights the counterregulatory role of TRIM31, which
promotes K48-linked ubiquitination and proteasomal degradation of
NLRP3 (62). Collectively, these
findings indicate that metabolic stress disrupts the normal
regulatory constraints on the NLRP3 inflammasome through a dual
mechanism, involving coordinately activating DUBs while
simultaneously suppressing specific E3 ubiquitin ligases (62–64).
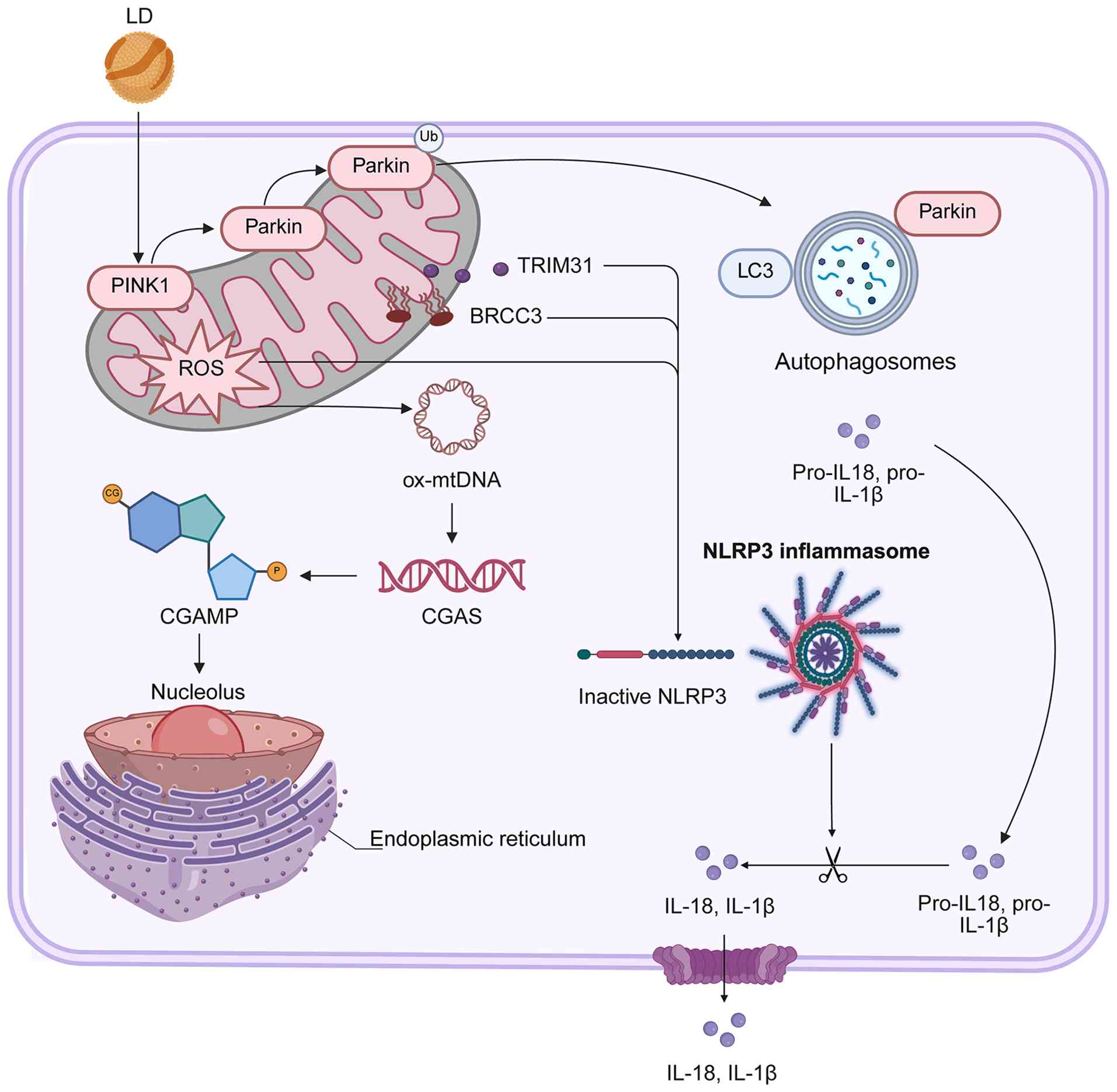 | Figure 3.Metabolic stress-driven dysregulation
of hepatic ubiquitin system nodes and pathogenic initiation,
including impaired PINK1-PARKIN mitochondrial quality control
(linked to mitochondrial ROS/oxmDNA release), BRCC3
(deubiquitinase) activation that relieves NLRP3 inflammasome
inhibition and suppressed protective E3 ligase MARCH5. These
altered ubiquitin-modifying enzyme nodes are repurposed from
homeostatic guardians to drivers of pathogenic events (such as
NLRP3-dependent IL-18/1β secretion) initiating hepatic pathology.
PINK1, PTEN-induced kinase 1; PARKIN, Parkin RBR E3 ubiquitin
ligase; TRIM31, tripartite motif-containing 31; BRCC3,
BRCA1-BRCA2-containing complex subunit 3; NLRP3, NOD-like receptor
family pyrin domain-containing 3; IL-1β, interleukin-1β; cGAMP,
cyclic GMP-AMP; STING, stimulator of interferon genes. |
Disruption of mitochondrial quality
control: Impairment of the PINK1-PARKIN pathway
While competent mitochondria constitute the hub of
cellular energy metabolism, dysfunctional organelles are notable
sources of reactive oxygen species (ROS) and mitochondrial DNA
(mtDNA) (65). These released
molecules serve as potent damage-associated molecular patterns
(DAMPs), capable of activating both the cyclic GMP-AMP synthase
(cGAS)-stimulator of interferon genes (STING) pathway and the NLRP3
inflammasome (66–68). Therefore, the selective removal of
damaged mitochondria via mitophagy is a critical homeostatic
mechanism. The canonical PINK1-PARKIN mitophagy pathway, which is
crucial for this process, has been reported to be susceptible to
disruption under lipotoxic conditions (69), although the precise mechanisms
remain incompletely defined and may vary by cell type or
experimental context. Fig. 3
captures the proposed consequences of this disruption, including
mitochondrial damage accumulation driven by PINK1-PARKIN
impairment, and the ensuring of pathological crosstalk with immune
signaling cascades, representing a potential vicious cycle that
links metabolic stress to sterile inflammation.
Under physiological conditions, the PINK1-PARKIN
pathway continuously monitors mitochondrial integrity (70). Following the dissipation of the
mitochondrial membrane potential, PINK1 accumulates on the outer
mitochondrial membrane and recruits cytosolic PARKIN through the
phosphorylation of ubiquitin molecules (71). The activated PARKIN
polyubiquitinates the mitochondrial surface, generating ubiquitin
chains that serve as recognition signals for the autophagy
machinery via specific autophagy receptors (such as p62, optineurin
and nuclear dot protein 52 kDa), thereby marking the damaged
organelle for sequestration and clearance (72).
In addition to the PINK1-PARKIN pathway, the liver
employs alternative mitochondrial quality control mechanisms that
contribute to cellular homeostasis. These include receptor-mediated
mitophagy, in which mitophagy receptors such as BCL2 interacting
protein 3 (BNIP3), NIP3-like protein X (NIX) and FUN14
domain-containing protein 1 (FUNDC1) directly recruit autophagic
machinery to damaged mitochondria independently of PARKIN (73). Additionally, mitochondrial-derived
vesicles represent an alternative route for selective removal of
oxidized mitochondrial proteins or mtDNA without requiring
whole-organelle degradation. While the functional relevance of
these pathways in NAFLD and alcoholic liver disease (ALD) remains
incompletely characterized, they may partially compensate when
PINK1-PARKIN function is compromised under lipotoxic stress.
Elucidating the interplay between these parallel quality control
systems represents an important direction for future
investigation.
In hepatocytes affected by NAFLD and ALD, emerging
evidence suggests that the PINK1-PARKIN pathway becomes
dysfunctional. Severe lipid overload directly impairs mitochondrial
function, leading to membrane depolarization and excessive ROS
production (74). This creates a
supra-physiological burden of damaged mitochondria, which may
overwhelm the PINK1-PARKIN clearance system. Furthermore, elevated
levels of free fatty acids have been proposed to interfere with the
effective translocation of PARKIN to impaired mitochondria,
potentially through post-translational modifications of PARKIN by
lipid metabolites or alterations in mitochondrial membrane lipid
composition. However, conflicting studies have reported that
PARKIN-independent mitophagy pathways (such as those mediated by
FUNDC1 or NIX) may partially compensate under lipotoxic conditions
(75,76), and it is not clear whether PARKIN
dysfunction is a primary driver or a secondary consequence of
mitochondrial stress.
These functional defects are considered to
contribute to the intracellular accumulation of dysfunctional
mitochondria. These organelles are not only bioenergetically
inefficient but also perpetually generate ROS, exacerbating
oxidative stress and inducing lipid peroxidation and DNA damage.
The mtDNA released from these mitochondria acts as a DAMP, which is
recognized by cGAS, thereby activating the cGAS-STING pathway and
inducing the expression of interferon-stimulated genes, including
type I interferons. This response aggravates hepatic inflammation
and immune cell infiltration. Simultaneously, mtDNA serves as a
potent activator of the NLRP3 inflammasome. Thus, it is
hypothesized that by impairing PINK1-PARKIN-mediated mitochondrial
quality control, metabolic stress could transform the hepatocyte
into a persistent source of oxidative stress and pro-inflammatory
signals, potentially providing a sustained driving force for
chronic hepatitis and subsequent fibrogenesis. It must be
acknowledged that these mechanistic conclusions are largely derived
from in vitro models using acute lipotoxic stimuli; further
studies using chronic in vivo models and human tissue are
needed to fully validate the causal role of PINK1-PARKIN impairment
in NAFLD or ALD pathogenesis.
Summary
This section systematically outlined the mechanisms
through which metabolic stress initiates hepatic pathology by
disrupting key regulatory nodes within the ubiquitination system.
Specifically, metabolic stress disrupts the normal inhibitory
constraints on the NLRP3 inflammasome by activating
deubiquitinating enzymes such as BRCC3 while concurrently
suppressing protective E3 ligases such as MARCH5. In parallel, it
disrupts mitochondrial quality control by impairing the function of
the PINK1-PARKIN pathway. The defining feature of this initial
pathogenic phase is the dysregulation of critical E3 ligases and
DUBs within the disease microenvironment, which may repurpose them
from regulators of cellular homeostasis to drivers of pathological
progression.
Understanding this early stage carries notable
therapeutic implications. It suggests that dysregulated
ubiquitin-modifying enzymes may represent candidate therapeutic
targets, and that intervening during the initial phase of disease
could potentially interfere with the progression from simple
steatosis to active steatohepatitis. However, given that multiple
ubiquitin-modifying enzymes are likely implicated at this early
stage, and given the inherent redundancy within the ubiquitin
system, it remains uncertain whether targeting any single enzyme or
combination would suffice to halt disease progression. Future
studies employing genetic and pharmacological approaches in
preclinical models are needed to evaluate the feasibility and
efficacy of such strategies. However, once this cascade is
initiated, it fosters a more complex and self-sustaining
pathological milieu. This culminates in a broader, more pervasive
reprogramming of the ubiquitin system. The subsequent stage,
characterized by the breakdown of immune tolerance and alterations
in cell fate decisions under conditions of persistent inflammation
and cell death, will be the primary focus of the following
section.
Systemic regulatory mechanisms of immune
tolerance disruption and altered cell fate
Evolution to a systemic pathological
landscape
The progressive and sustained impact of metabolic
stress drives fundamental reprogramming of the hepatic ubiquitin
system. This reprogramming extends beyond initial, focal
disruptions and culminates in a complex, self-perpetuating
pathological landscape within the liver. The microenvironment
evolves from one defined by metabolic disturbance and localized
inflammation to a state characterized by the pervasive accumulation
of cellular debris, elevated titers of pro-inflammatory cytokines,
sustained infiltration and activation of diverse immune cells and
excessive deposition of extracellular matrix components.
At this advanced stage, disease progression becomes
increasingly decoupled from the original exogenous metabolic
insults. Instead, it is critically driven and amplified by the
breakdown of intrinsic homeostatic and regulatory circuits. The
hepatic ubiquitin system undergoes systemic and maladaptive
reprogramming, with its most consequential manifestations being the
comprehensive breakdown of immune tolerance and the loss of precise
control over cell fate decisions. These cardinal dysfunctions are
molecularly instantiated through the impairment of adaptive immune
checkpoint pathways and the pathological rewiring of the regulatory
networks that govern hepatocyte survival, death and phenotypic
plasticity. This section will delineate the mechanisms through
which the reprogrammed ubiquitin system orchestrates this
transition to a state of chronic, unresolving inflammation and
tissue remodeling.
Disruption of B-cell tolerance via the
B-cell activating factor (BAFF)/TRAF3/NF-κB axis
Under homeostatic conditions, E3 ubiquitin ligases
serve pivotal roles in maintaining peripheral immune tolerance by
modulating the activation threshold of both T and B lymphocytes.
However, this regulatory mechanism becomes dysfunctional within the
inflammatory milieu characteristic of NASH or chronic viral
hepatitis.
In the context of B-cell homeostasis, the B-cell
activating factor (BAFF) signaling pathway is critical for the
survival of mature B-cells. Under physiological conditions, an E3
ligase complex composed of cellular inhibitor of apoptosis protein
(cIAP-)1, cIAP2, TRAF2 and TRAF3 constitutively suppresses
BAFF-receptor (BAFF-R) signaling. Central to this suppression,
TRAF3 continuously recruits NF-κB-inducing kinase (NIK) and targets
it for ubiquitin-mediated degradation, thereby maintaining the
non-canonical NF-κB pathway in a quiescent state (77).
Within the inflammatory microenvironment, cytokines
such as type I interferon and interleukin-6 upregulate BAFF
expression. The ensuing excess BAFF binds to BAFF-R, inducing
receptor multimerization (78).
This event triggers the recruitment of the TRAF2-cIAP1 and cIAP2
complex, which subsequently targets TRAF3 for proteasomal
degradation via K48-linked polyubiquitination (79). The elimination of TRAF3 relieves
the constitutive inhibition of NIK (80), and stabilized NIK phosphorylates
and activates IKKα, which in turn processes the NF-κB precursor
p100 into its mature form, p52 (81). The p52 subunit forms a heterodimer
with reticuloendotheliosis viral oncogene homolog B, and this
complex translocates to the nucleus, where it initiates the
transcription of genes that promote B-cell survival and
maturation.
In autoimmune liver diseases, including primary
biliary cholangitis and autoimmune hepatitis, elevated serum BAFF
promotes sustained non-canonical NF-κB2 activation through chronic
TRAF3 degradation (82). This
process subverts B-cell central tolerance, permitting the survival
and maturation of autoreactive B-cells. Their subsequent
differentiation into autoantibody-producing plasma cells drives
hepatic inflammation through complement activation and
immune-complex deposition.
Inflammatory suppression of E3 ligases
compromises T-cell tolerance
In T cells, the inflammatory microenvironment
disrupts the function of key E3 ubiquitin ligases, notably Cbl-b
and Itch, through multiple mechanisms. Cbl-b activity is
compromised via transcriptional repression and oxidative
inactivation under inflammatory conditions. The consequent loss of
Cbl-b function, combined with enhanced co-stimulatory signals,
markedly lowers the threshold for T-cell activation. This
dysregulation renders T cells prone to excessive responses against
self-antigens or persistent viral antigens. This notion is
supported by evidence from Cbl-b-deficient models, which develop
severe CD8+ T-cell-driven hepatitis, suggesting that
analogous mechanisms may contribute to immunopathology in human
chronic viral hepatitis (83).
Similarly, the activity of Itch is modulated by the
inflammatory milieu. Itch function is tightly regulated by its
phosphorylation status and conformational dynamics; inflammatory
signaling may alter the kinase/phosphatase balance, thereby
affecting Itch activation. Deficiency in Itch leads to the
accumulation of transcription factor JunB, which drives excessive
production of T helper (Th-)2-type cytokines and impairs the
negative regulation of TCR signaling. In the hepatic context,
downregulation or functional impairment of Itch may disrupt the
balance between Th17 cells and regulatory T cells, thereby
facilitating the activation of autoreactive T cells and promoting
inflammatory liver pathology (84).
Metabolic reprogramming and loss of
feedback control in hepatocytes
In the context of hepatocyte fate determination,
dysregulation occurs in metabolic control pathways. During the
steatotic phase, hepatocytes initiate a compensatory feedback
response through the upregulation of E3 ligases, such as Fbw7,
which targets the sterol regulatory element-binding protein (SREBP)
for degradation, thereby suppressing lipogenesis (85). However, as NASH progresses,
upregulation of the DUB USP20, either in expression or activity,
antagonizes Fbw7-mediated degradation and stabilizes the SREBP
protein (86). This disruption of
regulatory equilibrium leads to persistent activation of the
lipogenic program even under lipotoxic conditions, establishing a
self-sustaining cycle of metabolic dysregulation. Concurrently, the
transcription factor carbohydrate-responsive element-binding
protein, which mediates glucose-to-lipid conversion, may accumulate
aberrantly, potentially due to insufficient activity of its cognate
E3 ligase(s).
Deregulation of cell death pathways:
From survival signaling to necroptosis
Simultaneously, regulatory control over cell death
pathways becomes compromised. TNF-α serves as a pivotal cytokine
within the inflammatory hepatic microenvironment. Under
physiological conditions, TNF-α binding to TNF receptor 1 induces
the formation of Complex I, leading to recruitment of the linear
ubiquitin chain assembly complex (LUBAC) (87). LUBAC catalyzes the synthesis of
linear (M1-linked) ubiquitin chains on key signaling molecules,
including RIPK1 and NF-κB essential modulator (also termed KKγ).
This specific ubiquitination event is critical for productive NF-κB
pathway activation and subsequent expression of pro-survival
genes.
In the context of NASH or drug-induced liver injury,
this homeostatic balance is disrupted. Persistent inflammation and
oxidative stress can impair NF-κB transcriptional activity, while
concurrently activating the deubiquitinating enzyme CYLD. Under
pathological conditions, signaling inputs such as TNF-α, ROS or
feedback from necroptotic pathways can activate phosphatases that
dephosphorylate and thereby activate CYLD. Activated CYLD is
subsequently recruited to the TNF-R1 signaling complex, where it
catalyzes the removal of both K63-linked and linear (M1-linked)
ubiquitin chains from RIPK1 (88).
This deubiquitination promotes the release of RIPK1 kinase activity
and facilitates the stabilization of Complex II (also referred to
as the death-inducing signaling complex) (89). The resultant signaling outcome
shifts toward caspase-8-dependent apoptosis or, if caspase-8 is
inhibited, RIPK3/mixed lineage kinase domain-like protein-mediated
necroptosis. In contrast to apoptosis, necroptosis elicits a more
robust inflammatory response due to the release of intracellular
DAMPs. The critical role of CYLD activation as a key driver of
extensive hepatocyte death has been substantiated across multiple
experimental models of liver injury.
In addition to apoptosis and necroptosis,
ferroptosis, a form of regulated cell death driven by
iron-dependent lipid peroxidation, has emerged as a key contributor
to hepatocyte death in NAFLD and NASH (90). Ferroptosis is characterized by the
accumulation of lethal lipid peroxides resulting from glutathione
depletion and inactivation of glutathione peroxidase 4, a critical
enzyme that neutralizes phospholipid hydroperoxides (91). The lipotoxic environment in
steatotic livers, characterized by excess free fatty acids, iron
overload and mitochondrial dysfunction, creates a permissive
setting for ferroptosis (91).
Recent studies have demonstrated that pharmacological inhibition of
ferroptosis alleviates hepatic inflammation and fibrosis in
preclinical NASH models, highlighting its pathogenic relevance
(92,93). Notably, ferroptosis may intersect
with the aforementioned CYLD-driven cell death axis, as ROS
generated during ferroptosis can further potentiate necroptotic
signaling, and conversely, mitochondrial dysfunction, a hallmark of
both pathways, may serve as a common upstream trigger (94). While the precise interplay between
ferroptosis, apoptosis and necroptosis in the context of hepatic
ubiquitin system dysregulation remains to be fully elucidated,
emerging evidence suggests that ferroptosis represents an
additional, and potentially parallel, cell death mechanism that
warrants consideration in the pathogenesis of steatohepatitis
(95).
Summary
Understanding this phase of functional rewiring
reveals novel targets for therapeutic intervention. It suggests
that during intermediate and advanced disease stages, strategies
should aim to directly modulate these critical nodal points. Such
approaches may include enhancing Cbl-b activity, promoting targeted
degradation of hyperactive CYLD or employing BAFF-neutralizing
antibodies. When extensive fibrosis develops and hepatocytes
acquire pre-malignant transformative potential, the ubiquitin
system is further co-opted by emerging tumor cells, which is a
process that will form the central focus of the subsequent
section.
Functional reprogramming of the ubiquitin
system and construction of the tumor microenvironment in HCC
As chronic liver disease progresses to the cirrhotic
stage, the hepatic microenvironment undergoes profound alterations.
During this phase, the ubiquitin system, which has already
undergone functional adaptation in earlier disease stages, is
further reprogrammed. Under neoplastic selection pressure,
hepatocyte clones with a survival advantage establish a
microenvironment conducive to tumor growth, invasion and immune
evasion by subverting ubiquitination regulatory networks. The core
mechanisms driving this process include the systematic inactivation
of tumor suppressor pathways, aberrant stabilization of
oncoproteins and effective evasion of antitumor immune
responses.
Systematic inactivation of the p53
tumor suppressor pathway
The tumor suppressor p53 is primarily regulated at
the protein stability level by the E3 ubiquitin ligase MDM2
(96). Under physiological
conditions, p53 and MDM2 form an autoregulatory negative feedback
loop, ensuring that p53 is activated only in response to cellular
stress (97). In HCC, this balance
is disrupted through MDM2 gene amplification or overexpression
(98). The resulting excess of
MDM2 drives constitutive ubiquitin-mediated degradation of p53,
thereby preventing the initiation of p53-dependent programs such as
cell cycle arrest, DNA repair and apoptosis (99).
The DUB USP7 further reinforces suppression of the
p53 pathway by stabilizing both MDM2 and DNA methyltransferase 1
(DNMT1) (100,101). Through its deubiquitinating
activity, USP7 extends the half-life of MDM2 (102). Simultaneously, by stabilizing
DNMT1, it promotes hypermethylation of the promoter regions of p53
target genes, adding a layer of transcriptional repression to the
existing post-translational inhibition. These actions block p53 at
both the protein level and the transcriptional level.
In addition to post-translational regulation, the
p53 pathway is frequently inactivated through somatic mutations in
HCC. Missense mutations in the TP53 gene, particularly at hotspot
codons such as R249S, R273H and R175H, occur in 30–50% of human
HCCs, with the highest frequency observed in geographic regions
associated with aflatoxin B1 exposure (103). These mutations not only abolish
the tumor-suppressive functions of wild-type p53, but also confer
gain-of-function (GOF) properties that promote proliferation,
invasion and chemoresistance (104). Notably, numerous mutant p53
proteins exhibit increased stability due to impaired MDM2-mediated
degradation, leading to their accumulation in tumor cells (105). Thus, in a substantial subset of
HCCs, p53 inactivation is driven by genetic mutations rather than,
or in addition to, MDM2 overexpression. The coexistence of MDM2
amplification and TP53 mutations is typically mutually exclusive,
reflecting distinct molecular subtypes of HCC (106). Understanding the mutation status
of p53 is therefore critical for accurately assessing the status of
the tumor suppressor pathway and for guiding therapeutic strategies
that may differentially target wild-type vs. mutant p53.
Fig. 4 depicts the
mechanism by which this system operates in HCC, demonstrating that
MDM2 drives p53 degradation, while USP7 locks in repression by
stabilizing both enzymes.
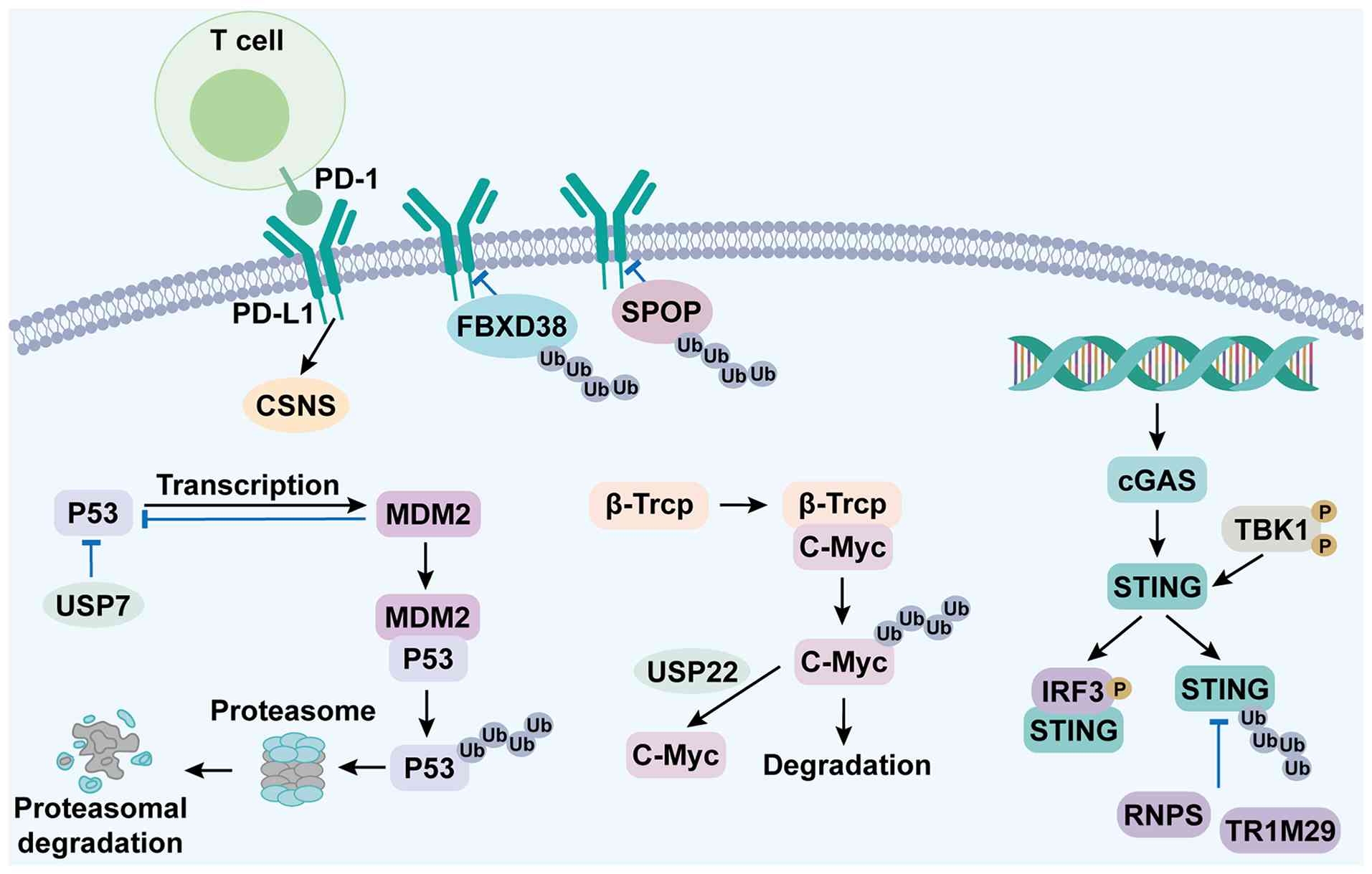 | Figure 4.Schematic of ubiquitin system
reprogramming in hepatocarcinogenesis and PROTAC therapeutic
potential. During hepatocarcinogenesis, ubiquitin system rewiring
drives tumorigenesis via four core alterations: i) CSN5-stabilized
PD-L1 engages PD-1 on T-cells for immune evasion; ii) MDM2
(p53-induced) + USP7 promote p53 ubiquitination and proteasomal
degradation; iii) impaired β-TrCP + enhanced USP22 block c-Myc
ubiquitination, causing its accumulation; and iv) TRIM29 mediates
STING ubiquitination, inhibiting the cGAS-STING pathway. These
changes foster a tumor-promoting microenvironment. PROTAC may
selectively degrade key nodes, although delivery/resistance
challenges remain. PROTACs (proteolysis-targeting chimeras) offer a
potential strategy to selectively degrade key oncogenic nodes
within this reprogrammed network. PD-1, programmed death 1; PD-L1,
programmed death-ligand 1; FBXO38, F-box only protein 38; SPOP,
Speckle-type POZ protein; CSN5, COP9 signalosome subunit 5; cGAS,
cyclic GMP-AMP synthase; STING, stimulator of interferon genes;
TBK1, TANK-binding kinase 1; IRF3, interferon regulatory factor 3;
MDM2, mouse double minute 2 homolog; USP7, ubiquitin-specific
protease 7; p53, tumor protein p53; β-TrCP, β-transducin
repeat-containing protein; c-Myc, cellular myelocytomatosis
oncogene; RNF5, ring finger protein 5; TRIM29, tripartite
motif-containing 29; PROTAC, proteolysis-targeting chimera; Ub,
ubiquitin. |
Impaired degradation and aberrant
stabilization of oncogenic drivers
β-Transducin repeat-containing protein (β-TrCP) acts
as the substrate-recognition subunit within the SCF family of E3
ubiquitin ligases, orchestrating the ubiquitin-mediated degradation
of numerous critical regulatory proteins (107,108). Within the Wnt signaling pathway,
β-TrCP serves as an essential component of the β-catenin
destruction complex, specifically targeting phosphorylated
β-catenin for proteasomal degradation (109). In HCC, this regulatory axis is
frequently subverted, either through inactivating mutations in
genes encoding core components of the destruction complex or
through activating mutations in the β-catenin gene itself.
The destruction complex is composed of several core
proteins, including adenomatous polyposis coli (APC), axin 1
(AXIN1), AXIN2, glycogen synthase kinase 3β and casein kinase 1. In
HCC, inactivating mutations in these genes are observed at varying
frequencies, with AXIN1 mutations occurring in ~8% of cases, AXIN2
mutations in ~3% and APC mutations in ~3% (110). Taken together, disruption of the
destruction complex through mutations in these core components
occurs in 10–15% of HCCs, complementing the 25–30% of cases with
activating mutations in the gene encoding β-catenin (CTNNB1)
(111,112). These mutations disrupt the
assembly or function of the destruction complex, leading to
impaired phosphorylation and subsequent ubiquitination of
β-catenin, thereby allowing its nuclear accumulation and
transcriptional activation of pro-proliferative target genes such
as c-Myc and cyclin D1 (111). In
numerous cases, these mutations are mutually exclusive with
activating mutations in CTNNB1, reflecting distinct molecular
subclasses of HCC with different clinical outcomes (113). Collectively, regardless of
whether the initiating event is a destruction complex mutation or a
CTNNB1 mutation, the common consequence is the stabilization and
nuclear accumulation of β-catenin. Consequently, β-catenin escapes
degradation, accumulates aberrantly and translocates to the
nucleus, where it drives the transcription of
proliferation-associated genes. Fig.
4 schematically outlines the coordinated disruption of
ubiquitin-dependent oncoprotein control in HCC, demonstrating the
loss of β-TrCP-mediated degradation and gain of USP22-driven
stabilization of β-catenin and c-Myc.
The regulatory function of β-TrCP toward the
oncoprotein c-Myc is also commonly impaired in HCC. Phosphorylated
c-Myc constitutes a physiological substrate for β-TrCP and its
degradation represents a crucial mechanism for restraining
c-Myc-driven oncogenicity. Tumor cells employ diverse strategies to
evade this β-TrCP-mediated degradation, including alteration of
c-Myc phosphorylation status or engagement of competitive binding
partners (114). Concurrently,
the frequent upregulation of the DUB USP22 in HCC further enhances
c-Myc stability by catalyzing the removal of its ubiquitin chains.
In addition to stabilizing c-Myc, USP22 also promotes the stability
of other pro-tumorigenic transcription factors, thereby
collectively fostering malignant progression in HCC.
Evasion of innate immune surveillance
via STING pathway degradation
In tumor immune evasion, cytosolic DNA released from
genomically unstable or damaged tumor cells is recognized by cGAS,
leading to activation of the cGAS-STING signaling pathway (115). Following STING activation,
TANK-binding kinase 1 and the transcription factor IRF3 are
recruited and phosphorylated, thereby driving the production of
type I interferons and initiating an antitumor immune response
(116).
HCC cells circumvent this innate immune surveillance
by upregulating the E3 ubiquitin ligases ring finger protein 5
(RNF5) (117) and TRIM29
(118), which target STING for
ubiquitination and subsequent proteasomal degradation, effectively
dampening pathway activation. Clinical analyses corroborate that
elevated expression of either RNF5 or TRIM29 is associated with
suppressed STING signaling, reduced intratumoral infiltration of
CD8+ T-cells and poorer patient prognosis (117,118). Together, these mechanisms enable
tumor cells to evade cell-intrinsic immune detection.
Promotion of adaptive immune evasion
through PD-L1 stabilization
PD-L1, a critical immune-regulatory molecule
expressed on the surface of tumor cells, functions to suppress
T-cell activity (119). The
disruption of PD-L1 homeostatic turnover in HCC via COP9
signalosome subunit 5 (CSN5)-dependent deubiquitination, and its
implications for antitumor immunity and therapeutic resistance, is
illustrated in Fig. 4. The
homeostatic turnover of PD-L1 is governed by several E3 ubiquitin
ligases, including Speckle-type POZ protein and F-box only protein
38, which mediate its ubiquitin-dependent degradation (120,121). However, in HCC, the frequent
overexpression of the DUB CSN5 counteracts this regulatory process.
By catalyzing the removal of ubiquitin chains from PD-L1, CSN5
stabilizes the protein, prolongs its half-life and elevates its
surface expression on tumor cells (122).
This stabilization mechanism holds considerable
clinical relevance. During treatment with immune checkpoint
inhibitors, tumor cells can upregulate CSN5 to maintain PD-L1
stability. Consequently, even under antibody-mediated blockade, the
high synthesis rate of PD-L1 coupled with its enhanced
stabilization sustains its surface expression, contributing to
acquired therapeutic resistance. A promising strategy to overcome
such resistance may involve combination therapy that co-administers
programmed cell death 1/PD-L1-blocking antibodies with a CSN5
inhibitor, thereby synergistically reducing PD-L1 stability and
restoring antitumor immunity.
Summary
During hepatocarcinogenesis, the ubiquitin system is
systematically reprogrammed to favor tumorigenesis. This
reprogramming is characterized by: i) Coordinated suppression of
the p53 pathway via MDM2 overexpression and USP7-mediated
stabilization, as well as through TP53 somatic mutations that
confer GOF properties and occur in 30–50% of HCCs; ii) aberrant
stabilization of oncoproteins such as β-catenin and c-Myc,
resulting from inactivating mutations in destruction complex
components (such as APC, ANIX1 and ANIX2) in 10–15% of HCCs,
activating CTNNB1 mutations in 25–30% of HCCs, impaired
β-TrCP-mediated degradation and enhanced USP22 activity; iii)
inhibition of innate immune surveillance through RNF5- and
TRIM29-dependent degradation of STING; and iv) promotion of
adaptive immune evasion via CSN5-stabilized PD-L1. Together, these
alterations establish a tumor-promoting microenvironment that fuels
the initiation and progression of HCC.
These mechanistic insights provide a rational basis
for therapeutic intervention. Conventional single-target inhibitors
typically show limited efficacy against the complex, rewired
network. By contrast, proteolysis-targeting chimera (PROTAC)
technology offers a promising strategy by enabling the selective
degradation of key nodal proteins within the ubiquitin-proteasome
system, thereby allowing precise intervention in the reprogrammed
network. Nevertheless, critical challenges remain, including the
development of liver-specific delivery systems and the need to
overcome intrinsic and acquired resistance mechanisms. In addition
to the asialoglycoprotein receptor (ASGPR)-targeted approach,
alternative liver-specific delivery strategies have been actively
explored, such as N-acetylgalactosamine (GalNAc)-conjugated PROTACs
that leverage ASGPR-mediated endocytosis for hepatocyte-selective
targeting (123,124). The successful clinical
translation of PROTAC-based therapies for HCC will likely require
the integration of such targeted delivery platforms with optimized
pharmacokinetic properties.
Therapeutic strategies targeting the
ubiquitin system: From mechanism to clinic
Paradigm shift: From pathway
inhibition to network restoration
Informed by a comprehensive analysis of the
functional evolution of the ubiquitin system throughout liver
disease progression, contemporary therapeutic strategies must now
pivot from merely inhibiting discrete signaling pathways toward
intervening in the dysregulated regulatory networks themselves.
This paradigm shift entails moving beyond the suppression of
aberrant signals to actively restoring the homeostatic function of
the ubiquitination machinery. Within this conceptual framework,
targeted protein degradation technologies emerge as a
transformative modality, offering novel therapeutic potential for
achieving this objective.
Limitations of conventional
therapeutic paradigms
Conventional small-molecule inhibitors typically
function by occupying the active site of a target protein, thereby
blocking its activity (125).
However, this approach faces inherent limitations when applied to
the complex regulatory networks governed by the ubiquitin system.
For targets such as transcription factors and scaffold proteins,
which often lack conventional, druggable active sites, developing
effective inhibitors remains a notable challenge (126). Furthermore, tumor cells can
develop resistance through feedback mechanisms and the activation
of compensatory pathways. Early therapeutics targeting the
ubiquitin-proteasome system have provided both proof-of-concept and
important lessons for the field. The proteasome inhibitors
bortezomib and carfilzomib were approved by the US Food and Drug
Administration (FDA) in 2003 and 2012, respectively, for the
treatment of multiple myeloma and mantle cell lymphoma (127,128). These agents demonstrated that
pharmacological manipulation of the UPS is clinically feasible.
However, their application in solid tumors such as HCC has been
limited by notable off-target toxicity, including peripheral
neuropathy and thrombocytopenia, as well as the development of
acquired resistance (127,129).
These limitations are largely attributable to their broad, systemic
impact on global proteostasis, highlighting the need for more
selective strategies that target specific E3 ligases or DUBs rather
than the proteasome itself (129).
Rise of targeted protein degradation:
PROTACs and molecular glues
PROTACs and molecular glues represent a novel class
of therapeutics that exploit the endogenous ubiquitin-proteasome
system to achieve selective degradation of target proteins
(130). A PROTAC molecule is a
heterobifunctional agent designed to simultaneously bind both a
protein of interest and an E3 ubiquitin ligase, thereby inducing
ubiquitination and subsequent proteasomal degradation of the target
(124). Unlike conventional
inhibitors, this strategy operates through a catalytic mechanism,
enables the targeting of proteins traditionally considered
‘undruggable’ and offers the potential for enhanced selectivity
through rational molecular design.
The clinical translation of targeted protein
degradation has advanced notably. The molecular glue degraders
lenalidomide and pomalidomide, which recruit the E3 ligase cereblon
(CRBN) to induce degradation of Ikaros (IKZF1) and Aiolos (IKZF3),
are FDA-approved for multiple myeloma and have provided clinical
validation for pharmacologically induced protein degradation
(124,131).
PROTAC technology has now entered clinical
evaluation. The most advanced PROTAC, vepdegestrant (ARV-471),
which targets the estrogen receptor, has completed Phase III trials
and is under regulatory review for ER+/HER2−
advanced breast cancer (132).
Other PROTACs in clinical development include ARV-110 targeting
androgen receptor for prostate cancer and KT-474 targeting
interleukin-1 receptor-associated kinase 4 for inflammatory
diseases (132). These
clinical-stage degraders have demonstrated proof-of-mechanism and
favorable safety profiles in patients (124,132).
Application prospects in liver
diseases
Targeted protein degradation holds considerable
therapeutic promise in the context of liver diseases (124,133,134). In metabolic-associated liver
diseases, PROTACs designed to target the TGF-β receptor or Smad2/3
proteins could potentially inhibit fibrosis progression by
eliminating key signaling molecules responsible for hepatic
stellate cell activation (134–138). Similarly, selective degraders
targeting the NLRP3 inflammasome may allow precise modulation of
the innate immune response. Furthermore, restoring hepatic
metabolic homeostasis could be achieved through the degradation of
USP20, which would normalize the stability and activity of the
transcription factor SREBP.
Potential in HCC therapy
In HCC, targeted protein degradation holds
considerable therapeutic promise. PROTAC molecules designed against
historically challenging oncoproteins, such as c-Myc and β-catenin,
have demonstrated notable efficacy in a preclinical study (138).
The therapeutic application of MDM2-targeted
degradation in HCC is critically dependent on p53 mutation status.
In p53 wild-type HCC, targeted degradation of MDM2 is expected to
restore p53-mediated tumor suppression by preventing MDM2 from
ubiquitinating and degrading p53.
However, this strategy is unlikely to be effective
in p53-mutated HCC, which accounts for 30–50% of cases (103). In these tumors, the mutant p53
protein lacks wild-type tumor-suppressive function and often
acquires oncogenic GOF properties that promote proliferation,
invasion and chemoresistance (104). Notably, numerous mutant p53
proteins exhibit increased stability due to impaired MDM2-mediated
degradation, leading to their accumulation in tumor cells (105). Therefore, in p53-mutated HCC,
MDM2 degradation would not restore functional p53 activity. For
these patients, alternative PROTAC-based strategies are being
actively explored, including direct degradation of the mutant p53
itself or targeting downstream effectors of mutant p53-driven
oncogenic pathways (105).
Beyond target selection, the pharmacokinetic
properties of PROTACs, particularly their in vivo half-life,
represent a critical determinant of therapeutic efficacy. The
catalytic mechanism of PROTACs, where one molecule can induce
degradation of multiple target proteins, partially compensates for
pharmacokinetic limitations, enabling sustained target suppression
even after drug clearance (139).
Furthermore, the development of liver-specific delivery systems
(such as GalNAc-conjugated PROTACs or lipid nanoparticles) has been
shown to improve tissue accumulation and prolong effective exposure
in the liver, as discussed previously (111,112). Notably, the turnover rates of
MDM2 and p53 also influence the duration of the therapeutic effect.
p53 is a short-lived protein (half-life, 20–40 min), whereas MDM2
has a longer half-life (2–4 h), suggesting that sustained MDM2
degradation may be required for prolonged p53 stabilization. p53 is
a short-lived protein (half-life, 20–40 min), whereas MDM2 has a
longer half-life (2–4 h), suggesting that sustained MDM2
degradation may be required for prolonged p53 stabilization
(140).
Furthermore, employing PROTAC technology to degrade
immune checkpoint molecules such as PD-L1 represents a novel
strategic approach to overcoming resistance mechanisms associated
with current immunotherapies.
Challenges in clinical
translation
The clinical translation of targeted degradation
technologies continues to face notable challenges. Tissue-specific
delivery is crucial to ensure both efficacy and safety, as systemic
administration may lead to off-target effects, highlighting the
need for liver-targeted delivery systems such as those utilizing
the ASGPR. Additionally, optimizing targeting specificity remains
an ongoing endeavor. A PROTAC molecule must exhibit high
selectivity not only for the target protein and the recruited E3
ligase, but must also account for the physiological roles of the
hijacked E3 ligase, as prolonged use could disrupt the homeostasis
of its endogenous substrates. Finally, the issue of drug resistance
cannot be overlooked. Tumor cells may develop resistance through
various mechanisms, including downregulation of the E3 ligase,
mutations in the target protein or alterations in proteasome
function.
Beyond these well-established obstacles, several
additional challenges have emerged from recent clinical experience.
First, the suboptimal pharmacokinetic properties of PROTACs,
including limited oral bioavailability and short half-lives, pose
formulation and dosing hurdles (124). Second, acquired resistance to
degraders has been documented clinically. For example, decreased
expression of the E3 ligase CRBN underlies resistance to
lenalidomide in multiple myeloma (124). Third, on-target off-tumor
toxicity remains a concern, as the targeted protein may also play
essential roles in normal tissues, necessitating careful evaluation
of therapeutic windows. Finally, robust biomarkers for patient
selection and pharmacodynamic monitoring are critical for
successful clinical deployment (124). Addressing these challenges will
require iterative optimization of degrader chemistry, innovative
delivery strategies and combination therapies.
Conclusion
The progression of liver disease, from steatosis
and hepatitis to fibrosis, and ultimately to HCC, cannot be
adequately described by a linear, additive model driven by distinct
etiologies. The present review advances a central thesis, whereby
this deleterious pathological cascade is fundamentally propelled by
a systemic functional reprogramming of the ubiquitin system,
initiated by persistent insults within the evolving hepatic
microenvironment. A pivotal molecular link connecting these
pathological stages is the transformation of key E3 ubiquitin
ligases and DUBs from guardians of cellular homeostasis under
physiological conditions into active drivers of disease
pathogenesis.
Functional reprogramming of the ubiquitin system
constitutes a core mechanism underlying disease progression. Under
physiological conditions, this system ensures precise control of
immune tolerance, metabolic equilibrium and quality control via key
regulators such as A20, Cbl-b, Fbw7 and the PINK1-PARKIN axis.
However, the pathological microenvironment induces a specific
rewiring of this regulatory network. For instance, metabolic stress
activates effectors such as BRCC3, while suppressing others such as
MARCH5, thereby lifting the inhibition of the NLRP3 inflammasome
and concurrently impairing mitophagy. Notably, alternative
mitophagy pathways mediated by FUNDC1, BNIP3 and NIX may partially
compensate when the PINK1-PARKIN axis is compromised under
lipotoxic stress. Within the inflammatory milieu, suppression of
Cbl-b and Itch activity, coupled with BAFF-mediated persistent
degradation of TRAF3, disrupts adaptive immune tolerance. In
hepatocytes, USP20 stabilizes SREBP to promote lipogenesis, whereas
CYLD activation drives programmed cell death, including apoptosis,
necroptosis and ferroptosis, the latter being a newly recognized
iron-dependent, lipid peroxidation-driven cell death mechanism
critically involved in steatohepatitis.
In HCC, the ubiquitin system is reconfigured to
support tumor survival and immune evasion. This is exemplified by
MDM2/USP7 axis-mediated functional inactivation of p53, which is
further compounded by TP53 somatic mutations (occurring in 30–50%
of HCCs) that confer GOF properties and exhibit increased stability
due to impaired MDM2-mediated degradation; aberrant accumulation of
β-catenin and c-Myc resulting from inactivating mutations in
destruction complex components (such as APC, AXIN1 and AXIN2 in
10–15% of HCCs) or activating CTNNB1 mutations (in 25–30% of
cases); RNF5/TRIM29-dependent degradation of STING to suppress
innate immune sensing; and CSN5-mediated stabilization of PD-L1,
which fosters an immunosuppressive microenvironment.
This insight necessitates a paradigm shift in the
understanding of liver diseases. Pathological stages previously
viewed as discrete entities are unified by a common framework of
staged reprogramming within the ubiquitin system. Diverse
initiating etiologies ultimately converge on perturbing critical
nodal points within this system, thereby accounting for both the
heterogeneity in clinical manifestations and the limitations of
single-pathway therapeutic interventions.
Consequently, therapeutic strategies must pivot
towards interventions that directly correct the dysregulated
ubiquitin system. Targeted protein degradation technologies, such
as PROTACs, offer a potent means to achieve this by enabling the
direct elimination of key pathological driver proteins. This
approach holds distinct advantages for addressing traditionally
‘undruggable’ targets and overcoming acquired treatment resistance.
Notably, the therapeutic application of MDM2 degradation is
effective primarily in p53 wild-type HCC; for p53-mutated tumors,
alternative strategies such as direct degradation of mutant p53
itself are being actively explored. Promising clinical applications
may include the degradation of inflammatory mediators (such as
NLRP3 or TGF-β signaling components), the elimination of
oncoproteins (such as c-Myc, β-catenin or MDM2) and the enhancement
of immunotherapies via degradation of immune checkpoint proteins
such as PD-L1. The ultimate objective of this strategic paradigm is
to achieve a functional reset of the pathological ubiquitinome.
The clinical translation of this therapeutic
paradigm faces distinct challenges. Achieving tissue-specific
delivery necessitates the development of sophisticated delivery
systems leveraging liver-specific receptors or carriers, including
GalNAc-conjugated PROTACs, lipid nanoparticles and antibody-drug
conjugates. Optimizing on-target specificity requires careful
consideration of the physiological functions of the recruited E3
ligase to avoid unintended consequences on its native substrates.
The suboptimal pharmacokinetic properties of PROTACs, such as
limited oral bioavailability and short half-lives, pose formulation
and dosing hurdles, although their catalytic mechanism partially
compensates for these limitations. Furthermore, proactively
understanding and addressing potential drug resistance mechanisms
is paramount, mandating the exploration of rational combination
strategies and the development of backup compounds. Future research
should be directed towards three key objectives: i) To
systematically map the functional landscape of E3 ligases and DUBs
across disease stages; ii) to advance cell-type-specific delivery
technologies; and iii) to propel degrader-based combination
therapies into clinical validation.
In conclusion, the present review establishes that
functional reprogramming of the ubiquitin system is a consistent
molecular hallmark throughout the spectrum of liver disease
progression. This understanding provides a compelling rationale for
developing therapies aimed at correcting these dysregulated core
networks. Targeted interventions against specific components of the
ubiquitin system signify a strategic shift in the therapeutic
paradigm for liver diseases towards approaches informed by
system-level biology.
Acknowledgements
BioRender (biorender.com) provided the platform to
create a subset of the mechanism-based illustrations included in
this review.
Funding
The present study was supported by the Clinical Research Project
of Investigator-Initiated Trials (IIT), Medical-Industry
Integration Program of the Pudong New Area Health Commission (grant
no. 2025-PWYC-04).
Availability of data and materials
Not applicable.
Authors' contributions
YJ conceptualized the present review, performed the
literature search and selection, reviewed the data and wrote the
manuscript. WN, YaS and YL provided supervision, contributed to the
conceptual framework and critically revised the manuscript for
important intellectual content. YJ, YiS and XC reviewed and edited
the manuscript. Data authentication is not applicable. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
A20
|
TNF-α-induced protein 3
|
|
ALD
|
alcoholic liver disease
|
|
BAFF
|
B-cell activating factor
|
|
BAFFR
|
BAFF receptor
|
|
BRCC3
|
BRCA1-BRCA2-containing complex
subunit 3
|
|
βTrCP
|
β-transducin repeat-containing
protein
|
|
Cbl-b
|
casitas B-lineage lymphoma-b
|
|
cIAP1
|
cellular inhibitor of apoptosis
protein 1
|
|
CSN5
|
COP9 signalosome subunit 5
|
|
CYLD
|
cylindromatosis
|
|
c-Myc
|
cellular myelocytomatosis
oncogene
|
|
CD28
|
cluster of differentiation 28
|
|
cGAS
|
cyclic GMP-AMP synthase
|
|
DAMPs
|
damage-associated molecular
patterns
|
|
DUBs
|
deubiquitinating enzymes
|
|
E3 ligases
|
E3 ubiquitin ligases
|
|
FBXO38
|
F-box only protein 38
|
|
Fbw7
|
F-box/WD repeat-containing protein
7
|
|
HCC
|
hepatocellular carcinoma
|
|
HECT
|
homologous to E6-AP carboxyl
terminus
|
|
IKKα
|
IκB kinase α
|
|
IRF3
|
interferon regulatory factor 3
|
|
IL-1β
|
interleukin-1β
|
|
Itch
|
itchy E3 ubiquitin ligase
|
|
K63
|
lysine 63
|
|
LUBAC
|
linear ubiquitin chain assembly
complex
|
|
MARCH5
|
membrane-associated RING-CH 5
|
|
Met1
|
N-terminal methionine 1
|
|
MDM2
|
mouse double minute 2 homolog
|
|
mtDNA
|
mitochondrial DNA
|
|
NAFLD
|
non-alcoholic fatty liver disease
|
|
NEDD4
|
neural precursor cell expressed
developmentally downregulated protein 4
|
|
NF-κB
|
nuclear factor κB
|
|
NEMO/IKKγ
|
NF-κB essential modulator/IκB kinase
γ
|
|
NIK
|
NF-κB-inducing kinase
|
|
NLRP3
|
NOD-like receptor family pyrin
domain-containing 3
|
|
PD-1
|
programmed death 1
|
|
PD-L1
|
programmed death-ligand 1
|
|
PARKIN
|
Parkin RBR E3 ubiquitin ligase
|
|
PINK1
|
PTEN-induced kinase 1
|
|
PLCγ1
|
phospholipase C γ1
|
|
PKCθ
|
protein kinase C θ
|
|
PPARγ
|
peroxisome proliferator-activated
receptor γ
|
|
PROTACs
|
proteolysis-targeting chimeras
|
|
RBR
|
RING-between-RING
|
|
RING
|
really interesting new gene
|
|
RIPK1
|
receptor-interacting
serine/threonine-protein kinase 1
|
|
RNF5
|
ring finger protein 5
|
|
ROS
|
reactive oxygen species
|
|
SCF
|
Skp1-Cullin-F-box
|
|
Smad2/3
|
mothers against decapentaplegic
homolog 2/3
|
|
SPOP
|
Speckle-type POZ protein
|
|
SREBP1c
|
sterol regulatory element-binding
protein 1c
|
|
STING
|
stimulator of interferon genes
|
|
TCR
|
T-cell receptor
|
|
TBK1
|
TANK-binding kinase 1
|
|
TGFβ
|
transforming growth factor β
|
|
Th2
|
type 2 T-helper cell
|
|
TNFα
|
tumor necrosis factor α
|
|
TRAF2
|
TNF receptor-associated factor 2
|
|
TRIM29
|
tripartite motif-containing 29
|
|
UCHs
|
ubiquitin C-terminal hydrolases
|
|
USP7
|
ubiquitin-specific protease 7
|
References
|
1
|
Brunt EM, Wong VW, Nobili V, Day CP,
Sookoian S, Maher JJ, Bugianesi E, Sirlin CB, Neuschwander-Tetri BA
and Rinella ME: Nonalcoholic fatty liver disease. Nat Rev Dis
Primers. 1:150802015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gan C, Yuan Y, Shen H, Gao J, Kong X, Che
Z, Guo Y, Wang H, Dong E and Xiao J: Liver diseases: Epidemiology,
causes, trends and predictions. Signal Transduct Target Ther.
10:332025. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stanley M and Virdee S: Chemical
ubiquitination for decrypting a cellular code. Biochem J.
473:1297–1314. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Satija YK, Bhardwaj A and Das S: A
portrayal of E3 ubiquitin ligases and deubiquitylases in cancer.
Int J Cancer. 133:2759–2768. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Schmidt CK, Galanty Y, Sczaniecka-Clift M,
Coates J, Jhujh S, Demir M, Cornwell M, Beli P and Jackson SP:
Systematic E2 screening reveals a UBE2D-RNF138-CtIP axis promoting
DNA repair. Nat Cell Biol. 17:1458–1470. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hrdinka M, Fiil BK, Zucca M, Leske D,
Bagola K, Yabal M, Elliott PR, Damgaard RB, Komander D, Jost PJ and
Gyrd-Hansen M: CYLD Limits Lys63- and Met1-Linked ubiquitin at
receptor complexes to regulate innate immune signaling. Cell Rep.
14:2846–2858. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jiang Y, Ni S, Xiao B and Jia L: Function,
mechanism and drug discovery of ubiquitin and ubiquitin-like
modification with multiomics profiling for cancer therapy. Acta
Pharm Sin B. 13:4341–4372. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Palazón-Riquelme P, Worboys JD, Green J,
Valera A, Martín-Sánchez F, Pellegrini C, Brough D and
López-Castejón G: USP7 and USP47 deubiquitinases regulate NLRP3
inflammasome activation. EMBO Rep. 19:e447662018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rao Z, Chen X, Wu J, Xiao M, Zhang J, Wang
B, Fang L, Zhang H, Wang X, Yang S and Chen Y: Vitamin D receptor
inhibits NLRP3 activation by impeding its BRCC3-mediated
deubiquitination. Front Immunol. 10:27832019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
When AC, Khalin I, Duering M, Hellal F,
Culmsee C, Vandenabeele P, Plesnila N and Terpolilli NA: RIPK1 or
RIPK3 deletion prevents progressive neuronal cell death and
improves memory function after traumatic brain injury. Acta
Neuropathol Commun. 9:1382021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Keir ME, Butte MJ, Freeman GJ and Sharpe
AH: PD-1 and its ligands in tolerance and immunity. Annu Rev
Immunol. 26:677–704. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Barac YD, Emrich F, Krutzwakd-Josefson E,
Schrepfer S, Sampaio LC, Willerson JT, Robbins RC, Ciechanover A,
Mohr FW, Aravot D and Taylor DA: The ubiquitin-proteasome system: A
potential therapeutic target for heart failure. J Heart Lung
Transplant. 36:708–714. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yuan T, Yan F, Ying M, Cao J, He Q, Zhu H
and Yang B: Inhibition of ubiquitin-specific proteases as a novel
anticancer therapeutic strategy. Front Pharmacol. 9:10802018.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Glickman MH and Ciechanover A: The
ubiquitin-proteasome proteolytic pathway: Destruction for the sake
of construction. Physiol Rev. 82:373–428. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Huang HT, Lumpkin RJ, Tsai RW, Su S, Zhao
X, Xiong Y, Chen J, Mageed N, Donovan KA, Fischer ES and Sellers
WR: Ubiquitin-specific proximity labeling for the identification of
E3 ligase substrates. Nat Chem Biol. 20:1227–1236. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Morreale FE and Walden H: Types of
ubiquitin ligases. Cell. 165:248–248.e1. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Asmamaw MD, Liu Y, Zheng YC, Shi XJ and
Liu HM: Skp2 in the ubiquitin-proteasome system: A comprehensive
review. Med Res Rev. 40:1920–1949. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bulatov E, Valiullina A, Sayarova R and
Rizvanov A: Promising new therapeutic targets for regulation of
inflammation and immunity: RING-type E3 ubiquitin ligases. Immunol
Lett. 202:44–51. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zheng N and Shabek N: Ubiquitin ligases:
Structure, function, and regulation. Annu Rev Biochem. 86:129–157.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Berndsen CE and Wolberger C: New insights
into ubiquitin E3 ligase mechanism. Nat Struct Mol Biol.
21:301–307. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Deshaies RJ and Joazeiro CA: RING domain
E3 ubiquitin ligases. Annu Rev Biochem. 78:399–434. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Harrigan JA, Jacq X, Martin NM and Jackson
SP: Deubiquitylating enzymes and drug discovery: Emerging
opportunities. Nat Rev Drug Discov. 17:57–78. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee MJ, Lee BH, Hanna J, King RW and
Finley D: Trimming of ubiquitin chains by proteasome-associated
deubiquitinating enzymes. Mol Cell Proteomics. 10:R110.003871.
2011. View Article : Google Scholar
|
|
24
|
Xian Y, Ye J, Tang Y, Zhang N, Peng C,
Huang W and He G: Deubiquitinases as novel therapeutic targets for
diseases. MedComm (2020). 5:e700362024. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
O'Donnell MA and Ting AT: NFκB and
ubiquitination: Partners in disarming RIPK1-mediated cell death.
Immunol Res. 54:214–226. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Patrick KL, Bell SL, Weindel CG and Watson
RO: Exploring the ‘multiple-hit hypothesis’ of neurodegenerative
disease: Bacterial infection comes up to bat. Front Cell Infect
Microbiol. 9:1382019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Diamond T, Burn TN, Nishiguchi MA,
Minichino D, Chase J, Chu N, Kreiger PA and Behrens EM: Familial
hemophagocytic lymphohistiocytosis hepatitis is mediated by IFN-γ
in a predominantly hepatic-intrinsic manner. PLoS One.
17:e02695532022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zheng M and Tian Z: Liver-mediated
adaptive immune tolerance. Front Immunol. 10:25252019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cheng ML, Nakib D, Perciani CT and
MacParland SA: The immune niche of the liver. Clin Sci (Lond).
135:2445–2466. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shih CC, Liu PY, Chen JH, Liao MH, Hsieh
CM, Ka SM, Wu CC, Lin HT, Wu TH and Chen YC: Macrophage expression
of E3 ubiquitin ligase Grail protects mice from
lipopolysaccharide-induced hyperinflammation and organ injury. PLoS
One. 13:e02082792018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Song G, Liu B, Li Z, Wu H, Wang P, Zhao K,
Jiang G, Zhang L and Gao C: E3 ubiquitin ligase RNF128 promotes
innate antiviral immunity through K63-linked ubiquitination of
TBK1. Nat Immunol. 17:1342–1351. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Komander D, Clague MJ and Urbé S: Breaking
the chains: Structure and function of the deubiquitinases. Nat Rev
Mol Cell Biol. 10:550–563. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kwon YT and Ciechanover A: The ubiquitin
code in the ubiquitin-proteasome system and autophagy. Trends
Biochem Sci. 42:873–886. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Min Y, Kim MJ, Lee S, Chun E and Lee KY:
Inhibition of TRAF6 ubiquitin-ligase activity by PRDX1 leads to
inhibition of NFKB activation and autophagy activation. Autophagy.
14:1347–1358. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhou Q, Wang H, Schwartz DM, Stoffels M,
Park YH, Zhang Y, Yang D, Demirkaya E, Takeuchi M, Tsai WL, et al:
Loss-of-function mutations in TNFAIP3 leading to A20
haploinsufficiency cause an early-onset autoinflammatory disease.
Nat Genet. 48:67–73. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Heyninck K, De Valck D, Vanden Berghe W,
Van Criekinge W, Contreras R, Fiers W, Haegeman G and Beyaert R:
The zinc finger protein A20 inhibits TNF-induced
NF-kappaB-dependent gene expression by interfering with an RIP- or
TRAF2-mediated transactivation signal and directly binds to a novel
NF-kappaB-inhibiting protein ABIN. J Cell Biol. 145:1471–1482.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shi CS and Kehrl JH: Traf6 and A20
differentially regulate TLR4-induced autophagy by affecting the
ubiquitination of Beclin 1. Autophagy. 6:986–987. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Korman AJ, Peggs KS and Allison JP:
Checkpoint blockade in cancer immunotherapy. Adv Immunol.
90:297–339. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nurieva RI, Liu X and Dong C: Molecular
mechanisms of T-cell tolerance. Immunol Rev. 241:133–144. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bao M, Hanabuchi S, Facchinetti V, Du Q,
Bover L, Plumas J, Chaperot L, Cao W, Qin J, Sun SC and Liu YJ:
CD2AP/SHIP1 complex positively regulates plasmacytoid dendritic
cell receptor signaling by inhibiting the E3 ubiquitin ligase Cbl.
J Immunol. 189:786–792. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sundqvist A, Bengoechea-Alonso MT, Ye X,
Lukiyanchuk V, Jin J, Harper JW and Ericsson J: Control of lipid
metabolism by phosphorylation-dependent degradation of the SREBP
family of transcription factors by SCF(Fbw7). Cell Metab.
1:379–391. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhao GN, Tian ZW, Tian T, Zhu ZP, Zhao WJ,
Tian H, Cheng X, Hu FJ, Hu ML, Tian S, et al: TMBIM1 is an
inhibitor of adipogenesis and its depletion promotes adipocyte
hyperplasia and improves obesity-related metabolic disease. Cell
Metab. 33:1640–1654.e8. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Matheoud D, Sugiura A, Bellemare-Pelletier
A, Laplante A, Rondeau C, Chemali M, Fazel A, Bergeron JJ, Trudeau
LE, Burelle Y, et al: Parkinson's disease-related proteins PINK1
and Parkin repress mitochondrial antigen presentation. Cell.
166:314–327. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Park YJ, Dodantenna N, Kim Y, Kim TH, Lee
HS, Yoo YS, Heo J, Lee JH, Kwon MH, Kang HC, et al:
MARCH5-dependent NLRP3 ubiquitination is required for mitochondrial
NLRP3-NEK7 complex formation and NLRP3 inflammasome activation.
EMBO J. 42:e1134812023. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li X, Lin Y, Niu R, Chen S, Pan J, Zhong
Y, Du J, Dong Q, Zhang H, Fang H, et al: Integrated lipidomics and
network pharmacology reveal the AMPK-mediated therapeutic mechanism
of 3,3′-diindolylmethane in hepatic lipid metabolism. Antioxidants
(Basel). 14:10932025. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Dowman JK, Tomlinson JW and Newsome PN:
Pathogenesis of non-alcoholic fatty liver disease. QJM. 103:71–83.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tilg H and Moschen AR: Evolution of
inflammation in nonalcoholic fatty liver disease: The multiple
parallel hits hypothesis. Hepatology. 52:1836–1846. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Celebi G, Kesim H, Ozer E and Kutlu O: The
effect of dysfunctional ubiquitin enzymes in the pathogenesis of
most common diseases. Int J Mol Sci. 21:63352020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Karki R, Lee E, Sharma BR, Banoth B and
Kanneganti TD: IRF8 regulates gram-negative bacteria-mediated NLRP3
inflammasome activation and cell death. J Immunol. 204:2514–2522.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Finamore C, De Marino S, Cassiano C,
Napolitano G, Rapacciuolo P, Marchianò S, Biagioli M, Roselli R, Di
Giorgio C, Festa C, et al: BAR502/fibrate conjugates: Synthesis,
biological evaluation and metabolic profile. Front Chem.
12:14258672024. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Swanson KV, Deng M and Ting JP: The NLRP3
inflammasome: Molecular activation and regulation to therapeutics.
Nat Rev Immunol. 19:477–489. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Py BF, Kim MS, Vakifahmetoglu-Norberg H
and Yuan J: Deubiquitination of NLRP3 by BRCC3 critically regulates
inflammasome activity. Mol Cell. 49:331–338. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Xu J and Núñez G: The NLRP3 inflammasome:
Activation and regulation. Trends Biochem Sci. 48:331–344. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
He Y, Hara H and Núñez G: Mechanism and
regulation of NLRP3 inflammasome activation. Trends Biochem Sci.
41:1012–1021. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Jo EK, Kim JK, Shin DM and Sasakawa C:
Molecular mechanisms regulating NLRP3 inflammasome activation. Cell
Mol Immunol. 13:148–159. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Pinzón-Fernández MV, Saavedra-Torres JS,
López Garzón NA, Pachon-Bueno JS, Tamayo-Giraldo FJ, Rojas Gomez
MC, Arias-Intriago M, Gaibor-Pazmiño A, López-Cortés A and
Izquierdo-Condoy JS: NLRP3 and beyond: Inflammasomes as central
cellular hub and emerging therapeutic target in inflammation and
disease. Front Immunol. 16:16247702025. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Yalcinkaya M, Liu W, Thomas LA, Olszewska
M, Xiao T, Abramowicz S, Papapetrou EP, Westerterp M, Wang N, Tabas
I and Tall AR: BRCC3-mediated NLRP3 deubiquitylation promotes
inflammasome activation and atherosclerosis in Tet2 clonal
hematopoiesis. Circulation. 148:1764–1777. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ren G, Zhang X, Xiao Y, Zhang W, Wang Y,
Ma W, Wang X, Song P, Lai L, Chen H, et al: ABRO1 promotes NLRP3
inflammasome activation through regulation of NLRP3
deubiquitination. EMBO J. 38:e1003762019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Gong Z, Li Y, Nie Y, Zhang S, Tang X, Hu
Y, Yang T, Zhu M, Tang W, Su Q, et al: USP50-mediated NLRP3
deubiquitination enhances NLRP3 inflammasome activation to suppress
HCC metastasis. J Pharm Anal. 15:1013802025. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Yu J, Li H, Wu Y, Luo M, Chen S, Shen G,
Wei X and Shao B: Inhibition of NLRP3 inflammasome activation by
A20 through modulation of NEK7. Proc Natl Acad Sci USA.
121:e23165511212024. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
D'Arcy MS: Mitophagy in health and
disease: Molecular mechanisms, regulatory pathways, and therapeutic
implications. Apoptosis. 29:1415–1428. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Li YT, Li KY, Tao SS, Wang T, Lu Y, Chen
H, Zhan YQ, Zhao K, Xiang SS, Li JJ, et al: USP13 stabilizes NLRP3
to facilitate inflammasome activation by preventing TRIM31-mediated
NLRP3 ubiquitination and degradation. Sci Adv. 11:eadx38272025.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Liu J, Qian C and Cao X:
Post-translational modification control of innate immunity.
Immunity. 45:15–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Jiang X and Chen ZJ: The role of
ubiquitylation in immune defence and pathogen evasion. Nat Rev
Immunol. 12:35–48. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Min-Wen JC, Jun-Hao ET and Shyh-Chang N:
Stem cell mitochondria during aging. Semin Cell Dev Biol.
52:110–118. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Chen Y, Wang J, An C, Bao S and Zhang C:
The role and research progress of macrophages after heart
transplantation. Heliyon. 10:e338442024. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Mun Y, Kim J, Choi YJ and Lee BH:
cGAS-STING-NF-κB axis mediates rotenone-induced NLRP3 inflammasome
activation through mitochondrial DNA release. Antioxidants (Basel).
14:12762025. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Xian H, Watari K, Sanchez-Lopez E,
Offenberger J, Onyuru J, Sampath H, Ying W, Hoffman HM, Shadel GS
and Karin M: Oxidized DNA fragments exit mitochondria via mPTP- and
VDAC-dependent channels to activate NLRP3 inflammasome and
interferon signaling. Immunity. 55:1370–1385.e8. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Nuzzo D, Galizzi G, Amato A, Terzo S,
Picone P, Cristaldi L, Mulè F and Di Carlo M: Regular intake of
pistachio mitigates the deleterious effects of a high fat-diet in
the brain of obese mice. Antioxidants (Basel). 9:3172020.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Eiyama A and Okamoto K:
PINK1/Parkin-mediated mitophagy in mammalian cells. Curr Opin Cell
Biol. 33:95–101. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
E Y, Lin Y, Yan G, Yang J, Jiao L, Wu R,
Yan Q, Chen Y, Chen Y, Yan X and Li H: Exogenous H2S alleviates
senescence of glomerular mesangial cells through up-regulating
mitophagy by activation of AMPK-ULK1-PINK1-parkin pathway in mice.
Biochim Biophys Acta Mol Cell Res. 1870:1195682023. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Yamano K and Youle RJ: PINK1 is degraded
through the N-end rule pathway. Autophagy. 9:1758–1769. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Florentino RM, Animasahun O, Haep N,
Nenwani M, Omoloja K, Altay LN, Achreja A, Morita K, Motomura T,
Diaz-Aragon R, et al: PNPLA3-I148M genetic variant rewires lipid
metabolism to drive programmed cell death in human hepatocytes. JCI
Insight. 10:e1938052025. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Xu X, Pang Y and Fan X: Mitochondria in
oxidative stress, inflammation and aging: From mechanisms to
therapeutic advances. Signal Transduct Target Ther. 10:1902025.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Chen L, Zhang Q, Meng Y, Zhao T, Mu C, Fu
C, Deng C, Feng J, Du S, Liu W, et al: Saturated fatty acids
increase LPI to reduce FUNDC1 dimerization and stability and
mitochondrial function. EMBO Rep. 24:e547312023. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
da Silva Rosa SC, Martens MD, Field JT,
Nguyen L, Kereliuk SM, Hai Y, Chapman D, Diehl-Jones W, Aliani M,
West AR, et al: BNIP3L/Nix-induced mitochondrial fission,
mitophagy, and impaired myocyte glucose uptake are abrogated by
PRKA/PKA phosphorylation. Autophagy. 17:2257–2272. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Shin CH and Choi DS: Essential roles for
the non-canonical IκB kinases in linking inflammation to cancer,
obesity, and diabetes. Cells. 8:1782019. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Warnatz K, Salzer U, Rizzi M, Fischer B,
Gutenberger S, Böhm J, Kienzler AK, Pan-Hammarström Q, Hammarström
L, Rakhmanov M, et al: B-cell activating factor receptor deficiency
is associated with an adult-onset antibody deficiency syndrome in
humans. Proc Natl Acad Sci USA. 106:13945–13950. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Grondona P, Bucher P, Schulze-Osthoff K,
Hailfinger S and Schmitt A: NF-κB activation in lymphoid
malignancies: Genetics, signaling, and targeted therapy.
Biomedicines. 6:382018. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Vallabhapurapu S, Matsuzawa A, Zhang W,
Tseng PH, Keats JJ, Wang H, Vignali DA, Bergsagel PL and Karin M:
Nonredundant and complementary functions of TRAF2 and TRAF3 in a
ubiquitination cascade that activates NIK-dependent alternative
NF-kappaB signaling. Nat Immunol. 9:1364–1370. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Xiao G, Harhaj EW and Sun SC:
NF-kappaB-inducing kinase regulates the processing of NF-kappaB2
p100. Mol Cell. 7:401–409. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Zhang B, Hu M, Zhang P, Cao H, Wang Y,
Wang Z and Su T: BAFF promotes regulatory T-cell apoptosis and
blocks cytokine production by activating B cells in primary biliary
cirrhosis. Braz J Med Biol Res. 46:433–439. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Aichele P, Neumann-Haefelin C, Ehl S,
Thimme R, Cathomen T, Boerries M and Hofmann M: Immunopathology
caused by impaired CD8+ T-cell responses. Eur J Immunol.
52:1390–1395. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Moser EK and Oliver PM: Regulation of
autoimmune disease by the E3 ubiquitin ligase Itch. Cell Immunol.
340:1039162019. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Aldaalis A, Bengoechea-Alonso MT and
Ericsson J: The SREBP-dependent regulation of cyclin D1 coordinates
cell proliferation and lipid synthesis. Front Oncol. 12:9423862022.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Shimizu K, Nihira NT, Inuzuka H and Wei W:
Physiological functions of FBW7 in cancer and metabolism. Cell
Signal. 46:15–22. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Han J, Zhong CQ and Zhang DW: Programmed
necrosis: Backup to and competitor with apoptosis in the immune
system. Nat Immunol. 12:1143–1149. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Lin X, Chen Q, Huang C and Xu X: CYLD
promotes TNF-α-induced cell necrosis mediated by RIP-1 in human
lung cancer cells. Mediators Inflamm. 2016:15427862016. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Tang J, Zhuang Y, Zhang Y, Hu H, Wang H,
Xu H, Li Y and Tu C: Necroptosis in cancer: Insight from
epigenetic, post-transcriptional and post-translational
modifications. J Hematol Oncol. 18:772025. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Yang Q, Shen X, Luo Y, Li R, Meng X, Xu P,
Liu X, Bian D, Wang J, Shi J and Chen J: ELANE enhances KEAP1
protein stability and reduces NRF2-mediated ferroptosis inhibition
in metabolic dysfunction-associated fatty liver disease. Cell Death
Dis. 16:2662025. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Yu Y, Wang Q, Huang X and Li Z: GA
receptor targeted chitosan oligosaccharide polymer nanoparticles
improve non-alcoholic fatty liver disease by inhibiting
ferroptosis. Int J Biol Macromol. 278((Pt 2)): 1347792024.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
El-Naby SMA, Khedr NF, El-Ashmawy NE and
Ibrahim AO: Proanthocyanidin and mitoglitazone suppress lipogenesis
by targeting ferroptosis in metabolic dysfunction-associated
steatohepatitis. Naunyn Schmiedebergs Arch Pharmacol.
398:15825–15836. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Vucur M, Kondylis V, Broz P and Luedde T:
Regulated necrosis at the crossroads of liver inflammation and
cancer development. Nat Rev Gastroenterol Hepatol. 23:331–354.
2026. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Cao P, Jaeschke H, Ni HM and Ding WX: The
ways to die: Cell death in liver pathophysiology. Semin Liver Dis.
45:397–419. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Hsu IC, Metcalf RA, Sun T, Welsh JA, Wang
NJ and Harris CC: Mutational hotspot in the p53 gene in human
hepatocellular carcinomas. Nature. 350:427–428. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Brooks CL and Gu W: New insights into p53
activation. Cell Res. 20:614–621. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Iwakuma T and Lozano G: MDM2, an
introduction. Mol Cancer Res. 1:993–1000. 2003.PubMed/NCBI
|
|
98
|
Onel K and Cordon-Cardo C: MDM2 and
prognosis. Mol Cancer Res. 2:1–8. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Chalhoub N and Baker SJ: PTEN and the
PI3-kinase pathway in cancer. Annu Rev Pathol. 4:127–150. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Tu Y, Wu W, Wu T, Cao Z, Wilkins R, Toh
BH, Cooper ME and Chai Z: Antiproliferative autoantigen CDA1
transcriptionally up-regulates p21(Waf1/Cip1) by activating p53 and
MEK/ERK1/2 MAPK pathways. J Biol Chem. 282:11722–11731. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Khoronenkova SV, Dianova II, Ternette N,
Kessler BM, Parsons JL and Dianov GL: ATM-dependent downregulation
of USP7/HAUSP by PPM1G activates p53 response to DNA damage. Mol
Cell. 45:801–813. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Fan YH, Cheng J, Vasudevan SA, Dou J,
Zhang H, Patel RH, Ma IT, Rojas Y, Zhao Y, Yu Y, et al: USP7
inhibitor P22077 inhibits neuroblastoma growth via inducing
p53-mediated apoptosis. Cell Death Dis. 4:e8672013. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Müller PA and Vousden KH: Mutant p53 in
cancer: New functions and therapeutic opportunities. Cancer Cell.
25:304–317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Zhao M, Wang T, Gleber-Netto FO, Chen Z,
McGrail DJ, Gomez JA, Ju W, Gadhikar MA, Ma W, Shen L, et al:
Mutant p53 gains oncogenic functions through a chromosomal
instability-induced cytosolic DNA response. Nat Commun. 15:1802024.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Zucman-Rossi J, Villanueva A, Nault JC and
Llovet JM: Genetic landscape and biomarkers of hepatocellular
carcinoma. Gastroenterology. 149:1226–1239.e4. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Xu C, Xu Z, Zhang Y, Evert M, Calvisi DF
and Chen X: β-Catenin signaling in hepatocellular carcinoma. J Clin
Invest. 132:e1545152022. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Jiang J and Struhl G: Regulation of the
Hedgehog and Wingless signalling pathways by the F-box/WD40-repeat
protein Slimb. Nature. 391:493–496. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Jiao S, Wang H, Shi Z, Dong A, Zhang W,
Song X, He F, Wang Y, Zhang Z, Wang W, et al: A peptide mimicking
VGLL4 function acts as a YAP antagonist therapy against gastric
cancer. Cancer Cell. 25:166–180. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Cucu I and Nicolescu MI: A synopsis of
signaling crosstalk of pericytes and endothelial cells in salivary
gland. Dent J (Basel). 12:1442021. View Article : Google Scholar
|
|
110
|
Perugorria MJ, Olaizola P, Labiano I,
Esparza-Baquer A, Marzioni M, Marin JJG, Bujanda L and Banales JM:
Wnt-β-catenin signalling in liver development, health and disease.
Nat Rev Gastroenterol Hepatol. 16:121–136. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Terra ML, Sant'Anna TBF, de Barros JJF and
de Araujo NM: Geographic and viral etiology patterns of TERT
promoter and CTNNB1 exon 3 mutations in hepatocellular carcinoma: A
comprehensive review. Int J Mol Sci. 26:28892025. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Schulze K, Imbeaud S, Letouzé E,
Alexandrov LB, Calderaro J, Rebouissou S, Couchy G, Meiller C,
Shinde J, Soysouvanh F, et al: Exome sequencing of hepatocellular
carcinomas identifies new mutational signatures and potential
therapeutic targets. Nat Genet. 47:505–511. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Yuan S, Nisar A, Chen C, Dong X, Pan Y, Zi
M, Wang Q, Khan S, Guo Y, Zhang X and He Y: Liver-targeted
degradation of BRD4 reverses hepatic fibrosis and enhances
metabolism in murine models. Theranostics. 15:7270–7290. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Popov N, Schülein C, Jaenicke LA and
Eilers M: Ubiquitylation of the amino terminus of Myc by
SCF(β-TrCP) antagonizes SCF(Fbw7)-mediated turnover. Nat Cell Biol.
12:973–981. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Gan Y, Li X, Han S, Liang Q, Ma X, Rong P,
Wang W and Li W: The cGAS/STING pathway: A novel target for cancer
therapy. Front Immunol. 12:7954012022. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Sun L, Wu J, Du F, Chen X and Chen ZJ:
Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates
the type I interferon pathway. Science. 339:786–791. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Zhong B, Zhang L, Lei C, Li Y, Mao AP,
Yang Y, Wang YY, Zhang XL and Shu HB: The ubiquitin ligase RNF5
regulates antiviral responses by mediating degradation of the
adaptor protein MITA. Immunity. 30:397–407. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Li Q, Lin L, Tong Y, Liu Y, Mou J, Wang X,
Wang X, Gong Y, Zhao Y, Liu Y, et al: TRIM29 negatively controls
antiviral immune response through targeting STING for degradation.
Cell Discov. 4:132018. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Francisco LM, Sage PT and Sharpe AH: The
PD-1 pathway in tolerance and autoimmunity. Immunol Rev.
236:219–242. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Gou Q, Dong C, Xu H, Khan B, Jin J, Liu Q,
Shi J and Hou Y: PD-L1 degradation pathway and immunotherapy for
cancer. Cell Death Dis. 11:9552020. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Meng X, Liu X, Guo X, Jiang S, Chen T, Hu
Z, Liu H, Bai Y, Xue M, Hu R, et al: FBXO38 mediates PD-1
ubiquitination and regulates anti-tumour immunity of T cells.
Nature. 564:130–135. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Lim SO, Li CW, Xia W, Cha JH, Chan LC, Wu
Y, Chang SS, Lin WC, Hsu JM, Hsu YH, et al: Deubiquitination and
stabilization of PD-L1 by CSN5. Cancer Cell. 30:925–939. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Peng Y, Li T, Liu D, Li W, Zhang Y, Zhao
Y, Jiang X, Liang Y, Chen P, Ma B, et al: Development of cathepsin
B-responsive GalNAc-PROTACs for hepatocyte-targeting protein
degradation. J Med Chem. 69:517–532. 2026.PubMed/NCBI
|
|
124
|
Békés M, Langley DR and Crews CM: PROTAC
targeted protein degraders: The past is prologue. Nat Rev Drug
Discov. 21:181–200. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Bouvier C, Lawrence R, Cavallo F, Xolalpa
W, Jordan A, Hjerpe R and Rodriguez MS: Breaking bad
proteins-discovery approaches and the road to clinic for degraders.
Cells. 13:5782024. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Toure M and Crews CM: Small-molecule
PROTACS: New approaches to protein degradation. Angew Chem Int Ed
Engl. 55:1966–1973. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Spano D and Catara G: Targeting the
ubiquitin-proteasome system and recent advances in cancer therapy.
Cells. 13:292023. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Fuchs O: Treatment of lymphoid and myeloid
malignancies by immunomodulatory drugs. Cardiovasc Hematol Disord
Drug Targets. 19:51–78. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Kim YJ, Lee Y, Shin H, Hwang S, Park J and
Song EJ: Ubiquitin-proteasome system as a target for anticancer
treatment-an update. Arch Pharm Res. 46:573–597. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Scholes NS, Mayor-Ruiz C and Winter GE:
Identification and selectivity profiling of small-molecule
degraders via multi-omics approaches. Cell Chem Biol. 28:1048–1060.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Chamberlain PP and Hamann LG: Development
of targeted protein degradation therapeutics. Nat Chem Biol.
15:937–944. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Garber K: The PROTAC gold rush. Nat
Biotechnol. 40:12–16. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Sakamoto KM, Kim KB, Kumagai A, Mercurio
F, Crews CM and Deshaies RJ: Protacs: chimeric molecules that
target proteins to the Skp1-Cullin-F box complex for ubiquitination
and degradation. Proc Natl Acad Sci USA. 98:8554–8559. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Burslem GM and Crews CM:
Proteolysis-targeting chimeras as therapeutics and tools for
biological discovery. Cell. 181:102–114. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Chirnomas D, Hornberger KR and Crews CM:
Protein degraders enter the clinic-a new approach to cancer
therapy. Nat Rev Clin Oncol. 20:265–278. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Paiva SL and Crews CM: Targeted protein
degradation: Elements of PROTAC design. Curr Opin Chem Biol.
50:111–119. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Yang Z, Zhao J, Xie K, Tang C, Gan C and
Gao J: MASLD development: From molecular pathogenesis toward
therapeutic strategies. Chin Med J (Engl). 138:1807–1824. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Shachaf CM, Kopelman AM, Arvanitis C,
Karlsson A, Beer S, Mandl S, Bachmann MH, Borowsky AD, Ruebner B,
Cardiff RD, et al: MYC inactivation uncovers pluripotent
differentiation and tumour dormancy in hepatocellular cancer.
Nature. 431:1112–1117. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Olson DC, Marechal V, Momand J, Chen J,
Romocki C and Levine AJ: Identification and characterization of
multiple mdm-2 proteins and mdm-2-p53 protein complexes. Oncogene.
8:2353–2360. 1993.PubMed/NCBI
|
|
140
|
Lawasut P, Chauhan D, Laubach J, Hayes C,
Fabre C, Maglio M, Mitsiades C, Hideshima T, Anderson KC and
Richardson PG: New proteasome inhibitors in myeloma. Curr Hematol
Malig Rep. 7:258–266. 2012. View Article : Google Scholar : PubMed/NCBI
|














